Embed Size (px)
DESCRIPTION
Oncology Practice Management
Citation preview

Chicago, IL—Wilshire Oncology’spatient-centered medical oncolo-gy home pilot has transformed care
by offering maximum support of patientsthroughout their treatments, includingattention to patients between office visitsto minimize side effects and symptoms.Details of the development and en-
gagement process, the plan to aggressive-ly manage symptoms to reduce cost, andthe payment methodology were offered by
Linda Bosserman, MD, at the2011 Cancer Center BusinessSummit.The pilot, launched
on June 1, 2011, was
From the publishers of
©2011 Engage Healthcare Communications, LLC
www.OncPracticeManagement.com
ONCOLOGY PRACTICEMANAGEMENT
™
DECEMBER 2011 VOLUME 1• NUMBER 4
PROCESS IMPROVEMENTS TO ENHANCE PATIENT CARE™
Wilshire’s Medical OncologyHome Project: Re-Engineeringthe Oncology PracticeBy Wayne Kuznar
For oncology to move forward, inno-vation is required. Science, technol-ogy, patient care, and communica-
tion are all components of this movetoward significant improvement. Withthe advent of government incentives toencourage the uptake of oncologists’ useof technology in a meaningful way, thepressure is on vendors to adapt standardsset by the government oversight body,the Office of the National Coordinator(ONC) for Health Information Tech-nology (HIT). This is development, butnot necessarily innovation. True innovation comes when the
MedPAC under Fire forProposed SGR FixOncologists Face Particular Challenges under This ModelBy Dawn Holcombe, MBA, FACMPE, ACHE
Continued on page 6
On October 6, 2011, the MedicarePayment Advisory Commission(MedPAC) proposed a way that
Congress, if it wanted to eliminate thesustainable growth rate (SGR) formulawithout raising the federal deficit andwithin the confines of Medicare, couldapproach an SGR fix. Since that proposal,the opposition has been deafening.
The MedPAC proposal would changethe ratio between primary care and spe-cialists for good. The plan is to freeze feesfor primary care services for 10 years,while payments for all other serviceswould be reduced by 5.9% for each of thefirst 3 years, followed by a freeze for theremaining 7 years. Before the final MedPAC proposal was
Continued on page 8
PATIE
NT AND PROVID
ER ACCESS
Brought to you by the Association of
Community Cancer Centers
Will t
he Su
nshin
e Ac
t Affe
ct
Physician
–Pha
rmac
eutic
al
Interac
tion?
.....45
Continued on page 3
From the Editor
10 Steps toAchieve“Meaningful Use”Innovation and Change forCommunity OncologyBy Carla C. Wood, CPC, MS

Cracking the Code
www.AmgenAssistOnline.com1-888-4ASSIST (1-888-427-7478)
© 2011 Amgen. All rights reserved. 63513-R1-V1 11/11
— XGEVA® and Prolia® will have a permanent HCPCS on January 1, 2012
— This new J0897 code will replace miscellaneous codes J3490, J3590, and C9272, which providers have been using to bill for XGEVA® and Prolia® to date
A N N O U N C I N G A N E W J - C O D E F O R X G E V A® A N D P R O L I A®
HCPCS CODE DESCRIPTION EFFECTIVE
J0897 Injection, denosumab, 1 mg January 1, 2012

Cancer Center Business Summit
3December 2011 I www.OncPracticeManagement.com I
Ronald Barkley, MS, JDPresidentCancer Center BusinessDevelopment GroupBedford, NH
Peggy Barton, RNPractice ManagerToledo ClinicToledo, OH
Risë Marie ClelandPresidentOplinc, IncLawton, OK
Bruce A. Cutter, MDPresidentCutter HealthCareConsultingSpokane, WA
Craig Deligdish, MDMedical DirectorFlorida ComprehensiveCancer NetworkMelbourne, FL
Patrick A.Grusenmeyer, ScD,FACHEPresidentChristiana Care HealthInitiativesNewark, DE
Teri U. Guidi, MBA,FAAMAPresident & CEOOncology ManagementConsulting GroupPipersville, PA
Ruth Lander, RN,FACMPEPractice AdministratorColumbus Oncology & Hematology Associates, IncColumbus, OH
Bonnie J. Miller, RN,BSN, OCN, FAAMAAdministrative DirectorWomen’s Cancer CenterFox Chase CancerCenterPhiladelphia, PA
Cindy C. Parman, CPC,CPC-H, RCCCSI Coding Strategies IncPowder Springs, GA
Jeffrey A. Scott, MDSenior Vice PresidentCardinal HealthDublin, OH
Carla C. Wood, CPC, MS PresidentAltos Solutions, IncLos Altos, CA
Dawn Holcombe, MBA, FACMPE, ACHEPresident
DGH ConsultingSouth Windsor, CT
Editorial Advisory Board
Editor-in-Chief
developed in conjunction with Anthem Blue Cross, CA,which makes it available to some of its members with can-cer living in Los Angeles, San Bernardino, and Riversidecounties. “We were able to move beyond breast, colon, andlung, to all of our cancer patients being part of this modelfor payment,” said Dr Bosserman, clinical oncologist andPresident of the Wilshire Oncology Medical Group.The medical oncology home pilot offers additional reim-
bursement for clinicians, symptom management support by nurses, advance care planning, and collaboration withurgent care centers to reduce the number of costly emer-gency department visits.Care transformation requires a re-engineering of the
practice “from the ground up,” and has resulted in partner-ships with payers and providers that have transformed payment methodology to support desired care, said DrBosserman. The development process with the health plantook 3 years.
“We worked for the past 6 to 10 years to re-engineerevery step of our practice…being a group, having groupmeetings, and agreeing to group standards…having data,feeding back data, continuous improvement, working atevery level of the staff to be efficient and fully engage themission,” she said.Wilshire has contracts to provide care in 2 counties in
California, where 26% of patients are uninsured. “We’re up450% in the patients we care for over 3 years,” she said.“They have never seen such efficiency.”
Engaging Payers Engaging payers was an important initial step in the
development process to build a medical oncology home.“We started out with our major payer in California, and wehad to build a personal relationship,” she said. “I got on theboard of Blue Cross of California—I’m on the physiciancommittee that passes all final decisions for the health planfor the state. Attending quarterly meetings gave me accessto decision makers.”Engagement of medical directors, contracting, actuaries,
Wilshire’s Medical…Continued from the cover
Continued on page 19
“We’re up 450% in the patients wecare for over 3 years. They havenever seen such efficiency.”
—Linda Bosserman, MD

4 I ONCOLOGY PRACTICE MANAGEMENT I December 2011
In This Issue
PUBLISHING STAFF
PublisherNicholas [email protected]
Associate PublisherMaurice [email protected]
Director, Client ServicesCristopher [email protected]
Editorial DirectorDalia [email protected]
Editorial AssistantJennifer [email protected]
Production ManagerMarie R. S. Borelli
Quality Control DirectorBarbara Marino
Business ManagerBlanche Marchitto
MISSION STATEMENT
Oncology healthcare requires providers tofocus attention on financial concerns andstrategic decisions that affect the bottomline. To continue to provide the high-quality care cancer patients deserve,providers must master the ever-changingbusiness of oncology. Oncology PracticeManagement will offer process solutionsfor members of the cancer care team—medical, surgical, and radiation oncolo-gists, as well as executives, administrators,and coders/billers—to assist them in reim-bursement, staffing, electronic healthrecords, REMS, and compliance withstate and federal regulations. VOPM4
Oncology Practice Management™ is published 4 times a year by Engage Healthcare Communications, LLC, 241 Forsgate Drive, Suite 205A, MonroeTownship, NJ 08831. Copyright © 2011 by Engage Healthcare Communications, LLC. All rights reserved. Oncology Practice Management™ is a registeredtrademark of Engage Healthcare Communications, LLC. No part of this publication may be reproduced or transmitted in any form or by any means now orhereafter known, electronic or mechanical, including photocopy, recording, or any informational storage and retrieval system, without written permissionfrom the publisher. Printed in the United States of America. ISSN: applied for.
The ideas and opinions expressed in Oncology Practice Management™ do not necessarily reflect those of the editorial board, the editors, or the publisher.Publication of an advertisement or other product mentioned in Oncology Practice Management™ should not be construed as an endorsement of the product orthe manufacturer’s claims. Readers are encouraged to contact the manufacturers about any features or limitations of products mentioned. Neither the editorsnor the publisher assume any responsibility for any injury and/or damage to persons or property arising out of or related to any use of the material mentionedin this publication.
Postmaster: Correspondence regarding subscriptions or change of address should be directed to CIRCULATION DIRECTOR, Oncology PracticeManagement™, 241 Forsgate Drive, Suite 205A, Monroe Township, NJ 08831. Fax: 732-992-1881. Yearly subscription rates: 1 year: $99.00 USD; 2 years:$149.00 USD; 3 years: $199.00 USD.
FEATURESCASE STUDY
Practice Changes in Management of PatientsReceiving Oral Chemotherapy ..................................13By Robert Mancini, PharmD, Dave Wilson, RPh, Colleen Powell, CPhT, andClementine Mehrens
CANCER CENTER BUSINESS SUMMIT
First Level III Oncology Medical Home ...................19
Accountable Care Organizations Paying Little Attention to Oncology Services..................................27
DRUG CODING
Medications Used for the Treatment of Breast Cancer..............................................................34
ONCOLOGY NETWORKS
Enhancing Oncology Networks..................................42By Gerard M. Nussbaum and Laura K. Rehfeld
DEPARTMENTS
Physician Wealth Managementwith Lawrence B. Keller, CLU, ChFC, CFP®
Selecting the Best Rider for Your Policy ........................40
Patient and Provider Accessbrought to you by the Association of Community Cancer Centers
Will the Sunshine Act Affect Physician-PharmaceuticalInteraction?…....................................................................45By Sydney Abbott, JD

Want integrated care? You need integrated technology.
© 2011 McKesson Specialty Health. All rights reserved.
At McKesson Specialty Health, we combine our in-depth software development experience
and oncology practice knowledge to create an integrated technology platform that delivers
the solutions you need for an enhanced level of comprehensive, patient-centered care.
Proven, market-leading solutions like our iKnowMed™ EHR and Lynx Mobile® provide
practice support while helping you increase your practice productivity, revenue and
clinical strength. The result? Even more effective patient care and practice success.
To learn more about McKesson Specialty Health, visit
mckessonspecialtyhealth.com/oncology or contact us at: 800.482.6700, Option 4

6 I ONCOLOGY PRACTICE MANAGEMENT I December 2011
MedPAC and SGR
revealed, a number ofmedical organizations,including the Amer -ican Medical Assoc -iation, the AmericanCollege of Cardiology,the American Col -lege of EmergencyPhysicians, the Amer -ican College of Physi -cians, the AmericanCollege of Surgeons,
and the American Psyc hiatric As-sociation, had already urged MedPACto revise the proposal as it was out-lined in an earlier, September draft. Concerns have ranged from the
impracticality of freezing reimburse-ments to physicians (many of whomoperate as small businesses) over 10years in the face of escalating operat-ing costs, staffing challenges, andtech nology demands to the needlesscreation of misaligned interests be -tween primary and specialty doctors,at a time when collaboration and con-tinuity of care is a harbinger of qualityand cost-effective care delivery. Two of the 17 MedPAC commis-
sioners also spoke against the recom-mendation: both are specialty physi-cians, expressing concerns aboutspecialty practice viability and theescalating impact from other payersthat peg payments to Medicare rates,such as Medicaid.Oncologists face particular chal-
lenges under this MedPAC model:changes in Medicare reimbursement;new edits and service bundling; andchanges in base rates of the values forphysician work, practice expenses,and geographic indicators have dra-matically reduced oncology-relatedprofessional fees, in addition to the
average selling price–related drugreimbursement reductions. In 2011, few oncologists are cover-
ing the costs of treating Medicarepatients from direct Medicare reim-bursement, and patients are finding it increasingly harder to fund theirown copay and coinsurance responsi-
bilities, particularly for MedicareAdvantage programs. Three years of5.9% reductions and a 7-year freezein payments are likely to dramatical-ly change the ability of physicians toprovide cancer services to Medicarepatients, whether the physician isself-employed or employed by a
hospital or by another organization.Fortunately, oncologists are not
alone in expressing concern aboutthis new MedPAC proposal; they canstand with their other physician col-leagues from many other specialties. Congress and the Centers for
Medicare & Medicaid Services(CMS) have often not acceptedMedPAC recommendations overthe years. By engaging in a strongand productive dialogue about themyriad of patient access issues, aswell as quality and continuity of careissues that would be created by thisMedPAC proposal, the medicalcommunity should be able to helpthose who are in a position to decideto accept or ignore this proposal.As an alternative, many believe
that physicians can be a valued partof the solution to healthcare costreduction. Traditionally, MedicarePart A (hospital costs) and Part B(physician services) do not commu-nicate or look at related costs ofcare. Reductions in physician reim-bursement quickly lead to increasesin more costly care from the non-physician (usually hospital-based)sector. It falls to everyone affected by the
MedPAC proposal to productivelycommunicate with Congress andCMS about the impact of focusingsolely on funding an SGR fix fromwithin the physician services buck-et, and to consider how changes incommunication and measurementof quality and care shifting may bebetter brought into the picture. It isnot enough to challenge MedPACfor its proposal; it is also necessaryto provide suggestions for alterna-tive solutions. l
MedPAC under Fire…Continued from the cover
It falls to everyoneaffected by theMedPAC proposal toproductivelycommunicate withCongress and CMSabout the impact offocusing solely onfunding an SGR fix fromwithin the physicianservices bucket, and toconsider how changesin communication andmeasurement of qualityand care shifting maybe better brought intothe picture.

Pharmacy / 877.662.6633 Fax / 877.662.6355 Website / OncoMed.net
CANCER DRUG REIMBURSEMENT IS COMPLEX.
FINANCIAL BARRIERS EXIST FOR
YOUR PATIENTS.
IS THE SOLUTION.
The OncoADVOCATE ProgramFOR HEALTH. FOR HELP. FOR YOU.The OncoAdvocate Program is dedicated to helping patients who need �nancial assistance and support achieve comprehensive access to their oncology drug treatments. In the process, OncoMed relieves your practice of the burden of �nding reimbursement solutions and lets you focus on your patient’s health.

8 I ONCOLOGY PRACTICE MANAGEMENT I December 2011
Innovation
enhanced technology is fully used andimplemented in an oncologist’s office. A well-run oncology office is a
beautiful sight to behold, but oneoften gets the impression it is atightrope walk every month. Thebalance of workload on humanresources, finances, and patienthealth is the primary concern of anyoncology practice. And technology,at its best, can help each of theseareas significantly. The Centers for Medicare &
Medicaid Services (CMS) Elec -tronic Health Record (EHR) Incen-tive Program requires HIT be at itsbest to allow physicians to achieveand successfully attest to “meaning-ful use,” defined as “ways that can bemeasured significantly in quality andin quantity.” As overwhelming asachieving meaningful use may seem,however, it can be done.
Implementation andAttestation Northern Hematology Oncology
is a community oncology practice inThornton, CO, that implemented a 2011/2012 ONC-certified EHRsystem in March 2011 and success-fully attested to meaningful use bythe end of July, within approximate-ly 120 days. To be clear, this groupwent from a paper medical chartenvironment to fully compliantmeaningful use workflow in a paper-less EHR system in less than 120days. Analysis of the successful attesta-
tions using this EHR indicates thatthe innovation was the practice’sadaptability and openness to changethroughout the entire team. Strong physician leadership clearly
set expectations for the staff, and alsoled by example. These doctors, busywith learning a new EHR system,took the time to implement the newtechnology in a way that set up suc-cess for receiving CMS incentives.
This impressive implementation—and resulting attestation and in -centive payments—can be replicat-ed in any oncology practice. Thetrainers and support staff of the EHRsystem offer the following 10-stepguide to successful meaningful useimplementation and attestation.
1Review all the required meaning-ful use criteria on the CMS web-
site (www.cms.gov/EHRIncentivePrograms/30_Meaningful_Use.asp).You may choose from Medicare orMedicaid incentive programs.
2Register each of your eligibleproviders (physicians) at www.cms.
gov/EHRIncentivePrograms/20_RegistrationandAttestation.asp. It is the
eligible provider’s responsibility tounderstand and achieve the incen-tive requirements.
3If you are not yet using an EHRsystem, implement an ONC-cer-
tified complete EHR. The ONC pro-vides a list of certified HIT products
(http://onc-chpl.force.com/ehrcert).
4Meet with the entire staff to pro-vide an overview of meaningful
use and its requirements for 2011/2012, known as Stage 1. Includemention of the eventual penalties if apractice isn’t compliant by 2015.
5Review criteria for meaningful useand the separate clinical quality
measures. Note: the meaningful usecriteria are all pass or fail. Eachprovider must pass each measure for aminimum of 90 days for the firstreporting year. Subsequent years re -quire reporting for the entire year. Ifyou fail one meaningful use item with -in the 90-day reporting period, you re -ceive no prize. The good news is that clinical qual-
ity measures do not require a pass/failthreshold. Each physician is simplyresponsible for re porting numeratorsand de nom inators for each chosenquality measure.
6Evaluate the holes in your prac-tice’s current workflow. If your
group currently does not assesspatient smoking status, indicate thisas a gap. If you do not provide accessto a patient portal, put this on thegap list. Do not entertain any aver-sion to change around these gapsthat must be filled. Change can bedifficult, but the processes your prac-tice puts in place now for Stage 1will make the subsequent 2 stagesless overwhelming.
7Assign staff members responsibil-ity for workflow changes required
for meaningful use. If your practice’schemotherapy nurses provide pa tienteducation, make each nurse responsi-ble for providing patients with a doc-umented patient education experi-ence. If the medical assistants usuallyenter vitals for the patients they
10 Steps…Continued from the cover
A well-run oncologyoffice is a beautifulsight to behold, butone often gets theimpression it is atightrope walk everymonth. The balance ofworkload on humanresources, finances,and patient health isthe primary concernof any oncologypractice.
Continued on page 9

Innovation
9December 2011 I www.OncPracticeManagement.com I
escort to the examination rooms, besure they also include height, weight,and blood pressure in the chart.
8Monitor progress each week. Runa meaningful use report, if your
EHR system includes this feature.Alternatively, create a worksheet toevaluate your progress for eachmeaningful use measure. Take actionto remediate any shortcomings asyour providers must pass all measuresfor a full, consecutive 90 days. Eachphysician may have a different 90-day reporting period if needed.
9Attest. The attestation is quickand easy. This must be done
through CMS’s EHR IncentiveProgram Registration and AttestationSystem (https://ehrincentives.cms.gov/hitech/login.action). The web-site is well designed, and it is fairlyeasy to follow onscreen instructions.For attestation, you will need yourmeaningful use report printed andready to reference, as well as yournumerators and denominators for theclinical quality measures. Sign in and answer all the ques-
tions. Each physician will need to
complete the attestation, because itrequires a statement of attestationand electronic signature. Upon suc-cessful attestation, make a note ofthe attestation number, which isbasically a tracking number to allowyou to track payment.
10Last, but not least, receivemoney.
Upon your successful attestation,the celebration of your healthcareinnovation should be enjoyed by all.Share your story, and assist others intheir quest to become meaningful. l
10 Steps…Continued from page 8
American College of Oncology Administrators
Annual Oncology Conference Addresses Oncology Management StrategiesChicago, June 20 – 22, 2012
Developed by and for oncology administrators with networking and exhibiting opportunities
AAMA/ACOA • 701 Lee St., Suite 600 • Des Plaines, IL 60016 • Phone 847-759-8601 • Fax 847-759-8602
ACOA is a national College of the American Academy of Medical Administrators (AAMA)
Professionals dedicated to providing the best careto oncology patients. Here’s where you belong.
Transform Your Leadership
Work Smarter
Access Solutions
Make Key Contacts
Strengthen Your Skills
Ensure Your Connections. Join Now.www.aameda.org • [email protected]

New Data: 5-Year Median Follow-up
W
P
H
P
N
T
In combination with MP* vs MP alone for previously untreated multiple myeloma
VELCADE DELIVERED 13-MONTH OVERALL SURVIVAL ADVANTAGE At 3-Year Median Follow-up, VELCADE® (bortezomib)+MP Provided an OS Advantage Over MP That Was Not Regained With Subsequent Therapies▼ Of the 69% of MP patients who received subsequent therapies,
50% received VELCADE or a VELCADE-containing regimen1
VELCADE is indicated for the treatment of patients with multiple myeloma.
VELCADE is contraindicated in patients with hypersensitivity to bortezomib, boron, or mannitol.
For Patient Assistance Information or Reimbursement Assistance, call 1-866-VELCADE (835-2233), Option 2, or visit VELCADE.com
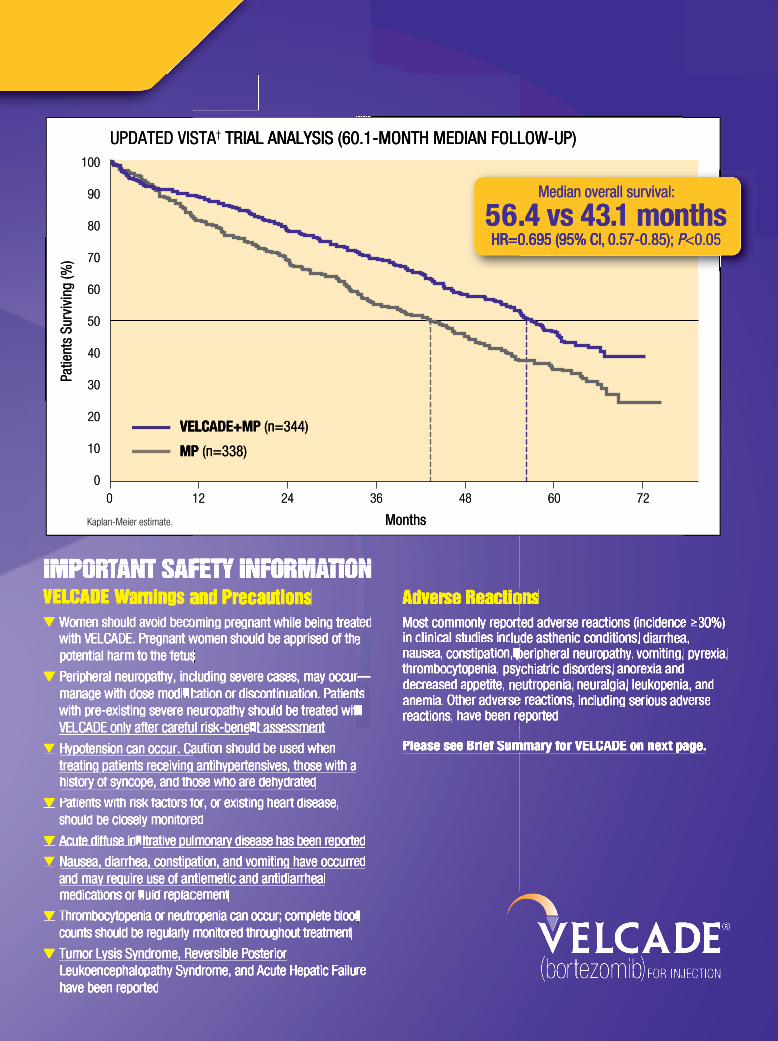
*Melphalan+prednisone.† VISTA: a randomized, open-label, international phase 3 trial (N=682) evaluating the efficacy and safety of VELCADE in combination with MP vs MP in previously untreated multiple myeloma. The primary endpoint was TTP. Secondary endpoints were CR, ORR, PFS, and OS. At a pre-specified interim analysis (median follow-up 16.3 months), VcMP‡ resulted in significantly superior results for TTP, PFS, OS, and ORR. Further enrollment was halted and patients receiving MP were offered VELCADE in addition.
‡VELCADE (Vc) in combination with MP.
Reference: 1. Mateos M-V, Richardson PG, Schlag R, et al. Bortezomib plus melphalan and prednisone compared with melphalan and prednisone in previously untreated multiple myeloma: updated follow-up and impact of subsequent therapy in the phase III VISTA trial. J Clin Oncol. 2010;28(13):2259-2266.
M
New Data: 5-Year Median Follow-upNew D New D New D N D
ea ea Data: 5-Y Data: 5-Y Year Median Follow-upN Ye Year Median Follow-up Data: 5 Y D 5 Y Y
ar Median ar Median M di
Follow-up Follow-up p F ll
p p p
Patie
nts
Surv
ivin
g (%
)
Months
604836 7224120
VELCADE+MP (n=344)
MP (n=338)
100
90
80
70
60
50
40
30
20
10
0
IMPORTANT SAFETY INFORMATIONVELCADE Warnings and Precautions ▼ Women should avoid becoming pregnant while being treated
with VELCADE. Pregnant women should be apprised of the potential harm to the fetus
▼ Peripheral neuropathy, including severe cases, may occur—manage with dose modification or discontinuation. Patients with pre-existing severe neuropathy should be treated with VELCADE only after careful risk-benefit assessment
▼ Hypotension can occur. Caution should be used when treating patients receiving antihypertensives, those with a history of syncope, and those who are dehydrated
▼ Patients with risk factors for, or existing heart disease, should be closely monitored
▼ Acute diffuse infiltrative pulmonary disease has been reported
▼ Nausea, diarrhea, constipation, and vomiting have occurred and may require use of antiemetic and antidiarrheal medications or fluid replacement
▼ Thrombocytopenia or neutropenia can occur; complete blood counts should be regularly monitored throughout treatment
▼ Tumor Lysis Syndrome, Reversible Posterior Leukoencephalopathy Syndrome, and Acute Hepatic Failure have been reported
Adverse Reactions Most commonly reported adverse reactions (incidence ≥30%) in clinical studies include asthenic conditions, diarrhea, nausea, constipation, peripheral neuropathy, vomiting, pyrexia, thrombocytopenia, psychiatric disorders, anorexia and decreased appetite, neutropenia, neuralgia, leukopenia, and anemia. Other adverse reactions, including serious adverse reactions, have been reported
Please see Brief Summary for VELCADE on next page.
b
Median overall survival:
56.4 vs 43.1 months HR=0.695 (95% CI, 0.57-0.85); P<0.05
N
80
90
100
UPDATED VISTA
TRIAL ANALYSIS (60.1-MONTH MEDIAN FOLLOW-UP)†UPDATED VISTA
TRIAL ANALYSIS (60.1-MONTH MEDIAN FOLLOW-UP)
HR=0.695 (95% CI,.65
TRIAL ANALYSIS (60.1-MONTH MEDIAN FOLLOW-UP)
P 0.57-0.85); HR=0.695 (95% CI,on m13..1 4 vs4.
vival:Median overall sur
<0 05PP<
shton
30
40
50
60
70
vivi
ng (%
)tie
nts
Sur
aP
HR=0.695 (95% CI,
P 0.57-0.85); HR=0.695 (95% CI,
<0.05PP<
0
MP 10
20
0
VELC
te.plan-Meier estimaKa
12
n=338)(MP
(n=344)+MPADEVELC
24
36 48
Months
7260

VELCADE, MILLENNIUM and are registered trademarks of Millennium Pharmaceuticals, Inc. Other trademarks are property of their respective owners.
Millennium Pharmaceuticals, Inc., Cambridge, MA 02139 Copyright © 2011, Millennium Pharmaceuticals, Inc.All rights reserved. Printed in USA
Brief Summary
INDICATIONS:
VELCADE® (bortezomib) for Injection is indicated for the treatment of patients with multiple myeloma. VELCADE® (bortezomib) for Injection is indicated for the treatment of patients with mantle cell lymphoma who have received at least 1 prior therapy.
CONTRAINDICATIONS:
VELCADE is contraindicated in patients with hypersensitivity to bortezomib, boron, or mannitol.
WARNINGS AND PRECAUTIONS:
VELCADE should be administered under the supervision of a physician experienced in the use of antineoplastic therapy. Complete blood counts (CBC) should be monitored frequently during treatment with VELCADE.
Peripheral Neuropathy: VELCADE treatment causes a peripheral neuropathy that is predominantly sensory. However, cases of severe sensory and motor peripheral neuropathy have been reported. Patients with pre-existing symptoms (numbness, pain or a burning feeling in the feet or hands) and/or signs of peripheral neuropathy may experience worsening peripheral neuropathy (including ≥Grade 3) during treatment with VELCADE. Patients should be monitored for symptoms of neuropathy, such as a burning sensation, hyperesthesia, hypoesthesia, paresthesia, discomfort, neuropathic pain or weakness. Patients experiencing new or worsening peripheral neuropathy may require change in the dose and schedule of VELCADE. Following dose adjustments, improvement in or resolution of peripheral neuropathy was reported in 51% of patients with ≥Grade 2 peripheral neuropathy in the relapsed multiple myeloma study. Improvement in or resolution of peripheral neuropathy was reported in 73% of patients who discontinued due to Grade 2 neuropathy or who had ≥Grade 3 peripheral neuropathy in the phase 2 multiple myeloma studies. The long-term outcome of peripheral neuropathy has not been studied in mantle cell lymphoma.
Hypotension: The incidence of hypotension (postural, orthostatic, and hypotension NOS) was 13%. These events are observed throughout therapy. Caution should be used when treating patients with a history of syncope, patients receiving medications known to be associated with hypotension, and patients who are dehydrated. Management of orthostatic/postural hypotension may include adjustment of antihypertensive medications, hydration, and administration of mineralocorticoids and/or sympathomimetics.
Cardiac Disorders: Acute development or exacerbation of congestive heart failure and new onset of decreased left ventricular ejection fraction have been reported, including reports in patients with no risk factors for decreased left ventricular ejection fraction. Patients with risk factors for, or existing heart disease should be closely monitored. In the relapsed multiple myeloma study, the incidence of any treatment-emergent cardiac disorder was 15% and 13% in the VELCADE and dexamethasone groups, respectively. The incidence of heart failure events (acute pulmonary edema, cardiac failure, congestive cardiac failure, cardiogenic shock, pulmonary edema) was similar in the VELCADE and dexamethasone groups, 5% and 4%, respectively. There have been isolated cases of QT-interval prolongation in clinical studies; causality has not been established.
Pulmonary Disorders: There have been reports of acute diffuse infiltrative pulmonary disease of unknown etiology such as pneumonitis, interstitial pneumonia, lung infiltration and Acute Respiratory Distress Syndrome (ARDS) in patients receiving VELCADE. Some of these events have been fatal. In a clinical trial, the first two patients given high-dose cytarabine (2 g/m2 per day) by continuous infusion with daunorubicin and VELCADE for relapsed acute myelogenous leukemia died of ARDS early in the course of therapy. There have been reports of pulmonary hypertension associated with VELCADE administration in the absence of left heart failure or significant pulmonary disease. In the event of new or worsening cardiopulmonary symptoms, a prompt comprehensive diagnostic evaluation should be conducted.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS): There have been reports of RPLS in patients receiving VELCADE. RPLS is a rare, reversible, neurological disorder which can present with seizure, hypertension, headache, lethargy, confusion, blindness, and other visual and neurological disturbances. Brain imaging, preferably MRI (Magnetic Resonance Imaging), is used to confirm the diagnosis. In patients developing RPLS, discontinue VELCADE. The safety of reinitiating VELCADE therapy in patients previously experiencing RPLS is not known.
Gastrointestinal Adverse Events: VELCADE treatment can cause nausea, diarrhea, constipation, and vomiting sometimes requiring use of antiemetic and antidiarrheal medications. Ileus can occur. Fluid and electrolyte replacement should be administered to prevent dehydration.
Thrombocytopenia/Neutropenia: VELCADE is associated with thrombocytopenia and neutropenia that follow a cyclical pattern with nadirs occurring following the last dose of each cycle and typically recovering prior to initiation of the subsequent cycle. The cyclical pattern of platelet and neutrophil decreases and recovery remained consistent over the 8 cycles of twice weekly dosing, and there was no evidence of cumulative thrombocytopenia or neutropenia. The mean platelet count nadir measured was approximately 40% of baseline. The severity of thrombocytopenia was related to pretreatment platelet count. In the relapsed multiple myeloma study, the incidence of significant bleeding events (≥Grade 3) was similar on both the VELCADE (4%) and dexamethasone (5%) arms. Platelet counts should be monitored prior to each dose of VELCADE. Patients experiencing thrombocytopenia may require change in the dose and schedule of VELCADE. There have been reports of gastrointestinal and intracerebral hemorrhage in association with VELCADE. Transfusions may be considered. The incidence of febrile neutropenia was <1%.
Tumor Lysis Syndrome: Because VELCADE is a cytotoxic agent and can rapidly kill malignant cells, the complications of tumor lysis syndrome may occur. Patients at risk of tumor lysis syndrome are those with high tumor burden prior to treatment. These patients should be monitored closely and appropriate precautions taken.
Hepatic Events: Cases of acute liver failure have been reported in patients receiving multiple concomitant medications and with serious underlying medical conditions. Other reported hepatic events include increases in liver enzymes, hyperbilirubinemia, and hepatitis. Such changes may be reversible upon discontinuation of VELCADE. There is limited re-challenge information in these patients.
Hepatic Impairment: VELCADE is metabolized by liver enzymes. VELCADE exposure is increased in patients with moderate or severe hepatic impairment. These patients should be treated with VELCADE at reduced starting doses and closely monitored for toxicities.
Use in Pregnancy: Pregnancy Category D. Women of childbearing potential should avoid becoming pregnant while being treated with VELCADE. Bortezomib administered to rabbits during organogenesis at a dose approximately 0.5 times the clinical dose of 1.3 mg/m2 based on body surface area caused post-implantation loss and a decreased number of live fetuses.
ADVERSE EVENT DATA:
Safety data from phase 2 and 3 studies of single-agent VELCADE 1.3 mg/m2/dose twice weekly for 2 weeks followed by a 10-day rest period in 1163 patients with previously treated multiple myeloma (N=1008, not including the phase 3, VELCADE plus DOXIL® [doxorubicin HCI liposome injection] study) and previously treated mantle cell lymphoma (N=155) were integrated and tabulated. In these studies, the safety profile of VELCADE was similar in patients with multiple myeloma and mantle cell lymphoma.
In the integrated analysis, the most commonly reported adverse events were asthenic conditions (including fatigue, malaise, and weakness); (64%), nausea (55%), diarrhea (52%), constipation (41%), peripheral neuropathy NEC (including peripheral sensory neuropathy and peripheral neuropathy aggravated); (39%), thrombocytopenia and appetite decreased (including anorexia); (each 36%), pyrexia (34%), vomiting (33%), anemia (29%), edema (23%), headache, paresthesia and dysesthesia (each 22%), dyspnea (21%), cough and insomnia (each 20%), rash (18%), arthralgia (17%), neutropenia and dizziness (excluding vertigo); (each 17%), pain in limb and abdominal pain (each 15%), bone pain (14%), back pain and hypotension (each 13%), herpes zoster, nasopharyngitis, upper respiratory tract infection, myalgia and pneumonia (each 12%), muscle cramps (11%), and dehydration and anxiety (each 10%). Twenty percent (20%) of patients experienced at least 1 episode of ≥Grade 4 toxicity, most commonly thrombocytopenia (5%) and neutropenia (3%). A total of 50% of patients experienced serious adverse events (SAEs) during the studies. The most commonly reported SAEs included pneumonia (7%), pyrexia (6%), diarrhea (5%), vomiting (4%), and nausea, dehydration, dyspnea and thrombocytopenia (each 3%).
In the phase 3 VELCADE + melphalan and prednisone study, the safety profile of VELCADE in combination with melphalan/prednisone is consistent with the known safety profiles of both VELCADE and melphalan/prednisone. The most commonly reported adverse events in this study (VELCADE+melphalan/prednisone vs melphalan/prednisone) were thrombocytopenia (52% vs 47%), neutropenia (49% vs 46%), nausea (48% vs 28%), peripheral neuropathy (47% vs 5%), diarrhea (46% vs 17%), anemia (43% vs 55%), constipation (37% vs 16%), neuralgia (36% vs 1%), leukopenia (33% vs 30%), vomiting (33% vs 16%), pyrexia (29% vs 19%), fatigue (29% vs 26%), lymphopenia (24% vs 17%), anorexia (23% vs 10%), asthenia (21% vs 18%), cough (21% vs 13%), insomnia (20% vs 13%), edema peripheral (20% vs 10%), rash (19% vs 7%), back pain (17% vs 18%), pneumonia (16% vs 11%), dizziness (16% vs 11%), dyspnea (15% vs 13%), headache (14% vs 10%), pain in extremity (14% vs 9%), abdominal pain (14% vs 7%), paresthesia (13% vs 4%), herpes zoster (13% vs 4%), bronchitis (13% vs 8%), hypokalemia (13% vs 7%), hypertension (13% vs 7%), abdominal pain upper (12% vs 9%), hypotension (12% vs 3%), dyspepsia (11% vs 7%), nasopharyngitis (11% vs 8%), bone pain (11% vs 10%), arthralgia (11% vs 15%) and pruritus (10% vs 5%).
DRUG INTERACTIONS:
Bortezomib is a substrate of cytochrome P450 enzyme 3A4, 2C19 and 1A2. Co-administration of ketoconazole, a strong CYP3A4 inhibitor, increased the exposure of bortezomib by 35% in 12 patients. Therefore, patients should be closely monitored when given bortezomib in combination with strong CYP3A4 inhibitors (e.g. ketoconazole, ritonavir). Co-administration of omeprazole, a strong inhibitor of CYP2C19, had no effect on the exposure of bortezomib in 17 patients. Co-administration of rifampin, a strong CYP3A4 inducer, is expected to decrease the exposure of bortezomib by at least 45%. Because the drug interaction study (n=6) was not designed to exert the maximum effect of rifampin on bortezomib PK, decreases greater than 45% may occur. Efficacy may be reduced when VELCADE is used in combination with strong CYP3A4 inducers; therefore, concomitant use of strong CYP3A4 inducers is not recommended in patients receiving VELCADE. St. John’s Wort (Hypericum perforatum) may decrease bortezomib exposure unpredictably and should be avoided. Co-administration of dexamethasone, a weak CYP3A4 inducer, had no effect on the exposure of bortezomib in 7 patients. Co-administration of melphalan-prednisone increased the exposure of bortezomib by 17% in 21 patients. However, this increase is unlikely to be clinically relevant.
USE IN SPECIFIC POPULATIONS:
Nursing Mothers: It is not known whether bortezomib is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from VELCADE, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use: The safety and effectiveness of VELCADE in children has not been established.
Geriatric Use: No overall differences in safety or effectiveness were observed between patients ≥age 65 and younger patients receiving VELCADE; but greater sensitivity of some older individuals cannot be ruled out.
Patients with Renal Impairment: The pharmacokinetics of VELCADE are not influenced by the degree of renal impairment. Therefore, dosing adjustments of VELCADE are not necessary for patients with renal insufficiency. Since dialysis may reduce VELCADE concentrations, the drug should be administered after the dialysis procedure. For information concerning dosing of melphalan in patients with renal impairment, see manufacturer’s prescribing information.
Patients with Hepatic Impairment: The exposure of VELCADE is increased in patients with moderate and severe hepatic impairment. Starting dose should be reduced in those patients.
Patients with Diabetes: During clinical trials, hypoglycemia and hyperglycemia were reported in diabetic patients receiving oral hypoglycemics. Patients on oral antidiabetic agents receiving VELCADE treatment may require close monitoring of their blood glucose levels and adjustment of the dose of their antidiabetic medication.
Please see full Prescribing Information for VELCADE at VELCADE.com.
V-11-0264 12/11

As developments in cancertreatment shift toward oralagents, new processes, pro-
cedures, and strategies must bedeveloped to help healthcare pro -viders maintain the same level ofinteraction with patients. Tradition -ally, patients are seen by their oncol-ogists and then sent to an infusioncenter for their intravenous chemo -therapy where they interact withnurses and/or pharmacists. Theseinteractions allow for early assess-ment and intervention of medica-tion-related issues, side effects, andtolerability.In the shifting paradigm, patients
see their oncologist, are given a writ-ten prescription, and then senthome. This transfers a bulk of theresponsibility onto the patient, withno true indicator of adherence. Inaddition, the patient has no interac-tion with other healthcare providersregarding adverse reactions andother medication-related concerns.One solution is to design and imple-ment a pharmacist-managed oralchemotherapy program, which willensure that patients can still interactwith practitioners between theironcologist visits.
The ProcessThe St. Luke’s Mountain States
Tumor Institute created a pharma-cist-managed program to help dealwith the emerging issues surroundingoral chemotherapy. When a newprescription is written, it is trans-ferred to the oral chemotherapypharmacist. The pharmacist thenevaluates it for accuracy of dosing, aswell as indication, drug interactions,and side-effect management. Each
patient is counseled directly by anexperienced oncology pharmacistabout the drug and the process forfilling the prescription. Next, the script is sent to a closed-
access pharmacy, which can be usedsolely by staff and patients of thehealth system. The prescription thenis run through insurance and filled.Once filled, it gets delivered to thepatient at the nearest infusion center(or mailed to a select subset of pa tientswith transportation restrictions).
If issues arise regarding high co pay,denial of coverage, or lack of insur-ance, the cancer center’s financialadvocates are brought into the pro cessto help with copay assistance or freedrug programs. Finally, after the pa -tient starts the medication, the phar-macist calls the patient on a weeklybasis for the first cycle, then monthly1 week before each refill date.
JustificationAn institution must be able to jus-
tify the staffing requirements of theprogram. Start by creating a break-even point analysis, in which you
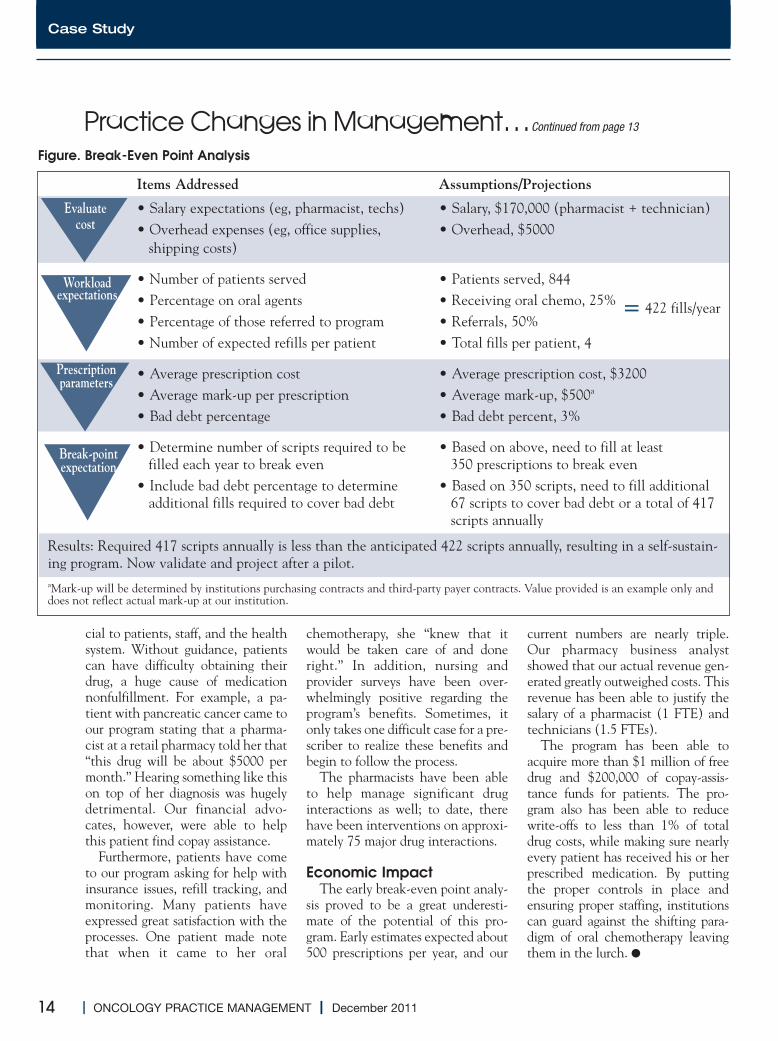
figure the costs andrev-enue potential.Then, evaluate thesalary requirements,estimated number ofpre scriptions expect-ed, and average costs/income of each pre-scription to show therequired break-evenpoint. Next, imple-ment a 2- to 3-month pilot to verify the assump-tions in the initial analysis. Pilotdata can then be annualized to justi-fy the long-term program (Figure).
Barriers to OvercomeOne of the biggest barriers is over-
coming your current processes.Someone must be able to meet withprescribing oncologists to ensure thatprescriptions pass through the oralchemotherapy pharmacist. We useregistered nurses who work with eachphysician to act as gatekeepers to oralchemotherapy prescriptions. In addi-tion, we use standardized order forms,which help remind prescribers thatthere is something special that needsto happen with these patients.Another barrier is insurance issues.
A variety of issues may need to beaddressed, such as prior authorizationforms and requirements to use mailorder pharmacies. We had our phar-macy billing specialist analyst set upcontracts with third-party payers,which has helped reduce our totalpopulation using mail order pharma-cies to approximately 10% to 15%.
Clinical ImpactThe program has proved benefi-
Case Study
13December 2011 I www.OncPracticeManagement.com I
Chemotherapy 2.0
Practice Changes in Management ofPatients Receiving Oral ChemotherapyBy Robert Mancini, PharmD, Dave Wilson, RPh, Colleen Powell, CPhT, and Clementine Mehrens,St. Lukes Mountain States Tumor Institute
Robert Mancini
One solution is to designand implement apharmacist-managedoral chemotherapyprogram, which willensure that patientscan still interact withpractitioners betweentheir oncologist visits.
Continued on page 14
cial to patients, staff, and the healthsystem. Without guidance, patientscan have difficulty obtaining theirdrug, a huge cause of medicationnonfulfillment. For example, a pa -tient with pancreatic cancer came toour program stating that a pharma-cist at a retail pharmacy told her that“this drug will be about $5000 permonth.” Hearing something like thison top of her diagnosis was hugelydetrimental. Our financial advo-cates, however, were able to helpthis patient find copay assistance. Furthermore, patients have come
to our program asking for help withinsurance issues, refill tracking, andmonitoring. Many patients haveexpressed great satisfaction with theprocesses. One patient made notethat when it came to her oral
chemotherapy, she “knew that itwould be taken care of and doneright.” In addition, nursing andprovider surveys have been over-whelmingly positive regarding theprogram’s benefits. Sometimes, itonly takes one difficult case for a pre-scriber to realize these benefits andbegin to follow the process. The pharmacists have been able
to help manage significant druginteractions as well; to date, therehave been interventions on approxi-mately 75 major drug interactions.
Economic ImpactThe early break-even point analy-
sis proved to be a great underesti-mate of the potential of this pro-gram. Early estimates expected about500 prescriptions per year, and our
current numbers are nearly triple.Our pharmacy business analystshowed that our actual revenue gen-erated greatly outweighed costs. Thisrevenue has been able to justify thesalary of a pharmacist (1 FTE) andtechnicians (1.5 FTEs). The program has been able to
acquire more than $1 million of freedrug and $200,000 of copay-assis-tance funds for patients. The pro-gram also has been able to reducewrite-offs to less than 1% of totaldrug costs, while making sure nearlyevery patient has received his or herprescribed medication. By puttingthe proper controls in place andensuring proper staffing, institutionscan guard against the shifting para-digm of oral chemotherapy leavingthem in the lurch. l
14 I ONCOLOGY PRACTICE MANAGEMENT I December 2011
Case Study
Items Addressed Assumptions/Projections
• Salary expectations (eg, pharmacist, techs) • Salary, $170,000 (pharmacist + technician)• Overhead expenses (eg, office supplies, • Overhead, $5000shipping costs)
• Number of patients served • Patients served, 844• Percentage on oral agents • Receiving oral chemo, 25%• Percentage of those referred to program • Referrals, 50%• Number of expected refills per patient • Total fills per patient, 4
• Average prescription cost • Average prescription cost, $3200• Average mark-up per prescription • Average mark-up, $500a
• Bad debt percentage • Bad debt percent, 3%
• Determine number of scripts required to be • Based on above, need to fill at least filled each year to break even 350 prescriptions to break even
• Include bad debt percentage to determine • Based on 350 scripts, need to fill additionaladditional fills required to cover bad debt 67 scripts to cover bad debt or a total of 417
scripts annually
Figure. Break-Even Point Analysis
= 422 fills/year
Results: Required 417 scripts annually is less than the anticipated 422 scripts annually, resulting in a self-sustain-ing program. Now validate and project after a pilot.aMark-up will be determined by institutions purchasing contracts and third-party payer contracts. Value provided is an example only anddoes not reflect actual mark-up at our institution.
Evaluatecost
Workloadexpectations
Prescriptionparameters
Break-pointexpectation
Practice Changes in Management…Continued from page 13

ADVERTISEMENT
To learn more about our united strength, visit usoncology.com

Injection, eribulin mesylate, 0.1 mg
J9179
Product coding does not guarantee payor coverage or payment.*
For more details visit www.halavenreimbursement.com
Effective January 1, 2012
Scan this code to visit www.halaven.com
Please see accompanying brief summary of Halaven full Prescribing Information.
IndicationHalaven is indicated for the treatment of patients with metastatic breast cancer who have previously received at least two chemotherapeutic regimens for the treatment of metastatic disease. Prior therapy should have included an anthracycline and a taxane in either the adjuvant or metastatic setting.
Important Safety InformationNeutropenia• Monitor complete blood counts prior to each dose,
and increase the frequency of monitoring in patients who develop Grade 3 or 4 cytopenias. Delay administration and reduce subsequent doses in patients who experience febrile neutropenia or Grade 4 neutropenia lasting longer than 7 days
• Severe neutropenia (ANC <500/mm3) lasting more than 1 week occurred in 12% (62/503) of patients. Patients with elevated liver enzymes >3 × ULN and bilirubin >1.5 × ULN experienced a higher incidence of Grade 4 neutropenia and febrile neutropenia than patients with normal levels
• Grade 3 and Grade 4 neutropenia occurred in 28% and 29%, respectively, of patients who received Halaven. Febrile neutropenia occurred in 5% of patients and two patients (0.4%) died from complications
Peripheral Neuropathy• Patients should be monitored closely for signs of
peripheral motor and sensory neuropathy • Grade 3 peripheral neuropathy occurred in 8%
of patients, and Grade 4 in 0.4% of patients who received Halaven. Delay administration of Halaven until resolution to Grade 2 or less
• Neuropathy lasting more than 1 year occurred in 5% of patients. Twenty-two percent of patients developed a new or worsening neuropathy that had
not recovered within a median follow-up duration of 269 days (range 25-662 days)
Pregnancy Category D • Halaven is expected to cause fetal harm when
administered to a pregnant woman and patients should be advised of these risks
QT Prolongation• In an uncontrolled ECG study in 26 patients, QT
prolongation was observed on Day 8, independent of eribulin concentration, with no prolongation on Day 1. ECG monitoring is recommended for patients with congestive heart failure; bradyarrhythmias; concomitant use of drugs that prolong QT interval, including Class Ia and III antiarrhythmics; and electrolyte abnormalities
• Correct hypokalemia or hypomagnesemia prior to initiating Halaven and monitor electrolytes periodically during therapy. Avoid in patients with congenital long QT syndrome
Hepatic and Renal Impairment • For patients with mild (Child-Pugh A) or moderate
(Child-Pugh B) hepatic and/or moderate (CrCl 30-50 mL/min) renal impairment, a reduction in starting dose is recommended
References: 1. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology™: Breast Cancer. Version 2.2011. http:NCCN.org. Published January 5, 2011. Accessed October 18, 2011.† 2. Halaven [package insert]. Woodcliff Lake, NJ: Eisai Inc; 2010. 3. Saad ED, Katz A, Buyse M. Overall survival and post-progression survival in advanced breast cancer: a review of recent randomized clinical trials. J Clin Oncol. 2010;28(11):1958-1962. 4. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783-792. 5. Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733-2743. 6. von Minckwitz G, du Bois A, Schmidt M, et al. Trastuzumab beyond progression in human epidermal growth factor receptor 2–positive advanced breast cancer: a German Breast Group 26/Breast International Group 03-05 study. J Clin Oncol. 2009;27(12):1999-2006. 7. Miller K, Wang M, Gralow J, et al. Paclitaxel
plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357(26):2666-2676. 8. Robert NJ, Dieras V, Glaspy J, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab (B) for ¥ rst-line treatment of HER2-negative locally recurrent or metastatic breast cancer (MBC). J Clin Oncol. 2009;27(suppl; abstr 1005). 9. Sparano JA, Vrdoljak E, Rixe O, et al. Randomized phase III trial of ixabepilone plus capecitabine versus capecitabine in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2010;28(20):3256-3263. 10. Jones SE, Erban J, Overmoyer B, et al. Randomized phase III study of docetaxel compared with paclitaxel in metastatic breast cancer. J Clin Oncol. 2005;23(24):5542-5551. 11. Cortes J, O’Shaughnessy J, Loesch D, et al. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet. 2011;377(9769):914-923.
† Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines™) for Breast Cancer V.2.2011. © 2011 National Comprehensive Cancer Network, Inc. All rights reserved. Accessed October 18, 2011. To view the most recent and complete version of the NCCN Guidelines, go online to NCCN.org. NATIONAL COMPREHENSIVE CANCER NETWORK®, NCCN®, NCCN GUIDELINES™,and all other NCCN Content are trademarks owned by the National Comprehensive Cancer Network, Inc.
O
CI=con¥ d *
The FIRST and ONLY third-line, single-agent therapy proven to signi�
* Eisai cannot guarantee payment of any claim. Coding, coverage, and reimbursement may vary signi¥ cantly by payor, plan, patient, and setting of care. Actual coverage and reimbursement decisions are made by individual payors following the receipt of claims. For additional information, customers should consult with their payors for all relevant coding, reimbursement, and coverage requirements. It is the sole responsibility of the provider to select the proper code and ensure the accuracy of all claims used in seeking reimbursement. All services must be medically appropriate and properly supported in the patient medical record.
HALAVEN® is a registered trademark used by Eisai Inc. under license from Eisai R&D Management Co., Ltd.© 2011 Eisai Inc. All rights reserved. Printed in USA/December 2011 ERI 81AR2
A
HalavenControl Arm
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
0 6 12 18 24 30 36
TIME (MONTHS)
PROP
ORTI
ON O
F PA
TIEN
TS A
LIVE
508 406 274 142 54 11 0254 178 106 61 26 5 0
HalavenTPC Arm
Number of patients at risk
TPC Arm (n=254)
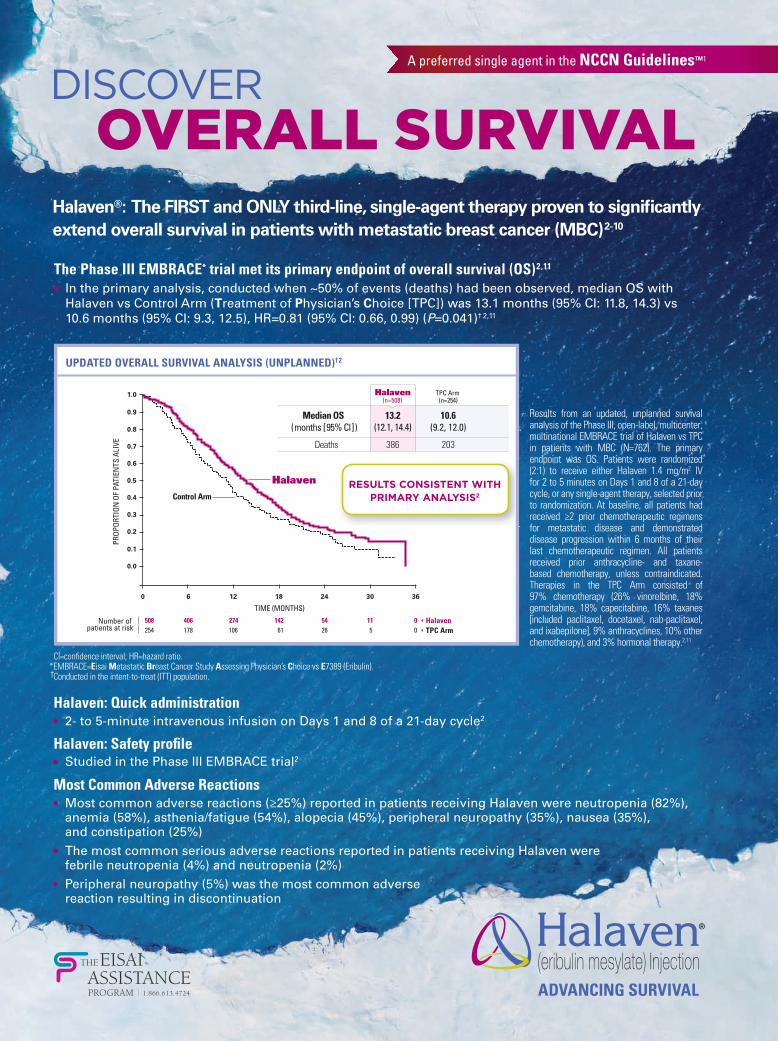
10.6(9.2, 12.0)
203
13.2(12.1, 14.4)
386
Median OS (months [95% Cl])
Deaths
Halaven(n=508)
RESULTS CONSISTENT WITH PRIMARY ANALYSIS2
experience febrile neutropenia or G
S
G
P P
• G
N
C
Breast Cancer. V
A
OVERALL SURVIVALDISCOVER
Halaven: Quick administration • 2- to 5-minute intravenous infusion on Days 1 and 8 of a 21-day cycle2
Halaven: Safety pro� le• Studied in the Phase III EMBRACE trial2
Most Common Adverse Reactions• Most common adverse reactions (≥25%) reported in patients receiving Halaven were neutropenia (82%),
anemia (58%), asthenia/fatigue (54%), alopecia (45%), peripheral neuropathy (35%), nausea (35%), and constipation (25%)
• The most common serious adverse reactions reported in patients receiving Halaven were febrile neutropenia (4%) and neutropenia (2%)
• Peripheral neuropathy (5%) was the most common adverse reaction resulting in discontinuation
The Phase III EMBRACE* trial met its primary endpoint of overall survival (OS) 2,11 • In the primary analysis, conducted when ~50% of events (deaths) had been observed, median OS with
Halaven vs Control Arm (Treatment of Physician’s Choice [TPC]) was 13.1 months (95% CI: 11.8, 14.3) vs 10.6 months (95% CI: 9.3, 12.5), HR=0.81 (95% CI: 0.66, 0.99) (P=0.041)† 2, 11
CI=con¥ dence interval; HR=hazard ratio. *EMBRACE=Eisai Metastatic Breast Cancer Study Assessing Physician’s Choice vs E7389 (Eribulin). †Conducted in the intent-to-treat (ITT) population.
Results from an updated, unplanned survival analysis of the Phase III, open-label, multicenter, multinational EMBRACE trial of Halaven vs TPC in patients with MBC (N=762). The primary endpoint was OS. Patients were randomized (2:1) to receive either Halaven 1.4 mg/m2 IV for 2 to 5 minutes on Days 1 and 8 of a 21-day cycle, or any single-agent therapy, selected prior to randomization. At baseline, all patients had received ≥2 prior chemotherapeutic regimens for metastatic disease and demonstrated disease progression within 6 months of their last chemotherapeutic regimen. All patients received prior anthracycline- and taxane-based chemotherapy, unless contraindicated. Therapies in the TPC Arm consisted of 97% chemotherapy (26% vinorelbine, 18% gemcitabine, 18% capecitabine, 16% taxanes [included paclitaxel, docetaxel, nab-paclitaxel, and ixabepilone], 9% anthracyclines, 10% other chemotherapy), and 3% hormonal therapy.2,11
Halaven®: The FIRST and ONLY third-line, single-agent therapy proven to signi� cantly extend overall survival in patients with metastatic breast cancer (MBC) 2-10
* E
All rights reserved. Printed in USA/December 2011 ERI 81AR2
A preferred single agent in the NCCN Guidelines™1
UPDATED OVERALL SURVIVAL ANALYSIS (UNPLANNED)†2

HALAVENTM (eribulin mesylate) Injection BRIEF SUMMARY – See package insert for full prescribing information.2.2 Dose Modi�cation Assess for peripheral neuropathy and obtain complete blood cell counts prior to each dose.Recommended dose delays• Do not administer HALAVEN on Day 1 or Day 8 for any of the following:
– ANC <1,000/mm3
– Platelets <75,000/mm3
– Grade 3 or 4 non-hematological toxicities• The Day 8 dose may be delayed for a maximum of 1 week.
– If toxicities do not resolve or improve to ≤ Grade 2 severity by Day 15, omit the dose. – If toxicities resolve or improve to ≤ Grade 2 severity by Day 15, administer HALAVEN at a reduced dose and
initiate the next cycle no sooner than 2 weeks later.Recommended dose reductions• If a dose has been delayed for toxicity and toxicities have recovered to Grade 2 severity or less, resume HALAVEN
at a reduced dose as set out in Table 1.• Do not re-escalate HALAVEN dose after it has been reduced.Table 1 Recommended Dose Reductions
Event Description Recommended HALAVEN Dose
Permanently reduce the 1.4 mg/m2 HALAVEN dose for any of the following:
1.1 mg/m2
ANC <500/mm3 for >7 days ANC <1,000 /mm3 with fever or infectionPlatelets <25,000/mm3
Platelets <50,000/mm3 requiring transfusionNon-hematologic Grade 3 or 4 toxicities Omission or delay of Day 8 HALAVEN dose in previous cycle for toxicityOccurrence of any event requiring permanent dose reduction while receiving 1.1 mg/m2 0.7 mg/m2
Occurrence of any event requiring permanent dose reduction while receiving 0.7 mg/m2 Discontinue HALAVEN ANC = absolute neutrophil count. Toxicities graded in accordance with National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) version 3.0. 5 WARNINGS AND PRECAUTIONS5.1 Neutropenia Severe neutropenia (ANC <500/mm3) lasting more than one week occurred in 12% (62/503) of patients in Study 1, leading to discontinuation in <1% of patients. Patients with alanine aminotransferase or aspartate aminotransferase >3 × ULN (upper limit of normal) experienced a higher incidence of Grade 4 neutropenia and febrile neutropenia than patients with normal aminotransferase levels. Patients with bilirubin >1.5 × ULN also had a higher incidence of Grade 4 neutropenia and febrile neutropenia.Monitor complete blood counts prior to each dose; increase the frequency of monitoring in patients who develop Grade 3 or 4 cytopenias. Delay administration of HALAVEN and reduce subsequent doses in patients who experience febrile neutropenia or Grade 4 neutropenia lasting longer than 7 days. Clinical studies of HALAVEN did not include patients with baseline neutrophil counts below 1,500/mm3.5.2 Peripheral Neuropathy Grade 3 peripheral neuropathy occurred in 8% (40/503) of patients, and Grade 4 in 0.4% (2/503) of patients in Study 1. Peripheral neuropathy was the most common toxicity leading to discontinuation of HALAVEN (5% of patients; 24/503). Neuropathy lasting more than one year occurred in 5% (26/503) of patients. Twenty-two percent (109/503) of patients developed a new or worsening neuropathy that had not recovered within a median follow-up duration of 269 days (range 25-662 days). Monitor patients closely for signs of peripheral motor and sensory neuropathy. Withhold HALAVEN in patients who experience Grade 3 or 4 peripheral neuropathy until resolution to Grade 2 or less.5.3 Embryo-Fetal ToxicityThere are no adequate and well-controlled studies of HALAVEN in pregnant women. HALAVEN is a microtubule inhibitor; therefore, it is expected to cause fetal harm when administered to a pregnant woman. Embryo-fetal toxicity and teratogenicity occurred in rats that received eribulin mesylate at approximately half of the recommended human dose based on body surface area. If this drug is used during pregnancy, or if a patient becomes pregnant while taking this drug, she should be apprised of the potential hazard to the fetus.5.4 QT ProlongationIn an uncontrolled open-label ECG study in 26 patients, QT prolongation was observed on Day 8, independent of eribulin concentration, with no QT prolongation observed on Day 1. ECG monitoring is recommended if therapy is initiated in patients with congestive heart failure, bradyarrhythmias, drugs known to prolong the QT interval, including Class Ia and III antiarrhythmics, and electrolyte abnormalities. Correct hypokalemia or hypomagnesemia prior to initiating HALAVEN and monitor these electrolytes periodically during therapy. Avoid HALAVEN in patients with congenital long QT syndrome. 6 ADVERSE REACTIONSThe following adverse reactions are discussed in detail in other sections of the labeling:• Neutropenia • Peripheral neuropathy • QT interval prolongationThe most common adverse reactions (≥25%) reported in patients receiving HALAVEN were neutropenia, anemia, asthenia/fatigue, alopecia, peripheral neuropathy, nausea, and constipation. The most common serious adverse reactions reported in patients receiving HALAVEN were febrile neutropenia (4%) and neutropenia (2%). The most common adverse reaction resulting in discontinuation of HALAVEN was peripheral neuropathy (5%). Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in other clinical trials and may not re§ect the rates observed in clinical practice. In clinical trials, HALAVEN has been administered to 1,222 patients with multiple tumor types, including 240 patients exposed to HALAVEN for 6 months or longer. The majority of the 1,222 patients were women (82%) with a median age of 58 years (range: 26 to 91 years). The racial and ethnic distribution was Caucasian (83%), Black (5%), Asian (2%), and other (5%).The adverse reactions described in Table 2 were identi©ed in 750 patients treated in Study 1. In Study 1, patients were randomized (2:1) to receive either HALAVEN (1.4 mg/m2 on Days 1 and 8 of a 21-day cycle) or single agent treatment chosen by their physician (control group). A total of 503 patients received HALAVEN, and 247 patients in the control group received therapy consisting of chemotherapy [total 97% (anthracyclines 10%, capecitabine 18%, gemcitabine 19%, taxanes 15%, vinorelbine 25%, other chemotherapies 10%)] or hormonal therapy (3%). The median duration of exposure was 118 days for patients receiving HALAVEN and 63 days for patients receiving control therapy. Table 2 reports the most common adverse reactions occurring in at least 10% of patients in either group.Table 2 Adverse Reactions with a Per-Patient Incidence of at Least 10% in Study 1
MedDRA ver 10.0 HALAVEN (n=503) Control Group (n=247)All Grades ≥ Grade 3 All Grades ≥ Grade 3
Blood and Lymphatic System Disordersa Neutropenia 82% 57% 53% 23%Anemia 58% 2% 55% 4%Nervous system disordersPeripheral neuropathyb 35% 8% 16% 2%Headache 19% <1% 12% <1%General disorders and administrative site conditionsAsthenia/Fatigue 54% 10% 40% 11%Mucosal in§ammation 9% 1% 10% 2%Pyrexia 21% <1% 13% <1%Gastrointestinal disordersConstipation 25% 1% 21% 1%Diarrhea 18% 0 18% 0Nausea 35% 1% 28% 3%Vomiting 18% 1% 18% 1%Musculoskeletal and connective tissue disordersArthralgia/Myalgia 22% <1% 12% 1%Back pain 16% 1% 7% 2%Bone pain 12% 2% 9% 2%Pain in extremity 11% 1% 10% 1%InvestigationsWeight decreased 21% 1% 14% <1%Metabolism and nutrition disordersAnorexia 20% 1% 13% 1%
Table 2 (cont’d)
Cytopenias: Grade 3 neutropenia occurred in 28% (143/503) of patients who received HALAVEN in Study 1, and 29% (144/503) of patients experienced Grade 4 neutropenia. Febrile neutropenia occurred in 5% (23/503) of patients; two patients (0.4%) died from complications of febrile neutropenia. Dose reduction due to neutropenia was required in 12% (62/503) of patients and discontinuation was required in <1% of patients. The mean time to nadir was 13 days and the mean time to recovery from severe neutropenia (<500/mm3) was 8 days. Grade 3 or greater thrombocytopenia occurred in 1% (7/503) of patients. G-CSF (granulocyte colony-stimulating factor) or GM-CSF (granulocyte–macrophage colony-stimulating factor) was used in 19% of patients who received HALAVEN. Peripheral Neuropathy: In Study 1, 17% of enrolled patients had Grade 1 peripheral neuropathy and 3% of patients had Grade 2 peripheral neuropathy at baseline. Dose reduction due to peripheral neuropathy was required by 3% (14/503) of patients who received HALAVEN. Four percent (20/503) of patients experienced peripheral motor neuropathy of any grade and 2% (8/503) of patients developed Grade 3 peripheral motor neuropathy.Liver Function Test Abnormalities: Among patients with Grade 0 or 1 ALT levels at baseline, 18% of HALAVEN-treated patients experienced Grade 2 or greater ALT elevation. One HALAVEN-treated patient without documented liver metastases had concomitant Grade 2 elevations in bilirubin and ALT; these abnormalities resolved and did not recur with re-exposure to HALAVEN.Less Common Adverse Reactions: The following additional adverse reactions were reported in ≥5% to <10% of the HALAVEN-treated group: • Eye Disorders: increased lacrimation• Gastrointestinal Disorders: dyspepsia, abdominal pain, stomatitis, dry mouth• General Disorders and Administration Site Conditions: peripheral edema• Infections and Infestations: upper respiratory tract infection• Metabolism and Nutrition Disorders: hypokalemia• Musculoskeletal and Connective Tissue Disorders: muscle spasms, muscular weakness• Nervous System Disorders: dysgeusia, dizziness• Psychiatric Disorders: insomnia, depression• Skin and Subcutaneous Tissue Disorders: rash8 USE IN SPECIFIC POPULATIONS8.1 Pregnancy Category DThere are no adequate and well-controlled studies with HALAVEN in pregnant women. HALAVEN is a microtubule inhibitor; therefore, it is expected to cause fetal harm when administered to a pregnant woman. Embryo-fetal toxicity and teratogenicity occurred in rats that received eribulin mesylate at approximately half of the recommended human dose based on body surface area. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. In a developmental toxicity study, pregnant rats received intravenous infusion of eribulin mesylate during organogenesis (Gestation Days 8, 10, and 12) at doses approximately 0.04, 0.13, 0.43 and 0.64 times the recommended human dose, based on body surface area (mg/m2). Increased abortion and severe external or soft tissue malformations were observed in offspring at doses 0.64 times the recommended human dose based on body surface area (mg/m2), including the absence of a lower jaw, tongue, stomach and spleen. Increased embryo-fetal death/resorption, reduced fetal weights, and minor skeletal anomalies consistent with developmental delay were also reported at or above doses of 0.43 times the recommended human dose. Maternal toxicity of eribulin mesylate was reported in rats at or above doses of 0.43 times the recommended human dose (mg/m²), and included enlarged spleen, reduced maternal weight gain and decreased food consumption.8.3 Nursing MothersIt is not known whether HALAVEN is excreted into human milk. No studies in humans or animals were conducted to determine if HALAVEN is excreted into milk. Because many drugs are excreted into human milk and because of the potential for serious adverse reactions in human milk fed infants from HALAVEN, a decision should be made whether to discontinue nursing or to discontinue HALAVEN taking into account the importance of the drug to the mother. 8.4 Pediatric UseThe safety and effectiveness of HALAVEN in pediatric patients below the age of 18 years have not been established.8.6 Hepatic ImpairmentA study evaluated the PK of eribulin in patients with mild (Child-Pugh A; n=7) and moderate (Child-Pugh B; n=5) hepatic impairment. Compared to patients with normal hepatic function (n=6), eribulin exposure increased 1.8-fold and 2.5-fold in patients with mild and moderate hepatic impairment, respectively. Administration of HALAVEN at a dose of 1.1 mg/m2 to patients with mild hepatic impairment and 0.7 mg/m2 to patients with moderate hepatic impairment resulted in similar exposure to eribulin as a dose of 1.4 mg/m2 to patients with normal hepatic function. A lower starting dose of 1.1 mg/m2 is recommended for patients with mild hepatic impairment (Child-Pugh A) and of 0.7 mg/m2 is recommended for patients with moderate hepatic impairment (Child-Pugh B). HALAVEN was not studied in patients with severe hepatic impairment (Child-Pugh C). 8.7 Renal ImpairmentNo formal PK trials were conducted with HALAVEN in patients with renal impairment. Available data suggests that no dose adjustment is necessary for patients with mild renal impairment (CrCl 50-80 mL/min). However, for patients with moderate renal impairment (CrCl 30-50 mL/min), the geometric mean dose-normalized systemic exposure increased 2-fold compared to patients with normal renal function. A lower starting dose of 1.1 mg/m2 is recommended for patients with moderate renal impairment. The safety of HALAVEN was not studied in patients with severe renal impairment (CrCl <30 mL/min). 10 OVERDOSAGE Overdosage of HALAVEN has been reported at approximately 4 times the recommended dose, which resulted in Grade 3 neutropenia lasting seven days and a Grade 3 hypersensitivity reaction lasting one day. There is no known antidote for HALAVEN overdose.12 CLINICAL PHARMACOLOGY 12.2 PharmacodynamicsCardiac ElectrophysiologyThe effect of HALAVEN on the QTc interval was assessed in an open-label, uncontrolled, multicenter, single-arm dedicated QT trial. A total of 26 patients with solid tumors received 1.4 mg/m2 of HALAVEN on Days 1 and 8 of a 21-day cycle. A delayed QTc prolongation was observed on Day 8, with no prolongation observed on Day 1. The maximum mean QTcF change from baseline (95% upper con©dence interval) was 11.4 (19.5) ms.13 NONCLINICAL TOXICOLOGY13.1 Carcinogenesis, mutagenesis, impairment of fertilityCarcinogenicity studies have not been conducted with eribulin mesylate. Eribulin mesylate was not mutagenic in in vitro bacterial reverse mutation assays (Ames test). Eribulin mesylate was positive in mouse lymphoma mutagenesis assays, and was clastogenic in an in vivo rat bone marrow micronucleus assay. The effects of HALAVEN on human fertility are unknown. Fertility studies have not been conducted with eribulin mesylate in humans or animals. However, nonclinical ©ndings in repeated-dose dog and rat toxicology studies suggest that male fertility may be compromised by treatment with eribulin mesylate. Rats exhibited testicular toxicity (hypocellularity of seminiferous epithelium with hypospermia/aspermia) following dosing with eribulin mesylate at or above 0.43 times the recommended human dose (mg/m2) given once weekly for 3 weeks, or at or above 0.21 times the recommended human dose (mg/m2) given once weekly for 3 out of 5 weeks, repeated for 6 cycles. Testicular toxicity was also observed in dogs given 0.64 times the recommended human dose (mg/m2) weekly for 3 out of 5 weeks, repeated for 6 cycles. 17 PATIENT COUNSELING INFORMATIONSee FDA-Approved Patient Labeling• Advise patients to contact their health care provider for a fever of 100.5°F or greater or other signs or symptoms of
infection such as chills, cough, or burning or pain on urination.• Advise women of childbearing potential to avoid pregnancy and to use effective contraception during treatment with HALAVEN.––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––
HALAVEN™ is a trademark used by Eisai Inc. under license from Eisai R&D Management Co., Ltd.© 2011 Eisai Inc. All rights reserved. Printed in USA / May 2011 ERI 161
MedDRA ver 10.0 HALAVEN (n=503) Control Group (n=247)All Grades ≥ Grade 3 All Grades ≥ Grade 3
Respiratory, thoracic, and mediastinal disordersCough 14% 0 9% 0Dyspnea 16% 4% 13% 4%Skin and subcutaneous tissue disordersAlopecia 45% NAc 10% NAc
Infections and InfestationsUrinary Tract Infection 10% 1% 5% 0
aBased upon laboratory data.b Includes neuropathy peripheral, neuropathy, peripheral motor neuropathy, polyneuropathy, peripheral sensory neuropathy, and paraesthesia.
cNot applicable; (grading system does not specify > Grade 2 for alopecia).

and pharmacy was also part of theprocess. Validation of the health plandata and comparators was required.The key impactable cost drivers in
cancer care identified include thecosts of therapy, supportive care,symptom management, site of care,and end-of-life care.
Supportive CareThe costs of supportive care iden-
tified were the management of nau-sea/vomiting, bone metastases, andwhite blood cell and red blood cellsupport. “We decided to focus onnausea and bone medicine support,”she said.Aggressive management of 7 com-
mon symptoms (nausea/vomiting,diarrhea, pain, fever, infection, neu-tropenic fever, blood clots) in theoffice or at local urgent care centerswas a goal to reduce the number ofhospitalizations and keep patientsfrom emergency department visits.A partnership was therefore begunwith local urgent care centers to seepatients who experienced thesesymptoms after hours.Better oversight of medications,
whether oral or intravenous (IV),
was determined to improve medica-tion compliance and adherence,leading to better outcomes.
New PaymentMethodologiesStandard fee-for-service is still
part of the payment methodology forwhich evaluation and managementvisits are paid, as are infusion codesand reimbursement for IV and oraldrugs, using the average sales priceplus 6% methodology.New payment methodologies
included the development of newcodes for care planning and caremanagement, “so that care planninggoes on for every single patient forevery type of treatment—adjuvant,neoadjuvant, prevention, mainte-nance, consolidation, first-line, sec-ond-line, and third-line,” said DrBosserman. “Everyone who is onactive management has a per cyclemanagement fee.” The entire care plan is paid for in
addition to the office visits. Wil -shire is committed to delivering 6reports of transparency: • Number of patients under care
and their clinical characteristics
• Therapies patients are taking andthe supportive therapy thataccompanies them (whether onor off guidelines)
• Potential cost-saving for the reg-imen chosen compared with aNational Comprehensive Can -cer Network–allowable regimen
• Interval care report (events thatrequired an extended office visitor urgent care visit vs emergencydepartment visit and hospitaladmission)
• End-of-life care report• Nationally validated quality mea -
s ures (ie, key American Society ofClinical Oncology QualityOncology Practice Initiativemeasures, meaningful use, andMedicare Physician QualityReporting Initiative).
Early ExperienceThe program has led to a 25%
increased rate overall in the con-tract. “We’re not dependent on thedrug margin,” said Dr Bosserman.“We’ll be doing all of the analytics asto what our costs are to deliver, andwhether the 25% increase isenough.” l
Cancer Center Business Summit
19December 2011 I www.OncPracticeManagement.com I
Wilshire’s Medical…Continued from page 3
First Level III Oncology Medical HomeWastes Fewer ResourcesBy Wayne Kuznar
Chicago, IL—Offering superiorquality of care at lower cost,Consultants in Medical On-
cology and Hematology (CMOH) isthe first oncology practice in thenation to achieve Level III recogni-tion by the National Committee forQuality Assurance.“The oncology-patient centered,
medical-home model has standard-ized approaches to care from assess-
ment to patient navigation and dis-ease management,” said John D.Sprandio, MD, oncologist and ownerof CMOH.“Four terms to remember in
achieving accountable cancer careare patient needs, quality, value, anddemonstration of results,” he said atthe 2011 Cancer Center BusinessSummit. “That is what healthcarereform is all about.”
Dr Sprandio and his colleagues atCMOH began transforming theirpractice in 2004 in preparation for achange in payment method thatwould reimburse the practice basedon the quality of care provided.As the practice was transformed
to electronic medical records, cus-tomized software was being devel-oped to better suit practice, patient
Continued on page 21

WillWeexhaust all possibilities.
Celgene Patient Support® provides free and personalized assistance with patients’ access and reimbursement needs.
With continual communication and consistent follow-through, your dedicated Celgene Patient Support® Specialist will streamline access to Celgene products by helping you and your patients with:
Benefits investigation
Prior authorization
Appeal support
Medicare
Co-pay assistance – Celgene Commercial Co-pay Program – Co-pay assistance through third-party organizations
Prescription status
Celgene free medication program
Celgene products and restricted distribution programs
To Contact Celgene Patient Support®:
Call: 1-800-931-8691
E-mail: [email protected]
Fax: 1-800-822-2496
Visit: www.CelgenePatientSupport.com
Monday through Friday, 8:00 AM to 7:00 PM ET
4 out of 5 patients who requested assistance from Celgene Patient Support ® received their medication.
We will…because patients are our priority.
Celgene Patient Support® is a registered trademark of Celgene Corporation.©2011 Celgene Corporation 11/11 US-CELG110061

needs, and data collection. Thesecustomized software applicationswere also designed to support com-prehensive processes of care thatwere required for the Level III,patient-centered, medical homerecognition, Dr Sprandio explained.
Care-Coordinated ModelEncourages ClinicalIntegrationThe care-coordinated model was
developed to “promote a value-basedagenda that facilitates physicianaccountability and encourages clini-cal integration among like-mindedmedical oncology practices,” he said.“It enhances communication andcoordination with primary care. Inthe past 4 months, it has promotedcollaboration with payers. It does allthis while focusing on patient needsand evidence-based care.”The primary care team addresses
all nononcologic issues. “We takeresponsibility for the coordination ofall oncologic-related services, fromthe time of diagnosis through thesurvivorship stage of care or to thetime of death,” said Dr Sprandio. Patient engagement is the focus
according to Dr Sprandio, especiallythe encouragement of patients to beproactive about reporting symptoms,as early treatment of symptoms oftenresults in fewer hospitalizations.
Standardized CareThe model promotes collabora-
tion between the caregiving team,adherence to evidence-based guide-lines, prevention of complications,and increasing access to care, “all ina standardized way.”“Everything is standardized…pa-
tient assessment, collection of data,documentation, patient naviga-tion….Telephone triage has been abig plus in directing patients away
from the ER [emergency room] andmore toward self-management,” hesaid. Potentially avoidable complica-tions have been defined, measured,and reduced in number. “We’re alsobig proponents of integrated pallia-tive care from the time of diagnosisand allowing physicians to track it.”
Triage Call Service The nurse-triage call service has
taken 3900 calls during the past year;of these calls: • 75% of patients’ symptoms weremanaged at home
• 4.3% of patients who wentthrough the phone triage werereferred to the emergency depart-
ment (5.8% overall since 2006)• 5.5% were seen the same day • 4.5% were seen the next day.“We encourage them to call early.
If they think they might have aproblem at 8:00 in the morning, weencourage them to call by 8:15.Calling us at 4:15 may result in anunnecessary ER evaluation,” said DrSprandio.The impact on emergency depart-
ment utilization has been significant,Dr Sprandio noted. The number ofemergency department visits perchemotherapy-receiving patient hasbeen reduced from 2.6 in 2004 to0.91 in 2010. The number of hospi-tal admissions per chemotherapy-receiving patient was 0.6 in 2010.Documentation turnaround time hasbeen reduced from 28 days to lessthan 1 day.Patient outcomes have not been
affected by the reductions in thenumber of emergency departmentvisits and hospitalizations, DrSprandio observed.End-of-life care offered by the
practice has resulted in an increasein the average hospice length of stayfrom 26 days in 2009 to 32 days in2010. The end-of-life discussionshave resulted in less use ofchemotherapy by the practice, andthe number of outpatient chemo -therapy visits has declined by 12%.The oncology medical home
model, coupled with a pathways pro-gram, has led to: • A 43% reduction in hospitaliza-
tions • A 65% reduction in emergency
department evaluations• An estimated savings between
6.6% and 12.7% of the total costof cancer care.
“If you plug in our numbers, itwas higher than 13%,” Dr Sprandiosaid. l
Cancer Center Business Summit
21December 2011 I www.OncPracticeManagement.com I
“We take responsibilityfor the coordination ofall oncologic-relatedservices, from the timeof diagnosis throughthe survivorship stageof care or to the timeof death.”
—John D. Sprandio, MD
First Level III Oncology…Continued from page 19






Chicago, IL—A survey con-ducted by the Cancer CenterBusiness Summit of 36 health -
care organizations on account ablecare initiatives involved in oncolo-gy-specific, nontraditional paymentmethodology showed a low priorityfor achieving cost-savings throughoncology care services, according toRonald Barkley, MS, JD, ManagingDirector, Cancer Center BusinessDevel opment Group, and the sur-vey’s principal investigator.Overall, survey responders indi-
cated that the concept of account-able care organizations (ACOs) hasfocused on chronic diseases, becausetheir costs are more predictable com-pared with oncology-related costs.The comments gathered from
direct phone interviews of these or -ganizations, conducted during June-August 2011, were identified aseither responding to ACO initia-tives or participating in nontradi-tional and innovative payment reim-bursement (payment other thanfee-for-service format). The interviews revealed that 10
of the organizations surveyed wereACO proactive, 13 were ACO ex -ploratory, 8 took a “wait-and-see”approach, and 2 were ignoring ACOsaltogether.One pattern that emerged from
the market profiles of these organiza-tions is that proactive ACO respon-ders tended to vary from somewhatto highly consolidated and highlycompetitive markets, according toMr Barkley.
Emerging PatternsKey comments about ACO readi-
ness showed that:
1The proposed rules were too oner-ous.
2Uncertainty over Medicare ACOreporting data requirements with
respect to cancer (immature data) istoo great.
3Determining the true cost of can-cer care is difficult, given the lack
of a consistent definition of cancercare.
4There is a need to “spend a lot ofpolitical capital” with physicians
to get ready for ACOs.
5Too much time and effort is spentdetermining which physicians are
fully aligned with the ACO concept.Of the organizations participating
in oncology-specific nontraditionalpayment methodologies, United-Healthcare instituted what it calledan episode-of-care payment system,in which a historic drug margin, plusa per-patient administrative fee, ispaid for practices complying withselected pathways for select cancersites (breast, colon, lung, ovarian).All other services are on a fee-for-service basis. As such, it is not a trueepisode-of-care payment but rather“locks in” the historic drug margin,said Barkley.Many state Blue Cross plans and
P4 Healthcare have models in whichpractices are paid premiums on drug
reimbursement for 80% pathwayscompliance. Within the ACO responder orga -
nizations, there was essentially novariation from traditional paymentmethods in oncology.Oncology as a health condition
seems to be of lesser priority in thecontext of ACO planning than ischronic diseases, such as heart dis-ease and chronic obstructive pul-monary disease. The ACO respon-der organizations tend to agree that
these chronic conditions have beencited as better candidates for cost-savings, noting that ACO conceptshave been developed around pri-mary care, and not as much thoughthas been given to subspecialty care,which tends to be fragmented. In general, the sheer volume of
chronic disease patients offers moreopportunity for cost-savings com-pared with cancer patients, said
Cancer Center Business Summit
27December 2011 I www.OncPracticeManagement.com I
Accountable Care Organizations PayingLittle Attention to Oncology Services, Not Seeing the Cost-SavingsCancer Center Business Summit’s Survey ResultsBy Wayne Kuznar
The survey of 36healthcareorganizations onaccountable careinitiatives found a lowpriority for achievingcost-savings throughoncology care services.
Continued on page 28

Chicago, IL—The accountablecare organization (ACO) con -cept is in the early stages of
being defined, our oncology prac-tices should keep their options open,“and remain independent while thedust settles,” said Matthew Brow,Vice President of Communication,Government Relations and PublicPolicy at McKesson Specialty CareSolutions/US Oncology at theCancer Center Business Summit.ACOs and the Medicare Shared
Savings Program (MSSP) are intend-ed to reduce healthcare spending byrealigning incentives for physicianswho manage care and better coordi-nating care across settings.A sustainable, value-based reim-
bursement approach for the care ofMedicare beneficiaries (instead of avolume-based approach) is the goalof ACOs and the MSSP, accordingto Mr Brow. An oncology-led ACO is not per-
missible under the proposed MSSPrules; under MSSP rules, only pa -tients of primary care physicians are
included in the program; a minimumof 5000 Medicare beneficiaries arerequired for ACO participation, andoncologist-only practices cannot
meet this minimum. It is permissibleunder the Medicare Pioneer ACOprogram, but no one applied in time.The current ACO model, which
revolves around primary care physi-cians, provides little incentive foroncologists to join. “If the only wayfor a patient to be assigned to anACO is through a primary carephysician, and patients are onlyassigned to a primary care physicianwho provided the plurality of evalu-ation and management services to a patient, oncologist-managed pa -tients don’t count toward targets orperformance,” Mr Brow said. “Thereis significant pressure to pay forvalue, and the private side is leadingthe way in my view.” The legislative framework for
ACOs sets benchmarks for expectedMedicare Part A and Part B spend-ing for beneficiaries based on his-toric cost in the 3 years preceding anACO contract, adjusted annually forthe trend in Medicare cost growth,said Michael L. Blau, Esq, Chair atVenture Health Practices, Foley &Lardner LLP.A comparison of current to histor-
ical costs, adjusted for inflation, doesnot work in oncology, according to
28 I ONCOLOGY PRACTICE MANAGEMENT I December 2011
Cancer Center Business Summit
Now Is Not the Right Time for OncologyPractices to Jump on the ACOBandwagonBy Wayne Kuznar
A comparison ofcurrent to historicalcosts, adjusted forinflation, does notwork in oncology,because expenditureon cancer drugs isexpected to increaseby more than 10%annually.
Accountable Care…Continued from page 27
ACO responder organizations, andoncology is too complex to tackle inACO planning, with greater costvariability and unpredictability. “Cancer is too broad to get disease
focus. Whereas hip, knee, and cardiacsurgery are more predictable,” replieda health system oncology serviceexecutive in the mid-Atlantic region. Too much money is spent on futile
care in the last 6 months of life,
and conversations need to revolvearound end-of-life care much earlierthan they do now, said executivesand medical directors in the ACOresponder organizations.
Trends to Watch in 2012According to Mr Barkley, some
trends to watch for include:
1Expanding beyond pathways toinclude programs that emphasize
cost reductions by reducing redun-dant hospitalizations and emergencydepartment visits.
2Oncology practices organizing to be specialist “neighbors” of pri-
mary care medical homes.
3Oncology “bundled” pricing as a new oncologist–hospital align-
ment strategy. l

Mr Brow, because expenditure oncancer drugs is expected to increaseby more than 10% annually, and“new technology and generics aresignificant in oncology and mandateconcurrent controls and benchmarksversus historical.”Most practices are opting not to
participate, expecting no rate ofreturn on their investment, accord-ing to Mr Blau. In addition, severalproposed ACO regulations are ofconcern to oncologists: • A more limited role in ACOgovernance.
• None of the 65 quality measuresare specific to oncology.
• Meeting quality measures is thedeterminant of shared savings.
Oncology patients carry highercosts and therefore are less likely to
contribute to cost-savings. Further -more, ACO cost and risks are sub-stantial, and there is no assurance
that oncologists will be allocated afair share of any savings realized, said
Mr Brow. “Each ACO must specifyits method of allocating and distrib-uting shared savings dollars, and allspecialties do not need to be includ-ed or treated equally.”A 3-year lock-in is also required,
during which time a practice has toincur downside risk. Before all is set-tled, oncologists are likely to haveadditional value-based options withMedicare, so a wait-and-see ap -proach is advised, said Mr Brow.“Don’t jump in if you’re an oncologypractice.”“The ability to deliver and docu-
ment high-quality, cost-effectivecare is vital regardless of whetherone joins an ACO,” he said. “Workwith an organization that leveragesyears of investment in technologyand physician-driven pathways.” l
Cancer Center Business Summit
29December 2011 I www.OncPracticeManagement.com I
“The ability to deliverand document high-quality, cost-effectivecare is vital regardlessof whether one joinsan ACO.”
—Matthew Brow
The One Conference You Can’t AFFORD to Miss!
Second Annual Association for Value-Based Cancer Care ConferenceStrategies for Optimizing Value in Cancer Care Delivery
March 29-31, 2012 • JW Marriott • Houston, Texas
REGISTER TODAY FOR ONLY $275 at www.regonline.com/avbcc2012
����������������
REGISTER TODAY
SAVE $100

IndicationYERVOY (ipilimumab) is indicated for the treatmentof unresectable or metastatic melanoma.1
REFERENCES 1. YERVOY (ipilimumab) [package insert]. Princeton, NJ: Bristol-Myers Squibb; March 2011.2. Alpha-numeric HCPCS. Centers for Medicare & Medicaid Services Web site. http://www.cms.gov/HCPCSReleaseCodeSetsDownloads/12anweb.zip. Accessed November 1, 2011.
Please see Important Safety Information, including Boxed WARNING regarding immune-mediated adverse reactions, continued on adjacent page.
WARNING: IMMUNE-MEDIATED ADVERSE REACTIONS YERVOY can result in severe and fatal immune-mediated adverse reactions due to T-cell activation and proliferation. These immune-mediated reactions may involve any organ system; however, the most common severe immune-mediated adverse reactions are enterocolitis, hepatitis, dermatitis (including toxic epidermal necrolysis), neuropathy, and endocrinopathy. The majority of these immune-mediated reactions initially manifested during treatment; however, a minority occurred weeks to months after discontinuation of YERVOY.
Assess patients for signs and symptoms of enterocolitis, dermatitis, neuropathy and endocrinopathy and evaluate clinical chemistries including liver function tests (LFTs) and thyroid function tests at baseline and before each dose.
Permanently discontinue YERVOY and initiate systemic high-dose corticosteroid therapy for severe immune-mediated reactions.
Important Safety Information
Announcing: J-code for YERVOY™ (ipilimumab) J9228
HCPCS Code
J9228a Injection, ipilimumab, 1 mg
Description Effective
January 1, 2012
J-code for YERVOY2
The accurate completion of reimbursement- or coverage-related documentation is the responsibility of the healthcare provider and patient. Bristol-Myers Squibb and its agentsmake no guarantee regarding reimbursement for any service or item. This coding guidance is not intended to provide speci� c directions on requesting prior authorization or submitting claims for YERVOY and does not provide a guarantee of receiving prior authorization or reimbursement. Oncology practices need to make coding decisions based on the diagnosis and treatment of each patient and the speci� c insurer requirements.
aReplaces J9999, J3490, J3590, and C9284.
www.destinationaccess.com1-800-861-0048 (phone) Monday through Friday, 8:00 AM to 8:00 PM ET1-888-776-2370 (fax)
ProductDescription
50-mg/10 mL (5 mg/mL),single-use vial of YERVOY
200-mg/40 mL (5 mg/mL),single-use vial of YERVOY
NDC Number
10-digit 0003-2327-11 0003-2328-22
11-digit 00003-2327-11 00003-2328-22
single-use vial of YERVOY
Replaces J9999, J3490, J3590, and C9284.
731US11AB18303 11/11 Printed in USA Y
1
M
6
U
Y
I
2 1 1:00 PM

© 2011 Bristol-Myers Squibb 731US11AB18303 11/11 Printed in USA YERVOY is a trademark of Bristol-Myers Squibb.
Please see brief summary of Full Prescribing Information, including Boxed WARNING regarding immune-mediated adverse reactions, on the following spread.
Recommended Dose Modifi cationsWithhold dose for moderate immune-mediated adverse reactions until return to baseline, improvement to mild severity, or complete resolution, and patient is receiving <7.5 mg prednisone or equivalent per day.
Permanently discontinue YERVOY™ (ipilimumab) for any of the following: Persistent moderate adverse reactions or inability to reduce
corticosteroid dose to 7.5 mg prednisone or equivalent per day. Failure to complete full treatment course within 16 weeks from
administration of � rst dose. Severe or life-threatening adverse reactions.
Immune-mediated Enterocolitis: In the pivotal Phase 3 study in YERVOY-treated patients, severe,
life-threatening or fatal (diarrhea of ≥7 stools above baseline, fever, ileus, peritoneal signs; Grade 3-5) immune-mediated enterocolitis occurred in 34 (7%) and moderate (diarrhea with up to 6 stools above baseline, abdominal pain, mucus or blood in stool; Grade 2) enterocolitis occurred in 28 (5%) patients.
Across all YERVOY-treated patients (n=511), 5 (1%) developed intestinal perforation, 4 (0.8%) died as a result of complications, and 26 (5%) were hospitalized for severe enterocolitis.
Monitor patients for signs and symptoms of enterocolitis (such as diarrhea, abdominal pain, mucus or blood in stool, with or without fever) and of bowel perforation (such as peritoneal signs and ileus).
– In symptomatic patients, rule out infectious etiologies and consider endoscopic evaluation for persistent or severe symptoms.
Immune-mediated Hepatitis: In the pivotal Phase 3 study in YERVOY-treated patients, severe,
life-threatening, or fatal hepatotoxicity (AST or ALT elevations >5X the upper limit of normal (ULN) or total bilirubin elevations >3X the ULN; Grade 3–5) occurred in 8 (2%), with fatal hepatic failure in 0.2% and hospitalization in 0.4%.
13 (2.5%) additional YERVOY-treated patients experienced moderate hepatotoxicity manifested by LFT abnormalities (AST or ALT elevations >2.5X but ≤5X the ULN or total bilirubin elevation >1.5X but ≤3X the ULN; Grade 2).
Monitor LFTs (hepatic transaminase and bilirubin levels) and assess patients for signs and symptoms of hepatotoxicity before each dose of YERVOY.
– In patients with hepatotoxicity, rule out infectious or malignant causes and increase frequency of LFT monitoring until resolution.
Immune-mediated Dermatitis: In the pivotal Phase 3 study in YERVOY-treated patients, severe,
life-threatening or fatal immune-mediated dermatitis (e.g., Stevens-Johnson syndrome, toxic epidermal necrolysis, or rash complicated by full thickness dermal ulceration, or necrotic, bullous, or hemorrhagic manifestations; Grade 3–5) occurred in 13 (2.5%) patients.
– 1 (0.2%) patient died as a result of toxic epidermal necrolysis. – 1 additional patient required hospitalization for severe dermatitis.
There were 63 (12%) YERVOY-treated patients with moderate (Grade 2) dermatitis.
Monitor patients for signs and symptoms of dermatitis such as rash and pruritus. Unless an alternate etiology has been identi� ed, signs or symptoms of dermatitis should be considered immune-mediated.
Immune-mediated Neuropathies: In the pivotal Phase 3 study in YERVOY-treated patients, 1 case
of fatal Guillain-Barré syndrome and 1 case of severe (Grade 3) peripheral motor neuropathy were reported.
Across the clinical development program of YERVOY, myasthenia gravis and additional cases of Guillain-Barré syndrome have been reported.
Monitor for symptoms of motor or sensory neuropathy such as unilateral or bilateral weakness, sensory alterations, or paresthesia.
Immune-mediated Endocrinopathies: In the pivotal Phase 3 study in YERVOY-treated patients, severe to
life-threatening immune-mediated endocrinopathies (requiring hospitalization, urgent medical intervention, or interfering with activities of daily living; Grade 3-4) occurred in 9 (1.8%).
– All 9 patients had hypopituitarism and some had additional concomitant endocrinopathies such as adrenal insuf� ciency, hypogonadism, and hypothyroidism.
– 6 of the 9 patients were hospitalized for severe endocrinopathies.
Moderate endocrinopathy (requiring hormone replacement or medical intervention; Grade 2) occurred in 12 (2.3%) YERVOY-treated patients and consisted of hypothyroidism, adrenal insuf� ciency, hypopituitarism, and 1 case each of hyperthyroidism and Cushing’s syndrome.
Median time to onset of moderate to severe immune-mediated endocrinopathy was 11 weeks and ranged up to 19.3 weeks after the initiation of YERVOY.
Monitor patients for clinical signs and symptoms of hypophysitis, adrenal insuf� ciency (including adrenal crisis), and hyper- or hypothyroidism.
– Patients may present with fatigue, headache, mental status changes, abdominal pain, unusual bowel habits, and hypotension, or nonspeci� c symptoms which may resemble other causes such as brain metastasis or underlying disease.
– Unless an alternate etiology has been identi� ed, signs or symptoms of endocrinopathies should be considered immune-mediated.
– Monitor thyroid function tests and clinical chemistries at the start of treatment, before each dose, and as clinically indicated based on symptoms.
– In a limited number of patients, hypophysitis was diagnosed by imaging studies through enlargement of the pituitary gland.
Other Immune-mediated Adverse Reactions, Including Ocular Manifestations:
In the pivotal Phase 3 study in YERVOY-treated patients, clinically signi� cant immune-mediated adverse reactions seen in <1% were: nephritis, pneumonitis, meningitis, pericarditis, uveitis, iritis, and hemolytic anemia.
Across the clinical development program for YERVOY, immune-mediated adverse reactions also reported with <1% incidence were: myocarditis, angiopathy, temporal arteritis, vasculitis, polymyalgia rheumatica, conjunctivitis, blepharitis, episcleritis, scleritis, leukocytoclastic vasculitis, erythema multiforme, psoriasis, pancreatitis, arthritis, and autoimmune thyroiditis.
Pregnancy & Nursing: YERVOY is classi� ed as pregnancy category C. There are no
adequate and well-controlled studies of YERVOY in pregnant women. Use YERVOY during pregnancy only if the potential bene� t justi� es the potential risk to the fetus.
Human IgG1 is known to cross the placental barrier and YERVOY is an IgG1; therefore, YERVOY has the potential to be transmitted from the mother to the developing fetus.
It is not known whether YERVOY is secreted in human milk. Because many drugs are secreted in human milk and because of the potential for serious adverse reactions in nursing infants from YERVOY, a decision should be made whether to discontinue nursing or to discontinue YERVOY.
Common Adverse Reactions: The most common adverse reactions (≥5%) in patients who
received YERVOY at 3 mg/kg were fatigue (41%), diarrhea (32%), pruritus (31%), rash (29%), and colitis (8%).
Important Safety Information (cont)

YERVOY™ (ipilimumab) Injection, for intravenous infusion
Brief Summary of Prescribing Information. For complete prescribing information consult official package insert.
WARNING: IMMUNE-MEDIATED ADVERSE REACTIONSYERVOY (ipilimumab) can result in severe and fatal immune-mediated adverse reactions due to T-cell activation and proliferation. These immune-mediated reactions may involve any organ system; however, the most common severe immune-mediated adverse reactions are enterocolitis, hepatitis, dermatitis (including toxic epidermal necrolysis), neuropathy, and endocrinopathy. The majority of these immune-mediated reactions initially manifested during treatment; however, a minority occurred weeks to months after discontinuation of YERVOY.
Permanently discontinue YERVOY and initiate systemic high-dose corticosteroid therapy for severe immune-mediated reactions. [See Dosage and Administration (2.2) in Full Prescribing Information]
Assess patients for signs and symptoms of enterocolitis, dermatitis, neuropathy, and endocrinopathy and evaluate clinical chemistries including liver function tests and thyroid function tests at baseline and before each dose. [See Warnings and Precautions]
INDICATIONS AND USAGE
YERVOY (ipilimumab) is indicated for the treatment of unresectable or metastatic melanoma.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
YERVOY can result in severe and fatal immune-mediated reactions due to T-cell activation and proliferation. [See Boxed Warning]
Immune-mediated Enterocolitis
In Study 1, severe, life-threatening, or fatal (diarrhea of 7 or more stools above baseline, fever, ileus, peritoneal signs; Grade 3–5) immune-mediated enterocolitis occurred in 34 (7%) YERVOY-treated patients, and moderate (diarrhea with up to 6 stools above baseline, abdominal pain, mucus or blood in stool; Grade 2) enterocolitis occurred in 28 (5%) YERVOY-treated patients. Across all YERVOY-treated patients (n=511), 5 (1%) patients developed intestinal perforation, 4 (0.8%) patients died as a result of complications, and 26 (5%) patients were hospitalized for severe enterocolitis.
The median time to onset was 7.4 weeks (range 1.6–13.4) and 6.3 weeks (range 0.3–18.9) after the initiation of YERVOY for patients with Grade 3–5 enterocolitis and with Grade 2 enterocolitis, respectively.
Twenty-nine patients (85%) with Grade 3–5 enterocolitis were treated with high-dose (≥40 mg prednisone equivalent per day) corticosteroids, with a median dose of 80 mg/day of prednisone or equivalent; the median duration of treatment was 2.3 weeks (ranging up to 13.9 weeks) followed by corticosteroid taper. Of the 28 patients with moderate enterocolitis, 46% were not treated with systemic corticosteroids, 29% were treated with <40 mg prednisone or equivalent per day for a median duration of 5.1 weeks, and 25% were treated with high-dose corticosteroids for a median duration of 10 days prior to corticosteroid taper. Infliximab was administered to 5 of the 62 patients (8%) with moderate, severe, or life-threatening immune-mediated enterocolitis following inadequate response to corticosteroids.
Of the 34 patients with Grade 3–5 enterocolitis, 74% experienced complete resolution, 3% experienced improvement to Grade 2 severity, and 24% did not improve. Among the 28 patients with Grade 2 enterocolitis, 79% experienced complete resolution, 11% improved, and 11% did not improve.
Monitor patients for signs and symptoms of enterocolitis (such as diarrhea, abdominal pain, mucus or blood in stool, with or without fever) and of bowel perforation (such as peritoneal signs and ileus). In symptomatic patients, rule out infectious etiologies and consider endoscopic evaluation for persistent or severe symptoms.
Permanently discontinue YERVOY in patients with severe enterocolitis and initiate systemic corticosteroids at a dose of 1 to 2 mg/kg/day of prednisone or equivalent. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least one month. In clinical trials, rapid corticosteroid tapering resulted in recurrence or worsening symptoms of enterocolitis in some patients.
Withhold YERVOY dosing for moderate enterocolitis; administer anti-diarrheal treatment and, if persistent for more than one week, initiate systemic corticosteroids at a dose of 0.5 mg/kg/day prednisone or equivalent. [See Dosage and Administration (2.2) in Full Prescribing Information]
Immune-mediated Hepatitis
In Study 1, severe, life-threatening, or fatal hepatotoxicity (AST or ALT elevations of more than 5 times the upper limit of normal or total bilirubin elevations more than 3 times the upper limit of normal; Grade 3–5) occurred in 8 (2%) YERVOY-treated patients, with fatal hepatic failure in 0.2% and hospitalization in 0.4% of YERVOY-treated patients. An additional 13 (2.5%) patients experienced moderate hepatotoxicity manifested by liver function test abnormalities (AST or ALT elevations of more than 2.5 times but not more than 5 times the upper limit of normal or total bilirubin elevation of more than 1.5 times but not more than 3 times the upper limit of normal; Grade 2). The underlying pathology was not ascertained in all patients but in some instances included immune-mediated hepatitis. There were insufficient numbers of patients with biopsy-proven hepatitis to characterize the clinical course of this event.
Monitor liver function tests (hepatic transaminase and bilirubin levels) and assess patients for signs and symptoms of hepatotoxicity before each dose of YERVOY. In patients with hepatotoxicity, rule out infectious or malignant causes and increase frequency of liver function test monitoring until resolution.
Permanently discontinue YERVOY in patients with Grade 3–5 hepatotoxicity and administer systemic corticosteroids at a dose of 1 to 2 mg/kg/day of prednisone or equivalent. When liver function tests show sustained improvement or return to baseline, initiate corticosteroid tapering and continue to taper over 1 month. Across the clinical development program for YERVOY, mycophenolate treatment has been administered in patients who have persistent severe hepatitis despite high-dose corticosteroids. Withhold YERVOY in patients with Grade 2 hepatotoxicity. [See Dosage and Administration (2.2) in Full Prescribing Information]
Immune-mediated Dermatitis
In Study 1, severe, life-threatening, or fatal immune-mediated dermatitis (eg, Stevens-Johnson syndrome, toxic epidermal necrolysis, or rash complicated by full thickness dermal ulceration, or necrotic, bullous, or hemorrhagic manifestations; Grade 3–5) occurred in 13 (2.5%) YERVOY-treated patients. One (0.2%) patient died as a result of toxic epidermal necrolysis and one additional patient required hospitalization for severe dermatitis. There were 63 (12%) patients with moderate (Grade 2) dermatitis.
The median time to onset of moderate, severe, or life-threatening immune-mediated dermatitis was 3.1 weeks and ranged up to 17.3 weeks from the initiation of YERVOY (ipilimumab).
Seven (54%) YERVOY-treated patients with severe dermatitis received high-dose corticosteroids (median dose 60 mg prednisone/day or equivalent) for up to 14.9 weeks followed by corticosteroid taper. Of these 7 patients, 6 had complete resolution; time to resolution ranged up to 15.6 weeks.
Of the 63 patients with moderate dermatitis, 25 (40%) were treated with systemic corticosteroids (median of 60 mg/day of prednisone or equivalent) for a median of 2.1 weeks, 7 (11%) were treated with only topical corticosteroids, and 31 (49%) did not receive systemic or topical corticosteroids. Forty-four (70%) patients with moderate dermatitis were reported to have complete resolution, 7 (11%) improved to mild (Grade 1) severity, and 12 (19%) had no reported improvement.
Monitor patients for signs and symptoms of dermatitis such as rash and pruritus. Unless an alternate etiology has been identified, signs or symptoms of dermatitis should be considered immune-mediated.
Permanently discontinue YERVOY in patients with Stevens-Johnson syndrome, toxic epidermal necrolysis, or rash complicated by full thickness dermal ulceration, or necrotic, bullous, or hemorrhagic manifestations. Administer systemic corticosteroids at a dose of 1 to 2 mg/kg/day of prednisone or equivalent. When dermatitis is controlled, corticosteroid tapering should occur over a period of at least 1 month. Withhold YERVOY dosing in patients with moderate to severe signs and symptoms. [See Dosage and Administration (2.2) in Full Prescribing Information]
For mild to moderate dermatitis, such as localized rash and pruritus, treat symptomatically. Administer topical or systemic corticosteroids if there is no improvement of symptoms within 1 week.
Immune-mediated Neuropathies
In Study 1, one case of fatal Guillain-Barré syndrome and one case of severe (Grade 3) peripheral motor neuropathy were reported. Across the clinical development program of YERVOY, myasthenia gravis and additional cases of Guillain-Barré syndrome have been reported.
Monitor for symptoms of motor or sensory neuropathy such as unilateral or bilateral weakness, sensory alterations, or paresthesia. Permanently discontinue YERVOY in patients with severe neuropathy (interfering with daily activities) such as Guillain-Barré-like syndromes. Institute medical intervention as appropriate for management of severe neuropathy. Consider initiation of systemic corticosteroids at a dose of 1 to 2 mg/kg/day prednisone or equivalent for severe neuropathies. Withhold YERVOY dosing in patients with moderate neuropathy (not interfering with daily activities). [See Dosage and Administration (2.2) in Full Prescribing Information]
Immune-mediated Endocrinopathies
In Study 1, severe to life-threatening immune-mediated endocrinopathies (requiring hospitalization, urgent medical intervention, or interfering with activities of daily living; Grade 3–4) occurred in 9 (1.8%) YERVOY-treated patients. All 9 patients had hypopituitarism and some had additional concomitant endocrinopathies such as adrenal insufficiency, hypogonadism, and hypothyroidism. Six of the 9 patients were hospitalized for severe endocrinopathies. Moderate endocrinopathy (requiring hormone replacement or medical intervention; Grade 2) occurred in 12 (2.3%) patients and consisted of hypothyroidism, adrenal insufficiency, hypopituitarism, and one case each of hyperthyroidism and Cushing’s syndrome. The median time to onset of moderate to severe immune-mediated endocrinopathy was 11 weeks and ranged up to 19.3 weeks after the initiation of YERVOY.
Of the 21 patients with moderate to life-threatening endocrinopathy, 17 patients required long-term hormone replacement therapy including, most commonly, adrenal hormones (n=10) and thyroid hormones (n=13).
Monitor patients for clinical signs and symptoms of hypophysitis, adrenal insufficiency (including adrenal crisis), and hyper- or hypothyroidism. Patients may present with fatigue, headache, mental status changes, abdominal pain, unusual bowel habits, and hypotension, or nonspecific symptoms which may resemble other causes such as brain metastasis or underlying disease. Unless an alternate etiology has been identified, signs or symptoms of endocrinopathies should be considered immune-mediated.
Monitor thyroid function tests and clinical chemistries at the start of treatment, before each dose, and as clinically indicated based on symptoms. In a limited number of patients, hypophysitis was diagnosed by imaging studies through enlargement of the pituitary gland.
Withhold YERVOY dosing in symptomatic patients. Initiate systemic corticosteroids at a dose of 1 to 2 mg/kg/day of prednisone or equivalent, and initiate appropriate hormone replacement therapy. [See Dosage and Administration (2.2) in Full Prescribing Information]
Other Immune-mediated Adverse Reactions, Including Ocular Manifestations
The following clinically significant immune-mediated adverse reactions were seen in less than 1% of YERVOY-treated patients in Study 1: nephritis, pneumonitis, meningitis, pericarditis, uveitis, iritis, and hemolytic anemia.
Across the clinical development program for YERVOY, the following likely immune-mediated adverse reactions were also reported with less than 1% incidence: myocarditis, angiopathy, temporal arteritis, vasculitis, polymyalgia rheumatica, conjunctivitis, blepharitis, episcleritis, scleritis, leukocytoclastic vasculitis, erythema multiforme, psoriasis, pancreatitis, arthritis, and autoimmune thyroiditis.
Permanently discontinue YERVOY for clinically significant or severe immune-mediated adverse reactions. Initiate systemic corticosteroids at a dose of 1 to 2 mg/kg/day prednisone or equivalent for severe immune-mediated adverse reactions.
Administer corticosteroid eye drops to patients who develop uveitis, iritis, or episcleritis. Permanently discontinue YERVOY for immune-mediated ocular disease that is unresponsive to local immunosuppressive therapy. [See Dosage and Administration (2.2) in Full Prescribing Information]
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling.
[see Warnings and Precautions].
[see Warnings and Precautions].
[see Warnings and Precautions].
[see Warnings and Precautions].
[see Warnings and Precautions].
[see Warnings and Precautions].

Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared with rates in other clinical trials or experience with therapeutics in the same class and may not reflect the rates observed in clinical practice.
The clinical development program excluded patients with active autoimmune disease or those receiving systemic immunosuppression for organ transplantation. Exposure to YERVOY (ipilimumab) 3 mg/kg for four doses given by intravenous infusion in previously treated patients with unresectable or metastatic melanoma was assessed in a randomized, double-blind clinical study (Study 1). [See Clinical Studies (14) in Full Prescribing Information] One hundred thirty-one patients (median age 57 years, 60% male) received YERVOY as a single agent, 380 patients (median age 56 years, 61% male) received YERVOY with an investigational gp100 peptide vaccine (gp100), and 132 patients (median age 57 years, 54% male) received gp100 peptide vaccine alone. Patients in the study received a median of 4 doses (range 1 to 4 doses). YERVOY was discontinued for adverse reactions in 10% of patients.
The most common adverse reactions (≥5%) in patients who received YERVOY at 3 mg/kg were fatigue, diarrhea, pruritus, rash, and colitis.
Table 1 presents selected adverse reactions from Study 1, which occurred in at least 5% of patients in the YERVOY-containing arms and with at least 5% increased incidence over the control gp100 arm for all-grade events and at least 1% incidence over the control group for Grade 3–5 events.
Table 1: Selected Adverse Reactions in Study 1
Percentage (%) of Patientsa
YERVOY 3 mg/kg n=131
YERVOY 3 mg/kg+gp100
n=380
gp100 n=132
System Organ Class/ Preferred Term
Any Grade
Grade3–5
Any Grade
Grade3–5
Any Grade
Grade 3–5
Gastrointestinal Disorders Diarrhea ColitisSkin and Subcutaneous Tissue Disorders Pruritus RashGeneral Disorders and Administration Site Conditions Fatigue
328
3129
41
55
02
7
375
2125
34
43
<12
5
202
118
31
10
00
3
a Incidences presented in this table are based on reports of adverse events regardless of causality.
Table 2 presents the per-patient incidence of severe, life-threatening, or fatal immune-mediated adverse reactions from Study 1.
Table 2: Severe to Fatal Immune-mediated Adverse Reactions in Study 1
Percentage (%) of Patients
YERVOY3 mg/kgn=131
YERVOY3 mg/kg+gp100
n=380
Any Immune-mediated Adverse ReactionEnterocolitisa,b
Hepatotoxicitya
Dermatitisa
Neuropathya
Endocrinopathy Hypopituitarism Adrenal insufficiencyOther Pneumonitis Meningitis Nephritis Eosinophiliac
Pericarditisa,c
157121440
00110
12723
<1111
<1<100
<1
a Including fatal outcome. b Including intestinal perforation. c Underlying etiology not established.
Across clinical studies that utilized YERVOY doses ranging from 0.3 to 10 mg/kg, the following adverse reactions were also reported (incidence less than 1% unless otherwise noted): urticaria (2%), large intestinal ulcer, esophagitis, acute respiratory distress syndrome, renal failure, and infusion reaction.
Based on the experience in the entire clinical program for melanoma, the incidence and severity of enterocolitis and hepatitis appear to be dose dependent.
Immunogenicity
In clinical studies, 1.1% of 1024 evaluable patients tested positive for binding antibodies against ipilimumab in an electrochemiluminescent (ECL) based assay. This assay has substantial limitations in detecting anti-ipilimumab antibodies in the presence of ipilimumab. Infusion-related or peri-infusional reactions consistent with hypersensitivity or anaphylaxis were not reported in these 11 patients nor were neutralizing antibodies against ipilimumab detected.
Because trough levels of ipilimumab interfere with the ECL assay results, a subset analysis was performed in the dose cohort with the lowest trough levels. In this analysis, 6.9% of 58 evaluable patients, who were treated with 0.3 mg/kg dose, tested positive for binding antibodies against ipilimumab.
Immunogenicity assay results are highly dependent on several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to YERVOY with the incidences of antibodies to other products may be misleading.
DRUG INTERACTIONS
No formal drug-drug interaction studies have been conducted with YERVOY (ipilimumab).
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Category C
There are no adequate and well-controlled studies of YERVOY in pregnant women. Use YERVOY during pregnancy only if the potential benefit justifies the potential risk to the fetus.
In a combined study of embryo-fetal and peri-postnatal development, severe toxicities including increased incidences of third-trimester abortion, stillbirth, premature delivery, low birth weight, and infant mortality occurred following intravenous administration of ipilimumab to pregnant cynomolgus monkeys every 21 days from the onset of organogenesis through parturition at doses of 2.6 or 7.2 times the recommended human dose of 3 mg/kg (by AUC). [See Nonclinical Toxicology (13.2) in Full Prescribing Information]
In genetically engineered mice in which the gene for CTLA-4 has been deleted (a “knockout mouse”), offspring lacking CTLA-4 were born apparently healthy, but died within 3–4 weeks due to multi-organ infiltration and damage by lymphocytes.
Human IgG1 is known to cross the placental barrier and ipilimumab is an IgG1; therefore, ipilimumab has the potential to be transmitted from the mother to the developing fetus.
Nursing Mothers
It is not known whether ipilimumab is secreted in human milk. Because many drugs are secreted in human milk and because of the potential for serious adverse reactions in nursing infants from YERVOY, a decision should be made whether to discontinue nursing or to discontinue YERVOY, taking into account the importance of YERVOY to the mother.
Pediatric Use
Safety and effectiveness of YERVOY have not been established in pediatric patients.
Geriatric Use
Of the 511 patients treated with YERVOY at 3 mg/kg, 28% were 65 years and over. No overall differences in safety or efficacy were reported between the elderly patients (65 years and over) and younger patients (less than 65 years).
Renal Impairment
No formal studies of YERVOY in patients with renal impairment have been conducted. [See Clinical Pharmacology (12.3) in Full Prescribing Information]
Hepatic Impairment
No formal studies of YERVOY in patients with hepatic impairment have been conducted. [See Clinical Pharmacology (12.3) in Full Prescribing Information]
OVERDOSAGE
There is no information on overdosage with YERVOY.
PATIENT COUNSELING INFORMATION
See MEDICATION GUIDE in Full Prescribing Information.
Manufactured by: Bristol-Myers Squibb Company Princeton, NJ 08543 USA
1281558A2 IP-B0001A-03-11 Issued: March 2011

34 I ONCOLOGY PRACTICE MANAGEMENT I December 2011
Drug Coding
Medications Used for the Treatment of Breast CancerThe following sections will assist healthcareprofessionals and payers by providing appro-priate coding information associated withthe management of breast cancer.
This information includes:• Associated ICD-9-CM codes used for
the classification of breast cancer• Drugs that have been FDA-approved
in the treatment of breast cancer• Drugs that are Compendia listed for
off-label use for breast cancer basedon clinical studies that suggest bene-ficial use in some cases.Please note: if a check mark appears inthe FDA column it will NOT appearin the Compendia off-label use col-umn
• Corresponding HCPCS/CPT® codesand code descriptions
• Possible CPT® admnistration codesfor each medication
Associated ICD-9-CM Codes for Breast Cancer174 Malignant neoplasm of female breast
Includes: breast (female)connective tissuesoft partsPaget’s disease of:
breastnipple
Use additional code to identify estrogen receptor status (V86.0, V86.1)
Excludes: skin of the breast (172.5, 173.5)174.0 Nipple and areola174.1 Central portion174.2 Upper-inner quadrant174.3 Lower-inner quadrant174.4 Upper-outer quadrant174.5 Lower-outer quadrant174.6 Axillary tail174.8 Other specified sites of female breast
Ectopic sitesInner breastLower breastMalignant neoplasm of contiguous or overlapping sites of breast whose point of origin cannot be determinedMidline of breastOuter breastUpper breast
174.9 Breast (female), unspecified
175 Malignant neoplasm of male breastUse additional code to identify estrogen receptor status (V86.0, V86.1)
Excludes: skin of breast (172.5, 173.5)175.0 Nipple and areola175.9 Other and unspecified sites of male breast
Ectopic breast tissue, male
Supplied by: RJ Health Systems
Drug Coding
35December 2011 I www.OncPracticeManagement.com I
Supplied by: RJ Health Systems
Compendia listed CPT®
Generic (brand) HCPCS code: FDA-approved off-label use for administration
name code description for breast cancer breast cancer codes
anastrozole (Arimidex) J8999aa: Prescription drug, oral, ✓ N/Achemotherapeutic, not otherwise specified
anastrozole (Arimidex) S0170: Anastrozole, oral, 1 mg ✓ N/A
Bacillus Calmette-Guerin 90585: Bacillus Calmette-Guerin vaccine ✓ 90471(Tice BCG, TheraCys) (BCG) for tuberculosis, live, for
percutaneous use
Bacillus Calmette-Guerin 90586: Bacillus Calmette-Guerin vaccine ✓ 51720(Tice BCG, TheraCys) (BCG) for bladder cancer, live, for
intravesical use
Bacillus Calmette-Guerin J9031: BCG (intravesical), per installation ✓ 51720(Tice BCG, TheraCys)
bevacizumab (Avastin) J9035: Injection, bevacizumab, 10 mg ✓ 96413, 96415
capecitabine (Xeloda) J8520: Capecitabine, oral, 150 mg ✓ N/A
capecitabine (Xeloda) J8521: Capecitabine, oral, 500 mg ✓ N/A
carboplatin (Paraplatin) J9045: Injection, carboplatin, 50 mg ✓ 96409, 96413, 96415
cisplatin (Platinol AQ) J9060: Injection, cisplatin, powder or ✓ 96409, 96413,solution, per 10 mg 96415
cyclophosphamide J8530: Cyclophosphamide, oral, 25 mg ✓ N/A(Cytoxan)
cyclophosphamide J9070: Cyclophosphamide, 100 mg ✓ 96409, 96413,(Cytoxan) 96415
dexamethasone J1100: Injection, dexamethasone sodium ✓ 11900, 11901,(Decadron) phosphate, 1 mg 20600, 20605,
20610, 96372, 96374
dexamethasone J8540: Dexamethasone, oral, 0.25 mg ✓ N/A(Decadron)
docetaxel (Taxotere) J9171: Injection, docetaxel, 1 mg ✓ 96413
doxorubicin HCl J9000: Injection, doxorubicin hydrochloride, ✓ 96409(Adriamycin) 10 mg
doxorubicin HCl J9001: Injection, doxorubicin hydrochloride, ✓ 96413liposome (Doxil) all lipid formulations, 10 mg
epirubicin (Ellence) J9178: Injection, epirubicin HCl, 2 mg ✓ 96409, 96413
eribulin (Halaven) J9179: Injection, eribulin mesylate, 0.1 mg ✓ 96409
estradiol (Estrace) J8499a: Prescription drug, oral, ✓ N/Anonchemotherapeutic,not otherwise specified
etoposide (Vepesid) J8560: Etoposide, oral, 50 mg ✓ N/A
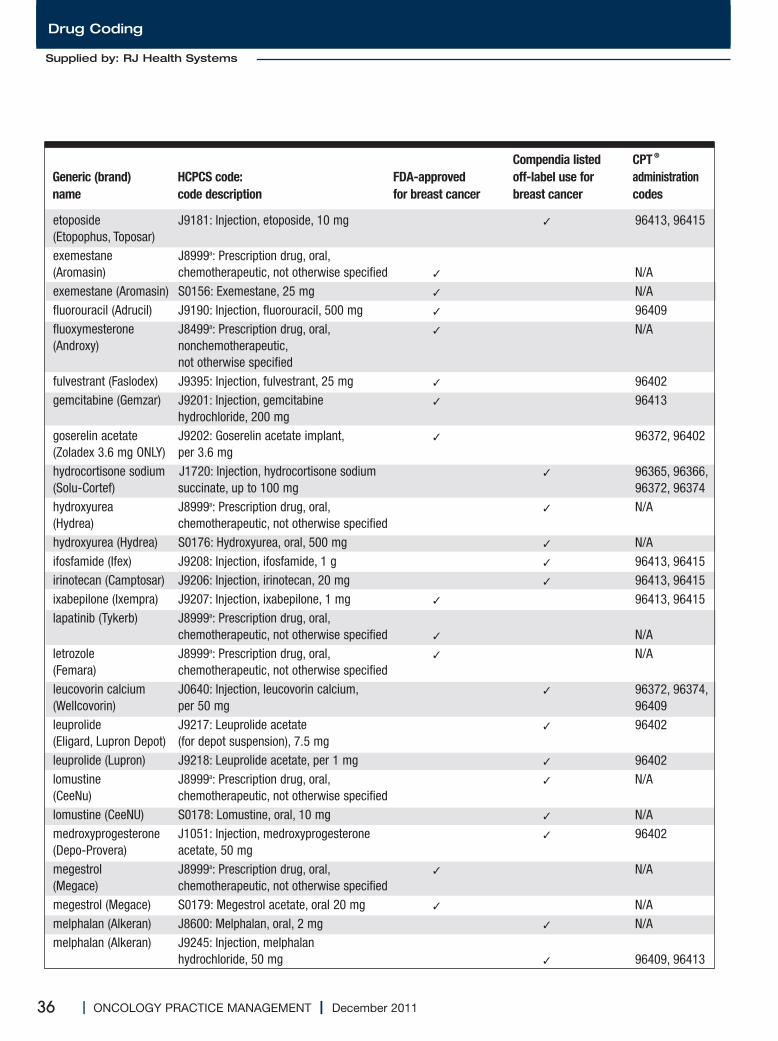
36 I ONCOLOGY PRACTICE MANAGEMENT I December 2011
Drug Coding
Compendia listed CPT®
Generic (brand) HCPCS code: FDA-approved off-label use for administration
name code description for breast cancer breast cancer codes
etoposide J9181: Injection, etoposide, 10 mg ✓ 96413, 96415(Etopophus, Toposar)exemestane J8999a: Prescription drug, oral, (Aromasin) chemotherapeutic, not otherwise specified ✓ N/Aexemestane (Aromasin) S0156: Exemestane, 25 mg ✓ N/Afluorouracil (Adrucil) J9190: Injection, fluorouracil, 500 mg ✓ 96409fluoxymesterone J8499a: Prescription drug, oral, ✓ N/A (Androxy) nonchemotherapeutic,
not otherwise specifiedfulvestrant (Faslodex) J9395: Injection, fulvestrant, 25 mg ✓ 96402gemcitabine (Gemzar) J9201: Injection, gemcitabine ✓ 96413
hydrochloride, 200 mggoserelin acetate J9202: Goserelin acetate implant, ✓ 96372, 96402(Zoladex 3.6 mg ONLY) per 3.6 mghydrocortisone sodium J1720: Injection, hydrocortisone sodium ✓ 96365, 96366, (Solu-Cortef) succinate, up to 100 mg 96372, 96374hydroxyurea J8999a: Prescription drug, oral, ✓ N/A(Hydrea) chemotherapeutic, not otherwise specifiedhydroxyurea (Hydrea) S0176: Hydroxyurea, oral, 500 mg ✓ N/Aifosfamide (Ifex) J9208: Injection, ifosfamide, 1 g ✓ 96413, 96415irinotecan (Camptosar) J9206: Injection, irinotecan, 20 mg ✓ 96413, 96415ixabepilone (Ixempra) J9207: Injection, ixabepilone, 1 mg ✓ 96413, 96415lapatinib (Tykerb) J8999a: Prescription drug, oral,
chemotherapeutic, not otherwise specified ✓ N/Aletrozole J8999a: Prescription drug, oral, ✓ N/A(Femara) chemotherapeutic, not otherwise specifiedleucovorin calcium J0640: Injection, leucovorin calcium, ✓ 96372, 96374,(Wellcovorin) per 50 mg 96409leuprolide J9217: Leuprolide acetate ✓ 96402(Eligard, Lupron Depot) (for depot suspension), 7.5 mgleuprolide (Lupron) J9218: Leuprolide acetate, per 1 mg ✓ 96402lomustine J8999a: Prescription drug, oral, ✓ N/A(CeeNu) chemotherapeutic, not otherwise specifiedlomustine (CeeNU) S0178: Lomustine, oral, 10 mg ✓ N/Amedroxyprogesterone J1051: Injection, medroxyprogesterone ✓ 96402(Depo-Provera) acetate, 50 mgmegestrol J8999a: Prescription drug, oral, ✓ N/A(Megace) chemotherapeutic, not otherwise specifiedmegestrol (Megace) S0179: Megestrol acetate, oral 20 mg ✓ N/Amelphalan (Alkeran) J8600: Melphalan, oral, 2 mg ✓ N/Amelphalan (Alkeran) J9245: Injection, melphalan
hydrochloride, 50 mg ✓ 96409, 96413
Supplied by: RJ Health Systems
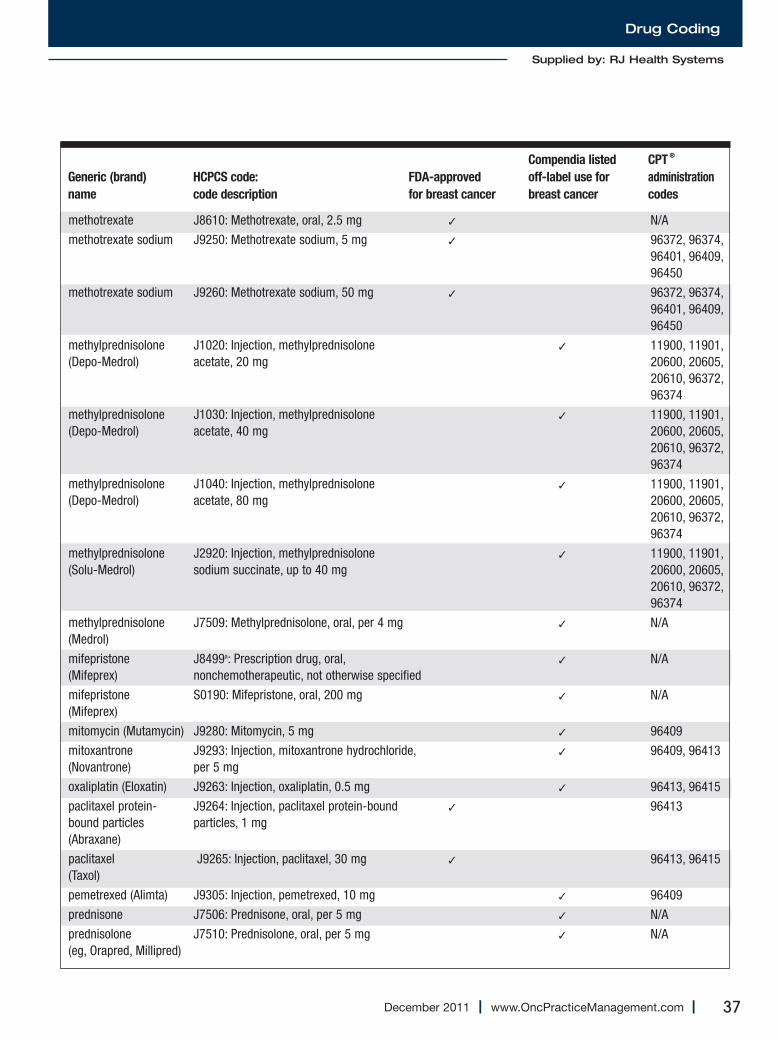
Drug Coding
37December 2011 I www.OncPracticeManagement.com I
Compendia listed CPT®
Generic (brand) HCPCS code: FDA-approved off-label use for administration
name code description for breast cancer breast cancer codes
methotrexate J8610: Methotrexate, oral, 2.5 mg ✓ N/A
methotrexate sodium J9250: Methotrexate sodium, 5 mg ✓ 96372, 96374, 96401, 96409, 96450
methotrexate sodium J9260: Methotrexate sodium, 50 mg ✓ 96372, 96374, 96401, 96409, 96450
methylprednisolone J1020: Injection, methylprednisolone ✓ 11900, 11901,(Depo-Medrol) acetate, 20 mg 20600, 20605,
20610, 96372, 96374
methylprednisolone J1030: Injection, methylprednisolone ✓ 11900, 11901,(Depo-Medrol) acetate, 40 mg 20600, 20605,
20610, 96372, 96374
methylprednisolone J1040: Injection, methylprednisolone ✓ 11900, 11901,(Depo-Medrol) acetate, 80 mg 20600, 20605,
20610, 96372, 96374
methylprednisolone J2920: Injection, methylprednisolone ✓ 11900, 11901,(Solu-Medrol) sodium succinate, up to 40 mg 20600, 20605,
20610, 96372, 96374
methylprednisolone J7509: Methylprednisolone, oral, per 4 mg ✓ N/A(Medrol)
mifepristone J8499a: Prescription drug, oral, ✓ N/A(Mifeprex) nonchemotherapeutic, not otherwise specified
mifepristone S0190: Mifepristone, oral, 200 mg ✓ N/A(Mifeprex)
mitomycin (Mutamycin) J9280: Mitomycin, 5 mg ✓ 96409
mitoxantrone J9293: Injection, mitoxantrone hydrochloride, ✓ 96409, 96413(Novantrone) per 5 mg
oxaliplatin (Eloxatin) J9263: Injection, oxaliplatin, 0.5 mg ✓ 96413, 96415
paclitaxel protein- J9264: Injection, paclitaxel protein-bound ✓ 96413bound particles particles, 1 mg(Abraxane)
paclitaxel J9265: Injection, paclitaxel, 30 mg ✓ 96413, 96415 (Taxol)
pemetrexed (Alimta) J9305: Injection, pemetrexed, 10 mg ✓ 96409
prednisone J7506: Prednisone, oral, per 5 mg ✓ N/A
prednisolone J7510: Prednisolone, oral, per 5 mg ✓ N/A(eg, Orapred, Millipred)
Supplied by: RJ Health Systems
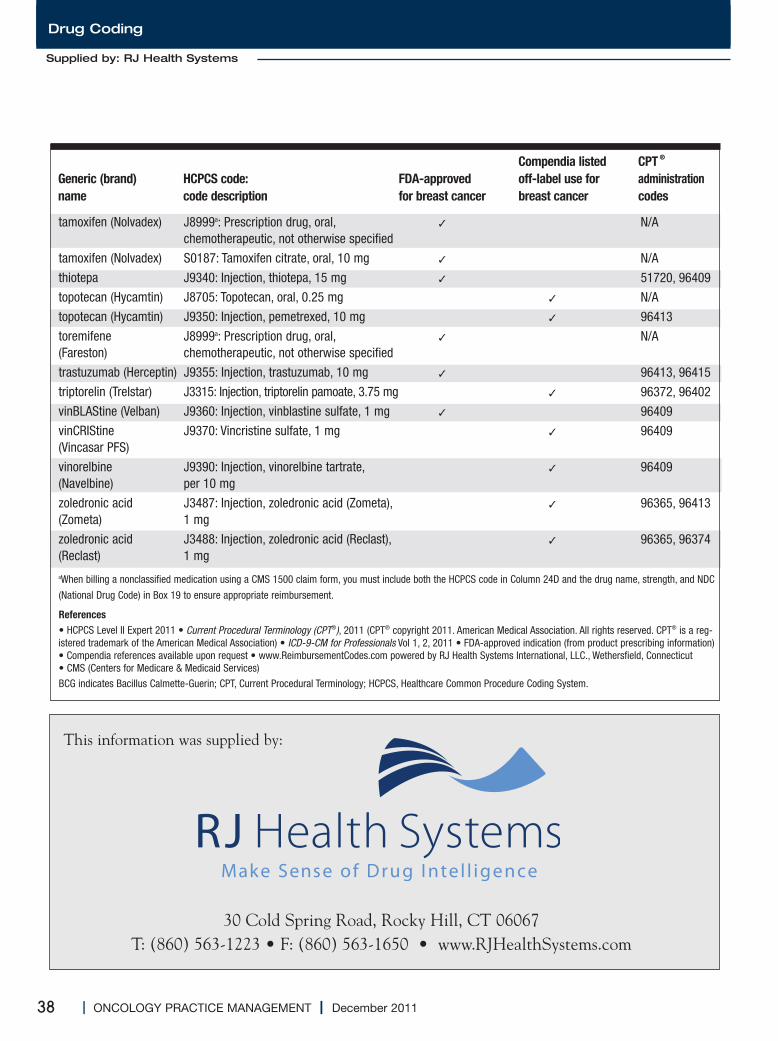
38 I ONCOLOGY PRACTICE MANAGEMENT I December 2011
Drug Coding
30 Cold Spring Road, Rocky Hill, CT 06067 T: (860) 563-1223 • F: (860) 563-1650 • www.RJHealthSystems.com
This information was supplied by:
Compendia listed CPT®
Generic (brand) HCPCS code: FDA-approved off-label use for administration
name code description for breast cancer breast cancer codes
tamoxifen (Nolvadex) J8999a: Prescription drug, oral, ✓ N/Achemotherapeutic, not otherwise specified
tamoxifen (Nolvadex) S0187: Tamoxifen citrate, oral, 10 mg ✓ N/A
thiotepa J9340: Injection, thiotepa, 15 mg ✓ 51720, 96409
topotecan (Hycamtin) J8705: Topotecan, oral, 0.25 mg ✓ N/A
topotecan (Hycamtin) J9350: Injection, pemetrexed, 10 mg ✓ 96413
toremifene J8999a: Prescription drug, oral, ✓ N/A(Fareston) chemotherapeutic, not otherwise specified
trastuzumab (Herceptin) J9355: Injection, trastuzumab, 10 mg ✓ 96413, 96415
triptorelin (Trelstar) J3315: Injection, triptorelin pamoate, 3.75 mg ✓ 96372, 96402
vinBLAStine (Velban) J9360: Injection, vinblastine sulfate, 1 mg ✓ 96409
vinCRIStine J9370: Vincristine sulfate, 1 mg ✓ 96409(Vincasar PFS)
vinorelbine J9390: Injection, vinorelbine tartrate, ✓ 96409(Navelbine) per 10 mg
zoledronic acid J3487: Injection, zoledronic acid (Zometa), ✓ 96365, 96413(Zometa) 1 mg
zoledronic acid J3488: Injection, zoledronic acid (Reclast), ✓ 96365, 96374(Reclast) 1 mgaWhen billing a nonclassified medication using a CMS 1500 claim form, you must include both the HCPCS code in Column 24D and the drug name, strength, and NDC
(National Drug Code) in Box 19 to ensure appropriate reimbursement.
References• HCPCS Level II Expert 2011 • Current Procedural Terminology (CPT®), 2011 (CPT® copyright 2011. American Medical Association. All rights reserved. CPT® is a reg-istered trademark of the American Medical Association) • ICD-9-CM for Professionals Vol 1, 2, 2011 • FDA-approved indication (from product prescribing information)• Compendia references available upon request • www.ReimbursementCodes.com powered by RJ Health Systems International, LLC., Wethersfield, Connecticut• CMS (Centers for Medicare & Medicaid Services)
BCG indicates Bacillus Calmette-Guerin; CPT, Current Procedural Terminology; HCPCS, Healthcare Common Procedure Coding System.
Supplied by: RJ Health Systems

RJ Health SystemsThe Creators of ReimbursementCodes.com
>> ReimbursementCodes.com>> Drug Diagnosis Coding>> CMAC>> PartBorPartD.com>> NDC Conversion Database>> Min/Max Dosing>> Advisories>> Notations>> Clinical Consulting
rjhealthsystems.com
RJ Health Systems — the pharmaceutical specialists that healthcare
professionals have turned to since 1983 for their drug information.
We work with over 170 Payor organizations that touch approximately
110 million lives.
RJ Health Systems has established and continuously maintains
a Library of Drug Intelligence that provides the most comprehensive,
trusted, and up-to-date coding and reimbursement information in
the industry.
ReimbursementCodes.com incorporates the CMS HCPCS and AMA
CPT Drug codes into a system that crosswalks each drug code with
the drug’s NDC and brand/generic name.
Please visit www.rjhealthsystems.com to learn more about our
products and services listed below:
10:36 PM

My last articlede sc r ibedhow to be -
gin to determine theamount of individualdisability insuranceavailable. This arti-cle is focused onwhat to look for in apolicy and help youdecide which riders
should be part of your policy.
Noncancellable andGuaranteed RenewableIf you purchase a policy that is
both noncancellable and guaranteedrenewable, you are ensured that thepremium rates and policy provisionswill not be changed. This combina-tion provides the greatest degree ofconsumer protection.
Definition of Total DisabilityWhether you are a physician, an
executive, a practice or hospitaladministrator, or working in anothercapacity, look for a policy that con-tains a true “own-occupation” defi-nition of total disability. This defini-tion pays benefits if you are disabledand are unable to perform the mate-rial and substantial duties of youroccupation, even if you are gainfullyemployed in another occupation.
Residual Disability RiderAlthough “own-occupation” is
the most liberal definition of disabil-ity, it is not the end all. What hap-pens if your physician states that youcan still work in your occupation buthe or she requires that you workfewer days per week or less hours perday? A residual disability rider pro-tects your income by providing ben-
efits proportionate to your loss ofincome in the event you are nottotally disabled. Generally, to qualifyfor residual disability benefits, youmust experience an income loss of15% to 20% or more compared withyour predisability earnings. In addi-tion, if your loss of earnings weregreater than 75% to 80%, then100% of your monthly disabilitybenefit would be paid.This rider is also extremely impor-
tant if you are totally disabled firstand then return to your occupationwith a limited schedule as you recov-er, or if you have a continued loss ofincome, because you were previouslydisabled, even if you are back towork on a full-time basis.Finally, it is imperative that you
do not purchase a policy thatrequires that you be totally disabledfirst to collect benefits under theresidual disability rider. Althoughthis is not the case with individualpolicies, this is very common ingroup policies offered by medical orother professional associations inwhich you may be a member.
Cost of Living AdjustmentRider A Cost of Living Adjustment
(COLA) rider is designed to helpyour benefits keep pace with infla-tion after your disability has lastedfor 12 months. This adjustment canbe a fixed percentage or tied to theConsumer Price Index. Ideally, youwant a COLA that is adjusted annu-ally on a compound interest basis,with no “cap” on the monthly bene-fit. Although expensive, this ridercan provide significant increases toyour monthly benefit if you are dis-abled young. However, if cutting the
cost of coverage is an issue, this maybe the first optional rider to considerexcluding from your policy. Al ter -natively you may use the additionalpremium associated with this rider topurchase a larger monthly benefit ifyou are not already contemplatingthe purchase of the maximummonthly benefit for which you areeligible, based on your income or anyother in-force coverage.
Future Increase Option RiderThis rider is a must for young pro-
fessionals. As your income rises, thisrider provides you with the ability toincrease your disability coverage,without providing evidence of goodhealth. This guarantees that anymedical conditions that developafter your original policy’s purchasewould be fully covered and not sub-ject to new medical underwriting.It is important to know when you
can increase your coverage, as well asby what increments, on any givenoption date. Some companies mayallow you to use your entire optionin one year as long as your then-cur-rent income warrants the increase;others, however, may limit theamount that you can purchase.
Catastrophic DisabilityBenefit RiderIf you become catastrophically
disabled under the terms of the poli-cy and lose the ability to perform 2or more activities of daily livingwithout assistance, become cogni-tively im paired, or become presump-tively disabled, you would receive amonthly benefit in addition to thebase monthly benefit purchased.This additional benefit can be as
40 I ONCOLOGY PRACTICE MANAGEMENT I December 2011
Physician Wealth Management
Disability Insurance Planning for theCancer Care TeamSelecting the Best Riders for Your Policy
With Lawrence B. Keller, CLU, ChFC, CFP®
Continued on page 44

TARGET AUDIENCEThis activity was developed for physicians, nurses, pharmacists, and managed care professionals who areinvolved in the care of patients with cancer.
CONFERENCE GOALThe Association for Value-Based Cancer Care will foster an open dialogue between providers, payers,and/or other members of the oncology team in order for attendees to gain a better understanding ofvarious points of view regarding cost, quality, and access in cancer care.
EDUCATIONAL OBJECTIVES• Discuss the current trends and challenges facing all stakeholders in optimizing value in cancer care
delivery• Define the barriers associated with cost, quality, and access as it relates to healthcare reform and
what solutions are currently being considered• Compare and contrast the different approaches/tools that providers and payers are utilizing to
manage and deliver care collaboratively• Examine the current trends in personalized care and companion diagnostics• Analyze the patient issues around cost, quality, and access to care
DESIGNATION OF CREDIT STATEMENTSPhysician Accreditation – Joint SponsorThe Medical Learning Institute, Inc. (MLI) designates this live activity for a maximum of 13.5 AMAPRA Category 1 Credits™. Physicians should claim only the credit commensurate with the extent oftheir participation in the activity. This activity has been planned and implemented in accordance withthe Essential Areas and policies of the Accreditation Council for Continuing Medical Education(ACCME) through the joint sponsorship of the Medical Learning Institute, Inc. and the Associationfor Value-Based Cancer Care, Inc. The Medical Learning Institute, Inc. is accredited by the ACCMEto provide continuing medical education for physicians.
Registered Nurse DesignationMedical Learning Institute, Inc. (MLI)Provider approved by the California Board of Registered Nursing, Provider Number 15106, for 13.5contact hours.
Registered Pharmacy DesignationMedical Learning Institute, Inc. (MLI) is accredited by the Accreditation Council for Pharmacy Education (ACPE) as a provider of continuing pharmacy education. Completion
of this activity provides for 13.5 contact hours (1.35 CEUs) of continuing education credit.
The One Conference You Can’t AFFORD to Miss!
Second Annual Association for Value-Based Cancer Care ConferenceStrategies for Optimizing Value in Cancer Care Delivery
REGISTER TODAY
SAVE $100
March 29-31, 2012 • JW Marriott • Houston, Texas
This activity is jointly sponsored by Medical Learning Institute, Inc., and theAssociation for Value-Based Cancer Care
REGISTER TODAY at www.regonline.com/avbcc2012
��������� �����
Gary Owens, MDPresident Gary Owens Associates
Al B. Benson III, MD, FACPProfessor of MedicineAssociate Director for Clinical InvestigationsRobert H. Lurie ComprehensiveCancer Center Northwestern University
Burt Zweigenhaft, BSPresident, CEO OncoMed
CONFERENCE CO-CHAIRS
CONFERENCE REGISTRATION
SAVE $100 off full Conference Tuition
REGISTER TODAY FOR ONLY $275at www.regonline.com/avbcc2012
For more information, please visit www.avbcconline.org/2012 or
e-mail [email protected].
� �� �� � �� �

42 I ONCOLOGY PRACTICE MANAGEMENT I December 2011
Oncology Networks
Arecent Kurt Salmon surveyof best practices among thenation’s leading oncology
networks suggests that ownershipcan have significant influence on anetwork’s ability to implement bestpractices, and provides a road mapfor each ownership type to enhanceits level of development.
The study included more than 132inpatient oncology sites withinregional oncology networks acrossthe United States. The participants,who requested confidentiality as acondition for participation, repre-sent some of the largest and mostrespected oncology centers in thecountry.The study found that the most
advanced oncology networks aregoverned through a central oncologynetwork leadership council, havedeveloped highly coordinated cen-tralized systems and resources, andhave implemented common clinicalcare protocol sets across sites andinstantaneous access to patientrecords at any point of care. Theownership model of an oncologynetwork can influence its ability toimplement best practices.
System NetworksSystem networks are perhaps best
organized to achieve an advancedlevel of development. Because thesenetworks have a single commonowner over all sites, they are betterpositioned to develop a centralizedgoverning council with clear ac -countability for goals and direction.Primary control over the networkallows systems to define administra-tive and physician leadership rolesand hire appropriate staff to supportnetwork planning and coordination.The system can mediate the differ-ent interests of individual sites andensure leadership incentives supportthe network, rather than encouragecompetition be tween network par-ticipants.One key benefit of common own-
ership is that the system can allocatefunding to support the oncology net-work. Within the system networks
interviewed, most were funded inthis way. As part of the system, theoncology network competes forresources with other initiatives, suchas other service line developmentand capital investments. To ensure itreceives adequate resources, theoncology network is encouraged toconduct robust planning, defineclear priorities, and articulate re -quired investments and benefits.System networks also have control
over all sites of care, operations, andresources, and thus greater ability tomandate care processes, monitorcompliance in a highly coordinatedfashion, and develop a strong centralgroup of oncology physicians. Systemnetworks can adopt common clinicalcare protocols and processes morerapidly than in other ownershipmodels, a key to improving qualityand consistency of care.
Collaborator NetworksCollaborator networks are driven
by the need to balance various own-ership interests, because they arecomprised of separate owners whocome together to advance oncologycare. As such, these networks requirestronger central governance modelsto coordinate collective directionand initiatives. Within the most advanced of these
networks, a dedicated, centralizedgoverning oncology leadership coun-cil (OLC) exists to ensure that net-work goals are achieved. The OLC isresponsible for defining administra-tive and physician leadership rolesand hiring appropriate staff to supportnetwork planning and coordination.This is typically an independent func-tion of the OLC, with minimal influ-ence from participating members.
Enhancing Oncology NetworksTips for Each Ownership ModelBy Gerard M. Nussbaum and Laura K. Rehfeld
Continued on page 44
Keys to SuccessfulEnhancement of YourOncology Network
• Install network-level leadership withrepresentation (clinical and adminis-trative) from member entities
• Institute multiyear planning withclearly defined priorities, requiredinvestments, and benefits to supportongoing funding and success of majorinitiatives
• In the absence of central, system-levelownership, obtain key site executivesponsorship to help advance networkcohesiveness
• Write agreements that detail roles andresponsibilities, funding commit-ments, and participation in the net-work leadership council to cement col-laborator and hybrid networks andenhance system networks.

VELCADE and Millennium are registered trademarks of Millennium Pharmaceuticals, Inc. Other trademarks are property of their respective owners.
Millennium Pharmaceuticals, Inc., Cambridge, MA 02139
Copyright © 2010, Millennium Pharmaceuticals, Inc. All rights reserved. Printed in USA V-10-0196 11/10

44 I ONCOLOGY PRACTICE MANAGEMENT I December 2011
Oncology Networks
Collaborator networks require addi-tional effort and attention to balancecompetition among sites and ensurenetwork strength.Collaborator networks rely on
funding contributions from each siteto support network operations. Thelevel of funding can be highly vari-able among sites and typically isdetermined by the leadership com-mittee on an annual basis within theprovisions of the affiliation agree-ment. The budget is based on theagreed-upon set of initiatives andresources required to support devel-opment and may be limited by thewillingness of members to fundmajor initiatives. Typically, these networks require
more advanced levels of systems(functional staff, information tech-nology [IT]) as is necessary to sup-port the higher complexity of theoperations within different hospitalsand sites, although actual levels varygreatly.Collaborator networks are chal-
lenged to mandate the adoption ofstandard treatment protocols andcare processes across sites. To addressthis area, some affiliation agreementsdefine the level of required participa-
tion from each member. However, because each site still
operates within its broader organiza-tional context, adopting commonprotocols requires complex, time-consuming processes. These are facil-itated by a central guiding body com-
prised of both network leadership andsite clinicians. Adding complexity tocoordinating clinical care, collabora-tor networks typically have a greaterdiversity of IT systems, because eachsite has adopted the electronic healthrecord of its organization. These networks face significant
challenge and expense in developingsystems (either IT interfaces or staffresources) that can facilitate informa-tion flow within the network, and will
need to dedicate additional leadershiptime and network re sources to miti-gate these barriers.
Hybrid NetworksHybrid networks have wider vari-
ability in their governance models,with success driven by a highly devel-oped, central decision-making coun-cil. These collaborator networks are acombination of a system network andnon-owned or joint venture affiliatesites. Similar to collaborator net-works, hybrid networks benefit fromstrong administrative and physicianleadership directed by an OLC. Theseleaders invest greater effort to balancecompetition within the network andensure overall success.Hybrid networks typically have a
“lead system” plus collaborator sitesthat rely on the lead system to devel-op and sustain re sources while con-tributing financially to ensure re -sources are available.Hybrid networks share the same
challenges as collaborator networkswhen adopting standard treatmentprotocols and care processes acrosssites. Both rely on the need to buildconsensus and protocol adoptionover time, and initially achieveagreement on an overarching qualityplatform as a basis for developingprotocols that become more consis-tent over time. Most hybrid net-works also have multiple IT systems,requiring time, staffing resources,technology, and expense to facilitateinformation flow. l
Gerard M. Nussbaum and Laura K.Rehfeld have more than 30 years ofexperience advising hospital and healthsystem leaders. Kurt Salmon offersstrategic planning for clinical pro-grams; developing cutting-edge, opera-tionally efficient facilities; and design-ing agile information managementenvironments.
Enhancing Oncology Networks…Continued from page 42
Disability Insurance…Continued from page 40
high as $12,000, depending on thespecific insurance carrier fromwhich you purchase the policy.This benefit amount plus the basepolicy, plus other in-force coverage,if any, generally cannot exceed100% of your income.
SummaryThe next, and final, disability
insurance article will offer tips onhow to compare one company’spolicy to another when shoppingfor coverage. This will help you
ultimately to choose the companyand policy that best meet yourindividual needs and goals.
Lawrence B. Keller, CLU, ChFC,CFP®, is the founder of PhysicianFinancial Services, a New York–based firm specializing in income pro-tection and wealth accumulationstrategies for physicians. He can bereached at 516-677-6211 or [email protected] forcomments or questions.
Adding complexity tocoordinating clinicalcare, collaboratornetworks typically havea greater diversity of IT systems.

45December 2011 I www.OncPracticeManagement.com I
We are just now beginning to see some of the lesserknown pieces of the 2010
Patient Protection and AffordableCare Act, such as the Federal Phy -sician Sunshine Act (Social SecurityAct § 1128G), which is designed toilluminate the relationship betweenphysicians and manufacturers. Start -ing by fall 2012, pharmaceutical anddevice manufacturers will be requiredto report payments to physicians andteaching hospitals. These records willbe made public from September 30,2013, onward.Regulating manufacturers. It is
important to note that the SunshineAct regulates manufacturers; it doesnot impose any obligations or re -strictions on healthcare providers. Itrequires manufacturers to report thevalue, nature, purpose, and recipientinformation for any payment to aphysician or teaching hospital. Ingeneral, disclosure is required for allmeals, gifts, consulting payments,and grants; however, there are a fewexceptions. The Sunshine Act calls for manu-
facturers to report any individualexpense of $10 or more, and if thetotal of all payments or gifts to a par-ticular healthcare provider exceeds$100 in a given year, then all pay-ments to that provider must bereported, regardless of the value.Reporting responsibility falls solely tothe pharmaceutical or device manu-facturer, not the individual provider.However, providers will have theopportunity to correct inaccuratereports before they are made public.ACCC members’ interaction
with manufacturers. Many mem-bers of the Association of Com -munity Cancer Centers (ACCC)
are curious how this new law willimpact their interaction with manu-facturers. Sales representatives canstill visit physicians in their offices,and they may still provide a cup ofcoffee or small snack to providers ata conference. However, because of
the relatively low spending limit, adrug or device company will have topublicly report taking a physicianout to dinner at a restaurant to dis-cuss a new product line. Somehealthcare providers have writtenagreements with pharmaceuticalcompanies to serve on scientificadvisory boards. Reporting reimbursement agree-
ments. Written agreements provid-ing reasonable compensation andreimbursement in connection withbona fide advisory board services arestill permissible, but they will bereported and made public. Similarly,payments and reimbursement relatedto clinical trials are subject to report-ing, but a provision in the Sunshine
Act delays the publicdisclosure of clinicaltrial payments for upto 4 years. A pharmaceutical
or device companymay make a grant tothe hospital where aphysician works tohost a continuingmed ical educationprogram, but that grant would bereported and disclosed to the publicif it were made to a teaching hospitalor to another entity at the request ofa teaching hospital.Similar laws already on the books
in some states.Although the federalSunshine Act may change health-care providers’ interactions withdrug and device man ufacturers, it iscertainly not the first law of its kind.Seven states and the District ofColumbia already have laws on thebooks that impose marketing restric-tions and disclosure requirements onmanufacturers’ interactions withproviders and institutions. The lawsvary in scope, but all place reportingresponsibility and restrictions onmanufacturers. They can be found inCalifornia, Connecticut, Massachu -setts, Minnesota, Nevada, Vermont,West Virginia, and the District ofColumbia. The federal SunshineAct, like the state laws, arose from ageneral commitment to transparen-cy in relationships between industryand physicians—shining a light onthese interactions. Further guidance on the Sunshine
Act was expected from the Centersfor Medicare & Medicaid Services inearly October, but as of the writing ofthis article in November 2011, thatfinal guidance has yet to be released. l
Patient and Provider Access
Brought to you by the Association of Community Cancer Centers
Will the Sunshine Act AffectPhysician–Pharmaceutical Interaction? By Sydney Abbott, JD
Starting by fall 2012,pharmaceutical anddevice manufacturerswill be required toreport payments tophysicians andteaching hospitals.These records will bemade public fromSeptember 30, 2013,onward.

DOXIL®(doxorubicin HCl liposome injection) for intravenous infusionBRIEF SUMMARY. Please see Full Prescribing Information.
INDICATIONS AND USAGE: Ovarian Cancer: DOXIL (doxorubicin HCl liposome injection) is indicated for thetreatment of patients with ovarian cancer whose disease has progressed or recurred after platinum-basedchemotherapy. AIDS-Related Kaposi’s Sarcoma: DOXIL is indicated for the treatment of AIDS-relatedKaposi’s sarcoma in patients after failure of prior systemic chemotherapy or intolerance to such therapy.Multiple Myeloma: DOXIL in combination with bortezomib is indicated for the treatment of patients withmultiple myeloma who have not previously received bortezomib and have received at least one prior therapy.
CONTRAINDICATIONS: DOXIL (doxorubicin HCl liposome injection) is contraindicated in patients whohave a history of hypersensitivity reactions to a conventional formulation of doxorubicin HCl or thecomponents of DOXIL [see Warnings and Precautions]. DOXIL is contraindicated in nursing mothers [seeFull Prescribing Information].
WARNINGS AND PRECAUTIONS: Cardiac Toxicity: Special attention must be given to the risk ofmyocardial damage from cumulative doses of doxorubicin HCl. Acute left ventricular failure may occurwith doxorubicin, particularly in patients who have received a total cumulative dosage of doxorubicinexceeding the currently recommended limit of 550 mg/m2. Lower (400 mg/m2) doses appear to cause heartfailure in patients who have received radiotherapy to the mediastinal area or concomitant therapy withother potentially cardiotoxic agents such as cyclophosphamide. Prior use of other anthracyclines oranthracenodiones should be included in calculations of total cumulative dosage. Congestive heart failureor cardiomyopathy may be encountered after discontinuation of anthracycline therapy. Patients with ahistory of cardiovascular disease should be administered DOXIL only when the potential benefit oftreatment outweighs the risk. Cardiac function should be carefully monitored in patients treated withDOXIL. The most definitive test for anthracycline myocardial injury is endomyocardial biopsy. Othermethods, such as echocardiography or multigated radionuclide scans, have been used to monitor cardiacfunction during anthracycline therapy. Any of these methods should be employed to monitor potentialcardiac toxicity in patients treated with DOXIL. If these test results indicate possible cardiac injuryassociated with DOXIL therapy, the benefit of continued therapy must be carefully weighed against therisk of myocardial injury. In a clinical study in patients with advanced breast cancer, 250 patients receivedDOXIL at starting dose of 50 mg/m2 every 4 weeks. At all cumulative anthracycline doses between 450-500 mg/m2, or between 500–550 mg/m2, the risk of cardiac toxicity for patients treated with DOXIL was11%. In this study, cardiotoxicity was defined as a decrease of >20% from baseline if the resting leftventricular ejection fraction (LVEF) remained in the normal range, or a decrease of >10% if the resting LVEFbecame abnormal (less than the institutional lower limit of normal). The data on left ventricular ejectionfraction (LVEF) defined cardiotoxicity and congestive heart failure (CHF) are in the table below.
Table 1: Number of Patients With Advanced Breast CancerDOXIL (n=250)
Patients who Developed Cardiotoxicity 10(LVEF Defined)
Cardiotoxicity (With Signs & Symptoms of CHF) 0Cardiotoxicity (no Signs & Symptoms of CHF) 10
Patients With Signs and Symptoms of CHF Only 2
In the randomized multiple myeloma study, the incidence of heart failure events (ventricular dysfunction,cardiac failure, right ventricular failure, congestive cardiac failure, chronic cardiac failure, acute pulmonaryedema and pulmonary edema) was similar in the DOXIL+bortezomib group and the bortezomib monotherapygroup, 3% in each group. LVEF decrease was defined as an absolute decrease of 15% over baseline or a
5% decrease below the institutional lower limit of normal. Based on this definition, 25 patients in thebortezomib arm (8%) and 42 patients in the DOXIL+bortezomib arm (13%) experienced a reduction in LVEF.
Infusion Reactions: Acute infusion-related reactions were reported in 7.1% of patients treated withDOXIL in the randomized ovarian cancer study. These reactions were characterized by one or more ofthe following symptoms: flushing, shortness of breath, facial swelling, headache, chills, chest pain, backpain, tightness in the chest and throat, fever, tachycardia, pruritus, rash, cyanosis, syncope,bronchospasm, asthma, apnea, and hypotension. In most patients, these reactions resolve over thecourse of several hours to a day once the infusion is terminated. In some patients, the reaction resolvedwhen the rate of infusion was slowed. In this study, two patients treated with DOXIL (0.8%) discontinueddue to infusion-related reactions. In clinical studies, six patients with AIDS-related Kaposi’s sarcoma(0.9%) and 13 (1.7%) solid tumor patients discontinued DOXIL therapy because of infusion-relatedreactions. Serious and sometimes life-threatening or fatal allergic/anaphylactoid-like infusion reactionshave been reported. Medications to treat such reactions, as well as emergency equipment, should beavailable for immediate use. The majority of infusion-related events occurred during the first infusion.Similar reactions have not been reported with conventional doxorubicin and they presumably representa reaction to the DOXIL liposomes or one of its surface components. The initial rate of infusion should be1 mg/min to help minimize the risk of infusion reactions [see Full Prescribing Information].
Myelosuppression: Because of the potential for bone marrow suppression, careful hematologicmonitoring is required during use of DOXIL, including white blood cell, neutrophil, platelet counts, andHgb/Hct. With the recommended dosage schedule, leukopenia is usually transient. Hematologic toxicitymay require dose reduction or delay or suspension of DOXIL therapy. Persistent severe myelosuppressionmay result in superinfection, neutropenic fever, or hemorrhage. Development of sepsis in the setting ofneutropenia has resulted in discontinuation of treatment and, in rare cases, death. DOXIL may potentiatethe toxicity of other anticancer therapies. In particular, hematologic toxicity may be more severe whenDOXIL is administered in combination with other agents that cause bone marrow suppression. In patientswith relapsed ovarian cancer, myelosuppression was generally moderate and reversible. In the threesingle-arm studies, anemia was the most common hematologic adverse reaction (52.6%), followed byleukopenia (WBC <4,000 mm3; 42.2%), thrombocytopenia (24.2%), and neutropenia (ANC <1,000; 19.0%). Inthe randomized study, anemia was the most common hematologic adverse reaction (40.2%), followed byleukopenia (WBC <4,000 mm3; 36.8%), neutropenia (ANC <1,000; 35.1%), and thrombocytopenia (13.0%)[see Adverse Reactions]. In patients with relapsed ovarian cancer, 4.6% received G-CSF (or GM-CSF) tosupport their blood counts [see Full Prescribing Information]. For patients with AIDS-related Kaposi’ssarcoma who often present with baseline myelosuppression due to such factors as their HIV disease orconcomitant medications, myelosuppression appears to be the dose-limiting adverse reaction at therecommended dose of 20 mg/m2 [see Adverse Reactions]. Leukopenia is the most common adversereaction experienced in this population; anemia and thrombocytopenia can also be expected. Sepsisoccurred in 5% of patients; for 0.7% of patients the event was considered possibly or probably related toDOXIL. Eleven patients (1.6%) discontinued study because of bone marrow suppression or neutropenia.Table 10 presents data on myelosuppression in patients with multiple myeloma receiving DOXIL andbortezomib in combination [see Adverse Reactions].
Hand-Foot Syndrome (HFS): In the randomized ovarian cancer study, 50.6% of patients treated with DOXILat 50 mg/m2 every 4 weeks experienced HFS (developed palmar-plantar skin eruptions characterized byswelling, pain, erythema and, for some patients, desquamation of the skin on the hands and the feet), with23.8% of the patients reporting HFS Grade 3 or 4 events. Ten subjects (4.2%) discontinued treatment dueto HFS or other skin toxicity. HFS toxicity grades are described in Dosage and Administration section [seeFull Prescribing Information]. Among 705 patients with AIDS-related Kaposi’s sarcoma treated with DOXILat 20 mg/m2 every 2 weeks, 24 (3.4%) developed HFS, with 3 (0.9%) discontinuing. In the randomizedmultiple myeloma study, 19% of patients treated with DOXIL at 30 mg/m2 every three weeks experiencedHFS. HFS was generally observed after 2 or 3 cycles of treatment but may occur earlier. In most patientsthe reaction is mild and resolves in one to two weeks so that prolonged delay of therapy need not occur.However, dose modification may be required to manage HFS [see Full Prescribing Information]. Thereaction can be severe and debilitating in some patients and may require discontinuation of treatment.
Radiation Recall Reaction: Recall reaction has occurred with DOXIL administration after radiotherapy.
Fetal Mortality: Pregnancy Category D: DOXIL can cause fetal harm when administered to a pregnantwoman. There are no adequate and well-controlled studies in pregnant women. If DOXIL is to be usedduring pregnancy, or if the patient becomes pregnant during therapy, the patient should be apprised ofthe potential hazard to the fetus. If pregnancy occurs in the first few months following treatment withDOXIL, the prolonged half-life of the drug must be considered. Women of childbearing potential shouldbe advised to avoid pregnancy during treatment with Doxil. [see Full Prescribing Information].
Toxicity Potentiation: The doxorubicin in DOXIL may potentiate the toxicity of other anticancer therapies.Exacerbation of cyclophosphamide-induced hemorrhagic cystitis and enhancement of the hepatotoxicityof 6-mercaptopurine have been reported with the conventional formulation of doxorubicin HCl. Radiation-induced toxicity to the myocardium, mucosae, skin, and liver have been reported to be increased by theadministration of doxorubicin HCl.
Monitoring: Laboratory Tests: Complete blood counts, including platelet counts, should be obtainedfrequently and at a minimum prior to each dose of DOXIL [see Warnings and Precautions].
ADVERSE REACTIONS: Overall Adverse Reactions Profile: The following adverse reactions are discussedin more detail in other sections of the labeling. • Cardiac Toxicity [see Warnings and Precautions] • Infusionreactions [see Warnings and Precautions] • Myelosuppression [see Warnings and Precautions] • Hand-Foot syndrome [see Warnings and Precautions]
The most common adverse reactions observed with DOXIL are asthenia, fatigue, fever, nausea, stomatitis,vomiting, diarrhea, constipation, anorexia, hand-foot syndrome, rash and neutropenia, thrombocytopeniaand anemia. The most common serious adverse reactions observed with DOXIL are described in SectionAdverse Reactions in Clinical Trials. The safety data described below reflect exposure to DOXIL in 1310patients including: 239 patients with ovarian cancer, 753 patients with AIDS-related Kaposi’s sarcoma and318 patients with multiple myeloma.
Adverse Reactions in Clinical Trials: Because clinical trials are conducted under widely varyingconditions, the adverse reaction rates observed cannot be directly compared to rates on other clinicaltrials and may not reflect the rates observed in clinical practice. The following tables present adversereactions from clinical trials of DOXIL in ovarian cancer, AIDS-Related Kaposi’s sarcoma, and multiplemyeloma.
Patients With Ovarian Cancer: The safety data described below are from 239 patients with ovariancancer treated with DOXIL (doxorubicin HCl liposome injection) at 50 mg/m2 once every 4 weeks for aminimum of 4 courses in a randomized, multicenter, open-label study. In this study, patients receivedDOXIL for a median number of 98.0 days (range 1-785 days). The population studied was 27-87 years ofage, 91% Caucasian, 6% Black and 3% Hispanic and other. Table 2 presents the hematologic adversereactions from the randomized study of DOXIL compared to topotecan.
Table 2: Ovarian Cancer Randomized Study Hematology Data Reported in Patients With Ovarian CancerDOXIL Topotecan
Patients Patients(n = 239) (n = 235)
Neutropenia500 - <1000/mm3 19 (7.9%) 33 (14.0%)<500/mm3 10 (4.2%) 146 (62.1%)
Anemia6.5 - <8 g/dL 13 (5.4%) 59 (25.1%)<6.5 g/dL 1 (0.4%) 10 (4.3%)
Thrombocytopenia10,000 - <50,000/mm3 3 (1.3%) 40 (17.0%)<10,000/mm3 0 (0.0%) 40 (17.0%)
Table 3 presents a comparative profile of the non-hematologic adverse reactions from the randomizedstudy of DOXIL compared to topotecan.
WARNING: INFUSION REACTIONS, MYELOSUPPRESSION, CARDIOTOXICITY, LIVER IMPAIRMENT,ACCIDENTAL SUBSTITUTION1. The use of DOXIL (doxorubicin HCl liposome injection) may lead to cardiac toxicity. Myocardialdamage may lead to congestive heart failure and may occur as the total cumulative dose of doxorubicinHCl approaches 550 mg/m2. In a clinical study in patients with advanced breast cancer, 250 patients received DOXIL at a starting dose of 50 mg/m2 every 4 weeks. At all cumulativeanthracycline doses between 450-500 mg/m2 or between 500-550 mg/m2, the risk of cardiac toxicity forpatients treated with DOXIL was 11%. Prior use of other anthracyclines or anthracenediones should beincluded in calculations of total cumulative dosage. Cardiac toxicity may also occur at lowercumulative doses in patients with prior mediastinal irradiation or who are receiving concurrentcyclophosphamide therapy [see Warnings and Precautions]. 2. Acute infusion-related reactionsincluding, but not limited to, flushing, shortness of breath, facial swelling, headache, chills, backpain, tightness in the chest or throat, and/or hypotension have occurred in up to 10% of patientstreated with DOXIL. In most patients, these reactions resolve over the course of several hours to aday once the infusion is terminated. In some patients, the reaction has resolved with slowing of theinfusion rate. Serious and sometimes life-threatening or fatal allergic/anaphylactoid-like infusionreactions have been reported. Medications to treat such reactions, as well as emergency equipment,should be available for immediate use. DOXIL should be administered at an initial rate of 1 mg/minto minimize the risk of infusion reactions [see Warnings and Precautions]. 3. Severemyelosuppression may occur [see Warnings and Precautions]. 4. Dosage should be reduced inpatients with impaired hepatic function [see Full Prescribing Information]. 5. Accidental substitutionof DOXIL for doxorubicin HCl has resulted in severe side effects. DOXIL should not be substituted fordoxorubicin HCl on a mg per mg basis [see Full Prescribing Information].
3:28 PM

Table 3: Ovarian Cancer Randomized StudyNon-Hematologic DOXIL (%) treated Topotecan (%) treatedAdverse Reaction 10% or Greater (n = 239) (n =235)
All Grades All Grades grades 3-4 grades 3-4
Body as a WholeAsthenia 40.2 7.1 51.5 8.1Fever 21.3 0.8 30.6 5.5Mucous Membrane 14.2 3.8 3.4 0DisorderBack Pain 11.7 1.7 10.2 0.9Infection 11.7 2.1 6.4 0.9Headache 10.5 0.8 14.9 0
DigestiveNausea 46.0 5.4 63.0 8.1Stomatitis 41.4 8.3 15.3 0.4Vomiting 32.6 7.9 43.8 9.8Diarrhea 20.9 2.5 34.9 4.2Anorexia 20.1 2.5 21.7 1.3Dyspepsia 12.1 0.8 14.0 0
Nervous Dizziness 4.2 0 10.2 0
RespiratoryPharyngitis 15.9 0 17.9 0.4Dyspnea 15.1 4.1 23.4 4.3Cough increased 9.6 0 11.5 0
Skin and AppendagesHand-foot syndrome 50.6 23.8 0.9 0Rash 28.5 4.2 12.3 0.4Alopecia 19.2 N/A 52.3 N/A
The following additional adverse reactions (not in table) were observed in patients with ovarian cancerwith doses administered every four weeks. Incidence 1% to 10%: Cardiovascular: vasodilation, tachycardia, deep thrombo phlebitis, hypotension,cardiac arrest. Digestive: oral moniliasis, mouth ulceration, esophagitis, dysphagia, rectal bleeding, ileus.Hemic and Lymphatic: ecchymosis. Metabolic and Nutritional: dehydration, weight loss,hyperbilirubinemia, hypokalemia, hypercalcemia, hyponatremia. Nervous: somnolence, dizziness,depression. Respiratory: rhinitis, pneumonia, sinusitis, epistaxis. Skin and Appendages: pruritus, skindiscoloration, vesiculobullous rash, maculopapular rash, exfoliative dermatitis, herpes zoster, dry skin,herpes simplex, fungal dermatitis, furunculosis, acne. Special Senses: conjunctivitis, taste perversion,dry eyes. Urinary: urinary tract infection, hematuria, vaginal moniliasis.Patients With AIDS-Related Kaposi’s Sarcoma: The safety data below is based on the experiencereported in 753 patients with AIDS-related Kaposi’s sarcoma enrolled in four studies. The median age ofthe population was 38.7 years (range 24-70 years), which was 99% male, 1% female, 88% Caucasian, 6%Hispanic, 4% Black, and 2% Asian/other/unknown. The majority of patients were treated with 20 mg/m2
of DOXIL every two to three weeks. The median time on study was 127 days and ranged from 1 to 811days. The median cumulative dose was 120 mg/m2 and ranged from 3.3 to 798.6 mg/m2. Twenty-sixpatients (3.0%) received cumulative doses of greater than 450 mg/m2. Of these 753 patients, 61.2% wereconsidered poor risk for KS tumor burden, 91.5% poor for immune system, and 46.9% for systemic illness;36.2% were poor risk for all three categories. Patients’ median CD4 count was 21.0 cells/mm3, with 50.8%of patients having less than 50 cells/mm3. The mean absolute neutrophil count at study entry wasapproximately 3,000 cells/mm3. Patients received a variety of potentially myelotoxic drugs in combinationwith DOXIL. Of the 693 patients with concomitant medication information, 58.7% were on one or moreantiretroviral medications; 34.9% patients were on zidovudine (AZT), 20.8% on didanosine (ddI), 16.5% onzalcitabine (ddC), and 9.5% on stavudine (D4T). A total of 85.1% patients were on PCP prophylaxis, most(54.4%) on sulfamethoxazole/trimethoprim. Eighty-five percent of patients were receiving antifungalmedications, primarily fluconazole (75.8%). Seventy-two percent of patients were receiving antivirals,56.3% acyclovir, 29% ganciclovir, and 16% foscarnet. In addition, 47.8% patients received colony-stimulating factors (sargramostim/filgrastim) sometime during their course of treatment. Adversereactions led to discontinuation of treatment in 5% of patients with AIDS related Kaposi’s sarcoma. Thosethat did so included bone marrow suppression, cardiac adverse reactions, infusion-related reactions,toxoplasmosis, HFS, pneumonia, cough/dyspnea, fatigue, optic neuritis, progression of a non-KS tumor,allergy to penicillin, and unspecified reasons.Table 4: Hematology Data Reported in Patients With AIDS-Related Kaposi’s Sarcoma
Patients With Total PatientsRefractory or Intolerant With
AIDS-Related AIDS-RelatedKaposi’s Sarcoma Kaposi’s Sarcoma
(n = 74) (n = 720)Neutropenia
1000/mm3 34 (45.9%) 352 (48.9%)500/mm3 8 (10.8%) 96 (13.3%)
Anemia10 g/dL 43 (58.1%) 399 (55.4%)8 g/dL 12 (16.2%) 131 (18.2%)
Thrombocytopenia150,000/mm3 45 (60.8%) 439 (60.9%)25,000/mm3 1 (1.4%) 30 (4.2%)
Table 5: Probably and Possibly Drug-Related Non-Hematologic Adverse Reactions Reported in 5%of Patients With AIDS-Related Kaposi’s Sarcoma
Adverse Patients With Total PatientsReactions Refractory or Intolerant With
AIDS-Related AIDS-RelatedKaposi’s Sarcoma Kaposi’s Sarcoma
(n = 77) (n = 705)Nausea 14 (18.2%) 119 (16.9%)Asthenia 5 (6.5%) 70 (9.9%)Fever 6 (7.8%) 64 (9.1%)Alopecia 7 (9.1%) 63 (8.9%)Alkaline Phosphatase 1 (1.3%) 55 (7.8%)Increase Vomiting 6 (7.8%) 55 (7.8%)Diarrhea 4 (5.2%) 55 (7.8%)Stomatitis 4 (5.2%) 48 (6.8%)Oral Moniliasis 1 (1.3%) 39 (5.5%)
The following additional (not in table) adverse reactions were observed in patients with AIDS-relatedKaposi’s sarcoma. Incidence 1% to 5%: Body as a Whole: headache, back pain, infection, allergicreaction, chills. Cardiovascular: chest pain, hypotension, tachycardia. Cutaneous: herpes simplex, rash,itching. Digestive: mouth ulceration, anorexia, dysphagia. Metabolic and Nutritional: SGPT increase,weight loss, hyperbilirubinemia. Other: dyspnea, pneumonia, dizziness, somnolence. Incidence Less Than 1%: Body As A Whole: sepsis, moniliasis, cryptococcosis. Cardiovascular: thrombophlebitis,cardiomyopathy, palpitation, bundle branch block, congestive heart failure, heart arrest, thrombosis,ventricular arrhythmia. Digestive: hepatitis. Metabolic and Nutritional Disorders: dehydration.Respiratory: cough increase, pharyngitis. Skin and Appendages: maculopapular rash, herpes zoster.Special Senses: taste perversion, conjunctivitis.Patients With Multiple Myeloma: The safety data below are from 318 patients treated with DOXIL (30 mg/m2 as a 1-hr i.v. infusion) administered on day 4 following bortezomib (1.3 mg/m2 i.v. bolus on days1, 4, 8 and 11) every three weeks, in a randomized, open-label, multicenter study. In this study, patients inthe DOXIL + bortezomib combination group were treated for a median number of 138 days (range 21-410days). The population was 28-85 years of age, 58% male, 42% female, 90% Caucasian, 6% Black, and 4%Asian and other. Table 6 lists adverse reactions reported in 10% or more of patients treated with DOXIL incombination with bortezomib for multiple myeloma. Table 6: Frequency of treatment emergent adverse reactions reported in 10% patients treated for
multiple myeloma with DOXIL in combination with bortezomib, by Severity, Body System, and MedDRA Terminology.
Adverse DOXIL + bortezomib BortezomibReaction (n=318) (n=318)
Any Grade Grade Any Grade Grade(%) 3 4 (%) 3 4
Blood and lymphatic system disordersNeutropenia 36 22 10 22 11 5Thrombocytopenia 33 11 13 28 9 8Anemia 25 7 2 21 8 2General disorders and administration site conditionsFatigue 36 6 1 28 3 0Pyrexia 31 1 0 22 1 0Asthenia 22 6 0 18 4 0Gastrointestinal disordersNausea 48 3 0 40 1 0Diarrhea 46 7 0 39 5 0Vomiting 32 4 0 22 1 0Constipation 31 1 0 31 1 0
Abdominal pain 11 1 0 8 1 0Infections and infestationsHerpes zoster 11 2 0 9 2 0Herpes simplex 10 0 0 6 1 0InvestigationsWeight decreased 12 0 0 4 0 0Metabolism and Nutritional disorders
Nervous system disorders
Neuralgia 17 3 0 20 4 1
Respiratory, thoracic and mediastinal disordersCough 18 0 0 12 0 0Skin and subcutaneous tissue disordersRash** 22 1 0 18 1 0
*Peripheral neuropathy includes the following adverse reactions: peripheral sensory neuropathy,neuropathy peripheral, polyneuropathy, peripheral motor neuropathy, and neuropathy NOS.
**Rash includes the following adverse reactions: rash, rash erythematous, rash macular, rash maculo-papular, rash pruritic, exfoliative rash, and rash generalized.
Post Marketing Experience: The following additional adverse reactions have been identified during postapproval use of DOXIL. Because these reactions are reported voluntarily from a population of uncertain size,it is not always possible to reliably estimate their frequency or establish a causal relationship to drugexposure. Musculoskeletal and Connective Tissue Disorders: rare cases of muscle spasms. Respiratory,Thoracic and Mediastinal Disorders: rare cases of pulmonary embolism (in some cases fatal). Hematologicdisorders: Secondary acute myelogenous leukemia with and without fatal outcome has been reported inpatients whose treatment included DOXIL. Skin and subcutaneous tissue disorders: rare cases of erythemamultiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis have been reported.DRUG INTERACTIONS: No formal drug interaction studies have been conducted with DOXIL. DOXIL mayinteract with drugs known to interact with the conventional formulation of doxorubicin HCl.USE IN SPECIFIC POPULATIONS: Pediatric Use: The safety and effectiveness of DOXIL in pediatricpatients have not been established.Geriatric Use: Of the patients treated with DOXIL in the randomized ovarian cancer study, 34.7% (n=83)were 65 years of age or older while 7.9% (n=19) were 75 years of age or older. Of the 318 patients treatedwith DOXIL in combination with bortezomib for multiple myeloma, 37% were 65 years of age or older and8% were 75 years of age or older. No overall differences in safety or efficacy were observed betweenthese patients and younger patients.OVERDOSAGE: Acute overdosage with doxorubicin HCl causes increases in mucositis, leucopenia, andthrombocytopenia. Treatment of acute overdosage consists of treatment of the severelymyelosuppressed patient with hospitalization, antibiotics, platelet and granulocyte transfusions, andsymptomatic treatment of mucositis.
0016716-5BManufactured by:Ben Venue Laboratories, Inc.Bedford, OH 44146Distributed by:Ortho Biotech Products, LPRaritan, NJ 08869-0670
STEALTH® and DOXIL® are registered trademarks of ALZA Corporation.
TM
An ALZA STEALTH®
Technology Product

Invite your patients to enroll in DOXIL® C.A.R.E.S.: 1-877-CARES-49 (1-877-227-3749) Monday to Friday, 9:30 AM to 10:00 PM (ET). To learn more, visit www.doxil.com.
DOXIL® C.A.R.E.S. Complements the Efforts of the Healthcare Team With:� Treatment follow-up
— Regularly scheduled telephone calls from oncology nurses to review lifestyle tips to help manage potential side effects of DOXIL® therapy such as hand-foot syndrome and stomatitis
� Patient education materials
Distributed by: Centocor Ortho Biotech Products, L.P., Horsham, Pennsylvania 19044-3607 © Centocor Ortho Biotech Products, L.P. 2010 9/10 08D10019B
For your patients receiving or about to start treatment with DOXIL® for recurrent ovarian cancer or relapsed/refractory multiple myeloma…
INDICATIONS� DOXIL® is indicated for the treatment of patients with ovarian cancer whose disease
has progressed or recurred after platinum-based chemotherapy� DOXIL® in combination with VELCADE® (bortezomib) is indicated for the treatment of
patients with multiple myeloma who have not previously received VELCADE and have received at least one prior therapy
IMPORTANT SAFETY INFORMATIONBOXED WARNINGSCardiotoxicity, infusion reaction, myelosuppression, liver impairment, substitution � The use of DOXIL® may lead to cardiac toxicity. Myocardial damage may lead
to congestive heart failure and may occur as the total cumulative dose of doxorubicin HCl approaches 550 mg/m2
— Prior use of other anthracyclines or anthracenediones should be included in calculations of total cumulative dose
— Cardiac toxicity may also occur at lower cumulative doses (400 mg/m2) in patients with prior mediastinal irradiation or who are receiving concurrent cyclophosphamide therapy
� Acute infusion-related reactions including, but not limited to, flushing, shortness of breath, facial swelling, headache, chills, back pain, tightness in the chest or throat, and/or hypotension have occurred in up to 10% of patients treated with DOXIL®. In most patients, these reactions have resolved within several hours to a day once the infusion is terminated. In some patients, reactions resolved with slowing of the infusion rate
— Serious and sometimes life-threatening or fatal allergic/anaphylactoid-like infusion reactions have occurred. Medications to treat such reactions, as well as emergency equipment, should be available for immediate use
— The initial rate of infusion should be 1 mg/min to minimize the risk of infusion reactions
� Severe myelosuppression may occur� DOXIL® dosage should be reduced in patients with impaired hepatic function� Accidental substitution has resulted in severe side effects. Do not substitute
for doxorubicin HCl on a mg per mg basis
CONTRAINDICATIONS� Patients with a history of hypersensitivity reactions to a conventional doxorubicin
formulation or the components of DOXIL®
� Nursing mothers
ADDITIONAL SAFETY INFORMATION� Cardiac function should be carefully monitored — Congestive heart failure or cardiomyopathy may occur after discontinuation of
anthracycline therapy — For patients with a history of cardiovascular disease, or if the results of cardiac
monitoring indicate possible cardiac injury, the benefit of therapy must be weighed against the risk of myocardial injury
— In the randomized multiple myeloma study, 25 patients (8%) in the VELCADE arm and 42 patients (13%) in the DOXIL® plus VELCADE arm experienced left ventricular ejection fraction decrease (defined as absolute decrease 15% over baseline or a
5% decrease below institutional lower limit of normal)
� Myelosuppression may occur; frequently monitor complete blood count (including platelet count), at least prior to each dose of DOXIL®
— In patients with recurrent ovarian cancer, hematologic toxicity (based on platelet count or absolute neutrophil count) may require dose reduction or delay in administration of DOXIL®
— In patients with multiple myeloma, hematologic toxicity (based on platelet count, absolute neutrophil count, hemoglobin level, or neutropenia with fever) may require dose reduction, delay in administration, or suspension of DOXIL® and/or VELCADE
— Persistent severe myelosuppression may result in superinfection, neutropenic fever, or hemorrhage
— Sepsis occurring during neutropenia has resulted in discontinuation of treatment and in rare cases of death
� DOXIL® may potentiate the toxicity of other anticancer therapies, especially hematologic toxicities, when used in combination with other therapies that suppress bone marrow
� Hand-foot syndrome (HFS) may occur during therapy with DOXIL®
— Based on HFS toxicity grade, dose reduction, delay in administration, or discontinuation of DOXIL® may be required
— HFS was generally observed after 2 to 3 cycles of treatment, but may occur earlier• The reaction was mild in most patients, resolving in 1 to 2 weeks• The reaction can be severe and debilitating in some patients, resulting in
discontinuation of therapy� DOXIL® is an irritant, not a vesicant; use precautions to avoid extravasation� DOXIL® can cause fetal harm when used during pregnancy� Recall reaction has occurred with DOXIL® administration after radiotherapy� DOXIL® may interact with drugs known to interact with the conventional formulation
of doxorubicin HCl� In patients with recurrent ovarian cancer, the most common all-grade adverse reactions
(ARs) 20% (DOXIL® vs topotecan, respectively) included: asthenia (40% vs 51%), fever (21% vs 31%), nausea (46% vs 63%), stomatitis (41% vs 15%), vomiting (33% vs 44%), diarrhea (21% vs 35%), anorexia (20% vs 22%), dyspnea (15% vs 23%), HFS (51% vs 1%), and rash (29% vs 12%)
— In addition, 19% vs 52.3% reported alopecia (all grades) — Grade 3/4 hematologic ARs reported in 5% (DOXIL® vs topotecan, respectively)
were neutropenia (12% vs 76%) and anemia (6% vs 29%)� In patients with multiple myeloma, the most common all-grade ARs 20% (DOXIL®
plus VELCADE vs VELCADE, respectively) included: neutropenia (36% vs 22%), thrombocytopenia (33% vs 28%), anemia (25% vs 21%), fatigue (36% vs 28%), pyrexia (31% vs 22%), asthenia (22% vs 18%), nausea (48% vs 40%), diarrhea (46% vs 39%), vomiting (32% vs 22%), constipation (31% vs 31%), mucositis/stomatitis (20% vs 5%), peripheral neuropathy (42% vs 45%), neuralgia (17% vs 20%), and rash (22% vs 18%)
— In addition, 19% vs <1% reported HFS
Please see brief summary of Prescribing Information, including Boxed WARNINGS, on adjacent pages.
VELCADE is a registered trademark of Millennium Pharmaceuticals, Inc.
DOXIL® C.A.R.E.S. Provides Help and Support
3:27 PM























![KOREA magazine [DECEMBER 2011 VOL. 8 NO. 12]](https://img.pdfslide.us/doc/110x75/577d23a51a28ab4e1e9a62aa/korea-magazine-december-2011-vol-8-no-12.jpg)





