Embed Size (px)
Citation preview

Data Management and Structure Determination at SDC/JCSG
Qingping XuSSRL/JCSG
Refinement
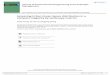
SDC activityYear
Crystals screened
Targets screened
Datasets collected
Targets collected
Targets in PDB
2000 33 15 0 0 0
2001 552 93 42 29 2
2002 2762 149 73 42 24
2003 6118 179 216 94 31
2004 4735 246 140 94 98
2005 6929 282 162 102 93
2006 19152 387 262 167 126
2007 29397 721 346 256 203
Total 69678 2072 1241 784 577
600 crystals / week 7 datasets / week 4 structures / week
26
67
180162 165
223
347
0
50
100
150
200
250
300
350
400
2001 2002 2003 2004 2005 2006 2007
Year
Dat
a co
llect
ed
1740
87 91106
139
252
0
50
100
150
200
250
300
2001 2002 2003 2004 2005 2006 2007
Year
Targ
ets
colle
cted
4/2007-4/2008
29,327 crystals screened from 721 targets335 datasets collected from 242 targets216 targets solved206 targets deposited
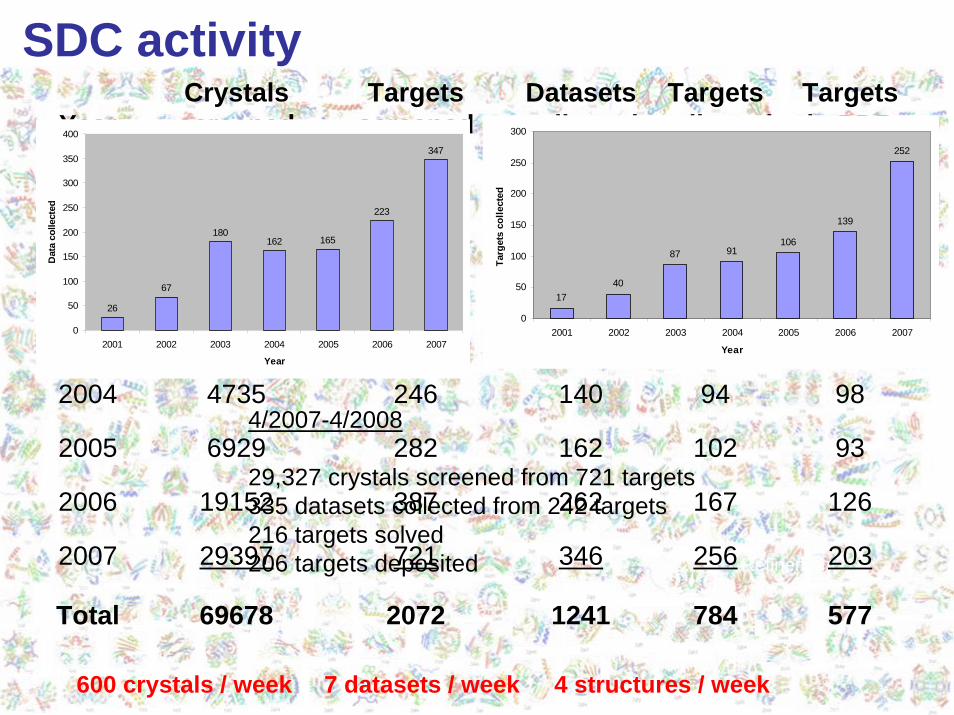
SDC Workflow1. Each crystal has a unique ID2. Essential information is captured in a central DB3. All data are online locally, also archived off-site4. Fixed data (directory) structures for all crystallographic data
Target info,crystal info
and storage ofanalysis data
for PDB
Diffraction images Data collection info
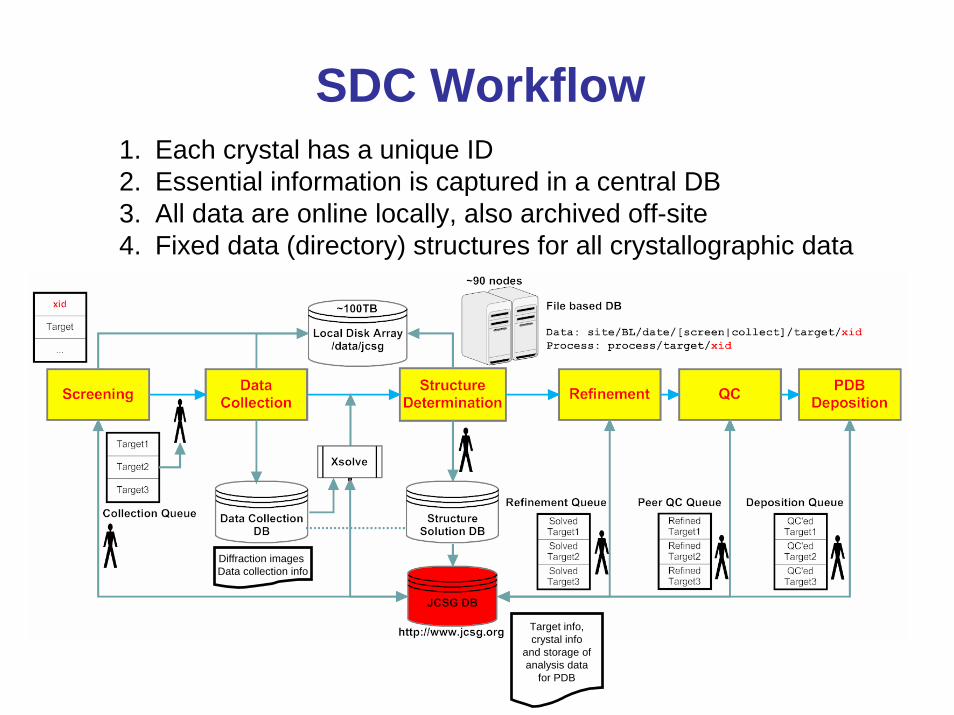
Crystal ScreeningBLU-ICE automatic screening interface
1. Co-developed by JCSG in 2000/2001
2. Robust and reliable crystal screening, 10 lost crystals in >150,000
3. Adopted by 85% of users; 75% of users collect data remotely
4. Implemented at other SR sources
Analysis of screening images
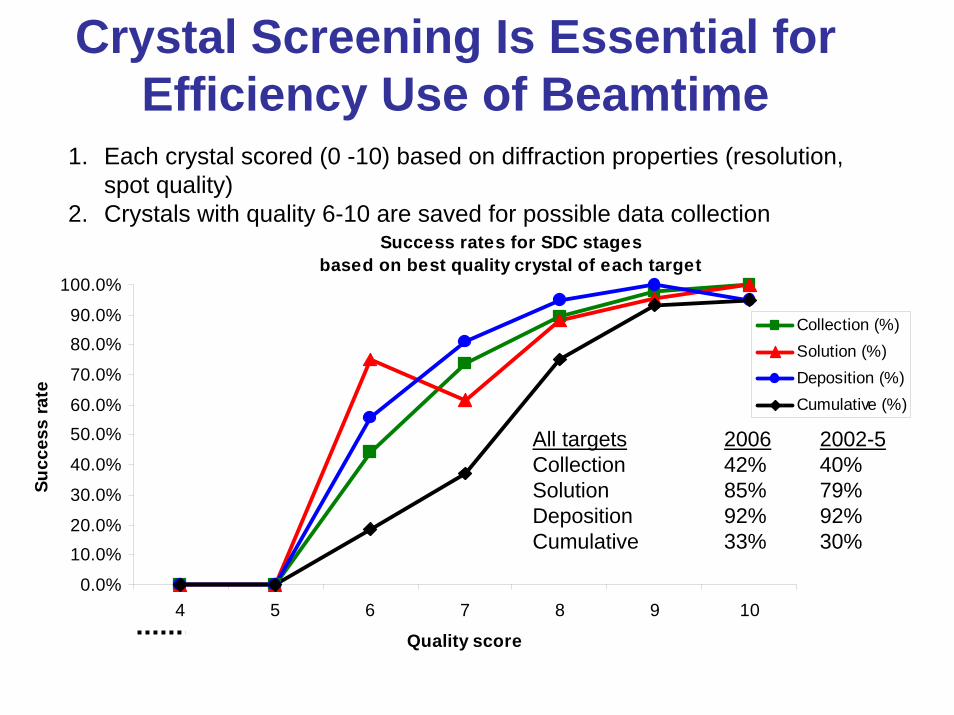
Crystal Screening Is Essential for Efficiency Use of Beamtime
1. Each crystal scored (0 -10) based on diffraction properties (resolution, spot quality)
2. Crystals with quality 6-10 are saved for possible data collectionSuccess rates for SDC stages
based on best quality crystal of each target
0.0%
10.0%20.0%
30.0%
40.0%
50.0%60.0%
70.0%
80.0%90.0%
100.0%
4 5 6 7 8 9 10
Quality score
Succ
ess
rate
Collection (%)Solution (%)Deposition (%)Cumulative (%)
All targets 2006 2002-5Collection 42% 40%Solution 85% 79%Deposition 92% 92%Cumulative 33% 30%

Structure Determination at SDC
• SDC structure determination strategy– Quick structure solution at the beamline by hand or
autoXDSp– Automated structure solution to systematically explore
application space, data space, and parameter space with Xsolve
– Human evaluation of automatic processing results– Manual inspection and resolution of unsolved/difficult
cases – Timely upload of inspected data to JCSG STSS
database

Development of Xsolve• Large scale operations call for integrated platforms
– No suitable third party platform from raw diffraction images to initial model for SDC needs
– SDC off-the-shelf PC Linux cluster– Many 3rd party crystallographic components to assemble the system
• Don’t always talk nice to each other, non-uniform data exchange• Different styles• Different capabilities
• Goal: To solve large number of MAD structures consistently and optimally with minimal human input– Automatically perform all processing steps without human intervention.– Provide reliable, high quality data processing for majority of datasets. – Provide best phases, optimal trace and processed data for refinement. – Exploit low cost modern computing for time-consuming data analysis.– Enforce uniform standards and provide data organization.
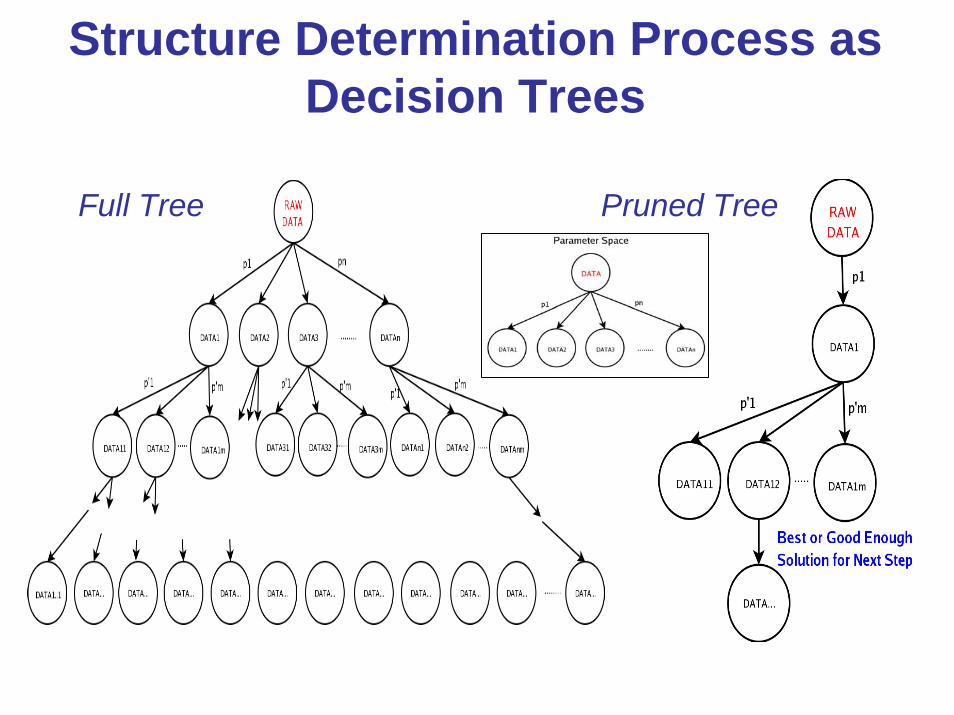
Structure Determination Process as Decision Trees
Full Tree Pruned Tree

Xsolve Implementation• Xsolve is developed as a distributed platform for wrapping third party
software• Xsolve adopts the “nearly” full tree approach by systematically
exploring parameters that are critical to structure solutions.• Methods: MAD/SAD and MR• Programs
– Data processing programs/strategies (MOSFLM, XDS & HKL ) – Phasing programs (SHELX, SOLVE, AUTOSHARP)– Density modification (DM, RESOLVE, SOLOMON)– Tracing programs (wARP, RESOVLE, RESOLVE_BUILD) – MR programs (MOLREP, PHASERS & EPMR)
• Combination of data/models• Space groups• Resolution• ASU content
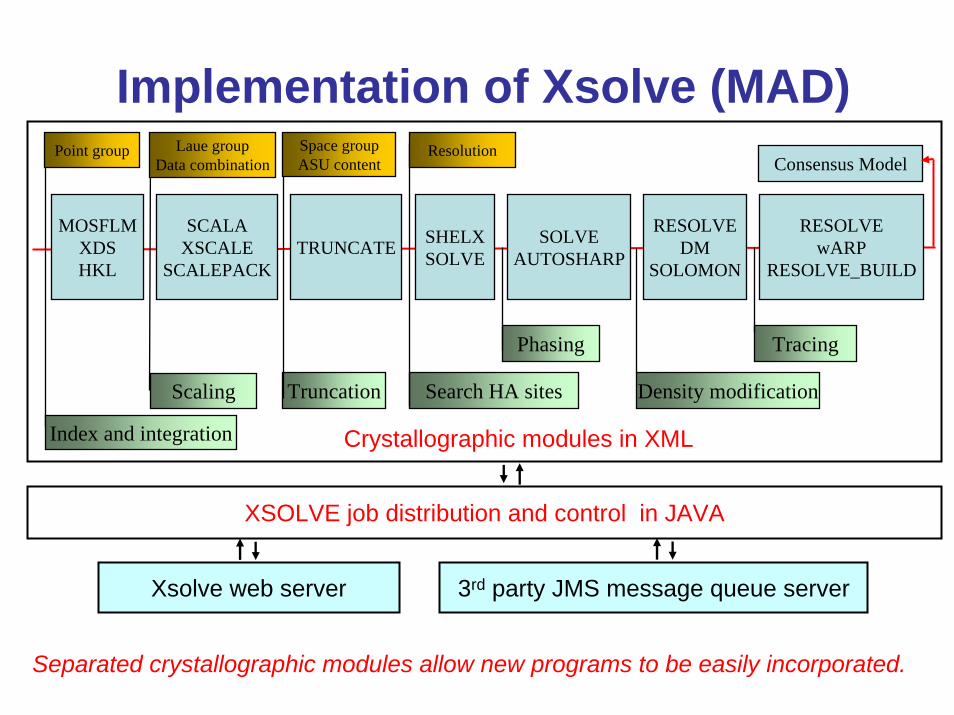
Implementation of Xsolve (MAD)
MOSFLMXDSHKL
SCALAXSCALE
SCALEPACKTRUNCATE SHELX
SOLVESOLVE
AUTOSHARP
RESOLVEDM
SOLOMON
RESOLVEwARP
RESOLVE_BUILD
Index and integration
Scaling Truncation Search HA sites
Phasing
Density modification
Tracing
Consensus ModelPoint group Space group
ASU contentResolutionLaue group
Data combination
XSOLVE job distribution and control in JAVA
Crystallographic modules in XML
3rd party JMS message queue serverXsolve web server
Separated crystallographic modules allow new programs to be easily incorporated.
Data collection
Structure Determination
Start XsolveEvaluation of results
JCSG STSS DB
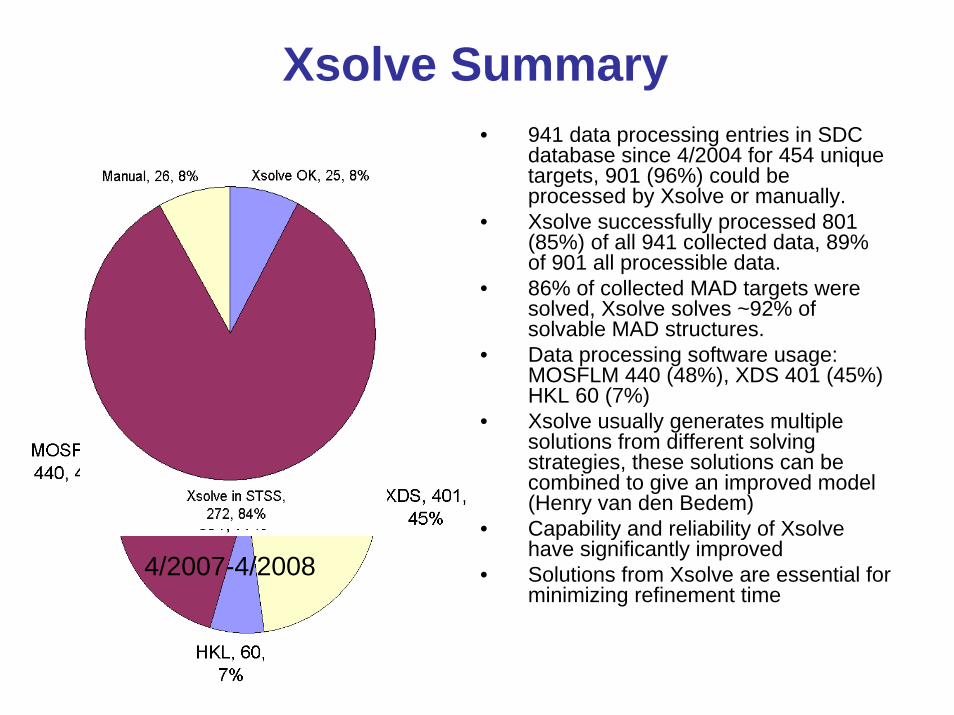
Xsolve Summary• 941 data processing entries in SDC
database since 4/2004 for 454 unique targets, 901 (96%) could be processed by Xsolve or manually.
• Xsolve successfully processed 801 (85%) of all 941 collected data, 89% of 901 all processible data.
• 86% of collected MAD targets were solved, Xsolve solves ~92% of solvable MAD structures.
• Data processing software usage: MOSFLM 440 (48%), XDS 401 (45%) HKL 60 (7%)
• Xsolve usually generates multiple solutions from different solving strategies, these solutions can be combined to give an improved model (Henry van den Bedem)
• Capability and reliability of Xsolve have significantly improved
• Solutions from Xsolve are essential for minimizing refinement time
4/2007-4/2008
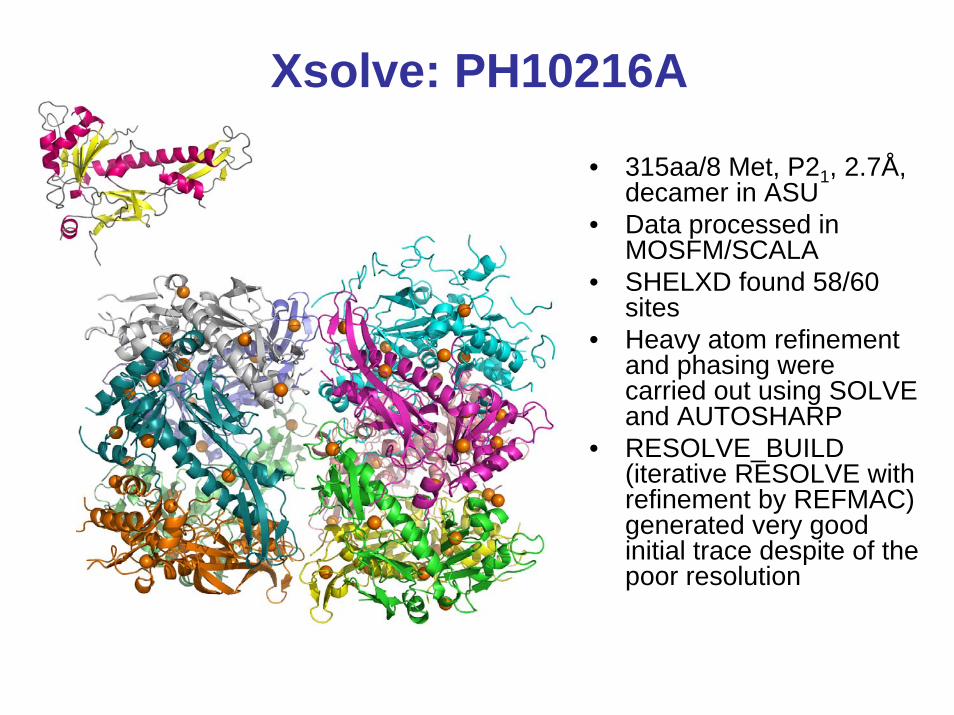
Xsolve: PH10216A
• 315aa/8 Met, P21, 2.7Å, decamer in ASU
• Data processed in MOSFM/SCALA
• SHELXD found 58/60 sites
• Heavy atom refinement and phasing were carried out using SOLVE and AUTOSHARP
• RESOLVE_BUILD (iterative RESOLVE with refinement by REFMAC) generated very good initial trace despite of the poor resolution
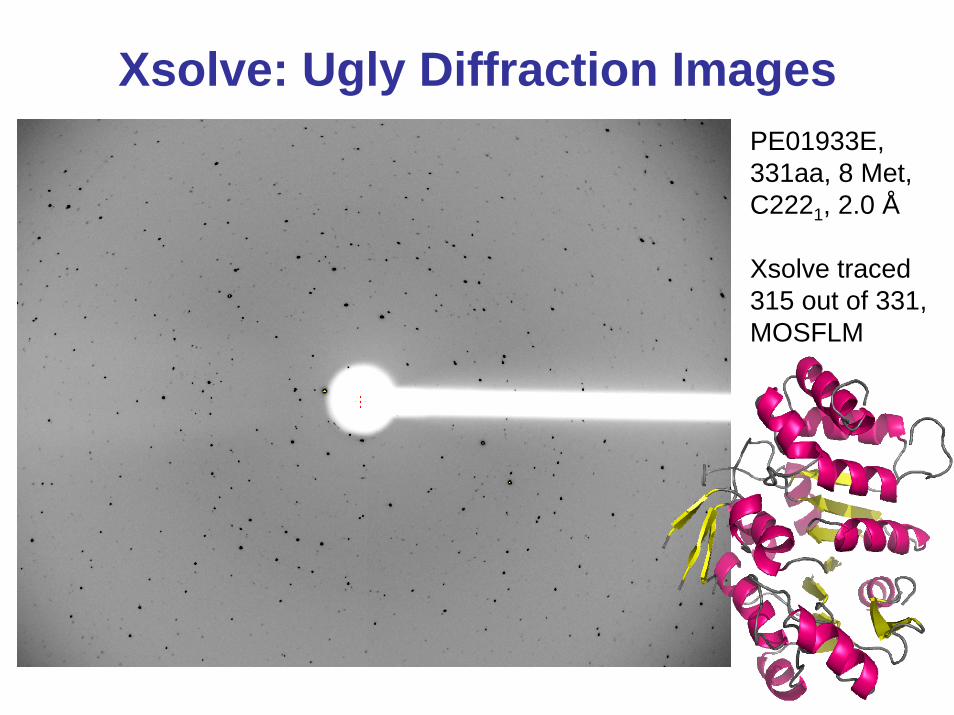
Xsolve: Ugly Diffraction ImagesPE01933E, 331aa, 8 Met, C2221, 2.0 Å
Xsolve traced 315 out of 331, MOSFLM
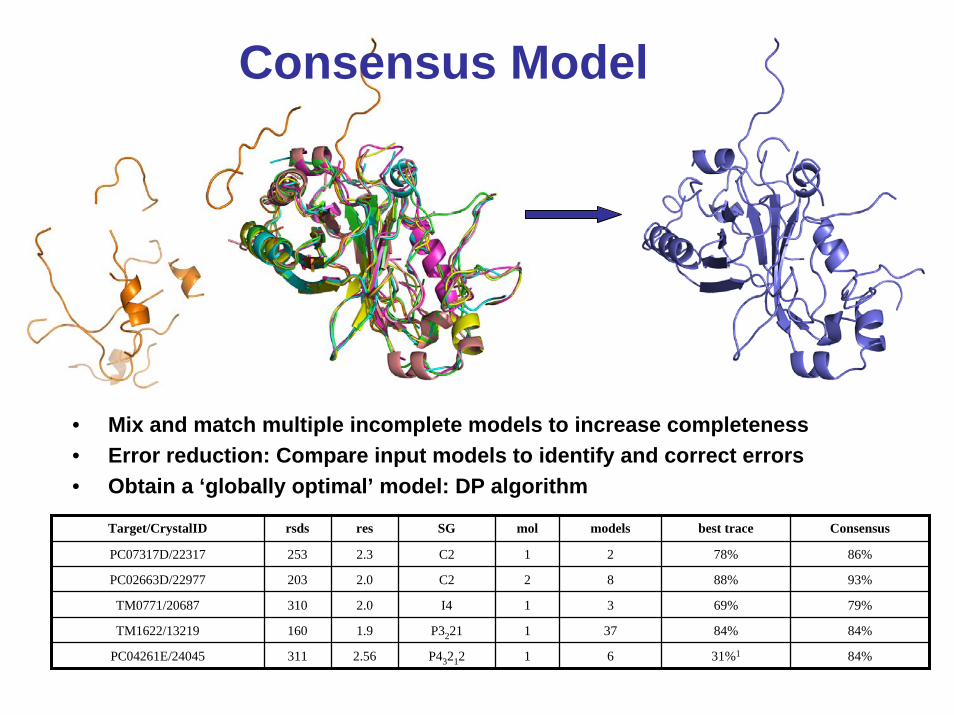
Consensus Model
• Mix and match multiple incomplete models to increase completeness• Error reduction: Compare input models to identify and correct errors• Obtain a ‘globally optimal’ model: DP algorithm
Target/CrystalID rsds res SG mol models best trace Consensus
PC07317D/22317 253
203
310
160
PC04261E/24045 311 2.56 P43212 1 6 31%1
2.3
84%
1 2 78% 86%
PC02663D/22977 2.0
C2
C2
I4
2 8 88% 93%
TM0771/20687 2.0 1 3 69% 79%
TM1622/13219 1.9 1 37 84% 84%P3221
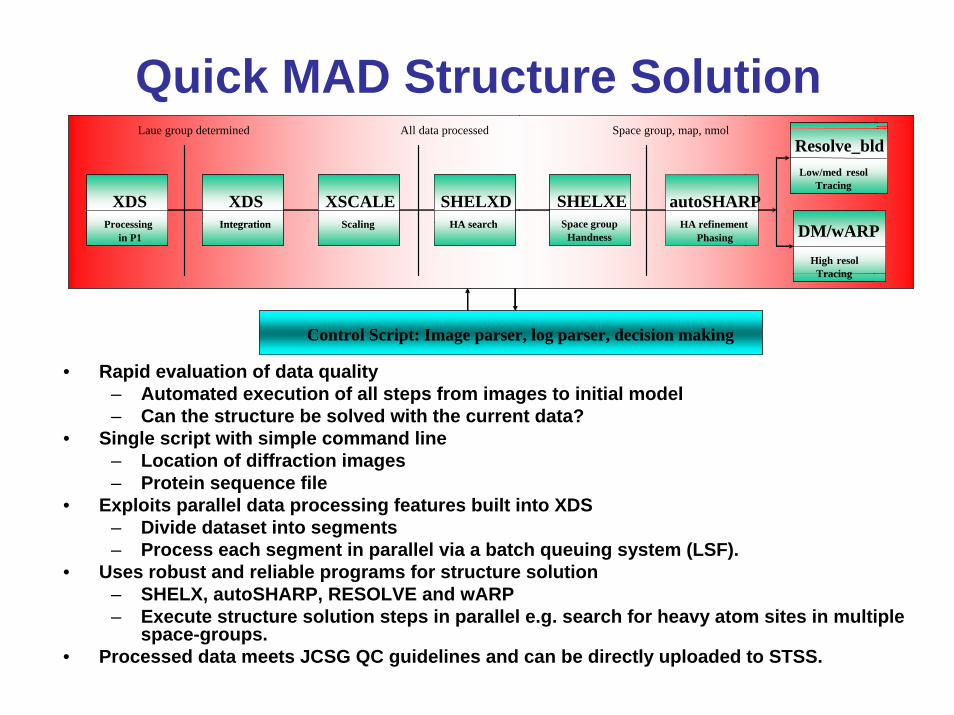
Quick MAD Structure Solution
XDSProcessing
in P1
XDSIntegration
XSCALEScaling
SHELXDHA search
SHELXESpace group
Handness
autoSHARPHA refinement
Phasing wARPHigh resol
Tracing
ResolveLow/med resol
Tracing
Laue group determined All data processed Space group, map, nmol
Control Script: Image parser, log parser, decision making
XDSProcessing
in P1
XDSProcessing
in P1
XDSIntegration
XDSIntegration
XSCALEScaling
XSCALEScaling
SHELXDHA search
SHELXDHA search
SHELXESpace group
Handness
SHELXESpace group
Handness
autoSHARPHA refinement
Phasing wARPHigh resol
Tracing
DM/wARPHigh resol
Tracing
ResolveLow/med resol
Tracing
Resolve_bldLow/med resol
Tracing
Laue group determined All data processed Space group, map, nmol
Control Script: Image parser, log parser, decision making
• Rapid evaluation of data quality– Automated execution of all steps from images to initial model– Can the structure be solved with the current data?
• Single script with simple command line– Location of diffraction images – Protein sequence file
• Exploits parallel data processing features built into XDS– Divide dataset into segments – Process each segment in parallel via a batch queuing system (LSF).
• Uses robust and reliable programs for structure solution– SHELX, autoSHARP, RESOLVE and wARP– Execute structure solution steps in parallel e.g. search for heavy atom sites in multiple
space-groups.• Processed data meets JCSG QC guidelines and can be directly uploaded to STSS.

autoXDSp: PC05995D
First interpretable map in less than 30 minutes (P41212, 1 monomer per asu, interpretable map). SHELXE map overall correlation to the final model is 0.73 (<φ>=51). The phases were subsequently improved in autoSHARP, CC=0.86, <φ>=36. 191 aa traced in wARP.
Step Time* (s)
Laue Group 361
Integration 906
Scale 941
MAD Scale 1063
SHELX 1519
207aa/3 Met, 3-wavelength MAD, each sweep consists of 90 MARCCD 325 images
* Measures time from start to the end of current step
%autoXDSp -data /data/jcsg/ssrl2/9_2/20060312/collection/PC05995D/23156 -seq seq.dat

FM11607A: Corrupted Sweeps
H32 3wav MAD, 36 sweeps
Applicable to small sweeps from multiple crystals

• Lots of data in short amount of time• 65 data sets, 90 frames each (MARCCD325)• Automatic processing:
– index-integrate-scale-truncate-refine– 59 out of 65 processed automatically in 2hrs
Parallel Processing of High-throughput Fragment Screening
Stout, Scripps

Manual Processing• Automatic processing allow human efforts on more difficult cases• Human intelligence to overcome program failures
– Benchmarks for automatic processing– Processing difficult cases– Interpreting Xsolve results, guiding/fine tuning Xsolve jobs toward
success– Feedbacks to programmers for future improvements
• Parallel fine sampling to solve large or difficult structures– Manually exploring large parameter space to find right combination of
parameters is time-consuming and frustrating. It could lead to prematurely abandonment a potentially solvable structure.
– Fine grid sampling (Xsolve strategy applied locally) to solve large or difficult structures
• Parallel exploration of parameter space by brute force is an effective approach to solve challenging structures efficiently and reliably
• Systematically explore parameter space• Speed up with parallel execution on cluster
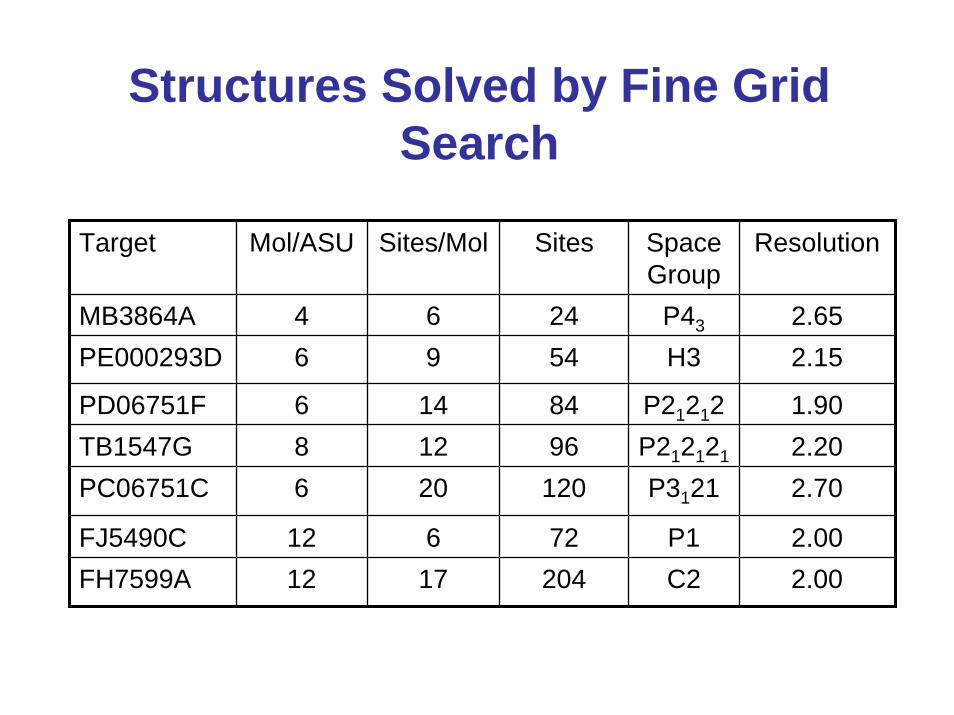
Structures Solved by Fine Grid Search
Target Mol/ASU Sites/Mol Sites Space Group
Resolution
MB3864A 4 6 24 P43
H3
P21212P212121
P3121
P1C2
2.65PE000293D 6 9 54 2.15
PD06751F 6 14 84 1.90TB1547G 8 12 96 2.20PC06751C 6 20 120 2.70
FJ5490C 12 6 72 2.00FH7599A 12 17 204 2.00

TB1547G: Mislabeled Target• 409aa/13 Met, P212121, 2 tetramers per
asu• Initially labeled as something else
(TB5131A, 179aa/2 Met)• POINTLESS and XPREP to narrow
down space group choices, XPREP to generate FA values
• Treated as an unknown target, SHELXD Grid search:
– Sites 20-120 in step of 10– Resolution cutoff 3.3-4.5 in step of 0.1– E value cutoff from 1.1-1.5 in step of 0.1
• 520 parallel SHELXD jobs, each SHELXD job attempts 200 trials
• The job order was randomized to uniformly sample the search space initially
• Solutions appeared in minutes (jobs could be terminated early)
• Each SHELXD job needed ~1hrs, ~2 hrs for all jobs to finish on SDC cluster (220 CPUs)
• Interpretation of density map gave correct identification of the target
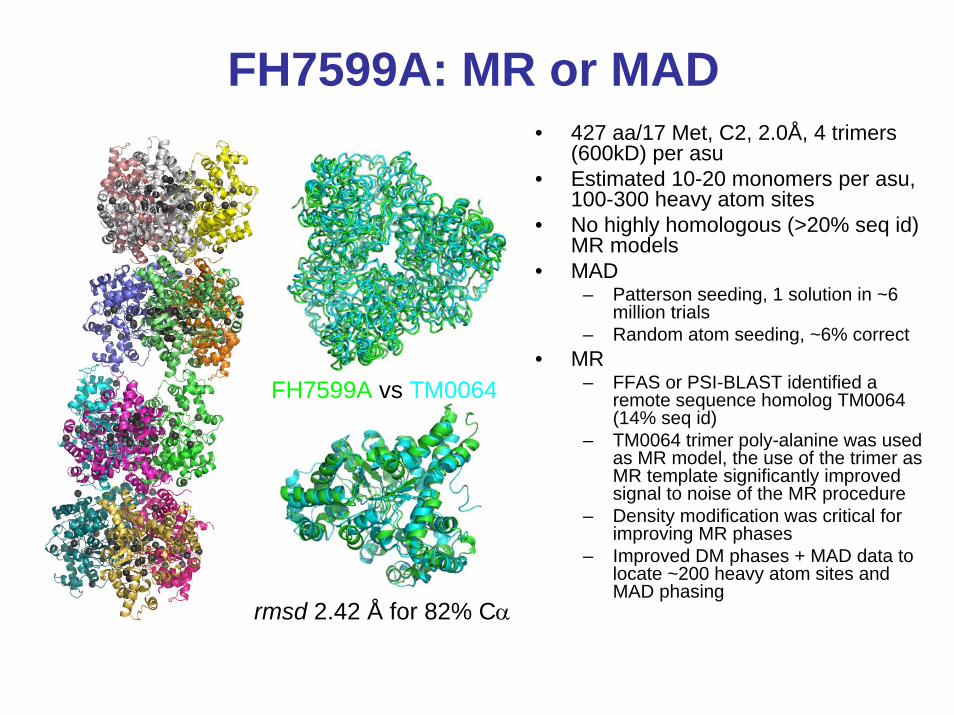
FH7599A: MR or MAD• 427 aa/17 Met, C2, 2.0Å, 4 trimers
(600kD) per asu• Estimated 10-20 monomers per asu,
100-300 heavy atom sites• No highly homologous (>20% seq id)
MR models• MAD
– Patterson seeding, 1 solution in ~6 million trials
– Random atom seeding, ~6% correct • MR
– FFAS or PSI-BLAST identified a remote sequence homolog TM0064 (14% seq id)
– TM0064 trimer poly-alanine was used as MR model, the use of the trimer as MR template significantly improved signal to noise of the MR procedure
– Density modification was critical for improving MR phases
– Improved DM phases + MAD data to locate ~200 heavy atom sites and MAD phasing
rmsd 2.42 Å for 82% Cα
FH7599A vs TM0064
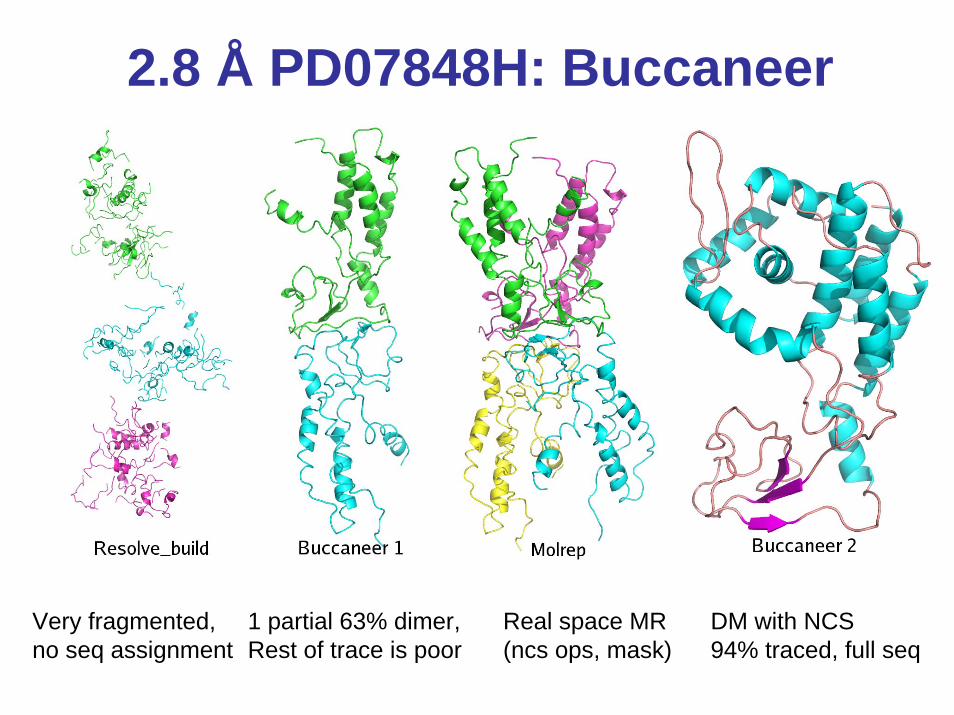
2.8 Å PD07848H: Buccaneer
Very fragmented,no seq assignment
1 partial 63% dimer,Rest of trace is poor
Real space MR(ncs ops, mask)
DM with NCS94% traced, full seq

Problems with Structure Solution• 20% MAD/SAD data sets (24% collected targets) not
solved 1st time collected– Data that can not be indexed and processed (~20% data sets), or
the processed data have poor quality (~30%) – Poor resolution (~20%)– Targets with limited or no anomalous signal (1 or 2 Se-Met) (10-
15%)– Twinning (10-15%)
• Improved detection of twinning in Xsolve with PHENIX tools• All current JCSG twinned structures are solved by ignoring
twinning at solve stage (i.e. the structure was solved in the apparent space group and later refined in the correct space group)
• Sometimes there may be no correlation between twinning fraction and whether it can be solved by MAD method
• Lack effective and reliable methods to solve twinned MAD data
– Many targets (~50%) were solved later by screening/collection more crystals
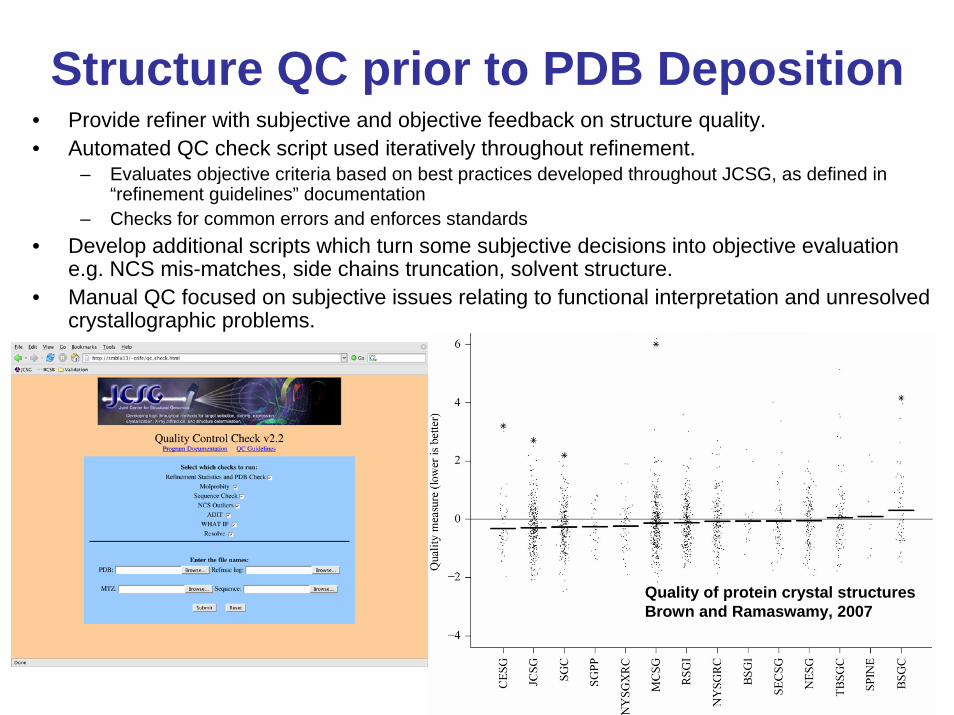
Structure QC prior to PDB Deposition• Provide refiner with subjective and objective feedback on structure quality.• Automated QC check script used iteratively throughout refinement.
– Evaluates objective criteria based on best practices developed throughout JCSG, as defined in “refinement guidelines” documentation
– Checks for common errors and enforces standards• Develop additional scripts which turn some subjective decisions into objective evaluation
e.g. NCS mis-matches, side chains truncation, solvent structure.• Manual QC focused on subjective issues relating to functional interpretation and unresolved
crystallographic problems.
Quality of protein crystal structuresBrown and Ramaswamy, 2007
Dissemination
http://www.topsan.org
• Complete and accurate PDB deposition– Experimental
details– Unmerged data– Experimental
phases – Coordinates
• Community based structure annotation TOPSAN
• Provide developers or educators with well documented datasets

Conclusions and Future Directions• Xsolve and other tools
– Automatically perform all processing steps without human intervention– Provide reliable, high quality data processing for majority of datasets– Exploit the strength of different program packages– Provide optimal trace and processed data ready for refinement– Trade-off between low cost modern computing and time-consuming data
analysis– Enforce uniformity in data organization and standards
• Future developments– Better tools for analysis of results– Make it available through a web server to SSRL users and make version
that can be installed at other large facilities/institutions– Incorporating new programs/features– Become more intelligent, more feedbacks, handle more difficult cases– Improve flexibility and consistency– Better usage of resources– Developing a fully distributable version

AcknowledgementsAshley Deacon
Günter Wolf Henry van den Bedem

UCSD & BurnhamBioinformatics Core
John WooleyAdam Godzik
Lukasz JaroszewskiSlawomir Grzechnik
Sri Krishna SubramanianAndrew Morse
Tamara AstakhovaLian Duan
Piotr KozbialDana Weekes
Natasha SefcovicPrasad Burra
Konstantina BakolitsaAndrei Istomin
Kyle EllrottJosie AlaoenCindy Cook
GNF & TSRICrystallomics Core
Scott LesleyMark KnuthHeath Klock
Dennis CarltonThomas Clayton
Marc DellerDaniel McMullan Polat Abdubek
Julie FeuerhelmJoanna C. Hale
Thamara JanaratneHope Johnson
Edward NigoghossianLinda Okach
Sebastian SudekGlen Spraggon
Bernhard GeierstangerSanjay AgarwallaAnna Grzechnik
Connie ChenDustin Ernst
Regina GorskiSachin Kale
Amanda NopakunChristina PuckettTiffany Wooten
Jessica CansecoMimmi Brown
Scientific Advisory BoardSir Tom BlundellUniv. CambridgeHomme Hellinga
Duke University Medical CenterJames Naismith
The Scottish Structural Proteomics FacilityUniv. St. AndrewsJames Paulson
Consortium for Functional Glycomics,The Scripps Research Institute
Robert StroudCenter for Structure of Membrane Proteins,
Membrane Protein Expression Center, UCSF Soichi Wakatsuki
Photon Factory, KEK, JapanJames Wells
UC San FranciscoTodd Yeates
UCLA-DOE, Inst. for Genomics and Proteomics
TSRI, NMR CoreKurt Wüthrich
Reto HorstMaggie Johnson
Amaranth ChatterjeeMichael Geralt
Wojtek AugustyniakPedro Serrano
Bill PedriniBiswaranjan Mohanty
Jin-Kyu Rhee
TSRI Administrative Core
Ian WilsonMarc ElsligerGye Won Han
David MarcianoHenry Tien
Lisa van Veen
Stanford /SSRL Structure Determination Core
Keith HodgsonAshley Deacon
Mitchell Miller Herbert Axelrod
Hsiu-Ju (Jessica) ChiuKevin Jin
Christopher RifeQingping Xu
Silvya OommachenHenry van den Bedem
Scott TalafuseRonald Reyes
Abhinav KumarChristine Trame
Debanu DasWinnie Lam
The JCSG is supported by the NIGMS Protein Structure Initiative Grant U54 GM074898
Ex officio founding members JCSG-1Raymond Stevens , TSRI
Susan Taylor, UCSDPeter Kuhn, SSRL/TSRI
Duncan McRee, TSRI/SyrrxPeter Schultz, TSRI/GNF







![SDC-12/SDC-15 · 2018-01-14 · Introduction 6 SDC-12/SDC-151080pD-ILA3D Front Projector2013 User’s Manual AdditionalOptionalAccessories Replacementlamps: •FormodelsSDC-12andSDC-15[2013productionandbeyond]orderlampWC-LPU230](https://img.pdfslide.us/doc/110x75/5f30eb9530d2254a2869f490/sdc-12sdc-15-2018-01-14-introduction-6-sdc-12sdc-151080pd-ila3d-front-projector2013.jpg)