Embed Size (px)
Citation preview

8/3/2019 Dan Peer et al- Nanocarriers as an emerging platform for cancer therapy
http://slidepdf.com/reader/full/dan-peer-et-al-nanocarriers-as-an-emerging-platform-for-cancer-therapy 1/10
nature nanotechnology | VOL 2 | DECEMBER 2007 | www.nature.com/naturenanotechnology 751
REVIEW ARTICLE
Nanocarriers as an emerging platform for
cancer therapy
Nanotechnology has the potential to revolutionize cancer diagnosis and therapy. Advances in protein
engineering and materials science have contributed to novel nanoscale targeting approaches that
may bring new hope to cancer patients. Several therapeutic nanocarriers have been approved for
clinical use. However, to date, there are only a few clinically approved nanocarriers that incorporate
molecules to selectively bind and target cancer cells. This review examines some of the approved
formulations and discusses the challenges in translating basic research to the clinic. We detail the
arsenal of nanocarriers and molecules available for selective tumour targeting, and emphasize the
challenges in cancer treatment.
Dan Peer1†, Jerey M. KarP2,3†,SeungPyo Hong4†, oMiD C. aroKHzaD5,riMona Margalit6 anD robert langer3,4*
1Immune Disease Institute and Department of Anesthesia, Harvard Medical
School, Boston, Massachusetts 02115, USA; 2HST Center for Biomedical
Engineering, Department of Medicine, Brigham and Women’s Hospital, Harvard
Medical School, Cambridge, Massachusetts 02139, USA; 3Harvard-MIT Division
of Health Sciences and Technology, Massachusetts Institute of Technology,Cambridge, Massachusetts 02139, USA; 4Department of Chemical Engineering,
Massachusetts Institute of Technology, Cambridge, Massachusetts 02139,
USA; 5Laboratory of Nanomedicine and Biomaterials and Department of
Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School,
Boston, Massachusetts 02115, USA; 6Department of Biochemistry, George
S. Wise Faculty of Life Sciences, and the Center for Nanoscience and
Nanotechnology, Tel Aviv University, Tel Aviv, 69978, Israel†These authors contributed equally to this review
*e-mail: [email protected]
Cancer remains one o the world’s most devastating diseases, withmore than 10 million new cases every year1. However, mortality has decreased in the past two years2 owing to better understanding
o tumour biology and improved diagnostic devices and treatments.Current cancer treatments include surgical intervention, radiationand chemotherapeutic drugs, which oen also kill healthy cells andcause toxicity to the patient. It would thereore be desirable to developchemotherapeutics that can either passively or actively target cancerouscells. Passive targeting exploits the characteristic eatures o tumourbiology that allow nanocarriers to accumulate in the tumour by theenhanced permeability and retention (EPR) eect2. Passively targetingnanocarriers rst reached clinical trials in the mid-1980s, and the rstproducts, based on liposomes and polymer–protein conjugates, weremarketed in the mid-1990s. Later, therapeutic nanocarriers based onthis strategy were approved or wider use (able 1) and methods o urther enhancing targeting o drugs to cancer cells were investigated.Active approaches achieve this by conjugating nanocarriers containingchemotherapeutics with molecules that bind to overexpressed antigens
or receptors on the target cells. Recent reviews provide perspective onthe use o nanotechnology as a undamental tool in cancer researchand nanomedicine3,4. Here we ocus on the potential o nanocarriersand molecules that can selectively target tumours, and highlight thechallenges in translating some o the basic research to the clinic.
PaSSive anD aCtive targeting
Nanocarriers encounter numerous barriers en route to their target,
such as mucosal barriers and non-specic uptake5,6
. o address thechallenges o targeting tumours with nanotechnology, it is necessary to combine the rational design o nanocarriers with the undamentalunderstanding o tumour biology (Box 1).
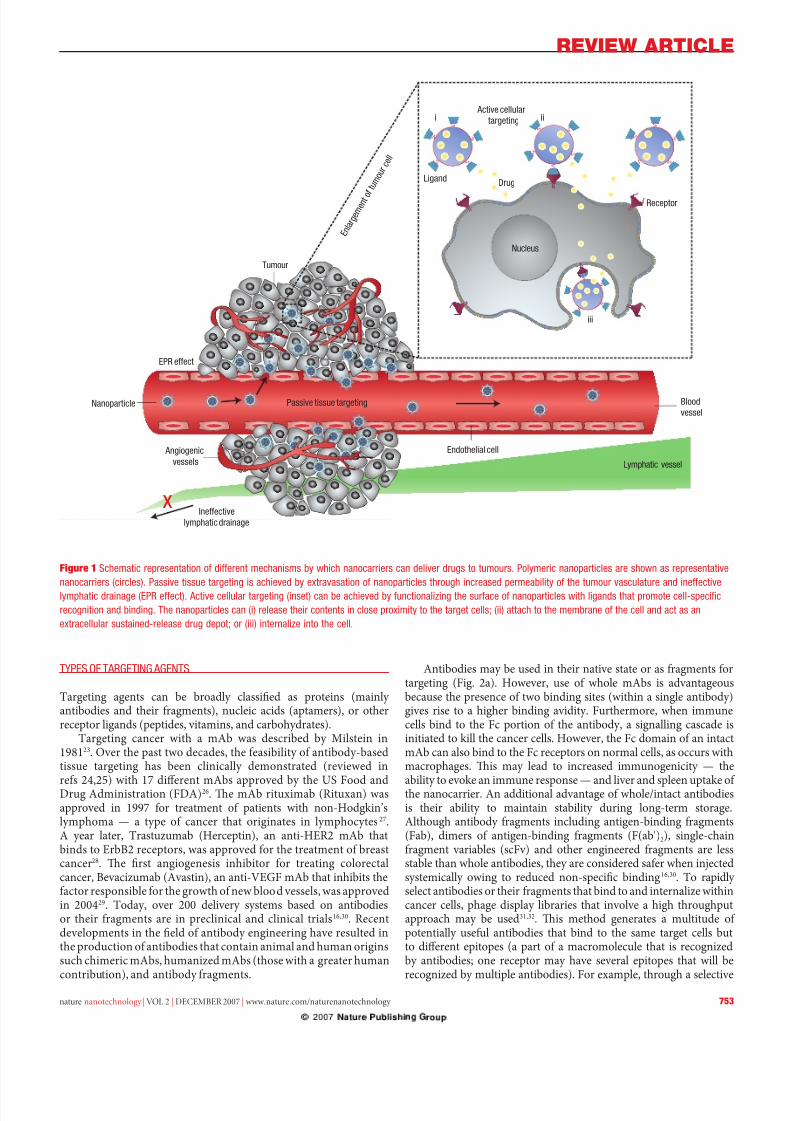
General eatures o tumours include leaky blood vessels andpoor lymphatic drainage. Whereas ree drugs may diuse non-specically, a nanocarrier can extravasate (escape) into the tumourtissues via the leaky vessels by the EPR eect7 (Fig. 1). Te increasedpermeability o the blood vessels in tumours is characteristic o rapidand deective angiogenesis (ormation o new blood vessels romexisting ones). Furthermore, the dysunctional lymphatic drainagein tumours retains the accumulated nanocarriers and allows themto release drugs into the vicinity o the tumour cells. Experimentsusing liposomes o dierent mean size suggest that the threshold
vesicle size or extravasation into tumours is ∼400 nm (re. 8), butother studies have shown that particles with diameters <200 nm aremore eective5,8–10.
Although passive targeting approaches orm the basis o clinicaltherapy, they suer rom several limitations. Ubiquitously targetingcells within a tumour is not always easible because some drugs cannotdiuse eciently and the random nature o the approach makesit dicult to control the process. Tis lack o control may inducemultiple-drug resistance (MDR) — a situation where chemotherapy treatments ail patients owing to resistance o cancer cells towardsone or more drugs. MDR occurs because transporter proteins thatexpel drugs rom cells are overexpressed on the surace o cancercells4,11,12. Expelling drugs inevitably lowers the therapeutic eect andcancer cells soon develop resistance to a variety o drugs. Te passivestrategy is urther limited because certain tumours do not exhibit

8/3/2019 Dan Peer et al- Nanocarriers as an emerging platform for cancer therapy
http://slidepdf.com/reader/full/dan-peer-et-al-nanocarriers-as-an-emerging-platform-for-cancer-therapy 2/10
REVIEW ARTICLE
752 nature nanotechnology | VOL 2 | DECEMBER 2007 | www.nature.com/naturenanotechnology
the EPR eect, and the permeability o vessels may not be the samethroughout a single tumour13.
One way to overcome these limitations is to programmethe nanocarriers so they actively bind to speciic cells aterextravasation. his binding may be achieved by attachingtargeting agents such as ligands — molecules that bind to speciicreceptors on the cell surace — to the surace o the nanocarrier by a variety o conjugation chemistries9. Nanocarriers will recognizeand bind to target cells through ligand–receptor interactions,and bound carriers are internalized beore the drug is released
inside the cell (Fig 1). In general, when using a targeting agentto deliver nanocarriers to cancer cells, it is imperative that theagent binds with high selectivity to molecules that are uniquely expressed on the cell surace. Other important considerationsare outlined below.
o maximize speciicity, a surace marker (antigen or receptor)should be overexpressed on target cells relative to normal cells.For example, to eiciently deliver liposomes to B-cell receptorsusing the anti-CD19 monoclonal antibody (mAb), the density o receptors should be in the range o 104–105 copies per cell. hosewith lower density are less eectively targeted14. In a breastcancer model, a receptor density o 10 5 copies o ErbB2 receptorsper cell was necessary to improve the therapeutic eicacy o ananti-ErbB2-targeted liposomal doxorubicin relative to its non-targeted counterpart15.
Te binding o certain ligands to their receptors may causereceptor-mediated internalization, which is oen necessary i nanocarriers are to release drugs inside the cell16–18. For example,a more signicant therapeutic outcome was achieved whenimmunoliposomes targeted to human blood cancer (B-celllymphoma) were labelled with an internalizing anti-CD19 ligandrather than a non-internalizing anti-CD20 ligand19. In contrast,targeting nanocarriers to non-internalizing receptors may sometimes be advantageous in solid tumours owing to the bystandereect, where cells lacking the target receptor can be killed through
drug release at the surace o the neighbouring cells, where carrierscan bind20.It is generally known that higher binding anity increases
targeting ecacy. However, or solid tumours, there is evidence thathigh binding anity can decrease penetration o nanocarriers dueto a ‘binding-site barrier’, where the nanocarrier binds to its target sostrongly that penetration into the tissue is prevented16,21. In additionto enhanced anity, multivalent binding eects (or avidity) may alsobe used to improve targeting. Te collective binding in a multivalentinteraction is much stronger than monovalent binding. For example,dendrimer nanocarriers conjugated to 3–15 olate molecules showeda 2,500–170,000-old enhancement in dissociation constants(KD) over ree olate when attaching to olate-binding proteinsimmobilized on a surace. Tis was attributed to the avidity o themultiple olic acid groups on the periphery o the dendrimers22.
Table 1 Representative examples of nanocarrier-based drugs on the market
Cmpd Cmmc m nc idcs
S mc hdd-cs
(SManCS)
zs/Smm Pm–p cj Hpc ccm
Peg-l-sps ocsp Pm–p cj ac mphsc kmPeg-c c-sm c
(g-CS)
ns/Pegsm Pm–p cj P chmhp-sscdp
il2 sd dphh x ok (Dk dx) immx (s p) Cs t-c mphm a-CD33 d cjd
cchmc
M Chm-mmcj ac ms km
a-CD20 cjd m-90
dm-111
z rd-mmcj rpsd c, w-d, c, smd -Hdk’s mphm
a-CD20 cjd d-131 bxx rd-mmcj rpsd c, w-d, c, smd -Hdk’s mphm
Dc DXm lpsms Kps’s scmDxc Mc lpsms Cm hp c s
cc, cc, Kps’s scm
Dxc Dx/Cx Peg-psms rc Kps’s scm, c scc, cc
vcs oc tCS lpsms rpsd ss -Hdk’smphm (nHl)
Pcx ax am-d pcx pcs Msc s cc
Nanocarriers can oer many advantages over ree drugs. Tey:
• protect the drug rom premature degradation;• prevent drugs rom prematurely interacting with the
biological environment;
• enhance absorption o the drugs into a selected tissue(or example, solid tumour);
• control the pharmacokinetic and drug tissuedistribution prole;
• improve intracellular penetration.
For rapid and eective clinical translation, the nanocarrier should:
• be made rom a material that is biocompatible, wellcharacterized, and easily unctionalized;
• exhibit high dierential uptake eciency in the target
cells over normal cells (or tissue);• be either soluble or colloidal under aqueous conditions
or increased eectiveness;• have an extended circulating hal-lie, a low rate o
aggregation, and a long shel lie.
Box 1 Rational design of nanocarriers for cancer therapy
8/3/2019 Dan Peer et al- Nanocarriers as an emerging platform for cancer therapy
http://slidepdf.com/reader/full/dan-peer-et-al-nanocarriers-as-an-emerging-platform-for-cancer-therapy 3/10
REVIEW ARTICLE
nature nanotechnology | VOL 2 | DECEMBER 2007 | www.nature.com/naturenanotechnology 753
tyPeS o targeting agentS
argeting agents can be broadly classied as proteins (mainly antibodies and their ragments), nucleic acids (aptamers), or otherreceptor ligands (peptides, vitamins, and carbohydrates).
argeting cancer with a mAb was described by Milstein in198123. Over the past two decades, the easibility o antibody-basedtissue targeting has been clinically demonstrated (reviewed in
res 24,25) with 17 dierent mAbs approved by the US Food andDrug Administration (FDA)26. Te mAb rituximab (Rituxan) wasapproved in 1997 or treatment o patients with non-Hodgkin’slymphoma — a type o cancer that originates in lymphocytes 27.A year later, rastuzumab (Herceptin), an anti-HER2 mAb thatbinds to ErbB2 receptors, was approved or the treatment o breastcancer28. Te rst angiogenesis inhibitor or treating colorectalcancer, Bevacizumab (Avastin), an anti-VEGF mAb that inhibits theactor responsible or the growth o new blood vessels, was approvedin 200429. oday, over 200 delivery systems based on antibodiesor their ragments are in preclinical and clinical trials16,30. Recentdevelopments in the eld o antibody engineering have resulted inthe production o antibodies that contain animal and human originssuch chimeric mAbs, humanized mAbs (those with a greater humancontribution), and antibody ragments.
Antibodies may be used in their native state or as ragments ortargeting (Fig. 2a). However, use o whole mAbs is advantageousbecause the presence o two binding sites (within a single antibody)gives rise to a higher binding avidity. Furthermore, when immunecells bind to the Fc portion o the antibody, a signalling cascade isinitiated to kill the cancer cells. However, the Fc domain o an intactmAb can also bind to the Fc receptors on normal cells, as occurs withmacrophages. Tis may lead to increased immunogenicity — the
ability to evoke an immune response — and liver and spleen uptake o the nanocarrier. An additional advantage o whole/intact antibodiesis their ability to maintain stability during long-term storage.Although antibody ragments including antigen-binding ragments(Fab), dimers o antigen-binding ragments (F(ab′)2), single-chainragment variables (scFv) and other engineered ragments are lessstable than whole antibodies, they are considered saer when injectedsystemically owing to reduced non-specic binding16,30. o rapidly select antibodies or their ragments that bind to and internalize withincancer cells, phage display libraries that involve a high throughputapproach may be used31,32. Tis method generates a multitude o potentially useul antibodies that bind to the same target cells butto dierent epitopes (a part o a macromolecule that is recognizedby antibodies; one receptor may have several epitopes that will berecognized by multiple antibodies). For example, through a selective
Active cellular
targeting
LigandDrug
Receptor
Nucleus
i ii
iii
Blood
vessel
Endothelial cell
Passive tissue targetingNanoparticle
EPR effect
Angiogenic
vessels
Ineffective
lymphatic drainage
Tumour
Lymphatic vessel
E n l a
r g e m
e n t o
f t u m
o u r
c e l l
x
Figure 1 Schmc ps d mchsms whch cs c d ds ms. Pmc pcs shw s ps
cs (ccs). Pss ss s chd xs pcs hh csd pm h m sc d c
mphc d (ePr c). ac c (s) c chd c h sc pcs wh ds h pm c-spcfc
c d d. th pcs c () s h cs cs pxm h cs; () ch h mm h c d c s
xc ssd-s d dp; () h c.

8/3/2019 Dan Peer et al- Nanocarriers as an emerging platform for cancer therapy
http://slidepdf.com/reader/full/dan-peer-et-al-nanocarriers-as-an-emerging-platform-for-cancer-therapy 4/10
REVIEW ARTICLE
754 nature nanotechnology | VOL 2 | DECEMBER 2007 | www.nature.com/naturenanotechnology
process, scFv antibodies have been identied or superior bindingand internalization properties or prostate cancer cells33.
It is possible to increase the ecacy o antibodies by conjugatinga therapeutic agent directly to it or targeted delivery. For example,in 2000, the chemotherapeutic drug, calicheamicin, which isconjugated with the anti-CD33 antibody (marketed under the tradename Mylotarg), was the rst clinically approved ormulation thattargets cancerous cells. Others include Zevalin and Bexxar, whichuse anti-CD20 antibodies to target radioisotopes to cancer cells(able 1). Although the ecacy o these therapies has been proven,lethal side eects have been observed, likely due to non-specicbinding34 between the targeting agent and non-target moietieson the cell surace. Another reason could be the interaction o thetargeting agent with its target expressed on non-cancerous cells. Forexample, BR96-doxorubicin — an immunoconjugate linked withdoxorubicin and comprising an antibody that targets and binds
to the Lewis-Y antigen (expressed on 75% o all breast cancers) —demonstrated signicant anti-tumour activity in mouse tumourmodels. BR96-doxorubicin showed lower toxicity than that resultingrom doxorubicin alone and it was ecacious in these animalmodels35. However, in dogs, an acute enteropathy (pathology o theintestine) was observed presumably due to binding o the conjugate toLewis-Y-related antigens expressed by non-targeted gastrointestinalepithelial cells. In Phase II human clinical studies, BR96-doxorubicinimmunoconjugates had limited anti-tumour activity and causedsevere gastrointestinal toxicity, leading to termination o the study 36.
Although using genomics and proteomics technology to chooseappropriate targets is an active area o research, to date no clinically eective targets have been identied. Creating new technologies toenhance selectivity and targeting ecacy with existing targets seemmore promising. For example, usion proteins can be created by
combining two or more genes to produce a new protein with desiredproperties. Antibodies can be engineered so they bind to their target
with high anity, and using molecular biology techniques, it is possibleto design protein-based ligand mimetics based on the structure o areceptor. Dimerization o proteins or peptides can increase ligandanity through divalency — two simultaneous binding events,usually involving concurrent binding o a protein or a peptide to thetwo Fc domains o an antibody (Fig 2b). For example, dimerization o a low-anity scFv (also known as diabody) against the ErbB2, led toenhanced tumour localization in a mouse tumour model37.
It is also possible to increase binding anity and selectivity to cell surace targets by engineering proteins that detect a specicconormation o a target receptor. In a recent in vivo study using ausion protein consisting o an scFv antibody ragment to target anddeliver small interering RNA (siRNA) to lymphocytes — a typeo white blood cell — a 10,000-old increased anity or the target
receptor, integrin LFA-1, was observed18
. Integrin LFA-1 is usually present in a low-anity non-adhesive orm on naïve leukocytes(white blood cells that are not activated by cancer cells or pathogensthat enter the body), but converts to the high-anity adhesive ormthrough conormational changes on activation o the immune system.Tereore, targeting the high-anity orm o LFA-1 enables drugs tobe selectively delivered to the activated and adhesive leukocytes. Newclasses o targeting molecules can be engineered to target specicconormations. Tese include small protein domains, known asabodies, that can be engineered to bind specically to dierenttarget proteins in a conormational-sensitive manner. Other smallproteins that act like antibodies — called avimers — are used to bindselectively to target receptors through multivalent eects. Nanobodies,which are heavy-chain antibodies engineered to one tenth o the sizeo an intact antibody with a missing light chain, have been used to
Antibody
F(ab')2 Fab' ScFv Diabody Non-antibody
ligand
Aptamer
Ligand
Liganddimerization
Binding
surface
Alpha helix
bundle
Binding
domain 1
Binding
domain 2
Binding
domain 3
Affibody
Avimer
Fc
-s-s-
-s-s-
- s - s -
- s - s -
- s - s -
- s - s -
CH
CL
VL
VH
FC
Figure 2 Cmm s d ws mp h f d sc. a, th p shws mcs sch s mc d
ds’ ms, -d ds, d pms. th d ms (′) 2 d ′ d mc c whs h ′, sc, d
sc (dd) ms cd mc chqs. vH: h ch; vl: h ch; CH: cs h ch; Cl: cs h
ch. n-d ds cd ms, chds, ppds, d h ps. apms c cmpsd h Dna rna. b, af d sc c
csd hh d dm sc cm-ss s sch s fds, ms d ds, s w s c ds
d h ms.

8/3/2019 Dan Peer et al- Nanocarriers as an emerging platform for cancer therapy
http://slidepdf.com/reader/full/dan-peer-et-al-nanocarriers-as-an-emerging-platform-for-cancer-therapy 5/10
REVIEW ARTICLE
nature nanotechnology | VOL 2 | DECEMBER 2007 | www.nature.com/naturenanotechnology 755
bind to carcinoembryonic antigen (CEA), a protein used as a tumour
marker38–40
(Fig. 2b).In addition to the rational design o antibodies, high-throughput approaches have been used to generate targeting agentssuch as aptamers, which are short single-stranded DNA or RNAoligonucleotides selected in vitro rom a large number o randomsequences (∼1014–1015). Aptamers are selected to bind to a wide variety o targets, including intracellular proteins, transmembrane proteins,soluble proteins, carbohydrates, and small molecule drugs. Severalaptamers have also been developed to bind specically to receptorson cancer cells, and thus may be suitable or nanoparticle-aptamerconjugate therapy 41. For example, docetaxel (Dtxl)-encapsulatednanoparticles whose surace is modied with an aptamer that targetsthe antigen on the surace o prostate cancer cells, were delivered withhigh selectivity and ecacy in vivo42.
Growth actor or vitamin interactions with cancer cells representa commonly used targeting strategy, as cancer cells oen overexpressthe receptors or nutrition to maintain their ast-growing metabolism.Epidermal growth actor (EGF) has been shown to block and reducetumour expression o the EGF receptor, which is overexpressedin a variety o tumour cells such as breast and tongue cancer 43.Additionally, based on the same idea, the vitamin olic acid (olate)has also been used or cancer targeting because olate receptors (FRs)are requently overexpressed in a range o tumour cells includingovarian, endometrial and kidney cancer44. ranserrin () interactswith receptors (Rs), which are overexpressed on a variety o tumour cells (including pancreatic, colon, lung, and bladder cancer)owing to increased metabolic rates45. Direct coupling o thesetargeting agents to nanocarriers containing chemotherapies such asdrugs has improved intracellular delivery and therapeutic outcome
in animal tumour models46–48. One challenge with targeting receptors
whose expression correlates with metabolic rate, such as olate and ,is that these receptors are also expressed in ast-growing healthy cellssuch as broblasts, epithelial and endothelial cells. Tis could lead tonon-specic targeting and subsequently decrease the eectiveness o the drug and increase toxicity 49.
Te use o peptides as targeting agents — including arginine–glycine–aspartic acid (RGD), which is the ligand o the cell adhesionintegrin α vβ3 on endothelial cells — results in increased intracellulardrug delivery in dierent murine tumour models50,51. However, RGDalso binds to other integrins such as α5β1 and α4β1 and thereore isnot specic to cancer cells, which may limit its use. In addition tocell surace antigens, extracellular matrices (ECMs) overexpressedin tumours, such as heparin sulphate, chondroitin sulphate, andhyaluronan (HA), may also serve as eective targets or specic
ECM receptors52,53
. Coating liposomes with HA improves circulationtime and enhances targeting to HA receptor-expressing tumoursin vivo54,55.
tHe arSenal o nanoCarrierS
Nanocarriers are nanosized materials (diameter 1–100 nm) that cancarry multiple drugs and/or imaging agents. Owing to their highsurace-area-to-volume ratio, it is possible to achieve high liganddensity on the surace or targeting purposes. Nanocarriers canalso be used to increase local drug concentration by carrying thedrug within and control-releasing it when bound to the targets.Currently, natural and synthetic polymers and lipids are typically used as drug delivery vectors; clinically approved ormulationsare listed in able 1. Te amily o nanocarriers includes polymer
Table 2 Examples of nano-based platforms and their current stage of development for use in cancer therapy
tp c d m dm (m) D ppd kd C s dpm tp cc ( cc s) rcs
Pm–d cjs (6–15) Dxc, Pcx, Cmphc,P, tnP-470
12 pdcs d cc s
(Phss i–iii) d in vivo
vs ms rwd 3, 61
lpsms (h Peg d -Peg cd)(85–100)
lc, pm cmpds,
amc
S pdcs cc s(Phss i–iii) d in vivo
Sd ms, c ccm,mshm, d cmphsc km
rwd 9
Pmc pcs
(50–200)
Dxc, Pcx, pm-
sd ds, Dcx
S pdcs cc s
(Phss i–iii) d in vivo
adccm h sphs,
msc s cc d cmphsc km
5, 91, 100, 101
Pmsms (~100) Dxc, Pcx In vivo 73, 74
Mcs (pd sd d pmc)(5–100)
Dxc Cc s (Phs i) Msc c sdms c cchmhp
77, 92, 102
Pcx Cc s (Phs i) Pcc, dc, sc dcc ccs
Pm-sd ds (cp/
csp), Cmphc, tmx,epc
In vivo d in vitro rwd 75
nshs (gd-sc) (~130) n d ( phhm hp) In vivo 37, 103gd pcs (10–40) n d ( phhm ) In vivo 104
ncs (30–40) n d Chms, sc ss din vitro 90, 105
Ddms (~ 5) Mhx In vitro / in vivo 46, 86imm-Peg-psms (100) Dxc Cc s (Phs i) Msc smch cc 76immpsms (100–150) Dxc, pm-sd ds,
vs, vcs, tpc,Pcx
In vivo rwd 9, 106
immxs, immpms, d
s ps (3–15)
vs ds, xs Cc s (Phss i–iii) vs ps cc rwd 16, 17, 61

8/3/2019 Dan Peer et al- Nanocarriers as an emerging platform for cancer therapy
http://slidepdf.com/reader/full/dan-peer-et-al-nanocarriers-as-an-emerging-platform-for-cancer-therapy 6/10
REVIEW ARTICLE
756 nature nanotechnology | VOL 2 | DECEMBER 2007 | www.nature.com/naturenanotechnology
conjugates, polymeric nanoparticles, lipid-based carriers such asliposomes and micelles, dendrimers, carbon nanotubes, and goldnanoparticles, including nanoshells and nanocages (Fig. 3a). Tesenanocarriers have been explored or a variety o applications suchas drug delivery, imaging, photothermal ablation o tumours,radiation sensitizers, detection o apoptosis, and sentinel lymph-node mapping3,4,56 (able 2).
o date, at least 12 polymer–drug conjugates have entered Phase Iand II clinical trials (able 2 and Fig. 3a) and are especially useul ortargeting blood vessels in tumours. Examples include anti-endothelialimmunoconjugates, usion proteins57–59, and caplostatin, the rstpolymer-angiogenesis inhibitor conjugates60. Polymers that arechemically conjugated with drugs are oen considered new chemicalentities (NCEs) owing to a distinct pharmacokinetic prole rom
Figure 3 exmps cs cc. a, a wh d s pss h m cmps pc cd c,
m cjd h c, d c (sch s h dsd chmhpc ds). b, Schmc dm h d cj d pm pcsss.
th chmhpcs cd d h c, s h s pm–d cjs, ddms d sm pc cs, h cd ppd
sd h c.
Immuno-toxin/drug
fusion protein
Carbon nanotube Micelles
Polymer-conjugate
drug/protein
Nanobased carriers
for cancer detection
and therapy
Dendrimers
Nanoshells Liposomes Polymeric carriers
Drug
conjugation
Drug
entrapmentLigand-bound
nanocarrier
Liposome
Biodegradable polymer
Chemotherapeutic
Surface functionality
Targeting molecule
(aptamers,antibodies and their fragments)
Spacer group/
long circulating agent
Inorganic particle
Metallic shell
Amphipathic molecule
Dendrimer
Carbon nanotube

8/3/2019 Dan Peer et al- Nanocarriers as an emerging platform for cancer therapy
http://slidepdf.com/reader/full/dan-peer-et-al-nanocarriers-as-an-emerging-platform-for-cancer-therapy 7/10
REVIEW ARTICLE
nature nanotechnology | VOL 2 | DECEMBER 2007 | www.nature.com/naturenanotechnology 757
that o the parent drug. Despite the variety o novel drug targets andsophisticated chemistries available, only our drugs (doxorubicin,camptothecin, paclitaxel, and platinate) and our polymers(N-(2-hydroxylpropyl)methacrylamide (HPMA) copolymer, poly-L-glutamic acid, poly(ethylene glycol) (PEG), and Dextran) have beenrepeatedly used to develop polymer–drug conjugates3,61.
Polymers are the most commonly explored materials orconstructing nanoparticle-based drug carriers. One o the earliest
reports o their use or cancer therapy dates back to 197962 whenadsorption o anticancer drugs to polyalkylcyanoacrylatenanoparticles was described. Couvreur et al. revealed the releasemechanism o the drugs rom the polymer in cal serum, ollowedby tissue distribution and ecacy studies in a tumour model63. Tiswork laid the oundation or the development o doxorubicin-loadednanoparticles that were tested in clinical trials in the mid-1980s64.Polymeric nanoparticles can be made rom synthetic polymers,including poly(lactic acid) (PLA) and poly(lactic co-glycolic acid)65,or rom natural polymers such as chitosan66 and collagen67 and may beused to encapsulate drugs without chemical modication. Te drugscan be released in a controlled manner through surace or bulk erosion,diusion through the polymer matrix, swelling ollowed by diusion,or in response to the local environment. Several multiunctional
polymeric nanoparticles are now in various stages o pre-clinical andclinical development4,56,68,69. Concerns arising rom the use o polymer-based nanocarriers include the inherent structural heterogeneity o polymers, reected, or example, in a high polydispersity index (theratio o the weight-and-number-average molecular weight ( M w/ M n)).Tere are, however, a ew examples o polymeric nanoparticles thatshow near-homogenous size distribution70.
Lipid-based carriers have attractive biological properties,including general biocompatibility, biodegradability, isolation o drugs rom the surrounding environment, and the ability to entrapboth hydrophilic and hydrophobic drugs. Trough the additiono agents to the lipid membrane or by the alteration o the suracechemistry, properties o lipid-based carriers, such as their size,charge, and surace unctionality, can easily be modied. Liposomes,
polymersomes, and micelles represent a class o amphiphile-basedparticles. Liposomes are spherical, sel-closed structures ormed by one or several concentric lipid bilayers with inner aqueous phases.oday, liposomes are approved by regulatory agencies to carry a rangeo chemotherapeutics26,71,72 (able 1).
Polymersomes have an architecture similar to that o liposomes,but they are composed o synthetic polymer amphiphiles, includingPLA-based copolymers73,74 (able 2). However, as with polymertherapeutics, there are still no clinically approved strategies that useactive cellular targeting or lipid-based carriers.
Micelles, which are sel-assembling closed lipid monolayerswith a hydrophobic core and hydrophilic shell, have beensuccessully used as pharmaceutical carriers or water-insolubledrugs (able 2)75. Tey belong to a group o amphiphilic colloids
that can be ormed spontaneously under certain concentrationsand temperatures rom amphiphilic or surace-active agents(suractants) (Fig. 3a). An example o a polymeric micelle underclinical evaluation is NK911, which is a block copolymer o PEG and poly(aspartic acid). NK911, which consists o a bounddoxorubicin raction (~45%) (Fig. 3b) and a ree drug76, wasevaluated or metastatic pancreatic cancer treatment. Anothercarrier is NK105, a micelle containing paclitaxel, was evaluatedor pancreatic, colonic and gastric tumour treatment77.
Lipid-based carriers pose several challenges, which representgeneral issues in the use o other targeted nanocarriers suchas polymeric nanoparticles. For example, upon intravenousinjection, particles are rapidly cleared rom the bloodstream by the reticuloendothelial deence mechanism, regardless o particlecomposition78,79. Moreover, instability o the carrier and burst drug
release, as well as non-specic uptake by the mononuclear phagocyticsystem (MPS), provides additional challenges or translating thesecarriers to the clinic.
Given their long history, liposome-based carriers serve as aclassic example o the challenges encountered in the development o nanocarriers and the solutions that have been attempted. For example,PEG has been used to improve circulation time by stabilizing andprotecting micelles and liposomes rom opsonization — a plasma
protein deposition process that signals Kuper cells in the liver toremove the carriers rom circulation75,80. However, Daunosomeand Myocet are examples o clinically used liposomes (80–90 nmin diameter) without PEG coating that have been reported toexhibit enhanced circulation times, although to a lesser degree thanPEGylated liposomes such as Doxil/Caelyx (able 1).
In addition to rapid clearance, another challenge is the astburst release o the chemotherapeutic drugs rom the liposomes.o overcome this phenomenon, doxorubicin, or example, may be encapsulated in the liposomal aqueous phase by an ammoniumsulphate gradient81. Tis method achieves a stable drug entrapmentwith negligible drug leakage during circulation, even aer prolongedresidence in the blood stream82. In clinical practice, liposomal systemshave shown preerential accumulation in tumours, via the EPR
eect, and reduced toxicity o their cargo (ables 1 and 2). However,long-circulating liposomes may lead to extravasation o the drug inunexpected sites. Te most commonly experienced clinical toxiceect rom the PEGylated liposomal doxorubicin is palmar-plantarerythrodysesthesia (PPE), also called the hand-oot syndrome.PPE — a dermatologic toxicity reaction seen with high doses o many types o chemotherapy — can be addressed by changing the dosingand scheduling o the treatment83. Other challenges acing the use o liposomes in the clinic include the high production cost, ast oxidationo some phospholipids, and lack o controlled-release properties o encapsulated drugs.
o achieve temporal release o two drugs, polymers andphospholipids can be combined as a single delivery agent (polymercore/lipid shell). Aer locating at a tumour site through the EPR
eect, the outer phospholipid shell releases an anti-angiogenesisagent, and the inner polymeric nanoparticle subsequently releases achemotherapy agent in response to local hypoxia — shortage o oxygen.Tis strategy led to reduced toxicity and enhanced anti-metastaticeects in two dierent mouse tumour models, emphasizing theadvantages o a mechanism-based design or targeted nanocarriers84.
Organic nanoparticles include dendrimers, viral capsids andnanostructures made rom biological building blocks such as proteins.Abraxane is an albumin-bound paclitaxel nanoparticle ormulationapproved by the FDA in 2005 as a second-line treatment or metastaticbreast cancer. Abraxane was designed to address insolubility problemsencountered with paclitaxel. Its use eliminates the need or toxicsolvents like Cremophor EL (polyoxyethylated castor oil), which hasbeen shown to limit the dose o axol that can be administered85.
Dendrimers are synthetic, branched macromolecules that orma tree-like structure whose synthesis represents a relatively new eldin polymer chemistry. Polyamidoamine dendrimers have shownpromise or biomedical applications because they (1) can be easily conjugated with targeting molecules, imaging agents, and drugs,(2) have high water solubility and well-dened chemical structures,(3) are biocompatible, and (4) are rapidly cleared rom the bloodthrough the kidneys, made possible by their small size (<5 nm), whicheliminates the need or biodegradability.In vivodelivery o dendrimer–methotrexate conjugates using multivalent targeting results in atenold reduction in tumour size compared with that achieved withthe same molar concentration o ree systemic methotrexate22,46.Tis work provided motivation or urther pre-clinical development,and a variety o dendrimers are now under investigation or cancertreatment and are extensively reviewed elsewhere86,87. Although

8/3/2019 Dan Peer et al- Nanocarriers as an emerging platform for cancer therapy
http://slidepdf.com/reader/full/dan-peer-et-al-nanocarriers-as-an-emerging-platform-for-cancer-therapy 8/10
REVIEW ARTICLE
758 nature nanotechnology | VOL 2 | DECEMBER 2007 | www.nature.com/naturenanotechnology
promising, dendrimers are more expensive than other nanoparticlesand require many repetitive steps or synthesis, posing a challenge orlarge-scale production.
Inorganic nanoparticles are primarily metal based and havethe potential to be produced with near monodispersity. Inorganicmaterials have been extensively studied or magnetic resonanceimaging and high-resolution superconducting quantum intererencedevices88. Inorganic particles may also be unctionalized to introduce
targeting molecules and drugs. Specic types o recently developedinorganic nanoparticles include nanoshells and gold nanoparticles.
Nanoshells (100–200 nm) may use the same carrier or bothimaging and therapy (able 2). Tey are composed o a silica coreand a metallic outer layer. Nanoshells have optical resonances thatcan be adjusted to absorb or scatter essentially anywhere in theelectromagnetic spectrum, including the near inrared region (NIR,820 nm, 4 W cm–2), where transmission o light through tissue isoptimal. Absorbing nanoshells are suitable or hyperthermia-basedtherapeutics, where the nanoshells absorb radiation and heat up thesurrounding cancer tissue. Scattering nanoshells, on the other hand,are desirable as contrast agents or imaging applications. Recently,a cancer therapy was developed based on absorption o NIR lightby nanoshells, resulting in rapid localized heating to selectively
kill tumours implanted in mice. issues heated above the thermaldamage threshold displayed coagulation, cell shrinkage and loss o nuclear staining, which are indicators o irreversible thermal damage,whereas control tissues appeared undamaged37,89.
A similar approach involves gold nanocages which are smaller(<50 nm) than the nanoshells. Tese gold nanocages (able 2) can beconstructed to generate heat in response to NIR light and thus may also be useul in hyperthermia-based therapeutics90. Unlike nanoshellsand nanocages, pure gold nanoparticles (able 2) are relatively easy to synthesize and manipulate. Non-specic interactions thatcause toxicity in healthy tissues may impede the use o many typeso nanoparticles, but using inorganic particles or photo-ablationsignicantly limits non-specic toxicity because light is locally directed. However, inorganic particles may not provide advantages
over other types o nanoparticles or systemic targeting o individualcancer cells because they are not biodegradable or small enough to becleared easily, resulting in potential accumulation in the body, whichmay cause long-term toxicity.
tHe CHallengeS o MultiDrug reSiStanCe
Te delivery o drugs through targeted nanocarriers that areinternalized by cells provides an alternative route to diusion o drugsinto cells. Tis approach may allow targeted carriers to bypass theactivity o integral membrane proteins, known as MDR transporters,which transport a variety o anticancer drugs out o the cancer cell andproduce resistance against chemotherapy 11. Te molecular basis o cancer drug resistance is complex and has been correlated to elevated
levels o enzymes that can neutralize chemotherapeutic drugs. Moreoen, however, it is due to the overexpression o MDR transportersthat actively pump chemotherapeutic drugs out o the cell and reducethe intracellular drug doses below lethal threshold levels. Becausenot all cancer cells express the MDR transporters, chemotherapy will kill only drug-sensitive cells that do not or only mildly expressMDR transporters, while leaving behind a small population o drug-resistant cells that highly express MDR transporters. With tumourrecurrence, chemotherapy may ail because residual drug-resistantcells dominate the tumour population.
Among the MDR transporters, the most widely investigatedproteins are: P-glycoprotein (also reerred to as MDR1 or ABCB1); themultidrug resistance associated proteins (MRPs), o which the moststudied is the MRP1 (or ABCC1); and the breast cancer resistanceprotein (ABCG2). Tese proteins have dierent structures, but they
share a similar unction o expelling chemotherapy drugs rom thecells12. Several studies have demonstrated the possibility o usingnanocarriers to bypass the MDR transporters. SP1049C is a non-ionic(pluronic or also known as poloxamer) block copolymer composedo a hydrophobic core and hydrophilic tail that contains doxorubicin.SP1049C has been shown to circumvent p-glycoprotein-mediated drugresistance in a mouse model o leukaemia and is now under clinicalevaluation91,92. Folate receptor-mediated cell uptake o doxorubicin–
loaded liposomes into an MDR cell line was shown to be unaected by P-glycoprotein (Pgp)-mediated drug efux, in contrast to the uptake o ree doxorubicin93. In an attempt to reverse MDR, vincristine-loadedlipid nanoparticles conjugated to an anti-Pgp mAb (MRK-16), showedgreater cytotoxicity in resistant human myelogenous leukaemia celllines than control non-targeted particles — a response attributed tothe inhibition o the Pgp-mediated efux o vincristine by MRK-1694.Additional reports have addressed the challenge o MDR using polymertherapeutics95, polymeric nanoparticles96, lipid nanocapsules97 andmicelles98 within cell lines or in mouse tumour models. Combinationtreatments with targeted nanocarriers or selective delivery o drugsand MDR pump inhibitors will likely address some o the problemsposed by resistant tumours.
into tHe uture
Te choice o an appropriate nanocarrier is not obvious, and theew existing comparative studies are dicult to interpret becauseseveral actors may simultaneously aect biodistribution andtargeting. In addition, developing suitable screening methodologiesor determining optimal characteristics o nanocarriers remainselusive. Tereore, successul targeting strategies must be determinedexperimentally on a case-by-case basis, which is laborious. Inaddition, systemic therapies using nanocarriers require methodsthat can overcome non-specic uptake by mononuclear phagocyticcells and by non-targeted cells. It is also not clear to what extent thisis possible without substantially increasing the complexity o thenanocarrier and without inuencing commercial scale-up. Improved
therapeutic ecacy o targeted nanocarriers has been established inmultiple animal models o cancer, and currently more than 120 clinicaltrials are underway with various antibody-containing nanocarrierormulations99. For the clinician, in addition to enhancing condencethrough the ability to image the type and location o the tumour, itis imperative to construct appropriate therapeutic regimens. Whentargeting cell surace markers presents a signicant challenge, asin the case or solid tumours, targeting tumour vasculature or theextracellular matrix surrounding the tumour microenvironment may be necessary. In the case o circulating cancer cells, as in leukaemiaand lymphoma, a therapy that targets surace antigens with highanity and includes a carrier with a long circulating hal-lie may be the most ecacious. Similar to combination drug strategies thatmay be personalized to optimize treatment regimens, oncologists in
the near uture may be presented with the ability to choose specicnanocarrier/targeting molecule combinations which could lead toimproved therapeutic outcomes and reduced costs.
Although we are still ar rom Nobel Prize winner Paul Ehrlich’s‘magic bullet’, many believe that we will soon enter an era in whichnanocarrier-based approaches will represent an important modality within therapeutic and diagnostic oncology.
d:10.1038/.2007.387
rcs1. Stewart, B. W. & Kleihues, P.World Cancer Report (World Health Organization Press, Geneva, 2003).
2. Cancer Facts & Figures 2007 (American Cancer Society, Atlanta, 2007).
3. Duncan, R. Polymer conjugates as anticancer nanomedicines. Nat. Rev. Cancer 6, 688–701 (2006).
4. Ferrari, M. Cancer nanotechnology : opportunitie s and challenges. Nat. Rev. Cancer 5,
161–171 (2005).

8/3/2019 Dan Peer et al- Nanocarriers as an emerging platform for cancer therapy
http://slidepdf.com/reader/full/dan-peer-et-al-nanocarriers-as-an-emerging-platform-for-cancer-therapy 9/10
REVIEW ARTICLE
nature nanotechnology | VOL 2 | DECEMBER 2007 | www.nature.com/naturenanotechnology 759
5. Couvreur, P. & Vauthier, C. Nanotechnology: Intelligent design to treat complex disease. Pharm. Res.
23, 1417–1450 (2006).
6. Alonso, M. J. Nanomedicines or overcoming biological barriers. Biomed. Pharmacother. 58,
168–172 (2004).
7. Matsumura, Y. & Maeda, H. A new concept or macromolecular therapeutics in cancer-
chemotherapy — Mechanism o tumoritropic accumulation o proteins and the antitumor agent
smancs. Cancer Res. 46, 6387–6392 (1986).
8. Yuan, F. et al. Vascular-permeability in a human tumor xenogra — Molecular-size dependence
and cuto size. Cancer Res. 55, 3752–3756 (1995).
9. orchilin, V. P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov.
4,145–160 (2005).10. Hobbs, S. K. et al. Regulation o transport pathways in tumor vessels: role o tumor type and
microenvironment. Proc. Natl Acad. Sci. USA 95, 4607–4612 (1998).
11. Gottesman, M. M., Fojo, . & Bates, S. E. Multidrug resistance in cancer : Role o AP-dependent
transporters. Nat. Rev. Cancer 2, 48–58 (2002).
12. Peer, D. & Margalit, R. Fluoxetine and reversal o multidrug resistance. Cancer Lett. 237,
180–187 (2006).
13. Jain, R. K. Barriers to drug-delivery in solid tumors. Sci. Am. 271, 58–65 (1994).
14. de Menezes, D. E. L., Pilarski, L. M. & Allen, . M. In vitro and in vivo targeting o
immunoliposomal doxorubicin to human B-cell lymphoma. Cancer Res. 58, 3320–3330 (1998).
15. Park, J. W. et al. Anti-HER2 immunoliposomes: enhanced ecacy attributable to targeted delivery.
Clin. Cancer Res. 8, 1172–1181 (2002).
16. Allen, . M. Ligand-targeted therapeutics in anticancer therapy. Nat. Rev. Cancer 2, 750–763 (2002).
17. Pastan, I., Hassan, R., FitzGeral d, D. J. & Kreitman, R. J. Immunotoxin therapy o cancer.
Nat. Rev. Cancer 6, 559–565 (2006).
18. Peer, D., Zhu, P., Carman, C. V., Lieberman, J. & Shimaoka, M. Selective gene silencing in activated
leukocytes by targeting siRNAs to the integrin lymphocyte unction-associated antigen-1.
Proc. Natl Acad. Sci. USA 104, 4095–4100 (2007).
19. Sapra, P. & Allen, . M. Internalizing antibodies are necessary or improved therapeutic ecacy o
antibody-targeted liposomal drugs. Cancer Res. 62, 7190–7194 (2002).
20. Allen, . M. Long-circulating (sterically stabilized) liposomes or targeted drug-delivery.
rends Pharmacol. Sci. 15, 215–220 (1994).
21. Adams, G. P. et al. High anity restricts the localization and tumor penetration o single-chain Fv
antibody molecules. Cancer Res. 61, 4750–4755 (2001).
22. Hong, S. et al. Te binding avidity o a nanoparticle-based multivalent targeted drug delivery
platorm. Chem. Biol. 14, 107–115 (2007).
23. Warenius, H. M., Galre, G., Bleehen, N. M. & Milstein, C. Attempted targeting o A monoclonal-
antibody in a human-tumor xenogra system. Eur. J. Cancer Clin. Oncology 17, 1009–1015 (1981).
24. von Mehren, A. G., Weiner L. M. Monoclonal antibody therapy or cancer. Annu. Rev. Med. 54,
343–369 (2003).
25. Weiner, L. M. & Adams, G. P. New approaches to antibody therapy. Oncogene 19, 6144–6151 (2000).
26. Gabizon, A. A. Pegylated liposomal doxorubicin: metamorphosis o an old drug into a new orm o
chemotherapy. Cancer Invest. 19, 424–436 (2001).
27. James, J. S. & Dubs, G. FDA approves new kind o lymphoma treatment. AIDS reat. News 284,
2–3 (1997).
28. Albanell, J. & Baselga, J. rastuzumab, a humanized anti-HER2 monoclonal antibody, or the
treatment o breast cancer. Drugs oday 35, 931–946 (1999).
29. Ferrara, N. VEGF as a therapeutic target in cancer. Oncology 69 (Suppl. 3), 11–16 (2005).
30. Carter, P. Improving the ecacy o antibody-based cancer therapies. Nat. Rev. Cancer 1, 118–129 (2001).
31. Marks, J. D. Selection o internalizing antibodies or drug delivery. Methods Mol. Biol. 248,
201–208 (2004).
32. Marks, J. D. et al. Human-antibody ragments specic or human blood-groups antigens rom a
phage display library. Bio-echnol. 11, 1145–1149 (1993).
33. Liu, B., Conrad, F., Cooperberg, M. R., Kirpotin , D. B. & Marks, J. D. Mapping tumor epitope space
by direct selection o single-chain Fv antibody libraries on prostate cancer cells. Cancer Res. 64,
704–710 (2004).
34. Arnold, D. M. et al. Systematic review: ecacy and saety o rituximab or adults with idiopathic
thrombocytopenic purpura. Ann. Intern. Med. 146, 25–33 (2007).
35. rail, P. A. et al. Cure o xenograed human carcinomas by Br96-doxorubicin immunoconjugates.
Science 261, 212–215 (1993).
36. olcher, A. W. et al. Randomized phase II study o BR96-doxorubicin conjugate in patients with
metastatic breast cancer. J. Clin. Oncology 17, 478–484 (1999).
37. Hirsch, L. R. et al. Nanoshell-mediated near-inrared thermal therapy o tumors under magnetic
resonance guidance. Proc. Natl Acad. Sci. USA 100, 13549–13554 (2003).
38. Silverman, J. et al. Multivalent avimer proteins evolved by exon shufing o a amily o human
receptor domains. Nat. Biotechnol. 23, 1556–1561 (2005).
39. Cortez-Re tamozo, V. et al. Ecient cancer therapy with a nanobody-based conjugate. Cancer Res.
64, 2853–2857 (2004).
40. Nord, K. et al. Binding proteins selected rom combinatorial libraries o an alpha-helical bacterial
receptor domain. Nat. Biotechnol. 15, 772–777 (1997).
41. White, R. R., Sullenger, B. A. & Rusconi, C. P. Developing aptamers into therapeutics. J. Clin. Invest.
106, 929–934 (2000).
42. Farokhzad, O. C. et al. argeted nanoparticle-aptamer bioconjugates or cancer chemotherapy in
vivo. Proc. Natl Acad. Sci. USA 103, 6315–6320 (2006).
43. Sanlippo, J. S. et al. Quantitative analyses o epidermal growth actor receptors, HER-2/neu
oncoprotein and cathepsin D in nonmalignant and malignant uteri. Cancer 77, 710–716 (1996).
44. Antony, A. C. Te biological chemistry o olate receptors. Blood 79, 2807–2820 (1992).
45. Prost, A. C. et al. Dierential transerrin receptor density in human colorectal cancer: A potential
probe or diagnosis and therapy. Int. J. Oncol. 13, 871–875 (1998).
46. Kukowska-Latal lo, J. F. et al. Nanoparticle targeting o anticancer drug improves therapeutic
response in animal model o human epithelial cancer. Cancer Res. 65, 5317–5324 (2005).
47. Iinuma, H. et al. Intracellular targeting therapy o cisplatin-encapsulated transerrin-polyethylene
glycol liposome on peritoneal dissemination o gastric cancer. Int. J. Cancer 99, 130–137 (2002).
48. Ishida, O. et al. Liposomes bearing polyethyleneglycol-coupled transerrin with intracellular
targeting property to the solid tumors in vivo. Pharm. Res. 18, 1042–1048 (2001).
49. Ekblom, P., Tesle, I., Lehto, V. P. & Virtanen, I. Distribution o the transerrin receptor in normal
human-broblasts and bro-sarcoma cells. Int. J. Cancer 31, 111–117 (1983).
50. Li, J. et al. Fusion protein rom RGD peptide and Fc ragment o mouse immunoglobulin G inhibits
angiogenesis in tumor. Cancer Gene Ter. 11, 363–370 (2004).
51. Ruoslahti, E. Cell adhesion and tumor metastasis. Princess akamatsu Symp. 24, 99–105 (1994).
52. Peer, D. & Margalit, R. umor-targeted hyaluronan nanoliposomes increase the antitumor activity
o liposomal Doxorubicin in syngeneic and human xenogra mouse tumor models. Neoplasia 6,
343–353 (2004).
53. Hu, Z., Sun, Y. & Garen, A. argeting tumor vasculature endoth elial cells and tumor cells orimmunotherapy o human melanoma in a mouse xenogra model. Proc. Natl Acad. Sci. USA 96,
8161–8166 (1999).
54. Peer, D. & Margalit, R. Loading mitomycin C inside long circulating hyaluronan targeted
nano-liposomes increases its antitumor activity in three mice tumor models. Int. J. Cancer 108,
780–789 (2004).
55. Eliaz, R. E. & Szoka, F. C. Jr. Liposome-encapsulated doxorubicin targeted to CD44: a strategy to
kill CD44-overexpressing tumor cells. Cancer Res. 61, 2592–2601 (2001).
56. LaVan, D. A., McGuire, . & Langer, R. Small-scal e systems or in vivo drug delivery.
Nat. Biotechnol. 21, 1184–1191 (2003).
57. Arap, W., Pasqualini, R. & Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor
vasculature in a mouse model. Science 279, 377–380 (1998).
58. Schraa, A. J. et al. argeting o RGD-modied proteins to tumor vasculature: A pharmacokinetic
and cellular distribution study. Int. J. Cancer 102, 469–475 (2002).
59. Halin, C. et al. Enhancement o the antitumor activity o interleukin-12 by targeted delivery to
neovasculature. Nat. Biotechnol. 20, 264–269 (2002).
60. Satchi-Fainaro, R. et al. argeting angiogenesis with a conjugate o HPMA copolymer and NP-
470. Nat. Med. 10, 255–261 (2004).
61. Satchi-Fainaro, R., Duncan, R. & Barnes, C. M. in Polymer Terapeutics II: Polymers as Drugs,
Conjugates and Gene Delivery Systems Vol. 193 (eds Satchi-Fainaro, R. & Duncan, R.) 1–65
(Springer-Verlag, Berlin, 2006).
62. Couvreur, P., Kante, B., Roland, M. & Speiser, P. Adsorption o anti-neoplast ic drugs to
polyalkylcyanoacrylate nanoparticles and their release in cal serum. J. Pharm. Sci. 68,
1521–1524 (1979).
63. Couvreur, P. et al. issue distribution o anti-tumor drugs associated with polyalkylcyanoacrylate
nanoparticles. J. Pharm. Sci. 69, 199–202 (1980).
64. Couvreur, P., Kante, B., Grislain, L., Roland, M. & Speiser, P. oxicity o polyalkylcyanoacry late
nanoparticles II: Doxorubicin-loaded nanoparticles. J. Pharm. Sci. 71, 790–792 (1982).
65. Hrkach, J. S., Peracchia, M. ., Domb, A., Lotan, N. & Langer, R. Nanotechnology or biomaterials
engineering: Structural characterization o amphiphilic polymeric nanoparticles by H-1 NMR
spectroscopy. Biomaterials 18, 27–30 (1997).
66. Calvo, P., RemunanLopez, C., VilaJato, J. L. & Alonso, M. J. Chitosan and chitosan ethylene oxide
propylene oxide block copolymer nanoparticles as novel carriers or proteins and vaccines.
Pharm. Res. 14, 1431–1436 (1997).
67. Elsamaligy, M. S. & Rohdewald, P. Reconstituted collagen nanoparticles, a novel drug carrier
delivery system. J. Pharm. Pharmacol. 35, 537–539 (1983).
68. Moses, M. A., Brem, H. & Langer, R. Advancing the eld o drug delivery : taking aim at cancer.
Cancer Cell 4, 337–341 (2003).
69. Farokhzad, O. C. & Langer, R. Nanomedicine: Developing smarter therapeutic and diagnostic
modalities. Adv. Drug Deliv. Rev. 58, 1456–1459 (2006).
70. Guo, R. et al. Synthesis o alginic acid-poly[2-(diethylamino)ethyl methacrylate] monodispersed
nanoparticles by a polymer-monomer pair reaction system. Biomacromolecules 8, 843–850 (2007).
71. Gabizon, A. A. Stealth liposomes and tumor targeting: one step urther in the quest or the magic
bullet. Clin. Cancer Res. 7, 223–225 (2001).
72. Sara, . et al. Pegylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients
reaching or exceeding cumulative doses o 500 mg/m2. Ann. Oncol. 11, 1029–1033 (2000).
73. Ahmed, F. et al. Shrinkage o a rapidly growing tumor by drug-loaded polymersomes: pH-triggered
release through copolymer degradation. Mol. Pharm. 3, 340–350 (2006).
74. Discher, D. E. & Ahmed, F. Polymersomes. Annu. Rev. Biomed. Eng. 8, 323–341 (2006).
75. Matsumura, Y. et al. Phase I clinical trial and pharmacokinetic evaluation o NK911, a micelle-
encapsulated doxorubicin.Brit. J. Cancer 91, 1775–1781 (2004).
76. Kato, K. et al. Phase I study o NK105, a paclitaxel-incorporating micellar nanoparticle, in patients
with advanced cancer. J. Clin. Oncol 24 (suppl.), 2018 (2006).
77. orchilin, V. P. Micellar nanocarriers: Pharmaceutical perspectiv es. Pharm. Res. 24,
1–16 (2007).
78. Brigger, I., Dubernet, C. & Couvreur, P. Nanoparticles in cancer therapy and diagnosis. Adv. Drug
Deliv. Rev. 54, 631–651 (2002).
79. Kreuter, J. & Higuchi, . Improved delivery o methoxsalen. J. Pharm. Sci. 68, 451–454 (1979).
80. Papahadjopoul os, D. et al. Sterically stabilized liposomes - improvements in pharmacokinetics and
antitumor therapeutic ecacy. Proc. Natl Acad. Sci. USA 88, 11460–11464 (1991).
81. Haran, G., Cohen, R., Bar, L. K. & Barenholz, Y. ransmembrane ammonium-sulate gradients in
liposomes produce ecient and stable entrapment o amphipathic weak bases. Biochim. Biophys.
Acta 1151, 201–215 (1993).
82. Gabizon, A. A., Shmeeda, H. & Zalipsky, S. Pros and cons o the liposome platorm in cancer drug
targeting. J. Liposome Res. 16, 175–183 (2006).
83. Lorusso, D. et al. Pegylated liposomal doxorubicin-related palmar-plantar erythrodysesthesia
(‘hand-oot’ syndrome). Ann. Oncol. (2007).
84. Sengupta, S. et al. emporal targeting o tumour cells and neovasculature with a nanoscale delivery
system. Nature 436, 568–572 (2005).
85. Damascelli, B. et al. Intraarterial chemotherapy with polyoxyethylated castor oil ree paclitaxel,
incorporated in albumin nanoparticles (ABI-007). Cancer 92, 2592–2602 (2001).
86. Gillies, E. R. & Frechet, J. M. J. Dendrimers and dendritic polymers in drug delivery. Drug Discov.
oday 10, 35–43 (2005).

8/3/2019 Dan Peer et al- Nanocarriers as an emerging platform for cancer therapy
http://slidepdf.com/reader/full/dan-peer-et-al-nanocarriers-as-an-emerging-platform-for-cancer-therapy 10/10
REVIEW ARTICLE
760 nature nanotechnology | VOL 2 | DECEMBER 2007 | www.nature.com/naturenanotechnology
87. Malik, N. et al. Dendrimers: Relationship between structure and biocompatibility in vitro, and
preliminary studies on the biodistribution o I-125-labelled polyamidoamine dendrimers in vivo.
J. Control. Release 65, 133–148 (2000).
88. Morawski, A. M., Lanza, G. A. & Wickline, S. A. argeted contrast agents or magnetic resonance
imaging and ultrasound. Curr. Opin. Biotechnol. 16, 89–92 (2005).
89. Loo, C., Lowery, A., Halas, N., West, J., Drezek, R. Immunotargeted nanoshells or integrated
cancer imaging and therapy. Nano Lett. 5, 709–711 (2005).
90. Chen, J. et al. Gold nanocages: Bioconjugation and their potential use as optical imaging contrast
agents. Nano Lett. 5, 473–477 (2005).
91. Danson, S. et al. Phase I dose escalation and pharmacokinetic study o pluronic polymer-bound
doxorubicin (SP 1049C) in patients with advanced cancer. Brit. J. Cancer 90,
2085–2091 (2004).92. Batrakova, E. V. et al. Anthracycline antibiotics non-covalently incorporated into the block
copolymer micelles: In vivo evaluation o anti-cancer activity. Brit. J. Cancer 74, 1545–1552 (1996).
93. Goren, D. et al. Nuclear delivery o doxorubicin via olate-targeted liposomes with bypass o
multidrug-resistance efux pump. Clin. Cancer Res. 6, 1949–1957 (2000).
94. Matsuo, H. et al. Possibility o the reversal o multidrug resistance and the avoidance o side eects
by liposomes modied with MRK-16, a monoclonal antibody to P-glycoprotein. J. Control. Release
77, 77–86 (2001).
95. Duncan, R., Vicent, M. J., Greco, F. & Nicholson, R. I. Polymer-drug conjugates: towards a novel
approach or the treatment o endrocine-related cancer. Endocrine-Relat. Cancer 12, S189–S199 (2005).
96. Wong, H. L. et al. A new polymer-lipid hybrid nanoparticle system increases cytotoxicity o doxorubicin
against multidrug-resistant human breast cancer cells. Pharm. Res. 23,1574–1585 (2006).
97. Garcion, E. et al. A new generation o anticancer, drug-loaded, colloidal vectors reverses multidrug
resistance in glioma and reduces tumor progression in rats. Mol. Cancer Ter. 5, 1710–1722 (2006).
98. Lee, E. S., Na, K. & Bae, Y. H. Doxorubicin loaded pH-sensitive polymeric micelles or reversal o
resistant MCF-7 tumor. J. Control. Release 103, 405–418 (2005).
99. Sapra, P. & Allen, . M. Ligand-targeted liposomal anti cancer drugs. Prog. Lipid Res. 42,
439–462 (2003).
100. Moghimi, S. M. Recent developments in polymeric nanoparticle engineering and their
applications in experimental and clinical oncology. Anticancer Agents Med. Chem. 6,
553–561 (2006).
101. Lee, K. S. et al. Multicenter phase II trial o Genexol-PM, a Cremophor-ree, polymeric
micelle ormulation o paclitaxel, in patients with metastatic breast cancer.
Breast Cancer Res. reat. (2007).
102. Nakanishi, . et al. Development o the polymer micelle carrier system or doxorubicin. J. Control.
Release 74, 295–302 (2001).
103. Hirsch, L. R. et al. Metal nanoshells. Ann. Biomed. Engin. 34,
15–22 (2006).104. Sokolov, K. et al. Real-time vital optical imaging o precancer using anti-epidermal growth actor
receptor antibodies conjugated to gold nanoparticles. Cancer Res. 63, 1999–2004 (2003).
105. Chen, J. Y. et al. Facile synthesis o gold-silver nanocages with controllable pores on the surace.
J. Am. Chem. Soc. 128, 14776–14777 (2006).
106. Kontermann, R. E. Immunolliposomes or cancer therapy. Curr. Opin. Mol. Ter. 8, 39–45 (2006).
ackwdmsWe would like to acknowledge Shiladitya Sengupta or critically reviewing the manuscript and
Maeve Cullinane or helpul discussions. This work was supported by ederal unds NIH/NCI
CA119349, NIH/NIBIB EB 003647, and NIH R01-EB000244. The content is solely the responsibility o
the authors and does not necessarily represent the ofcial view o the NIH.
Cmp fc ssThe authors declare competing fnancial interests: details accompany the ull-text HTML version o the
paper at www.nature.com/naturenanotechnology.





























