Embed Size (px)
Citation preview

CYSTIC FIBROSIS
by
[Caroline Karunya Ponnarasi Kanagaraj]
Group-IV
Tbilisi state medical university
Date:14-12-2011


IntroductionI like to start with the story of "65 Roses" :is what little children suffering from Cystic Fibrosis call their disease. As the following story illustrates, the words are much easier for children to pronounce.Mary G. Weiss became a volunteer for the Cystic Fibrosis Foundation in 1965 after learning that her three little boys had Cystic Fibrosis. Her duty was to call every civic club, social and service organization seeking financial support for CF research. Mary's four year old son, Richard, listened closely to his mother as she made each call. After several calls, Richard came into the room and told his mom, "I know what you are working for." . With some trepidation, Mary posed the question back to Richard, "What am I working for, Richard?" "You are working for '65 Roses'," he answered so sweetly. Mary was speechless. She went over to him and tenderly pressed his tiny body to hers. He could not see the tears running down Mary's cheeks as she stammered, "Yes, Richard, I'm working for '65 Roses'." So that is the diseases we are going to see below:
DefinitionCystic fibrosis is a disease passed down through families that causes thick, sticky mucus to build up in the lungs, digestive tract, and other areas of the body. It is one of the most common chronic lung diseases in children and young adults. It is a life-threatening disorder.
Causes, incidence and risk factors
Cystic fibrosis (CF) is caused by a defective gene which causes the body to produce abnormally thick and sticky fluid, called mucus. This mucus builds up in the breathing passages of the lungs and in the pancreas, the organ that helps to break down and absorb food.
This collection of sticky mucus results in life-threatening lung infections and serious digestion problems. The disease may also affect the sweat glands and a man's reproductive system.
Millions of Americans carry the defective CF gene, but do not have any symptoms. That's because a person with CF must inherit two defective CF genes -- one from each parent. An estimated 1 in 29 Caucasian Americans have the CF gene. The disease is the most common, deadly, inherited disorder affecting Caucasians in the United States. It's more common among those of Northern or Central European descent.
Most children with CF are diagnosed by age 2. A small number, however, are not diagnosed until age 18 or older. These patients usually have a milder form of the disease.
Epidemiology
• About 1,000 new cases of cystic fibrosis are diagnosed each year.
• More than 70% of patients are diagnosed by age two.
• More than 45% of the CF patient population is age 18 or older.

• The predicted median age of survival for a person with CF is in the late 30s.
• Most prevalent among the Caucasians:1 in 25-30 Caucasians is a carrier making disease prevalance 1 in 3300
• Mutation of ∆F508 allele is the most common cause of the disorder among the Caucasians
• Almost 2500 children with CF are born each year
• The disease is less frequent in other ethnic groups
Types of cystic fibrosis
Cystic fibrosis is classified in to:
Classic
Non classic
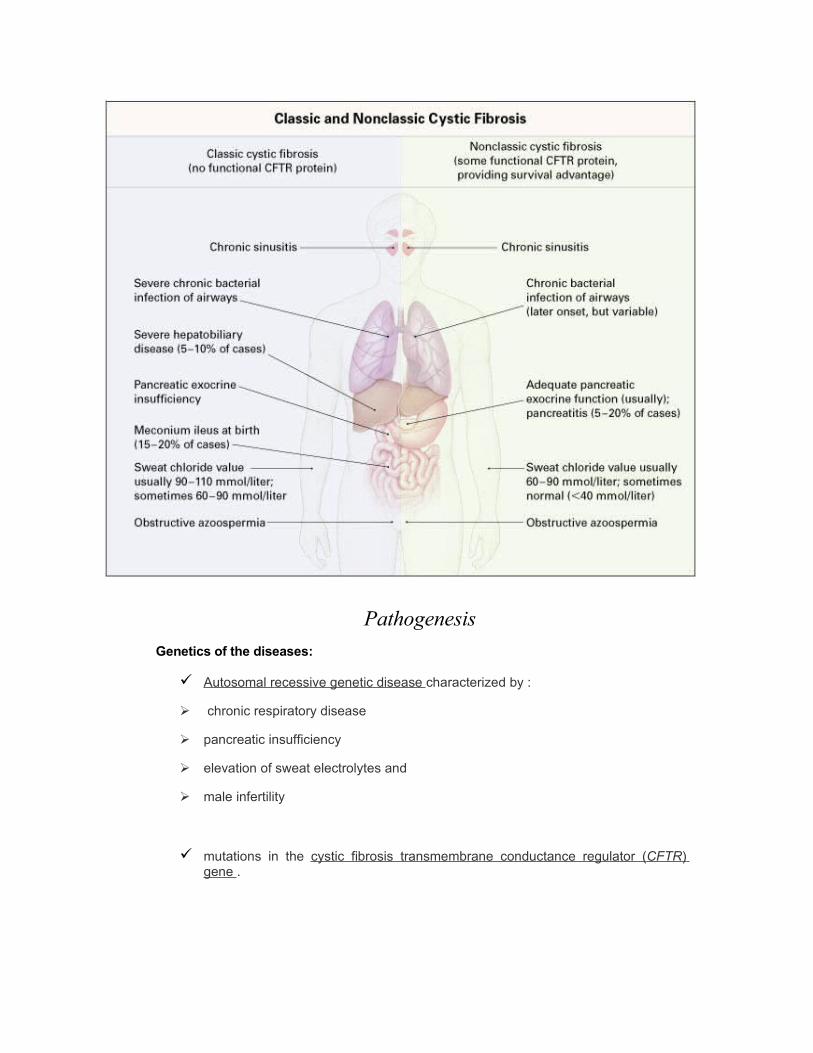
Pathogenesis
Genetics of the diseases:
Autosomal recessive genetic disease characterized by :
chronic respiratory disease
pancreatic insufficiency
elevation of sweat electrolytes and
male infertility
mutations in the cystic fibrosis transmembrane conductance regulator ( CFTR ) gene .

CFTR gene:
Locus : 7q31.2 - The CFTR gene is found in region q31.2 on the long (q) arm of human chromosome 7.
Gene Structure : The normal allelic variant for this gene is about 250,000 bp long and contains 27 exons.
Gene Function : It is predicted that it codes a Integral membrane protein of about 170kb know as CFTRANSMEMBRANE CONDUCTANCE REGULATOR(CFTR).
CFTR Protein:CFTR protein is a single polypeptide chain, containing 1480 amino acids, that appears to function both as a cyclic AMP-regulated Cl-channel and, as its name implies, a regulator of other ion channels. CFTR transports chloride ions (Cl-) ions across the membranes of cells in the lungs,
liver, pancreas, digestive tract, reproductive tract, and skin.
The CFTR chloride channels has FIVE DOMAINS:
1. Two membrane –spanning domains : Each with six transmembrane sequences;
2. Two nucleotide(ATP)-binding domains;
3. Regulatory domain with multiple phosphorylation site.
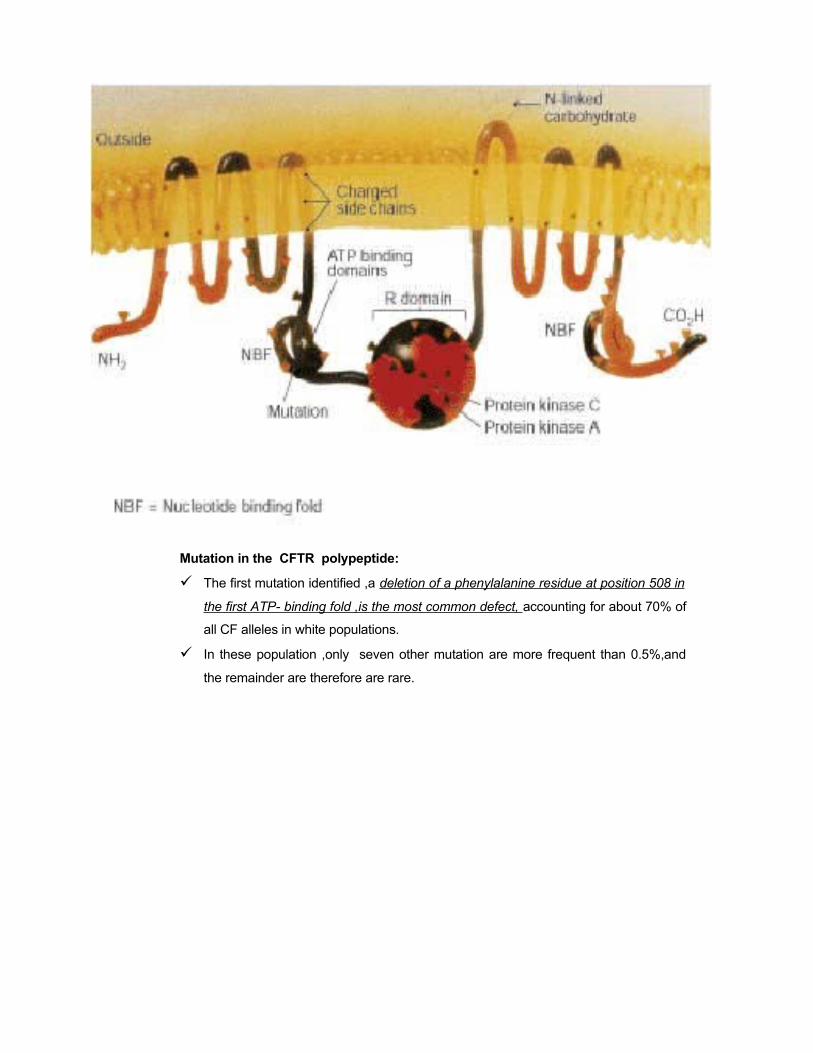
Mutation in the CFTR polypeptide:
The first mutation identified ,a deletion of a phenylalanine residue at position 508 in
the first ATP- binding fold ,is the most common defect, accounting for about 70% of
all CF alleles in white populations.
In these population ,only seven other mutation are more frequent than 0.5%,and
the remainder are therefore are rare.

Classes of CF gene:
Class 1 mutations : Defective protein production with premature termination of
CFTR protein production. Class 1 mutations produce few or no functioning CFTR
chloride channels
Class 2 mutations : Defective trafficking of CFTR so that it does not reach the apical
surface membrane where it is intended to function
Class 3 mutations : Defective regulation (opening and closing) of the CFTR
chloride channel which allows movement of chloride in and out of the cell even
though the CFTR protein is able to reach the apical cell surface
Class 4 mutations: CFTR reaches the apical surface but conduction (passage of
chloride ions through the channel) is defective
Class 5 mutations : Associated with reduced synthesis of functional CFTR
Class 6 mutations :proteins are synthesized normally but are unstable at the cell
surface.
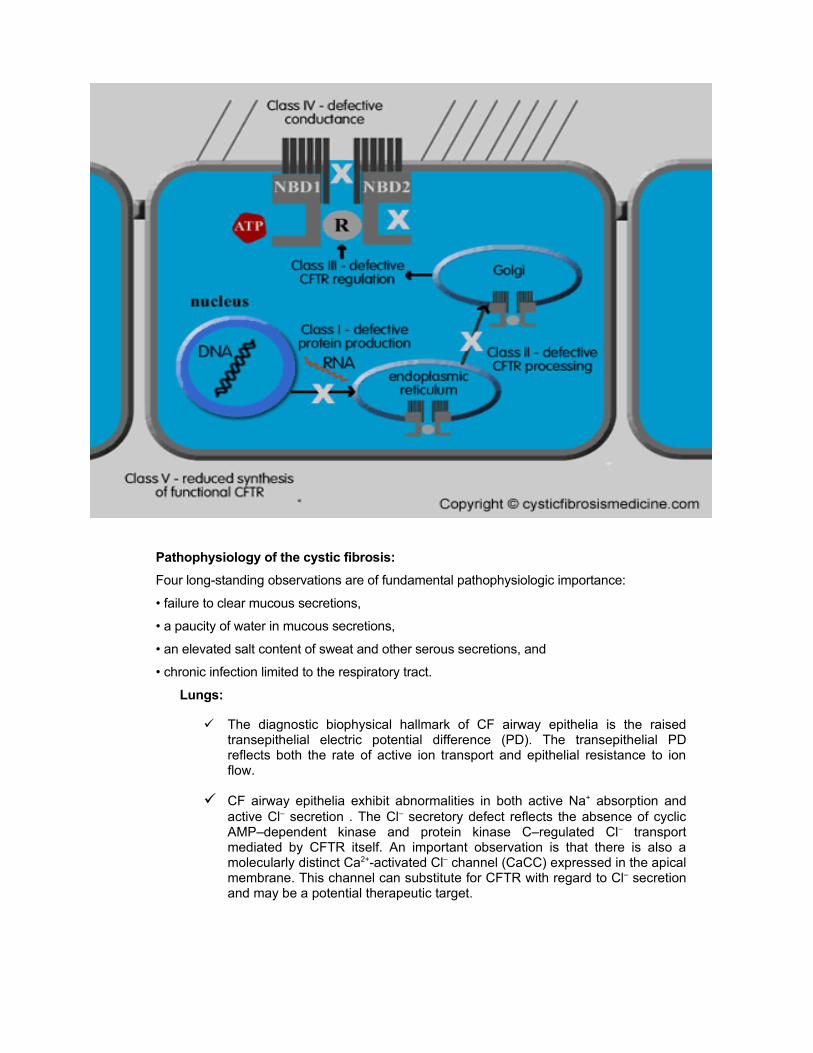
Pathophysiology of the cystic fibrosis:
Four long-standing observations are of fundamental pathophysiologic importance:
• failure to clear mucous secretions,
• a paucity of water in mucous secretions,
• an elevated salt content of sweat and other serous secretions, and
• chronic infection limited to the respiratory tract.
Lungs:
The diagnostic biophysical hallmark of CF airway epithelia is the raised transepithelial electric potential difference (PD). The transepithelial PD reflects both the rate of active ion transport and epithelial resistance to ion flow.
CF airway epithelia exhibit abnormalities in both active Na+ absorption and active Cl– secretion . The Cl– secretory defect reflects the absence of cyclic AMP–dependent kinase and protein kinase C–regulated Cl– transport mediated by CFTR itself. An important observation is that there is also a molecularly distinct Ca2+-activated Cl– channel (CaCC) expressed in the apical membrane. This channel can substitute for CFTR with regard to Cl– secretion and may be a potential therapeutic target.

Abnormal regulation of Na+ absorption is a key feature of CF airway epithelia. This abnormality reflects a second function of CFTR, its function as a tonic inhibitor of the epithelial Na+ channel. The molecular mechanisms mediating this action of CFTR remain unknown.
Mucus clearance is the primary innate airways defense mechanism against infection by inhaled bacteria. Normal airways vary the rates of active Na+
absorption and Cl– secretion to adjust the volume of liquid (water), i.e., "hydration," on airway surfaces for efficient mucus clearance. The central hypothesis of CF airways pathophysiology is that the faulty regulation of Na+
absorption and inability to secrete Cl– via CFTR reduce the volume of liquid on airway surfaces; i.e., they are "dehydrated." Both the thickening of mucus and the depletion of the periciliary liquid lead to adhesion of mucus to the airway surface. Mucus adhesion leads to a failure to clear mucus from the airways both by ciliary and airflow-dependent (cough) mechanisms. The absence of a strict correspondence between gene mutation class and severity of lung disease suggests important roles for modifier genes and gene-environmental interactions.
The infection that characterizes CF airways involves the mucus layer rather than epithelial or airway wall invasion. The predisposition of CF airways to chronic infection by Staphylococcus aureus and Pseudomonas aeruginosa is consistent with failure to clear mucus. Recently it has been demonstrated that the O2 tension is very low in CF mucus, and adaptations to hypoxia are important determinants of the physiology of bacteria in the CF lung. Indeed, both mucus stasis and mucus hypoxia may contribute to the propensity for Pseudomonas to grow in biofilm colonies within mucus plaques adherent to CF airway surfaces.
Gastrointestinal tract:
The gastrointestinal effects of CF are diverse. In the exocrine pancreas, the absence of the CFTR Cl– channel in the apical membrane of pancreatic ductal epithelia limits the function of an apical membrane Cl–-HCO3
– exchanger to secrete bicarbonate and Na+ (by a passive process) into the duct. The failure to secrete Na+
HCO3– and water leads to retention of enzymes in the pancreas and
ultimately destruction of virtually all pancreatic tissue.
The CF intestinal epithelium, because of the lack of Cl– and water secretion, fails to flush secreted mucins and other macromolecules from intestinal crypts. The diminished CFTR-mediated secretion of liquid may be exacerbated by excessive absorption of liquid, reflecting abnormalities of CFTR-mediated regulation of Na+
absorption (both mediated by Na+ channels and possibly other Na+
transporters, e.g., Na+-H+ exchangers). Both dysfunctions lead to desiccated intraluminal contents and obstruction of both the small and large intestine.
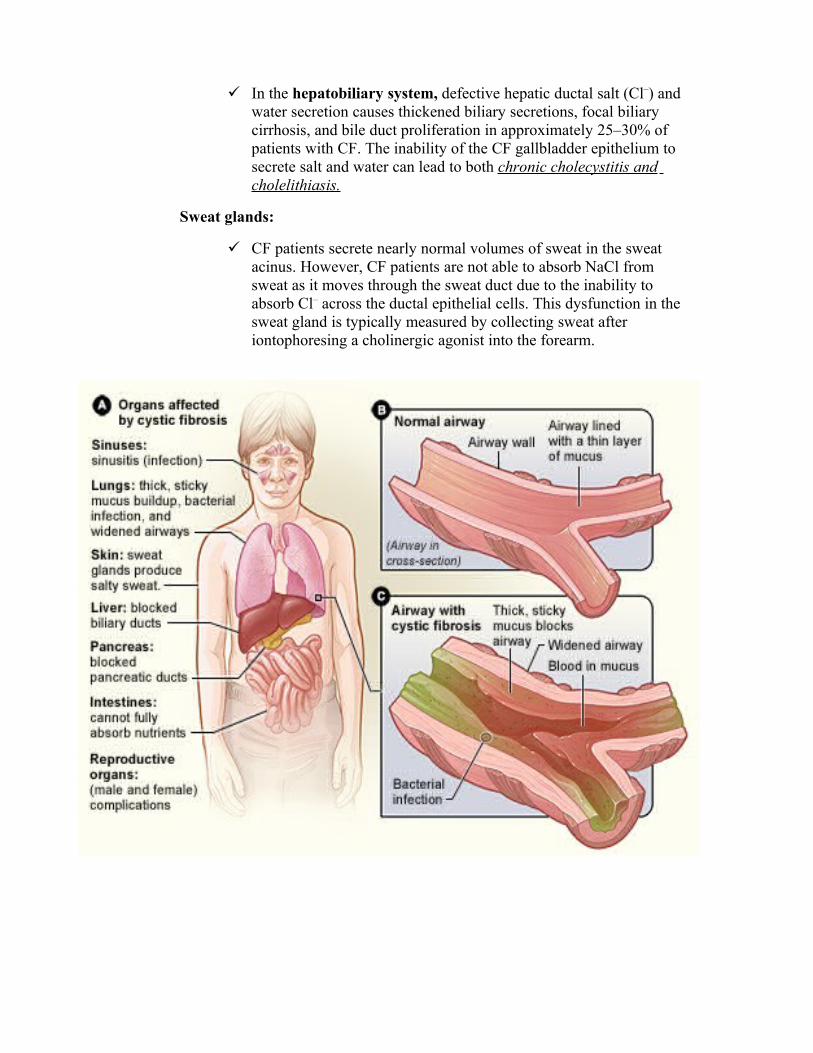
In the hepatobiliary system, defective hepatic ductal salt (Cl–) and water secretion causes thickened biliary secretions, focal biliary cirrhosis, and bile duct proliferation in approximately 25–30% of patients with CF. The inability of the CF gallbladder epithelium to secrete salt and water can lead to both chronic cholecystitis and cholelithiasis.
Sweat glands:
CF patients secrete nearly normal volumes of sweat in the sweat acinus. However, CF patients are not able to absorb NaCl from sweat as it moves through the sweat duct due to the inability to absorb Cl– across the ductal epithelial cells. This dysfunction in the sweat gland is typically measured by collecting sweat after iontophoresing a cholinergic agonist into the forearm.

Clinical features
Approximately 20% of patients present within the first 24 h of life with gastrointestinal obstruction, termed meconium ileus .
within the first year or two of life include respiratory tract symptoms, most prominently cough and/or recurrent pulmonary infiltrates, and failure to thrive.
Respiratory tract:
Upper respiratory:
Chronic sinusitis is common in childhood
nasal obstruction and rhinorrhea.
The occurrence of nasal polyps approaches 25% and often requires treatment with topical steroids and/or surgery
Lower respiratory:
cough -persistent and produces viscous, purulent, often greenish-colored sputum.
"exacerbations," defined by increased cough, weight loss, low-grade fever, increased sputum volume, and decrements in pulmonary function.
Over the course of years, the exacerbations become more frequent and the recovery of lost lung function incomplete, leading to respiratory failure.
CF patients exhibit a characteristic sputum microbiology.
Haemophilus influenzae
S. aureus
. P. aeruginosa,
Burkholderia
The earliest chest x-ray change in CF lungs is hyperinflation, reflecting small-airways obstruction. Later, signs of luminal mucus impaction, bronchial cuffing, and finally bronchiectasis, e.g., ring shadows, are noted. For reasons that remain speculative, the right upper lobe displays the earliest and most severe changes.

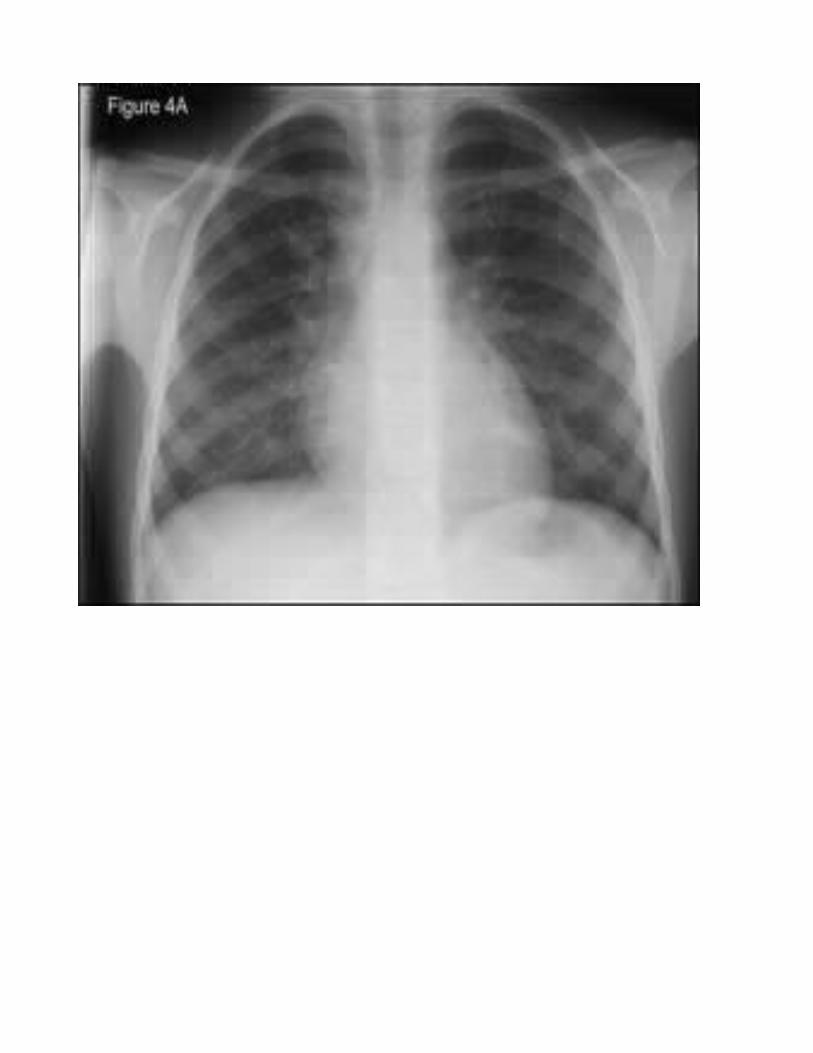

Gastrointestinal tract:
Intestine:
meconium ileus in infants -abdominal distention, failure to pass stool, and emesis. The abdominal flat plate can be diagnostic, with small-intestinal air-fluid levels, a granular appearance representing meconium, and a small colon
Pancreas:
Exocrine pancreatic insufficiency occurs in >90% of patients with CF.

Insufficient pancreatic enzyme secretion yields the typical pattern of protein and fat malabsorption, with frequent, bulky, foul-smelling stools.
Pancreatic beta cells are spared early, but function decreases with age. This effect, plus inflammation-induced insulin resistance, causes hyperglycemia and a requirement for insulin in >15% of older patients with CF (>35 years).
Symptoms in newborns may include:
Delayed growth Failure to gain weight normally during childhood No bowel movements in first 24 to 48 hours of life Salty-tasting skin
Symptoms related to bowel function may include:
Belly pain from severe constipation
Increased gas, bloating, or a belly that appears swollen (distended)
Nausea and loss of appetite
Stools that are pale or clay colored, foul smelling, have mucus, or that float
Weight loss
Symptoms related to the lungs and sinuses may include:
Coughing or increased mucus in the sinuses or lungs
Fatigue
Nasal congestion caused by nasal polyps
Recurrent episodes of pneumonia. Symptoms in someone with cystic fibrosis include:
Fever
Increased coughing

Increased shortness of breath
Loss of appetite
More sputum
Sinus pain or pressure caused by infection or polyps
Symptoms that may be noticed later in life:
Infertility (in men)
Repeated inflammation of the pancreas (pancreatitis)
Respiratory symptoms
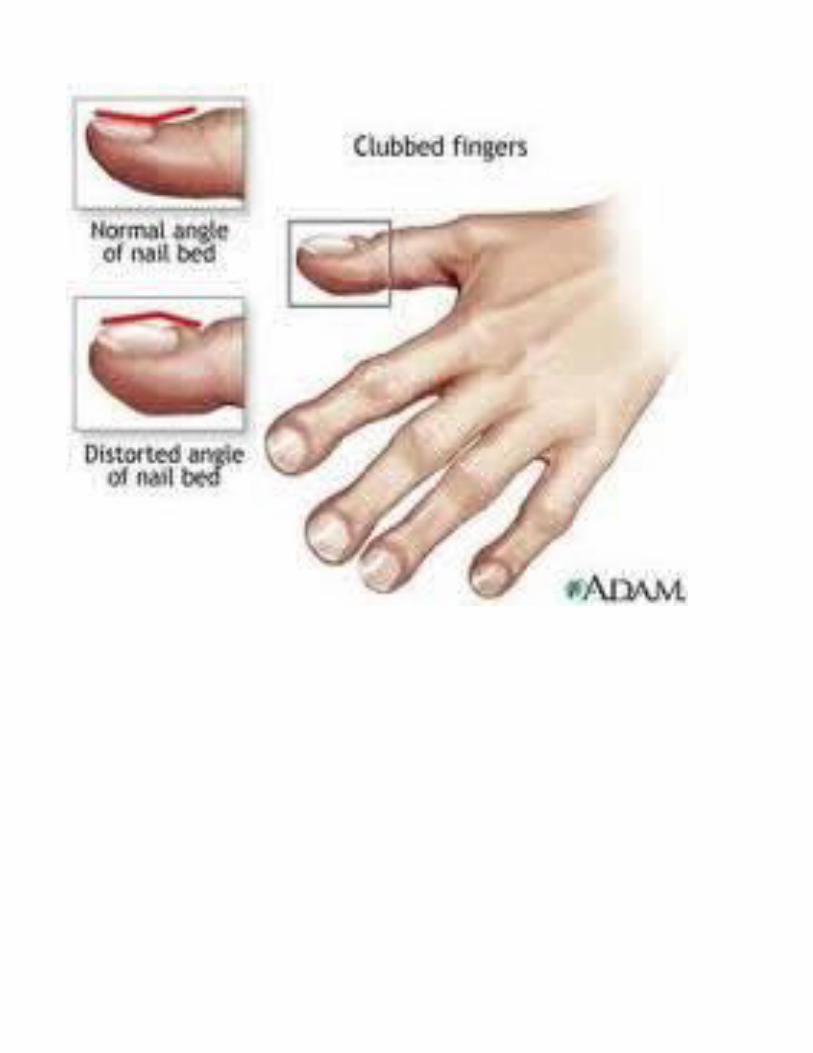

DiagnosisNewborn ScreeningSweat TestGenetic tests A chest x-rayLung function testsPrenatal ScreeningCystic Fibrosis Carrier Testing
Treatment
Gene therapyThe time-honored techniques for clearing pulmonary secretions are breathing exercises, flutter valves, and chest percussionPharmacologic agents for increasing mucus clearance are in use and in development.(eg:Human DNAses)Antibiotics are used for treating lung infectionpancreatic enzyme replacement
PrognosisLife expectancy depends on the severity of disease and the parts of the body involved.
Prognosis for patients with cystic fibrosis has improved dramatically over the past three decades. In the United States, median survival age is now 28.9 years.
Lung disease has the strongest impact on illness severity. Death usually is caused by lung complications.
The average life span of a person with cystic fibrosis has increased to 35 years. Some people with mild symptoms live much longer.
Conclusion“Woe is the child who tastes salty from a kiss on the brow, for he is cursed , and soon must die” After coming to know about these diseases and the worst thinking's of the mankind about a child ,I think you would have got the motivation to live long and stay strong for anything and you will realize that your life is not that tough compare to those who are suffering from an incurable diseases. Till now doctors all around through world are still finding medicine and ways to cure this diseases
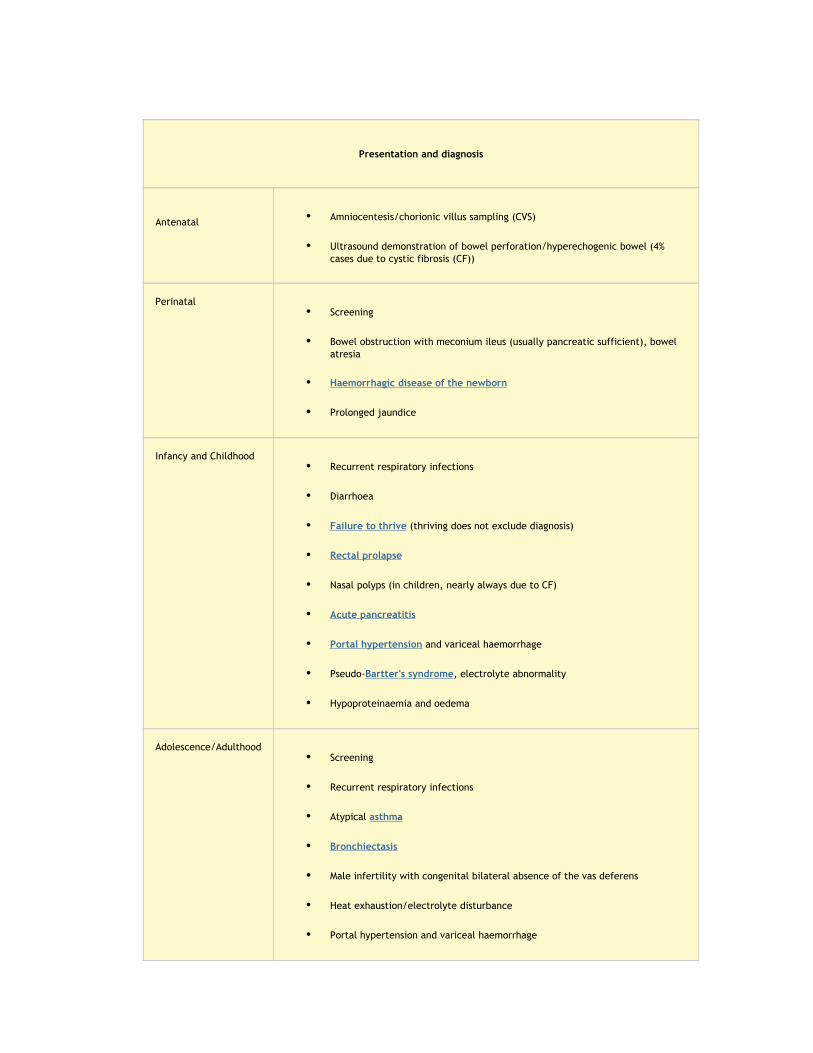
Presentation and diagnosis
Antenatal • Amniocentesis/chorionic villus sampling (CVS)
• Ultrasound demonstration of bowel perforation/hyperechogenic bowel (4% cases due to cystic fibrosis (CF))
Perinatal• Screening
• Bowel obstruction with meconium ileus (usually pancreatic sufficient), bowel atresia
• Haemorrhagic disease of the newborn
• Prolonged jaundice
Infancy and Childhood• Recurrent respiratory infections
• Diarrhoea
• Failure to thrive (thriving does not exclude diagnosis)
• Rectal prolapse
• Nasal polyps (in children, nearly always due to CF)
• Acute pancreatitis
• Portal hypertension and variceal haemorrhage
• Pseudo-Bartter's syndrome, electrolyte abnormality
• Hypoproteinaemia and oedema
Adolescence/Adulthood• Screening
• Recurrent respiratory infections
• Atypical asthma
• Bronchiectasis
• Male infertility with congenital bilateral absence of the vas deferens
• Heat exhaustion/electrolyte disturbance
• Portal hypertension and variceal haemorrhage

Reference
Harrison text book
Davidson text book
http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001167/
http://easypediatrics.com/pathophysiology-of-cystic-fibrosis
http://www.mayoclinic.com/health/cystic-fibrosis/DS00287













