Embed Size (px)
Citation preview

CXCL9, but not CXCL10, Promotes CXCR3-DependentImmune-Mediated Kidney Disease
Julia Menke,* Geraldine C. Zeller,* Eriya Kikawada,* Terry K. Means,† Xiao R. Huang,‡
Han Y. Lan,‡ Bao Lu,§ Joshua Farber,� Andrew D. Luster,† and Vicki R. Kelley*
*Laboratory of Molecular Autoimmune Disease, Renal Division, Brigham and Women’s Hospital, †Center forImmunology and Inflammatory Diseases, Division of Rheumatology, Allergy and Immunology, MassachusettsGeneral Hospital, Harvard Medical School, and §Perlmutter Laboratory, Children’s Hospital and Harvard MedicalSchool, Boston, Massachusetts; ‡Department of Medicine, University of Hong Li Ka Shing Facility of Medicine, HongKong, China; and �Inflammation Biology Section, Laboratory of Molecular Immunology, National Institute of Allergyand Infectious Disease, National Institute of Child Health and Human Development, National Institute of Health,Bethesda, Maryland
ABSTRACTChemokines are instrumental in macrophage- and T cell–dependent diseases. The chemokine CCL2promotes kidney disease in two models of immune-mediated nephritis (MRL-Faslpr mice and thenephrotoxic serum nephritis model), but evidence suggests that multiple chemokines are involved. Foridentification of additional therapeutic targets for immune-mediated nephritis, chemokine ligands andreceptors in CCL2�/� and wild-type (WT) MRL-Faslpr kidneys were profiled. The focus was on intrarenalchemokine ligand/receptor pairs that were highly upregulated downstream of CCL2; the chemokineCXCL10 and its cognate receptor, CXCR3, stood out as potential therapeutic targets. However, renaldisease was not suppressed in CXCL10�/� MRL-Faslpr mice, and CXCL10�/� C57BL/6 mice were notprotected from nephrotoxic serum nephritis compared with WT mice. Because CXCR3 engages with theligand CXCL9, CXCR3�/�, CXCL9�/�, and CXCL10�/� B6 mice were compared with WT mice withnephrotoxic serum nephritis. Kidney disease, measured by loss of renal function and histopathology, wassuppressed in both CXCR3�/� and CXCL9�/� mice but not in CXCL10�/� mice. With nephrotoxic serumnephritis, CXCR3�/� and CXCL9�/� mice had fewer intrarenal activated T cells and activated macro-phages. Both IgG glomerular deposits and antigen-specific IgG in serum were reduced in these mice,suggesting that although CXCR3 and CXCL9 initiate nephritis through cell-mediated events, renalinflammation may be sustained by their regulation of IgG. It is concluded that specific blockade of CXCL9or CXCR3 may be a potential therapeutic target for human immune-mediated kidney diseases.
J Am Soc Nephrol 19: 1177–1189, 2008. doi: 10.1681/ASN.2007111179
Chemokines are instrumental in the recruitment,migration, and effector functions of immune cellsduring inflammation.1– 4 Chemokine ligands en-gaging with their cognate receptors promote the in-flux of leukocytes into the kidney, a hallmark ofnephritis.5,6 Although multiple chemokines sharethe same receptor, they are not necessarily redun-dant.7 Because chemokine ligand/receptors are in-duced during inflammation, these molecules areappealing therapeutic targets for nephritis.
Macrophages (Mø) and T cells in the kidney me-diate inflammation.8 MRL-Faslpr mice and nephro-
toxic serum nephritis (NSN) are two distinct T cell–and Mø-dependent mouse models of immune-me-diated nephritis.9 We previously determined thatCCL2 (monocyte chemoattractant protein-1) is
Received November 8, 2007. Accepted January 9, 2008.
Published online ahead of print. Publication date available atwww.jasn.org.
Correspondence: Dr. Vicki Rubin Kelley, Harvard Institutes ofMedicine, 4 Blackfan Circle, Boston, MA 02115. Phone: 617-525-5915; Fax: 617-525-5830; E-mail: [email protected]
Copyright � 2008 by the American Society of Nephrology
BASIC RESEARCH www.jasn.org
J Am Soc Nephrol 19: 1177–1189, 2008 ISSN : 1046-6673/1906-1177 1177

pivotal in promoting renal disease in these models. UsingCCL2�/� MRL-Faslpr mice, we established that tubular/inter-stitial and glomerular disease is suppressed.10 By comparison,tubular/interstitial but not glomerular disease is suppressed inCCL2�/� mice during NSN.10,11 In each model, Mø and T cellsare no longer recruited to sites in the interstitium adjacent totubular epithelial cells (TEC), the major source of CCL2 in WTmice with nephritis10,11; however, Mø and T cells remain inperivascular areas lacking CCL2 but rich in CCL5 (RANTES), achemokine capable of inciting local renal inflammation inMRL-Faslpr mice.12 This suggests that multiple chemokinesdictate the tempo and locale of kidney disease.
To identify the chemokines along with CCL2 that are in-strumental in T cell– and Mø-mediated nephritis, we exten-sively profiled kidneys of CCL2�/� and wild-type (WT) MRL-Faslpr mice during the development of lupus nephritis. Weidentified a highly expressed chemokine ligand/receptor pairinstrumental in attracting T cells during inflammation,CXCL10 (IP-10)/CXCR3.13–16 We report that CXCR3 is ex-pressed on intrarenal activated Mø in addition to T cells duringexperimental immune-mediated kidney disease. Furthermore,the expression of CXCR3 on T cells and Mø seems to mediatetheir recruitment into the kidneys expressing CXCL9 duringNSN. Finally, we determined that CXCR3 and one, CXCL9, butnot another, CXCL10, of its ligands promote T cell– and Mø-dependent nephritis. Thus, CXCL9 and CXCR3 are potentialtherapeutic targets for immune-mediated kidney illnesses.
RESULTS
Multiple Chemokine Ligand/Receptors AreUpregulated in MRL-Faslpr Nephritic KidneysTo identify potential therapeutic chemokine ligand/receptortargets in MRL-Faslpr mice, we compared intrarenal chemo-kine ligand/receptor transcripts in mice before (2 mo of age)and after (5 mo of age) onset of nephritis. The majority ofintrarenal chemokine ligands (18 of 23) and chemokine recep-tors (10 of 16) we evaluated increased with advancing nephritis(Supplemental Figure 1). We focused on the groups with thehighest increase in intrarenal transcript expression. Withinthis group of chemokine ligands, we detected an increase inCXCL10 (nine-fold), CXCL9 (45-fold), CXCL11 (10-fold),CXCL13 (133-fold), CCL5 (16-fold), CCL20 (seven-fold), andCX3CL1 (two-fold), and within the group of chemokine re-ceptors, we detected an increase in CXCR3 (nine-fold),CXCR4 (six-fold), CXCR5 (42-fold), CCR2 (10-fold), andCX3CR1 (six-fold; Supplemental Figure 1). Of note, the che-mokine ligand/receptor transcript levels in the MRL-Faslpr andB6 kidneys at 2 mo of age were similar.
Identifying Chemokine Ligand/Receptors Other thanCCL2 that Are Expressed in Nephritic MRL-Faslpr KidneysCCL2 promotes lupus nephritis in MRL-Faslpr mice.10 Toidentify chemokines other than CLL2 that are central to MRL-
Faslpr nephritis, we compared chemokine ligand/receptor tran-scripts in CCL2�/� and WT MRL-Faslpr kidneys. Most(�80%) chemokine ligand/receptor transcripts that were up-regulated in WT MRL-Faslpr nephritic kidneys were sup-pressed in CCL2�/� MRL-Faslpr kidneys (Supplemental Figure2). One possible interpretation is that within the hierarchicalpattern of chemokine ligand/receptor expression regulatingimmune responses, CCL2 is proximal in the chemokine cas-cade leading to nephritis in MRL-Faslpr mice; therefore, ourgoal was to focus on the chemokine ligand/receptors thatmaybe expressed “downstream” of CCL2 in MRL-Faslpr kid-neys during nephritis.
Mø and TEC Are Sources of Intrarenal CXCL10Expression during Lupus Nephritis in MRL-Faslpr MiceCXCL10 engaging with its receptor CXCR3 is a potent T cellchemoattractant.15 Because CXCL10 is among the most highlyupregulated chemokines and may be downstream of CCL2, weexplored the role of CXCL10 in MRL-Faslpr mice. Alveolar Møexpress CXCL10,17 and TEC are a rich source of multiple che-mokines in MRL-Faslpr mice10; therefore, Mø and TEC wereprime candidates in our attempt to identify sources ofCXCL10. We detected CXCL10 in TEC (Figure 1A), leukocytes(Figure 1A, inset), and endothelial cells (data not shown) inMRL-Faslpr mice with nephritis. CXCL10�/� MRL-Faslpr
kidneys served as negative controls. The frequency ofCXCL10� Mø increased (two-fold) in MRL-Faslpr kidneysfrom 3 to 6 mo of age with advancing nephritis as deter-mined by FACS analysis (Figure 1A). By comparison, thefrequency of CXCL10� T cells (CD4�, CD8�) in MRL-Faslpr kidneys, before and during nephritis, was minimal(approximately 2%; data not shown). Similarly, the expres-sion of CXCL10 in primary TEC derived from MRL-Faslpr
mice increased (more than nine-fold) as determined by re-al-time PCR (Figure 1A). Of note, CXCL10� Mø and TEC inMRL-Faslpr kidneys were similar to B6 kidneys (3 mo ofage). Thus, Mø and TEC are intrarenal sources of CXCL10during nephritis in MRL-Faslpr mice.
Frequency of CXCR3� T Cells and Mø Is Increased inMRL-Faslpr Nephritic KidneysBecause there is an increase in the frequency of intrarenalCXCL10� Mø and TEC, we probed for the expression ofCXCR3. The frequency of CD4�, CD8�, and B220� (uniquedouble-negative) T cells and Mø (CD68�) expressing CXCR3in the kidneys of MRL-Faslpr mice increased with nephritis (6mo of age) as compared with non-nephritic MRL-Faslpr and B6mice (2 mo of age; Figure 1B). The increased frequency ofCXCR3� T cells and Mø was not limited to the kidney; theseleukocytes increased in their spleens (p � 0.05; data notshown). Taken together, an increase in intrarenal and extrare-nal CXCR3� T cell subsets and Mø is associated with a rise inintrarenal CXCL10 in nephritic MRL-Faslpr mice.
BASIC RESEARCH www.jasn.org
1178 Journal of the American Society of Nephrology J Am Soc Nephrol 19: 1177–1189, 2008
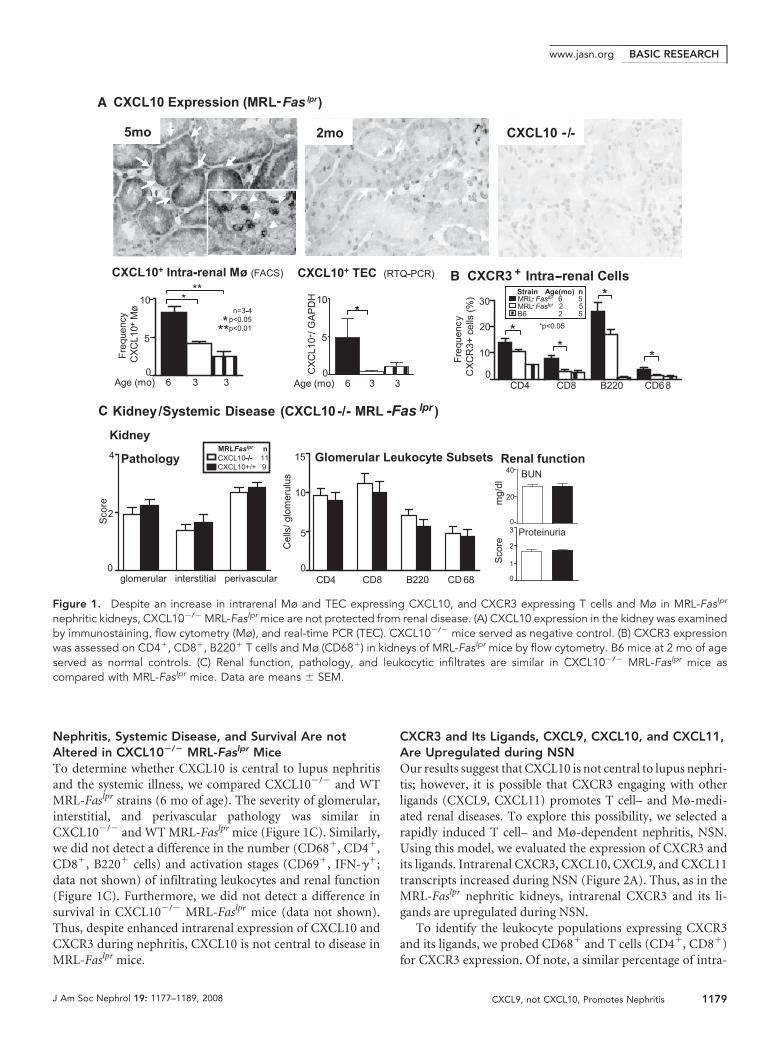
Nephritis, Systemic Disease, and Survival Are notAltered in CXCL10�/� MRL-Faslpr MiceTo determine whether CXCL10 is central to lupus nephritisand the systemic illness, we compared CXCL10�/� and WTMRL-Faslpr strains (6 mo of age). The severity of glomerular,interstitial, and perivascular pathology was similar inCXCL10�/� and WT MRL-Faslpr mice (Figure 1C). Similarly,we did not detect a difference in the number (CD68�, CD4�,CD8�, B220� cells) and activation stages (CD69�, IFN-��;data not shown) of infiltrating leukocytes and renal function(Figure 1C). Furthermore, we did not detect a difference insurvival in CXCL10�/� MRL-Faslpr mice (data not shown).Thus, despite enhanced intrarenal expression of CXCL10 andCXCR3 during nephritis, CXCL10 is not central to disease inMRL-Faslpr mice.
CXCR3 and Its Ligands, CXCL9, CXCL10, and CXCL11,Are Upregulated during NSNOur results suggest that CXCL10 is not central to lupus nephri-tis; however, it is possible that CXCR3 engaging with otherligands (CXCL9, CXCL11) promotes T cell– and Mø-medi-ated renal diseases. To explore this possibility, we selected arapidly induced T cell– and Mø-dependent nephritis, NSN.Using this model, we evaluated the expression of CXCR3 andits ligands. Intrarenal CXCR3, CXCL10, CXCL9, and CXCL11transcripts increased during NSN (Figure 2A). Thus, as in theMRL-Faslpr nephritic kidneys, intrarenal CXCR3 and its li-gands are upregulated during NSN.
To identify the leukocyte populations expressing CXCR3and its ligands, we probed CD68� and T cells (CD4�, CD8�)for CXCR3 expression. Of note, a similar percentage of intra-
CXCL10 Expression (MRL-Fas lpr)
5mo 2mo CXCL10-/-5mo 2mo CXCL10 -/-
CXCL10+ Intra-renal Mø (FACS)
Age (mo) 6 3 3
Fre
quen
cy
CX
CL1
0+M
ø
10
5
0
***
n=3-4p<0.05p<0.01*
**
-
+
--
Age (mo) 6 3 3
CX
CL1
0+/
GA
PD
H 10
5
0
*
CXCL10+ TEC (RTQ-PCR)
*
- CXCR3 + Intra -renal Cells
-
CD4 CD8 B220 CD680
10
20
30
**
*
*p<0.05
Fre
quen
cy
CX
CR
3+ c
ells
(%
) *-
C Kidney/Systemic Disease (CXCL10-/- MRL -Fas lpr )
Glomerular Leukocyte Subsets
Cel
ls/ g
lom
erul
us
Pathology
Sco
re
MRLFaslpr nCXCL10-/- 11CXCL10+/+ 9
4
2
0glomerular interstitial perivascular
15
10
5
0CD4 CD8 B220 CD 68
2
3
1
0
Proteinuria
40
20
0
BUN
Renal function
Sco
rem
g/dl
Kidney
- - -
-/-- -
Strain Age(mo) nMRL Faslpr 6 5MRL FaslprB6 2 5
2 5-
A
B
Figure 1. Despite an increase in intrarenal Mø and TEC expressing CXCL10, and CXCR3 expressing T cells and Mø in MRL-Faslpr
nephritic kidneys, CXCL10�/� MRL-Faslpr mice are not protected from renal disease. (A) CXCL10 expression in the kidney was examinedby immunostaining, flow cytometry (Mø), and real-time PCR (TEC). CXCL10�/� mice served as negative control. (B) CXCR3 expressionwas assessed on CD4�, CD8�, B220� T cells and Mø (CD68�) in kidneys of MRL-Faslpr mice by flow cytometry. B6 mice at 2 mo of ageserved as normal controls. (C) Renal function, pathology, and leukocytic infiltrates are similar in CXCL10�/� MRL-Faslpr mice ascompared with MRL-Faslpr mice. Data are means � SEM.
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 19: 1177–1189, 2008 CXCL9, not CXCL10, Promotes Nephritis 1179
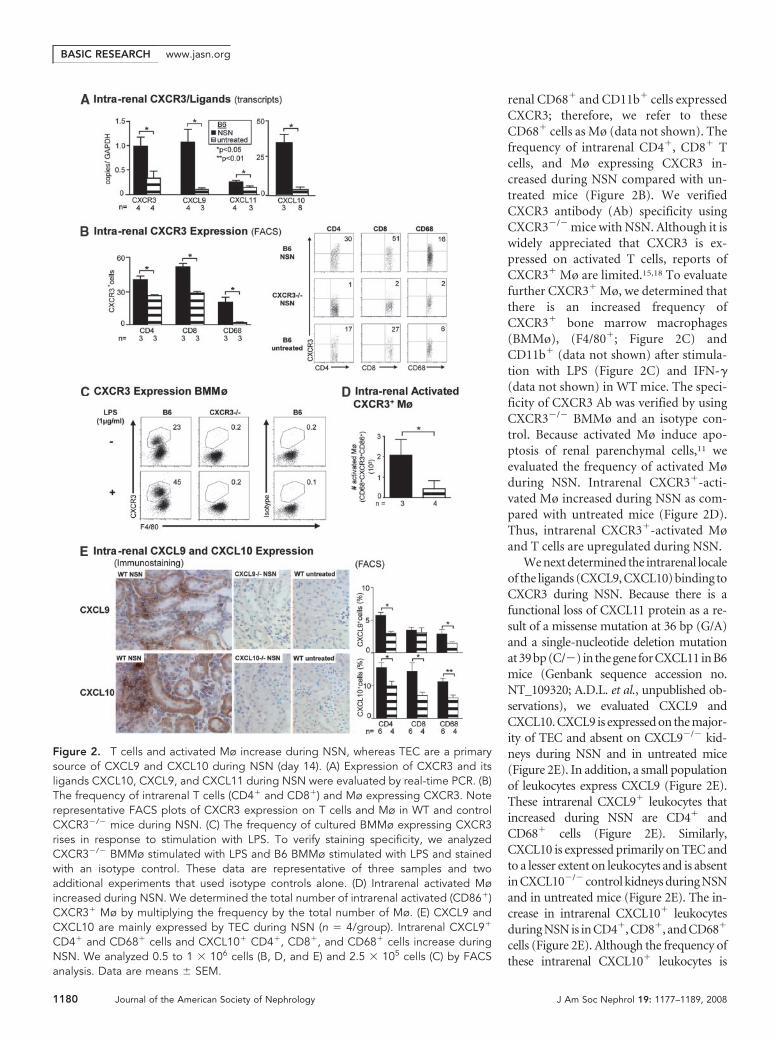
renal CD68� and CD11b� cells expressedCXCR3; therefore, we refer to theseCD68� cells as Mø (data not shown). Thefrequency of intrarenal CD4�, CD8� Tcells, and Mø expressing CXCR3 in-creased during NSN compared with un-treated mice (Figure 2B). We verifiedCXCR3 antibody (Ab) specificity usingCXCR3�/� mice with NSN. Although it iswidely appreciated that CXCR3 is ex-pressed on activated T cells, reports ofCXCR3� Mø are limited.15,18 To evaluatefurther CXCR3� Mø, we determined thatthere is an increased frequency ofCXCR3� bone marrow macrophages(BMMø), (F4/80�; Figure 2C) andCD11b� (data not shown) after stimula-tion with LPS (Figure 2C) and IFN-�(data not shown) in WT mice. The speci-ficity of CXCR3 Ab was verified by usingCXCR3�/� BMMø and an isotype con-trol. Because activated Mø induce apo-ptosis of renal parenchymal cells,11 weevaluated the frequency of activated Møduring NSN. Intrarenal CXCR3�-acti-vated Mø increased during NSN as com-pared with untreated mice (Figure 2D).Thus, intrarenal CXCR3�-activated Møand T cells are upregulated during NSN.
We next determined the intrarenal localeof the ligands (CXCL9, CXCL10) binding toCXCR3 during NSN. Because there is afunctional loss of CXCL11 protein as a re-sult of a missense mutation at 36 bp (G/A)and a single-nucleotide deletion mutationat 39 bp (C/�) in the gene for CXCL11 in B6mice (Genbank sequence accession no.NT_109320; A.D.L. et al., unpublished ob-servations), we evaluated CXCL9 andCXCL10. CXCL9 is expressed on the major-ity of TEC and absent on CXCL9�/� kid-neys during NSN and in untreated mice(Figure 2E). In addition, a small populationof leukocytes express CXCL9 (Figure 2E).These intrarenal CXCL9� leukocytes thatincreased during NSN are CD4� andCD68� cells (Figure 2E). Similarly,CXCL10 is expressed primarily on TEC andto a lesser extent on leukocytes and is absentin CXCL10�/� control kidneys during NSNand in untreated mice (Figure 2E). The in-crease in intrarenal CXCL10� leukocytesduring NSN is in CD4�, CD8�, and CD68�
cells (Figure 2E). Although the frequency ofthese intrarenal CXCL10� leukocytes is
Figure 2. T cells and activated Mø increase during NSN, whereas TEC are a primarysource of CXCL9 and CXCL10 during NSN (day 14). (A) Expression of CXCR3 and itsligands CXCL10, CXCL9, and CXCL11 during NSN were evaluated by real-time PCR. (B)The frequency of intrarenal T cells (CD4� and CD8�) and Mø expressing CXCR3. Noterepresentative FACS plots of CXCR3 expression on T cells and Mø in WT and controlCXCR3�/� mice during NSN. (C) The frequency of cultured BMMø expressing CXCR3rises in response to stimulation with LPS. To verify staining specificity, we analyzedCXCR3�/� BMMø stimulated with LPS and B6 BMMø stimulated with LPS and stainedwith an isotype control. These data are representative of three samples and twoadditional experiments that used isotype controls alone. (D) Intrarenal activated Møincreased during NSN. We determined the total number of intrarenal activated (CD86�)CXCR3� Mø by multiplying the frequency by the total number of Mø. (E) CXCL9 andCXCL10 are mainly expressed by TEC during NSN (n � 4/group). Intrarenal CXCL9�
CD4� and CD68� cells and CXCL10� CD4�, CD8�, and CD68� cells increase duringNSN. We analyzed 0.5 to 1 � 106 cells (B, D, and E) and 2.5 � 105 cells (C) by FACSanalysis. Data are means � SEM.
BASIC RESEARCH www.jasn.org
1180 Journal of the American Society of Nephrology J Am Soc Nephrol 19: 1177–1189, 2008
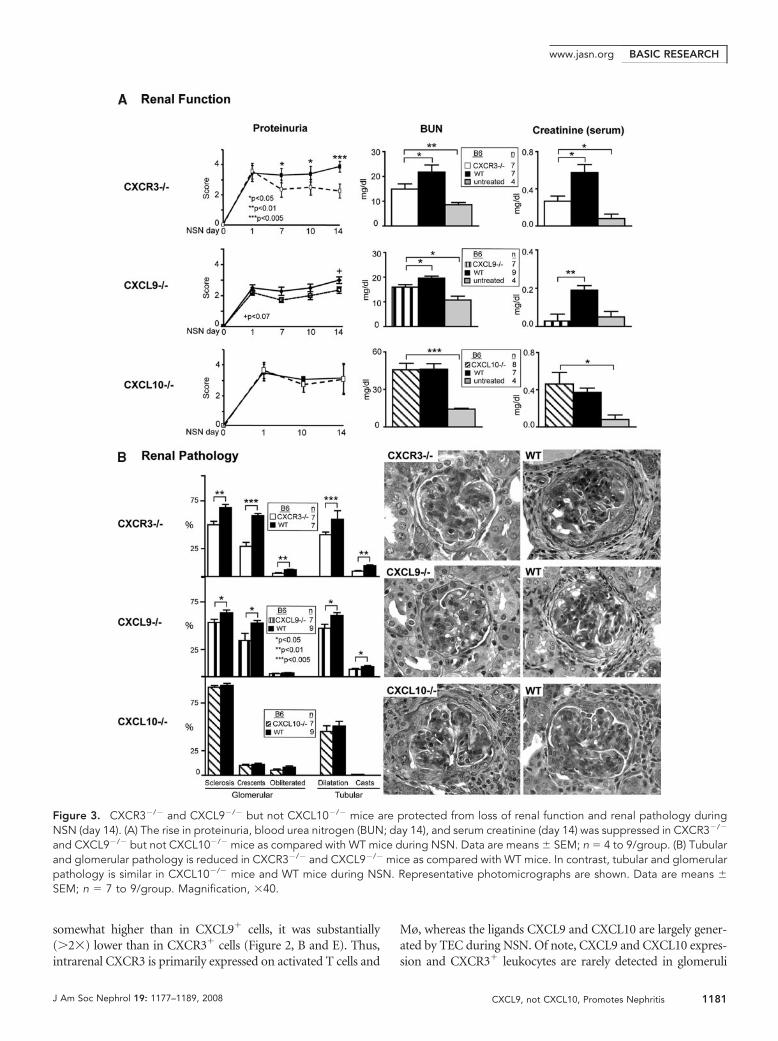
somewhat higher than in CXCL9� cells, it was substantially(�2�) lower than in CXCR3� cells (Figure 2, B and E). Thus,intrarenal CXCR3 is primarily expressed on activated T cells and
Mø, whereas the ligands CXCL9 and CXCL10 are largely gener-ated by TEC during NSN. Of note, CXCL9 and CXCL10 expres-sion and CXCR3� leukocytes are rarely detected in glomeruli
Figure 3. CXCR3�/� and CXCL9�/� but not CXCL10�/� mice are protected from loss of renal function and renal pathology duringNSN (day 14). (A) The rise in proteinuria, blood urea nitrogen (BUN; day 14), and serum creatinine (day 14) was suppressed in CXCR3�/�
and CXCL9�/� but not CXCL10�/� mice as compared with WT mice during NSN. Data are means � SEM; n � 4 to 9/group. (B) Tubularand glomerular pathology is reduced in CXCR3�/� and CXCL9�/� mice as compared with WT mice. In contrast, tubular and glomerularpathology is similar in CXCL10�/� mice and WT mice during NSN. Representative photomicrographs are shown. Data are means �SEM; n � 7 to 9/group. Magnification, �40.
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 19: 1177–1189, 2008 CXCL9, not CXCL10, Promotes Nephritis 1181
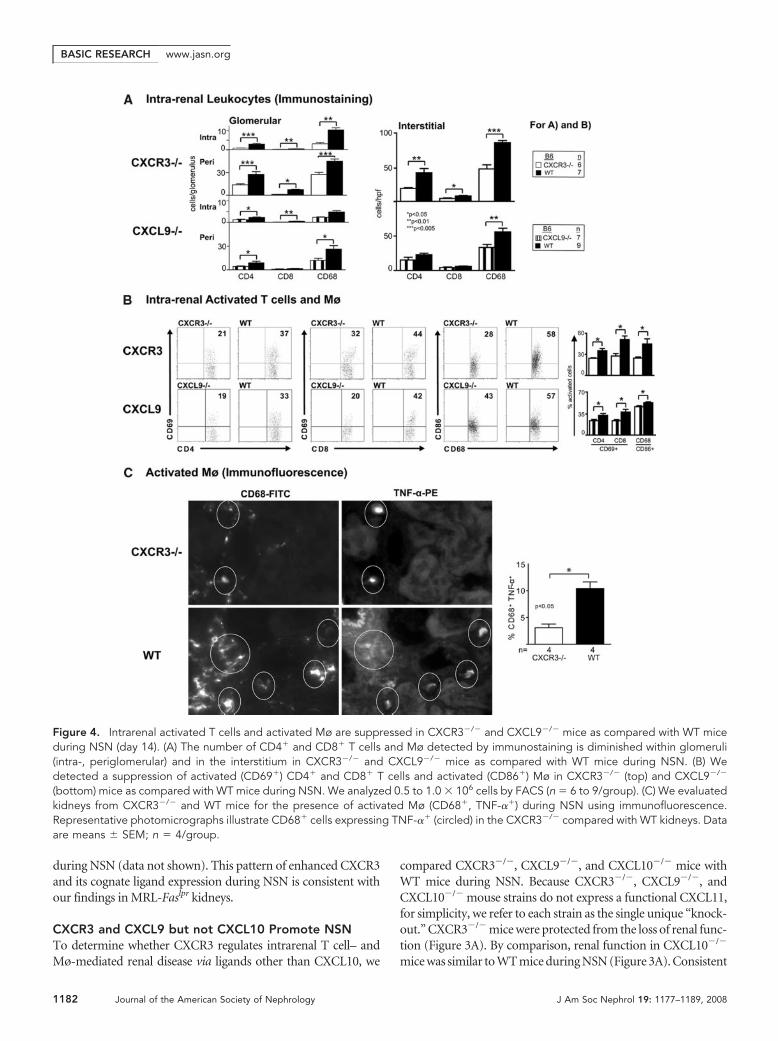
during NSN (data not shown). This pattern of enhanced CXCR3and its cognate ligand expression during NSN is consistent withour findings in MRL-Faslpr kidneys.
CXCR3 and CXCL9 but not CXCL10 Promote NSNTo determine whether CXCR3 regulates intrarenal T cell– andMø-mediated renal disease via ligands other than CXCL10, we
compared CXCR3�/�, CXCL9�/�, and CXCL10�/� mice withWT mice during NSN. Because CXCR3�/�, CXCL9�/�, andCXCL10�/� mouse strains do not express a functional CXCL11,for simplicity, we refer to each strain as the single unique “knock-out.” CXCR3�/� mice were protected from the loss of renal func-tion (Figure 3A). By comparison, renal function in CXCL10�/�
mice was similar to WT mice during NSN (Figure 3A). Consistent
Figure 4. Intrarenal activated T cells and activated Mø are suppressed in CXCR3�/� and CXCL9�/� mice as compared with WT miceduring NSN (day 14). (A) The number of CD4� and CD8� T cells and Mø detected by immunostaining is diminished within glomeruli(intra-, periglomerular) and in the interstitium in CXCR3�/� and CXCL9�/� mice as compared with WT mice during NSN. (B) Wedetected a suppression of activated (CD69�) CD4� and CD8� T cells and activated (CD86�) Mø in CXCR3�/� (top) and CXCL9�/�
(bottom) mice as compared with WT mice during NSN. We analyzed 0.5 to 1.0 � 106 cells by FACS (n � 6 to 9/group). (C) We evaluatedkidneys from CXCR3�/� and WT mice for the presence of activated Mø (CD68�, TNF-��) during NSN using immunofluorescence.Representative photomicrographs illustrate CD68� cells expressing TNF-�� (circled) in the CXCR3�/� compared with WT kidneys. Dataare means � SEM; n � 4/group.
BASIC RESEARCH www.jasn.org
1182 Journal of the American Society of Nephrology J Am Soc Nephrol 19: 1177–1189, 2008
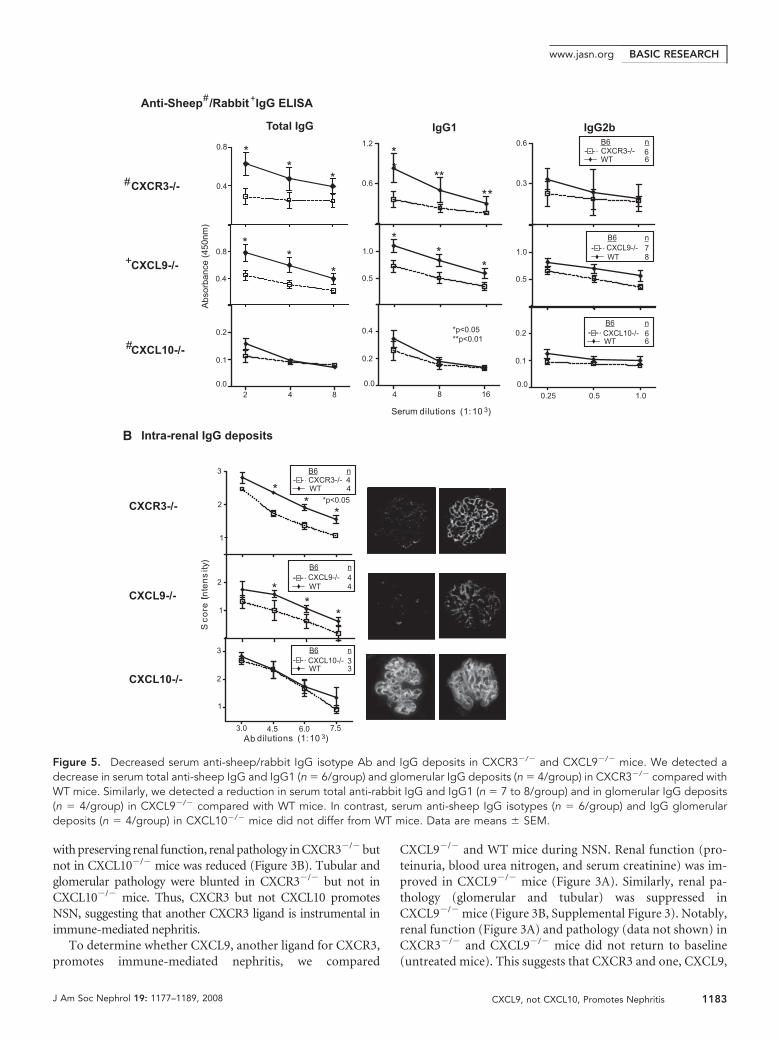
with preserving renal function, renal pathology in CXCR3�/� butnot in CXCL10�/� mice was reduced (Figure 3B). Tubular andglomerular pathology were blunted in CXCR3�/� but not inCXCL10�/� mice. Thus, CXCR3 but not CXCL10 promotesNSN, suggesting that another CXCR3 ligand is instrumental inimmune-mediated nephritis.
To determine whether CXCL9, another ligand for CXCR3,promotes immune-mediated nephritis, we compared
CXCL9�/� and WT mice during NSN. Renal function (pro-teinuria, blood urea nitrogen, and serum creatinine) was im-proved in CXCL9�/� mice (Figure 3A). Similarly, renal pa-thology (glomerular and tubular) was suppressed inCXCL9�/� mice (Figure 3B, Supplemental Figure 3). Notably,renal function (Figure 3A) and pathology (data not shown) inCXCR3�/� and CXCL9�/� mice did not return to baseline(untreated mice). This suggests that CXCR3 and one, CXCL9,
Anti-Sheep /Rabbit IgG ELISA
Total IgG IgG1 IgG2b
Ab
sorb
an
ce (
45
0n
m)
0.4
0.8
0.6
1.2
0.3
0.6*
*
**
** **
* CXCR3-/- WT
B6 n
66
CXCL9-/- WT
B6 n
87
CXCL10-/- WT
B6 n
66
**
*
**
*0.4
0.8
0.5
1.0
0.5
1.0
0.1
0.2
0.1
0.2
0.2
0.4
0.0 0.0 0.02 4 8 4 8 16 0.25 0.5 1.0
CXCR3-/-
CXCL9-/-
CXCL10-/-
Intra-renal IgG deposits
dilutions (1:10 3)Serum
Ab dilutions (1:10 3)3.0 4.5 6.0 7.5
*p<0.05**p<0.01
Sc
ore
(In
ten
sit
y)
1
2
3
*p<0.05*
**
CXCR3-/- WT
B6 n
44
CXCR3-/-
CXCL9-/-
CXCL10-/-
**
*2
2
1
3
1
CXCL9-/- WT
B6 n
44
B6CXCL10-/- WT
n
33
# +
#
#
+
B
Figure 5. Decreased serum anti-sheep/rabbit IgG isotype Ab and IgG deposits in CXCR3�/� and CXCL9�/� mice. We detected adecrease in serum total anti-sheep IgG and IgG1 (n � 6/group) and glomerular IgG deposits (n � 4/group) in CXCR3�/� compared withWT mice. Similarly, we detected a reduction in serum total anti-rabbit IgG and IgG1 (n � 7 to 8/group) and in glomerular IgG deposits(n � 4/group) in CXCL9�/� compared with WT mice. In contrast, serum anti-sheep IgG isotypes (n � 6/group) and IgG glomerulardeposits (n � 4/group) in CXCL10�/� mice did not differ from WT mice. Data are means � SEM.
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 19: 1177–1189, 2008 CXCL9, not CXCL10, Promotes Nephritis 1183

but not another, CXCL10, of its ligands promote immune-mediated nephritis.
Intrarenal Activated T Cells and Mø Are Decreased inCXCR3�/� and CXCL9�/� Mice during NSNBecause CXCR3 is expressed on intrarenal T cells (CD4�,CD8�) and Mø during NSN, we hypothesized these intrarenalleukocytes decrease in CXCR3�/� as compared with WT mice.Fewer CD4�, CD8�, and Mø were detected in the glomeruli(intra-, periglomerular) and interstitium in CXCR3�/� mice(Figure 4A). A similar decrease in CD4� T cells and Mø oc-curred in the CXCL9�/� kidneys, whereas we did not detect areduction in these leukocytic populations in CXCL10�/� mice(data not shown).
CXCR3 is expressed on activated T cells19 and Mø; there-fore, we compared these intrarenal leukocytes in CXCR3�/�
mice with WT mice during NSN. We detected a decreasedfrequency of activated (CD69�) CD4� and CD8� T cells andactivated (CD86�) Mø in CXCR3�/� mice during NSN (Fig-ure 4B). In support of this decrease in activated Mø, the fre-quency of TNF-�� Mø was diminished (Figure 4C, circled,immunofluorescence). Similarly, CXCL9�/� mice had a re-duced frequency of activated T cells and Mø (Figure 4B). Thus,CXCR3 engaging with CXCL9 fosters an intrarenal accumula-tion of activated CD4�, CD8� T cells and Mø during NSN.
Serum Antigen-Specific IgG Isotypes and GlomerularIgG Deposits Are Suppressed in CXCR3�/� andCXCL9�/� Mice during NSNElevated Ig and Ig class switching are hallmarks of autoimmunedisease20,21; therefore, we determined whether the attenuated se-verity of kidney disease in CXCR3�/� and CXCL9�/� mice wasrelated to a reduction in circulating levels of antigen-specific IgGisotypes during NSN. For this purpose, we measured the anti-sheep and -rabbit IgG isotypes in the sera of CXCR3�/� andCXCL9�/� mice, respectively, in comparison with WT mice dur-ing NSN. We detected a suppression of IgG and IgG1 and notIgG2b at multiple titers (2.0 to 8.0 � 103) in CXCR3�/� andCXCL9�/� mice (Figure 5A). Because serum isotypes were atten-uated in CXCR3�/� and CXL9�/� mice, we hypothesized that thereduced renal pathology resulted from decreased deposition ofIgG within glomeruli of CXCR3�/� and CXCL9�/� mice. Wedetected a reduction in IgG deposits at multiple dilutions (3.0 to7.5 � 103) in CXCR3�/� and CXCL9�/� glomeruli (Figure 5B).Contrastingly, circulating anti-sheep IgG isotypes and glomerularIgG deposits in CXCL10�/� did not differ from WT mice. Takentogether, this suggests CXCR3 engaging with CXCL9 may pro-mote renal disease by increasing systemic IgG and IgG depositswithin glomeruli.
DISCUSSION
Chemokines are tempting therapeutic targets for a wide rangeof diseases, because they are specifically upregulated during
inflammation. Mø and T cells are instrumental in mediatingrenal inflammation; thus, chemokines that regulate Mø and Tcells within the kidney are potential therapeutic targets for ne-phritis. Therefore, we extensively profiled MRL-Faslpr kidneysbefore and during Mø- and T cell– dependent lupus nephritis.Although previous reports claimed that a few chemokine li-gands (four of nine) and chemokine receptors (three of six) areupregulated in MRL-Faslpr kidneys, we now report that themajority of chemokine ligands (18 of 23) and chemokine re-ceptors (10 of 16) that we profiled were upregulated in MRL-Faslpr mice with lupus nephritis.6,22 This difference in our find-ings may be related to the larger panel of chemokine ligand/receptors in our experiments and the detection system(ribonuclease protection assay22 versus real-time PCR, ourstudy). We previously established that CCL2 is required topromote Mø- and T cell– dependent lupus nephritis in MRL-Faslpr mice.10 We now report that the rise in the panel of che-mokine ligand/receptors that increase in MRL-Faslpr mice issuppressed in CCL2�/� MRL-Faslpr mice. This may suggestthat CCL2 is a “proximal master switch” in the chemokinecascade; however, increased expression of a specific chemokineduring disease does not necessarily indicate that it promotesinflammation; it may not have an impact or even thwart in-flammation. Therefore, we investigated the impact of CXCR3and its ligands, CXCL9 and CXCL10, during Mø and T cellimmune–mediated nephritis (NSN). Of note, we could notanalyze the impact of CXCL11 during NSN because the proteinfor this remaining CXCR3 ligand is not expressed in B6 mice.We now report that CXCR3 and CXCL9 but not CXCL10 arecentral to Mø- and T cell– dependent kidney disease.
The impact of CXCL10 in autoimmune and kidney diseaseis controversial. Whereas Ab blockade of CXCL10 in experi-mental insulin-dependent diabetes23 and adjuvant arthritis24
abrogates disease, deleting CXCL10 does not diminish experi-mental autoimmune encephalomyelitis, although it lowersdisease threshold.25 Thus, the consequences of CXCL10 engag-ing with CXCR3 in autoimmune diseases are complex and yetnot fully understood. Furthermore, the impact of CXCL10 onglomerular and tubulointerstitial renal disease is unclear. Ininduced forms of renal injury, puromycin aminonucleoside–and anti-nephrin Ab–induced nephropathy,26 Ab blockade ofCXCL10 exacerbated proteinuria and podocyte injury. Con-versely, Ab blockade of CXCL10 in a model of renal endothelialinjury reduced interstitial T cell infiltration and improved re-nal function; however, pathology was not evaluated.27 We nowreport that despite the abundant protein expression of intrarenalCXCL10 on TEC and leukocyte infiltrates, CXCL10 is not instru-mental in two distinct models of immune-mediated renal disease,MRL-Faslpr mice and NSN. This indicates that CXCL10 alone isnot central to immune-mediated kidney disease.
Because it is possible that other ligands alone or togetherwith CXCL10 promote immune-mediated kidney disease, wecompared CXCR3�/� and CXCL9�/� mice during NSN withWT mice. CXCR3 and CXCL9 but not CXCL10 are central toimmune-mediated kidney disease. This is the first report indi-
BASIC RESEARCH www.jasn.org
1184 Journal of the American Society of Nephrology J Am Soc Nephrol 19: 1177–1189, 2008

cating that CXCL9�/� mice are protected from nephritis. Ourfindings are in agreement with recent studies reporting thatCXCR3�/� mice develop less severe nephritis than WT miceduring NSN.28 In fact, CXCR3 has been shown to incite non-immune-induced renal inflammation because CXCR3 orches-trates the recruitment of T cells instrumental in mediating re-nal ischemic injury.16 Thus, CXCR3 may be a potentialtherapeutic target for a broad array of kidney diseases. Ourfindings are intriguing; although CXCL9 and CXCL10 proteinexpression is similar in locale and intensity during NSN, thefunctional impact of these two CXCR3 ligands differ. This dif-ferential impact on nephritis may be related to distinct bio-availability of CXCL9 and CXCL10. The preferential contribu-tion of one CXCR3 ligand to disease pathology has been seen inother inflammatory models of disease, such as CXCL10 inmouse hepatitis virus and Dengue virus encephalitis29,30 andCXCL9 in herpes simplex virus-1 ocular infection and cyto-megalovirus infection.31,32 Regardless of the exact mecha-nisms, our findings indicate that selective blockade of CXCL9and CXCR3 is a potential therapeutic strategy to combat im-mune-mediated renal diseases.
Although it is well appreciated that activated T cells expressCXCR3, we now report that activated and resting Mø expressCXCR3 during kidney disease. The frequency of intrarenalCD68� cells along with CD4� and CD8� T cells expressingCXCR3 increased during NSN. This rise in CXCR3� Mø in thekidney is consistent with studies reporting that a low percentage ofCXCR3� monocytes in the normal circulation increases in pa-tients with rheumatoid arthritis.33 Furthermore, we determinedthat whereas some primary cultured BMMø express CXCR3, thefrequency increases after IFN-� stimulation and is even more ro-bust after LPS stimulation. This suggests that whereas low num-bers of resting Mø express CXCR3, the increase in CXCR3� Mø isa reflection of a rise in activated Mø during renal inflammation.Consistent with this concept, the total number of intrarenal acti-vated CXCR3� Mø increases during NSN as compared with un-treated mice. Furthermore, CXCR3 is responsible for recruitingthese activated Mø, because there is a decrease in intrarenal acti-vated Mø, along with activated CD4� and CD8� T cells, inCXCR3�/� during NSN. This suggests that activated Mø bearingCXCR3 are central to inducing renal injury, because activated Mørelease mediators that initiate TEC apoptosis.11 Thus, we providethe first evidence that CXCR3 is instrumental in recruiting acti-vated Mø, as well as T cells, into the kidney, that are responsible forinciting renal injury.
Humoral and cell-driven immune mechanisms mediate kid-ney disease.20 We investigated whether the suppression in renaldisease in the CXCR3�/� and CXCL9�/� mice during NSN wasrelated to a decrease in pathogenic Ab within the kidney. We de-tected a reduction in total serum antigen-specific IgG and IgG1Ab in CXCR3�/� and CXCL9�/� mice during NSN. Our findingis reminiscent of reduced Ab against a bacterial pathogen (Fran-cisella tularensis) in CXCL9�/� mice34; however, our data are notin agreement with the recent study of CXCR3�/� mice, indicatingthat there is no difference in total mouse anti-sheep IgG or anti-
sheep IgG1 during NSN (day 8).28 This difference may be relatedto the assay methods, the day of analysis, or other variables relatedto inducing NSN. Nevertheless, our data support the concept thatCXCR3 and CXCL9 enhance Ab production during immune re-sponses. The regulation of serum IgG and glomerular deposits byCXCR3 and CXCL9 remains unclear. We speculate that distinctpatterns of CXCR3�CD4� T cells and CXCL9 expression in ger-minal centers during inflammation and/or the expression ofCXCR3 on a subset of memory B cells and plasma cells may reg-ulate serum IgG and, in turn, glomerular deposits.35,36 Notably,the decline in antigen-specific IgG in CXCR3�/� and CXCL9�/�
serum correlates with diminished IgG deposits within glomeruli;therefore, suppressed renal pathology and improved renal func-tion may, in part, result from a reduced intrarenal deposition ofpathogenic Ab in CXCR3�/� and CXCL9�/� mice during NSN.In addition, we did not detect CXCL9 and CXCL10 expressionand CXCR3� leukocytes in glomeruli of WT mice duringNSN. Thus, the reduction in proteinuria in CXCR3�/� andCXCL9�/� mice during the later stages of NSN may be re-lated to the suppression of humoral immune responses.This is consistent with findings indicating that although thegeneration of autologous murine IgG against the nephro-toxic Ab is not essential for disease initiation, it may con-tribute to sustaining renal dysfunction.37 This may indicatethat although CXCR3 and CXCL9 promote the initiation ofNSN via cell-mediated events, their regulation of the pro-duction and deposition of pathogenic Ab may be responsi-ble for sustaining immune-mediated renal disease.
In conclusion, CXCR3 and one, CXCL9, but not another,CXCL10, of its ligands mediate activated T cells and Mø re-cruitment into the kidney and enhance Ab deposition in glo-meruli that may, in turn, promote immune-mediated kidneydisease. This suggests that blocking CXCR3 and CXCL9 is apotential therapeutic target for human immune-mediated kid-ney diseases.
CONCISE METHODS
MiceWe purchased MRL/MpJ-Faslpr/Faslpr (MRL-Faslpr) and C57BL/6
(B6) from the Jackson Laboratory (Bar Harbor, ME). Mice were
housed and bred in our pathogen-free animal facility. CXCL9 null
(�/�), CXCL10�/�, CXCR3�/� (B6 background), and CCL2�/�
(MRL-Faslpr background) mice were generated as described previous-
ly.4,10,34,38 We generated CXCL10�/� MRL-Faslpr mice using a back-
cross-intercross scheme. MRL-Faslpr mice were mated with
CXCL10�/� (B6 background) mice to yield heterozygous F1 off-
spring. We intercrossed F1 mice and screened the progeny for dis-
rupted and intact CXCL10 and the Faslpr mutation in tail genomic
DNA. The DNA was assessed by PCR using oligonucleotide primers
that recognized the normal CXCL10 gene: Sense (5�-TCC CTC CCG
TAA CCA CAC AGT AAA T-3�) and antisense (5�-GCG GAT AGA
CTC TGC TTT CAC TTT GG-3�) and Neo gene sense (5�-TGG ATG
TGG AAT GTG TGC GAG-3�) and antisense (5-TTT CAC TTT GG-
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 19: 1177–1189, 2008 CXCL9, not CXCL10, Promotes Nephritis 1185

3�). Gel analysis of the PCR products identified the CXCL10 and Neo
gene fragments at 342 and 640 bp, respectively. The Faslpr mutation
was identified as previously reported.39 After five generations of back-
cross matings (N5), we analyzed and compared CXCL10�/� MRL-
Faslpr mice with age- and gender-matched WT MRL-Faslpr litter-
mates. The number of mice analyzed is specified in each figure. We
compared these strains at the N5 generation, because we previously
established that there are sufficient MRL-Faslpr background genes to
result in consistent phenotypic changes characteristic of MRL-Faslpr
mice.40 Use of mice in our study was reviewed and approved by the
Standing Committee on Animals in the Harvard Medical School in
adherence to the National Institutes of Health Guide for the Care and
Use of Laboratory Animals.
Inducing NSNWe prepared nephrotoxic serum by immunizing sheep with a partic-
ulate fraction of mouse glomerular basement membrane from B6
mice kidneys, as described previously.41 To induce NSN, we primed
CXCL10�/�, CXCR3�/�, and WT male mice (6 wk of age) by inject-
ing 0.5 mg of sheep IgG in Freund’s complete adjuvant (Sigma Chem-
ical Co., St. Louis, MO) subcutaneously in each flank. We challenged
these mice 7 d later with an intravenous injection of nephrotoxic
serum (15 �l/g body wt). We used nephrotoxic serum from rabbits to
immunize CXCL9�/� and WT mice as described previously.42 Mice
were killed 14 d after challenge and analyzed. The number of mice in
each group is specified within each figure.
Intrarenal Chemokine and Chemokine ReceptorTranscriptsWe analyzed the expression of chemokines (CXCL10, CXCL9,
CXCL11, CXCL12, CCL13, CCL5, CCL28, CCL22, CCL24, CCL1,
CCL17, CX3CL1, CCL25, CCL2, CCL8, CCL7, CCL3, CXCL2,
CCL28�, CCL4, CCL28�, CCL27, CCL21, CXCL16, and CXCL1) and
chemokine receptors (CXCR2, CXCR3, CXCR4, CXCR5, CXCR6,
CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9,
CCR10, and CX3CR1) in MRL-Faslpr mice by using real-time, two-
step, quantitative PCR, as described previously.43 Primers were de-
signed, as previously reported.44 To detect CXCL10 expression during
NSN, we used primers designed by Applied Biosystems (Foster City,
CA). To detect CXCL9, CXCL11, and CXCR3 expression during
NSN, we used the following primers: glyceraldehyde-3-phosphate de-
hydrogenase sense 5�-CAT GGC CTC CAA GGA GTA AG-3� and
antisense 5�-CCT AGG CCC CTC CTG TTA TT-3�; CXCL9 sense
5�-TCC TTT TGG GCA TCA TCT TC-3� and antisense 5�-TTC CCC
CTC TTT TGC TTT TT-3�; CXCL11 sense 5�-AGT AAC GGC TGC
GAC AAA GT-3� and antisense 5�-GCA TGT TCC AAG ACA GCA
GA-3�; and CXCR3 sense 5�-TGA GAC AAC TGA GGC CTC CTA-3�
and antisense 5�-TCT TGC TCC CCA GTT GAT G-3� (Invitrogen,
Carlsbad CA). We evaluated expression as previously reported.45
Isolation of TEC and BMMøWe isolated and cultured TEC from B6 and MRL-Faslpr mice as pre-
viously reported,46 and we isolated and cultured BMMø from B6 mice
as described previously.47
Flow CytometryWe prepared and stained single-cell suspensions from kidneys,
spleens, or primary cultured TEC/BMMø as described previously.48
We collected 0.5 to 1.0 � 106 total kidney or spleen cells and 0.5 to
1.0 � 105 of cultured cells using a FACSCalibur (Becton Dickinson,
San Jose, CA) and analyzed data using Flowjo software (Tree Star,
Palo Alto, CA).
AntibodiesWe used the following Ab from eBioscience (San Diego, CA) for FACS
analysis: FITC-conjugated anti-CD4 (L3T4), anti-CD8 (53-6.7), anti-
B220 (RA3-6B2), and anti-CD45.2 (104); PE-conjugated anti-CD4,
anti-CD45.2, anti-CD69 (H1.2F3), and anti-CD86 (GL1); PE-Cy5–
conjugated anti-CD8 and anti–TCR-� chain (H57-597); and allophy-
cocyanin-conjugated anti-CD4, anti-CD45.2, and anti-F4/80 (BM8).
We used FITC- and allophycocyanin-conjugated anti-CD68 Ab
(FA11; Serotec, Oxford, UK). We used purified rabbit anti-mouse
CXCR3 Ab (Zymed, San Francisco, CA), purified goat anti-mouse
CXCL9 Ab (R&D Systems, Minneapolis, MN), and rabbit anti-mouse
CXCL10 Ab (PeproTech, Rocky Hill, NJ). The following isotype-spe-
cific Ab were used for controls: Rat-IgG2a (BR2a), rat-IgG2b (KLH/
G2b-1-2), and rabbit-IgG (eBioscience). As secondary PE- or allophy-
cocyanin-conjugated Ab, we used goat anti-rabbit (Jackson
ImmunoResearch Laboratories; West Grove, PA) and biotin-conju-
gated rabbit anti-goat Ab (Vector Laboratories, Burlingame, CA). To
detect biotin-conjugated secondary Ab, we used streptavidin PE or
allophycocyanin (Jackson ImmunoResearch Laboratories). We iden-
tified renal proximal tubules by binding of fluorescein-conjugated
lotus lectin (Vector Laboratories).49,50
HistopathologyWe fixed kidneys in 10% formalin and prepared and stained paraffin
sections with the periodic acid Schiff reagent. Slides were coded before
grading the renal pathology.
MRL-Faslpr Mice.We evaluated renal (glomerular, tubular, intersti-
tial, and perivascular) pathology on a scale of 0 (normal) to 3 (severe)
as described previously.45
NSN. We evaluated CXCL9�/�, CXCL10�/�, CXCR3�/�, and
B6 kidneys for glomerular and tubular damage as described previ-
ously.11
Gross PathologyWe evaluated skin lesions monthly from 2 to 6 mo of age using a
scoring system previously described.10
ImmunohistochemistryWe stained cryostat-cut kidney sections for the presence of Mø with
anti-CD68 Ab (FA-11; Serotec) and for T cells with anti-CD4 Ab
(RM4-5), anti-CD8 Ab (53-6.7), and anti-B220 (RA3– 6B2) rat anti-
mouse Ab (Pharmingen, San Diego, CA) according to a previously
described immunoperoxidase method.51 To evaluate CXCL9 and
CXCL10 expression in the kidney, we stained frozen sections with
goat anti-mouse CXCL9 and rabbit anti-mouse CXCL10. The follow-
BASIC RESEARCH www.jasn.org
1186 Journal of the American Society of Nephrology J Am Soc Nephrol 19: 1177–1189, 2008

ing isotype-specific Ab were used for controls: Rat-IgG2a (R35–95),
rat IgG2b (R35-38), rabbit IgG (eBioscience), and goat IgG (Southern
Biotechnology, Birmingham, AL). The secondary Ab for immuno-
staining was biotin-conjugated rabbit anti-rat IgG (Vector Laborato-
ries). The immunostaining was analyzed by counting for the presence
of CD68�, CD4�, CD8�, and B220� cells within and surrounding
glomeruli and in the interstitium in 10 randomly selected high-power
fields. Of note, we have determined that the B220� cells in the kidney
of MRL-Faslpr mice are the unique double-negative (CD4�CD8�
double-negative) T cells and are not B cells.52
To determine the number of activated Mø in the kidney during
NSN, we fixed frozen kidney sections in paraformaldehyde, stained
them with rat anti-mouse TNF-�–PE (MP6-XT22; eBioscience) and
anti–CD68-FITC (FA-11; Serotec), and analyzed these sections using
a fluorescence microscope. The frequency (%) of activated Mø was
assessed by enumerating the number of CD68�TNF-�� cells within
the total number of Mø (CD68�) in five high-power fields.
Renal FunctionWe assessed urine proteins semiquantitatively by dipstick analysis as
described previously.45 We measured blood urea nitrogen levels using
a colorimetric analysis kit (Infinity; Thermo Electron, Melbourne,
Australia) and serum creatinine using the creatinine reagent kit and
Creatinine Analyzer 2 (Beckman Coulter, Galway, Ireland) according
to the manufacturer’s instructions.
SurvivalWe compared survival of the CXCL10�/� MRL-Faslpr and WT MRL-
Faslpr mice using similar numbers of males and females from birth to
10 mo of age.
Serum Anti-Sheep and Anti-Rabbit IgG IsotypesWe measured the levels of mouse anti-sheep IgG Ab (Sigma Chemical
Co.) and anti-rabbit IgG (Jackson ImmunoResearch Laboratories) by
ELISA using sera collected at day 14 during NSN as described previ-
ously.53 We detected bound mouse anti-sheep/anti-rabbit IgG, IgG1,
and IgG2b using peroxidase-conjugated rabbit anti-mouse IgG Ab
(1:5000; Southern Biotechnology).
IgG Deposits within the KidneyWe evaluated IgG deposits within renal glomeruli as described previ-
ously.54 We evaluated stained sections by scoring 20 glomeruli as ei-
ther positive or negative and graded the amount (severity) of deposits
in 20 positive glomeruli per specimen on a scale of 0 to 3 using mul-
tiple dilutions (1:3000, 1:4500, 1:6000, and 1:7500) of the detection
Ab (fluorescein-conjugated goat anti-mouse IgG; MP Biomedicals,
Aurora, OH).40
Statistical AnalysesThe data are means � SEM and were analyzed by GraphPad Prism 4.0
(GraphPad, San Diego, CA). We used the nonparametric Mann-
Whitney U test to determine differences among groups. Survival
curves were compared and analyzed using the log-rank test.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants DK
52369 (V.R.K.), DK 56848 (V.R.K.), DK 36149 (V.R.K.), KO1 AR
051367 (T.K.M.), and RO1 CA 069212 (A.D.L.); by Deutsche For-
schungsgemeinschaft grants ME-3194/1-1 (J.M.) and ZE-711/1
(G.Z.); and the Research Grants Council of Hong Kong HKU 7592/06
(H.Y.L.).
We thank Dr. Craig Gerard for providing CXCR3�/� mice, Dr.
Tanya Mayadas for the rabbit anti– glomerular basement membrane
anti-serum, and Dr. Julie Lucas for editorial assistance.
DISCLOSURESNone.
REFERENCES
1. Sallusto F, Mackay CR, Lanzavecchia A: The role of chemokine recep-tors in primary, effector, and memory immune responses. Annu RevImmunol 18: 593–620, 2000
2. Rossi D, Zlotnik A: The biology of chemokines and their receptors.Annu Rev Immunol 18: 217–242, 2000
3. Segerer S, Cui Y, Hudkins KL, Goodpaster T, Eitner F, Mack M,Schlondorff D, Alpers CE: Expression of the chemokine monocytechemoattractant protein-1 and its receptor chemokine receptor 2 inhuman crescentic glomerulonephritis. J Am Soc Nephrol 11: 2231–2242, 2000
4. Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD:IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice re-veal a role for IP-10 in effector T cell generation and trafficking.J Immunol 168: 3195–3204, 2002
5. Anders HJ, Vielhauer V, Schlondorff D: Chemokines and chemokinereceptors are involved in the resolution or progression of renal dis-ease. Kidney Int 63: 401–415, 2003
6. Vielhauer V, Anders HJ, Schlondorff D: Chemokines and chemokinereceptors as therapeutic targets in lupus nephritis. Semin Nephrol 27:81–97, 2007
7. Premack BA, Schall TJ: Chemokine receptors: Gateways to inflamma-tion and infection. Nat Med 2: 1174–1178, 1996
8. Hooke DH, Gee DC, Atkins RC: Leukocyte analysis using monoclonalantibodies in human glomerulonephritis. Kidney Int 31: 964–972,1987
9. Kelley VE, Roths JB: Interaction of mutant lpr gene with backgroundstrain influences renal disease. Clin Immunol Immunopathol 37: 220–229, 1985
10. Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR: Monocytechemoattractant protein 1-dependent leukocytic infiltrates are re-sponsible for autoimmune disease in MRL-Fas(lpr) mice. J Exp Med190: 1813–1824, 1999
11. Tesch GH, Schwarting A, Kinoshita K, Lan HY, Rollins BJ, Kelley VR:Monocyte chemoattractant protein-1 promotes macrophage-medi-ated tubular injury, but not glomerular injury, in nephrotoxic serumnephritis. J Clin Invest 103: 73–80, 1999
12. Moore KJ, Wada T, Barbee SD, Kelley VR: Gene transfer of RANTESelicits autoimmune renal injury in MRL-Fas(lpr) mice. Kidney Int 53:1631–1641, 1998
13. Luster AD, Ravetch JV: Biochemical characterization of a gammainterferon-inducible cytokine (IP-10). J Exp Med 166: 1084–1097,1987
14. Xie JH, Nomura N, Lu M, Chen SL, Koch GE, Weng Y, Rosa R, Di Salvo
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 19: 1177–1189, 2008 CXCL9, not CXCL10, Promotes Nephritis 1187

J, Mudgett J, Peterson LB, Wicker LS, DeMartino JA: Antibody-medi-ated blockade of the CXCR3 chemokine receptor results in diminishedrecruitment of T helper 1 cells into sites of inflammation. J Leukoc Biol73: 771–780, 2003
15. Liu L, Callahan MK, Huang D, Ransohoff RM: Chemokine receptorCXCR3: An unexpected enigma. Curr Top Dev Biol 68: 149–181, 2005
16. Fiorina P, Ansari MJ, Jurewicz M, Barry M, Ricchiuti V, Smith RN, SheaS, Means TK, Auchincloss H Jr, Luster AD, Sayegh MH, Abdi R: Role ofCXC chemokine receptor 3 pathway in renal ischemic injury. J Am SocNephrol 17: 716–723, 2006
17. Shiozawa F, Kasama T, Yajima N, Odai T, Isozaki T, Matsunawa M,Yoda Y, Negishi M, Ide H, Adachi M: Enhanced expression of inter-feron-inducible protein 10 associated with Th1 profiles of chemokinereceptor in autoimmune pulmonary inflammation of MRL/lpr mice.Arthritis Res Ther 6: R78–R86, 2004
18. Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, KovacAD, Gerard C, Boddeke HW, Nitsch R, Kettenmann H: CXCR3-depen-dent microglial recruitment is essential for dendrite loss after brainlesion. J Neurosci 24: 8500–8509, 2004
19. Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M,Koch AE, Moser B, Mackay CR: The chemokine receptors CXCR3 andCCR5 mark subsets of T cells associated with certain inflammatoryreactions. J Clin Invest 101: 746–754, 1998
20. Dean EG, Wilson GR, Li M, Edgtton KL, O’Sullivan KM, Hudson BG,Holdsworth SR, Kitching AR: Experimental autoimmune Goodpas-ture’s disease: A pathogenetic role for both effector cells and anti-body in injury. Kidney Int 67: 566–575, 2005
21. Izui S, Fossati-Jimack L, da Silveira SA, Moll T: Isotype-dependent patho-genicity of autoantibodies: Analysis in experimental autoimmune hemo-lytic anemia. Springer Semin Immunopathol 23: 433–445, 2001
22. Perez de Lema G, Maier H, Nieto E, Vielhauer V, Luckow B, MampasoF, Schlondorff D: Chemokine expression precedes inflammatory cellinfiltration and chemokine receptor and cytokine expression duringthe initiation of murine lupus nephritis. J Am Soc Nephrol 12: 1369–1382, 2001
23. Christen U, McGavern DB, Luster AD, von Herrath MG, Oldstone MB:Among CXCR3 chemokines, IFN-gamma-inducible protein of 10 kDa(CXC chemokine ligand (CXCL) 10) but not monokine induced byIFN-gamma (CXCL9) imprints a pattern for the subsequent develop-ment of autoimmune disease. J Immunol 171: 6838–6845, 2003
24. Salomon I, Netzer N, Wildbaum G, Schif-Zuck S, Maor G, Karin N:Targeting the function of IFN-gamma-inducible protein 10 suppressesongoing adjuvant arthritis. J Immunol 169: 2685–2693, 2002
25. Klein RS, Izikson L, Means T, Gibson HD, Lin E, Sobel RA, Weiner HL,Luster AD: IFN-inducible protein 10/CXC chemokine ligand 10-inde-pendent induction of experimental autoimmune encephalomyelitis.J Immunol 172: 550–559, 2004
26. Han GD, Suzuki K, Koike H, Yoneyama H, Narumi S, Shimizu F,Kawachi H: IFN-inducible protein-10 plays a pivotal role in maintainingslit-diaphragm function by regulating podocyte cell-cycle balance.J Am Soc Nephrol 17: 442–453, 2006
27. Panzer U, Steinmetz OM, Reinking RR, Meyer TN, Fehr S, Schneider A,Zahner G, Wolf G, Helmchen U, Schaerli P, Stahl RA, Thaiss F: Com-partment-specific expression and function of the chemokine IP-10/CXCL10 in a model of renal endothelial microvascular injury. J Am SocNephrol 17: 454–464, 2006
28. Panzer U, Steinmetz OM, Paust HJ, Meyer-Schwesinger C, Peters A,Turner JE, Zahner G, Heymann F, Kurts C, Hopfer H, Helmchen U,Haag F, Schneider A, Stahl RA: Chemokine receptor CXCR3 mediatesT cell recruitment and tissue injury in nephrotoxic nephritis in mice.J Am Soc Nephrol 18: 2071–2084, 2007
29. Liu MT, Keirstead HS, Lane TE: Neutralization of the chemokineCXCL10 reduces inflammatory cell invasion and demyelination andimproves neurological function in a viral model of multiple sclerosis.J Immunol 167: 4091–4097, 2001
30. Hsieh MF, Lai SL, Chen JP, Sung JM, Lin YL, Wu-Hsieh BA, Gerard C,
Luster A, Liao F: Both CXCR3 and CXCL10/IFN-inducible protein 10are required for resistance to primary infection by dengue virus. J Im-munol 177: 1855–1863, 2006
31. Wuest T, Farber J, Luster A, Carr DJ: CD4� T cell migration into thecornea is reduced in CXCL9 deficient but not CXCL10 deficient micefollowing herpes simplex virus type 1 infection. Cell Immunol 243:83–89, 2006
32. Salazar-Mather TP, Hamilton TA, Biron CA: A chemokine-to-cytokine-to-chemokine cascade critical in antiviral defense. J Clin Invest 105:985–993, 2000
33. Katschke KJ Jr, Rottman JB, Ruth JH, Qin S, Wu L, LaRosa G, PonathP, Park CC, Pope RM, Koch AE: Differential expression of chemokinereceptors on peripheral blood, synovial fluid, and synovial tissuemonocytes/macrophages in rheumatoid arthritis. Arthritis Rheum 44:1022–1032, 2001
34. Park MK, Amichay D, Love P, Wick E, Liao F, Grinberg A, Rabin RL,Zhang HH, Gebeyehu S, Wright TM, Iwasaki A, Weng Y, DeMartinoJA, Elkins KL, Farber JM: The CXC chemokine murine monokineinduced by IFN-gamma (CXC chemokine ligand 9) is made by APCs,targets lymphocytes including activated B cells, and supports anti-body responses to a bacterial pathogen in vivo. J Immunol 169:1433–1443, 2002
35. Rabin RL, Alston MA, Sircus JC, Knollmann-Ritschel B, Moratz C, NgoD, Farber JM: CXCR3 is induced early on the pathway of CD4� T celldifferentiation and bridges central and peripheral functions. J Immu-nol 171: 2812–2824, 2003
36. Muehlinghaus G, Cigliano L, Huehn S, Peddinghaus A, LeyendeckersH, Hauser AE, Hiepe F, Radbruch A, Arce S, Manz RA: Regulation ofCXCR3 and CXCR4 expression during terminal differentiation of mem-ory B cells into plasma cells. Blood 105: 3965–3971, 2005
37. Rosenkranz AR, Knight S, Sethi S, Alexander SI, Cotran RS, MayadasTN: Regulatory interactions of alphabeta and gammadelta T cells inglomerulonephritis. Kidney Int 58: 1055–1066, 2000
38. Hancock WW, Gao W, Csizmadia V, Faia KL, Shemmeri N, Luster AD:Donor-derived IP-10 initiates development of acute allograft rejection.J Exp Med 193: 975–980, 2001
39. Wu J, Zhou T, He J, Mountz JD: Autoimmune disease in mice due tointegration of an endogenous retrovirus in an apoptosis gene. J ExpMed 178: 461–468, 1993
40. Kikawada E, Lenda DM, Kelley VR: IL-12 deficiency in MRL-Fas(lpr)mice delays nephritis and intrarenal IFN-gamma expression, and di-minishes systemic pathology. J Immunol 170: 3915–3925, 2003
41. Tipping PG, Cornthwaite LJ, Holdsworth SR: Beta 2 integrin indepen-dent neutrophil recruitment and injury in anti-GBM glomerulonephritisin rabbits. Immunol Cell Biol 72: 471–479, 1994
42. Vielhauer V, Stavrakis G, Mayadas TN: Renal cell-expressed TNF re-ceptor 2, not receptor 1, is essential for the development of glomer-ulonephritis. J Clin Invest 115: 1199–1209, 2005
43. Abdi R, Means TK, Ito T, Smith RN, Najafian N, Jurewicz M, Tchipach-vili V, Charo I, Auchincloss H Jr, Sayegh MH, Luster AD: Differentialrole of CCR2 in islet and heart allograft rejection: Tissue specificity ofchemokine/chemokine receptor function in vivo. J Immunol 172: 767–775, 2004
44. Means TK, Hayashi F, Smith KD, Aderem A, Luster AD: The Toll-likereceptor 5 stimulus bacterial flagellin induces maturation and chemokineproduction in human dendritic cells. J Immunol 170: 5165–5175, 2003
45. Lenda DM, Stanley ER, Kelley VR: Negative role of colony-stimulatingfactor-1 in macrophage, T cell, and B cell mediated autoimmunedisease in MRL-Fas(lpr) mice. J Immunol 173: 4744–4754, 2004
46. Wuthrich RP, Glimcher LH, Yui MA, Jevnikar AM, Dumas SE, Kelley VE:MHC class II, antigen presentation and tumor necrosis factor in renaltubular epithelial cells. Kidney Int 37: 783–792, 1990
47. Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH: Macrophages instreptozotocin-induced diabetic nephropathy: Potential role in renalfibrosis. Nephrol Dial Transplant 19: 2987–2996, 2004
48. Lenda D, Kikawada E, Stanley ES, Kelley VR: Reduced macrophage
BASIC RESEARCH www.jasn.org
1188 Journal of the American Society of Nephrology J Am Soc Nephrol 19: 1177–1189, 2008

recruitment, proliferation, and activation in colony-stimulating factor-1-deficient mice results in decreased tubular apoptosis during renalinflammation. J Immunol 170: 3254–3262, 2003
49. Faraggiana T, Malchiodi F, Prado A, Churg J: Lectin-peroxidase con-jugate reactivity in normal human kidney. J Histochem Cytochem 30:451–458, 1982
50. Swinford AE, Bernstein J, Toriello HV, Higgins JV: Renal tubular dys-genesis: Delayed onset of oligohydramnios. Am J Med Genet 32:127–132, 1989
51. Moore KJ, Naito T, Martin C, Kelley VR: Enhanced response of mac-rophages to CSF-1 in autoimmune mice: A gene transfer strategy.J Immunol 157: 433–440, 1996
52. Schwarting A, Wada T, Kinoshita K, Tesch G, Kelley VR: IFN-gammareceptor signaling is essential for the initiation, acceleration, and
destruction of autoimmune kidney disease in MRL- Fas(lpr) mice.J Immunol 161: 494–503, 1998
53. Topham PS, Csizmadia V, Soler D, Hines D, Gerard CJ, Salant DJ,Hancock WW: Lack of chemokine receptor CCR1 enhances Th1 re-sponses and glomerular injury during nephrotoxic nephritis. J ClinInvest 104: 1549–1557, 1999
54. Kinoshita K, Tesch G, Schwarting A, Maron R, Sharpe AH, Kelley VR:Costimulation by B7–1 and B7–2 is required for autoimmune diseasein MRL-Faslpr mice. J Immunol 164: 6046–6056, 2000
Supplemental information for this article is available online at http://www.jasn.org/.
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 19: 1177–1189, 2008 CXCL9, not CXCL10, Promotes Nephritis 1189







![Role of CXCR3/CXCL10 Axis in Immune Cell Recruitment into ... fileIFNc [2]. This abundance of LP Th1 cells is largely responsible for the maintenance of an appropriate environment](https://img.pdfslide.us/doc/110x75/5ce8f8fd88c993e8488de8e4/role-of-cxcr3cxcl10-axis-in-immune-cell-recruitment-into-2-this-abundance.jpg)





















