Embed Size (px)
Citation preview



#152201 Cust: LWW Au: Webb Pg. No. i Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services
High-Resolution CT of the Lung

#152201 Cust: LWW Au: Webb Pg. No. ii Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services

#152201 Cust: LWW Au: Webb Pg. No. iii Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services
High-Resolution CT of the Lung
W. Richard Webb, MDProfessor Emeritus of Radiology and Biomedical ImagingEmeritus Member, Haile Debas Academy of Medical EducatorsUniversity of California San FranciscoSan Francisco, California
Nestor L. Müller, MD, PhDProfessor Emeritus of RadiologyDepartment of Radiology, University of British ColumbiaVancouver, British Columbia, Canada
David P. Naidich, MD, FACR, FAACPProfessor of Radiology and MedicineNew York UniversityLangone Medical CenterNew York, New York
F I F T H E D I T I O N

#152201 Cust: LWW Au: Webb Pg. No. iv Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services
Senior Executive Editor: Jonathan W. Pine, Jr.Acquisitions Editor: Ryan ShawProduct Development Editor: Amy G. DinkelProduction Project Manager: David OrzechowskiSenior Manufacturing Coordinator: Beth WelshMarketing Manager: Dan DresslerSenior Designer: Joan WendtProduction Service: S4Carlisle Publishing Services
Copyright © 2015 Wolters Kluwer Health
Two Commerce Square2001 Market StreetPhiladelphia, PA 19103 USALWW.com
4th edition © 2009 by LIPPINCOTT WILLIAMS & WILKINS, a WOLTERS KLUWER business
All rights reserved. This book is protected by copyright. No part of this book may be reproduced in any form by any means, including photocopying, or utilized by any information storage and retrieval sys-tem without written permission from the copyright owner, except for brief quotations embodied in critical articles and reviews. Materials appearing in this book prepared by individuals as part of their official duties as U.S. government employees are not covered by the above-mentioned copyright.
Printed in China
Library of Congress Cataloging-in-Publication Data
Webb, W. Richard (Wayne Richard), 1945- author.High-resolution CT of the lung / W. Richard Webb, Nestor L. Müller, David P. Naidich. — Fifth edition. p. ; cm.Includes bibliographical references and index.ISBN 978-1-4511-7601-8 (alk. paper)I. Müller, Nestor Luiz, 1948- , author. II. Naidich, David P., author. III. Title.[DNLM: 1. Lung—radiography. 2. Tomography, X-Ray Computed. 3. Lung Diseases—pathology. WF 600]RC734.T64616.2’407572—dc23
2014003388
Care has been taken to confirm the accuracy of the information presented and to describe generally accepted practices. However, the authors, editors, and publisher are not responsible for errors or omissions or for any consequences from application of the information in this book and make no warranty, expressed or implied, with respect to the currency, completeness, or accuracy of the contents of the publication. Application of the information in a particular situation remains the professional responsibility of the practitioner.
The authors, editors, and publisher have exerted every effort to ensure that drug selection and dosage set forth in this text are in accordance with current recommendations and practice at the time of publication. However, in view of ongoing research, changes in government regulations, and the constant flow of infor-mation relating to drug therapy and drug reactions, the reader is urged to check the package insert for each drug for any change in indications and dosage and for added warnings and precautions. This is particularly important when the recommended agent is a new or infrequently employed drug.
Some drugs and medical devices presented in the publication have Food and Drug Administration (FDA) clearance for limited use in restricted research settings. It is the responsibility of the health care providers to ascertain the FDA status of each drug or device planned for use in their clinical practice.
To purchase additional copies of this book, call our customer service department at (800) 638-3030 or fax orders to (301) 223-2320. International customers should call (301) 223-2300.
Visit Lippincott Williams & Wilkins on the Internet: at LWW.com. Lippincott Williams & Wilkins customer service representatives are available from 8:30 am to 6 pm, EST.
10 9 8 7 6 5 4 3 2 1

#152201 Cust: LWW Au: Webb Pg. No. v Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services
DEDICATION
To my father, who encouraged my curiosity and taught
me to figure things out
––WRW
To my wife, Isabela, and my children—Alison, Phillip,
and Noah Müller
––NLM
To Jocelyn, whose constant love and support has always
been my greatest inspiration
––DPN

#152201 Cust: LWW Au: Webb Pg. No. vi Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services

#152201 Cust: LWW Au: Webb Pg. No. vii Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services
vii
Contributing Authors
Brett M. Elicker, MDAssociate Professor of Clinical Radiology and Biomedical ImagingChief, Cardiac and Pulmonary ImagingUniversity of California San FranciscoSan Francisco, California
Myrna C. B. Godoy, MD, PhDAssistant Professor of RadiologyUniversity of TexasMD Anderson Cancer CenterHouston, Texas
C. Isabela S. Müller, MD, PhDDepartment of RadiologyDelfin ClinicSalvador, Bahia, Brazil

#152201 Cust: LWW Au: Webb Pg. No. viii Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services

#152201 Cust: LWW Au: Webb Pg. No. ix Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services
ix
Preface
During the past 25 years, high-resolution CT (HRCT) has become established as an indispensable tool in the evalu-ation of patients with diffuse lung disease. HRCT is now commonly used in clinical practice to detect and char-acterize a variety of lung abnormalities. In the approxi-mately 5 years since our fourth edition was published, considerable progress has taken place in the understand-ing of diffuse lung diseases and the recognition of new entities and their nature, causes, and characteristics. Without doubt, HRCT has played a fundamental role in contributing to this progress and has become essential to the diagnosis of a number of diffuse diseases.
This fifth edition continues what the three of us, in-dependently, in conjunction, and with each other’s en-couragement and support, began some 30 years ago. The photograph of the three of us below was taken by a local resident at the 1989 Diagnostic Course in Davos, on a walk we took on the promenade above the Sweitzerhof on the day of our arrival, when as junior faculty, we were more than a little anxious about teaching along with such im-portant and impressive chest radiologists as Fraser, Felson, Greenspan, Milne, Flowers, Heitzman, and many others.
of an inch thick, and, to our knowledge, referenced every known paper on HRCT. From our perspective, it was the most important thing we had ever done.
That is how things start. Maybe that is the best way things should start. It was certainly fun and rewarding for each of us. And we three have stuck together over the years, out of our combined respect, admiration, friend-ship, and good humor. Each one of us believes that we learned more from our collaboration than we taught.
In this edition, we have incorporated an update and review of numerous recent advances in the classifica-tion and understanding of diffuse lung diseases and their HRCT features. Recent technical modifications in obtain-ing HRCT have also been reviewed, most notably the use of helical HRCT and dose-reduction techniques. We hope the reader will find these changes and updates helpful. As is our wont, we have reorganized our discussions into new sections and chapters, which we feel best presents the most important topics in HRCT diagnosis for reference and learning.
A new section has been added at the end of the book to provide a general review of HRCT, including an illus-trated glossary of HRCT terms and a chapter providing a compilation of the common and typical appearances of the most common diffuse lung diseases encountered in clinical practice. These sections are intended to provide an illustrated index to the detailed descriptions of dis-eases found elsewhere in the book.
It is with a great deal of pride that we complete our fifth edition of this book, which has occupied so much of our thoughts, efforts, and time over the years. This task is accomplished in the hope that this book will encour-age future generations of thoracic imagers to develop mutually productive relationships with friends and col-leagues, in order to explore important questions in our understanding of the role of imaging in the assessment of thoracic disease.
To this end, we acknowledge the contributions of three esteemed colleagues, our former fellows, who have au-thored parts of this book. Their efforts have greatly in-spired our own enthusiasm for the considerable task of bringing this edition to fruition.
W. RichaRd Webb NestoR L. MüLLeR david P. Naidich
At this meeting, we each spoke about the use of HRCT, which, at the time, was a little-known technique that was regarded with skepticism by many radiologists. We learned from each other as we spoke, compared slides in the speaker-ready room, and gained confidence from our shared opinions. At this meeting, we began thinking about a collaboration that would combine our experience and thoughts about this new modality and its potential uses. Our first edition of this book was published in late 1991, with a grand total of 159 pages. It was a quarter

#152201 Cust: LWW Au: Webb Pg. No. x Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services

#152201 Cust: LWW Au: Webb Pg. No. xi Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services
xi
and prior editions of this book. Although they are too numerous to mention here, they are recognized through-out the following pages.
We wish to gratefully acknowledge the many colleagues who have provided us with insights and inspiration over the years, and allowed us to use their illustrations for this
Acknowledgments

#152201 Cust: LWW Au: Webb Pg. No. xii Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services

#152201 Cust: LWW Au: Webb Pg. No. xiii Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services
xiii
Contents
S E C T I O N IHIgH-REsOLuTION CT TECHNIquEs AND NORmAL ANATOmy 1
1 Technical Aspects of High-Resolution CT 2
2 Normal Lung Anatomy 47
S E C T I O N I IAPPROACH TO HRCT DIAgNOsIs AND FINDINgs OF LuNg DIsEAsE 71
3 HRCT Findings: Linear and Reticular Opacities 74
4 HRCT Findings: multiple Nodules and Nodular Opacities 106
5 HRCT Findings: Parenchymal Opacification 140
6 HRCT Findings: Air-Filled Cystic Lesions 165
7 HRCT Findings: Decreased Lung Attenuation 187
S E C T I O N I I I HIgH-REsOLuTION CT DIAgNOsIs OF DIFFusE LuNg DIsEAsE 207
8 The Idiopathic Interstitial Pneumonias, Part I: usual Interstitial Pneumonia/Idiopathic Pulmonary Fibrosis and Nonspecific Interstitial Pneumonia 208
9 The Idiopathic Interstitial Pneumonias, Part II: Cryptogenic Organizing Pneumonia, Acute Interstitial Pneumonia, Respiratory Bronchiolitis- Interstitial Lung Disease, Desquamative Interstitial Pneumonia, Lymphoid Interstitial Pneumonia, and Pleuroparenchymal Fibroelastosis 232
10 Collagen-Vascular Diseases 256
11 Diffuse Pulmonary Neoplasms and Pulmonary Lymphoproliferative Diseases 280
12 sarcoidosis 312
13 Pneumoconiosis, Occupational, and Environmental Lung Disease 342

#152201 Cust: LWW Au: Webb Pg. No. xiv Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services
xiv Contents
14 Hypersensitivity Pneumonitis and Eosinophilic Lung Diseases 376
15 Drug-Induced Lung Diseases and Radiation Pneumonitis 397
16 miscellaneous Infiltrative Lung Diseases 411
17 Infections 429
18 Pulmonary Edema and Acute Respiratory Distress syndrome 481
19 Cystic Lung Diseases 492
20 Emphysema and Chronic Obstructive Pulmonary Disease 517
21 Airways Diseases 552
22 Pulmonary Hypertension and Pulmonary Vascular Disease 622
S E C T I O N IV HIgH-REsOLuTION CT REVIEw 659
23 Illustrated glossary of High-Resolution CT Terms 660
24 Appearances and Characteristics of Common Diseases 678

256
RHEUMATOID ARTHRITIS 257
PROGRESSIVE SYSTEMIC SCLEROSIS (SCLERODERMA) 261
SYSTEMIC LUPUS ERYTHEMATOSUS 266
I M P O R T A N T T O P I C S
10 Collagen-Vascular Diseases
lung disease (ILD) and pulmonary hypertension, which together account for most of the morbidity and mortality in these patients (1,2).
The CVDs can cause a variety of ILDs, identical histo-logically to the idiopathic interstitial pneumonias (IIPs), including nonspecific interstitial pneumonia (NSIP), usual interstitial pneumonia (UIP), organizing pneumonia (OP), and lymphoid interstitial pneumonia (LIP) (Table 10-1) (5–7). The ILD may precede the clinical and laboratory manifestations of the CVD for up to several years, pres-ent together with systemic manifestations at the time of diagnosis of CVD or, more commonly, manifest later in the course of the disease (2,8,9). It is estimated that up to 20% of patients who present with a chronic ILD either have an occult CVD or subsequently develop a clinically overt CVD (9,10). The most common pattern of inter-stitial fibrosis seen in CVDs is NSIP (6,7,11). Therefore, CVDs tend to be associated with a finer reticular pattern and less honeycombing than typically seen in patients who have idiopathic pulmonary fibrosis (IPF), and ground-glass opacity is more common as a predominant abnormality. Furthermore, pleural thickening or effusion may be present in patients who have collagen diseases, but neither is a feature of IPF. Also, CVD may be asso-ciated with other abnormalities, such as bronchiectasis, bronchiolitis obliterans, and follicular bronchiolitis, not seen in patients who have IPF and having distinct high-resolution computed tomography (HRCT) appearances. It is important to note that pulmonary abnormalities in these patients may be due to the underlying CVD or may result from complications of treatment, such as oppor-tunistic infection and drug toxicity (2,12). Methotrexate, commonly used in the treatment of RA, may result in a variety of ILDs, most commonly NSIP (13). In recent years, there has been a major increase in the use of bio-logic disease-modifying agents in the treatment of CVDs
ATS American Thoracic Society
BOOP bronchiolitis obliterans organizing pneumonia
CVD collagen-vascular disease
DAD diffuse alveolar damage
DLCO carbon monoxide diffusing capacity
ERS European Respiratory Society
FVC forced vital capacity
IIP idiopathic interstitial pneumonia
ILD interstitial lung disease
IPF idiopathic pulmonary fibrosis
LIP lymphoid interstitial pneumonia
MALT mucosa-associated lymphoid tissue
MCTD mixed connective tissue disease
NSIP nonspecific interstitial pneumonia
OP organizing pneumonia
PFT pulmonary function test
PM-DM polymyositis-dermatomyositis
PSS progressive systemic sclerosis
RA rheumatoid arthritis
SLE systemic lupus erythematosus
UIP usual interstitial pneumonia
Abbreviations Used in This Chapter
POLYMYOSITIS-DERMATOMYOSITIS 268
MIXED CONNECTIVE TISSUE DISEASE 270
SJÖGREN SYNDROME 272
ANKYLOSING SPONDYLITIS 274
The collagen-vascular diseases (CVDs) are acquired immunologically mediated inflammatory disorders that affect many organs (1–3). The CVDs that most commonly involve the lungs are rheumatoid arthritis (RA), progres-sive systemic sclerosis (PSS), systemic lupus erythema-tosus (SLE), polymyositis-dermatomyositis (PM-DM), mixed connective tissue disease (MCTD), and Sjögren syndrome (1–3). The CVDs can affect all components of the lung, including the interstitium, the large and small airways, the pleura, and the pulmonary vasculature (4). The most important manifestations are diffuse interstitial

ChAPTER 10 Collagen-Vascular Diseases 257
+, uncommon; ++, common; +++, most common pattern.
pattern, and two (11%) patients had an OP pattern. RA preceded ILD in 12 patients; in 3 patients, ILD preceded RA, and in 3 patients, both conditions were diagnosed simultaneously (30). Yoshinouchi et al. (31) reported the HRCT and histologic findings in 16 patients with RA and ILD. Seven patients had UIP, seven had NSIP, and two had both UIP and NSIP (31). In the study by Tansey et al. (6) of 15 patients with RA and ILD, 7 had NSIP, 6 had follicular bronchiolitis and a minor component of NSIP, and 2 had UIP. Despite this controversy, perhaps because of the greater prevalence of the UIP pattern on HRCT and the fact that patients with characteristic findings of UIP seldom undergo lung biopsy, most recent reviews and studies consider UIP to be the most common pattern of ILD seen in patients with RA (1–4,18).
high-Resolution Computed Tomography FindingsThe HRCT findings of ILD in patients with RA are most commonly those of UIP (Figs. 10-1 to 10-3, Table 10-2) and less frequently NSIP (Fig. 10-4) or OP
(14,15). These medications, particularly agents blocking tumor necrosis factor-α such as etanercept and inflix-imab, may result in interstitial pneumonia, sarcoid-like disease, and vasculitis (14) (see Chapter 15).
RhEUMATOID ARThRITISRA is commonly associated with thoracic abnormalities, including interstitial fibrosis, OP (bronchiolitis obliterans organizing pneumonia [BOOP]), bronchiectasis, bronchiol-itis obliterans, necrobiotic nodules, and pleural effusion or pleural thickening (2,3,16). Other common complications include pulmonary infection and drug toxicity (12,17). The reported prevalence of ILD in patients with RA is highly variable, depending on the method of detection (e.g., pul-monary function test [PFT], radiograph, HRCT) and the population selected (asymptomatic, symptomatic, autopsy), ranging from as low as 4% and as high as 68% (18,19). Population-based studies have shown, however, that clini-cally significant ILD occurs in 5% to 10% of patients with RA (20,21) and fewer than 10% of patients die of respira-tory failure (20,22). Findings consistent with ILD are de-tectable on the chest radiograph in approximately 10% of patients (23–25). The reported prevalence of ILD on HRCT in patients with RA ranges from 19% to 56% (26–29). In many of these cases, the ILD was not associated with any pulmonary symptoms. Asymptomatic ILD is common in patients with RA, and its significance and implications for therapy is not clear (12). Symptomatic ILD in RA usually follows the onset of joint symptoms by up to several years; however, it may occasionally precede joint disease (12).
There is controversy in the literature about the relative prevalence of UIP and NSIP on surgical biopsy in patients with RA. Some investigators have reported a greater prev-alence of UIP (18,30), some a similar prevalence (31,32), and some a higher prevalence of NSIP (6). Lee et al. (30) reviewed the histologic findings in 18 patients with RA who underwent surgical biopsy for ILD using the Ameri-can Thoracic Society/European Respiratory Society (ATS/ERS) consensus classification (15). Ten (55%) patients had a UIP pattern, six (33%) patients had an NSIP
TABLE 10-1 Relative Frequency of Patterns of Abnormality in Collagen-Vascular Diseases
Pulmonary disease RA PSS SLE PM-DM MCTDSjögren syndrome
Ankylosing spondylitis
NSIP ++ +++ + +++ ++ +++UIP +++ + + + + +OP + + +++ + +LIP Rare + ++DAD + + ++ +Hemorrhage +++Mosaic perfusion and air trapping +++ ++ +Pleural effusion or thickening ++ +++
FIGURE 10-1 UIP in rheumatoid arthritis. HRCT shows reticular pattern and mild honeycombing in the subpleural lung regions.

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease258
FIGURE 10-2 A–C: Prone HRCT at three levels in a patient with RA and lung disease. Subpleural opacities in the mid-lung (A and B) have a small nodular or branching appearance, consistent with that of follicular bronchiolitis. At a lower level (C), findings of intralobular interstitial thickening and traction bronchiectasis are typical of fibrosis.
AB
C
FIGURE 10-3 RA and end-stage UIP with honeycombing. A: HRCT at the level of the tracheal carina shows subpleural honeycombing and interlobular septal thickening indistinguishable from IPF. B: HRCT through the right lung base shows diffuse honeycombing and septal thickening.
A B

ChAPTER 10 Collagen-Vascular Diseases 259
aMost common finding(s).bFinding(s) most helpful in differential diagnosis.
TABLE 10-2 hRCT Findings in Rheumatoid Arthritis
Bronchiectasis without fibrosisa
Findings of fibrosis (i.e., traction bronchiectasis and bronchiolectasis, intralobular interstitial thickening, irregular interlobular septal thickening, irregular interfaces)a
Honeycombing
Ground-glass opacitya
Peripheral and subpleural predominance of fibrosis or ground-glass opacitya,b
Lower lung zone and posterior predominancea,b
Pleural thickening or effusiona,b
Small centrilobular nodules (follicular bronchiolitis)
Large (rheumatoid) nodules
Findings of bronchiolitis obliterans (i.e., air trapping, mosaic perfusion)
of multiple patchy areas of airspace consolidation (80%) and ground-glass opacities (100%), usually with subpleu-ral or peribronchial distribution (33). In all but 2 of the 16 patients who underwent lung biopsy, the CT findings reflected the pathologic findings (33). Biederer et al. (34) correlated HRCT and PFTs in 53 patients with suspected ILD associated with RA. The most common finding was reticulation seen in 40 of 53 (75%) patients, presenting as a mixed pattern with ground-glass opacities in 15 of 40. A pure reticular pattern was most common in patients with long-standing ILD. The extent of interstitial abnormali-ties was highly variable and correlated with a decrease in carbon monoxide diffusing capacity (DLCO) (34). Patients with a definite UIP pattern on HRCT seldom undergo lung biopsy, but patients with RA and an NSIP pattern or indeterminate findings on HRCT may have UIP or NSIP on surgical biopsy. Kim et al. (35) reviewed the HRCT findings in 82 patients with RA who had ILD. The HRCT scans were interpreted as definite UIP in 20 (24%), likely NSIP in 19 (23%), and indeterminate in 43 (52%). Definite UIP was considered present when there was basilar predominant reticulation, traction bronchiec-tasis, and honeycombing, with limited ground-glass opac-ity. Predominantly bibasilar ground-glass opacities with limited (or no) reticulation and absent honeycombing was interpreted as likely NSIP. Of the 19 patients with a likely NSIP pattern on HRCT, 6 underwent surgical lung biopsy, which showed UIP in 4 and NSIP in 2 patients. Six out of 43 patients with an indeterminate pattern on HRCT underwent surgical lung biopsy, which showed UIP in 5 patients and NSIP in 1 (35).
Overall, patients with CVD-related ILD, including pa-tients with RA-related ILD, have a better prognosis than patients with IPF (4,36,37), but patients with RA and a definite UIP pattern on HRCT have a prognosis similar to that seen in patients with IPF (35,37). In one study of 362 patients (269 with IIP and 93 with interstitial pneu-monia associated with CVD), the patients with interstitial pneumonia associated with CVD survived longer (mean, 177 months) than patients with IIP (mean, 66.9 ± 6.5 months; p = 0.001) (36). A multivariate analysis showed that younger age, better pulmonary function, and the presence of a CVD were independent prognostic factors. No significant differences were found between CVD- associated NSIP and idiopathic NSIP in survival, clinical features, or lung function. The mean survival of patients with UIP associated with CVD was 177 months compared to 70 months in patients with IPF (36). However, the study showed a trend toward worse survival in RA-UIP patients than in RA-ILD patients with an NSIP pattern (p = 0.08) (36). Kim et al. (35) compared the prognosis in 82 pa-tients with RA-associated ILD with that in 51 patients with IPF. Twenty (24%) out of 82 patients with RA had HRCT findings interpreted as definite UIP. These patients showed worse survival than those without this pattern (median survival, 3.2 vs. 6.6 years), and a survival simi-lar to that shown by those with IPF. Analysis of specific HRCT features demonstrated that traction bronchiectasis
(27,33,34). Tanaka et al. (33) reviewed the HRCT find-ings in 63 patients with RA seen at an ILD clinic. The most common abnormalities evident on HRCT were reticulation (98% patients) and ground-glass opacities (90% patients). The authors identified four major CT patterns: UIP (41%), NSIP (30%), bronchiolitis (17%), and OP (8%). The UIP pattern was characterized by the presence of irregular linear opacities (100% of patients) and honeycombing (96%), involving predominantly the basal and subpleural lung regions with mild associated ground-glass opacities (92%) (Figs. 10-1 to 10-3). Trac-tion bronchiectasis and architectural distortion, when present, were always observed concomitantly with re-ticulation and honeycombing. NSIP was characterized by bilateral ground-glass opacities (100%), with some predominance of subpleural and basal regions, associated with fine reticulation (100%) and minor honeycombing (53%) (Fig. 10-4). OP was characterized by the presence
FIGURE 10-4 NSIP in rheumatoid arthritis. HRCT shows extensive bilateral ground-glass opacities and mild reticulation.

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease260
bronchiectasis on HRCT (48). Perez et al. (46) reviewed the prevalence and characteristics of airways involve-ment in 50 RA patients who did not have ILD. They found HRCT to be more sensitive in detecting airway ab-normalities than were PFTs. HRCT demonstrated bron-chial or lung abnormalities, or both, in 35 cases (70%), consisting of air trapping (n = 16; 32%), cylindrical bronchiectasis (n = 15; 30%), and mild heterogeneity in lung attenuation (i.e., mosaic perfusion) (n = 10; 20%). In contrast, PFTs demonstrated airway obstruction (i.e., reduced forced expiratory volume in 1 second/forced vi-tal capacity [FVC]) in only nine patients (18%) and evi-dence of small airways disease in only four (8%). PFT findings of airway obstruction and small airways disease correlated with the presence of bronchiectasis and bron-chial wall thickening (p = 0.003) (46). RA is a common cause of bronchiectasis in the general population. In a
and honeycomb fibrosis were associated with worse sur-vival. Female sex and a higher baseline DLCO were as-sociated with better survival (35).
Patients with RA-associated UIP or NSIP may occa-sionally develop acute exacerbation, i.e., rapid deterio-ration of respiratory symptoms without any identifiable cause and new parenchymal opacities due to diffuse alve-olar damage (DAD) or, less commonly, OP superimposed on the ILD (Fig. 10-5) (38–41). The HRCT findings of acute exacerbation consist of extensive bilateral ground-glass opacities with or without associated dependent ar-eas of consolidation superimposed on a background of UIP or NSIP (38,42). The prognosis of acute exacerbation in ILD in CVD is better than that of acute exacerbation of IPF (43). In one study (43), the 90-day mortality of acute exacerbation in 15 patients with CVD-associated inter-stitial pneumonias, including 6 with RA, was 33% com-pared to 69% in 13 patients who had acute exacerbation of IPF (43). Rarely, DAD may be the initial pulmonary manifestation of RA (44).
The most common abnormalities seen on HRCT in patients with RA are bronchiectasis and findings con-sistent with bronchiolitis (Fig. 10-6, Table 10-2). Bron-chiectasis has been reported on HRCT in approximately 30% of patients with RA (45,46). In one study of 84 patients with RA, 38 (49%) had abnormal HRCT scans (45). The findings included (a) bronchiectasis and/or bronchiolectasis (30%), (b) pulmonary nodules (22%), (c) subpleural micronodules and/or pseudoplaques (17%), (d) nonseptal linear attenuation (18%), (e) areas of ground-glass attenuation (14%), and (f) honeycomb-ing (10%) (45). Bronchiectasis and airways disease in RA can be associated with chronic infection, which has an increased incidence in rheumatoid patients, or bron-chiolitis obliterans (46,47). For example, in a study of 20 nonsmoking RA patients who had normal chest radio-graphs, 5 (25%) were found to have unsuspected basal
FIGURE 10-6 Bronchiectasis and obliterative bronchiolitis in RA. HRCT shows extensive bilateral bronchiectasis and areas of decreased attenuation and vascularity (mosaic perfusion), mainly in the left lung.
FIGURE 10-5 Acute exacerbation of ILD in rheumatoid arthritis. A: HRCT shows mild peripheral reticulation, irregular thickening of the interlobular septa, and minimal ground-glass opacities. The findings are consistent with UIP. B: HRCT 6 months later when the patient developed acute respiratory failure demonstrates extensive bilateral ground-glass opacities with associated linear opacities (crazy-paving pattern) and traction bronchiectasis consistent with DAD. The diagnosis of acute exacerbation of UIP was made after exclusion of other potential causes of DAD.
A B

ChAPTER 10 Collagen-Vascular Diseases 261
FIGURE 10-7 Necrobiotic nodules in RA. HRCT shows bilateral subpleural nodules. Also noted are several irregular linear opacities consistent with mild interstitial fibrosis.
Utility of high-Resolution Computed TomographyHRCT is more sensitive than chest radiography in the diagnosis of lung disease in patients who have RA. Fujii et al. (57) reviewed the chest radiographic and HRCT findings of 91 patients who had RA. On HRCT, 43 patients had findings of UIP with fibrosis, 5 had find-ings consistent with bronchiolitis obliterans, and 43 had a normal HRCT. In approximately half of these 91 patients, chest radiographic findings were similar to those shown on HRCT. However, 17 of 46 (37%) patients believed to have normal chest radiographs had HRCT abnormalities consistent with rheumatoid lung disease. Furthermore, 14 of 43 (33%) patients believed to have abnormal chest radiographs had no evidence of significant lung disease on HRCT (57). Also, HRCT can be useful in demonstrat-ing lung disease in RA patients who have normal chest radiographs but have pulmonary function abnormalities (59,60). Some HRCT findings are more frequent in symp-tomatic patients who have rheumatoid lung disease (45). These include honeycombing, bronchiectasis, nodules, and ground-glass opacity.
PROGRESSIVE SYSTEMIC SCLEROSIS (SCLERODERMA)PSS has a higher prevalence of pulmonary involvement than the other CVDs. The most common manifestations and leading causes of death are interstitial fibrosis, which occurs eventually in up to 75% of patients, and pulmo-nary arterial hypertension (61,62). ILD most often com-plicates the diffuse cutaneous form of PSS but can also be associated with the limited form of the disease or with PSS without cutaneous involvement (20,61). Initial stud-ies using echocardiography suggested a prevalence of PA hypertension of up to 49% in PSS, but more recent pro-spective studies using cardiac catheterization as the gold standard for diagnosis have shown a prevalence of 8 to 12% (61,63,64). Approximately 80% of patients with PSS and ILD have a histologic pattern of NSIP (11,65). Bouros et al. (11) reviewed the histologic findings in 80 patients with PSS and ILD. Approximately 78% had NSIP, 8% had UIP, 7% had end-stage lung disease, and the remain-ing had other patterns. The most common abnormality on chest radiography is a symmetric basal reticulonodular pattern. The chest radiograph, however, may be normal in patients with abnormal PFTs and abnormal HRCT (66). The incidence of radiographically recognizable interstitial disease is probably around 25%, although various studies quote an incidence ranging from 10% to 80% (67).
high-Resolution Computed Tomography FindingsThe HRCT findings of interstitial fibrosis in PSS usually resemble those of idiopathic NSIP and consist mainly of ground-glass opacities frequently with superimposed fine
recent study of 106 patients with bronchiectasis con-firmed by HRCT, RA was the cause of bronchiectasis in 29% of African American patients and 6% of European American patients (49). The occurrence of bronchiol-itis obliterans in patients who have RA is discussed in Chapter 20.
An uncommon abnormality seen in patients who have RA or other CVDs is follicular bronchiolitis (50,51). This is a benign condition characterized by prominent hyper-plasia of lymphoid follicles around the bronchioles and, to a lesser extent, bronchi (51). HRCT findings of fol-licular bronchiolitis consist of multiple small nodules in a predominantly centrilobular, subpleural, and peribron-chial distribution (see Fig. 11-23) (51,52). The nodules usually measure 1 to 4 mm in diameter but may occasion-ally be 1 cm or more in diameter (52). Follicular bron-chiolitis may also be associated with lung cysts similar to those seen in LIP (see Fig. 11-24).
Single or multiple lung nodules seen in patients who have RA may represent necrobiotic (rheumatoid) nod-ules. Rheumatoid nodules may range from a few mil-limeters to several centimeters in size, are frequently subpleural, and are usually multiple and asymptomatic (Fig. 10-7) (53–55). Cavitation occurs in approximately 50% and calcification is uncommon (55). Occasionally, rheumatoid nodules may result in bronchopleural fistula with associated pneumothorax (54,56).
Pleural thickening occurs in 20% to 33% of patients with RA (20,57). In a study by Fujii et al. (57), pleural thickening was visible on HRCT in 33% of the 91 pa-tients studied and in 44% of the patients who had HRCT findings of interstitial pneumonia. Pleural effusion has an incidence of 3% to 5% (20,58). The pleural effusion is usually small and resolves spontaneously (54). Enlarged central pulmonary arteries are seen on HRCT in approxi-mately 46% of patients with ILD and mediastinal lymph-adenopathy is seen in 20%.

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease262
FIGURE 10-8 NSIP in progressive systemic sclerosis. A: HRCT performed on a multidetector CT scanner shows extensive bilateral ground-glass opacities and mild superimposed reticulation. B: Coronal reformation demonstrates predominantly peripheral and lower lung zone distribution of the ground-glass opacities and reticulation.
A B
reticulation and traction bronchiectasis (Figs. 10-8 and 10-9, Table 10-3) (5,65). Associated focal areas of consoli-dation are seen in some cases (Fig. 10-10) (68). Desai et al. (65) compared the HRCT findings in 225 patients with ILD associated with PSS with the findings in 40 consecutive patients with IPF and 27 patients with idiopathic NSIP. Approximately two-thirds of patients with PSS had pre-dominant ground-glass opacities or a mixed pattern with ground-glass opacities and reticulation, and one-third of patients had a predominant reticular pattern. This was sim-ilar to patients with idiopathic NSIP. The only difference was the overall extent of abnormalities, which was smaller in patients with PSS (median extent of ILD was 13% of the lung parenchyma compared to 30% for idiopathic NSIP). Although NSIP is the most common pattern of abnormality seen in patients with PSS, reticulation may sometimes be the predominant abnormality on HRCT and result in an appearance similar to that of IPF (Figs. 10-11 and 10-12).
Remy-Jardin et al. (69) reviewed the HRCT, PFT, and bronchoalveolar lavage results of 53 patients who had PSS, emphasizing the frequency of ground-glass opac-ity and honeycombing in these subjects. Among the
32 patients who had abnormal HRCT findings, 26 (81%) had ground-glass opacities and 19 (59%) had honeycomb-ing. The honeycombing in PSS is usually of limited extent and associated with areas of ground-glass opacity. In the study by Remy-Jardin et al. (69), all patients who had honeycombing also showed ground-glass opacity. These abnormalities had a distinct basal, posterior, and periph-eral predominance. Goldin et al. (70) reviewed the HRCT scans in 162 patients with symptomatic PSS-related ILD. The main findings consisted of ground-glass opacities (90%) including areas of ground-glass attenuation with-out evidence of fibrosis (49%), evidence of fibrosis (93%), and honeycombing (37%). All findings involved mainly the lower lung zones. The authors concluded that in the majority of cases the findings were consistent with NSIP but that the presence of honeycombing in 37% of HRCT scans suggests that some patients may have a mixture or overlap of NSIP and UIP patterns (70). The extent of pul-monary fibrosis seen on HRCT scans was significantly negatively correlated with FVC and TLC, i.e., associated with restrictive physiologic impairment, and negatively correlated with DLCO, i.e., associated with impairment
FIGURE 10-9 A and B: Subpleural ground-glass opacity in a young patient with scleroderma. A subtle but distinct increase in opacity is visible in the posterior lungs on prone scans.
A B
ChAPTER 10 Collagen-Vascular Diseases 263
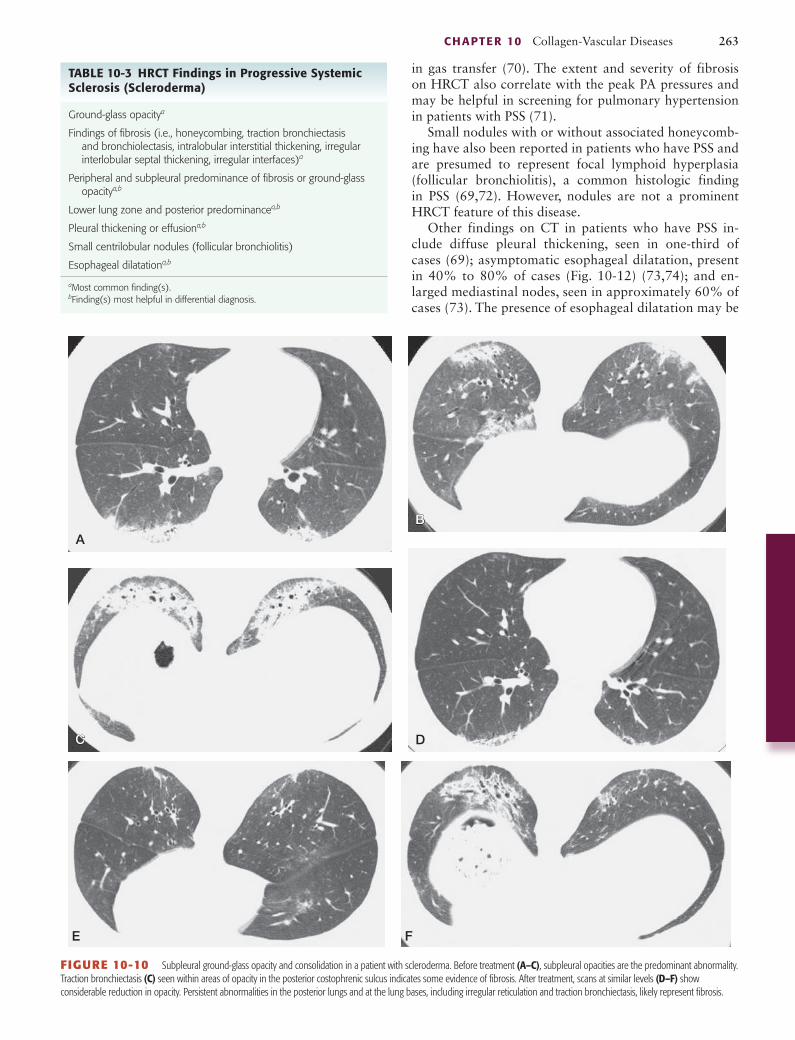
FIGURE 10-10 Subpleural ground-glass opacity and consolidation in a patient with scleroderma. Before treatment (A–C), subpleural opacities are the predominant abnormality. Traction bronchiectasis (C) seen within areas of opacity in the posterior costophrenic sulcus indicates some evidence of fibrosis. After treatment, scans at similar levels (D–F) show considerable reduction in opacity. Persistent abnormalities in the posterior lungs and at the lung bases, including irregular reticulation and traction bronchiectasis, likely represent fibrosis.
A
C
B
D
E F
aMost common finding(s).bFinding(s) most helpful in differential diagnosis.
TABLE 10-3 hRCT Findings in Progressive Systemic Sclerosis (Scleroderma)
Ground-glass opacitya
Findings of fibrosis (i.e., honeycombing, traction bronchiectasis and bronchiolectasis, intralobular interstitial thickening, irregular interlobular septal thickening, irregular interfaces)a
Peripheral and subpleural predominance of fibrosis or ground-glass opacitya,b
Lower lung zone and posterior predominancea,b
Pleural thickening or effusiona,b
Small centrilobular nodules (follicular bronchiolitis)
Esophageal dilatationa,b
in gas transfer (70). The extent and severity of fibrosis on HRCT also correlate with the peak PA pressures and may be helpful in screening for pulmonary hypertension in patients with PSS (71).
Small nodules with or without associated honeycomb-ing have also been reported in patients who have PSS and are presumed to represent focal lymphoid hyperplasia (follicular bronchiolitis), a common histologic finding in PSS (69,72). However, nodules are not a prominent HRCT feature of this disease.
Other findings on CT in patients who have PSS in-clude diffuse pleural thickening, seen in one-third of cases (69); asymptomatic esophageal dilatation, present in 40% to 80% of cases (Fig. 10-12) (73,74); and en-larged mediastinal nodes, seen in approximately 60% of cases (73). The presence of esophageal dilatation may be

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease264
PSS and severity of ILD. Overall, the pattern and distribu-tion of abnormalities were similar to those seen in adults. However, whereas in adults honeycombing predominates in the lower lung zones, in children the honeycombing was most severe in the upper lung zones (75).
Follow-up of patients with PSS and ILD may show initial improvement (Fig. 10-10), but in the majority of cases the fibrosis progresses on long-term follow-up (Fig. 10-13). Kim et al. (68) reviewed the findings on se-rial HRCT scans in 40 patients with PSS and ILD over a mean follow-up period of 39 months. On the initial HRCT, all patients had ground-glass opacities (mean extent 18%), 36 (90%) had irregular linear opacities (mean extent 4%), 13 (33%) had consolidation (mean extent 1.9%), and 12
helpful in the differential diagnosis of PSS from other dif-fuse ILDs. The coronal luminal diameter of the esopha-gus in patients who have PSS, as shown on CT, has been reported to range from 12 to 40 mm (mean, 23 mm), a finding that was not seen in a control group of 13 pa-tients who had a variety of other parenchymal and air-way abnormalities (73).
Seely et al. (75) assessed the radiographic and HRCT findings in 11 children (mean age, 11 years) with PSS. ILD was evident on the radiograph in 2 patients and on HRCT in 8. The HRCT findings consisted of ground-glass opacity, seen in all 8 patients; linear opacities, seen in 6; honeycombing, seen in 5; and small subpleural nodules, seen in 7. There was no correlation between duration of
FIGURE 10-11 A and B: Findings of fibrosis in a patient with scleroderma. Subpleural honeycombing, traction bronchiectasis, and some irregular interlobular septal thickening are the predominant features. These abnormalities closely mimic the appearance of IPF.
A B
FIGURE 10-12 A and B: Prone HRCT in a patient with scleroderma with mild interstitial fibrosis and esophageal dilatation. Irregular reticular opacities are visible in the peripheral lung, consistent with fibrosis. The esophagus (e) is dilated and contains an air-fluid level.
A B

ChAPTER 10 Collagen-Vascular Diseases 265
ILD may also represent fibrosis particularly when there is admixed reticulation or traction bronchiectasis (76–78). The current evidence in the literature is that ground-glass opacities in patients with PSS-associated NSIP are most commonly associated with irreversible disease (79,80). Shah et al. (79) performed sequential HRCT in 41 patients with PSS over a mean follow-up period of 27 months (range, 6–60 months). Ground-glass opacity was the most common imaging finding, present in 66% of patients, and usually associated with other signs of interstitial disease, including nonfibrotic interstitial opacities in 27% and fi-brotic interstitial opacities in 32%. Improvement was only documented in two (5%) patients with ground-glass opac-ities and nonfibrotic interstitial opacities (79).
The prognosis of NSIP associated with PSS is similar to that of idiopathic NSIP and considerably better than that of IPF (36). The prognosis of ILD in PSS is influenced more by the severity of lung involvement than the pat-tern of parenchymal disease. Bouros et al. (11) correlated the initial histologic findings with prognosis in 80 patients with PSS-associated ILD. The histologic appearances in-cluded cellular NSIP (n = 15), fibrotic NSIP (n = 47), UIP (n = 6), end-stage lung disease (n = 6), and other patterns (n = 6). Five-year survival differed little between NSIP (91%) and UIP/end-stage lung disease (82%); mortality was associated with lower initial DLCO and FVC levels (p = 0.004 and p = 0.007, respectively). Survival and se-rial FVC and DLCO did not differ between cellular and fibrotic NSIP. Increased mortality in NSIP was associated with lower initial DLCO (p = 0.04) and deterioration in DLCO levels during the next 3 years (p < 0.005). The au-thors concluded that outcome in PSS-associated ILD is linked more strongly to disease severity at presentation and serial DLCO than to histopathologic findings (11). Goldin et al. (70) reviewed the baseline HRCT scan im-ages of 162 patients with PSS-associated ILD randomized into a prospective clinical trial and assessed the extent and distribution of pure ground-glass opacity (i.e., increased lung attenuation in the absence of reticular interstitial
(30%) had honeycombing (mean extent 2%). Follow-up HRCT showed increase in overall extent of disease in 24 patients and showed no change in 16 patients. The worsen-ing was due mainly to an increased extent of ground-glass opacity and honeycombing. The increase in the extent of honeycombing on CT correlated significantly with the de-crease in DLCO (68). There was no significant difference in the extent of ground-glass opacity, irregular linear opacity, and honeycombing on the initial HRCT between patients who showed progression of disease or remained stable.
Utility of high-Resolution Computed TomographyHRCT plays a major role in the detection and charac-terization of lung involvement in PSS patients (70,76). HRCT is commonly performed to identify the presence of ILD in patients with PSS because the clinical symptoms and PFTs are nonspecific and the chest radiographic find-ings are often equivocal or falsely negative (76). Dyspnea may result from ILD, pulmonary vascular disease, or car-diac impairment and PFTs are limited by the wide range of normal, typically 80% to 120% of normal values (76). Schurawitzki et al. (66) studied 23 patients who had PSS using chest radiographs and HRCT. Chest radiographs were abnormal in only 15 (65%), but HRCT showed evidence of ILD in 21 (91%); the authors concluded that HRCT was clearly superior to chest radiographs for de-tecting minimal lung disease.
Wells et al. (77) assessed the potential role of HRCT as a predictor of lung histology on open-lung biopsy specimens in patients who had PSS. In this study, CT discriminated correctly between inflammatory and fibrotic histologic appearances in 16 of 20 (80%) biopsy specimens. Pre-dominant ground-glass opacities often correlated with the presence of inflammation, and the presence of a pre-dominantly reticular pattern on HRCT correlated closely with the presence of fibrosis on the pathologic specimens (77). However, ground-glass on HRCT in patients with
FIGURE 10-13 Disease progression in PSS. A: HRCT shows bilateral ground-glass opacities, reticulation, traction bronchiectasis, and minimal honeycombing. B: HRCT 2 years later demonstrates progression of fibrosis with more extensive reticulation and honeycombing. Also noted is a fluid level within the dilated esophagus.
A B

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease266
cases. The staging system was predictive of mortality for all scorers. The authors concluded that that discrimina-tory prognostic information in PSS-associated ILD can be obtained using a staging system based on combined evaluation with HRCT and PFTs (82).
SYSTEMIC LUPUS ERYThEMATOSUSSLE is a multisystem autoimmune CVD that typically af-fects young women (female-to-male ratio of 9:1) (83,84). The age at diagnosis is usually between 15 and 45 years (84). SLE is commonly associated with pleural and pul-monary abnormalities. An autopsy study of 90 patients with SLE found pleuropulmonary involvement in 98%, the most common being pleuritis (78%), bacterial infec-tions (58%), primary and secondary alveolar hemorrhages (26%), followed by distal airway alterations (21%), op-portunistic infections (14%), and pulmonary thrombo-embolism, both acute and chronic (8%) (85). Sepsis was considered the major cause of death (85). Pleural effusion is seen on chest radiographs in 30% to 50% of patients during the course of disease (3,84). The pleural effusion may be uni- or bilateral, and is usually small to moderate in size. Pleural involvement may be the first manifestation of SLE and is commonly associated with pericarditis (86).
More than 50% of patients who have SLE have lung disease at some time (87). Pulmonary parenchymal com-plications of SLE include pneumonia, acute lupus pneu-monitis, diffuse pulmonary hemorrhage, and ILD (86). The most common pulmonary complication of SLE is pneumonia (86,87). Patients with SLE are more suscep-tible to bacterial and opportunistic infections due to im-munosuppressive therapy with corticosteroids or other agents as well immunologic dysfunction related to SLE (86). Acute lupus pneumonitis occurs in 1% to 4% of patients with SLE and usually manifests with sudden on-set of fever, cough and dyspnea (86). It is characterized histologically by a combination of DAD, edema, and al-veolar hemorrhage (5,86). Diffuse alveolar hemorrhage is an uncommon but severe manifestation of SLE, with a prevalence ranging from 0.5% to 6% (86).
Clinically significant chronic ILD develops in 3% to 8% of patients with SLE, there being a progressive in-crease in prevalence with disease duration (86,88,89). Trivial interstitial abnormalities have been reported on HRCT in up to 38% of patients with SLE and no clini-cal evidence of lung involvement (4,90). Because signifi-cant ILD is uncommon in SLE, there is limited data on the pathologic pattern. However, as with other CVDs, the most common pattern appears to be NSIP followed by UIP (4,6,86). OP (BOOP) is also seen with increased frequency in patients who have SLE (86,91) and LIP has been described in a few patients (86).
high-Resolution Computed Tomography FindingsHRCT findings in patients who have SLE include (a) findings of fibrosis, although they are less common than in patients
thickening or architectural distortion), pulmonary fibrosis (i.e., reticular intralobular interstitial thickening, traction bronchiectasis, and bronchiolectasis), and honeycomb cysts. HRCT scan findings included evidence of pulmo-nary fibrosis (93% of patients), areas of pure ground-glass opacity (49%) and areas of honeycombing (37%). The extent of pulmonary fibrosis on baseline HRCT scans was predictive of the progression rate in the absence of active immunosuppressive therapy as well as the response to cyclophosphamide therapy, which was greatest in those with the most extensive pulmonary fibrosis seen on base-line HRCT scans (70). Pure ground-glass opacity and the extent of honeycombing at the baseline HRCT did not significantly affect the FVC at 12 months (70).
Densitometry may be more reproducible than visual assessment of lung changes on HRCT and may correlate better with functional impairment in patients with PSS. Camiciottoli et al. (81) assessed the intra- and interopera-tor reproducibility of visual and densitometric lung CT analysis in 48 PSS patients. The intra- and interoperator reproducibility of mean lung attenuation (intraobserver weighted kappa = 0.97; interobserver weighted kappa = 0.96) were higher than those of visual assessment (intrao-bserver weighted kappa = 0.71; interobserver weighted kappa = 0.69). In univariate analysis, only densitomet-ric measurements correlated with exercise and quality of life questionnaire parameters. In multivariate analysis, mean lung attenuation, skewness, and kurtosis correlated significantly with FRC, FVC, DLCO, exercise test, and quality of life questionnaire parameters, while visual as-sessment was associated only with FRC and FVC (81).
The best estimate of prognosis in PSS-ILD is proba-bly obtained by using a semi-quantitative assessment of extent of disease on CT, integrated, if necessary, with lung function. Goh et al. (82) evaluated the prognostic value of baseline HRCT and PFT variables in 215 patients with PSS-ILD. Increasingly extensive disease on HRCT was a powerful predictor of mortality (p < 0.0005), with an optimal extent threshold of 20%. Patients with disease extent less than or equal to 10% on HRCT were clas-sified as having limited disease and those with extent greater than or equal to 30% as having extensive disease. In patients with HRCT extent of 10% to 30% (termed indeterminate disease), a FVC threshold of 70% was an adequate prognostic substitute. On the basis of these observations, Systemic sclerosis associated interstitial lung disease (SSc-ILD) was staged as limited disease (min-imal disease on HRCT or, in indeterminate cases, FVC ⩾ 70%) or extensive disease (extensive disease on HRCT or, in indeterminate cases, FVC < 70%). This system (hazards ratio [HR], 3.46; 95% confidence interval [CI], 2.19–5.46; p < 0.0005) was more discriminatory than an HRCT threshold of 20% (HR, 2.48; 95% CI, 1.57–3.92; p < 0.0005) or an FVC threshold of 70% (HR, 2.11; 95% CI, 1.34–3.32; p = 0.001). The system was evaluated by four trainees and four practitioners, in PSS patients with minimal and severe disease on HRCT defined as clearly less than 20% or clearly more than 20%, respectively, and the use of an FVC threshold of 70% in indeterminate

ChAPTER 10 Collagen-Vascular Diseases 267
and 10-15) (90,92,93). These findings tend to be mild in-volving a small percentage of the lung parenchyma and usually are not associated with clinical symptoms or ab-normal pulmonary function (4,90,92). Diffuse interstitial disease with a pattern characteristic of NSIP or UIP is considerably less common being seen in approximately 4% of patients (Figs. 10-14 and 10-15) (94).
Ground-glass opacity and consolidation in patients with SLE may be associated with pneumonia, lupus pneu-monitis (Fig. 10-16), pulmonary hemorrhage (Fig. 10-17), or, occasionally organizing pneumonia (BOOP) (5,94). Pulmonary infection, acute lupus pneumonitis, and dif-fuse alveolar hemorrhage may result in clinical and ra-diologic findings of ARDS (84,95). Occasionally, hazy or fluffy centrilobular perivascular opacities may be seen, likely related to vasculitis (96).
Findings of airways disease such as bronchial wall thickening and bronchiectasis have been reported in 18% to 20% of patients who have SLE (90,92). Pleuropericar-dial abnormalities were seen in 15% to 17% of cases in two studies (92,93).
Utility of high-Resolution Computed TomographySeveral studies have shown that interstitial fibrosis is seen more frequently on HRCT than on chest radio-graphs (90,92,93,97). Bankier et al. (90) performed a
who have RA or scleroderma; (b) ground-glass opacity; (c) small nodules; (d) bronchial wall thickening or bronchi-ectasis; and (e) pleural thickening or effusion (Table 10-4).
The most common HRCT findings of interstitial fi-brosis in patients with SLE include interlobular septal thickening, intralobular interstitial thickening, ground-glass opacities, and architectural distortion (Figs. 10-14
aMost common finding(s).bFinding(s) most helpful in differential diagnosis.
TABLE 10-4 hRCT Findings in Systemic Lupus Erythematosus
Ground-glass opacitya
Findings of fibrosis (i.e., intralobular interstitial thickening, irregular interlobular septal thickening, irregular interfaces, bronchiectasis and bronchiolectasis)a
Honeycombing (less common than with IPF)
Peripheral and subpleural predominance of fibrosis or ground-glass opacitya,b
Lower lung zone and posterior predominancea,b
Bronchiectasis
Pleural thickening or effusiona,b
A
B
FIGURE 10-14 NSIP pattern in SLE. A: HRCT at the level of the aortic arch shows patchy bilateral ground-glass opacities and mild reticulation. B: HRCT at the level of the lung bases demonstrates more extensive abnormalities.
A
B
FIGURE 10-15 Supine (A) and prone (B) HRCT in a 34-year-old woman with SLE, ILD, and progression of PFT abnormalities. Reticular opacities in the peripheral lung are consistent with fibrosis.

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease268
POLYMYOSITIS-DERMATOMYOSITISPolymyositis and dermatomyositis are chronic autoim-mune disorders characterized by weakness in the proxi-mal limb muscles (98,99). Approximately 50% of patients have a characteristic skin rash, which enables distinction of dermatomyositis from polymyositis. PM-DM is a rare disease, with an overall incidence ranging from 2 to 10 new cases per million persons at risk per year (99). Pul-monary complications occur in more than 40% of pa-tients, and are associated with significant morbidity and mortality (99). Common complications include ILD, aspi-ration, pneumonia, and drug-induced lung diseases (99). The risk of malignancy is also increased, particularly in DM, the standardized index ratio for lung cancer being 5.9 for DM and 2.8 for PM (100).
It is estimated that 35% to 40% of patients with PM-DM develop ILD during the course of their disease (101). The pattern of involvement is most commonly NSIP (6,101–103). Other histologic patterns are OP, UIP, and DAD. LIP is uncommon (99,101). Patients with PM-DM may have more than one pattern of abnormality on lung biopsy, the most common combination being NSIP and OP (6). The radiographic findings of PM-DM-associated ILD usually consist of reticular opacities and/or areas of
prospective study in 48 patients who had SLE and no prior clinical evidence of lung involvement. Three (6%) patients had evidence of fibrosis on radiographs. Of the 45 patients who had normal chest radiographs, 17 (38%) had abnormalities evident on HRCT. These consisted of interlobular septal thickening in 15 patients (33%), intra-lobular interstitial thickening in 15 (33%), architectural distortion in 10 (22%), small nodules in 10 (22%), bron-chial wall thickening in 9 (20%), bronchial dilatation in 8 (18%), areas of ground-glass opacity in 6 (13%), and areas of airspace consolidation in 3 (7%) (90).
Fenlon et al. (92) assessed the radiographic and HRCT findings in 34 patients who had SLE. Abnormalities were identified on chest radiographs in 8 (24%) patients and on HRCT in 24 (70%). The most common findings on HRCT were interstitial fibrosis seen in 11 (32%) patients, bronchiectasis in 7 (20%), mediastinal or axillary lymph-adenopathy in 6 (18%), and pleuropericardial abnormali-ties in 5 (15%) (92). In another study of 29 patients who had SLE, the chest radiograph was abnormal in 10 (34%) and the HRCT in 20 (72%) patients. The most frequently detected abnormality on HRCT was ILD, which was seen in 11 (38%) patients. Of 15 patients who had normal clinical examination, normal PFTs, and normal chest ra-diographs, 4 (26%) had HRCT features of ILD (93).
FIGURE 10-16 HRCT in a 29-year-old woman with SLE and shortness of breath. A: Patchy ground-glass opacities are visible. B: Four months later, there has been progression of the abnormalities. Lung biopsy revealed lupus pneumonitis.
BA
FIGURE 10-17 A and B: HRCT in a 19-year-old woman with newly diagnosed SLE and pulmonary hemorrhage. Patchy areas of ground-glass opacity, which appear centrilobular and lobular, are visible.
A B

ChAPTER 10 Collagen-Vascular Diseases 269
airspace consolidation (52%), parenchymal micronod-ules (28%), and honeycombing (16%). A relatively high prevalence of airspace consolidation (52%) and a low prevalence of honeycombing (16%) were observed. Cor-relation of HRCT with pathologic findings showed that 2 patients who had extensive consolidation had DAD; 8 patients who had subpleural bandlike opacities or air-space consolidation, or both, had OP; and 4 patients who had honeycombing had UIP (104). Cottin et al. (103) as-sessed the HRCT and histologic findings in 17 patients with PM-DM. The most common HRCT findings were reticular and ground-glass opacities. Histologic patterns included NSIP in 11 (65%) patients, UIP in 2, OP (BOOP) in 2, LIP in 1, and unclassifiable interstitial pneumonia in 1 patient (60). Survival at 5 years was 50%. Douglas et al. (102) reviewed the HRCT findings in 30 patients with PM-DM-associated ILD. The findings included irregular linear opacities seen in 19 of 30 (63%) patients, consoli-dation in 16 of 30 (53%), and ground-glass opacities in 13 of 30 (43%). In the majority of patients the findings had a lower lobe predominance. None of the patients had honeycombing (102). Surgical lung biopsies available in
consolidation. In one study of 57 patients with PM-DM-associated ILD, reticular opacities were seen in 95% of cases and areas of consolidation in 25% (102). In more than 90% of patients, the findings involved mainly the lower lobes (102).
high-Resolution Computed Tomography FindingsHRCT findings of PM-DM include (a) ground-glass opacity; (b) findings of fibrosis, although honeycomb-ing is uncommon; and (c) consolidation (Figs. 10-18 to 10-20, Table 10-5). These findings are consistent with NSIP being the most common histologic pattern fol-lowed by OP, UIP, and DAD. Ikezoe et al. (104) reviewed the HRCT findings in 25 patients who had PM-DM; 23 had abnormal HRCT scans. The most common findings seen in these 23 patients included ground-glass opacities (92%), linear opacities (92%), irregular interfaces (88%),
FIGURE 10-20 OP pattern in polymyositis. HRCT shows bilateral peribronchial and subpleural areas of consolidation and patchy ground-glass opacities.
FIGURE 10-19 NSIP and OP in polymyositis. HRCT shows patchy bilateral ground-glass opacities consistent with NSIP. Also noted are perilobular opacity (white arrow) and areas of ground-glass opacity surrounded by a ring of consolidation (reversed halo sign) (black arrow) consistent with OP (BOOP). Surgical biopsy demonstrated characteristic features of NSIP and OP.
FIGURE 10-18 NSIP pattern in polymyositis. HRCT shows patchy bilateral ground-glass opacities and minimal reticulation.
aMost common finding(s).bFinding(s) most helpful in differential diagnosis.
TABLE 10-5 hRCT Findings in Polymyositis-Dermatomyositis
Ground-glass opacitya
Findings of fibrosis (i.e., traction bronchiectasis and bronchiolectasis, intralobular interstitial thickening, irregular interlobular septal thickening, irregular interfaces)a
Honeycombing (less common than with IPF)
Consolidation (secondary to OP)a,b
Peripheral and subpleural predominance of fibrosis or ground-glass opacitya,b
Lower lung zone and posterior predominancea,b

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease270
2 had mixed inflammatory and fibrotic changes, and 4 remained unchanged at the last examination; 1 patient had died and 1 had no follow-up examination. Three of the 4 patients with normal HRCT at the first examina-tion underwent follow-up investigation; 1 had developed fibrotic changes, 1 inflammatory changes, and 1 linear opacity changes. The authors concluded that the course of ILD in patients with PM-DM could not be predicted on the first examination (108).
MIXED CONNECTIVE TISSUE DISEASEMCTD is a condition characterized by clinical and labo-ratory findings overlapping those of PSS, SLE, and PM-DM (109). A prerequisite for diagnosis is the presence of high titers of circulating autoantibodies to uridine-rich small nuclear ribonucleoprotein (anti-U1 RNP) (4,109). MCTD is much more common in women (female-to-male ratio about 9:1) (110). Pulmonary abnormalities occur in 25% to 85% of MCTD patients during the course of the disease (111,112). The most common pul-monary complication is ILD, which has been reported in 21% to 67% of patients (3,112). The most frequent interstitial pattern in MCTD is NSIP; less common pat-terns include UIP, LIP, and OP (3,94). HRCT is superior to the chest radiograph in demonstrating the presence of ILD in MCTD and in distinguishing ILD from other parenchymal abnormalities (112). Other common com-plications of MCTD are pulmonary hypertension and pleural effusion. Pulmonary arterial hypertension oc-curs in 10% to 45% of patients and is associated with a poor prognosis (3). Pleural effusions, frequently tran-sient in nature, are seen in approximately 50% of pa-tients (109,113). Less common complications associated with MCTD include aspiration due to esophageal dys-motility; diffuse pulmonary hemorrhage and pulmonary thromboembolism (3,109).
high-Resolution Computed Tomography FindingsHRCT findings of MCTD include (a) ground-glass opaci-ties, (b) subpleural micronodules, (c) reticulation, (d) septal lines, and (e) honeycombing (Figs. 10-21 to 10-23, Table 10-6). Kozuka et al. (114) reviewed the HRCT find-ings in 41 patients with MCTD-associated ILD. The pre-dominant abnormalities included ground-glass opacities seen in all patients, subpleural micronodules seen in 98%, and nonseptal linear opacities seen in 80%. Other find-ings included intralobular reticular opacities (61%), ar-chitectural distortion (49%), and traction bronchiectasis (44%), and, less commonly, interlobular septal thicken-ing, ill-defined centrilobular nodules, and honeycombing. The abnormalities usually had a peripheral distribution and lower lobe predominance (114). Saito et al. (115) reviewed the HRCT scans of 35 patients with MCTD and ILD. Septal thickening was seen in 100% of patients, subpleural micronodules in 94%, honeycombing in 51%,
22 patients showed NSIP in 18, organizing DAD in 2, BOOP in 1, and UIP in 1 patient.
Mino et al. (105) assessed the HRCT findings before and after treatment with corticosteroids and immunosup-pressants in 19 patients who had PM-DM. Findings on the initial HRCT scans included pleural irregularities and prominent interlobular septa (n = 19), ground-glass opac-ity (n = 19), patchy consolidation (n = 19), parenchymal bands (n = 15), irregular peribronchovascular thickening (n = 15), and subpleural lines (n = 7). Honeycombing was not detected on any CT images. These findings were more severe in the basal and subpleural regions of the lungs. In 16 of the 17 patients who underwent sequential CT, areas of consolidation, parenchymal bands, and irreg-ular peribronchovascular thickening improved, becoming pleural irregularities and prominent interlobular septa, ground-glass opacity, and subpleural lines on follow-up CT scans (105).
Akira et al. (106) performed sequential HRCT scans in seven patients who had PM-DM, five of whom had his-tologic confirmation of pulmonary involvement. The pre-dominant finding on the initial CT scans in four patients was subpleural consolidation, which was shown to be due to OP (BOOP). One patient with bilateral patchy areas of ground-glass opacity and consolidation was shown to have DAD. In most cases, consolidation improved with use of corticosteroid or immunosuppressive therapy, or both; in two patients, however, consolidation evolved into honeycombing. In one patient with subpleural linear opacities, parenchymal abnormalities slowly progressed, and linear opacities had evolved into honeycombing at the patient’s 8-year follow-up.
Bonnefoy et al. (107) assessed the changes in the pattern, distribution, and extent of ILD on HRCT in 20 patients with PM-DM after a mean follow-up of 38 months. The patients were classified into four groups according to the dominant pattern of abnormality on the initial HRCT: ground-glass opacity and reticulation (45%), airspace consolidation (20%), honeycombing (20%), and nor-mal or almost normal lung (15%). Under medical treat-ment, the ground-glass opacities and consolidation most commonly improved, while the extent of honeycombing usually increased. Some patients showed marked clini-cal deterioration with development of extensive consoli-dation on HRCT and DAD histologically, regardless of the initial pattern on HRCT (107). In another follow-up study of 19 patients who had PM-DM and HRCT, the ini-tial HRCT showed findings consistent with predominant inflammation in 11 patients, mixed inflammation and fi-brosis in 2, fibrosis in 2, and was normal in 4 (108). All patients were treated with high-dose glucocorticoids and other immunosuppressive agents. Follow-up examination 12 to 238 weeks after the initial examination was per-formed in 15 patients. None of the patients with changes consistent with ILD in the initial HRCT experienced a complete resolution at follow-up investigation. Of the 11 patients with predominantly inflammatory findings at the first examination, 3 had developed fibrotic changes,

ChAPTER 10 Collagen-Vascular Diseases 271
FIGURE 10-21 MCTD with pulmonary fibrosis. A and B: HRCT at two levels shows a fine reticular pattern posteriorly, which reflects septal thickening and intralobular interstitial fibrosis.
A B
FIGURE 10-22 A–C: HRCT in a 26-year-old woman with MCTD, basilar crackles on physical examination, and restrictive disease on PFTs. Intralobular interstitial thickening results in a very fine reticular pattern in the subpleural lung and lower lobes. Traction bronchiectasis (A, arrows) is also visible.
A
B
C
subpleural linear opacities in 37%, and ground-glass opacities in 11%. The predominant HRCT pattern was interlobular septal thickening in 83% of patients, hon-eycombing in 11%, subpleural micronodules in 3%, and consolidation in 3% (115). The differences between the results of the study by Saito et al. (115) and the one by
Kozuka et al. (114) are presumably related to different patient populations and the different interval between on-set of disease and the HRCT. The patients in the report by Kozuka et al. (114) underwent HRCT of the chest within 1 year (mean, 4.5 months) of the diagnosis of MCTD, compared to a mean interval of 49.5 months in the study

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease272
SJÖGREN SYNDROMESjögren syndrome is an autoimmune disease character-ized by the clinical triad of keratoconjunctivitis sicca, xerostomia, and recurrent swelling of the parotid gland caused by lymphocytic infiltration of the exocrine glands (116). The prevalence (annual incidence) of pri-mary Sjögren syndrome in North America is 320 per 100,000 population (117), which is greater than that of SLE. It has a female-to-male ratio of 9:0. Second-ary Sjögren syndrome occurs in association with other autoimmune diseases, most commonly RA (116,118). More than half the patients have respiratory symp-toms, most commonly hoarseness (from dry larynx) and cough (from xerotrachea) (4). However, clinically sig-nificant respiratory disease was present in only 11% of patients in a study of 1,010 Spanish patients with pri-mary Sjögren syndrome (119). The majority of patients with ILD have a histologic pattern of NSIP (4,120,121). Less common patterns include LIP, UIP, and OP (BOOP) (4,121). Occasionally, the interstitial disease may repre-sent primary pulmonary lymphoma or diffuse interstitial amyloidosis (121). Airway abnormalities include bron-chiectasis, chronic bronchiolitis, and follicular bronchi-olitis (6,52,94). A relatively common finding in patients with Sjögren syndrome is the development of lymphoma, the prevalence being 40 times that in the general popula-tion (116). The prevalence of primary pulmonary lym-phoma is estimated to be 1% to 2% in patients with Sjögren syndrome (118). The most common type is non-Hodgkin lymphoma, most frequently mucosa-associated lymphoid tissue (MALT) lymphoma (118).
The frequency of reported radiographic abnormalities ranges from 2% to 34% (122). The most common radio-graphic finding consists of a reticular or reticulonodular pattern, usually with a basal predominance (122,123). This pattern may be caused by LIP, interstitial fibrosis, or, occasionally, lymphoma (123,124).
by Saito et al. (115). Although pleural effusion or pleural thickening had been previously considered to be seen in fewer than 10% of cases of MCTD (67), pleural thicken-ing was evident on HRCT in 66% of patients reviewed by Saito et al. (115).
Bodolay et al. (112) assessed the clinical findings, PFTs, and HRCT scans in 144 consecutive patients with MCTD. Ninety-six of the 144 patients (67%) had ac-tive ILD. HRCT demonstrated ground-glass opacities in 75 of the 96 (78%) patients and ground-glass opacities with mild fibrosis (interlobular septal thickening and in-tralobular linear opacities) in 22%. The patients who had active ILD received corticosteroids or corticosteroids in combination with cyclophosphamide. After 6 months of therapy, the HRCT scans in 67 of 75 (89%) patients with ground-glass opacities were normal. However, ground-glass opacity with mild fibrosis developed in 15 of 96 (16%) patients, mild fibrosis in 13 of 96 (13.5%) pa-tients, and the HRCT showed subpleural honeycombing in 1 case (1%) (112).
aMost common finding(s).bFinding(s) most helpful in differential diagnosis.
TABLE 10-6 hRCT Findings in Mixed Connective Tissue Disease
Ground-glass opacitya
Findings of fibrosis (i.e., honeycombing, traction bronchiectasis and bronchiolectasis, intralobular interstitial thickening, irregular interlobular septal thickening, irregular interfaces)a
Peripheral and subpleural predominance of fibrosis or ground-glass opacitya,b
Lower lung zone and posterior predominancea,b
Pleural thickening or effusiona,b
Subpleural micronodulesa
Septal linesa
FIGURE 10-23 HRCT in a patient with MCTD, findings of interstitial fibrosis (architectural distortion, honeycombing, and reticulation), and associated lung carcinoma. A: The findings of interstitial fibrosis predominate in the peripheral lung regions (A and B). A peripheral adenocarcinoma with infiltration of the pleura (B, arrows) is also present.
A B

ChAPTER 10 Collagen-Vascular Diseases 273
high-Resolution Computed Tomography FindingsCommon HRCT findings in Sjögren syndrome include (a) ground-glass opacity, (b) findings of fibrosis, (c) centrilobu-lar nodular opacities, and (d) lung cysts (Figs. 10-24 and 10-25, Table 10-7). Franquet et al. (122) assessed the HRCT findings in 50 patients who had Sjögren syndrome for a mean of 12 years (range, 2–37 years) after the onset of dis-ease. Abnormalities were detected in 17 patients (34%) on HRCT compared with 7 (14%) on chest radiographs. The most common findings consisted of bronchiolectasis and poorly defined centrilobular nodular or branching linear opacities (seen in 11 patients), areas of ground-glass opac-ity (in 7), and honeycombing (in 4). The latter was bilateral, asymmetric, and present almost exclusively in the periphery of the lower lobes (122). A tree-in-bud appearance related to infection or follicular bronchiolitis was visible in 3.
Uffmann et al. (125) performed HRCT in 37 consecutive patients with primary Sjögren syndrome and normal chest radiographs. Abnormal HRCT findings were seen in 24 of 37 patients (65%) and consisted mainly of interlobular septal thickening (n = 9), micronodules (n = 9), lung cysts (n = 5), and ground-glass opacities (n = 4). Intralobular opacities, honeycombing, and bronchiectasis were less frequent.
Lohrmann et al. (126) reviewed the HRCT scans of 24 patients with primary Sjögren syndrome. Nineteen patients (79%) had abnormal HRCT findings, including bronchiectasis, thin-walled cysts, and small pulmonary nodules (seen in 46% of patients), ground-glass opacities and emphysema (38%), interlobular septal thickening (29%), honeycombing (25%), tree-in-bud pattern (21%), and mosaic perfusion (17%) (126).
Ito et al. (120) correlated the HRCT and histologic findings in 33 patients with primary Sjögren syndrome. The most common histologic pattern was NSIP, seen in 61% of patients; less common findings included bronchi-olitis, LIP, MALT lymphoma, and amyloidosis. A charac-teristic HRCT pattern of NSIP was defined according to
aMost common finding(s).bFinding(s) most helpful in differential diagnosis.
TABLE 10-7 hRCT Findings in Sjögren Syndrome
Ground-glass opacitya
Findings of fibrosis (i.e., traction bronchiectasis or bronchiolectasis, intralobular interstitial thickening, irregular interlobular septal thickening, irregular interfaces)a
Honeycombing
Peripheral and subpleural predominance of fibrosis or ground-glass opacitya,b
Lower lung zone and posterior predominance
Small centrilobular nodules (follicular bronchiolitis)
Cysts or small subpleural nodules (LIP)a,b
FIGURE 10-24 NSIP in Sjögren syndrome. HRCT shows bilateral ground-glass opacities and mild reticulation.
FIGURE 10-25 LIP in Sjögren syndrome. A: HRCT at the level of the right upper lobe bronchus demonstrates bilateral thin-walled cystic lesions in a random distribution. Also noted are small foci of ground-glass opacity in the left lung. B: HRCT at the level of the lung bases shows several thin-walled cysts and patchy bilateral ground-glass opacities.
A
B
the ATS/ERS classification of IIPs as consisting predomi-nantly of ground-glass opacities, usually with associated irregular linear opacities in a predominantly peripheral and basal distribution (120,127). HRCT-pathologic cor-relation resulted in a 94% positive predictive value of this

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease274
The prevalence of bronchiectasis was evaluated in a cohort of 507 patients with primary Sjögren syndrome (132). Forty one (8%) patients were classified as having bronchiectasis associated with primary Sjögren syndrome (40 women, mean age of 64 years). The bronchiectasis was cylindrical and, in 29 cases (71%), located in the lower lobes. During follow-up, patients with bronchi-ectasis had a higher frequency of respiratory infections (56% vs. 3%, p < 0.001) and pneumonia (29% vs. 3%, p = 0.002) than did patients without (132).
ANKYLOSING SPONDYLITISAnkylosing spondylitis is a spondyloarthropathy charac-terized by sacroiliitis, spinal stiffness, and loss of spinal mobility (133). It is estimated to affect approximately 0.1% of the population and to have a male-to-female ratio ranging from 10:1 to 15:1 (134). The most charac-teristic pulmonary complication is upper-zonal fibrosis, which usually manifests 15 years or more after the onset of arthritic manifestations (134). Occasionally, however, pulmonary involvement may occur before any skeletal symptoms, or in asymptomatic persons with early-stage ankylosing spondylitis (134,135). An early review of the records of 2,080 patients with ankylosing spondylitis dis-closed 28 (1.3%) who had pleuropulmonary manifesta-tions, including 25 with apical fibrobullous lesions, 2 with pleural effusions, and 1 with apical fibrosis and pleural effusion (136). A more recent study of 1,028 ankylosing spondylitis patients seen over a 10-year period found 22 (2.1%) with apical lung fibrosis detected on chest radi-ography (137). Radiologically, the process begins as api-cal pleural involvement, and then apical opacities develop and progress to cyst formation. Generally, the disease be-gins unilaterally and becomes bilateral. The chest radio-graphic findings may closely mimic those of tuberculosis. Symptoms are usually absent, unless the cavities become secondarily infected. The histologic lesions consist of nonspecific inflammation and fibrosis. Mycobacterial or fungal superinfection of the upper lobe cysts and cavities, most commonly by Aspergillus fumigatus with formation of fungus balls, has been reported in up to one-third of the patients (134). Such infection may lead to recurrent and sometimes massive hemoptysis (134). Since the ad-vent of HRCT several studies have shown that upper lobe fibrosis is more common than previously evident on the radiograph and that ILD, beyond apical fibrosis, is also a feature of ankylosing spondylitis (138). The cause of ILD in ankylosing spondylitis is unclear and the histologic findings have rarely been described.
high-Resolution Computed Tomography FindingsCommon HRCT findings in ankylosing spondylitis in-clude (a) apical fibrosis, (b) septal lines, (c) bronchiectasis, and (d) pleural thickening (Fig. 10-27, Table 10-8). Apical fibrosis in ankylosing spondylitis is frequently associated
HRCT pattern for a pathologic diagnosis of NSIP; how-ever, the diagnostic value of HRCT was low (15%) for patterns other than NSIP (120).
Parambil et al. (121) correlated the histologic patterns with the radiographic and HRCT features in 18 patients with primary Sjögren syndrome and ILD. The main HRCT findings in NSIP were ground-glass and reticu-lar opacities distributed peripherally in both lower lung zones. HRCT in patients with OP (BOOP) showed bilat-eral patchy consolidation, mainly in the subpleural and peribronchovascular regions, and patchy ground-glass opacities. UIP was characterized by reticular opacities and traction bronchiectasis in a predominantly basal and peripheral distribution and commonly associated with subpleural honeycombing. The main findings in LIP and primary pulmonary lymphoma consisted of patchy areas of consolidation, ground-glass opacities, and nod-ules. Multiple cysts were seen in all three patients with LIP and one of the two patients with primary pulmonary lymphoma. Prominent interlobular septal thickening with small randomly distributed nodules were present in the patient who had diffuse interstitial amyloidosis (121).
The HRCT manifestations of LIP in Sjögren syndrome are similar to those seen in LIP associated with other condi-tions (see Chapter 9) (128,129). The predominant findings consist of extensive areas of ground-glass opacity and ran-domly distributed thin-walled cysts (Fig. 10-25) (128,129). The presence of cystic lesions and focal regions of air trap-ping on expiratory HRCT have been associated with fol-licular bronchiolitis in a patient with this disease (130). Other common findings in LIP include interlobular septal thickening, intralobular linear opacities, areas of consolida-tion, centrilobular nodules, and subpleural nodules (129). Amyloidosis occurring in relation to Sjögren syndrome may appear as multiple nodules and cystic lesions (131) or as septal thickening with small randomly distributed nod-ules; the nodules may calcify (see Fig. 9-38) (121).
Lymphoma in patients who have Sjögren syndrome may result in patchy areas of consolidation, ground-glass opacities, and multiple nodules (Fig. 10-26) (118,121).
FIGURE 10-26 Maltoma in Sjögren syndrome. HRCT shows bilateral focal areas of consolidation and patchy ground-glass opacities. Bronchial dilatation is present within the areas of consolidation, a finding commonly seen in patients with MALT lymphoma.

ChAPTER 10 Collagen-Vascular Diseases 275
(67%) with normal CT findings had mild restrictive lung function. Senocak et al. (144) reviewed the HRCT find-ings in 18 patients with ankylosing spondylitis. The most common findings in nonsmokers were septal and pleural thickening seen in 40% of nonsmokers. Apical fibrosis was present in 15% of patients; the fibrosis was unilateral and right sided in all cases and was associated with em-physema even in nonsmokers. Souza et al. (140) assessed the chest radiographs and inspiratory and expiratory HRCT features in 17 patients with ankylosing spondy-litis, 8 of which were smokers. Pulmonary abnormalities were seen on chest radiography in 2 (12%) patients and on CT in 15 (88%) patients. The abnormalities on CT included evidence of airway disease in 14 (82%), inter-stitial abnormalities in 11 (65%), and emphysema in 6 (35%) patients. Airway abnormalities included bronchial wall thickening in 7 (41%), mosaic perfusion pattern in 3 (18%), centrilobular nodules in 3 (18%), bronchiolecta-sis in 2 (12%), and air trapping on expiratory HRCT in 7 (41%) patients. Interstitial abnormalities included paren-chymal bands in 7 (41%), intralobular linear opacities in 2 (12%), and 1 patient each with irregular thickening of interlobular septa, subpleural lines, and honeycombing. Two (12%) patients had coarse irregular linear opacities predominantly in the upper lobes associated with focal lucencies consistent with bullae or thin-walled cavities. One of the patients had an intracavitary soft-tissue mass consistent with an aspergilloma. Two of the six patients with emphysema were lifetime nonsmokers (140).
Sampaio-Barros et al. (145) performed a prospective study of 52 consecutive asymptomatic patients with an-kylosing spondylitis, using chest radiography, PFTs, and
with apical bullae and cavities and may be complicated by aspergilloma formation or necrotizing Aspergillus pneumonia (Fig. 10-27) (139–141). Fenlon et al. (142) prospectively assessed the chest radiographic and HRCT findings in 26 patients who had ankylosing spondylitis. Abnormalities were evident on the radiograph in 4 (15%) patients and on HRCT in 18 (69%). The most common findings on HRCT consisted of ILD, seen in 4 patients; bronchiectasis, seen in 6; mediastinal lymphadenopathy, seen in 3; paraseptal emphysema, seen in 3; tracheal dila-tation, seen in 2; and apical fibrosis, seen in 2 (87). Chest radiography failed to identify any of the patients who had ILD.
Turetschek et al. (143) reviewed the HRCT findings in 25 patients with ankylosing spondylitis who had a normal chest radiographs and no history of smoking. Fif-teen of 21 patients (71%) had abnormalities on HRCT, including interlobular septal thickening (33%), mild bronchial wall thickening (29%), and pleural thickening and pleuropulmonary irregularities (both 29%). Eight of 15 patients (53%) with abnormal CT and 4 of 6 patients
aMost common finding(s).bFinding(s) most helpful in differential diagnosis.
TABLE 10-8 hRCT Findings in Ankylosing Spondylitis
Apical fibrosisa,b
Septal lines
Bronchiectasis
Mosaic perfusion and air trappinga
FIGURE 10-27 Apical fibrosis, cavitation, and aspergilloma in ankylosing spondylitis. A: HRCT performed on a multidetector CT scanner shows left upper lobe cavity containing a large aspergilloma and a crescent of air (air crescent sign). Also noted are patchy reticulation, focal ground-glass opacities, and mild emphysema in the right upper lobe. B: Coronal reformation demonstrates large left upper lobe cavity and aspergilloma and right upper lobe fibrosis with superior retraction of the right hilum. Mild reticulation and focal ground-glass opacities are present in the remaining lung.
A B

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease276
and their relationship to outcome. Am J Respir Crit Care Med 2002;165(12):1581–1586.
12. Amital A, Shitrit D, Adir Y. The lung in rheumatoid arthritis. Presse Med 2011;40(1, pt 2):e31–e48.
13. Myers JL, Limper AH, Swensen SJ. Drug-induced lung disease: a pragmatic classification incorporating HRCT appearances. Semin Respir Crit Care Med 2003;24(4):445–454.
14. Ramos-Casals M, Perez-Alvarez R, Perez-de-Lis M, et al. Pul-monary disorders induced by monoclonal antibodies in pa-tients with rheumatologic autoimmune diseases. Am J Med 2011;124(5):386–394.
15. Hadjinicolaou AV, Nisar MK, Bhagat S, et al. Non-infectious pul-monary complications of newer biological agents for rheumatic diseases—a systematic literature review. Rheumatology (Oxford) 2011;50(12):2297–2305.
16. Lynch DA. Lung disease related to collagen vascular disease. J Thorac Imaging 2009;24(4):299–309.
17. Nakajima R, Sakai F, Mimura T, et al. Acute- or subacute-onset lung complications in treating patients with rheumatoid arthritis. Can Assoc Radiol J 2013;64(3):200–207.
18. Kim EJ, Collard HR, King TE Jr. Rheumatoid arthritis-associated interstitial lung disease: the relevance of histopathologic and ra-diographic pattern. Chest 2009;136(5):1397–1405.
19. Kim DS. Interstitial lung disease in rheumatoid arthritis: recent advances. Curr Opin Pulm Med 2006;12(5):346–353.
20. Antin-Ozerkis D, Evans J, Rubinowitz A, et al. Pulmo-nary manifestations of rheumatoid arthritis. Clin Chest Med 2010;31(3):451–478.
21. Myasoedova E, Crowson CS, Turesson C, et al. Incidence of ex-traarticular rheumatoid arthritis in Olmsted County, Minnesota, in 1995–2007 versus 1985–1994: a population-based study. J Rheumatol 2011;38(6):983–989.
22. Suzuki A, Ohosone Y, Obana M, et al. Cause of death in 81 autopsied patients with rheumatoid arthritis. J Rheumatol 1994;21(1):33–36.
23. Frank ST, Weg JG, Harkleroad LE, et al. Pulmonary dysfunction in rheumatoid disease. Chest 1973;63:27–34.
24. Laitinen O, Nissila M, Salorinne Y, et al. Pulmonary involve-ment in patients with rheumatoid arthritis. Scand J Respir Dis 1975;56:297.
25. Shannon TM, Gale ME. Noncardiac manifestations of rheumatoid arthritis in the thorax. J Thorac Imaging 1992;7(2):19–29.
26. Gabbay E, Tarala R, Will R, et al. Interstitial lung disease in re-cent onset rheumatoid arthritis. Am J Respir Crit Care Med 1997;156(2, pt 1):528–535.
27. Dawson JK, Fewins HE, Desmond J, et al. Fibrosing alveolitis in patients with rheumatoid arthritis as assessed by high resolution computed tomography, chest radiography, and pulmonary func-tion tests. Thorax 2001;56(8):622–627.
28. Bilgici A, Ulusoy H, Kuru O, et al. Pulmonary involvement in rheu-matoid arthritis. Rheumatol Int 2005;25(6):429–435.
29. Gochuico BR, Avila NA, Chow CK, et al. Progressive preclinical interstitial lung disease in rheumatoid arthritis. Arch Intern Med 2008;168(2):159–166.
30. Lee HK, Kim DS, Yoo B, et al. Histopathologic pattern and clinical features of rheumatoid arthritis-associated interstitial lung disease. Chest 2005;127(6):2019–2027.
31. Yoshinouchi T, Ohtsuki Y, Fujita J, et al. Nonspecific interstitial pneumonia pattern as pulmonary involvement of rheumatoid ar-thritis. Rheumatol Int 2005;26(2):121–125.
32. Nakamura Y, Suda T, Kaida Y, et al. Rheumatoid lung disease: prognostic analysis of 54 biopsy-proven cases. Respir Med 2012;106(8):1164–1169.
33. Tanaka N, Kim JS, Newell JD, et al. Rheumatoid arthritis-related lung diseases: CT findings. Radiology 2004;232(1):81–91.
34. Biederer J, Schnabel A, Muhle C, et al. Correlation between HRCT findings, pulmonary function tests and bronchoalveolar lavage cytology in interstitial lung disease associated with rheumatoid arthritis. Eur Radiol 2004;14(2):272–280.
HRCT. Of the 52 patients, 33 (63%) had never smoked, 14 (27%) were current smokers, and 5 (10%) were ex-smokers. The chest radiograph showed pulmonary ab-normalities in four patients (8%) and the PFT presented a restrictive pattern in 52% of the patients. Thoracic HRCT demonstrated abnormalities in 21 patients (40%), consisting predominantly of linear parenchymal opaci-ties (19%), lymphadenopathy (12%), emphysema (10%), bronchiectasis (8%), and pleural involvement (8%). The authors concluded that nonspecific subclinical pulmo-nary involvement is frequent in AS.
In one study, the authors performed HRCT and PFTs in 20 patients with ankylosing spondylitis, including 10 with less than 10 years’ disease duration and 10 with 10 or more years’ disease duration. HRCT revealed ab-normalities in 14 patients (70%) (146). The most com-mon findings were apical fibrosis (45%) and emphysema (25%). HRCT findings were more prominent in late ankylosing spondylitis patients (disease duration ≥ 10 years) (p = 0.015). PFTs were considered as abnormal in four patients (20%). Three of these patients had con-comitant HRCT abnormalities. On the other hand, 10 patients with normal PFTs had abnormalities on HRCT. The authors concluded that HRCT is superior to PFTs in detecting lung abnormalities in patients with ankylosing spondylitis.
R E F E R E N C E S 1. Woodhead F, Wells AU, Desai SR. Pulmonary complications
of connective tissue diseases. Clin Chest Med 2008;29(1): 149–164, vii.
2. Silva CI, Muller NL. Interstitial lung disease in the setting of col-lagen vascular disease. Semin Roentgenol 2010;45(1):22–28.
3. Capobianco J, Grimberg A, Thompson BM, et al. Thoracic manifestations of collagen vascular diseases. Radiographics 2012;32(1):33–50.
4. de Lauretis A, Veeraraghavan S, Renzoni E. Review series: aspects of interstitial lung disease: connective tissue disease-associated in-terstitial lung disease: how does it differ from IPF? How should the clinical approach differ? Chron Respir Dis 2011;8(1):53–82.
5. Tanaka N, Newell JD, Brown KK, et al. Collagen vascular disease-related lung disease: high-resolution computed tomogra-phy findings based on the pathologic classification. J Comput As-sist Tomogr 2004;28(3):351–360.
6. Tansey D, Wells AU, Colby TV, et al. Variations in histologi-cal patterns of interstitial pneumonia between connective tissue disorders and their relationship to prognosis. Histopathology 2004;44(6):585–596.
7. Hwang JH, Misumi S, Sahin H, et al. Computed tomographic fea-tures of idiopathic fibrosing interstitial pneumonia: comparison with pulmonary fibrosis related to collagen vascular disease. J Comput Assist Tomogr 2009;33(3):410–415.
8. Sato T, Fujita J, Yamadori I, et al. Non-specific interstitial pneumo-nia; as the first clinical presentation of various collagen vascular disorders. Rheumatol Int 2006;26(6):551–555.
9. Tzelepis GE, Toya SP, Moutsopoulos HM. Occult connective tissue diseases mimicking idiopathic interstitial pneumonias. Eur Respir J 2008;31(1):11–20.
10. Strange C, Highland KB. Interstitial lung disease in the patient who has connective tissue disease. Clin Chest Med 2004;25(3):549–559, vii.
11. Bouros D, Wells AU, Nicholson AG, et al. Histopathologic sub-sets of fibrosing alveolitis in patients with systemic sclerosis

ChAPTER 10 Collagen-Vascular Diseases 277
59. Gamsu G. Radiographic manifestations of thoracic involvement by collagen vascular diseases. J Thorac Imaging 1992;7(3):1–12.
60. Meziane MA. High-resolution computed tomography scanning in the assessment of interstitial lung diseases. J Thorac Imaging 1992;7(3):13–25.
61. Hassoun PM. Lung involvement in systemic sclerosis. Presse Med 2011;40(1, pt 2):e3–e17.
62. Bussone G, Mouthon L. Interstitial lung disease in systemic sclero-sis. Autoimmun Rev 2011;10(5):248–255.
63. Mukerjee D, St George D, Coleiro B, et al. Prevalence and out-come in systemic sclerosis associated pulmonary arterial hy-pertension: application of a registry approach. Ann Rheum Dis 2003;62(11):1088–1093.
64. Hachulla E, Gressin V, Guillevin L, et al. Early detection of pul-monary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005;52(12):3792–3800.
65. Desai SR, Veeraraghavan S, Hansell DM, et al. CT features of lung disease in patients with systemic sclerosis: comparison with idio-pathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology 2004;232(2):560–567.
66. Schurawitzki H, Stiglbauer R, Graninger W, et al. Interstitial lung disease in progressive systemic sclerosis: high-resolution CT versus radiography. Radiology 1990;176:755–759.
67. Taorimina VJ, Miller WT, Gefter WB, et al. Progressive systemic sclerosis subgroups: variable pulmonary features. AJR Am J Roentgenol 1981;137:277–285.
68. Kim EA, Johkoh T, Lee KS, et al. Interstitial pneumonia in pro-gressive systemic sclerosis: serial high-resolution CT findings with functional correlation. J Comput Assist Tomogr 2001;25(5): 757–763.
69. Remy-Jardin M, Remy J, Wallaert B, et al. Pulmonary involvement in progressive systemic sclerosis: sequential evaluation with CT, pulmonary function tests, and bronchoalveolar lavage. Radiology 1993;188:499–506.
70. Goldin JG, Lynch DA, Strollo DC, et al. High-resolution CT scan findings in patients with symptomatic scleroderma-related intersti-tial lung disease. Chest 2008;134(2):358–367.
71. Pandey AK, Wilcox P, Mayo JR, et al. Predictors of pulmonary hy-pertension on high-resolution computed tomography of the chest in systemic sclerosis: a retrospective analysis. Can Assoc Radiol J 2010;61(5):291–296.
72. Harrison NK, Myers AR, Corrin B, et al. Structural features of interstitial lung disease in systemic sclerosis. Am Rev Respir Dis 1991;144:706–713.
73. Bhalla M, Silver RM, Shepard JO, et al. Chest CT in patients with scleroderma: prevalence of asymptomatic esophageal dila-tation and mediastinal lymphadenopathy. AJR Am J Roentgenol 1993;161:269–272.
74. Grenier P, Chevret S, Beigelman C, et al. Chronic diffuse infiltra-tive lung disease: determination of the diagnostic value of clinical data, chest radiography, and CT with Bayesian analysis. Radiology 1994;191:383–390.
75. Seely JM, Jones LT, Wallace C, et al. Systemic sclerosis: using high-resolution CT to detect lung disease in children. AJR Am J Roent-genol 1998;170(3):691–697.
76. Wells AU. High-resolution computed tomography and scleroderma lung disease. Rheumatology (Oxford) 2008;47(suppl 5):v59–v61.
77. Wells AU, Hansell DM, Corrin B, et al. High resolution computed tomography as a predictor of lung histology in systemic sclerosis. Thorax 1992;47:508–512.
78. Remy-Jardin M, Giraud F, Remy J, et al. Importance of ground-glass attenuation in chronic diffuse infiltrative lung disease: pathologic-CT correlation. Radiology 1993;189:693–698.
79. Shah RM, Jimenez S, Wechsler R. Significance of ground-glass opacity on HRCT in long-term follow-up of patients with systemic sclerosis. J Thorac Imaging 2007;22(2):120–124.
80. Strollo D, Goldin J. Imaging lung disease in systemic sclerosis. Curr Rheumatol Rep 2010;12(2):156–161.
35. Kim EJ, Elicker BM, Maldonado F, et al. Usual interstitial pneumo-nia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J 2010;35(6):1322–1328.
36. Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007;175(7):705–711.
37. Fischer A, du Bois R. Interstitial lung disease in connective tissue disorders. Lancet 2012;380(9842):689–698.
38. Silva CI, Muller NL, Fujimoto K, et al. Acute exacerbation of chronic interstitial pneumonia: high-resolution computed tomography and pathologic findings. J Thorac Imaging 2007;22(3):221–229.
39. Park IN, Kim DS, Shim TS, et al. Acute exacerbation of intersti-tial pneumonia other than idiopathic pulmonary fibrosis. Chest 2007;132(1):214–220.
40. Suda T, Kaida Y, Nakamura Y, et al. Acute exacerbation of intersti-tial pneumonia associated with collagen vascular diseases. Respir Med 2009;103(6):846–853.
41. Churg A, Wright JL, Tazelaar HD. Acute exacerbations of fibrotic interstitial lung disease. Histopathology 2011;58(4):525–530.
42. Horoupian N, English J, Muller NL. Accelerated deteriora-tion of usual interstitial pneumonia with acute development of honeycomb cysts in rheumatoid arthritis. J Thorac Imaging 2004;19(2):127–130.
43. Tachikawa R, Tomii K, Ueda H, et al. Clinical features and outcome of acute exacerbation of interstitial pneumonia: collagen vascular diseases-related versus idiopathic. Respiration 2012;83(1):20–27.
44. Parambil JG, Myers JL, Ryu JH. Diffuse alveolar damage: uncom-mon manifestation of pulmonary involvement in patients with connective tissue diseases. Chest 2006;130(2):553–558.
45. Remy-Jardin M, Remy J, Cortet B, et al. Lung changes in rheuma-toid arthritis: CT findings. Radiology 1994;193:375–382.
46. Perez T, Remy-Jardin M, Cortet B. Airways involvement in rheu-matoid arthritis: clinical, functional, and HRCT findings. Am J Respir Crit Care Med 1998;157(5, pt 1):1658–1665.
47. Aquino SL, Webb WR, Golden J. Bronchiolitis obliterans associ-ated with rheumatoid arthritis: findings on HRCT and dynamic expiratory CT. J Comput Assist Tomogr 1994;18:555–558.
48. Hassan WU, Keaney NP, Holland CD, et al. High resolution com-puted tomography of the lung in lifelong non-smoking patients with rheumatoid arthritis. Ann Rheum Dis 1995;54(4):308–310.
49. McShane PJ, Naureckas ET, Strek ME. Bronchiectasis in a diverse US population: effects of ethnicity on etiology and sputum culture. Chest 2012;142(1):159–167.
50. Kinoshita M, Higashi T, Tanaka C, et al. Follicular bron-chiolitis associated with rheumatoid arthritis. Intern Med 1992;31(5):674–677.
51. Hayakawa H, Sato A, Imokawa S, et al. Bronchiolar dis-ease in rheumatoid arthritis. Am J Respir Crit Care Med 1996;154(5):1531–1536.
52. Howling SJ, Hansell DM, Wells AU, et al. Follicular bron-chiolitis: thin-section CT and histologic findings. Radiology 1999;212(3):637–642.
53. Brown KK. Rheumatoid lung disease. Proc Am Thorac Soc 2007;4(5):443–448.
54. Manjunatha YC, Seith A, Kandpal H, et al. Rheumatoid arthritis: spectrum of computed tomographic findings in pulmonary dis-eases. Curr Probl Diagn Radiol 2010;39(6):235–246.
55. Sidhu HS, Bhatnagar G, Bhogal P, et al. Imaging features of the pleuropulmonary manifestations of rheumatoid arthritis: pearls and pitfalls. J Clin Imaging Sci 2011;1:32.
56. Kobayashi T, Satoh K, Ohkawa M, et al. Multiple rheumatoid nodules with rapid thin-walled cavity formation producing pneu-mothorax. J Thorac Imaging 2005;20(1):47–49.
57. Fujii M, Adachi S, Shimizu T, et al. Interstitial lung disease in rheumatoid arthritis: assessment with high-resolution computed tomography. J Thorac Imaging 1993;8:54–62.
58. Avnon LS, Abu-Shakra M, Flusser D, et al. Pleural effusion associ-ated with rheumatoid arthritis: what cell predominance to antici-pate? Rheumatol Int 2007;27(10):919–925.

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease278
105. Mino M, Noma S, Taguchi Y, et al. Pulmonary involvement in polymyositis and dermatomyositis: sequential evaluation with CT. AJR Am J Roentgenol 1997;169(1):83–87.
106. Akira M, Hara H, Sakatani M. Interstitial lung disease in associa-tion with polymyositis-dermatomyositis: long-term follow-up CT evaluation in seven patients. Radiology 1999;210(2):333–338.
107. Bonnefoy O, Ferretti G, Calaque O, et al. Serial chest CT findings in interstitial lung disease associated with polymyositis-dermato-myositis. Eur J Radiol 2004;49(3):235–244.
108. Fathi M, Vikgren J, Boijsen M, et al. Interstitial lung disease in polymyositis and dermatomyositis: longitudinal evalua-tion by pulmonary function and radiology. Arthritis Rheum 2008;59(5):677–685.
109. Hant FN, Herpel LB, Silver RM. Pulmonary manifestations of scleroderma and mixed connective tissue disease. Clin Chest Med 2010;31(3):433–449.
110. Venables PJ. Mixed connective tissue disease. Lupus 2006;15(3):132–137.
111. Prakash UB, Luthra HS, Divertie MB. Intrathoracic manifes-tations in mixed connective tissue disease. Mayo Clin Proc 1985;60(12):813–821.
112. Bodolay E, Szekanecz Z, Devenyi K, et al. Evaluation of interstitial lung disease in mixed connective tissue disease (MCTD). Rheuma-tology (Oxford) 2005;44(5):656–661.
113. Bull TM, Fagan KA, Badesch DB. Pulmonary vascular manifesta-tions of mixed connective tissue disease. Rheum Dis Clin North Am 2005;31(3):451–464, vi.
114. Kozuka T, Johkoh T, Honda O, et al. Pulmonary involvement in mixed connective tissue disease: high-resolution CT findings in 41 patients. J Thorac Imaging 2001;16(2):94–98.
115. Saito Y, Terada M, Takada T, et al. Pulmonary involvement in mixed connective tissue disease: comparison with other collagen vascular diseases using high resolution CT. J Comput Assist To-mogr 2002;26(3):349–357.
116. Fox RI. Sjögren’s syndrome. Lancet 2005;366(9482):321–331. 117. Shapira Y, Agmon-Levin N, Shoenfeld Y. Geoepidemiol-
ogy of autoimmune rheumatic diseases. Nat Rev Rheumatol 2010;6(8):468–476.
118. Kokosi M, Riemer EC, Highland KB. Pulmonary involvement in Sjögren syndrome. Clin Chest Med 2010;31(3):489–500.
119. Ramos-Casals M, Solans R, Rosas J, et al. Primary Sjögren syn-drome in Spain: clinical and immunologic expression in 1010 pa-tients. Medicine (Baltimore) 2008;87(4):210–219.
120. Ito I, Nagai S, Kitaichi M, et al. Pulmonary manifestations of pri-mary Sjögren’s syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med 2005;171(6):632–638.
121. Parambil JG, Myers JL, Lindell RM, et al. Interstitial lung disease in primary Sjögren syndrome. Chest 2006;130(5):1489–1495.
122. Franquet T, Giménez A, Monill JM, et al. Primary Sjögren’s syn-drome and associated lung disease: CT findings in 50 patients. AJR Am J Roentgenol 1997;169(3):655–658.
123. Kadota J, Kusano S, Kawakami K, et al. Usual interstitial pneu-monia associated with primary Sjögren’s syndrome. Chest 1995;108(6):1756–1758.
124. Tanoue LT. Pulmonary involvement in collagen vascular disease: a review of the pulmonary manifestations of the Marfan syndrome, ankylosing spondylitis, Sjögren’s syndrome, and relapsing poly-chondritis. J Thorac Imaging 1992;7(2):62–77.
125. Uffmann M, Kiener HP, Bankier AA, et al. Lung manifestation in asymptomatic patients with primary Sjögren syndrome: assess-ment with high resolution CT and pulmonary function tests. J Thorac Imaging 2001;16(4):282–289.
126. Lohrmann C, Uhl M, Warnatz K, et al. High-resolution CT imag-ing of the lung for patients with primary Sjögren’s syndrome. Eur J Radiol 2004;52(2):137–143.
127. American Thoracic Society/European Respiratory Society. American Thoracic Society/European Respiratory Society inter-national multidisciplinary consensus classification of the idio-pathic interstitial pneumonias. Am J Respir Crit Care Med 2002; 165:277–304.
81. Camiciottoli G, Orlandi I, Bartolucci M, et al. Lung CT densitom-etry in systemic sclerosis: correlation with lung function, exercise testing, and quality of life. Chest 2007;131(3):672–681.
82. Goh NS, Desai SR, Veeraraghavan S, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med 2008;177(11):1248–1254.
83. D’Cruz DP, Khamashta MA, Hughes GR. Systemic lupus erythe-matosus. Lancet 2007;369(9561):587–596.
84. Kamen DL, Strange C. Pulmonary manifestations of systemic lu-pus erythematosus. Clin Chest Med 2010;31(3):479–488.
85. Quadrelli SA, Alvarez C, Arce SC, et al. Pulmonary involvement of systemic lupus erythematosus: analysis of 90 necropsies. Lupus 2009;18(12):1053–1060.
86. Torre O, Harari S. Pleural and pulmonary involvement in systemic lupus erythematosus. Presse Med 2011;40(1, pt 2):e19–e29.
87. Kim JS, Lee KS, Koh EM, et al. Thoracic involvement of systemic lupus erythematosus: clinical, pathologic, and radiologic findings. J Comput Assist Tomogr 2000;24:9–18.
88. Haupt HM, Moore GW, Hutchins GM. The lung in systemic lupus erythematosus. Analysis of the pathologic changes in 120 patients. Am J Med 1981;71(5):791–798.
89. Jacobsen S, Petersen J, Ullman S, et al. A multicentre study of 513 Danish patients with systemic lupus erythematosus. I. Disease manifestations and analyses of clinical subsets. Clin Rheumatol 1998;17(6):468–477.
90. Bankier AA, Kiener HP, Wiesmayr MN, et al. Discrete lung in-volvement in systemic lupus erythematosus: CT assessment. Radi-ology 1995;196(3):835–840.
91. Gammon RB, Bridges TA, al-Nezir H, et al. Bronchiolitis obliter-ans organizing pneumonia associated with systemic lupus erythe-matosus. Chest 1992;102(4):1171–1174.
92. Fenlon HM, Doran M, Sant SM, et al. High-resolution chest CT in systemic lupus erythematosus. AJR Am J Roentgenol 1996;166(2):301–307.
93. Sant SM, Doran M, Fenelon HM, et al. Pleuropulmonary abnormal-ities in patients with systemic lupus erythematosus: assessment with high resolution computed tomography, chest radiography and pul-monary function tests. Clin Exp Rheumatol 1997;15(5):507–513.
94. Kim EA, Lee KS, Johkoh T, et al. Interstitial lung diseases associ-ated with collagen vascular diseases: radiologic and histopatho-logic findings. Radiographics 2002;22(spec no):S151–S165.
95. Kim WU, Kim SI, Yoo WH, et al. Adult respiratory distress syn-drome in systemic lupus erythematosus: causes and prognostic fac-tors: a single center, retrospective study. Lupus 1999;8(7):552–557.
96. Connolly B, Manson D, Eberhard A, et al. CT appearance of pulmonary vasculitis in children. AJR Am J Roentgenol 1996;167(4):901–904.
97. Ooi GC, Ngan H, Peh WC, et al. Systemic lupus erythematosus pa-tients with respiratory symptoms: the value of HRCT. Clin Radiol 1997;52(10):775–781.
98. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lan-cet 2003;362(9388):971–982.
99. Kalluri M, Oddis CV. Pulmonary manifestations of the idiopathic inflammatory myopathies. Clin Chest Med 2010;31(3):501–512.
100. Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet 2001;357(9250):96–100.
101. Connors GR, Christopher-Stine L, Oddis CV, et al. Interstitial lung disease associated with the idiopathic inflammatory myopa-thies: what progress has been made in the past 35 years? Chest 2010;138(6):1464–1474.
102. Douglas WW, Tazelaar HD, Hartman TE, et al. Polymyositis- dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med 2001;164(7):1182–1185.
103. Cottin V, Thivolet-Bejui F, Reynaud-Gaubert M, et al. Interstitial lung disease in amyopathic dermatomyositis, dermatomyositis and polymyositis. Eur Respir J 2003;22(2):245–250.
104. Ikezoe J, Johkoh T, Kohno N, et al. High-resolution CT findings of lung disease in patients with polymyositis and dermatomyositis. J Thorac Imaging 1996;11(4):250–259.

ChAPTER 10 Collagen-Vascular Diseases 279
128. Carignan S, Staples CA, Müller NL. Intrathoracic lymphoprolif-erative disorders in the immunocompromised patient: CT findings. Radiology 1995;197(1):53–58.
129. Johkoh T, Müller NL, Pickford HA, et al. Lymphocytic intersti-tial pneumonia: thin-section CT findings in 22 patients. Radiology 1999;212(2):567–572.
130. Meyer CA, Pina JS, Taillon D, et al. Inspiratory and expiratory high-resolution CT findings in a patient with Sjögren’s syndrome and cystic lung disease. AJR Am J Roentgenol 1997;168(1):101–103.
131. Desai SR, Nicholson AG, Stewart S, et al. Benign pulmonary lymphocytic infiltration and amyloidosis: computed tomo-graphic and pathologic features in three cases. J Thorac Imaging 1997;12(3):215–220.
132. Soto-Cardenas MJ, Perez-De-Lis M, Bove A, et al. Bronchiecta-sis in primary Sjögren’s syndrome: prevalence and clinical signifi-cance. Clin Exp Rheumatol 2010;28(5):647–653.
133. Braun J, Sieper J. Ankylosing spondylitis. Lancet 2007;369(9570): 1379–1390.
134. Kanathur N, Lee-Chiong T. Pulmonary manifestations of ankylos-ing spondylitis. Clin Chest Med 2010;31(3):547–554.
135. Ferdoutsis M, Bouros D, Meletis G, et al. Diffuse interstitial lung disease as an early manifestation of ankylosing spondylitis. Respi-ration 1995;62(5):286–289.
136. Rosenow EC, Strimlan CV, Muhm JR, et al. Pleuropulmo-nary manifestations of ankylosing spondylitis. Mayo Clin Proc 1977;52:641–649.
137. Lee CC, Lee SH, Chang IJ, et al. Spontaneous pneumothorax as-sociated with ankylosing spondylitis. Rheumatology (Oxford) 2005;44(12):1538–1541.
138. Momeni M, Taylor N, Tehrani M. Cardiopulmonary manifestations of ankylosing spondylitis. Int J Rheumatol 2011;2011:728471.
139. Franquet T, Muller NL, Flint JD. A patient with ankylosing spondy-litis and recurrent haemoptysis. Eur Respir J 2004;23(3):488–491.
140. Souza AS Jr, Muller NL, Marchiori E, et al. Pulmonary abnor-malities in ankylosing spondylitis: inspiratory and expiratory high-resolution CT findings in 17 patients. J Thorac Imaging 2004;19(4):259–263.
141. Pamuk ON, Harmandar O, Tosun B, et al. A patient with ankylos-ing spondylitis who presented with chronic necrotising aspergillo-sis: report on one case and review of the literature. Clin Rheumatol 2005;24(4):415–419.
142. Fenlon HM, Casserly I, Sant SM, et al. Plain radiographs and tho-racic high-resolution CT in patients with ankylosing spondylitis. AJR Am J Roentgenol 1997;168(4):1067–1072.
143. Turetschek K, Ebner W, Fleischmann D, et al. Early pulmonary in-volvement in ankylosing spondylitis: assessment with thin-section CT. Clin Radiol 2000;55(8):632–636.
144. Senocak O, Manisali M, Ozaksoy D, et al. Lung parenchyma changes in ankylosing spondylitis: demonstration with high res-olution CT and correlation with disease duration. Eur J Radiol 2003;45(2):117–122.
145. Sampaio-Barros PD, Cerqueira EM, Rezende SM, et al. Pulmo-nary involvement in ankylosing spondylitis. Clin Rheumatol 2007;26(2):225–230.
146. Ozdemir O, Gulsun Akpinar M, Inanici F, et al. Pulmonary abnor-malities on high-resolution computed tomography in ankylosing spondylitis: relationship to disease duration and pulmonary func-tion testing. Rheumatol Int 2012;32(7):2031–2036.

492
#152201 Cust: LWW Au: Webb Pg. No. 492 Title: High-Resolution CT of the Lung 5ed Server:
CMYK Short / Normal/Long
DESIGN SERVICES OF
S4carliSlePublishing Services
PULMONARY LANGERHANS CELL HISTIOCYTOSIS 492
LYMPHANGIOMYOMATOSIS AND TUBEROUS SCLEROSIS COMPLEX 500
I M P O R T A N T T O P I C S
19 cystic lung Diseases
As defined by the Fleischner Society glossary of terms, lung cysts are round parenchymal lucencies or low- attenuation areas with well-defined interfaces with normal lung. Cysts are characteristically thin-walled structures with walls less than 3 mm in size, usually containing air, al-though occasionally containing fluid or solid material (1). This is in distinction to lung cavities, which, by definition, demonstrate thicker walls, and have a distinctly different differential diagnosis.
A wide variety of disease states, ranging from com-mon to rare, can result in diffuse lung cysts (Table 19-1) (2,3). Of these, Langerhans cell histiocytosis (LCH) (4),
lymphangiomyomatosis (LAM) and tuberous sclerosis complex (TSC), lymphoid interstitial pneumonia (LIP) and follicular bronchiolitis, and different types of emphy-sema represent those entities most likely to be encoun-tered in routine clinical practice. Cystic lung disease is also reviewed in Chapter 6, and emphysema is discussed in the next chapter.
Other causes of lung cysts include Birt-Hogg-Dubé (BHD) syndrome (Fig. 6-13) (5–7), amyloidosis (which may be associated with lymphoproliferative disease in Sjögren syndrome) (Chapter 16) (8,9), light chain deposition disease (LCDD) (Chapter 16) (Fig. 6-14) (10,11), cystic metastases, benign metastasizing leiomy-oma (12), tracheobronchial papillomatosis (3), neurofi-bromatosis, barotrauma in deep sea divers (Fig. 6-15), and Proteus syndrome (Fig. 6-16) (13). Cysts may also be seen in association with other high-resolution com-puted tomography (HRCT) abnormalities in hyper-sensitivity pneumonitis (HP), desquamative interstitial pneumonia (DIP), interstitial pneumonias with honey-combing (e.g., idiopathic pulmonary fibrosis [IPF]), and small airways disease (14).
PULMONARY LANGERHANS CELL HISTIOCYTOSISCollectively, LCH refers to a group of diseases of un-known etiology often recognized in childhood, in which there is a proliferation and infiltration of CD1+ histio-cytes or Langerhans cells (LCs) in one or more body systems, including bone, lung, pituitary gland, mucous membranes, skin, lymph nodes, and liver (4,15–19). LCs are specialized immune cells belonging to a fam-ily of dendritic cells derived both from CD34+ bone marrow stem cells and CD11c+ blood precursor cells. These form a network of antigen-presenting sentinel mi-gratory cells throughout the body, typically including
BHD Birt-Hogg-Dubé syndrome
DIP desquamative interstitial pneumonia
DLCO carbon monoxide diffusing capacity
FEV1 forced expiratory volume in 1 second
FLCN folliculin
FVC forced vital capacity
HP hypersensitivity pneumonitis
IPF idiopathic pulmonary fibrosis
LAM lymphangiomyomatosis
LC Langerhans cell
LCDD light chain deposition disease
LCH Langerhans cell histiocytosis
LIP lymphoid interstitial pneumonia
mTOR mammalian target of rapamycin
PFT pulmonary function test
PLCH pulmonary Langerhans cell histiocytosis
QCT quantitative computed tomography
RB respiratory bronchiolitis
RB-ILD respiratory bronchiolitis-interstitial lung disease
S-LAM sporadic lymphangiomyomatosis
TSC tuberous sclerosis complex
Abbreviations Used in This Chapter
LYMPHOID INTERSTITIAL PNEUMONIA 510
BIRT-HOGG-DUBÉ SYNDROME 510

CHAPTER 19 Cystic Lung Diseases 493
PathologyAlthough PLCH in adults typically occurs in heavy smokers, its etiology remains unknown. The typical peribronchiolar location of lesions is consistent with a response to inhaled antigens; however, PLCH occurs in only a small minority of smokers, and little improve-ment occurs with smoking cessation. It has been sug-gested that this disease may represent a response to an unknown viral infection (19).
In its earliest stages, PLCH is characterized histo-logically by a cellular interstitial infiltrate composed of LCs, lymphocytes, macrophages, eosinophils, plasma cells, and fibroblasts (15,19,20). The infiltrate typically extends into the adjacent lung, resulting in a stellate le-sion. With progression, the infiltrate evolves into granu-lomatous nodules that subsequently cavitate (Fig. 19-1). Unlike other causes of LCH, PLCH nodules are typically nonclonal (4,15–19). Recently, it has been shown by Kambouchner et al. (23) that PLCH primarily involves the respiratory bronchioles with disease subsequently extending proximally to involve terminal bronchioles and distally to involve alveolar ducts; this results in linear rather than spherical lesions, and accounts for the macro-scopic appearance of nodules that are irregular in shape.
In its later stages, the cellular granulomas and cavitary nodules are replaced by fibrosis and the formation of lung cysts (Figs. 19-2 and 6-5) (4,15–19). Cystic lung lesions may be either thick or thin walled, appearances that have been attributed, respectively, to the presence of cavitary nodules or peribronchiolar, cicatricial emphysema. It has been suggested that cavitary nodules result from bronchi-olar dilatation resulting from inflammation and fibrosis of the bronchiolar wall (23).
Histologic findings are marked by temporal hetero-geneity with nodules in varying stages of development found in association with cystic fibrotic scars (24). As cystic spaces evolve, it is not unusual for pigmented mac-rophages to fill the resulting airspaces, resulting in a so-called DIP-like reaction (25). In fact, given the underlying association between LCH and smoking, it is not uncom-mon for other smoking-related diseases to be simultane-ously identified. This includes, in particular, respiratory bronchiolitis/respiratory bronchiolitis-interstitial lung disease (RB/RB-ILD) and DIP (26–28).
It should also be emphasized that as many as 80% of patients who have late-stage PLCH develop a form of pul-monary vasculitis leading to severe pulmonary hyperten-sion (19,25). In one study of 21 patients with end-stage PLCH and severe pulmonary hypertension, histopatho-logic evaluation in 60% of the cases revealed a prolifera-tive vasculopathy involving muscular arteries and veins, with evidence of medial hypertrophy and intimal and subintimal fibrosis with resulting arterial obliteration (29). Strikingly, these findings occurred in regions unaf-fected by LCs. Furthermore, follow-up histologic evalua-tion showed progression of vascular lesions in the absence of progression of granulomatous disease. In comparison
the skin, lymph nodes, bronchial mucosa, and the thy-mus, which primarily function to facilitate antigen-specific immune reactions (15,19,20). Although studies have shown that LCH represents a clonal proliferation of cells (21), it is not believed that this disease is neo-plastic. It is more likely that LCH in adults represents an uncontrolled or abnormal immune response initiated in situ by LCs in response to an unidentified antigenic stimulus (19,22). Evidence that the lesions in LCH result from cellular recruitment rather than neoplastic cell proliferation include a lack of cellular atypia, absence of focal tissue invasion, and near-complete absence of LCs in late lesions. LCH is classified as resulting either in single-organ disease, as occurs in patients with pul-monary Langerhans cell histiocytosis (PLCH), or in multiorgan or multisystem disease, as occurs in patients with Hand-Schüller-Christian disease or Letterer-Siwe disease, respectively (19).
PLCH is an uncommon disorder typically occurring in young adults between the ages of 20 and 40 years, although it may occur at any age (15). Previously referred to as pulmonary eosinophilic granuloma or pulmonary histiocytosis X, PLCH typically presents with isolated, single-organ involvement, although it may also be a man-ifestation of both multiorgan and multisystemic disease (15,19,20). In a review of 314 patients who had histologi-cally proven LCH, PLCH was identified in 129 (40.8%) patients, with isolated pulmonary involvement occurring in 87 (28%) patients (4). In patients who had multisys-tem disease in addition to pulmonary involvement, sites most often affected included the skeleton and the pitu-itary gland (4).
Common
Emphysema (e.g., bullae)
Cystic bronchiectasis
Honeycombing
Less Common
PLCH
LAM (isolated LAM or associated with the TSC)
LIP
Follicular bronchiolitis
Rare
BHD syndrome
Amyloidosis and LCDD
Cysts associated with DIP
Benign metastasizing leiomyoma
Cystic pulmonary metastases
Tracheobronchial papillomatosis
Proteus syndrome
Neurofibromatosis
TABLE 19-1 Causes of Diffuse Thin-Walled Lung Cysts

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease494
symptoms include cough and dyspnea (38,39). Up to 20% of patients present with pneumothorax (36). Pulmonary function tests (PFTs) in patients with PLCH prove to be highly variable, with obstructive, restrictive, and mixed patterns all being described (40).
Compared with patients who have multisystem disease, the prognosis in patients who have isolated pulmonary involvement is good; the disease regresses spontaneously in 25% of patients and stabilizes clinically and radio-graphically in 50%. In the remaining 25% of cases, the disease follows a progressive downhill course, resulting in diffuse cystic lung destruction. In a small minority of cases, death results from respiratory insufficiency, pul-monary hypertension, or both (4,18). For example, in a review of 87 patients who had isolated pulmonary dis-ease, 74 (85%) patients ultimately became disease free, and 3 patients had progressive disease resulting in severe pulmonary fibrosis and pulmonary hypertension (4).
Although spontaneous regression of disease is common, disease recurrence has been documented to occur up to 7.5 years after the initial presentation (41). Furthermore, there is no clear correlation between smoking history and recrudescence of disease, with nodules recurring in some patients after smoking cessation. PLCH may also recur
with patients who have pulmonary hypertension due to end-stage IPF or emphysema, pulmonary hypertension in patients with PLCH is significantly worse despite better expiratory function; this suggests that pulmonary hyper-tension in PLCH represents a specific entity and not sim-ply the result of chronic hypoxemia. In addition to arterial disease, patients who have severe LCH are also predis-posed to develop pulmonary veno-occlusive disease (30).
Clinical FindingsPLCH is an uncommon disease. Gaensler and Carrington (31) found LCH in only 3.4% of 502 patients who had open-lung biopsy for chronic, diffuse infiltrative lung disease.
More than 90% of patients who have PLCH are smok-ers, and this disease is considered to be related to smoking in most patients (25,32–37). In a review of 87 patients who had isolated pulmonary involvement by LCH, only 3 were nonsmokers (4). The majority of patients who have pulmonary LCH are young or middle-aged adults (average age, 32 years). Although previous reports have stressed a male preponderance, studies confirm that men and women are equally affected, likely the result of in-creasing tobacco use by women. Common presenting
FIGURE 19-1 Nodular LCH in a 30-year-old man. A: HRCT at the level of the right upper lobe bronchus shows several nodules (arrows). Some are solid, others are cavitated; some have smooth margins, others have irregular or poorly defined margins. B: HRCT through the right lower lung zone shows relative sparing of the lung bases. C: Histologic section from an open-lung biopsy shows a characteristic appearance of a poorly marginated granuloma surrounding a terminal bronchiole.
A B
C

CHAPTER 19 Cystic Lung Diseases 495
FIGURE 19-2 Pulmonary Langerhans cell histiocytosis. A: Autopsy specimen from a patient who had PLCH. Fine cystic spaces/diffuse reticulation predominate in the upper and middle lung zones. The lung bases are relatively spared. (From Müller NL, Miller RR. Computed tomography of chronic diffuse infiltrative lung disease. Part 2. Am Rev Respir Dis 1990;142:1440–1448, with permission.) B: Pathologic section from another patient with PLCH showing multiple cysts throughout the lungs, frequently exhibiting unusual branching configurations. Note relative sparing of the lung bases. (Courtesy of Dr. Carlos RR, de Carvalho, University of Säo Paulo, Säo Paulo, Brazil.) C: HRCT at the level of the middle lobe bronchus in a different patient than shown in A and B shows bizarrely shaped and branching cysts in the superior segments of the lower lobes, associated with a few tiny lung nodules, an appearance characteristic of LCH.
A B
C
following lung transplantation. Although it is considered a nonneoplastic disorder, PLCH has been noted to develop following treatment for Hodgkin disease. There is also an apparent association with non-small cell lung cancer, which has been reported to occur prior to, simultaneous with, and following the development of PLCH (15,19).
Radiographic FindingsThe radiographic findings of LCH include reticular, nodu-lar, and reticulonodular patterns, and honeycombing, often in combination (32,33,35,36,42). Abnormalities are usu-ally bilateral, predominantly involving the middle and up-per lung zones, with relative sparing of the costophrenic angles (35,36). Lung volumes are characteristically normal
or increased. Nodules when present have irregular borders and vary from 1 to 10 mm in size (15). With progression, PLCH results first in a predominantly reticulonodular pattern, with subsequent development of cystic lung dis-ease. Pneumothoraces occur and are frequently the initial presentation of recurrent disease. Additional findings that have been reported include mediastinal adenopathy, pulmo-nary consolidation, and a solitary pulmonary nodule (15).
High-Resolution Computed Tomography FindingsHRCT findings of pulmonary PLCH have been reported by a number of authors (26–28,43–45). In almost all pa-tients, HRCT demonstrates cystic airspaces, which are

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease496
FIGURE 19-3 HRCT in a 44-year-old man who has LCH, a history of cigarette smoking, and shortness of breath. A: HRCT through the upper lobes shows numerous thick- and thin-walled lung cysts. Some cysts are very irregular in shape. The intervening lung parenchyma appears normal, and there is no evidence of diffuse fibrosis. B: Near the lung bases, cysts are smaller and less numerous. The presence of irregularly shaped cysts with an apical predominance is typical of LCH.
A B
FIGURE 19-4 PLCH with cysts. A–D: Select 1-mm sections from the upper through the lower lobes in a patient with documented PLCH show scattered clusters of predominantly thin-walled cysts associated with ill-defined nodular opacities. Note relative sparing of the lung bases, characteristic of PLCH.
A B
C D
usually less than 10 mm in diameter (Figs. 6-5 to 6-8 and 19-2 to 19-4); these cysts are characteristic of PLCH (43,46–48) and were seen in 17 of 18 patients studied by Brauner et al. (44) and all 12 patients studied by Giron et al. (46) (Table 19-2).
On HRCT, the lung cysts have walls that range from being thin and barely perceptible (Figs. 6-5, 6-7, and 19-4) to being several millimeters in thickness (Figs. 6-6, 19-2, and 19-3). In a study by Grenier et al. (49), 88% of 51 pa-tients who had LCH showed thin-walled (less than 2 mm)

CHAPTER 19 Cystic Lung Diseases 497
FIGURE 19-5 A and B: PLCH with cavitary nodules. One-millimeter sections through the upper lobes show scattered well-defined cavitary nodules in both lungs.
A
B
FIGURE 19-6 Nodular LCH. Section below the carina shows diffuse, centrilobular thick-walled cavitary nodules, more extensive than shown in Figures 19-1 and 19-5.
cysts on HRCT, whereas 53% of patients showed thick-walled (greater than 2 mm) cysts. The presence of dis-tinct walls allows differentiation of these cysts from areas of emphysema, which can also be seen in some pa-tients. Although many cysts appear round, they can also have bizarre shapes, being bilobed, cloverleaf shaped, or branching in appearance (Figs. 6-5 to 6-7 and 19-2 to 19-4) (26–28,43–45). These unusual shapes are pos-tulated to occur because of fusion of several cysts, or perhaps because the cysts sometimes represent ectatic and thick-walled bronchi (36); in the series reported by Brauner et al. (43), confluent or joined cysts with per-sisting septations were seen in more than two-thirds of patients. Upper lobe predominance in the size and num-ber of cysts is common (Figs. 6-5 and 19-2 to 19-4). Large cysts or bullae (larger than 10 mm in diameter) are also seen in more than half of cases; some cysts are larger than 20 mm (26–28,43–45).
In some patients, cysts are the only abnormality visi-ble on HRCT, but in the majority of cases, small nodules (usually smaller than 5 mm in diameter) are also pres-ent (Figs. 4-20, 19-1, 19-5, and 19-6) (43,48); nodules were seen in 14 of 18 patients in Brauner’s series, and in 14 of 17 patients in Moore’s series (43,48). Larger nodules, sometimes exceeding 1 cm, may also be seen, but they are less common. In the study by Grenier et al. (50), 47% of 51 patients who had LCH showed nodules smaller than 3 mm in diameter, whereas 45% of patients showed nodules ranging between 3 mm and 1 cm in di-ameter, and 24% of patients showed nodules exceeding 1 cm in size. Nodules can vary considerably in number in individual cases, probably depending on the activity of the disease; nodules can be few in number or myriad (43,48). The margins of nodules are often irregular, par-ticularly when there is surrounding cystic or reticular disease. On HRCT, many nodules can be seen to be peri-bronchial or peribronchiolar and therefore centrilobu-lar in location; in this disease, there is a tendency for
granulomas to form around the bronchioles (43). HRCT may be valuable in directing lung biopsy to areas show-ing lung nodules (43).
The nodules are usually homogeneous in appearance, but some nodules, particularly those larger than 1 cm in diameter, may show lucent centers, presumably corre-sponding to small cavities (51). These cavities, however,
aFindings most helpful in differential diagnosis.bMost common findings.
TABLE 19-2 HRCT Findings in Pulmonary Langerhans Cell Histiocytosis
Thick-walleda,b or thin-walledb cysts
Nodules, usually smaller than 1 to 5 mm, centrilobular and peribronchiolar, may be cavitary, may be seen in association with cystsa,b
Progression over time from nodules to thick-walled cysts to thin-walled cystsb
Upper lobe predominance in size and number of nodules or cysts, costophrenic angles spareda,b
Fine reticular opacities
Ground-glass opacity
Mosaic perfusion or air trapping
Pulmonary hypertension

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease498
with a lower lung predominance. An upper lobe predomi-nance was reported in 57% of Grenier’s 51 patients (50), whereas a middle lung or basal predominance was never observed.
The evolution of lesions has been assessed using CT. As documented by Brauner et al. (44) in a study of 212 patients who had PLCH, although nodular lesions were twice as frequent as cysts on initial CT studies, follow-up CT examinations showed cystic lesions twice as often as nodular disease. Nodular opacities and thick-walled cysts typically underwent regression with time, while thin-walled cysts, linear densities, and emphysema either remained unchanged or progressed. These data support the previously noted conjecture that lesions in LCH un-dergo a predictable pathologic and radiologic evolution, beginning with centrilobular nodules (Fig. 19-1) and fol-lowed by cavitation (Figs. 19-5 and 19-6), the formation of thick-walled cysts (Figs. 6-6 and 19-3), and, finally, the development of thin-walled cysts (Figs. 6-5, 19-2, 19-4, and 19-8). Whereas nodular lesions may regress sponta-neously or be replaced by cysts, once formed, cystic le-sions persist, eventually becoming indistinguishable from diffuse emphysema.
Evidence of mosaic attenuation on inspiratory scans and air trapping on expiratory scans may also be seen in patients who have LCH and show nodular opacities or lung cysts (Fig. 19-8) (53). This may reflect the presence of bronchiolar obstruction or air trapping in cystic lung regions.
may sometimes represent a dilated bronchiole surrounded by granulomas and thickened interstitium (43). In the study by Grenier et al. (50), 25% of 51 patients who had PLCH showed cavitary nodules. In some patients, pro-gression of cavitary nodules to cystic lesions has been ob-served (44,46); this progression is characteristic as later described.
In many patients who have cysts or nodules, the intervening lung parenchyma appears normal on HRCT, without evidence of fibrosis or septal thickening (26–28,43–45). However, in a small percentage of cases, irregular interfaces (the interface sign) are present, or a fine reticular network of opacities is visible (Fig. 19-7) (26–28,43–45). These fine reticular opacities may cor-relate with intralobular fibrosis or early cyst formation, or with the progression and confluence of cysts (46). Ground-glass opacity is also sometimes seen but is not a prominent feature of this disease. This appearance needs to be differentiated from typical findings of basilar sub-pleural reticulation and fibrosis characteristic of usual interstitial pneumonitis, a disorder that has also been associated with smoking history (26–28,45).
HRCT shows no consistent central or peripheral pre-dominance of lesions (43,50), but in nearly all cases, the lung bases and the costophrenic sulci are relatively spared (42,52). In Brauner’s series (43) of 18 patients, 2 had ab-normalities localized to the upper lobes and 9 had disease that was predominant in the upper or middle lung zones; 2 patients had diffuse disease, but no patient had disease
FIGURE 19-7 Pulmonary Langerhans cell histiocytosis. A: Volumetric HRCT shows numerous thin-walled cysts (straight arrows) and small nodules (curved arrows) in the upper lobes. Also noted are irregular linear opacities consistent with fibrosis, focal ground-glass opacities, and emphysema (arrowheads). B: Coronal reformation demonstrates the characteristic distribution of disease mainly in the upper and middle lung zones with relative sparing of the lung bases. On both the cross-sectional and coronal images, it is difficult to distinguish some of the cysts due to LCH from emphysema. The patient was a 30-year-old woman. (Courtesy of Dr. Eduardo Sabbagh, Santiago, Chile.)
A B

CHAPTER 19 Cystic Lung Diseases 499
Utility of High-Resolution Computed TomographyHRCT is superior to chest radiographs in demonstrating the morphology and distribution of lung abnormalities in patients who have PLCH (26–28,43–45) and in making a specific diagnosis of this disease (50). In fact, in many pa-tients who have PLCH and plain radiographic findings of reticular abnormalities, HRCT shows that the plain film findings reflect the presence of numerous superimposed lung cysts. As compared with chest radiographs, HRCT is significantly more sensitive in detecting small and large cysts and nodules smaller than 5 mm in diameter (43,50).
FIGURE 19-8 LCH with small nodular opacities and mosaic perfusion due to air trapping. HRCT at three levels (A–C) shows small nodular opacities associated with areas of relative lucency (arrows), representing mosaic perfusion due to air trapping.
A
B
C
As previously noted, PLCH is not associated with any consistent pattern of PFT abnormalities, although air-ways obstruction is common (54) and probably related to peribronchiolar and bronchiolar luminal fibrosis (25). In a study by Moore et al. (48), the extent of disease on HRCT correlated better with impairment in gas ex-change, as assessed by the percent predicted carbon mon-oxide diffusing capacity (DLCO) (r = –0.71), than did plain radiographic findings (r = –0.57). In another study (54), significant correlation (r = 0.8) between HRCT and reduced diffusing capacity was also found. However, no correlation has been shown between CT findings and PFT findings of obstruction (48,54). Air trapping in as-sociation with lung cysts has been reported on expiratory HRCT in a patient who had LCH, despite the absence of evidence of airways obstruction on PFT (53).
Although HRCT may allow an accurate diagno-sis of this disease when typical findings are present (15,19,26–28,43–45), HRCT is limited in its ability to predict histopathologic activity. As documented by Soler et al. (55), there is excellent correlation between the ex-tent of nodules identified by HRCT and the density of nodules identified histopathologically. In distinction, in patients with a predominant pattern of cystic disease, histologic correlation is less accurate. As reported in this study of 13 patients with documented PLCH, inflam-matory granulomas could still be identified in most pa-tients with a predominant cystic pattern on HRCT; these investigators thus advise continued long-term follow-up in patients with findings otherwise suggestive of inactive fibrotic disease (55).
It is interesting to note that in a recent survey of pulm-onologists, regarding the role of HRCT in obviating lung biopsy in patients with diffuse interstitial lung disease, most of the 237 responders would not accept a diagno-sis of PLCH, despite the presence of characteristic HRCT findings (56). As is discussed in the following section, in this same study, a similar reluctance was also expressed by most responders for accepting HRCT findings as diag-nostic in patients with suspected LAM.
Differential DiagnosisIn patients who show nodules as the only HRCT ab-normality, the differential diagnosis is extensive; differ-entiation from sarcoidosis, silicosis, metastatic tumor, and tuberculosis may be impossible, although a typical distribution of the nodules can be valuable in diagnosis (57). Nodules in LCH tend to be centrilobular (Figs. 4-20, 19-1, and 19-5 to 19-7), whereas perilymphatic (septal, subpleural, and peribronchovascular) nodules are typi-cally seen in sarcoidosis, silicosis, and lymphangitic carci-nomatosis (57). Sparing of the costophrenic angles should raise the possibility of pulmonary LCH, but this finding can be seen in other nodular diseases as well.
Centrilobular emphysema and DIP, two diseases also related to smoking, can result in HRCT appearances

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease500
LCH can sometimes be identified (2,3). However, in dis-tinction to the parenchymal changes identified in patients with LCH, the lung bases are characteristically involved in patients with LAM or TSC.
The cysts in pulmonary LCH, when adjacent to blood vessels, can mimic the signet ring sign of bronchiectasis. However, distinction from bronchiectasis is straightfor-ward because the cysts in LCH lack the characteristic continuity of dilated bronchi seen on contiguous slices in patients who have airways disease (48).
As discussed in Chapter 6 and in this chapter, multiple thin-walled lung cysts are also seen in some patients who have LIP (Fig. 6-12) (59–61). These tend to have a lower lobe predominance. Other findings in patients who have LIP include small subpleural nodules, centrilobular nodules, in-terlobular septal thickening, and ground-glass opacity (61).
Despite the similar appearances of various cystic lung diseases, the accuracy of HRCT in distinguishing the most common causes of cystic lung disease (PLCH, pulmonary LAM, and emphysema) has been well documented (62). Recently, in a retrospective study of 92 patients with chronic cystic lung disease, including 18 patients with PLCH, 18 with pulmonary LAM, 17 with usual intersti-tial pneumonia, 16 with LIP, 15 with emphysema, and 8 with RB-ILD, two readers made a correct first-choice diagnosis in 148 of 184 (80%) readings. Although the correct diagnosis of PLCH was made in just 72% of pa-tients, in those cases in which the diagnosis was suggested with a high degree of certainty, a correct diagnosis was established in 88% (58).
LYMPHANGIOMYOMATOSIS AND TUBEROUS SCLEROSIS COMPLEXLAM, also known as lymphangioleiomyomatosis, is a rare multisystem disease characterized by infiltration of immature-appearing smooth muscle cells (LAM cells) into the lungs, airways (Fig. 19-10), and along axial lymphatic
similar to those of PLCH. Centrilobular emphysema re-sults in focal rounded lucencies, typically with an upper lobe predominance. However, in most patients who have centrilobular emphysema, focal areas of lung destruction lack visible walls, distinguishing them from the lung cysts typical of PLCH. On the other hand, in some patients with centrilobular emphysema, areas of emphysema show thin walls on HRCT, mimicking the appearance of PLCH; furthermore, in patients with late-stage PLCH, cyst walls may sometimes be very thin or imperceptible, closely mimicking centrilobular emphysema (Fig. 6-5). In the latter case, there would be little clinical significance in a misdiagnosis, as treatment would be similar.
In patients with DIP, the presence of underlying em-physema or focal areas of air trapping may result in an appearance of multiple thin-walled cysts associated with patchy ground-glass opacity (Fig. 19-9). Although the presence of cysts may cause confusion between PLCH and DIP, the finding of patchy ground-glass opacity seen in DIP usually allows an accurate differentiation (58). However, as noted previously, patients with documented PLCH may also develop DIP or RB-ILD.
Cystic lesions in PLCH, in contrast, can be easily dis-tinguished from honeycombing and larger air cysts in end-stage IPF (26–28,43–45). Pulmonary LCH character-istically involves the upper two-thirds of the lungs, with relative sparing of the costophrenic angles (Figs. 6-5, 19-1, 19-2, 19-4, 19-7, and 19-8) (16,17). IPF and other causes of honeycombing primarily involve the subpleural lung regions and the lung bases. Also, in patients who have IPF, the honeycomb cysts are surrounded by abnormal parenchyma, which shows findings of extensive fibrosis, whereas most of the cysts in LCH are surrounded by nor-mal lung. Lung volumes are normal or increased in cystic LCH, whereas they are generally reduced in patients who have IPF and show honeycombing.
In a woman with LAM, or patients with tuberous scle-rosis, cystic lesions identical to those seen in pulmonary
FIGURE 19-9 Desquamative interstitial pneumonitis. HRCT sections through the trachea (A) and middle lung (B), respectively, show ill-defined foci of ground-glass opacity in a patient with documented DIP. In this case, the presence of underlying emphysema gives a false impression of diffuse cystic lung disease. (Case courtesy of Dr. Ami Rubinowitz, Yale-New Haven Hospital, New Haven, CT.)
A B

CHAPTER 19 Cystic Lung Diseases 501
FIGURE 19-10 Low-power microscopic view of an open-lung biopsy specimen from a patient who has LAM. Characteristic cystic spaces with atypical spindle cells lining their walls are seen throughout the specimen.
FIGURE 19-11 Lymphangiomyomatosis. A: Open-lung biopsy specimen shows large cysts. (From Templeton PA, McLoud TC, Müller NL, et al. Pulmonary LAM: CT and pathological findings. J Comput Assist Tomogr 1989;13:54–57, with permission.) B: Whole lung specimen from a patient who had extensive cystic changes. (Case courtesy of Peter Kullnig, MD, University of Graz, Graz, Austria.)
A B
and S-LAM are linked to tuberous sclerosis gene (TSC1 and TSC2) mutations involving both TSC1 or TSC2 in the case of TSC-LAM, or TSC2 in the case of S-LAM, located on chromosomes 9q34 and 16p13 (68). LAM ap-pears to result from the metastatic dissemination of LAM cells bearing mutations that result in inactivation or loss of heterozygosity in TSC1 and TSC2, leading to hyper-activation of the mammalian (or mechanistic) target of rapamycin (mTOR). mTOR is a serine/threonine protein kinase that regulates cell growth, motility, and survival; protein synthesis and transcription.
The result of these mutations is inappropriate prolif-eration of LAM cells, migration, and tissue invasion. In patients with TSC-LAM, mutations are identified in all cell lines, whereas in patients with S-LAM, mutations are identified only in the lung, kidney, and lymph nodes. LAM cells have been shown to disseminate either through hematogenous or lymphatic pathways, raising the possi-bility that S-LAM actually represents a form of metastatic disease with cells arising from angiomyolipomatosis tis-sue, akin to benign metastasizing leiomyoma (66,68). In distinction to S-LAM, TSC is an autosomal-dominant dis-order with highly variable expressivity, most often occur-ring as a result of sporadic mutations.
The lesions of pulmonary LAM can be divided into early (active) and late phases (69,70). In its early phases, LAM results in a proliferation of cells primarily in ter-minal bronchioles and alveolar walls (Fig. 19-10). Find-ings of proximal acinar and irregular emphysema are identified adjacent to the periphery of smooth muscle infiltrates, findings due to the destructive effects of me-talloproteinases expressed by the LAM cells. Similar changes may be seen from more distal smooth muscle proliferation with normal-appearing proximal bronchi-oles (71). Evidence of fibrogenesis, including the presence of abundant fibronectin, may also be identified.
vessels in the chest and abdomen (63–68). The hallmark of this disease is cystic destruction of the lung parenchyma (Fig. 19-11). Spindle cell infiltration can also involve the hilar, mediastinal, and extrathoracic lymph nodes, some-times resulting in dilatation of intrapulmonary lymphat-ics and the thoracic duct. Involvement of the lymphatics can lead to chylous pleural effusions or ascites. Infiltra-tion of cells in the walls of pulmonary veins may rarely cause venous obstruction and lead to pulmonary venous hypertension with resultant hemoptysis.
PathologyLAM occurs either in association with tuberous scle-rosis complex (TSC-LAM) or in a sporadic form (spo-radic lymphangiomyomatosis [S-LAM]). Both TSC-LAM

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease502
(63,68), this likely represents an underestimation, as many cases are at least initially misdiagnosed as either asthma or chronic obstructive pulmonary disease. S-LAM affects approximately 1 in 400,000 women (78), while TSC-LAM affects 30% to 40% of adult female patients with TSC. TSC-LAM is five to tenfold more common than the isolated form of LAM (2,66).
The prevalence of LAM occurring in male TSC patients remains controversial. In one study of 186 patients with TSC having undergone abdominal CT, while pulmonary cysts were identified in 40 of 95 (42%) female patients, cysts were considerably less common in male patients, seen in only 12 of 91 (13%) patients (p < 0.001), and cysts were less numerous and smaller in men (79). In dis-tinction, recent data based on whole lung imaging suggests that LAM may occur with equal frequency in both sexes, being identified in 11 of 29 (38%) men with documented TSC (80). In this study, the diagnosis of cystic lung disease required that only four discrete cysts be identified (80,81).
The majority of patients with LAM present with dyspnea, pneumothorax, and/or cough (63,66,68,81). Patients with S-LAM are more likely to develop signs and symptoms than patients with TSC-LAM (81).
As documented in a cohort of 230 LAM patients, evi-dence of airway obstruction was the most common spiro-metric abnormality, occurring in 57% of individuals, with an average forced expiratory volume in 1 second (FEV1) of approximately 70%. About one-fourth of patients with obstructive airway disease exhibit reversible airflow obstruction after bronchodilator inhalation. In one-third of patients, spirometric results proved normal (81).
In this same population, pneumothorax was documented to have occurred in 55.5% of patients, with a mean of 4.4 pneumothoraces occurring in subjects with at least one prior episode (81). As a result, pleurodesis is now recommended with the initial episode of pneumothorax (66). Not surpris-ingly, a history of prior pleurodesis affects both pulmonary function and the appearance of the pleura on CT (82).
The disease follows an insidious course with a variable rate of progression and is generally first diagnosed because of pneumothoraces or chylous effusions in a woman of childbearing age. The mean time interval from the onset of symptoms to diagnosis is typically between 3 and 5 years (51,63). Sixty percent develop chylous pleural effusions, up to 80% develop pneumothorax, and 30% to 40% develop blood-streaked sputum or frank hemoptysis at some time in the course of the disease (64,65,83). In select cases, bi-opsy confirmation may be required for diagnosis, with ad-equate tissue generally available by transbronchial biopsy, with immunohistochemical staining for actin, desmin, and HMC-45; however, open-lung biopsy remains the gold standard (68). In one recent study of 80 women with LAM, the diagnosis was established by open-lung biopsy in 50, transbronchial biopsy in 14, retroperitoneal lymph node biopsy in 6, and characteristic HRCT findings in 10 (84).
Traditional therapy has relied on hormonal manipula-tion by oophorectomy or administration of progesterone with high dose medroxyprogesterone, tamoxifen, or lutein-izing hormone-releasing hormone analogs. Unfortunately,
These changes result in dilated emphysema-like spaces, within which may be identified hyperplastic type II pneu-mocytes and hemosiderin-laden macrophages, presum-ably the result of hemorrhage (70,71). Later in the course of disease, cellular infiltrates regress, leaving markedly dilated alveolar spaces associated with smooth muscle hyperplasia and diffuse collagen deposition.
Proliferation of spindle cells causes circumferential narrowing and obstruction of bronchioles, leading to airways obstruction and air trapping. Airways obstruc-tion has also been shown to result from loss of alveolar support occurring in association with lung cysts. Using detailed morphometric analysis of postmortem lungs ob-tained from two patients who had LAM, Sobonya et al. (72) showed that of these two mechanisms, it is likely that the loss of parenchymal interdependence resulting from diffuse cystic disease and consequent loss of alveolar sup-port may be the more important.
The smooth muscle cells found in LAM have been shown to be phenotypically heterogeneous, smooth muscle, actin and desmin-positive cells derived from myoid precursors. Immunohistochemical studies show that approximately 80% of proliferating cells in LAM patients stain positively for estrogen receptors, whereas nearly all stain positively for progesterone receptors, in distinction to normal smooth muscle cells (71). LAM cells also differ from normal smooth muscle cells be-cause they react with HMB-45, a monoclonal antibody that identifies a 100-kDa glycoprotein (gp 100) that is located in premelanosome antigens in the cytoplasm of melanoma cell lines (63,70–72). HMB-45 staining is also found in patients who have angiomyolipomas of the kidney, multifocal micronodular pneumocyte hyper-plasia (73,74), and clear cell tumors of the lung (75). Although the significance of immunohistochemical staining with HMB-45 remains uncertain, this finding has proved useful in improving the accuracy of trans-bronchial biopsy for diagnosing LAM. More recently, it has also been shown that LAM cells express lymphatic endothelial markers, including vascular endothelial growth factors of which VEGF-D has been shown to be elevated in the serum of patients with LAM, represent-ing a potentially useful means for monitoring response to therapeutic interventions (76).
Clinical FindingsLAM is thought to occur almost exclusively in women of childbearing age, but recent data have shown that increas-ing numbers of older women are being diagnosed with this disease (77). Using data recently derived from two large patient registries, Cohen et al. (77) found that the mean age of diagnosis was 46.7 years, with 50% of older women presenting without a history of pneumothorax. This likely reflects a growing awareness of LAM occurring in women with the TSC and the increasing use of HRCT to diagnose otherwise unexplained pulmonary symptoms (77).
While the incidence of LAM has been estimated to be between 1 and 2.6 cases per 100,000 women worldwide

CHAPTER 19 Cystic Lung Diseases 503
Chu et al. (63), chest radiographs were interpreted as normal in 9 of 35 (26%) patients who had proven LAM.
High-Resolution Computed Tomography FindingsOn HRCT, patients with LAM characteristically show nu-merous thin-walled lung cysts, surrounded by relatively normal lung parenchyma (Figs. 6-9 to 6-11 and 19-12 to 19-16, Table 19-3) (2,3,63,68,92–101). These cysts usu-ally range from 2 mm to 5 cm in diameter, but they can be larger. Their size tends to increase with progression of the disease (98). In patients who have mild disease, the cysts usually measure smaller than 5 mm in diameter. In patients who have more extensive disease, in which 80% or more of the lung parenchyma is involved, the cysts tend to be larger, most being larger than 1 cm in diameter. The walls of the lung cysts are usually thin and faintly percep-tible, but they may range up to 4 mm in thickness (96,98). Irregularly shaped lung cysts, as are seen in patients who have LCH, are uncommon. Lung cysts seen on HRCT cor-relate with the presence of the lung cysts that are common pathologically in this disease; these cysts are partially sur-rounded by the abnormal spindle cells typical of LAM.
LAM occurring in patients with TSC has the same ap-pearance as seen in individuals with isolated LAM. In one retrospective study of 186 adult TSC patients, including 91 (49%) male subjects, in whom as a minimum evalu-ated with abdominal CT including the lung bases, cysts were identified in 52 of 186 (28%) cases, with the size of lesions varying from 2 mm to greater than 2 cm (79).
Some patients with TSC have small lung nodules, rang-ing from a few millimeters to 8 mm in diameter, with a random distribution, representing micronodular pneumo-cyte hyperplasia (Fig. 19-17) (102,103), an entity never seen in patients with S-LAM. Franz et al. (103) performed genotyping and CT in 23 asymptomatic women with
there is little evidence of consistent efficacy. For example, in one study, Taviera-DaSilva et al. (85) showed that pro-gesterone therapy failed to slow the progression of lung function decline. In patients with reversible airway ob-struction, treatment with bronchodilators is of value, and in patients with angiomyolipomas larger than 4 cm in size, treatment with embolization is indicated. Unfortu-nately, mortality still occurs in 10% to 20% of patients with symptoms and 30% having lung biopsy (66). As a consequence, LAM is now listed as an indication for lung transplantation (70). This may be performed safely, de-spite a history of prior pleurodesis (66). Disease recur-rence in transplanted lungs has been reported (86).
Although lung transplantation is currently the only defin-itive treatment, it is reserved for individuals with advanced disease. It is anticipated that a role for HRCT screening will follow the advent of treatment methods focused on specific molecular targets (66). These include, among others, metal-loproteinase inhibitors, statins, interferon, VEGF inhibitors, and rapamycin (sirolimus). Of these, rapamycin (sirolimus), an mTOR inhibitor has shown the greatest promise in sta-bilizing pulmonary function and potentially decreasing the size of renal angiomyolipomas, although drug resistance has proved potentially problematic (87–90).
Radiographic FindingsThe plain radiographic manifestations of LAM include reticular, reticulonodular, miliary, and honeycomb patterns (68,83,91). More than 50% of patients have radiographic evidence of pneumothorax at the time of first presentation (70). Lung volumes can be increased in patients who have this disease. Radiologic abnormalities may precede, accompany, or postdate other manifestations of the disease, such as pneu-mothorax and chylous pleural effusion. Not infrequently, radiographs often fail to reveal the presence of diffuse lung cysts subsequently verified surgically (83). As documented by
FIGURE 19-12 Lymphangiomyomatosis. A and B: HRCT targeted to the left lung shows multiple cystic airspaces of varying sizes. These have walls ranging from being barely perceptible to 2 mm in thickness. The lung parenchyma between the cystic airspaces is normal. The cysts are primarily round in shape, but a few appear confluent.
A B

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease504
FIGURE 19-13 Tuberous sclerosis complex/lymphangioleiomyomatosis. A and B: Targeted HRCT sections through the left lung in a patient with documented tuberous sclerosis show typical appearance of diffuse thin-walled cysts randomly distributed throughout the lungs nearly replacing normal lung. Note that this appearance is indistinguishable from the diffuse cystic changes seen in isolated cases of LAM.
A B
FIGURE 19-14 Lymphangiomyomatosis. A and B: HRCT images through the upper and lower lobes, respectively, show the characteristic appearance of similar size, thin-walled cysts, uniformly distributed throughout both lungs. Note that in this case there is a small left-sided pneumothorax. C: Coronal reformatted image shows to good advantage the uniform distribution of cysts characteristic of LAM. Note the left-sided basilar pneumothorax.
A B
C

FIGURE 19-16 LAM in a patient with tuberous sclerosis. A–D: Select sections in a young woman with documented tuberous sclerosis show diffuse thin-walled cysts throughout both lungs, typical of patients with LAM. Note the presence of a left-sided chest tube, being used to treat an associated pneumothorax. E: Contrast-enhanced CT through the upper abdomen in this patient shows the characteristic appearance of a fat-containing angiomyolipoma in the right kidney (arrow). A left renal mass is also seen.
FIGURE 19-15 LAM with tuberous sclerosis. A–C: LAM in a 35-year-old woman with tuberous sclerosis. HRCT shows numerous discrete, round, thin-walled lung cysts. Cysts are thinner walled and more regular in size and shape than those seen in patients who have LCH. Intervening lung parenchyma appears normal. Cysts are diffusely distributed, and cysts at the lung bases (C) are similar in size and number to those seen at the lung apices (A). These abnormalities were associated with adenoma sebaceum, shortness of breath, airway obstruction on PFTs, and low diffusing capacity.
A
C
B
505
(Continued )
A B
C D

Thin-walled lung cysts, usually round in shapea,b
Diffuse distribution, costophrenic angles involveda,b
Mild septal thickening or ground-glass opacity
Lymph node enlargement
Small nodules in patients with TSC
Pleural effusionb
Pneumothoraxa
aMost common findingsbFindings most helpful in differential diagnosis
TABLE 19-3 HRCT Findings in Lymphangiomyomatosis
FIGURE 19-17 Micronodular pneumocyte hyperplasia in TSC. A–D: Target-reconstructed HRCT images through the middle and lower lungs show innumerable tiny lung nodules in a patient with tuberous sclerosis and micronodular pneumocyte hyperplasia. A few scattered lung cysts are present. E: Non-contrast CT image through the upper abdomen shows a fat-containing angiomyolipoma in the right kidney.
BA
506
C D
FIGURE 19-16 (Continued)
E

CHAPTER 19 Cystic Lung Diseases 507
FIGURE 19-17 (Continued)
FIGURE 19-18 A and B: Transplantation in LAM. LAM before and after lung transplantation. A: Targeted reconstruction through the left lung shows diffuse lung cysts associated with a pneumothorax. Recurrent pneumothoraces are a hallmark of this disease. B: Section in the same patient obtained following unilateral lung transplantation. Note subtle narrowing of the left upper lobe bronchus at the site of anastomosis. An occult right-sided pneumothorax is also identifiable anteriorly. C and D: Recurrent LAM after bilateral lung transplantation in a different patient than in A and B. HRCT images at the level of the upper lobes and coronal reconstruction, respectively, show diffuse lung cysts consistent with recurrent disease associated with a pneumothorax. Diffuse recurrence is a well-described potential complication of lung transplantation.
A
B
TSC. Pulmonary cysts consistent with LAM were found in nine (39%) patients. Ten (43%) patients had pulmonary parenchymal nodules. Pulmonary nodules were more common in women with cysts (78% vs. 21%, p < 0.05), and 52% of all patients had either cysts or nodules. TSC2 mutations were identified in all cyst-positive patients who were tested (n = 8), whereas both TSC1 and TSC2 mutations were found in patients with nodular disease.
In the majority of patients, the cysts are distributed dif-fusely throughout the lungs, and no lung zone is spared (Figs. 19-12 to 19-16); diffuse lung involvement is seen even in patients who have mild disease. In reported series
(96,98), there is no evidence of lower lung zone, central, or peripheral predominance on CT scans. Thus, the HRCT findings do not support the previous impression that the le-sions initially have a predominantly basal distribution (65).
In most patients, the lung parenchyma between the cysts appears normal on HRCT (Figs. 19-12 to 19-15). In some cases, however, a slight increase in linear interstitial markings (94,99), interlobular septal thickening (94,96), or patchy areas of ground-glass opacity (98) are also seen. The latter probably represent areas of pulmonary hemorrhage. Pneu-mothorax may be seen to be associated with cysts in patients who have this disease (Figs. 19-14, 19-16, and 19-18).
(Continued)
E

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease508
Other features of LAM include hilar, mediastinal, ret-rocrural lymph node enlargement and lymphedema. In one review of 228 patients with documented LAM, 8 (3.5%) had LAM-associated peripheral lymphedema, with lymphedema representing the initial presenting fea-ture in 5 (104). Adenopathy was visible in four of the seven patients who had complete chest CT scans reported by Sherrier et al. (100). Air trapping on expiratory scans may also be seen (53).
Not surprisingly, pleural effusion, pneumothorax, or both are frequently identified and can be helpful in distin-guishing LAM from PLCH. In one large series, they were identified in five (14%) and two (6%) patients, respec-tively (63). It should be emphasized that the appearance of the pleura on HRCT will vary, depending on whether there is a history of prior pleurodesis. As documented by Avila et al. (105), pleural abnormalities are more likely to be identified in patients following pleurodesis. Of 258 patients with documented LAM, pleural abnormalities were present in 76% of patients following pleurodesis, as compared with 38% of cases without prior pleurode-sis, with marked differences in the occurrence of pleural thickening (65% vs. 26%), loculated effusions (11% vs. 2.4%), pneumothoraces (10% vs. 1.6%), foci of high at-tenuation (23% vs. 1.6%), and masses (14% vs. 0.8%), respectively (105). In this same study, although pleural masses were noted to enhance following intravenous con-trast administration, they were characterized by a lack of interval growth.
Even though pulmonary and pleural findings clearly predominate, abdominal and pelvic findings are also fre-quent. In one study of 80 patients with LAM, 61 (76%) had positive findings at abdominal/pelvic CT, including renal angiomyolipomas in 54%, enlarged abdominal lymph nodes in 39%, and lymphangiomyomas in 16%, respectively (Fig. 19-16) (84). Angiomyolipomas are es-pecially frequent, occurring in more than 90% of patients
DC
FIGURE 19-18 (Continued)
with TSC-LAM and 30% to 50% of patients with S-LAM (66). Chu et al. (63) detected a total of 31 solid renal masses in 18 of 35 (51%) patients, including 6 (17%) pa-tients who had multiple angiomyolipomas and 4 (11%) who had bilateral involvement. Typically asymptomatic when small, these have a well-known tendency to cause retroperitoneal hemorrhage, hematuria, and loss of renal function, resulting when greater than 4 cm in diameter. Once this size, periodic screening with CT or ultrasound is generally recommended. Therapeutic options include embolization, radioablation, or nephron-sparing partial nephrectomy (66). Patients with TSC-LAM are more likely to develop hepatic and renal angiomyolipomas, while patients with S-LAM are more likely to develop pleural effusion, ascites, and abdominal lymphangioleio-myomas (81).
Utility of High-Resolution Computed TomographyHRCT is superior to chest radiography in determining the extent and distribution of air cysts in this disease, and it can demonstrate extensive abnormalities in patients who have normal radiographic findings (96,98,100). Also, cysts are commonly visible in asymptomatic patients with TSC (103). The cystic abnormalities of LAM are also much easier to assess and are better defined on HRCT than on conventional CT. Cysts visible on HRCT were rarely seen on chest radiographs, unless they were larger than 1 cm. Because HRCT findings closely mirror the gross pathologic appearances of this disease, HRCT con-stitute a major diagnostic criterion for establishing LAM (Table 19-4).
Disease extent as assessed on CT correlates better than do radiographic findings with clinical and functional im-pairment in patients who have LAM (70). Typically, PFTs reveal decreased diffusing capacity, and, less commonly,

CHAPTER 19 Cystic Lung Diseases 509
units to obtain a QCT index in 10 patients who had docu-mented LAM. Defined as the amount of cystic lung ex-pressed as a percent of total lung area for the two slices combined, a good correlation was found between the QCT index and the FEV1 (r = –0.9; p = 0.0005), residual volume (r = 0.7; p = 0.02), DLCO (r = –0.76; p = 0.01), and exercise performance measured as maximum work-load (r = –0.84; p = 0.002), among other measurements (108). These data are significant given previously noted correlations between measurements of airflow obstruc-tion and prognosis (106).
As shown by Avila et al. (82) in a study of 37 patients with LAM, qualitative ratings of the severity of cystic lung disease compared favorably with quantitative evalu-ation over a wide range of pulmonary dysfunction, with good agreement noted between two observers (k = 0.75). More recently, CT has been used to grade the severity of LAM using advanced textural analysis and feature cor-relation to identify and quantify cystic areas as well as to assess lung distal to obvious cysts. On the basis of this ap-proach, these investigators could identify damage in the form of emphysema-like spaces in portions of the lung separate from cysts, with declined lung function correlat-ing with these features (109).
Although quantitative assessment of HRCT data pro-vides good correlation with spirometric measures of lung function, in fact, quantitative CT is rarely obtained in clinical practice.
Given the characteristic nature of imaging findings, it has been proposed by several investigators that a specific diagnosis of LAM can be established by HRCT (66,110). As a consequence, the LAM Foundation has suggested that annual HRCT screening be performed in all women with unexplained recurrent pneumothoraces or emphy-sema in the absence of a history of tobacco use, as well as women older than 18 years with tuberous sclerosis (66). The potential for HRCT to identify otherwise as-ymptomatic women with TSC, in particular, may allow identification of a subset of patients with early disease (78,111–113). In one recent retrospective study of 101 female women with TSC in whom CT studies were avail-able for review, 48 (47.5%) had CT evidence of cystic lung disease (114). In this study the risk of LAM increased with age, with a prevalence of 27% in women below 21 years of age, increasing to 81% in women over the age of 40. Ultimately, 63% of subjects developed symptoms, while a total of 12.5% died of LAM.
Important for the potential use of CT to screen patients with TSC, among asymptomatic subjects with LAM, 84% had cysts identified on a single CT section at the level of the carina. This suggests a possible role for acquiring a limited, low-dose CT screening study to minimize ra-diation exposure (114). Also, a limited number (3 to 10) of CT slices has been reported as an effective means of screening patients with TSC, both men and women (80).
Patients who have lung transplantation for LAM have increased morbidity and mortality due to complications related to their underlying disease (Fig. 19-18) (115);
airflow obstruction with reduced FEV1 and FEV1/forced vital capacity (FVC) ratio, accompanied by a reduction in elastic recoil. Significant correlations have been doc-umented between a reduced FEV1/FVC ratio, increased total lung capacity, and prognosis (106). On CT, the best correlations have been observed between the extent of disease and impairment in gas transfer as assessed by the DLCO (92,96,98). Although significant correlations have also been demonstrated between extent of cystic dis-ease and severity of airways obstruction (92,96), this has proved more controversial. In the study by Aberle et al. (92), for example, good correlation was reported between CT scores and measures of airways obstruction, in par-ticular, the FEV1/FVC ratio (r = –0.92; p < 0.002). Simi-larly, Lenoir et al. (96), in a study of 11 patients who had LAM (n = 9) and tuberous sclerosis (n = 2), found good correlation between CT findings and FEV1/FVC ratios and DLCO. In distinction, although good correlations be-tween CT scores and DLCO have also been reported by Müller et al. (98) in their study of 14 patients who had LAM, similar good correlation was not seen with lung volumes or airflow parameters.
In an early study, Crausman et al. (107,108) assessed the use of quantitative computed tomography (QCT) measurements as a means to predict prognosis in LAM patients. Using two end-expiratory HRCT images (at the carina and just above the diaphragm), these authors used a density mask program with a threshold of –900 Hounsfield
aHRCT features characteristic of LAM are multiple (>10) thin-walled round well-defined air-filled cysts with preserved or increased lung volume, with no other significant pulmonary involvement, and specifically, no interstitial lung disease with the exception of possible features of multifocal micronodular pneumocyte hyperplasia in patients with TSC.bHRCT features are compatible with pulmonary LAM when only few (>2 and ≤10) cysts, as described above, are present.Modified from Johnson SR, Cordier JF, Lazor R, et al. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyo-matosis. Eur Respir J 2010;35:14–26.
TABLE 19-4 Proposed European Respiratory Society Criteria for Diagnosis of LAM
Definite LAM
1. Characteristica or compatibleb HRCT and lung biopsy fitting the pathologic criteria for LAM; or
2. Characteristica HRCT and any of the following:
Angiomyolipoma (kidney)
Thoracic or abdominal chylous effusion
Lymphangioleiomyoma or lymph node involved by LAM
Definite or probable TSC
Probable LAM
1. Characteristica HRCT and compatible clinical history; or
2. Compatibleb HRCT and any of the following:
Angiomyolipoma (kidney)
Thoracic or abdominal chylous effusion
Possible LAM
Characteristica or compatibleb HRCT

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease510
with AIDS, especially children, as well as in patients with common variable hypogammaglobulinemia (118); it is also frequently idiopathic. Previously classified as an id-iopathic interstitial pneumonia, LIP is now considered a lymphoproliferative disorder.
LIP results in diffuse infiltration of the lung interstitium by monotonous sheets of polyclonal lymphocytes in associ-ation with type II pneumocyte hyperplasia and the accumu-lation of mononuclear cells that result in the development of noncaseating granulomas and well-formed lymphoid germinal centers. HRCT findings include a variety of ab-normalities, including patchy ground-glass opacity, bibasi-lar reticulation, scattered parenchymal nodules frequently subpleural in location, and thickened interlobular septae.
Thin-walled lung cysts have been described in up to two-thirds of patients with LIP and appear randomly distributed (Figs. 6-12, 9-36, 9-37, 11-26, and 19-19 to 19-21) (119). In one study of 22 patients with LIP, CT findings most commonly identified included areas of ground-glass opacity and poorly defined centrilobular nodules in all 22 patients. Cystic airspaces were seen in 15 of 22 (68%) (61). Except for lung cysts, all CT find-ings in patients with LIP are potentially reversible (60).
Cysts in LIP are thin walled, up to 3 cm in diameter, often predominate in the lower lobes, and are usually lim-ited in number. In many cases, they tend to be associated with vessels, seen in relation to the cyst walls. A history of collagen-vascular disease, and particularly Sjögren syndrome, is very helpful in differential diagnosis.
BIRT-HOGG-DUBÉ SYNDROMEA number of review articles have described the range of features associated with BHD (120–122). BHD is a rare autosomal-dominant disorder clinically characterized by diffuse thin-walled lung cysts, cutaneous fibrofolliculo-mas predominantly involving the face, neck, and upper trunk, and most importantly, renal tumors, ranging from benign oncocytomas to malignant renal cell carcinomas. The latter occur in up to 70% of patients by age 70.
The underlying defect in patients with BHD consists of mutations in the folliculin (FLCN) gene located on chromosome 17p11.2, leading to a loss of the tumor suppressor, FLCN. FLCN, in turn, interacts with signal-ing molecules such as 5’-AMP activated protein kinase and similar to LAM, and mTOR (123). FLCN occurs in normal cells of the skin, kidney and lungs, among others: there is no gender predilection.
Clinical FindingsCysts may appear in isolation without the other stigmata of the syndrome and may be identified in patients as young as 20 to 30 years. In distinction, renal carcinoma tends to occur in older patients, typically those over 40 years of age. Given concerns over the development of renal cancer, it has been suggested that screening with MRI should be performed in patients with known BHD over the age of 20 (122).
these LAM-related complications can be diagnosed or suggested using CT. In a review of 13 patients who had unilateral (n = 8) or bilateral (n = 5) lung transplantation for LAM, complications found during and after transplan-tation included excessive pleural adhesions (n = 4), native lung pneumothorax (n = 3), chylous effusion (n = 1), chylous ascites (n = 3), complications from renal angio-myolipomas (n = 4), and recurrent LAM (n = 1). One patient died as a result of complications of LAM (115).
Differential DiagnosisA number of entities may be confused with LAM (Table 19-1). For example, lung cysts very similar to those seen in LAM have also been described in patients who have pulmonary PLCH (43,48). However, three findings usually allow the differentiation of these two diseases. In many patients who have pulmonary PLCH, a nodular component is also present (Figs. 19-5 and 19-6); this is less uncommon with LAM. However, it should be noted that in patients with tuberous sclerosis, similar micronodular opacities may be the result of micronodular pneumocyte hyperplasia (Fig. 19-17). This entity may also be associated with airflow obstruction and mosaic attenuation (116).
Irregularly shaped cysts commonly seen in patients who have PLCH (Figs. 19-2 and 19-3) are much less fre-quent with LAM. Furthermore, PLCH characteristically involves the upper two-thirds of the lungs and spares the costophrenic angles (Fig. 19-4), whereas LAM involves the lungs diffusely (2,3,68). Finally, it should be emphasized that PLCH is one of a number of diseases characteristically associated with tobacco use, while no such connection be-tween cigarette smoking and LAM has been demonstrated.
Additional entities that may be mistaken for LAM include LIP (Figs. 9-36, 9-37, and 6-12), follicular bron-chiolitis, subacute HP with cyst formation, amyloidosis (Fig. 16-17), LCDD (Figs. 6-14, 16-19, and 16-20), low-grade leiomyosarcomas and benign metastasizing leio-myoma, and BHD syndrome (66). Randomly distributed thin-walled lung cysts with similar morphology to those seen in LAM, for example, have been reported to occur in up to 13% of patients with subacute HP, although this en-tity is typically associated with a range of additional find-ings including poorly defined centrilobular nodules and later in the course of disease, extensive lung fibrosis (117).
Of those entities most likely to present with similar appearing thin-walled cysts, two entities in particular stand out as differential possibilities: LIP and the BHD syndrome.
LYMPHOID INTERSTITIAL PNEUMONIALIP is a rare interstitial lung disease that typically occurs in association with either underlying dysproteinemia or autoimmune diseases. It is also discussed in Chapters 6, 9, and 11. LIP is most often associated with Sjögren syn-drome or other collagen diseases, but LIP has also been reported in patients’ immunosuppression, such as those

FIGURE 19-19 Lymphoid interstitial pneumonia. A–D: Select axial HRCT sections from the upper lobes to the lung bases, respectively in a patient with Sjögren syndrome and documented LIP show variable size, thin-walled randomly distributed cysts, in the absence of lung nodules or reticulation. E and F: Coronal maximum and minimum intensity images in the same case as A–D. Note that the maximum intensity projection is of value by confirming a lack of nodules in this case, while the maximum intensity projection shows the extent and severity of cystic changes to good advantage. In this case, a combination of clinical history and involvement of the lower lobes allows differentiation of this appearance from LCH.
A B
C D
E F
511

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease512
FIGURE 19-20 Lymphoid interstitial pneumonia. High-resolution images through two different patients with documented LIP showing the range of cystic changes that may be identified, from a few scattered thin-walled cysts (arrows in A) to a diffuse profusion of tiny cysts superficially mimicking the appearance of LAM (B).
A
B
FIGURE 19-21 Lymphoid interstitial pneumonia. A and B: Sections through the lower lobes in a patient with Sjögren syndrome and documented LIP show variable size, thin-walled cysts, some of which appear coalescent, resulting in bizarre shapes. In this case, a combination of clinical history and involvement of the lower lobes allows differentiation of this appearance from LCH. (Case courtesy of Dr. Ami Rubinowitz, Yale-New Haven Hospital, New Haven, CT.)
A B

CHAPTER 19 Cystic Lung Diseases 513
be partially incorporated into the pleura and interlobular septa (123). Cyst walls are lined by alveolar cells without evidence of atypia or neoplastic transformation (123). While unruptured cysts show no evidence of inflam-mation, histologic evaluation of ruptured cysts shows evidence of inflammation, making these lesions indistin-guishable from emphysematous bullae or blebs.
High-Resolution Computed Tomography FindingsCysts in BHD appear thin walled; they can often be distin-guished from those of LCH and LAM by appearance and distribution (Table 19-5). In a study (6) of 17 patients with BHD syndrome, CT showed cystic lung disease in 15; the cysts varied in size up to 7.8 cm; such large cysts are uncom-mon with LCH and LAM. In another study, although most cysts (76%) were less than 1 cm in diameter, 6% were larger than 2 cm (7). Large cysts were frequently multiseptated (6).
Overall, multiple pulmonary cysts occur in 80% of members of affected families. Clinically up to 75% of patients with BHD present with spontaneous, frequently recurrent pneumothorax. Thin-walled cysts are nearly ubiquitous in patients with BHD, although, as docu-mented by Toro et al. (124) in a study of 198 patients with BHD, while 178 (89%) proved to have lung cysts, pneumothoraces occurred in only 24% of cases. Pneumo-thoraces tend to occur more often in younger patients, the incidence decreasing with age. Not surprisingly, the diagnosis is often delayed, with pneumothoraces mistak-enly attributed most often to underlying emphysema. In distinction to LAM, most patients with BHD and cystic lung disease have near-normal pulmonary function (125).
The cause of pulmonary cysts in BHD is unclear, (7) and their histology have been incompletely character-ized. However, cysts predominate in the medial and sub-pleural lung, and in the mid- and lower lung zones. In one study, cyst walls have been shown histologically to
FIGURE 19-22 The BHD syndrome. A–C: Select HRCT images through the mid- and lower thorax, respectively, show several large thin-walled cysts that predominantly involve the lower lung fields. Note the lack of uniform distribution and relative variability in the size of these lesions, findings that help differentiate this entity from LAM/TSC. Histologically, cysts in BHD characteristically extend into both the visceral pleura and interlobular septae. D: Section through the upper abdomen in a different patient than in A–C shows a right renal tumor (posterior arrows). Renal neoplasia is the most important complication occurring in patients with BHD syndrome. Incidentally noted is pericolonic edema anteriorly in this patient with mild diverticulosis.
A B
C D

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease514
5. Souza CA, Finley R, Müller NL. Birt-Hogg-Dubé syndrome: a rare cause of pulmonary cysts. AJR Am J Roentgenol 2005;185:1237–1239.
6. Agarwal PP, Gross BH, Holloway BJ, et al. Thoracic CT findings in Birt-Hogg-Dubé syndrome. AJR Am J Roentgenol 2011;196:349–352.
7. Tobino K, Gunji Y, Kurihara M, et al. Characteristics of pulmo-nary cysts in Birt-Hogg-Dubé syndrome: thin-section CT findings of the chest in 12 patients. Eur J Radiol 2011;77:403–409.
8. Jeong YJ, Lee KS, Chung MP, et al. Amyloidosis and lymphop-roliferative disease in Sjögren syndrome: thin-section computed tomography findings and histopathologic comparisons. J Comput Assist Tomogr 2004;28:776–781.
9. Desai SR, Nicholson AG, Stewart S, et al. Benign pulmonary lympho-cytic infiltration and amyloidosis: computed tomographic and patho-logic features in three cases. J Thorac Imaging 1997;12:215–220.
10. Colombat M, Mal H, Copie-Bergman C, et al. Primary cystic lung light chain deposition disease: a clinicopathologic entity derived from unmutated B cells with a stereotyped IGHV4-34/IGKV1 re-ceptor. Blood 2008;112:2004–2012.
11. Colombat M, Stern M, Groussard O, et al. Pulmonary cystic disor-der related to light chain deposition disease. Am J Respir Crit Care Med 2006;173:777–780.
12. Wongsripuemtet J, Ruangchira-urai R, Stern EJ, et al. Benign me-tastasizing leiomyoma. J Thorac Imaging 2012;27:W41–W43.
13. Irion KL, Hocchegger B, Marchiori E, et al. Proteus syndrome: high-resolution CT and CT pulmonary densitovolumetry findings. J Thorac Imaging 2009;24:45–48. doi:10.1097/RTI.0b013e31818b20cd
14. Rowan C, Hansell DM, Renzoni E, et al. Diffuse cystic lung dis-ease of unexplained cause with coexistent small airway disease: a possible causal relationship? Am J Surg Pathol 2012;36:228–234.
15. Abbott GF, Rosado-de-Christenson ML, Franks TJ, et al. From the archives of the AFIP: pulmonary Langerhans cell histiocytosis. Radiographics 2004;24:821–841.
16. Allen TC. Pulmonary Langerhans cell histiocytosis and other pulmonary histiocytic diseases: a review. Arch Pathol Lab Med 2008;132:1171–1181.
17. Rao RN, Moran CA, Suster S. Histiocytic disorders of the lung. Adv Anat Pathol 2010;17:12–22.
18. Soler P, Tazi A, Hance AJ. Pulmonary Langerhans cell granuloma-tosis. Curr Opin Pulm Med 1995;1:406–416.
19. Sundar KM, Gosselin MV, Chung HL, et al. Pulmonary Langer-hans cell histiocytosis: emerging concepts in pathobiology, radiol-ogy, and clinical evolution of disease. Chest 2003;123:1673–1683.
20. Hidalgo A, Franquet T, Gimenez A, et al. Smoking-related inter-stitial lung diseases: radiologic-pathologic correlation. Eur Radiol 2006;16:2463–2470.
21. Willman CL, Busque L, Griffith BB, et al. Langerhans’-cell histio-cytosis (histiocytosis X)—a clonal proliferative disease. N Engl J Med 1994;331:154–160.
22. Brabencova E, Tazi A, Lorenzato M, et al. Langerhans cells in Langerhans cell granulomatosis are not actively proliferating cells. Am J Pathol 1998;152:1143–1149.
23. Kambouchner M, Basset F, Marchal J, et al. Three-dimensional characterization of pathologic lesions in pulmonary Langerhans cell histiocytosis. Am J Respir Crit Care Med 2002;166:1483–1490.
24. Myers JL, Aubry MC. Pulmonary Langerhans cell histiocytosis: what was the question? Am J Respir Crit Care Med 2002;166:1419–1421.
25. Travis WD, Borok Z, Roum JH, et al. Pulmonary Langerhans cell granulomatosis (histiocytosis X). A clinicopathologic study of 48 cases. Am J Surg Pathol 1993;17:971–986.
26. Attili AK, Kazerooni EA, Gross BH, et al. Smoking-related intersti-tial lung disease: radiologic-clinical-pathologic correlation. Radio-graphics 2008;28:1383–1398.
27. Vassallo R, Ryu JH. Smoking-related interstitial lung diseases. Clin Chest Med 2012;33:165–178.
28. Caminati A, Cavazza A, Sverzellati N, et al. An integrated ap-proach in the diagnosis of smoking-related interstitial lung disease. Eur Respir Rev 2012;21:207–217.
29. Fartoukh M, Humbert M, Capron F, et al. Severe pulmonary hypertension in histiocytosis X. Am J Respir Crit Care Med 2000;161:216–223.
Cysts may be limited in number in some patients, but in a study by Agarwal et al. 6 of 15 patients, 33% had more than 20 cysts (6); Tobino et al. (7) found that cysts num-bered from 29 to 407 in a study of 12 patients with BHD.
Bilateral cysts are typical (6,7). In distinction to LCH and LAM, cysts predominate at the lung bases; 85% and 87% of cysts in two studies were in the lower lungs (6,7), and cysts tend to be largest in this location. Tobino et al. (7) found the majority of cysts to be visible in the medial (58%) rather than lateral (27%) lower lungs (Figs. 6-13 and 19-22).
Cysts often contact a pleural surface, and in some cases may appear to be within or related to a fissure. Tobino et al. (7) found that 40.5% of cysts were subpleural in location. Cysts can be round, oval, lenticular, or irregu-lar in shape; in one study, 77% of cysts were irregular in shape, rather than round or oval. Cysts in the medial lower lobes, those subpleural in location, and those with irregular shapes tended to be larger than cysts in other locations (p = 0.001) (7).
Differences between the appearance and distribution of cysts associated with BHD and LAM have recently been reported. In a study of 14 patients with BHD syn-drome and 52 with LAM, the extent, number, size, and circularity of cysts were calculated, and the distribution of cysts within six separate lung zones was also evalu-ated (125). As compared to LAM, BHD showed fewer cysts, which were more limited in extent, and more of-ten had non-rounded shapes. Cysts associated with BHD more frequently involved the lower and medial lung, and were larger than those seen in patients with LAM (both p < 0.001). A family history of pneumothorax also pre-dicted BHD (p < 0.001) (125).
R E F E R E N C E S 1. Hansell DM, Bankier AA, MacMahon H, et al. Fleischner
Society: glossary of terms for thoracic imaging. Radiology 2008;246:697–722.
2. Clarke BE. Cystic lung disease. J Clin Pathol 2013;66:904–908. 3. Seaman DM, Meyer CA, Gilman MD, et al. Diffuse cys-
tic lung disease at high-resolution CT. AJR Am J Roentgenol 2011;196:1305–1311.
4. Howarth DM, Gilchrist GS, Mullan BP, et al. Langerhans cell his-tiocytosis: diagnosis, natural history, management, and outcome. Cancer 1999;85:2278–2290.
aMost common findings.bFindings most helpful in differential diagnosis.
TABLE 19-5 HRCT Findings in Birt-Hogg-Dubé Syndrome
Thin-walled lung cysts, usually rounda, ovala, lenticulara,b or irregulara in shape
Lower lung predominance of cystsa,b
Medial predominancea,b
Subpleural (and fissural) cysts commona,b
Cysts often large (and less uniform in size than LAM)a,b
Pneumothoraxa,b

CHAPTER 19 Cystic Lung Diseases 515
55. Soler P, Bergeron A, Kambouchner M, et al. Is high-resolution computed tomography a reliable tool to predict the histopatho-logical activity of pulmonary Langerhans cell histiocytosis? Am J Respir Crit Care Med 2000;162:264–270.
56. Diette GB, Scatarige JC, Haponik EF, et al. Do high-resolution CT findings of usual interstitial pneumonitis obviate lung biopsy? Views of pulmonologists. Respiration 2005;72:134–141.
57. Gruden JF, Webb WR, Naidich DP, et al. Multinodular disease: anatomic localization at thin-section CT—multireader evaluation of a simple algorithm. Radiology 1999;210:711–720.
58. Koyama M, Johkoh T, Honda O, et al. Chronic cystic lung disease: diagnostic accuracy of high-resolution CT in 92 patients. AJR Am J Roentgenol 2003;180:827–835.
59. Ichikawa Y, Kinoshita M, Koga T, et al. Lung cyst formation in Lymphocytic interstitial pneumonia: CT features. J Comput Assist Tomogr 1994;18:745–748.
60. Johkoh T, Ichikado K, Akira M, et al. Lymphocytic intersti-tial pneumonia: follow-up CT findings in 14 patients. J Thorac Imaging 2000;15:162–167.
61. Johkoh T, Müller NL, Pickford HA, et al. Lymphocytic intersti-tial pneumonia: thin-section CT findings in 22 patients. Radiology 1999;212:567–572.
62. Bonelli FS, Hartman TE, Swensen SJ, et al. Accuracy of high-resolution CT in diagnosing lung diseases. AJR Am J Roentgenol 1998;170:1507–1512.
63. Chu SC, Horiba K, Usuki J, et al. Comprehensive evalua-tion of 35 patients with lymphangioleiomyomatosis. Chest 1999;115:1041–1052.
64. Colby TV, Carrington CB. Infiltrative lung disease. In: Thurlbeck WM, ed. Pathology of the lung. New York, NY: Thieme Medical; 1988:425–518.
65. Corrin B, Liebow AA, Friedman PJ. Pulmonary lymphangiomyo-matosis. A review. Am J Pathol 1975;79:348–382.
66. McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest 2008;133:507–516.
67. Mavroudi M, Zarogoulidis P, Katsikogiannis N, et al. Lymphangi-oleiomyomatosis: current and future. J Thorac Dis 2013;5:74–79.
68. Hohman DW, Noghrehkar D, Ratnayake S. Lymphangioleiomyo-matosis: a review. Eur J Intern Med 2008;19:319–324.
69. Beck GJ, Sullivan EJ, Stoller JK, et al. Lymphangioleiomyomatosis: new insights. Am J Respir Crit Care Med 1997;156:670.
70. Kalassian KG, Doyle R, Kao P, et al. Lymphangioleiomyomatosis: new insights. Am J Respir Crit Care Med 1997;155:1183–1186.
71. Sullivan EJ. Lymphangioleiomyomatosis: a review. Chest 1998;114:1689–1703.
72. Sobonya RE, Quan SF, Fleishman JS. Pulmonary lymphangioleio-myomatosis: quantitative analysis of lesions producing airflow limitation. Hum Pathol 1985;16:1122–1128.
73. Guinee D, Singh R, Azumi N, et al. Multifocal micronodular pneu-mocyte hyperplasia: a distinctive pulmonary manifestation of tu-berous sclerosis. Mod Pathol 1995;8:902–906.
74. Lantuejoul S, Ferretti G, Negoescu A, et al. Multifocal alveolar hy-perplasia associated with lymphangioleiomyomatosis in tuberous sclerosis. Histopathology 1997;30:570–575.
75. Flieder DB, Travis WD. Clear cell “sugar” tumor of the lung: asso-ciation with lymphangioleiomyomatosis and multifocal micronod-ular pneumocyte hyperplasia in a patient with tuberous sclerosis. Am J Surg Pathol 1997;21:1242–1247.
76. McCormack F, Brody A, Meyer C, et al. Pulmonary cysts con-sistent with lymphangioleiomyomatosis are common in women with tuberous sclerosis: genetic and radiographic analysis. Chest 2002;121:61S.
77. Cohen MM, Pollock-BarZiv S, Johnson SR. Emerging clinical pic-ture of lymphangioleiomyomatosis. Thorax 2005;60:875–879.
78. Johnson SR, Cordier JF, Lazor R, et al. European Respiratory So-ciety guidelines for the diagnosis and management of lymphangi-oleiomyomatosis. Eur Respir J 2010;35:14–26.
79. Adriaensen ME, Schaefer-Prokop CM, Duyndam DA, et al. Radiolog-ical evidence of lymphangioleiomyomatosis in female and male pa-tients with tuberous sclerosis complex. Clin Radiol 2011;66:625–628.
30. Hamada K, Teramoto S, Narita N, et al. Pulmonary veno-occlusive disease in pulmonary Langerhans’ cell granulomatosis. Eur Respir J 2000;15:421–423.
31. Gaensler EA, Carrington CB. Open biopsy for chronic diffuse infil-trative lung disease: clinical, roentgenographic, and physiological correlations in 502 patients. Ann Thorac Surg 1980;30:411–426.
32. Basset F, Corrin B, Spencer H, et al. Pulmonary histiocytosis X. Am Rev Respir Dis 1978;118:811–820.
33. Friedman PJ, Liebow AA, Sokoloff J. Eosinophilic granuloma of lung. Clinical aspects of primary histiocytosis in the adult. Medi-cine (Baltimore) 1981;60:385–396.
34. Hance AJ, Basset F, Saumon G, et al. Smoking and interstitial lung dis-ease. The effect of cigarette smoking on the incidence of pulmonary histiocytosis X and sarcoidosis. Ann N Y Acad Sci 1986;465:643–656.
35. Lacronique J, Roth C, Battesti JP, et al. Chest radiological features of pulmonary histiocytosis X: a report based on 50 adult cases. Thorax 1982;37:104–109.
36. Lewis JG. Eosinophilic granuloma and its variants with special reference to lung involvement. A report of 12 patients. Q J Med 1964;33:337–359.
37. Winterbauer RH, Dreis DF, Jolly PC. Clinical correlation. In: Dail DH, Hammar SP, eds. Pulmonary pathology. New York, NY: Springer-Verlag; 1988:1148–1151.
38. Colby TV, Lombard C. Histiocytosis X in the lung. Hum Pathol 1983;14:847–856.
39. Marcy TW, Reynolds HY. Pulmonary histiocytosis X. Lung 1985;163:129–150.
40. Tazi A, Soler P, Hance AJ. Adult pulmonary Langerhans’ cell his-tiocytosis. Thorax 2000;55:405–416.
41. Tazi A, Montcelly L, Bergeron A, et al. Relapsing nodular lesions in the course of adult pulmonary Langerhans cell histiocytosis. Am J Respir Crit Care Med 1998;157:2007–2010.
42. Prophet D. Primary pulmonary histiocytosis-X. Clin Chest Med 1982;3:643–653.
43. Brauner MW, Grenier P, Mouelhi MM, et al. Pulmonary histiocytosis X: evaluation with high-resolution CT. Radiology 1989;172:255–258.
44. Brauner MW, Grenier P, Tijani K, et al. Pulmonary Langerhans cell histiocytosis: evolution of lesions on CT scans. Radiology 1997;204:497–502.
45. Galvin JR, Franks JT. Smoking-related lung disease. J Thorac Im-aging 2009;24:274–284.
46. Giron J, Tawil A, Trussard V, et al. Contribution of high resolution x-ray computed tomography to the diagnosis of pulmonary his-tiocytosis X. Apropos of 12 cases [Article in French]. Ann Radiol (Paris) 1990;33:31–38.
47. Kulwiec EL, Lynch DA, Aguayo SM, et al. Imaging of pulmonary histiocytosis X. Radiographics 1992;12:515–526.
48. Moore AD, Godwin JD, Müller NL, et al. Pulmonary histiocyto-sis X: comparison of radiographic and CT findings. Radiology 1989;172:249–254.
49. Grenier P, Chevret S, Beigelman C, et al. Chronic diffuse infiltra-tive lung disease: determination of the diagnostic value of clinical data, chest radiography, and CT and Bayesian analysis. Radiology 1994;191:383–390.
50. Grenier P, Valeyre D, Cluzel P, et al. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991;179:123–132.
51. Taylor JR, Ryu J, Colby TV, et al. Lymphangioleiomyomatosis. Clinical course in 32 patients. N Engl J Med 1990;323:1254–1260.
52. Müller NL, Miller RR. Computed tomography of chronic diffuse infiltrative lung disease. Part 2. Am Rev Respir Dis 1990;142:1440–1448.
53. Stern EJ, Webb WR, Golden JA, et al. Cystic lung disease asso-ciated with eosinophilic granuloma and tuberous sclerosis: air trapping at dynamic ultrafast high-resolution CT. Radiology 1992;182:325–329.
54. Kelkel E, Pison C, Brambilla E, et al. Value of high resolution to-modensitometry in pulmonary histiocytosis X. Radiological, clini-cal and functional correlations [Article in French]. Rev Mal Respir 1992;9:307–311.

section i i i High-Resolution CT Diagnosis of Diffuse Lung Disease516
in women with tuberous sclerosis. Am J Respir Crit Care Med 2001;164:661–668.
104. Hoshika Y, Hamamoto T, Sato K, et al. Prevalence and clinical fea-tures of lymphedema in patients with lymphangioleiomyomatosis. Respir Med 2013;107:1253–1259.
105. Avila NA, Dwyer AJ, Rabel A, et al. CT of pleural abnormalities in lymphangioleiomyomatosis and comparison of pleural find-ings after different types of pleurodesis. AJR Am J Roentgenol 2006;186:1007–1012.
106. Kitaichi M, Nishimura K, Itoh H, et al. Pulmonary lymphangioleiomy-omatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med 1995;151:527–533.
107. Crausman RS, Jennings CA, Mortenson RL, et al. Lymphangi-oleiomyomatosis: the pathophysiology of diminished exercise ca-pacity. Am J Respir Crit Care Med 1996;153:1368–1376.
108. Crausman RS, Lynch DA, Mortenson RL, et al. Quantitative CT predicts the severity of physiologic dysfunction in patients with lymphangioleiomyomatosis. Chest 1996;109:131–137.
109. Yao J, Taveira-DaSilva AM, Colby TV, et al. CT grading of lung disease in lymphangioleiomyomatosis. AJR Am J Roentgenol 2012; 199:787–793.
110. Ryu JH, Doerr CH, Fisher SD, et al. Chylothorax in lymphangi-oleiomyomatosis. Chest 2003;123:623–627.
111. Costello LC, Hartman TE, Ryu JH. High frequency of pulmonary lymphangioleiomyomatosis in women with tuberous sclerosis complex. Mayo Clin Proc 2000;75:591–594.
112. Hancock E, Tomkins S, Sampson J, et al. Lymphangioleiomyoma-tosis and tuberous sclerosis. Respir Med 2002;96:7–13.
113. Moss J, Avila NA, Barnes PM, et al. Prevalence and clinical character-istics of lymphangioleiomyomatosis (LAM) in patients with tuberous sclerosis complex. Am J Respir Crit Care Med 2001;164:669–671.
114. Cudzilo CJ, Szczesniak RD, Brody AS, et al. Lymphangioleio-myomatosis screening in women with tuberous sclerosis. Chest 2013;144:578–585.
115. Collins J, Müller NL, Kazerooni EA, et al. Lung transplantation for lymphangioleiomyomatosis: role of imaging in the assessment of complications related to the underlying disease. Radiology 1999;210:325–332.
116. Adams H, Brack T, Kestenholz P, et al. Diffuse idiopathic neuro-endocrine cell hyperplasia causing severe airway obstruction in a patient with a carcinoid tumor. Respiration 2006;73:690–693.
117. Franquet T, Hansell DM, Senbanjo T, et al. Lung cysts in sub-acute hypersensitivity pneumonitis. J Comput Assist Tomogr 2003;27:475–478.
118. Davies CW, Juniper MC, Gray W, et al. Lymphoid interstitial pneu-monitis associated with common variable hypogammaglobulinae-mia treated with cyclosporin A. Thorax 2000;55:88–90.
119. Lee KH, Lee JS, Lynch DA, et al. The radiologic differential diag-nosis of diffuse lung diseases characterized by multiple cysts or cavities. J Comput Assist Tomogr 2002;26:5–12.
120. Furuya M, Tanaka R, Koga S, et al. Pulmonary cysts of Birt-Hogg-Dubé syndrome: a clinicopathologic and immunohistochemical study of 9 families. Am J Surg Pathol 2012;36:589–600.
121. Gupta N, Seyama K, McCormack FX. Pulmonary manifestations of Birt-Hogg-Dubé syndrome. Fam Cancer 2013;12:387–396.
122. Menko FH, van Steensel MA, Giraud S, et al.; European BHD Consortium. Birt-Hogg-Dubé syndrome: diagnosis and manage-ment. Lancet Oncol 2009;10:1199–1206.
123. Furuya M, Nakatani Y. Birt-Hogg-Dubé syndrome: clinicopatho-logic features of the lung. J Clin Pathol 2013;66:178–186.
124. Toro JR, Pautler SE, Stewart L, et al. Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med 2007;175: 1044–1053.
125. Tobino K, Hirai T, Johkoh T, et al. Differentiation between Birt-Hogg-Dubé syndrome and lymphangioleiomyomatosis: quantita-tive analysis of pulmonary cysts on computed tomography of the chest in 66 females. Eur J Radiol 2012;81:1340–1346.
80. Ryu JH, Sykes A-M, Lee AS, et al. Cystic lung disease is not un-common in men with tuberous sclerosis complex. Respir Med 2012;106:1586–1590.
81. Ryu J, Moss J, Beck GJ, et al. The NHLBI lymphangioleiomyoma-tosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med 2006;173:105–111.
82. Avila NA, Kelly JA, Dwyer AJ, et al. Lymphangioleiomyomato-sis: correlation of qualitative and quantitative thin-section CT with pulmonary function tests and assessment of dependence on pleurodesis. Radiology 2002;223:189–197.
83. Carrington CB, Cugell DW, Gaensler EA, et al. Lymphangioleio-myomatosis. Physiologic-pathologic-radiologic correlations. Am Rev Respir Dis 1977;116:977–995.
84. Avila NA, Kelly JA, Chu SC, et al. Lymphangioleiomyomatosis: ab-dominopelvic CT and US findings. Radiology 2000;216:147–153.
85. Taveira-Dasilva AM, Stylianou MP, Hedin CJ, et al. Decline in lung function in patients with lymphangioleiomyomatosis treated with or without progesterone. Chest 2004;126:1867–1874.
86. Nine JS, Yousem SA, Paradis IL, et al. Lymphangioleiomyomato-sis: recurrence after lung transplantation. J Heart Lung Transplant 1994;13:714–719.
87. Cai X, Pacheco-Rodriguez G, Haughey M, et al. Sirolimus de-creases circulating lymphangioleiomyomatosis cells in patients with lymphangioleiomyomatosis. Chest 2014;145:108–112.
88. McCormack FX, Inoue Y, Moss J, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med 2011;364:1595–1606.
89. Taveira-Dasilva AM, Moss J. Optimizing treatments for lymphan-gioleiomyomatosis. Expert Rev Respir Med 2012;6:267–276.
90. Ando K, Kurihara M, Kataoka H, et al. The efficacy and safety of low-dose sirolimus for treatment of lymphangioleiomyomatosis. Respir Investig 2013;51:174–183.
91. Silverstein EF, Ellis K, Wolff M, et al. Pulmonary lymphan-giomyomatosis. Am J Roentgenol Radium Ther Nucl Med 1974;120:832–850.
92. Aberle DR, Hansell DM, Brown K, et al. Lymphangiomyomato-sis: CT, chest radiographic, and functional correlations. Radiology 1990;176:381–387.
93. Rationale and design of the National Emphysema Treatment Trial (NETT): a prospective randomized trial of lung volume reduction surgery. J Thorac Cardiovasc Surg 1999;118:518–528.
94. Kirchner J, Stein A, Viel K, et al. Pulmonary lymphangioleiomyo-matosis: high-resolution CT findings. Eur Radiol 1999;9:49–54.
95. Kullnig P, Melzer G, Smolle-Juttner F. High-resolution computer tomographie des thorax bei lymphangioleiomyomatose and tube-roser sklerose. Rofo 1989;151:32–35.
96. Lenoir S, Grenier P, Brauner MW, et al. Pulmonary lymphangio-myomatosis and tuberous sclerosis: comparison of radiographic and thin-section CT findings. Radiology 1990;175:329–334.
97. Merchant RN, Pearson MG, Rankin RN, et al. Computerized to-mography in the diagnosis of lymphangioleiomyomatosis. Am Rev Respir Dis 1985;131:295–297.
98. Müller NL, Chiles C, Kullnig P. Pulmonary lymphangiomyomato-sis: correlation of CT with radiographic and functional findings. Radiology 1990;175:335–339.
99. Rappaport DC, Weisbrod GL, Herman SJ, et al. Pulmonary lymphangioleiomyomatosis: high-resolution CT findings in four cases. AJR Am J Roentgenol 1989;152:961–964.
100. Sherrier RH, Chiles C, Roggli V. Pulmonary lymphangioleiomyo-matosis: CT findings. AJR Am J Roentgenol 1989;153:937–940.
101. Templeton PA, McLoud TC, Müller NL, et al. Pulmonary lymphan-gioleiomyomatosis: CT and pathologic findings. J Comput Assist Tomogr 1989;13:54–57.
102. Ristagno RL, Biddinger PW, Pina EM, et al. Multifocal micronod-ular pneumocyte hyperplasia in tuberous sclerosis. AJR Am J Roentgenol 2005;184:S37–S39.
103. Franz DN, Brody A, Meyer C, et al. Mutational and radio-graphic analysis of pulmonary disease consistent with lymphan-gioleiomyomatosis and micronodular pneumocyte hyperplasia





























