Embed Size (px)
Citation preview

CRISPR/Cas9 system implementation in Torulaspora delbrueckii
Master thesis by Lev Koval
Supervisors:
César Simões da Fonseca
Lei Yang
AALBORG UNIVERSITY COPENHAGEN
SECTION OF SUSTAINABLE BIOTECHNOLOGY
June 2019

2
Semester: 9-10th semester
Semester theme: Master thesis
Title: Crispr/Cas9 system implementation in Torulaspora delbrueckii
Project period: 03.09.2018-10.06.2019
Supervisors: César Simões da Fonseca
Lei Yang
Group name: BBIO10
Group member: Lev Koval
Abstract: For the long time Saccharomyces cerevisiae has been used as the main
microorganism which in industrial and food biotechnology. It was
proven that it is an efficient ethanol producer and can tolerate high
amounts of ethanol produced during fermentative processes. S.
cerevisiae, as well as the other yeasts, also produces fusel alcohols
such as: 2-phenylethanol, tryptophol and tyrosol. Importance of fusel
alcohols is undeniable since they have a large application in different
industries. Torulaspora delbrueckii is the perfect candidate for the
2-phenylethanol production since it can produce the same or even
more amounts of 2-phenylethanol under certain conditions. Numerous
genetic manipulations were applied in S. cerevisiae in order to
increase 2-phenylethanol production. Mostly genes involved in
Ehrlich pathway were either disrupted or overexpressed from which
overexpression of ARO10 and disruption of PDC genes were
highlighted. On the other hand, T. delbrueckii could serve as a
potential ethanol producing organism if specific modifications such as
RIM15 gene disruption are considered, where RIM15 is the gene
responsible for ethanol production inhibition. However, unlike S.
cerevisiae, T. delbrueckii lacks of sufficient genetic manipulation
tools for efficient metabolic engineering. During this project it was
aimed to construct a toolkit with the possibility of utilization of
CRISPR/Cas9 system to obtain desired modifications of PDC gene
disruption and ARO10 overexpression for better 2-phenylethanol
production in one case and RIM15 disruption to increase ethanol
production in another case. For this purpose, EasyClone - Markerfree
toolkit constructed for S. cerevisiae was used as a model. It was also
aimed to investigate the requirement of integration cassette
homologous sites size for successful integration of the gene of interest
into desired genomic location. For this purpose 3 integration cassettes
containing 50, 200 and 500 base pairs of homology to URA3 gene in
T. delbrueckii were constructed. It was shown that the highest chance
of integrating the gene of interest requires 500 base pairs of homology
to the desired genomic location. Moreover, it was shown that
supposedly 70 base pair homology and/or low concentration of
supplied integration cassette was not enough to introduce ARO10 and
disruption cassette to desired location for PDC gene disruption.
Further investigation is required in order to understand how and where
RIM15 integration cassette containing 200 base pair homology was
integrated into genome.

3

4
Acknowledgement: I would like to thank my supervisors César Simões da Fonseca and Lei Yang, for help, guidance and support during the whole project period.

5
Table of Contents
1. Introduction ............................................................................................................................ 7
1.1 Yeasts and flavor and fragrance active compounds ...................................................................... 8
1.2 Ehrlich pathway overview ........................................................................................................... 10
1.3 Production and metabolism of 2-phenylethanol .......................................................................... 13
1.3.1 Influence of the key enzymes in the Erchlich pathway .................................................................................. 14
1.4 Improvement of the alcohol fermentation performance in yeast by gene deletions .................... 15
1.5 Tools for genetic engineering of conventional and non-conventional yeasts ............................... 16
1.5.1 CRISPR/Cas9 system .................................................................................................................................... 16 1.5.2 Selection system and relevance of the promoter efficiency on genes expression .......................................... 18 1.5.3 Genetic techniques and transformation methods ........................................................................................... 21
1.6 Aim and approaches .................................................................................................................... 23
2. Materials and methods .......................................................................................................... 25
2.1 Strain and cultivation media ....................................................................................................... 25
2.2 Selection of the genes and gRNA site selection ............................................................................ 25
2.2.1 Intergenic regions .......................................................................................................................................... 26
2.3 Preparation of the primers, plasmids and integration cassettes .................................................. 26
2.4 Preparation of the T. delbrueckii frozen competent cells ............................................................. 29
2.5 Yeast transformation ................................................................................................................... 30
2.5.1 Antibiotic resistance test ................................................................................................................................ 30 2.5.2 Introduction of integration cassettes for PDC disruption ............................................................................... 31 2.5.3 Introduction of integration cassettes for RIM15 disruption ........................................................................... 32 2.5.4 Introduction of integration cassettes for URA3 disruption ............................................................................. 33
3. Results .................................................................................................................................. 34
3.1 Implementation of the transformation protocol in T. delbrueckii ................................................ 34
3.2 Test of the required homologous sites by disruption of URA3 .................................................... 35
3.3 Disruption of RIM15 .................................................................................................................... 38
3.4 Detection of the PDC gene in T. delbrueckii and its disruption .................................................... 39

6
3.5 Construction of integration plasmids targeting IV, V and VI chromosomal regions .................. 40
4. Discussion ............................................................................................................................ 43
4.1 Utilized transformation methodology .......................................................................................... 44
4.2 Influence on the antibiotics on selection and yeast morphology .................................................. 44
4.3 Establishing the requirements for the size of homologous sites of the integration cassette by
URA3 disruption ............................................................................................................................... 45
4.4 RIM15 disruption ........................................................................................................................ 46
4.5 PDC disruption ............................................................................................................................ 47
4.6 Construction of the integrative plasmids ..................................................................................... 48
4.7 Further perspectives .................................................................................................................... 49
5. Conclusion ........................................................................................................................... 50
Reference list ............................................................................................................................ 51

7
1. Introduction
For the past decades Saccharomyces cerevisiae has been one of the preferred microorganisms used
in food and industrial biotechnology because it is an efficient ethanol producer and highly tolerant to
this fermentation product. Along with the ethanol, S. cerevisiae, as well as other yeasts, produces
fusel alcohols: tryptophol, 2-phenylethanol and tyrosol. Fusel alcohols have a large application in
different industries. For example, 2-phenylethanol is widely used in the food, cosmetics and
perfumery industries because of its rose-like fragrance. Since 2-phenylethanol can be produced by
conventional yeast (S. cerevisiae) and non-conventional yeasts (e.g. Kluyveromyces marxianus,
Starmerella bacillaris, Metschnikowia pulcherrima and Torulaspora delbrueckii), its microbial
production could be considered an alternative solution instead chemical synthesis or extraction from
essential oils of plant. Several genetic manipulations on the genes connected to 2-phenylethanol
synthesis were applied in S. cerevisiae in order to increase its production. From those genetic
manipulations disruption of pyruvate decarboxylases (PDC) with the overexpression of ARO10 gene
should be highlighted, since above genes are taking the major part in 2-phenylethanol metabolism. It
should be remarked that from non-conventional yeasts which are taking part in flavor compounds
metabolism the case of genetic manipulations for 2-phenylethanol production improvement was
present only in K. marxianus (Kim, et al., 2014). To fully understand how to increase flavor active
compounds production, the inside look onto the gene involvement in their metabolism is required,
thus the importance of Ehrlich pathway investigation is undoubtful.
Another important event which is taking part in S. cerevisiae sake strain during the ethanol
fermentation is that this yeast strain is capable of producing high amounts of ethanol. This strain is
capable of doing so because of the loss-function mutation in RIM15 gene which is encoding Great-
wall family protein kinase. Disruption of this gene in the non-conventional yeast (including T.
delbrueckii) could lead to the higher ethanol production.
All the above-mentioned genetic manipulations require convenient tool with which help cells can be
modified fast and efficiently. EasyClone – Markerfree toolkit is the perfect option since it utilizes
CRISPR/Cas9 plasmid system together with the easy possibility of the removal of selection marker.
However, this toolkit is designed and tested only in S. cerevisiae and it is required to design this
plasmid-based system for non-conventional yeast with the usage of the above-mentioned toolkit as a
model.

8
1.1 Yeasts and flavor and fragrance active compounds
As it was mentioned above, S. cerevisiae was involved in alcoholic fermentations for a long time, but
with the undeniable ability of it to produce ethanol it is capable of producing another, not least
important compounds for the industry – flavor and fragrance active compounds. Flavor active
compounds group which includes aldehydes, higher alcohols, esters, organic acids, carbonyl
compounds and organic sulfides has a serious influence on the product quality (Hazelwood, et al.,
2008). Formation of aromatic alcohols, also known as fusel alcohols, is unavoidable if yeast is taking
part in the food fermentation. Moreover, if the high concentrations of fusel alcohols are having an
impact on off-flavors, in low concentrations those compounds are making a tremendous contribution
in aromas and flavours of fermented bavareges and food, thus some of the companies are using it as
their own inimitible fingerprint in the bevarages and food (Hazelwood, et al., 2008). The beer industry
shows the highest interest in the developing production of flavor active compounds in which the final
taste of the beer is influenced in several hundrets of flavor active compunds, produced on the each
step of brewing (Pires, et al., 2014). For each type of beer the specific yeast strain should be chosen
in order to obtain desired beer. In addition, esters and higher alcohols should be kept in a perfect
balance, under certain threshold number in order to not ruin the flavor of the final product. At the
same time as balancing the concentrations of flavor compound formation, there is an interest in yeast
Figure 1 Aromatic alcohols production in the conventional and non-conventional yeast in low-glucose must. 1x and 5x represent the 1- and 5-fold increase in aromatic alcohol precursors supplied to the media (González, et al., 2018).

9
modification to increase one specific compound production, as for example 2-phenylethanol. Those
attempts would be discussed in further sections.
An alternative suitable solution of producing flavor compounds is the application of non-conventional
yeast in the industry, which are perfect candidates for the industrial production and are capable to
produce the same amount or even more of above mentioned compounds under certain conditions
(Figure 1) (González, et al., 2018). The worthy examples of non-conventional yeasts following those
specifications are: T. delbrueckii and K. marxianus. In case of K. marxianus suitable characteristics
for application in industry such as: temperature tolerance up to 52 degrees, short doubling time, high
acid tolerance and usage of various carbon sources including xylose and lactose (Kim, et al., 2014).
The bottleneck in usage and improvement of K. marxianus in industry is that unlike
S. cerevisiae it lacks the high variety of genetic manipulation tools. On the other hand, T. delbruecki
is mostly used in the wine making industry, since it has the ability of producing the certain flavor
compounds (González, et al., 2018). But the bottleneck in usage and improvement of T. delbruecki is
the same as for K. marxianus – lack of high variety of genetic manipulation tools.
In overall perspective, to improve production of the flavor active compounds through genetic
manipulations, first, their metabolism should be understood which can be done by investigating
Ehrlich pathway more in details.

10
1.2 Ehrlich pathway overview
The Ehrlich pathway was established by the German biochemist Felix Ehrlich in the early 20th century
who was working with the alcoholic fermentations in yeast. The first key stone in establishing the
Ehrlich pathway was the discovery of
structural similarities of leucine and
isoamyl alcohol after isolation and
characterization of isoleucine by Ehrlich
himself. This finding led Ehrlich to
investigate if isoamyl alcohol formation is
linked to presence of amino acids via
supplementing yeast fermentation
mixtures with either isoleucine or leucine
what led to the increasing amyl alcohol
production (Hazelwood, et al., 2008).
Several years after the general scheme for
the pathway was suggested (Figure 2) and
is used until today.
Ehrlich pathway can be generally
described as convertion of aromatic amino
acids (phenylalanine, tryptophan, and
tyrosine), sulfur containing amino acids (methionine) and branch-chained amino acids (valine,
isoleucine and leucine) to specific fusel alcohols or fusel acids. Table 1 represents products obtained
from the certain amino acids. It is devided into 4 main reaction steps: Transamination,
decarboxylation, oxidation and export (the reactions were examined in S. cerevisiae).
In the transamination, the transfer of amines between the amino acids and their respective a-keto
acids, while using 2-oxoglutarate or glutamate as donor/acceptor is catalized by four main enymes:
Figure 2: The Ehrlich pathway representation which includes four major steps: Transamination, decarboxylation, oxidation and export. The list of the enzymes regulating each step of the pathway are represented as well (Hazelwood, et al., 2008).

11
Tabl
e 1
Repr
esen
tatio
n of
the
prod
ucts
obta
ined
from
the
spec
ific
amin
o ac
ids (
Haz
elwo
od, e
t al.,
200
8)

12
BAT1, BAT2, ARO8 and ARO9. BAT1 is known as brached-chained amino acid transfarase and BAT2
is cytosolic enzyme, they are encrypted enzymes and take part in branched-chain amino acid
transamination (Pires, et al. 2014). First, ARO8 and ARO9 were described as amino acid
aminotransferases I and II. Later, it was shown that ARO8 and ARO9 have wider substrate specificities
as for example ARO8 demonstrated in vitro activity with phenylpyruvate as amino acceptor
(Urrestarazu, et al. 1998).
In the decarboxylation step, irreversibale reaction of decarboxylation of a-keto acids occurs leading
to formation of particular fusel aldehyde. This reaction is conducted by five main genes: pyruvate
decarboxylases 1, 5 and 6 (PDC1, PDC5 and PDC6), amino acid aminotransferase 10 (ARO10) and
thiamine phosphate-dependent decarboxylase 3 (THI3).
Typically, the final step of Ehrlich pathway is reduction of the fusel aldehyde to their respective
alcohol and the reaction is regulated by alcohol dehydrogenases. In S. cerevisiae, any of the alcohol
dehydrogenases (ADH1-5) or formaldehyde dehydrogenase (SFA1) are capable to catalyze the
conversion of fusel aldehyde to respective fusel alcohols (Pires, et al. 2014). However, it was shown
that the balance between the oxidation and reduction of fusel aldehydes depends on cultivation
conditions. For example: under oxygen and glucose limited conditions with the presence of various
amino acids, mostly fusel acids are formed (Hazelwood, et al., 2008). The process of fusel alcohol
export to the culture medium is still remains unknown. Nevertheless, in the export of fusel acids, at
least one of the membrane transporters in involved – PDR12.
Even though functions several above listed enzymes were described, those studies did not consider
their functions in the frames of overall Ehrlich pathway. One of the regulatory examples which was
described is the activity of ARO80. It was identified that ARO80 transcriptional activator induces
ARO9 and ARO10 genes by phenylalanine, tryptophan and tyrosine (Iraqui, et al., 1998). It was also
shown that nitrogen catabolite repression is mediated by GATA transcription factors, which include
GLN3 and GAT1 activators together with DAL80 and GZF3 repressors which are activated under
nitrogen limited conditions (Hofman-Bang, 1999). It means, that ARO80 requires commotion of the
activators in order to bind to ARO9-10 promoter region and not only presence of aromatic amino
acids.

13
1.3 Production and metabolism of 2-phenylethanol
2-phenylethanol or 2-PE is a volitile compound which, as it was mentioned above, is widely used in
several industries. There is no doubt in the importance of 2-PE production as its global production
reaches 10,000 metric tons per year which makes it one of the most valuable volitile compounds.
(Hua, et al., 2011). The main methodology of obtaining 2-phenylethanol for the major part of the
industry is derived from the chemical synthesis, but issue with this method is that a lot of toxic waste
is resulted afterwards (Chreptowicz, et al., 2016). Another existing method which is considered as
more environmentally friendly is extraction of 2-PE from the essential oils of the plants (e.g. rose
petals), however, this method is considered costly and inefficient (Chen, et al, 2017). It is undeniable
that as a result yeast became a crucial microorganism for 2-phenylethanol production (Garavaglia, et.
al 2007). Moreover, more research is being focused on genetic engineering of conventional and
non-concention yeast in order to increase 2-PE production. One of the stricking features aspects of
this research is the importance of understanding metabolism of this compound in the yeasts. Figure 3
crudely represents 2-phenylethanol metabolism and key enzymes which are taking part in this
process. First of all, fundamental compound for 2-PE production is L-phenylalanine, which is
aromatic alpha amino acid and it is derived from L-alanine biosynthesis (conversion of pyruvate to
L-alanine) with the activity of ARO9 or from the L-phenylalanine biosynthesis, where
phosphoenolpyruvate (compound derived from the glycolysis) is converted through several reactions
to chorismate which is converted to phenylpyruvate. Phenylpyruvate is later converted to
L-phenylalanine with the activities of either ARO9 or ARO8. 2-phenylethanol biosynthesis, which is
basically part of Ehrlichs pathway, starts with the L-phenylalanine synthesis. Then it is converted to
phenylpyruvate trough the activity of either ARO8 or ARO9. Phenylpyruvate is then converted to
phenylacetaldehyde with the activiry of ARO10 and pyruvate decarboxylases 1,5 and 6. The final step
is convertion of phenylacetaldehyde to 2-phenylethanol which is regulated by the hydrogen transfer
by NADH.

14
Figure 3: Simplified representation of 2-phenylethanol metabolism with the representation of the crucial steps, key enzymes and
co-factor activities. Glycolysis is marked with the green solid line, L-phenylalanine biosynthesis with the yellow dashed line and the
2-phenylethanol biosynthesis is marked by the violet dashed line. Important note: 2-phenylethanol is not produced directly from the
reaction where phenylpyruvate is obtained from chorismate. First, L-phenylalanine should be produced, for 2-phenylethanol
biosynthesis.
It should also be remarked that 2-phenylethanol could be metabolized trough the different pathway
because phenylpyruvate is reversibly converted to L-phenylalanine by the catalysis of transaminase.
It leads the generation of 2-phenylethanol trough phenylalanine conversion to 2-phenylacetamide
(Shen, et al., 2016).
1.3.1 Influence of the key enzymes in the Erchlich pathway
Several key enzymes could be highlighted in the Ehrlich pathway that picked the attention of the
researches in the sense of genetic modifications for increasing the yield of 2-PE production. Enzyme
such as ARO10 has high identity in between conventional and non-conventional yeast species and is
a worthy target for genetic manipulations (Bolat, et al., 2013). ARO9 and ARO8 are also suitable
candidates for the genetic manipulations, since they are taking part in the major process of converting
L-phenylalanine to phenylpyruvate. Moreover, ARO8, ARO9 and ARO10 were identified as the main

15
active ones present in the Ehrlich pathway, thus highlighting their importance to fusel alchohol
methabolism (Hazelwood, et al., 2008). On the other hand, PDC1, 5 and 6 disruption can serve as the
another tool for shifting the metabolism in the favour 2-PE. But it also raises the question if those
genes for S. cerevisiae have high homology to other non-conventional yeasts.
The essential matter is the application of the genetic modification tools and which of them could
efficiently work in the frames of the yeast cells.
1.4 Improvement of the alcohol fermentation performance in yeast by gene deletions
In contrast with the production of flavor compounds, one of the major products for which either
brewing or energy industry is aiming for is ethanol. S. cerevisiae is most commonly used yeast for
ethanol production. On the other hand, non-conventional yeasts, for example in brewry, are mostly
used for production of flavoring compounds (Holt, et al., 2018). Different genetic manipulations are
done in order to increase ethanol content in the fermentation broth. Mostly genetic changes were
performed in S. cerevisiae because it is a well studied organism (Recek, et al., 2018). One of the
options for achieveving high levels of ethanol production is genetic deactivation of the expression of
the enzymes by native gene. Perfect example for gene deletion is deletion of the GAL80, MIG1,
MIG2 genes which are transcriptional regulators of glucose repression (Nevoigt, 2008).
One of the latest options for improving the ethanol fermentation by selecting the desired
modifications were found in the sake production indistry. Sake yeast strains (including S. cerevisiae)
were selected based on their fermentation performance, counting the ability of the balanced
production of flavour and aroma compounds. The common trait of a loss-of-function mutation in
RIM15 is carried in sake yeasts and its relatives (Watanabe, et al., 2018). RIM15 codes protein kinase
of a highly conserved Greatwall-family which is involved in cell proliferation in response to nutriens.
If disruption of RIM15 is performed in non-sake yeasts, the fermentation rate is usually increased,
meaning that this gene is responsible for alcoholic fermentation inhibation (Watanabe, et al., 2018).

16
1.5 Tools for genetic engineering of conventional and non-conventional yeasts Speed, simplicity and efficiency are the major
factors when it comes to the genetic
modifications. The procedures such as
PCR-based gene deletion, which requires
laborious screening and selection processes
after replacement of targeted allele with a
selection marker cassette are highly time
consuming and sometimes even impractical
(Stovicek, et al., 2015). Recently, several
approaches such as application of
transcription activator-like effector nucleases
(TALENs) and Zinc finger nucleases (ZFNs) causing double strand breaks and thus stimulating cell
repair mechanisms (homologous recombination and non-homologous end joining), initiated a
revolution in genome editing in S. cerevisiae. Those mechanisms are based on the specific
endonucleases, as in TALENs, which contain FokI endonuclease cleavage domain and customizable
DNA-binding (Figure 4 B) domain to recognize any sequence of choice (Li, et al., 2011). ZFNs
consist of a DNA binding zinc finger domain and cleavage domain (Figure 4 A).
1.5.1 CRISPR/Cas9 system The alternative genome editing in S. cerevisiae
approach to those stated above is CRISPR-Cas9,
which stands for clusters of regulatory
interspaced palindromic repeats-CRISPR-
associated nuclease 9. On the other hand,
T. delbrueckii lacks of sufficient genetic tools
and usually genes are deleted by the
transformation of the integration cassette
targeting specific gene containing homologous
sites to this gene (Pacheco, et al., 2009). At this point CRISPR/Cas9 approach is only being developed
and eventually it could be adapted in T. delbrueckii (Mans, et al., 2018).
Figure 4 A) Representation of the performance of double stranded break (DSB) by ZFN B) Representation of the performance of DSB by TALENs (Gupta, et al., 2014).
Figure 5 Representation of the performance of double stranded break by CRISPR-Cas9 (Gupta, et al., 2014).

17
The mechanism of CRISPR-Cas9 system is originating from Streptococcus pyogenes which acts like
an immune system from bacteria against viral DNA or RNA. In order to perform double stranded
break in the target sequence of the DNA, Cas9 endonuclease forms a complex of
CRISPR RNA (crRNA) and trans-activating RNA (tracrRNA) (Stovicek, et al., 2015). crRNA is a
20 base pair target sequence (TS) guiding Cas9 to the desired genomic location and tracrRNA serves
as a structural part in the CRISPR-Cas9 complex. Moreover, protospacer adjacent motif (PAM) site,
located downstream the TS and usually consistent of 3 nucleotides is required to perform the double
stranded break (Figure 5). Eventually, crRNA and tracrRNA can be combined into the singe guide
RNA molecule (gRNA) with the target sequence on its end (Stovicek, et al., 2015). Redesigning the
TS located at the end of the gRNA molecule can lead to targeting the desired DNA sequences to
perform double stranded break. It has the advantage compared to the TALENs and ZFNs, since it is
less costly and time consuming to design.
It is important to mention that homologous recombination (HR) plays an important role in
collaboration with CRISPR/Cas9 system because of its efficiency of double-stranded breaks repair in
S. cerevisiae (Jessop-Fabre, et al., 2016). However, in T. delbrueckii success of fragment integration
with the help synergetic work of HR and CRISPR/Cas9 depends on the fragment flanking regions
size, thus efficiency of the transformation of the integration cassette with short flanking regions is
quite low (Pacheco, et al., 2009).
As the matter of fact, the CRISPR-Cas9 system has on- and off-target activities. On-target activity is
when Cas9 complex is cleaving DNA at the desired location due to high homology of gRNA target
sequences. Off-target activities happen are when DNA cleavage occurs at undesired locations due to
Cas9 being guided by the gRNA, which target sequences have high homology to unintended genomic
locations (Kleinstiver, et al., 2015). As a result, unwanted changes can occur in the microorganism.
To decrease off-target activity, several measures can be applied. One of the methods is usage of
shortened or truncated gRNA target sequences and/or lowering Cas9 expression. On the other hand,
this method could be inconvenient, since usage of truncated gRNA target sequence can result
sometimes increase in off-target and decrease in on-target efficiency (Kleinstiver et al., 2016).
Another solution for decreasing off-target activity of CRISPR/Cas9 can be the change nuclease
activity of Cas9 nuclease (Slaymaker, et al., 2015).

18
1.5.2 Selection system and relevance of the promoter efficiency on genes expression
Selection of the successfully transformed organisms is an important part of yeast modification. There
are two types of selection which are utilized in the molecular biology: auxotrophic and antibiotic.
Auxotrophic selection involves deactivation of the functional gene in the wild type such as URA3
gene which is encoding an essential enzyme in pyrimidine biosynthesis. This obtained strain could
be modified further and propagated on the selection media with the presence of appropriate growth
factor. Nutritional complementation is also important and could be reached by including growth
factors in the defined synthetic media or by using complex media constituents like peptone or yeast
extract that contains relevant growth factors (Pronk, 2002). Nevertheless, auxotrophic selection has
several disadvantages, e.g. engineering autotrophies in prototrophic strains first (industrial strains are
polyploid prototroph) (Vickers, et al., 2013). Antibiotic selection is based on introduction to the cell
genome or on the plasmid marker genes which provide resistance against antibiotics or other toxic
compounds. To select the modified cells, selection media should contain corresponding toxic
compounds. The disadvantages are the price of antibiotics which prohibits their usage on the
industrial scale and that the specific promoters and terminators should be considered in order to
express marker efficiently (Pronk, 2002).
However, the selection marker removal is a vital issue, since its presence in the cell can cause many
drawbacks. As a matter of fact, presence of the selection marker, either antibiotic or auxotrophic,
could negatively influence further modifications and cell physiology itself (Alam et al., 2016).
Utilization of dominant or auxotrophic selection markers and integration of up to three plasmids
Figure 6 Inversion of the DNA region which is located between LoxP sites is done when LoxP sites are on the same DNA strand and are in the opposite orientations. Deletions occurring when the LoxP sites are facing the same orientation, the fragment is cut our as a circular fragment. Translocation happens when LoxP sites are located on the separate DNA molecules (Chen, et al., 2019)

19
which can contain them is possible with the usage of EasyClone tool kit (Jensen, et al., 2014). The
sites in the genome where genes of interest (GOI) should be introduced are located in the intergenic
regions and placed in the interspaced manner to essential genes. Integrated DNA fragments with this
toolkit are at the low risk of removal by homologous recombination and are located in the highly
expressed area of the genome (Jessop-Fabre, et al., 2016). The difficulty which could be face with
the usage of this method is the removal of selection marker. Application of Cre-LoxP-mediated
recombination can be used for selection markers removal. Cre-LoxP-mediated recombination works
Figure 7 EasyClone – MarkerFree toolkit, explanation of the plasmids part and the process behind the gene insertion (A)Centromeric plasmid carrying gene coding Cas9 protein. Cas9 is expressed by the constitutive TEF1 promoter (green color). It is a low copy vector which contains CEN/ARS site for replication in the yeast and origin of replication ori (both in blue color) for replication on bacteria. Antibiotic selection markers present on the plasmid: Ampicillin also marked as amp (orange color) for bacterial selection and geneticin (KanMX) for selection in yeast (orange color). Episomal or high copy plasmid carrying gRNA cassette. gRNA cassette includes: SNR52 promoter, SUP4 terminator, target sequence) for targeting specific DNA location also called crRNA and gRNA structural part (tracrRNA). The other components of the plasmid: Nourceotricin or NatMX (orange color) for selection in yeast and ampicillin or amp (orange color) for selection in bacteria. Vector also contains 2-micron site (blue) site and pUC\ori(blue) for replication in yeast and bacteria respectively. (B) Representation of the target site of the GOI. TS is represented in red colored N letters and the PAM site in NGG (green color) (Stovicek, et al., 2015).

20
in the way, that Cre recombinase recognizes 34 base pairs LoxP sites and their location or orientation
will determine how the DNA material will be rearranged (Sternberg, et al., 1981). Three types of
rearrangements can be highlighted with this type of recombination: inversion, deletion, translocation
(Figure 6). Nonetheless, this method requires insertion of CreA containing plasmid, additional
cultivation for CreA expression activation and colony selection for marker loss confirmation (Jessop-
Fabre et al., 2016).
EasyClone - Markerfree toolkit is another option which can be used in the organism modification
(Jessop-Fabre, et al., 2016). This toolkit utilizes CRISPR/Cas9 system, where Cas9 coding gene is
located on one plasmid with specific selection marker and gRNA complex is encoded on another
plasmid. The advantage of it is that it is not required to integrate the selection marker into the genome,
since the selection markers are present on the plasmids. Moreover, importance of the removal of
gRNA is crucial if further modification should be performed and it is easy to perform by usage of this
toolkit because gRNA can be easily removed by growing transformed yeast on the non-selection
medium. It should also be remarked, that the advantages of EasyClone - Markerfree toolkit are
simplified cloning, reduced time for the strain modification, easier removal of the selection markers
(Figure 7). The reason behind usage of the two-plasmid system is the influence on the efficient
expression of the vectors in yeast. gRNA could be inhibited if Cas9 is replicated in high copies, that
is the reason why Cas9 containing plasmid is centromeric and gRNA containing plasmid is a high
copy plasmid. Centromeric plasmids are considered as low-copy plasmids and contain autonomously
replicated sequence (ARS) together with the centromere sequence, they replicate as a small
independent chromosome and usually found in a single copy. Episomal or high copy plasmids contain
2-micron replication site and allow for plasmid to replicate in the 50 and more copies (Patrick, 2014).
Promoters play a crucial role in the gene expression because the initiate the transcription of particular
genes. It is important to select the proper promoter for either heterologous gene expression in the
yeast genome or expression of the selection marker on the plasmid integrated to the yeast cell. In
Biotechnology, there are several types of promoters used regarding their type of transcriptional
activity (Roa-Rodriguez, 2003). Promoters can be divided into two types: inducible and constitutive
promoters. Inducible promoters can be regulated in positive, negative and multiregulated fashion.
Positive inducible promoters are inactive until activated after inducer binds to the activation protein,
thus it can bind to promoter and initiate transcription. Negative inducible promoter is not active
because of the presence of repressor, the transcription is initiated after inducer is bind to the repressor

21
and it is removed. Multiregulated promoters can be regulated by either presence of repressor or by
binding of the activation protein. Regulation of constitutive promoters is not affected by any
transcriptional factors and it allows to transcribe the gene continuously, however, it can be affected
by RNA polymerase levels or σ factors (proteins altering specificity of RNA polymerase for specific
sequences). The question of the choice of promoters is vital for expression of any gene in
Saccharomycetaceae family. Commonly used constitutive promoters provide different gene
expression level in more distinct species in non-conventional yeast compared to conventional yeast
(Lee, et al., 2013).
1.5.3 Genetic techniques and transformation methods
Expression of the heterologous genes, disruption of the genes of interest and its confirmation requires
the application of several genetic techniques. EasyClone - Markerfree toolkit requires some
modification as well. The provided plasmids containing gRNA are targeting specific regions in
S. cerevisiae and in order to change target sequence (Figure 7) or another specific nucleotide
sequence, PCR directed mutagenesis is used. This method consists of 3 steps: PCR amplification,
vector treatment (e.g. application of kinase, ligase and DpnI enzymes) and bacterial transformation
for cloning the obtained vector. The principle of the first steps is to anneal forward and reverse
primers containing homologous sites to an exact vector sequence and extend them with DNA
polymerase and dNTPs. In case of changing target site in gRNA containing plasmid, forward primer
contains a 20 base pair overhang with the new target sequence for the original sequence replacement.
Then the linear fragment is treated by kinase to phospholerate the end sites and ligase in order to
make it circular and DpnI restriction endonuclease digests the template vector. Then the treated vector
is transformed to Escherichia coli for cloning.
Integrative vectors take and important part in molecular biology. In principle, they contain a gene of
interest (GOI) with promoter and terminator for integration into the genome which is placed between
homologous sites corresponding to the specific genome location. This fragment is, for example, used
by yeast as a template to repair double stranded brake cause by CRISPR/Cas9 system by the activity
of homologous recombination, thus integrating GOI into genome. If the new GOI should be
introduced into the plasmid it could be done by combining PCR directed mutagenesis with Gibson
assembly. First both GOI and integrative vector are amplified with the primers containing the sites
homologous to each other. The difference is that plasmid is opened up by the primers containing
homologous sites (HS) and the GOI is amplified with the primers containing HS for their integration

22
into the fragment. All the above is the preparation for the Gibson assembly. Gibson assembly is the
method used for annealing up to four fragments. When the above-mentioned procedures are done,
those fragments are place into the Gibson assembly master mix containing exonucleases which create
3’ single stranded overhangs for annealing, DNA polymerase which filling the gaps within the
fragments and DNA ligase which serves for sealing the nicks of assembled DNA. Then, constructed
vector is transferred to E. coli for cloning.
The fastest way to confirm if the integration of the gene into the plasmid was successful is confirming
its size. Restriction digest (RD) is used for this purpose. RD is the process when DNA is cleaved to
the smaller pieces with restriction enzymes (RE). RE recognize definite DNA sequences and cut
them. RE are used to linearize the vector of interest and then their size is confirmed by gel
electrophoresis technique.
For the introduction of any kind of DNA to the yeast cell for its integration or expression on the
plasmids transformation methods applied. Three main transformation methods can be highlighted
which are used in yeast genomic engineering: chemical transformation, electroporation and
protoplast.
Treatment of the yeast cells with alkali citations in order to transform it was first reported in 1983
and since then this technique was broadly modified ending up with 106 transformants compared to
400 when the methodology was first implemented (Gietz, et al., 2003). This technique is also known
lithium acetate/single-stranded DNA/polyethylene glycol protocol or as LiAc/SS-DNA/PEG
protocol. The main methodology of this yeast transformation includes:
1. Mixing yeast cells with LiAc/SS-DNA/PEG together with the DNA which should be
introduced to the cell.
2. Heat shock the mixture at 42 OC for certain amount of time.
3. Recovery of the cells in the liquid medium (usually yeast nitrogen base (YNB) or yeast malt
(YM) media).
4. Selection of the modified cells by auxotrophic or antibiotic selection.
In terms of the compounds used, lithium acetate is used for increasing the permeability of the cell
wall of yeast because of its ability of denaturing DNA and RNA proteins (Kawai, et al., 2010). It was
hypothesized that PEG increases transformation frequency and efficiency by acting on the cell

23
membrane. Heat shock as well as lithium acetate makes the cell membrane more permeable. There
are two main theories how does single-stranded carrier DNA act in the transformation mixture.
Because of the presence of the multiple nucleases in the yeast cell, addition of bulk DNA increases
the chances of the plasmid or cassette being transferred to the cell nucleus without being degraded or
because of the yeast wall chemical composition SS-DNA binds to the wall allowing plasmid or
cassette get to the yeast cell (Gietz, et al., 1995; Burgers, et al., 1987).
Electroporation is the technique when the electrical field is applied to the cell in order to increase its
permeability (Chang, 2006). Electroporation works in the way that specific amounts of volts are
passing across of one or two millimeters of suspended cells in the electroporation cuvette. Then the
cells are recovered in the growth medium and plated to the medium with the selection markers or
auxotrophic selection method is used. However, electric impulse should be chosen wisely, because
of the danger of cell necrosis (Weaver, et al., 2012). First electroporation method was including the
resuspension of S. cerevisiae cells in liquid YPD medium, performing the electroporation at the
voltage of 900 V of the cells at the early log phase with less than 0.1 µg of DNA supplied. After that
first experiment protocols were constantly modified as for example 1M sorbitol was used in the
further protocols because it was providing continuous osmotic support (Kawai, et al., 2010).
The last but not least is the method for yeast transformation which implements the digesting of the
yeast cell wall creating protoplast. In order to prepare protoplast, the yeast cell wall can be degraded
by the several digestive enzymes or chemicals. When the protoplast is prepared either electroporation
or PEG-mediated transformation is used to transform the DNA into the cell (Rehman, et al., 2016).
The lack of the cell wall should allow DNA to enter the cell.
1.6 Aim and approaches This project has several aims:
1. Investigation on the insights of T. delbrueckii potential on producing specific compounds via
fermentation with higher yields compared to S. cerevisiae.
2. Construction of the convenient genetic tool for T. delbrueckii modification.
3. Research on the possibility of 2-phenylethanol production increase via PDC gene disruption
and ARO10 gene overexpression compared to the wild type strain
4. Investigation on the possibility of increase of the ethanol production in T. delbrueckii through
RIM15 gene disruption

24
The first aim is focused on the potential compounds findings through the literature which can be
possibly produced at higher yields compared to S. cerevisiae.
The second aim can be reached by considering EasyClone - Markerfree kit used to modify
S. cerevisiae as a template and adjust it in order to implement CRISPR/Cas9 modification system in
T. delbrueckii. Moreover, the plasmid-based system and the dependency of homologous sites size on
the integration of the disruption cassette and/or heterologous gene should be tested in T. delbrueckii
by URA3 disruption.
The third aim is the obtainment of insights in 2-phenylethanol production pathway in S. cerevisiae
and relevant non-conventional yeasts and improve its production in T. delbrueckii production through
metabolic engineering approaches performing several genetic modifications (Figure 8). The
modifications include: finding and disrupting PDC gene and overexpressing ARO10. PDC gene
disruption was chosen because it was hypothesized that removing ethanol production fermentation
could be shifted towards 2-PE production. In addition, it was hypothesized that ARO10 gene
overexpression could increase 2-phenylethanol production yield.
The fourth aim is to track the influence of RIM15 disruption on ethanol production in T. delbrueckii.
Figure 8 Planned modifications of Torulaspora delbrueckii. It includes: Disruption of the PDC gene by the stop codon containing cassette in the IDT and Benchling locations, overexpression of ARO10 by introduction of the ARO10 gene in the highly expressed intergenic region, disruption of the PDC gene and overexpression of ARO10 by introduction of the ARO10 containing cassette to the PDC gene and overexpression of the ARO10 gene by its introduction into highly expressed intergenic location and PDC disruption by the stop codon containing cassette.

25
2. Materials and methods
2.1 Strain and cultivation media
Haploid T. delbrueckii (PYCC 2477), S. cerevisiae S288c strain and K. marxianus (PYCC 3886) were
used in this project, provided by Portuguese yeast culture collection. This yeast was cultivated at
30 OC on the standard YPDA (yeast peptone dextrose agar) medium containing: 20 g/L glucose,
10 g/L yeast extract, 20 g/L peptone and 20 g/L agar. Media was sterilized at 121 OC in the autoclave,
spread on the petri dishes and stored at 4 OC. Modified yeast strains were selected on the plates with
addition of 150 µg/ml geneticin, 100 µg/ml nourseothricin or 100 µg/ml geneticin and 100 µg/ml
nourseothricin depending on the modification.
E. coli NEB10b strain was used for the plasmid cloning and cryopreservation at -80 OC. NEB10b
strain was cultivated on LB (Luria broth) media containing: 5 g/L yeast extract, 5 g/L sodium chloride
and 10 g/L tryptone. It was recovered after transformations in SOC medium containing: 0.5 % yeast
extract, 2% peptone, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4 and 20 mM glucose.
Moreover, after transformation NEB10b was selected on the LB medium with the addition of 15 g/L
of agar and 100 µg/ml of ampicillin. E. coli was cultivated and selected at 37 OC.
5-Fluoroorotic acid (5-FOA) plates were prepared by mixing solution containing: 20 g/L of glucose
and 15 g/L agar with 2 times concentrated YNB solution mixed with 80 mg/ml of uracil and 2 mg/L
of 5-FOA in order to select auxotrophic strains. Another batch of 5-FOA containing plates was
prepared in the same fashion as described above but only 40 mg/ml of uracil was used and 1 g/L
5-FOA was supplied
2.2 Selection of the genes and gRNA site selection
PDC and ARO10 gene sequences of T. delbrueckii were obtained from EnsemblFungi database
(Ensembl Fungi, 2009) through BLASTp of the protein sequences of PDC1, PDC5, PDC6 and
ARO10 of S. cerevisiae respectively. RIM15 gene sequence was kindly provided by supervisor Lei
Yang. URA3 gene sequence was obtained from EnsemblFungi database through BLASTp of the
protein sequence of URA3 from S. cerevisiae.
gRNA sequences were selected by using both tools for gRNA design from Benchling
(Wickramasekara, 2016) and IDT (Walder, 1987) The criteria of selecting most suitable gRNA sites

26
were: highest on-target score, region for double stranded break no longer than 100 bp from the active
site (For Ura3).
2.2.1 Intergenic regions
The intergenic regions in T. delbrueckii were obtained from EnsemblFungi database (Ensembl Fungi,
2009) through BLASTn of highly expressed regions with the specific chromosomal coordinates from
S. cerevisiae were used (Table 2). After confirmation of the gene synteny between above regions in
S. cerevisiae and T. delbrueckii, suitable highly expressed regions were located in T. delbrueckii
(Table 2). gRNA sequences for T. delbrueckii were selected by using tools for gRNA design from
Benchling (Wickramasekara, 2016). The requirements for the highly expressed region selection in
T. delbrueckii were: presence of tRNA coding regions, the selected region should have been located
1000 base pairs up or downstream from ORF (open reading frame) of the surrounded genes. Table 2 Chromosomal coordinates of the highly expressed regions
2.3 Preparation of the primers, plasmids and integration cassettes
Two plasmids: pCfB2312 and pCfB3044 were kindly provided by Irina Borodina, Novo Nordisk
Foundation Center for Biosustainability, from which pCfB2312 is the plasmid containing Cas9
coding sequence and pCfB3044 is the gRNA containing plasmid targeting XI chromosome in
S. cerevisiae. First, pCfB3044 was prepared for the transformation efficiency and antibiotic resistance
test. It treated by PvuII restriction enzyme at 37 OC for 1 hour in order to remove gRNA sequence.
Then the linear plasmid was ligated with T4 ligase and transformed to E. coli for cloning. Removal
of gRNA sequence was confirmed by amplifications with pScCRISPR-gRNA_FOR and
pScCRISPR-gRNA_REV, followed by the electrophoresis.
Chromosomal coordinates (chromosome number: location in in the chromosome)
Name of the organism
Reference
Chromosome X: 209709-239096 Saccharomyces cerevisiae Jessop-Fabre, et al., 2016 Chromosome XI: 74360-109361 Saccharomyces cerevisiae Jessop-Fabre, et al., 2016 Chromosome XII: 786102-832103 Saccharomyces cerevisiae Jessop-Fabre, et al., 2016 Chromosome IV: 519124 - 520579 Torulaspora delbrueckii current report Chromosome V: 70053 - 75722 Torulaspora delbrueckii current report Chromosome VI: 626066 - 627107 Torulaspora delbrueckii current report

27
For PDC gene disruption, three types of integration cassettes were prepared. First type of integration
cassette (BIC) was prepared by amplification with TdPDC-Ko-B_FOR and TdPDC-Ko-B_REV
primers of integration cassette containing stop codons, barcode sequence and homologous sites to
GRE3 gene which was kindly provided by the colleague. Second integration cassette (IIC) was
prepared in the same fashion but only amplified with different primers: TdPDC-Ko-IDT_FOR and
TdPDC-Ko-IDT_REV. Third integration cassette was prepared in the way described further. ARO10
was amplified from the purified T. delbrueckii genomic DNA by TdARO10_FOR and
TdARO10_REV primers. pJET1.2-TdHR-GFP was amplified by TdARO10-CYCt_FOR and
TdARO10-TEFp_REV primers containing overhangs for introduction of ARO10 in between of TEF1
promoter and CYC1 terminator by removing green fluorescent protein coding gene. ARO10 was
introduced to pJET1.2-TdHR-GFP via Gibson assembly. Finally, ARO10 containing promoter and
terminator was amplified by BC1-TEFp_FOR and BC2-CYCt_REV primers for introduction into
PDC gene locations selected via Benchling and IDT respectively. Moreover, pCfB3044 plasmid was
amplified by two sets of primers: TdPDC-sgRNA-IDT_FOR, pSc_gRNA_REV and TdPDC-sgRNA-
B_FOR, pSc_gRNA_REV. It was done in order to change gRNA sequence from targeting XI
chromosomal location in S. cerevisiae to target PDC gene in the location selected via Benchling and
IDT tools in T. delbrueckii.
Integration cassette containing 200 base pair long homologous sites and 44 base pair sequence
required for disruption of RIM15 was kindly provided by supervisor Lei Yang. GRE3 integration
cassette was amplified with the 3 sets of primers: LYy59, LYy60; LYy53, LYy54 and LYy51, LYy52
in order to construct 3 integration cassettes with homologous sites of 50, 200 and 500 base pairs
respectively to be integrated to URA3 region. pCfB3044 was amplified with two sets of primers: LYy
49, pSc_gRNA_REV and LYy50, pSc_gRNA_REV in order to target RIM15 and URA3 respectively.
The gRNA containing plasmid (pCfB3044) was amplified with three different forward primers:
TdChrIV-gRNA_FOR, TdChrV-gRNA_FOR, TdChrVI-gRNA_FOR and one common forward
primer: pSc_gRNA_REV for construction of the three gRNA containing plasmids targeting 3
different chromosomal locations in T. delbrueckii. Melting temperature of the primers for
amplification of the chromosomal regions in T. delbrueckii and primers required to open up the
plasmid with the integrated chromosomal regions was set up in the range between 55-57 OC. First,
specific regions were amplified by TdChIV-HR-5’_FOR, TdChIV-HR-3’_REV, TdChV-HR-
5’_FOR, TdChV-HR-3’_REV, TdChVI-HR-5’_FOR and TdChVI-HR-3’_REV primers and PCR
purified. Then those amplified fragments were integrated into pJet2.1 plasmid by using CloneJet PCR

28
cloning kit resulting: pJet1.2-ChrIV:2, pJet1.2-ChrV:1 and pJet1.2-ChrVI:1 plasmids. Later, those
plasmids were linearized with the BC1-TdChrIV-HR-5’_REV, BC2-TdChrIV-HR-3’_FOR, BC1-
TdChrV-HR-5’_REV, BC2-TdChrV-HR-3’_FOR, BC1-TdChrVI-HR-5’_REV and BC2-TdChrVI-
HR-3’_FOR in order to have gene of interest integrated in between of homologous sites via Gibson
assembly.
Table 3 Plasmids used in the project. This table consists of Cas9 and gRNA containing plasmids together with integrative plasmids
Plasmid name Relevant features Reference pCfB2312 KanMX, amp’, ori, CEN/ARS, Cas9
coding gene Jessop-Fabre, et al., 2016
pCfB3044 NatMX, amp’, ori, 2-micron, gRNA site targeting XI chromosome in Saccharomyces cerevisiae
Jessop-Fabre, et al., 2016
pCfB3044-PDC-IDT NatMX, amp’, ori, 2-micron, gRNA site targeting PDC gene location selected trough IDT in T. delbrueckii
Current report
pCfB3044-Rim15 NatMX, amp’, ori, 2-micron, gRNA site targeting Rim15 gene in T. delbrueckii
Current report
pCfB3044-Ura3 NatMX, amp’, ori, 2-micron, gRNA site targeting Ura3 gene in T. delbrueckii
Current report
pCfB3044-ChrIV:2 NatMX, amp’, ori, 2-micron, gRNA site targeting highly expressed IV chromosomal region in T. delbrueckii
Current report
pCfB3044-ChrV:1 NatMX, amp’, ori, 2-micron, gRNA site targeting highly expressed V chromosomal region in T. delbrueckii
Current report
pCfB3044-ChrVI:1 NatMX, amp’, ori, 2-micron, gRNA site targeting highly expressed VI chromosomal region in T. delbrueckii
Current report
pJet1.2 ori, amp’ Current report pJet1.2-ChrIV:2 Homologous sites to IV chromosomal
region in T. delbrueckii Current report
pJet1.2-ChrV:1 Homologous sites to V chromosomal region in T. delbrueckii
Current report
pJET1.2-ChrVI:1 Homologous sites to VI chromosomal region in T. delbrueckii
Current report
pJET1.2-TdHR-GFP Contains TEF1 promoter and CYC1 terminator, GFP coding gene
pCfB3044-gRNA NatMX, amp’, ori, 2-micron, removed gRNA site
Current report
pCfB3044-PDC-B NatMX, amp’, ori, 2-micron, gRNA site targeting PDC gene location selected trough Benchling in T. delbrueckii
Current report
Table 4 Primers used in the project. Homologous sites are marked in italic, barcode sequences are marked in bold.
Primer Sequence (5´-3) TdPDC-Ko-B_FOR TCCCATCCTTCGTCACCCCAATGGGTAAGGGTTCTATCGATGAACAAAACCCAAGATTCG
GTGGTGTTTAGTTAGCTAACTGAGGTCACG
TdPDC-Ko-B_REV ACCAACAGACAAGATCAAGTCAGCGGATTCAACAGCTTCCTTGACTTCTGGAG AGGACAAAGTACCGACGCTAACTAGGTGACCTACAGC
TdPDC-Ko-IDT_FOR TACTCTAGGTCGTTATCTATTCGAAAGATTAAAGCAAGTTGACACCAACACCATCTTTGGTTT GCCAGGTGTTAGCTAACTGAGGTCACG

29
TdPDC-Ko-IDT_REV CGTTAGCGTTACCAGCCCATCTCATACCTGGCACTTCGTAAATCT TGTCCAACAAGGACAAGTTGAAGTCCTAACTAGGTGACCTACAGC
TdARO10_FOR ATGAGAGAAACCTTTCATAT TdARO10_REV TTAGTTCTTAACTATTGCGT TdARO10-CYCt_FOR GATTGACGCAATAGTTAAGAACTAAATCCGCTCTAACCGAAAAGGAAGGA TdARO10-TEFp_REV ATTTTATATGAAAGGTTTCTCTCATCTAGAAAACTTAGATTAGATTGCTA BC1-TEFp_FOR GTTAGCTAACTGAGGTCACGCATAGCTTCAAAATGTTTCTACTCC BC2-CYCt_REV CTAACTAGGTGACCTACAGCCTTCGAGCGTCCCAAAACCTTCTCA TdPDC-sgRNA-IDT_FOR AAGGACAAGTTGAAGTCACCGTTTTAGAGCTAGAAATAGC TdPDC-sgRNA-B_FOR AGATTCGGTGTTTACGTGTTTTAGAGCTAGAAATAGC pScCRISPR-gRNA_FOR TCGCGCCATTCGCCATTCAGGCTG pScCRISPR-gRNA_REV AGTGAGCGAGGAAGCGGAAGAGCGC TdChIV-HR-5’_FOR GATAGATCGGCGAGCGACTT TdChIV-HR-3’_REV GGAATGGAACTCCACTCTACCT TdChV-HR-5’_FOR GGGCTCAAGAAACATAGGCTG TdChV-HR-3’_REV AAAATTTTCAGCTCCCATACTAAACCT TdChVI-HR-5’_FOR GCATTGTACCTAATGCTGGTCAT TdChVI-HR-3’_REV ACACAGCTGGCTGGTTATGA BC1-TdChrIV-HR-5’_REV GTTAGCTAACTGAGGTCACGGGGATCTGTCAGATCTTGAATCAGC BC2-TdChrIV-HR-3’_FOR GCTGTAGGTCACCTAGTTAGCGGGGTTAAGAAGAATGTTGCC BC1-TdChrV-HR-5’_REV GTTAGCTAACTGAGGTCACGTGCGGAGTTCGATCACCG BC2-TdChrV-HR-3’_FOR GCTGTAGGTCACCTAGTTAGCGCAGTATCTCATACAACTTCTAAAAATCT BC1-TdChrVI-HR-5’_REV GTTAGCTAACTGAGGTCACGATCGAGGCTTTATAACAATTTTTACAAGATC BC2-TdChrVI-HR-3’_FOR GCTGTAGGTCACCTAGTTAGGATCCTTTCTCGGTCACAGC TdChrIV-gRNA_FOR AGATCTGACAGATCCCAACGGTTTTAGAGCTAGAAATAGCAAGTT TdChrV-gRNA_FOR TGGTGGATGTCTTGGTGACAGTTTTAGAGCTAGAAATAGCAAGTT TdChrVI-gRNA_FOR CGATGATCCTTTCTCGGTCAGTTTTAGAGCTAGAAATAGCAAGTT LYy49 TGGAAGGGCCCAATTACTGGGTTTTAGAGCTAGAA LYy50 ACCTTGGGTGTATTCACCATGTTTTAGAGCTAGAA LYy51 ATATTTCGCTTCAGTACTATA LYy52 AATTCTTGCTGTTCTCTACT LYy53 CTTCAGGTGTCTACAGAATT LYy54 AGACAACTTCATCGACAGT LYy55 AAAATTCGATCATGGAAGAG LYy56 AGAAGATACAGCCCACAG LYy57 AGCTAACTGAGTCACCTAGT LYy58 TCCTCATCTTCTGGAGAGGT LYy59 CCAAGAGGTCTATTAATGTT LYy60 CTTTGGGCAATGAATCCA pSc_gRNA_REV GATCATTTATCTTTCACTGCGGAGAAG
2.4 Preparation of the T. delbrueckii frozen competent cells
T. delbrueckii cells containing pCfB2312 were inoculated to the 10 ml of liquid YPD medium
containing 100 µg/ml of geneticin, (concentration) adenine hemisulfate in 50 ml falcon tube and
incubated at 30 OC with the agitation speed of 200 rpm. Meanwhile, 50 ml of 2x YPD medium were
pre-incubated in 250 ml baffled flasks at 30 OC overnight. On the next day, 2.5 * 108 yeast cells were
inoculated into 50 ml pre-warmed medium (which gave the concentration of the cells of 5 * 106
cells/ml) with the addition of 100 µg/ml of geneticin and incubated at 30 OC with the agitation of 200

30
rpm for 4 hours. After the optical density (OD) reached 2.0 which equals to 2 * 107 cells/ml yeast
cells were spin down by the centrifugation at 3000 g for 5 minutes at the room temperature. Once the
supernatant was removed, cells were resuspended in 25 ml of sterile water, centrifuged at 3000 g for
5 minutes and water was discarded afterwards. This procedure was repeated one more time and then
cells were resuspended in frozen competent cell solution (5% v/v glycerol, 10% v/v DMSO). Later,
100 µL cell suspension was distributed to Eppendorf tubes and placed into the freezing container
filled with 250 ml of isopropanol. The container was stored at -80 oC in styrofoam box.
2.5 Yeast transformation 2.5.1 Antibiotic resistance test
For nourseothricin resistance test the modified high-efficiency yeast transformation using the
LiAc/SS carrier DNA/PEG method (Gietz, et al., 2007) was used. Three yeasts: T. delbrueckii (PYCC
2477), S. cerevisiae S288c, K. marxianus and S. cerevisiae Ethanol red were pre-grown on the YPD
plates for 17 hours. On the next day 50 µL of cells from each yeast were harvested by the loop and
resuspended in 950 ml of water in 1.5 ml Eppendorf tube. Then the tubes were centrifuged at
13,000 g for 1 minute and the water was discarded afterwards. Cells were washed twice with 500 µL
and 200 µL respectively of lithium buffer. Meanwhile single stranded (SS) DNA was boiled for 5
minutes. Subsequently cells were mixed with the compounds listed below and in the exact order
shown in the table 5.
Table 5 Compounds mixed for the transformation
Compound name Amount (µL) PEG3350 (50%) 260 Lithium Acetate 1M 36 SS DNA (10 mg/ml) 10 pCfB3044-gRNA (500 ng/µL) 1 Water 53
After addition of all the compounds the tubes were vortexed vigorously and incubated for 30 minutes
at 30 OC. Incubation was followed by heat shock of the cells, where the tubes containing
transformation mixture were placed to heating block at 42 OC for 40 minutes. Then the mixture was
centrifuged at 10,000 g, pipetted out and treated cells were resuspended in 1 ml of YPD medium. The
two groups of yeasts were recovered for 6h and overnight and plated out on YPD plates containing

31
either 50 µg/ml or 100 µg/ml of nourseothricin with the 1:10 and 1:2 dilutions respectively. When
the colonies were obtained on the selection medium, the transformation efficiency was calculated
according to the formula:
𝑇𝐸 = 𝑐𝑜𝑙𝑜𝑛𝑖𝑒𝑠𝑓𝑜𝑟𝑚𝑒𝑑 ∗ µ𝑔𝑜𝑓𝐷𝑁𝐴𝑠𝑢𝑝𝑝𝑙𝑖𝑒𝑑
𝑑𝑖𝑙𝑢𝑡𝑖𝑜𝑛𝑓𝑎𝑐𝑡𝑜𝑟
Another antibiotic resistance test with both geneticin and nourseothricin present on the selection plate
was performed. Four different sets of YPD plates were prepared containing: 0 µg/ml geneticin,
0 µg/ml nourseothricin; 50 µg/ml geneticin, 50 µg/ml nourseothricin; 50 µg/ml geneticin, 100 µg/ml
nourseothricin; 100 µg/ml geneticin, 50 µg/ml nourseothricin. For the test two type of T. delbrueckii
were used: wild type strain without any plasmids introduced, modified strain containing two separate
plasmids with geneticin and nourseothricin resistance genes respectively. Cells were first
resuspended in water, concentration of the cells was adjusted to OD600 = 0.1 and 30 µl of the cell
suspension for each type of strain was plated to the respective plate.
2.5.2 Introduction of integration cassettes for PDC disruption
First, pCfB2312 containing Cas9 was transformed into T. delbrueckii under the same conditions listed
above, but instead of pCfB3044-gRNA, pCfB2312 was added. For further transformation
T. delbrueckii with the Cas9 containing plasmids was used.
Two types of transformations in order to disrupt PDC were performed: introduction of the integration
cassettes containing stop codons and introduction of ARO10. The preparation of the cells was
performed in the same way as it was mentioned above but different types of DNA was supplemented
to the transformation mixture:
Table 6 Introduction of the stop codon containing integration cassette targeting location in the PDC selected trough Benchling
Compound name Amount (µL) PEG3350 (50%) 260 Lithium Acetate 1M 36 SS DNA (10 mg/ml) 10 BIC (50 ng/µL) 2 pCfB3044-PDC-B (550 ng/µL) 1,8 Water 50.2

32
Table 7 Introduction of the stop codon containing integration cassette targeting location in the PDC selected trough IDT
Compound name Amount (µL) PEG3350 (50%) 260 Lithium Acetate 1M 36 SS DNA (10 mg/ml) 10 IIC (60 ng/µL) 1.6 pCfB3044-PDC-IDT (600 ng/µL) 1.6 Water 50.8
Table 8 Introduction of the ARO10 containing integration cassette targeting location in the PDC selected trough Benchling
Compound name Amount (µL) PEG3350 (50%) 260 Lithium Acetate 1M 36 SS DNA (10 mg/ml) 10 ARO10 integration cassette (100 ng/µL) 5 pCfB3044-PDC-B (550 ng/µL) 1,8 Water 47.2
After addition of all the compounds the tubes were treated in the same way stated in 2.5.1. The yeast
was recovered for 6h and plated out on YPD plates containing both 100 µg/ml of geneticin and 100
µg/ml of nourseothricin with the 1:10 and 1:2 dilutions respectively.
2.5.3 Introduction of integration cassettes for RIM15 disruption
RIM15 integration cassette was also introduced into the T. delbrueckii containing pCfB2312. Frozen
competent cells were used for this transformation. After taking them out from the -80 OC freezer,
cells were unfrozen and spin down in the centrifuge at 10,000 g to remove supernatant. After, the
compounds listed above were added to the cells as followed:
Table 9 Introduction of the stop codon containing integration cassette targeting location in the RIM15
Compound name Amount (µL) PEG3350 (50%) 260 Lithium Acetate 1M 36 SS DNA (10 mg/ml) 10 RIM15 integration cassette (110 ng/µL) 9 pCfB3044-Rim15 (400 ng/µL) 2.5 Water 42.5

33
After mixing all the components, transformation mixture was incubated for 30 minutes at 30 OC,
followed by heat shock for 40 minutes at 40 OC. The mixture was centrifuged at 10,000 g and pipetted
out. Cells were resuspended in 1 ml of YPD medium and recovered for 17 hours. Recovered cells
were plated on the YPD plates containing 100 µg/ml of geneticin and 100 µg/ml of nourseothricin
with the 1:10 and 1:2 dilutions.
2.5.4 Introduction of integration cassettes for URA3 disruption
Before the transformation, the test of the required addition of 5-FOA to the plates was performed.
Four different concentrations of 5-FOA were tested: 0, 1, 2 and 5 mg/L. Three provided URA3 mutant
yeast strains and one wild type T. delbrueckii strain were used for the test.
For URA3 disruption frozen competent cells were used and they were prepared for the transformation
as described in 2.5.3. Solutions for the transformation were added in the same manner as it was
described previously, with the difference of adding 1 µg of pCfB3044-Ura3 and three different
amounts (in terms of nanograms) of distinctive fragments (Table 10). The supplemented amounts (in
terms of moles) of fragments were equimolar across the transformations.
Table 10 The amounts of supplemented URA3 disruption cassettes to the transformation mixture
When the transformation components were mixed, incubated for 30 minutes at 30 OC and heat
shocked for 40 minutes at 40 OC, they were recovered for 17 hours and plated out on YPD plates
containing 100 µg/ml of geneticin and 100 µg/ml of nourseothricin. Later obtained colonies were
selected on 5-FOA plates.
188 base pair fragment
488 base pair fragment
1088 base pair fragment
Nanograms added
pmol added Nanograms added
pmol added Nanograms added
pmol added
150 0.0012 400 0.0012 870 0.0012 340 0.0027 900 0.0027 2000 0.0027 860 0.0069 2250 0.0069 5000 0.0069

34
3. Results
3.1 Implementation of the transformation protocol in T. delbrueckii
In this section the transformation of the pCfB3044-gRNA plasmid containing nourseothricin resistant
marker was performed to the different yeast strains from the upper branch of Saccharomycetaceae
family including: S. cerevisiae S288c and Ethanol red, T. delbrueckii PYCC 2477 and K. marxianus
PYCC 3886 strains. The second test included growth of T. delbrueckii containing geneticin and
nourseothricin resistant markers on the selection plates with different combinations of the above
antibiotics. Those tests were done to understand T. delbrueckii antibiotic resistance ability for further
utilization of the plasmids included in the EasyClone-Markerfree toolkit. The results of the first test
transformation efficiency after 6- and 17-hours recovery for different yeast strains calculated in cfu
(colony forming units)/µg (of DNA supplied) are provided in the table 11 and 12.
Table 11 Transformation efficiency after 6 hours of recovery. Dilution factors represented in the brackets, show in which ratio samples were diluted before plating, where 0.11 and 0.5 dilution factors correspond to 1:10 and 1:2 dilution ratios respectively.
Yeast name (dilution factor) Nourseothricin concentration of 50 µg/ml
Nourseothricin concentration of 100 µg/ml
Saccharomyces cerevisiae S288c (0,11)
3.1 * 104 cfu/µg 3.2 * 104 cfu/µg
Saccharomyces cerevisiae S288c (0,5)
4.1 * 103 cfu/µg 3.2 * 103 cfu/µg
Saccharomyces cerevisiae Ethanol red (0,11)
8 * 102 cfu/µg 7.63 * 102 cfu/µg
Saccharomyces cerevisiae Ethanol red (0,5)
5 * 102 cfu/µg 4.88 * 102 cfu/µg
Torulaspora delbrueckii (0,11) 2.18 * 102 cfu/µg 1.27 * 102 cfu/µg Torulaspora delbrueckii (0,5) 1.4 * 102 cfu/µg 1.04 * 102 cfu/µg
Table 12 Transformation efficiency after 17 hours of recovery. Dilution factors represented in the brackets, show in which ratio samples were diluted before plating, where 0.11 and 0.5 dilution factors correspond to 1:10 and 1:2 dilution ratios respectively.
Yeast name (dilution factor) Nourseothricin Concentration of 50 µg/ml
Nourseothricin Concentration of 100 µg/ml
Saccharomyces cerevisiae S288c (0,11)
4.3 * 104 cfu/µg 1.2 * 104 cfu/µg
Saccharomyces cerevisiae S288c (0,5)
7.8 * 103 cfu/µg 6.4 * 103 cfu/µg
Klyveromyces marxianus (0,11) 8.72 * 102 cfu/µg 8.9 * 102 cfu/µg Klyveromyces marxianus (0,5) 2.64 * 102 cfu/µg 8.8 * 101 cfu/µg Torulaspora delbrueckii (0,11) 4 * 102 cfu/µg 9 * 101 cfu/µg Torulaspora delbrueckii (0,5) 5 * 102 cfu/µg 4.16 * 102 cfu/µg
S. cerevisiae strains showed the highest transformation efficiency. In case of S288c 6 hours and 17
hours recovery time did not show major difference in transformation efficiency. T. delbrueckii strain

35
showed lower transformation efficiency and
recovery for 17 hours did not show
significant difference in transformation
efficiency compared to 6 hours recovery.
K. marxianus in main terms showed the
lowest transformation efficiency, even after
17 hours of incubation. The main difference
between the tested yeast strains and
K. marxianus strain was the colonies size. If
for all the other yeast strains the colony sizes
were more or less equal in size, for
K. marxianus colonies were differing in sizes
drastically. With the presence of the big
colonies, a large number of the colonies were
small in size. In the second test plates which were not containing any antibiotics were expectedly full
off cells. Wild type strain, which did not contain any plasmids with antibiotic resistance markers did
not show any growth under different concentrations of the antibiotics. Growth for the strain
containing plasmids with antibiotic resistance markers (nourseothricin and geneticin) was observed
on the plates containing all the types of concentrations of antibiotics. Most growth was observed on
the plates containing 50 µg/ml geneticin and 50 µg/ml nourseothricin. Least growth was observed on
the plates containing 100 µg/ml geneticin and 50 µg/ml nourseothricin (Figure 8).
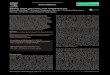
3.2 Test of the required homologous sites by disruption of URA3 This section focuses on the understanding what size of the homologous sites is required to use on the
integration cassette for its successful integration to the desired location. To understand it, integration
cassettes containing 50, 200, 500 base pairs flanking regions and targeting URA3 gene in
T. delbrueckii were transformed to the cell. After transformation of the fragments with different ratios
was performed as stated in 2.5.4 it was possible to obtain colonies with the introduced fragments
containing 50 base pairs and 500 base pairs of flanking regions. It was not possible to obtain
substantial amounts of the colonies with the introduced fragment containing 200 base pair flanking
Figure 8 Antibiotic resistance test. Two rows of plates contain strains of T. delbrueckii. Upper row represents the plates containing wild type strain containing no antibiotic resistance and the lower row represents plasmids containing cells. Concentration of antibiotics from left to right: 50 µg/ml geneticin, 50 µg/ml nourseothricin; 50 µg/ml geneticin, 100 µg/ml nourseothricin; 100 µg/ml geneticin, 50 µg/ml nourseothricin

36
However, when the colony PCR screening was performed, it was possible distinguish that not all of
Figure 9 Representation of the cell growth on the antibiotic containing plates (1.) and 5-FOA containing plates (2.). Upper row of the part 1. denotes the plates containing the colonies with the transformed 50 base pair flanking region fragments with different concentration Lower row shows the plates containing the colonies with the transformed 500 base pair flanking region fragments with different concentrations. Upper row of 2. represents the selection of the 20 colonies transferred from the plates containing 50 base pair flanking region fragments. Lower row represents the selection of the 20 colonies transferred from the plates containing 500 base pair flanking region fragments. Concentration of the integration cassettes increasing from left to right.
Figure 10 Colony PCR method used for confirmation of the URA3 disruption. A) Includes groups of 4 samples (from left to right) 50bpf with the lowest molar concentration selected on the 5-FOA plates, 50bpf with the lowest molar concentration selected on the antibiotic plates, 50bpf with the medium molar concentration selected on the 5-FOA plate. 1. represents the sample from the next group of 50bpf with the medium molar concentration selected on the antibiotic plate. P. stands for positive control, amplification of the URA3 region from the wild type for size comparison of modified and non-modified regions. B) Includes groups of 4 samples(from left to right) 50bpf with the medium molar concentration selected on the antibiotic plates, 50bpf with the highest molar concentration selected on the 5-FOA plates, 50bpf with the highest molar concentration selected on the antibiotic plate, first part of the 500bpf group with the lowest molar concentration selected on the 5-FOA C) Includes second part of the 500bpf group with the lowest molar concentration selected on the 5-FOA plate, 500bpf selected on the antibiotic plate with the lowest molar concentration, 500bpf with the medium molar concentration selected on the 5-FOA and antibiotic plate, D) Includes samples of 500bpf group with the highest molar concentration selected on 5-FOA plate and 500bpf group with the highest molar concentration selected on antibiotic plate. Size of the band successful modification – 200 base pairs, size of the band with no cassette integration – 150 base pairs. 100 base pair ladder was used for band size confirmation

37
regions. As a matter of fact, the amount of the colonies obtained from introduction of 500 base pair
flanking region fragment decreased compared to 50 base pair flanking region fragment. However,
when the colony PCR gene screening was performed, it was possible distinguish that not all of the
colonies were having URA3 disruption cassette integrated. The second test of transferring 20 colonies
from the antibiotic containing plate to 5-FOA plate showed that several colonies were containing
URA3 gene disruption (Figure 9).
Screening of the 4 colonies from the 5-FOA plates to the corresponding colonies from the plates
containing antibiotics showed that all the samples excluding the samples containing 500 base pair
flanking region cassette with the highest concentration transformed had a random mutation present
in the URA3 gene, allowing them to grow on 5-FOA plate (Figure 10). One targeted mutation from
the selected samples was present with the usage of 50 base pair flanking region fragment (50bpf) and
the lowest concentration. Even though with the second case of the usage of 50bpf one on-target
mutation
was shown on the antibiotic containing plate, with the 5-FOA selection cells containing random
mutations were preferred. Third case with the usage of 50bpf with the highest concentration of the
fragment added no on-target mutation was obtained. By the usage of the 500 base pair flanking
regions fragment (500bpf) with the lowest concentration it was possible to obtain 2 successful on
target mutations. Finally, 500bpf with the highest concentrations showed 4 out of 4 on-target
mutations (Figure 11).
25%
0% 0%
50%
0%
100%
0%
20%
40%
60%
80%
100%
120%
50 bp (0.0012 pmol)
50 bp (0.0027 pmol)
50 bp (0.0069 pmol)
500 bp (0.0012 pmol)
500 bp (0.0027 pmol)
500 bp (0.0069 pmol)
Percentage of the successful integration of the cassette into URA3 region
Figure 11 Representation of the percentage of successful integration of cassettes containing different flanking region sizes. The y-axis represents the percentage of successful cassette integration and the x-axis represents the sizes of the cassettes flanking regions (50 bp and 500 bp meaning 50 and 500 base pair flanking regions), and the amount of integration cassette supplied in 0.0012, 0.0027 and 0.0069 pmol respectively. Those results are based on the 4 colonies picked and compared from 5-FOA and antibiotic containing plates.

38
Additionally, the test for establishing the concentration of 5-FOA (Figure 12) was performed to
understand which concentration of 5-FOA to use for auxotrophic strain selection. Expectedly, it was
possible to see no growth on the plate with the concentration of 5-FOA matching to 0 mg/L. On the
plates containing 5-FOA no growth of wild type strain was observed and some/no growth of the
provided URA3 mutant strains. However, after transferring the mutant cells with URA3 gene
disrupted to the plates with the selected end concentration of 2 mg/L of 5-FOA no growth was
observed. The resulted transformants only showed growth when the concentration of the 5-FOA was
increased to 1 g/L.
3.3 Disruption of RIM15
In this section the aim was to disrupt RIM15 gene in T. delbrueckii by the cassette containing 200
base pairs of homologous sites. The cassette for RIM15 consisting of 44 base pair region which
Figure 13 A) Amplification of the modified strain with LYy57 and LYy58. 1, 2, 3 correspond to the tested colonies and P. correspond to positive control (amplification of non-modified region from the T. delbrueckii wild type). B) Amplification of the modified strain with LYy55 primers. C) Amplification of the modified strain and wild type with the LYy55, LYy57. M1 and M2 stand for mutated strains and WT stands for wild type strain.
Figure 12 Test of the yeast growth ability on 5-FOA plates. Concentrations of 5-FOA are corresponding to (from left to right): 5 mg/L, 2 mg/L, 1 mg/L, 0 mg/L. Provided URA3 mutants are marked as 1,2 and 3. Wild type strain is marked as WT

39
includes stop codon sequence and 200 base pair flanking region sequences from both sides of the
cassette was constructed and provided by the colleague from the lab. After the introduction of the
cassette and pCfB3044-Rim15 plasmid to the pCfB2312 containing cell, it was possible to see the
growth of the cells, indicating successful insertion of the plasmid. However, it was not possible to
confirm the disruption of the gene in the desired location. After amplification with the primers LYy57
and LYy58 which were targeting 44 base pair region and the region 95 base pairs away from the
downstream flanking region the multiple bands were observed, from which one of them was
corresponding to the successful integration size (Figure 13A). For additional tests it was decided to
try to amplify the fragment with only LYy55 or LYy56 primers which were targeting upstream and
downstream flanking regions respectively. It was possible to obtain the amplification with only
LYy55 primer but not with the LYy56 primer (Figure 13B). In addition, one more test was performed,
where the fragment was amplified by the LYy55 and LYy57 primers. It was possible to obtain single
band, but it was not possible to distinguish the difference between wild type and modified strain,
since the length of the integrated fragment was relatively small (Figure 13C). The amplified region
which were sent out for sequencing showed that cassette was not integrated to the desired region.
3.4 Detection of the PDC gene in T. delbrueckii and its disruption
The aim of this section was finding the PDC gene in T. delbrueckii and disrupt it with the integration
cassette containing 70 base pairs of homology to the desired region. Since genes were not annotated
in T. delbrueckii it was required to use BLASTp of PDC1, 5, 6 genes from S. cerevisiae to find the
locations of PDC genes in T. delbrueckii. As a result of BLASTp it was possible to conclude that
PDC5 and 6 regions from S. cerevisiae lacked any gene synteny to T. delbrueckii. The only synteny
was found to PDC1 gene region (Figure 14). This exact region was used as a target for further
modifications.
Figure 14 Representation of gene synteny of PDC gene regions between chromosome XII and chromosome V in S. cerevisiae and T.
delbrueckii respectively. Synteny between three genes annotated in S. cerevisiae as TRX1, PDC1 and STU2 is represented with the
yellow lines.

40
After choosing the site where double stranded brake should occur, the integration cassette was
constructed with the specific homologous sites (HS). It was possible to construct the cassette with
two different sets of primers containing HS resulting integration cassettes with 70 base pairs of
homologies to the selected locations (Figure 15).
Figure 15 Integration cassette for PDC disruption. HS (70bp) stands for flanking regions with the size of 70 base pairs homologous
to either IDT or Benchling target location. Barcode is the sequence present in all the integrative cassettes for required re-
amplification and stop codons (88bp) stands for the 88 base pair regions containing 6 stop codon sequences and multiple cloning
site.
Integration of the pCfB3044-PDC-IDT and pCfB3044-PDC-B with the respective cassettes resulted
sufficient amount of the colonies in both cases and with the presence of both antibiotics required for
selection. However, screening of the colonies via colony PCR did not show conclusive results of
PDC gene disruption by the integration cassettes.
The same circumstances occurred after the attempts of integrating ARO10 gene into the location
selected via Benchling. Though, ARO10 cassette was constructed successfully (Figure 16) and
according to the presence of the colonies on the plates with two selection markers (which shows the
integration of two plasmids) it was not possible to have any certain conclusions connected to
ARO10 gene insertion after performing colony PCR because of the more than 2 bands were present
on the gel.
Figure 16 Integration cassette for PDC disruption. HS (70bp) stands for flanking regions with the size of 70 base pairs homologous
to Benchling target location. Barcode is the sequence present in all the integrative cassettes for required re-amplification and
ARO10 with P and T stands for the ARO10 gene sequence with constitutive TEF1 promoter and CYC1 terminator respectively.
3.5 Construction of integration plasmids targeting IV, V and VI chromosomal regions
The first step of constructing the plasmids containing homologous sites to the specific chromosomal
location was finding the highly expressed regions in T. delbrueckii. After using
BLASTn of the highly expressed regions of S. cerevisiae against T. delbrueckii it was

41
possible to find a distinctive synteny of the genes between both yeasts. For the intergenic region (IR)
located on the XI chromosome in S. cerevisiae it was not possible to find the same region (Figure 17)
in the exact location on chromosome V which would follow conditions listed in 2.2.1
Figure 17 Gene synteny between specific chromosomal locations of S. cerevisiae and T. delbrueckii. Yellow lines show gene synteny. Agenda in the right corner shows the DNA length (blue line), intergenic region which follows listed conditions (red line) and RNA coding regions (black line).
Figure 18 Gene synteny between specific chromosomal locations of S. cerevisiae and T. delbrueckii.
8
Figure 19 Gene synteny between specific chromosomal locations of S. cerevisiae and T. delbrueckii. Two IR found on the chromosome IV in T. delbrueckii are marked in red and labelled as ChrIV:1, ChrVI:2.

42
The same situation was observed with the distinguishing IR chromosome IV in T. delbrueckii after
finding synteny with the genes of chromosome X in S. cerevisiae (Table 18). The IR location was not
following the conditions listed in 2.2.1.
After finding synteny between genes of the region of chromosome XII and chromosome IV in
S. cerevisiae and T. delbrueckii respectively, it was possible to find two regions which were following
the conditions listed in 2.2.1 (Figure 19). Although, it was not the exact IR location which should
have been used. For constructing proper modification kit for T. delbrueckii, at least two more regions
for gene integration were required. It was decided to search for the IR locations downstream the
chromosome V and include IR location from chromosome VI which would follow required
conditions. As a result, three regions were found on both chromosomes (one on the chromosome VI
and two on the chromosome V) (Figure 20).
After final selection of the regions to be used in the project were assigned (Table 13) Table 13 Intergenic regions in T. delbrueckii with the representation of the name of the region, chromosomal location (number and region) and length
After the amplification of the fragments from the genomic DNA and their integration into the
pJet 1.2 plasmid it resulted three plasmids which were ready for linearizing and introduction of the
any of the genes of interest (Figure 21).
Name of the region Chromosome number Region Length (base pairs) ChrIV:2 Chromosome IV 519124 - 520579 1456 ChrVI:1 Chromosome VI 626066 - 627107 1042 ChrV:1 Chromosome V 70053 - 75722 5670
Figure 20 Intergenic regions found on chromosome VI and chromosome V in Torulaspora delbrueckii. The regions are marked in red color and labelled as ChrVI:1, ChrV:2, ChrV:1.

43
4. Discussion The main goal of the project was development of the EasyClone - Markerfree plasmid kit which
would allow to specifically modify T. delbrueckii with the assist of CRISRP/Cas9 system. To achieve
this, succsesfully developed EasyClone - Markerfree plasmid kit showed in Jessop-Fabre, et al.,
(2016) was used as the reference model. For designing the kit, first, it was decided to test the influence
of the antibiotic selection and the concentrations required to select modified strains. Secondly, it was
vital to see the influence of the size of flanking regions on the integration cassettes transformation.
Having the certain sizes tested was required to uderstand how efficiently homologous recombination
Figure 21 Plasmids constructed for the further linearization and introduction of the genes of interests. Each plasmid has homologous sites corresponding to the specific chromosomal location marked as UP and DOWN (yellow), ampicillin resistance marker marked as AmpR (blue) and origin of replication marked as Ori (green) (Basu, 2001)

44
can recognize them and allow to integrate the gene of interest into the double stranded brake area.
Designed cassettes with 50, 200 and 500 base pair flanking sites which were homologous to URA3
gene in T. delbrueckii were used for testing. Third part of the project was investigation of the specific
cases of either gene disruption or overexpression. As in cases of RIM15 and PDC it was planned to
disrupt the genes and in case of ARO10 the decision was made to overexpress it. The final sets of the
modifications which was planned to obtain is represented in figure 8. The last but no least, was the
construction of the integration plasmids, which should have been based on the previous obtained
results in terms of the size of homologous sites
4.1 Utilized transformation methodology
From the all used transformation methods described in 1.6, LiAc/SS-DNA/PEG transformation is a
tested method in T. delbrueckii compared to electroporation or protoplast methods. Moreover, it is
possible to find a defined protocol of chemical transformation in T. delbrueckii. This is the reason
behind, why LiAc/SS-DNA/PEG transformation method was used in this project. Also, important
part of the chemical transformation is that it is performed in the single Eppendorf tube and does not
require a lot of performance time compared to expensive electroporation cuvettes used in
electroporation or time consuming procedure performed in protoplast (Yarimizu, et al., 2017).
4.2 Influence on the antibiotics on selection and yeast morphology
The concentrations of the antibiotics for the test were taken from Jessop-Fabre, et al. (2016) and
altered in case T. delbreckii can be selected with lower or higher concentrations of the antibiotics. It
was established that if the yeast strain contains one of the plasmids which should be either selected
on geneticin or nourseothricin it was optimal to use the concentration of 100 µg/ml in both cases.
However, when the synergetic use of the antibiotics used for selection with the concentration of
100 µg/ml at the same time it was observed that colonies shape was changing drastically compared
to the case when one antibiotic was used. It was hypothesized that cells are experiencing the high
level of stress. For that reason, it was decided to perform the test to determine with what antibiotic
concentrations it would be possible to still select the modified yeast and do not have it under high
stress conditions. It was possible to determine that it is enough to have 50 µg/ml nourseothricin and
50 µg/ml geneticin in the selection medium to select modified yeast and not to get the background

45
growth or contamination. However, it was observed that the cells morphology was still indicating
that they were present under stressed conditions.
4.3 Establishing the requirements for the size of homologous sites of the integration cassette
by URA3 disruption
It was possible to define that homologous recombination could work with no dependency on the size
of the flanking regions, since it was possible to obtain the disruption with both 50 and 500 base pair
flanking regions, confirming that it is not a requirement to use more that 500 base pairs of homology
to integrate the gene. Nevertheless, the amount of the colonies on the selection plates is decreasing
depending on the integration fragment size. It can be explained that the bigger the size of the fragment
is, the lower amount of the DNA can be transformed to the cell via chemcal transformation method.
Moreover, it was possible to show that the integration cassette can be introduced to the desired
location with the shortest possible flanking regions and with the lowest molar concentration (0.0012
pmol) of the cassette added. However, for the successful and high chance integration of the integration
cassette the longest flanking regions (500 base pairs) and the highest concentration (0.0069 pmol)
used is preferable. It should also be remarked, that the stability of the integration made by the cassette
with 50 base pair homology is much lower compared to 500 base pair homology and it has higher
chances of random mutations to occur in the URA3 region. It was shown in Hernandez-Lopez et al.,
(2003) that even when the high amounts (1-5 µg) of the fragment with 45 and 42 flanking regions
was used, the transformation efficiency obtained was extremely small – 2-5 transformants per
microgram of DNA added. The random mutations in yeast cell can be explained by environmental
stress cause by the presence of 5-FOA on the plate which is the compound toxic to the cell when the
URA3 gene is inactivated. As it was shown in S. cerevisiae it could be the event of single nucleotide
polymorphisms when the single nucleotide in the DNA is altered in the beneficial ways for the cell
(Steele, et al., 1992). The presence of the double bands after gel electrophoresis in the samples from
the antibiotic containing plates (Figure 8) can be explained of either integration of the cassette into
the random location of the yeast genome and its amplification by primers or by amplification of the
integration cassette present on the plate. In the second case the inwanted amplification can be avoided
by replating the cells on the new plates and it was done by replating the cells to the 5-FOA containing
plates, resulting no visible double bands after colony PCR. Another possible solution for avoiding

46
unwanted amplification is to use the primers which are targeting both upstream and downstream
locations outside of the modified region.
4.4 RIM15 disruption
In the context of fermentation, RIM15 has the key impact on the production of alcohol as it was shown
Watanabe, et al. (2018) on the example of S. cerevisiae and Schizosaccharomyces pombe sake strains.
It was represented that RIM15 deficient strains indicated higher carbon dioxide emission levels,
meaning the higher alcohol production rates (Watanabe, et al., 2018). So far, the disruption of RIM15
and its influence on alcoholic fermentation was not tested in T. delbreuckii making it a relevant case
of studying, especially in terms of testing CRISPR/Cas9 system. Even though, after performing the
transformation it was not possible to disrupt the gene in the desired location, the RIM15 disruption
cassette could have integrated into the different genome location or it could have been integrated in
the different fashion and still disrupt the gene. The several possibilities were examined in order to
understand how and where the disruption cassette was integrated. First theory was suggested, that the
cassette was integrated in the multiple copies, but it was not possible to obtain the clarity after trying
the different combinations of the primers since several bands appeared after the gel electrophoresis.
Then it was hypothesized that integration cassette could have been integrated in the reverse fashion
(Figure 22).
After the amplification with the single LYy56 reverse and single LYy55 forward primers it was only
possible to obtain the amplification with the reverse primers, but no expected size of the band was
observed. Next step was to try to combine the primer LYy58 which is located in the region outside
Figure 22 Possible integration of the integration cassette into the RIM15 gene. Blue color shows RIM15 gene, green color shows the 200 base pair homologous sites and red color shows the 44 base pair sequence containing stop codons for gene disruption

47
of the homologous region with the forward primer, since it did not give any amplification in the
previous experiment. It was finally possible to obtain the single band, but it was not possible to tell
from the gel electrophoresis if RIM15 gene was disrupted with the cassette or not, so the next step of
the region sequencing was performed. Despite the theory that integration cassette was able to be
integrated as mentioned above, sequencing showed that it was not integrated to the desired location
and it can indicate that cassette was possibly introduced to the different location of the genome.
4.5 PDC disruption
The reason behind why T. delbrueckii was chosen as the organism for 2-phenylethanol production is
its ability of producing higher amounts of this compound under certain conditions (González, et al.,
2018). It was shown in González, et al. (2018) under certain conditions such as: growth of the yeast
in the low-glucose must with the increased supply of aromatic alcohol precursors. Synergistic
influence of the fermentative conditions and genetic manipulations could lead to radical increase of
2-PE production (Figure 22). To shift fermentation towards production of the flavor compounds and
2-phenyethanol specifically, removal of ethanol production through PDC genes disruption seemed as
an option. It was shown in Kondo, et al. (2012) on the example of another compound (isobutanol)
produced via Ehrlich pathway the influence of the PDC disruption on the desired compound yield.
PDC disruption and several other modification allowed to increase isobutanol production 13 fold
from 11 mg/l to 143 mg/l (Kondo, et al., 2012). In Stovicek, et al. (2015) PDC1 and PDC5 were
disruption in S. cerevisiae by the use of EasyClone – Markerfree kit, but it was shown that with the
disruption of these genes, cells were loosing the ability of growing on the glucose as sole carbon
source due to redox cofactor imbalance. To avoid that and balance NADH, L-lactate dehydrogenase
from Lactobacillus plantarum was introduced to the cells. This problem does not occur, if the goal is
to produce flavor compounds, since redox cofactor imbalance is not present. In terms of
2-phenylethanol production reaction of convertion of phenylacetaldehyde to 2-phenylethanol
balances the cofactors (Figure 2). The issue with finding the sequences of PDC in T. delbrueckii was
that after using BLASTp of the PDC1, PDC5 and PDC6 protein sequences, it was possible to find
only one alignment with the high score, which was alignment of PDC1 gene from S. cerevisiae to
T. delbruecki gene annotated as TDEL0E5080-1. It was hypothesised that due to evolution
T. delbruecki genes packed more together and other PDC genes were discarded. In relations of ARO10
as was demonstrated in Shen, et al. (2016) that overexpression of this gene leads to increased

48
production of 2-phenhylethanol. Moreover, in Chen, et al., 2017 it was demostrated that ARO10 is a
crucial gene in the production of 2-phenylethanol. It was also shown that under glucose limited
conditions and supplementation of phenylalanine, leucine or methionine as the sole nitrogen source,
ARO9 and BAT2 are consistently upregulated (Hazelwood, et al., 2008). Moreover, it was shown in
Godard, et al., 2007 that not only expression of ARO9 and BAT2 is highly induced by the presence of
aromatic amino acids but also ARO10 is highly induced. The other reason for overexpressing ARO10
was that this gene together with PDC genes are responsible for conversion of phenylpyruvate to
phenylacetaldehyde which then leads to 2-phenylethanol conversion and overxpression of ARO10
could shift the increase of 2-phenylethanol production (Figure 1).
Problematics occurred with the tries of integrating two different types of integration cassettes. The
multiple bands were occuring after gel electrophoresis of the samples obtained via colony PCR. The
main factor which could be considered in this case, is the low amount of the donor DNA supplied to
the transformation mixture and possibility of the integration cassette to be introduced to the different
genomic location. As it was represented in the experement in 3.4, another factor of not having the
integration of the cassette can be short homologous sites. They were altering only on 20 base pairs
compared to the experiment in 3.4 and that is why their size can be considered as one of the factors
of not having the cassette integrated to the desired location. The possible solution can be the drastic
increase of the DNA supplied to the transformation mix in order to increase the chance of the cassette
integration.
4.6 Construction of the integrative plasmids
The advantages of using this method of the yeast modification are: single-step targeted integration of
one or multiple expression cassettes and the convenient way of the removal of antibiotic selection
marker by growing modified cells with no presence of selection markers (Jessop-Fabre, et al., 2016).
Another advantage of this kit is the pre-designed Cas9 containing and gRNA containing vectors.
Moreover, gRNA containing vectors are complementary integrative vectors which makes this kit easy
to use. As it was shown in Mikkelsen et al., (2012) the chosen integration sites had the high expression
of the introduced gene and did not have any interference with the cellular growth. The regions for the
integration of the genes of interest into S. cerevisiae were taken as the model from
EasyClone – MarkerFree toolkit for finding the integration sites corresponding to the above
specifications in T. delbrueckii. The reason behind having the 1,000 base pairs down and upstream

49
from the ORF of the surrounded genes was to avoid unwanted interference in gene expression. The
presence of RNA expression regions was crucial since it was hypothesized that it can influence the
expression of the integrated gene in a positive matter. gRNA containing vectors were also constructed
in the complementary manner to integrative plasmids. It should also be noted, that barcode sequences
used in the plasmid are required for simplification of the further integrative plasmids modification. If
it is mandatory to introduce different gene of interest to the desired location it is possible to amplify
it with the primers containing barcodes and use Gibson assembly to introduce this amplified fragment
into desired plasmid.
4.7 Further perspectives
Future perspective connected to this project can be assigned to the several groups: RIM15 disruption,
2-phenylethanol production and the modification kit construction related.
Considering plasmid kit construction gene of interest such as ARO10 should be introduced to the
constructed integrative plasmids and the transformed to the yeast with the corresponding gRNA
plasmid to see the gene integration efficiency of EasyClone – Markerfree system constructed
specifically for T. delbrueckii
In the case of 2-phenylethanol production, it is important to investigate further the transformation
conditions and perform transformation with either higher concentration of integration cassette
supplied or increase the size of the homologous sites to 500 base pairs. After the obtainment of desired
mutations, fermentation tests should be performed under certain conditions in order to see influence
of the modifications on 2-phenylethanol production.
Finally, RIM15, the future perspectives are the further understanding of transformation problematics
and obtainment of the desired mutation in RIM15 gene. Then, the fermentation test should be
performed in order to see the impact of this gene disruption on the ethanol production

50
5. Conclusion All in all, it was possible to confirm that 500 base pair homologous sites and the highest molar
concentration is required for the successful introduction of the fragment into desired location of
T. delbrueckii. It is still possible if the homologous sites are 50 base pair long but not efficient. It was
not possible to disrupt RIM15 gene and more investigation are required to confirm what did happen
in the genetic region. It was not possible to disrupt PDC gene and introduce ARO10 into the desired
location and the reason for that can be short homologous sites (70 base pairs) and not sufficient
amount of the fragment added. Even though in all the cases it was possible to successfully introduce
plasmids and select the cells on the selection medium, further investigation is required for
understanding why the fragments were not integrated into desired locations. It was possible to identify
regions in T. delbrueckii for their utilization in the EasyClone – Markerfree toolkit, amplify 200 base
pairs of those regions and introduce them to the pJet plasmid. It was also achievable to linearize those
plasmids and prepare them for further introduction of desired genes.

51
Reference list (April 2009). Retrieved from Ensembl Fungi : https://fungi.ensembl.org/index.html (Accessed: 10.
June 2019)
Alam, M. T., Zelezniak, A., Mülleder, A., Shliaha, R., Schwarz, P., Capuano, F., Ralser, M. (2016).
The metabolic background is a global player in Saccharomyces gene expression epistasis.
Nature Microbiology, Volume: 1, Article number: 15030.
Basu, M. K. (2001). Scalable vector graphics plasmid map. Retrieved from Savvy version 0.1:
http://www.bioinformatics.org/savvy/ (Accessed: 10. June 2019)
Bolat, I., Romagnoli, G., Pronk, J. T., & Daran, J. M. (2013). Functional analysis and transcriptional
regulation of two orthologs of ARO10, encoding broad-substrate-specificity 2-oxo-acid
decarboxylases, in the brewing yeast Saccharomyces pastorianus CBS1483. FEMS yeast
research, Volume 13, Issue 6, Pages: 505-517.
Burgers, P. M., & Percival, K. J. (1987). Transformation of yeast spheroplasts without cell fusion.
Analytical biochemistry, Pages: 391-397.
Chang, D. C. (2006). Electroporation and Electrofusion, doi:
https://doi.org/10.1002/3527600906.mcb.200300026. I R. A. Meyers, Reviews in Cell
Biology and Molecular Medicine. Wiley-Blackwell.
Chen, B., Fan, K., & Fan, M.. Addgene: Cre-lox Plasmids. Retrieved from Addgene: The nonprofit
plasmid repository: https://www.addgene.org/cre-lox/ (Accessed: 10. June 2019)
Chen, X., Wang, Z., Guo, X., Liu, S., & He, X. (2017). Regulation of general amino acid permeases
Gap1p, GATA transcription factors Gln3p and Gat1p on 2-phenylethanol biosynthesis via
Ehrlich pathway. Journal of Biotechnology, Volume 242, Pages: 83-91.
Chreptowicz, K., Wielechowska, M., Główczyk-Zubek, J., Rybak, E., & Mierzejewska, J. (2016).
Production of natural 2-phenylethanol: From biotransformation to purified product. Food and
Bioproducts Processing, Volume: 100, Pages: 275-281.
Garavaglia, J., Flores, S. H., Pizzolato, T. M., Peralba, M. d., & Ayub, M. A. (2007). Bioconversion
of L-phenylalanine into 2-phenylethanol by Kluyveromyces marxianus in grape must cultures.
World Journal of Microbiology and Biotechnology, Volume: 23, Pages: 1273–1279.
Gietz, R. D., & Schiestl, R. H. (2007). High-efficiency yeast transformation using the LiAc/SS carrier
DNA/PEG method. Nature Protocols, Pages: 31-34.
Gietz, R. D., & Woods, R. A. (2003). Yeast Transformation by the LiAc/SS Carrier DNA/PEG
Method. Methods in Enzymology, doi: 10.1016/S0076-6879(02)50957-5.

52
Gietz, R. D., Schiestl, R. H., Willems, A. R., & Woods, R. A. (1995). Studies on the transformation
of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast, Volume: 11, Issue: 4, Pages:
355-360.
Godard, P., Urrestarazu, A., Vissers, S., Kontos, K., Bontempi, G., van Helden, J., & André, B.
(2007). Effect of 21 different nitrogen sources on global gene expression in the yeast
Saccharomyces cerevisiae. Molecular and cellular biology, Volume: 27, Issue: 8, Pages:
3065-3068.
González, B., Vázquez, J., Morcillo-Parra, M. Á., Mas, A., Torija, M. J., & Beltran, G. (2018). The
production of aromatic alcohols in non-Saccharomyces wine yeast is modulated by nutrient
availability. Food Microbiology, Volume: 74, Pages: 64-74.
Gupta, R. M., & Musunuru, K. (2014). Expanding the genetic editing tool kit: ZFNs, TALENs, and
CRISPR-Cas9. The Journal of Clinical investigation, Volume: 124, Issue: 10, Pages: 4154-
4161.
Hazelwood, L. A., Daran, J.-M., van Maris, A. J., Pronk, J. T., & Dickinson, J. R. (2008). The Ehrlich
Pathway for Fusel Alcohol Production: a Century of Research on Saccharomyces cerevisiae
Metabolism. Applied and Environmental Microbiology, Pages: 2259 - 2266.
Hernandez-Lopez, M. J., Blasco, A., Prieto, J. A., & Randez-Gil, F. (2003). Ura− host strains for
genetic manipulation and heterologous expression of Torulaspora delbrueckii. International
Journal of Food Microbiology, Volume: 86, Issues: 1-2, Pages: 79-86.
Hofman-Bang, J. (1999). Nitrogen catabolite repression in Saccharomyces cerevisiae. Molecular
Biotechnology, Volume 12, Issue 1, Pages: 35-73.
Holt, S., Mukhrjee, V., Lievens, B., Verstrepen, K. J., & Thevelein, J. M. (2018). Bioflavoring by
non-conventional yeasts in sequential beer fermentations. Food Microbiology, Volume: 72,
Pages: 55-66.
Hua, D., & Xu, P. (2011). Recent advances in biotechnological production of 2-phenylethanol.
Biotechnology Advances, Volume: 29, Pages: 654–660.
Iraqui, I., Vissers , S., Cartiaux, M., & Urrestarazu, A. (1998). Characterisation of Saccharomyces
cerevisiae ARO8 and ARO9 genes encoding aromatic aminotransferases I and II reveals a new
aminotransferase subfamily. Molecular and General Genetics, Volume 257, Issue 2, Pages:
238-248.

53
Jensen, Strucko, Kildegaard, David, Maury, Mortensen, & Borodina. (2014). EasyClone: method for
iterative chromosomal integration of multiple genes in Saccharomyces cerevisiae. FEMS
Yeast Research, Volume: 14, Pages: 238-248.
Jessop-Fabre, M. M., Jakočiūnas, T., Stovicek, V., Dai, Z., Jensen, M., Keasling, J., & Borodina, I.
(2016). EasyClone-MarkerFree: A vector toolkit for marker-less integration of genes into
Saccharomyces cerevisiae via CRISPR-Cas9. Biotechnology Journal, Volume: 11, Issue: 8,
Pages: 1110 - 1117.
Kawai, S., Hashimoto, W., & Murata, K. (2010). Transformation of Saccharomyces cerevisiae and
other fungi. Methods and possible underlying mechanism. Bioengineered bugs, Volume: 1,
Issue:6, Pages: 395–403.
Kim, T.-Y., Lee, S.-W., & Oh, M.-K. (2014). Biosynthesis of 2-phenylethanol from glucose with
genetically engineered Kluyveromyces marxianus. Enzyme and Microbial Technology 61-62,
Pages: 44-47.
Kondo, T., Tezuka, H., Ishii, J., Matsuda, F., Ogino, C., & Kondo, A. (2012). Genetic engineering to
enhance the Ehrlich pathway and alter carbon flux for increased isobutanol production from
glucose by Saccharomyces cerevisiae. Journal of Biotechnology, Pages: 32-37.
Lee, K.-S., Kim, J.-S., Heo, P., Yang, T.-J., Sung, Y.-J., Cheon, Y., Kweon, D.-H. (2013).
Characterization of Saccharomyces cerevisiae promoters for heterologous gene expression in
Kluyveromyces marxianus. Applied Microbiology and Biotechnology, Volume: 97, Issue: 5,
Pages: 2029-2041.
Li, T., Huang, S., Zhao, X., Wright, D. A., Carpenter, S., Spalding, M. H., Yang, B. (2011). Modularly
assembled designer TAL effector nucleases for targeted gene knockout and gene replacement
in eukaryotes. Nucleic Acids Research; Volume: 39, Issue: 14, Pages: 6315-6325.
Mans, R., Wijsman, M., Daran-Lapujade, P., & Daran, J.-M. (2018). A protocol for introduction of
multiple genetic modifications in Saccharomyces cerevisiae using CRISPR/Cas9. FEMS
Yeast Research, Volume: 18, Issue: 7, doi: https://doi.org/10.1093/femsyr/foy063.
Mikkelsen, M. D., Buron, L. D., Salomonsen, B., Olsen, C. E., Hansen, B. G., Mortensen, U. H., &
Halkier, B. A. (2012). Microbial production of indolylglucosinolate through engineering of a
multi-gene pathway in a versatile yeast expression platform. Metabolic engineering, Volume:
14, Issue: 2, Pages: 104-111.
Nevoigt, E. (2008). Progress in Metabolic Engineering of Saccharomyces cerevisiae. Microbiology
and molecular biology reviews, Volume: 72, Issue: 3, Pages: 379–412.

54
Pacheco, A., Almeida, M. J., & Sousa, J. M. (2009). Improved gene disruption method for
Torulaspora delbrueckii. FEMS Yeast Research, Volume: 9, Issue: 1, Pages: 158–160.
Patrick, M. (2014). Chapter 3: Eukaryotic Expression Vectors. I Plasmids 101: A Desktop Resource
(3rd Edition), Pages: 78-79.
Pires, E. J., Teixeira , J. A., Brányik, T., & Vicente, A. A. (2014). Yeast: the soul of beer’s aroma —
a review of flavour-active esters and higher alcohols produced by the brewing yeast. Applied
Microbiological Biotechnology, Pages: 1937–1949.
Pronk, J. T. (2002). Auxotrophic yeast strains in fundamental and applied research. Applied
Environmental Microbiology, Volume: 68, Issue: 5, Pages: 2095–2100.
Recek, N., Zhou, R., Zhou, R., Te’o, V. S., Speight, R. E., Mozetič, M., Ostrikov, K. (2018).
Improved fermentation efficiency of S. cerevisiae by changing glycolytic metabolic
pathways with plasma agitation, Volume: 8. Scientific reports, doi: 10.1038/s41598-018-
26227-5.
Rehman, L., Su, X., Guo, H., Qi, X., & Cheng, H. (2016). Protoplast transformation as a potential
platform for exploring gene function in Verticillium dahliae. BMC biotechnology, Volume:16,
Issue: 57, doi: 10.1186/s12896-016-0287-4.
Roa-Rodriguez, C. (2003). Promoters Used to Regulate Gene Expression. CAMBIA (Center for the
Application of Molecular Biology to International Agriculture).
Shen, L., Nishimura, Y., Matsuda, F., Ishii, J., & Kondo, A. (2016). Overexpressing enzymes of the
Ehrlich pathway and deleting genes of the competing pathway in Saccharomyces cerevisiae
for increasing 2-phenylethanol production from glucose. Journal of Bioscience and
Bioengineering, Volume 122, No. 1, Pages: 34-39.
Steele, D. F., & Jinks-Robertson, S. (1992). An examination of adaptive reversion in Saccharomyces
cerevisiae. Genetics, Volume: 132, Issue: 1, Pages: 9-21.
Sternberg, N., & Hamilton, D. (1981). Bacteriophage P1 site-specific recombination. I.
Recombination between loxP sites. Journal of Molecular Biology, Volume: 150, Issue: 4,
Pages: 467-486.
Stovicek, V., Borodina, I., & Forster, J. (2015). CRISPR–Cas system enables fast and simple genome
editing of industrial Saccharomyces cerevisiae strains. Metabolic Engineering
Communications, Volume: 2, Pages: 13-22.

55
Urrestarazu, A., Vissers, S., Iraqui, I., & Grenson, M. (1998). Phenylalanine- and tyrosine-
auxotrophic mutants of Saccharomyces cerevisiae impaired in transamination. Molecular and
General Genetics, Volume 257, Issue 2, Pages: 230-237.
Vickers, C. E., Bydder, S. F., Zhou, Y., & Nielsen, L. K. (2013). Dual gene expression cassette
vectors with antibiotic selection markers for engineering in Saccharomyces cerevisiae.
Microbial Cell Factories, Volume: 12, Issue: 96, doi: https://doi.org/10.1186/1475-2859-12-
96.
Walder, J. A. (1987). CRISPR-Cas9 guide RNA. Retrieved from Integrated DNA technologies, Inc.:
https://eu.idtdna.com/site/order/designtool/index/CRISPR_SEQUENCE (Accessed: 27. May
2019)
Watanabe, D., Kajihara, T., Sugimoto, Y., Takagi, K., Mizuno, M., Zhou, Y., Takag. (2018).
PP2AB55δ responsible for the high initial rates of alcoholic fermentation in sake yeast strains
of Saccharomyces cerevisiae. Applied And Environmental Microbiology, doi:
10.1128/AEM.02083-18.
Weaver, J. C., Smith, K. C., Esser, A. T., Son, R. S., & Gowrishankara, T. R. (2012). A brief overview
of electroporation pulse strength-duration space: A region where additional intracellular
effects are expected. Bioelectrochemistry, Volume: 87, Pages: 236–243.
Wickramasekara, S. (2016). Retrieved from Banchling Inc.: https://www.benchling.com (Accessed:
27. May 2019)
Yarimizu, T., Shimoi, H., Sameshima‐Yamashita , Y., Morita, T., Koike, H., Watanabe, T., &
Kitamoto, H. (2017). Targeted gene replacement at the URA3 locus of the basidiomycetous
yeast Pseudozyma antarctica and its transformation using lithium acetate treatment. Yeast,
Vulome: 32, Issue: 12, Pages: 483-494.










![Generation of Targeted Knockout Mutants in Arabidopsis ... · Keywords: CRISPR/Cas9, Genome editing, Arabidopsis thaliana, Plants, Knockout [Background] The CRISPR/Cas9 system (Cas9)](https://img.pdfslide.us/doc/110x75/5fcbdfb69ddbe939ee10f004/generation-of-targeted-knockout-mutants-in-arabidopsis-keywords-crisprcas9.jpg)