Embed Size (px)
Citation preview

Acta Oto-Laryngologica. 2013; 133: 1242–1249
ORIGINAL ARTICLE
Correlation analysis of genotypes, auditory function, and vestibular sizein Chinese children with enlarged vestibular aqueduct syndrome
FEI-FAN ZHAO*, LAN LAN*, DA-YONG WANG, BING HAN, YUE QI, YALI ZHAO,LIANG ZONG, QIAN LI & QIU-JU WANG
Department of Otorhinolaryngology/Head and Neck Surgery, Chinese People’s Liberation Army Institute ofOtolaryngology, Chinese People’s Liberation Army General Hospital, Beijing, China
AbstractConclusion: In children with enlarged vestibular aqueduct syndrome (EVAS), their hearing was more related to genotype thanVA size, and VA size was related to genotype. Objective: To study genotypes of the SLC26A4 gene, types and levels of hearingloss, and vestibular aqueduct (VA) size in children with EVAS. Methods: A total of 271 children with nonsyndromicsensorineural hearing loss and EVA underwent SLC26A4 gene screening. According to genotype typing, the phenotypesincluding pure tone average (PTA), distribution of subjects, and diameters of the external aperture and middle portion of theVA, were compared by t test or Pearson’s c2 tests. Further, divided by the dilated level of the VA, subject distribution indifferent hearing loss levels was compared by Pearson’s c2 test. Results: In all, 66 types of mutations were identified and 2 werenovel (c.665G >T and c.1639G >A). Biallelic genotype was found in 207 subjects, monoallelic in 56, and nomutation in 8. Thehearing loss was more stable in the subjects with monoallelic mutation than in other genotype groups. An air–bone gap wasmore frequently found in subjects with biallelic missense mutations than in other groups. The patients with no mutation hadthe most slightly enlarged VA. There was no dominant correlation between hearing loss level and VA size, and between VA sizeand different genotypes.
Keywords: SLC26A4, nonsyndromic sensorineural hearing loss, vestibular aqueduct
Introduction
Valvassori and Clemis first described the associationbetween enlargement of the vestibular aqueduct (EVA)and sensorineural hearing impairment (SHI) and sug-gested the term enlarged vestibular aqueduct syn-drome (EVAS, MIM 600791). It was considered tobe one of the most common forms of deafness and wasestimated to account for about 10–20% of all hered-itary hearing impairment (HI) [1]. Molecular etiologyresearch mapped the locus to 7q31, SLC26A4 gene.The audiological features of patients with EVAS
have been described in detail: the hearing can benormal or impaired from mild to profound, the hear-ing loss can be from fluctuant to progressive, andconductive hearing loss in low frequencies is present
in some patients. The radiological features have beenreported to range from simple EVA to abnormalitycomplexes comprising deformed cochlea, narrowinternal auditory canal, and large vestibular canal.To date, more than 240 mutations have been found
in the encoding region of the SLC26A4 gene inpatients with EVAS. The mutation spectra are distinctin different populations and regions.Many investigators have attempted to discover the
correlations between genotypes and phenotypes.Some conclude that EVA is usually associated withprofound hearing loss [2]. Some studies suggestedthat it was the number of mutations, but not the type,that influenced the phenotype, which meant biallelicmutations were more frequently found in patientswith bilateral EVA [3].
Correspondence: Qiu-Ju Wang MD PhD, Department of Otorhinolaryngology/Head and Neck Surgery, Chinese People’s Liberation Army Institute ofOtolaryngology, Chinese People’s Liberation Army General Hospital, Beijing, China. Fax: +86 10 55499143. E-mail: [email protected]*These authors contributed equally to this work.
(Received 7 May 2013; accepted 30 June 2013)
ISSN 0001-6489 print/ISSN 1651-2251 online � 2013 Informa HealthcareDOI: 10.3109/00016489.2013.822555
Act
a O
tola
ryng
ol D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y U
nive
rsity
of
Tex
as a
t Aus
tin o
n 11
/25/
14Fo
r pe
rson
al u
se o
nly.

Respecting the special mutation spectrum with apredominant mutation (c.919-2A >G) in Chinesepatients with EVAS, it is necessary to study thecorrelations between genotype and phenotype. Inthis paper, we describe the mutation spectrum andclinical, audiological, radiological, and geneticfeatures of a group of 271 children with EVAS.
Material and methods
Subject recruitment
Subjects with nonsyndromic SHI diagnosed withEVAS by temporal bone CT scan were sequentiallyaccrued from the otolaryngology department andgenetics unit from 2003 to 2008. Enlargement ofthe VA was defined when its antero-posterior diameterwas 1.5 mm or greater, as measured via CT scansmidway between the outer aperture and common crus.Informed consent, blood samples, and clinical evalua-tions were obtained from all of the participants accord-ing to the protocols approved by the InstitutionalReview Board of the Ethics Committee of the ChinesePeople’s Liberation Army General Hospital. Thepersonal information comprised a complete history,physical examinations, radiological examinations, andaudiometric examinations.A total of 271 subjects with EVAS from 16 pro-
vinces underwent SLC26A4 gene screening. Thefollow-up time was from 11 months to 4 years, andthe mean was 19 months.
Audiological examination and grouping criteria
In 271 subjects that underwent SLC26A4 genescreening, 266 subjects underwent pure tone orbehavioral audiometry, acoustic impedance admit-tance measurements using a Madsen 622-typepure-tone audiometer in a soundproof room. Audi-tory brainstem response (ABR) was determined usinga Smart EP-type evoked potential instrument (Intel-ligent Hearing Systems, USA) in an electric shieldingroom, as were the distortion product otoacousticemissions (DPOAEs); the DPOAEs of 95 childrenwere measured. The infants’ auditory steady-stateresponses (ASSRs) were evaluated to estimate thehearing threshold. According to the guidelines ofEuropean working group, Gendeaf recommenda-tions, average thresholds in the range of 21–40 dBon frequencies from 0.5 to 4 kHz (PTA0.5–4kHz) weredefined as mild hearing impairment (HI), in the rangeof 41–70 dB as moderate HI, in the range of 71–95 dBas severe HI, and > 95 dB as profound HI. Accordingto auditory examination, HI was described as stable orfluctuating. Fluctuation of hearing was defined as a
change in threshold of 15 dB or more at any frequencyor a 20 dB threshold change in the ABR, with afollow-up of at least 3 months. Stable state wasdefined as a decline in the audiometric thresholdsof less than 5 dB at more than three frequencies over afollow-up period of at least 3 months. Based on puretone audiometry at each frequency, a symmetricalaudiogram was defined if the difference betweenthe left and right ears was less than 10 dB at morethan three frequencies.
CT temporal bone measurements
Among 266 subjects who underwent SLC26A4 genescreening and the complete set of auditory evalua-tions, detailed measurement by high-resolutioncomputed tomography (HRCT) was applied in152 patients. Measurements were made with imagesenlarged 10–15 times with the use of currentworkstation software (Synapse; Fuji Film MedicalSystems, Stamford, CT, USA). The width of theVA was measured at the operculum and the midpoint.Measurements were taken using the electroniccalipers and recorded in millimeters. The largestopercular width or midpoint width on CT imagewas used for analyses.
SLC26A4 gene screening
Genomic DNA was extracted using the phenolextraction method. All of the 21 exons of theSLC26A4 gene, including the flanking sequences,were amplified by the polymerase chain reaction(PCR) using genomic DNA, and the primers weredesigned using online PRIMER 3.0 software. ThePCR amplified products were purified with Milliporeplate and then sequenced with an ABI 3730Sequencer (Applied Biosystems). Sequence datawere analyzed by aligning with the National Centerfor Biotechnology Information reference sequence ofSLC26A4 (NT_007933) using DNAStar 5.0 andBioEdit software. Mutations or polymorphismswere identified per the reference sequence.
Statistical analysis
The correlations of data were investigated using thePearson correlation coefficient, r. As metering data,PTA was compared by Student’s t test. For all anal-yses, a two-sided p value of 0.05 was consideredsignificant. All statistical calculations were performedusing SPSS for Windows, version 13.0 (LeadTechnologies Inc., Charlotte, NC, USA).Using the HI classification of mild, moderate,
severe, and profound, Pearson’s c2 testing was
Genotype and phenotype correlations in children with EVAS 1243
Act
a O
tola
ryng
ol D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y U
nive
rsity
of
Tex
as a
t Aus
tin o
n 11
/25/
14Fo
r pe
rson
al u
se o
nly.

performed to determine whether the frequency dis-tribution of HI was randomized in different genotypegroups (biallelic mutations, monoallelic mutation,and no mutation). Then, Fisher’s exact probabilitytesting with 2 � 2 contingency tables of appropriatelydichotomized data was performed to determine themost frequent class of HI in each group.
Results
Identification of mutations and genotype typing
In all, 66 types of mutations were found in 271 sub-jects, and 2 were novel. There were 24 mutations thatappeared more than twice (Figure 1) and 42 withallelic frequency of only once (data not shown). The66 mutations comprised missense mutations, non-sense mutations, and frameshift and splice sitemutations.Two heterozygotes carried the genotype of c.665G
>T and one heterozygote carried c.1639A >G. Thesetwo mutations were not found in dbSNP and notreported. c.665G >T caused replacement of glycine atposition 222 with valine (p.G222V), and c.1639A >Gcaused replacement of isoleucine at position 547 withvaline (p.I547V). The SIFT software predicted thefunctional changes of p.G222V to be damaging andp.I547V to be tolerated. Both the sequences areevolutionarily highly conserved in the pendrin proteinof humans, mice, rats, rhesus, dog, chick, opossum,zebrafish, and X_tropicalis based on the University ofCalifornia Santa Cruz Genome Bioinformatics(http://genome.ucsc.edu).Among 271 subjects, there were 207 with biallelic
mutations in the SLC26A4 gene. These 207 subjectswere further classified into those with 2 alleles oftruncating mutations (n = 82), with 2 alleles of het-erozygous truncating mutations (n = 91), and with2 alleles of missense mutations (n = 31). There were
56 monoallelic mutation carriers, and no mutationwas found in 8 subjects.
Results of audiological and radiological examinations
In most subjects, hearing loss level was unsymmetri-cal; the better laterality was taken as the reference, andhearing thresholds were evaluated in 266 subjects byPTA or auditory stable stimulated reaction (ASSR).The average hearing thresholds of 32 subjects withmild hearing loss, 73 subjects with moderate hearingloss, 102 subjects with severe hearing loss, and 59 sub-jects with profound hearing loss were 30, 67, 84, and103 dB, respectively. The mean value of the averagePTA was compared among biallelic, monoallelic, andno mutation groups.Among the 266 subjects, the width of the VA was
measured at the operculum and the midpoint on axialCT scan in 152 subjects. The midpoint mean was2.3 mm, ranging from 1 to 5.8 mm, and the opercu-lum mean was 3.5 mm, ranging from 1.9 to 8 mm.EVA with normal cochlea was found in 109 subjectsand EVA combined with Mondini’s deformity in thecochlea was found in 43 subjects.
Correlation between genotype and hearing impairment
There was no significant difference of PTA mean ingenotype groups (all p values > 0.05). The hearing losswas more stable in the subjects with monoallelicmutation than in other groups (p < 0.05). A total of76 subjects were tested by ASSR. An air–bone gapwas found in 149 of 190 subjects who underwent puretone audiometry, ranging from 5 to 95 dB, and themean was 55 dB. The air–bone gap was more frequentin subjects with biallelic missense mutations thanthose with biallelic truncating, heterozygous truncat-ing, or monoallelic mutation (all p values < 0.05).p values were calculated by c2 or Fisher’s exact test for
281C > T (5) 589G > A (3)
349delC (2) 626G > A (2) 1174A > T (9) 1343C > T (4) 1586T > G (2) 1746delG (2)
1707 + 5G > A (4)1594A > C (3)1226G > A (10)
1229C > T (11)
665G > T (2) 919-2A > G (266)
916dupG (4) 1336C > T (5)
1991C > T (2)
1990G > A (2)
1975G > C (16)
2027T > A (10) 2168A > G (57)1318A > T (2) 1692duA (3)907G > C (3)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
Figure 1. The allelic frequencies of mutations in Chinese children with enlarged vestibular aqueduct (EVA). This indicates the distribution ofmutations in 21 exons of the SLC26A4 gene. The 26 mutations were present more than twice in the homozygous state or in the heterozygousstate combined with other mutations, and the numbers in brackets are the allelic frequencies in 271 subjects.
1244 F.-F. Zhao et al.
Act
a O
tola
ryng
ol D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y U
nive
rsity
of
Tex
as a
t Aus
tin o
n 11
/25/
14Fo
r pe
rson
al u
se o
nly.
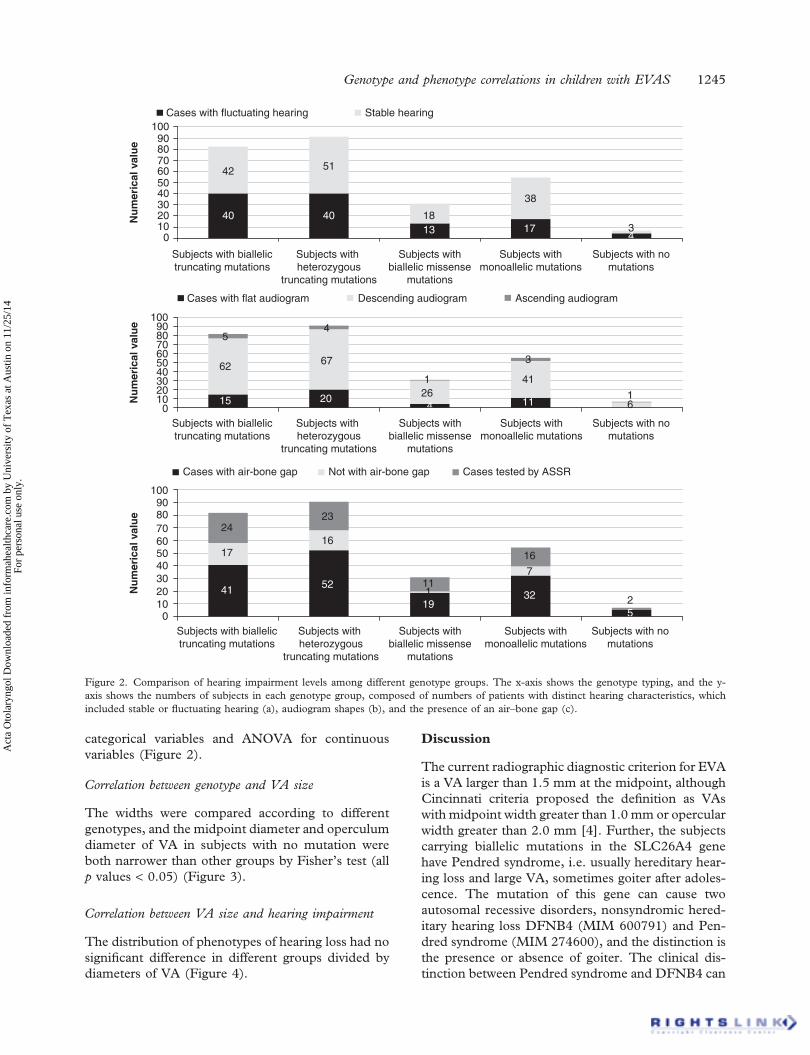
categorical variables and ANOVA for continuousvariables (Figure 2).
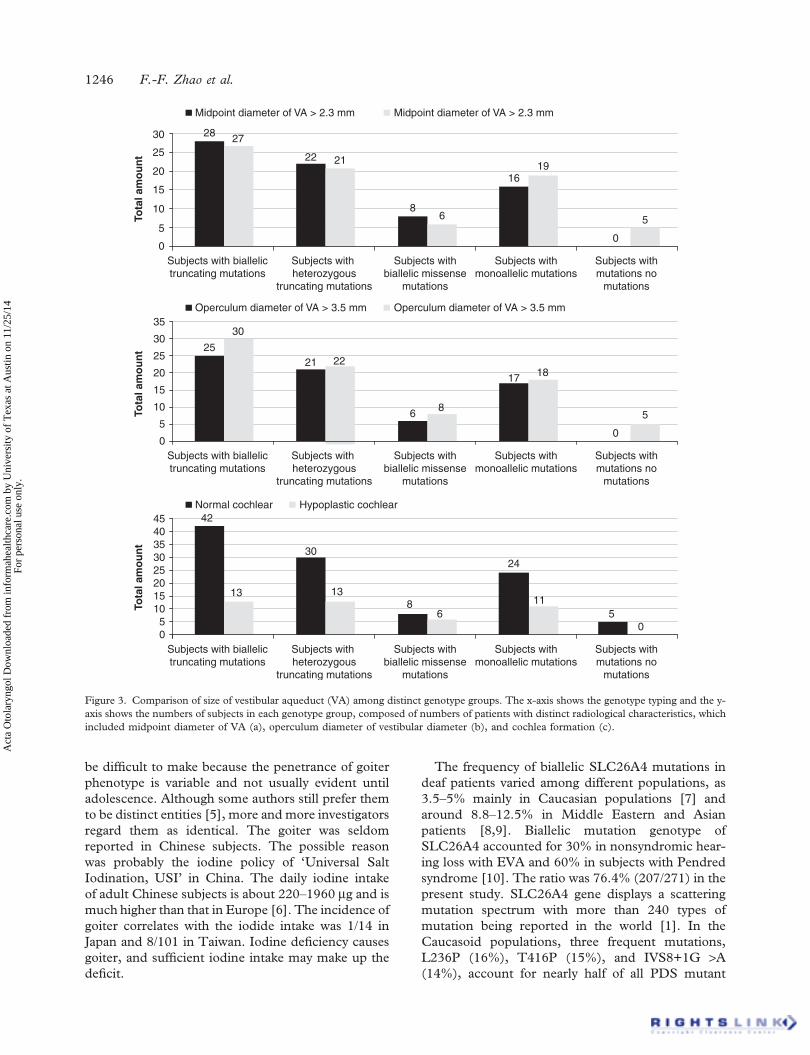
Correlation between genotype and VA size
The widths were compared according to differentgenotypes, and the midpoint diameter and operculumdiameter of VA in subjects with no mutation wereboth narrower than other groups by Fisher’s test (allp values < 0.05) (Figure 3).
Correlation between VA size and hearing impairment
The distribution of phenotypes of hearing loss had nosignificant difference in different groups divided bydiameters of VA (Figure 4).
Discussion
The current radiographic diagnostic criterion for EVAis a VA larger than 1.5 mm at the midpoint, althoughCincinnati criteria proposed the definition as VAswith midpoint width greater than 1.0 mm or opercularwidth greater than 2.0 mm [4]. Further, the subjectscarrying biallelic mutations in the SLC26A4 genehave Pendred syndrome, i.e. usually hereditary hear-ing loss and large VA, sometimes goiter after adoles-cence. The mutation of this gene can cause twoautosomal recessive disorders, nonsyndromic hered-itary hearing loss DFNB4 (MIM 600791) and Pen-dred syndrome (MIM 274600), and the distinction isthe presence or absence of goiter. The clinical dis-tinction between Pendred syndrome and DFNB4 can
Cases with fluctuating hearing
Cases with air-bone gap Cases tested by ASSRNot with air-bone gap
Cases with flat audiogram
Nu
mer
ical
val
ue
Nu
mer
ical
val
ue
Nu
mer
ical
val
ue
54
126
41
3
11416
62
100908070605040
1009080706050403020100
3020100
2423
16
52
17
4111
16
32 25
7
191
67
15 20
Descending audiogram Ascending audiogram
42
40
51
40 18
38
13 17 34
1009080706050403020100
Subjects with biallelictruncating mutations
Subjects with biallelictruncating mutations
Subjects with biallelictruncating mutations
Subjects with heterozygous
truncating mutations
Subjects with heterozygous
truncating mutations
Subjects with heterozygous
truncating mutations
Subjects withbiallelic missense
mutations
Subjects withbiallelic missense
mutations
Subjects withbiallelic missense
mutations
Subjects withmonoallelic mutations
Subjects withmonoallelic mutations
Subjects withmonoallelic mutations
Subjects with nomutations
Subjects with nomutations
Subjects with nomutations
Stable hearing
Figure 2. Comparison of hearing impairment levels among different genotype groups. The x-axis shows the genotype typing, and the y-axis shows the numbers of subjects in each genotype group, composed of numbers of patients with distinct hearing characteristics, whichincluded stable or fluctuating hearing (a), audiogram shapes (b), and the presence of an air–bone gap (c).
Genotype and phenotype correlations in children with EVAS 1245
Act
a O
tola
ryng
ol D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y U
nive
rsity
of
Tex
as a
t Aus
tin o
n 11
/25/
14Fo
r pe
rson
al u
se o
nly.
be difficult to make because the penetrance of goiterphenotype is variable and not usually evident untiladolescence. Although some authors still prefer themto be distinct entities [5], more and more investigatorsregard them as identical. The goiter was seldomreported in Chinese subjects. The possible reasonwas probably the iodine policy of ‘Universal SaltIodination, USI’ in China. The daily iodine intakeof adult Chinese subjects is about 220–1960 mg and ismuch higher than that in Europe [6]. The incidence ofgoiter correlates with the iodide intake was 1/14 inJapan and 8/101 in Taiwan. Iodine deficiency causesgoiter, and sufficient iodine intake may make up thedeficit.
The frequency of biallelic SLC26A4 mutations indeaf patients varied among different populations, as3.5–5% mainly in Caucasian populations [7] andaround 8.8–12.5% in Middle Eastern and Asianpatients [8,9]. Biallelic mutation genotype ofSLC26A4 accounted for 30% in nonsyndromic hear-ing loss with EVA and 60% in subjects with Pendredsyndrome [10]. The ratio was 76.4% (207/271) in thepresent study. SLC26A4 gene displays a scatteringmutation spectrum with more than 240 types ofmutation being reported in the world [1]. In theCaucasoid populations, three frequent mutations,L236P (16%), T416P (15%), and IVS8+1G >A(14%), account for nearly half of all PDS mutant
Midpoint diameter of VA > 2.3 mm
Tota
l am
ou
nt
27
22 21
86
1619
0
5
2830
20
5
10
15
25
0Subjects with biallelictruncating mutations
Subjects with heterozygous
truncating mutations
Subjects withbiallelic missense
mutations
Subjects withmonoallelic mutations
Subjects withmutations no
mutations
Midpoint diameter of VA > 2.3 mm
Operculum diameter of VA > 3.5 mm
Tota
l am
ou
nt
30
21 22
6 8
17 18
0
5
25
35
30
20
5
10
15
25
0Subjects with biallelictruncating mutations
Subjects with heterozygous
truncating mutations
Subjects withbiallelic missense
mutations
Subjects withmonoallelic mutations
Subjects withmutations no
mutations
Operculum diameter of VA > 3.5 mm
Normal cochlear
Tota
l am
ou
nt
13
30
13
68
24
115
0
42
354045
30
20
51015
25
0Subjects with biallelictruncating mutations
Subjects with heterozygous
truncating mutations
Subjects withbiallelic missense
mutations
Subjects withmonoallelic mutations
Subjects withmutations no
mutations
Hypoplastic cochlear
Figure 3. Comparison of size of vestibular aqueduct (VA) among distinct genotype groups. The x-axis shows the genotype typing and the y-axis shows the numbers of subjects in each genotype group, composed of numbers of patients with distinct radiological characteristics, whichincluded midpoint diameter of VA (a), operculum diameter of vestibular diameter (b), and cochlea formation (c).
1246 F.-F. Zhao et al.
Act
a O
tola
ryng
ol D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y U
nive
rsity
of
Tex
as a
t Aus
tin o
n 11
/25/
14Fo
r pe
rson
al u
se o
nly.

alleles [11]. p.H723R was a common allele in theKorean population, while p.L676Q was common inthe Mongolian population [1]. H723R accounted for53% of PDSmutations in Japanese subjects [12]. Thiswas the secondmost frequent mutation in the Chinesepopulation, which was present mostly in heterozygousand homozygous c.919-2A >G forms. However, theincidence of this mutation was found to be low inWestern countries and has been identified in only fourfamilies [13]. This difference suggested the existenceof a common ancestor (founder effect) rather than amutational hot spot [1].Two novel mutations were found in the present
study. The mutation c.665G >T (p.G222V) waspredicted to be damaging to pendrin by SIFT soft-ware, while the effect of p.I547V (c.1639A >G) on
pendrin was tolerated. In consideration of both thesequences involving the two mutations being highlyconserved in multiple species, both changes weresupposed to jeopardize the protein function. More-over, c.665G >T was found in two unrelated subjects,while c.1639A >G was found only once. Thus, basedon the combined evidence, c.665G >T was thought tobe a mutation, while c.1639A >G was more like avariation.Some studies showed that different kinds of muta-
tions could produce different phenotypes [14]. Butthere were at least 19 shared mutations betweenPendred syndrome, and autosomal recessive hearingloss associated with EVA has been reported [15].Wu et al. found hearing loss in subjects with the
biallelic genotype to be more severe than in subjects
Cases with fluctuating hearing
25
19
41
27 18
8113
80706050403020100
1.5−2 (44) 2−3 (68)
Mid-portion diameter (mm)
3−4 (26) > 4 (14)
Stable hearing
35
4
5
16
3
14
1
6
19 1
112
70Cases with flat audiogram
Cases with air-bone gap
Cases with flat audiogram Ascending audiogram
Not with air-bone gap Cases tested by ASSR
60
50
40
30
20
10
01.5−2 (44) 2−3 (68)
Mid-portion diameter (mm)
3−4 (26) > 4 (14)
611
27
8
14
46 12
113
2
122
7080
6050403020100
1.5−2 (44) 2−3 (68)
Mid-portion diameter (mm)
3−4 (26) > 4 (14)
Figure 4. Comparison of levels of hearing impairment among distinct VA size groups. The x-axis shows the vestibular size typing by midpointdiameter, and the y-axis shows the numbers of subjects in different size groups, composed of numbers of patients with distinct hearingcharacteristics, which included stable or fluctuating hearing (a), audiogram shapes (b), and the presence or absence of an air–bone gap (c).
Genotype and phenotype correlations in children with EVAS 1247
Act
a O
tola
ryng
ol D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y U
nive
rsity
of
Tex
as a
t Aus
tin o
n 11
/25/
14Fo
r pe
rson
al u
se o
nly.

with the monoallelic genotype [16]. The present studyalso showed no distinct difference of hearing loss levelin different genotypes, not only among subjects withbiallelic mutation genotype, monoallelic, and nomutation, but also among subjects with biallelic trun-cating mutation genotype, biallelic heterozygous trun-cating, and biallelic missense. However, we foundthat the subjects with monoallelic missense mutationsshowed more stable hearing loss than those with othergenotypes, which could be explained by the truegenotype effect or the interference of unstable hearingstatus because of young ages.Some studies found a significant difference in
mutation distribution between patients with EVAand those with Mondini’s deformity. In addition,bilateral malformations are also more frequently asso-ciated with the Mondini’s deformity phenotype groupas compared with the EVA phenotype [17], but moststudies did not show correlations between hearing lossand aqueduct size [18]. In addition, there were morethan 31 shared mutations [8,19]. We found that therewas no significant difference in the distribution ofhearing loss level among distinct genotypes. Also, themidpoint diameter and operculum diameter of VA insubjects with no mutation were narrower than othergenotype groups, which implied an effect ofSLC26A4 gene mutations on VA size.The air–bone conduction gap in low frequencies
was reported to be discovered in subjects with EVAS.A large VA may act as a third mobile window in theinner ear, resulting in an air–bone gap at low frequen-cies [20]. Our data showed that 149 of 190 subjectswho underwent pure tone audiometry had an air-bone gap, but no difference in distribution of subjectswith air–bone gap was identified according to thedilated level of the VA. In addition, the air–bonegap was more frequent in subjects with biallelic mis-sense mutations than in those with other genotypes.This finding implies that the air–bone conduction gapwas more related to genotypes than the VA size. Thisresult suggests that the membranous structures in theVA should be explored rather than bone structures.In other words, the endolymphatic sac and theintracellular ion concentration in the formation ofthe air–bone gap should not be ignored.Although we found some correlations between
genotype and phenotype, this is far from provingdirect correlation between the mutation genotypeand hearing loss level or EVA size.
Acknowledgments
We thank the patients and their families for theircooperation during this work. This work was sup-ported by grants from the National Natural Science
Foundation of China, Major Project, No.81120108009 and National Natural Science Foun-dation of Youth Science Foundation No.81100719.
Declaration of interest: The authors report noconflicts of interest. The authors alone are responsiblefor the content and writing of the paper.
References
[1] Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, etal. Origins and frequencies of SLC26A4 (PDS) mutations ineast and south Asians: global implications for the epidemi-ology of deafness. J Med Genet 2003;40:242–8.
[2] Harnsberger HR, Dahlen RT, Shelton C, Gray SD,Parkin JL. Advanced techniques in magnetic resonance imag-ing in the evaluation of the large endolymphatic duct and sacsyndrome. Laryngoscope 1995;105:1037–42.
[3] King KA, Choi BY, Zalewski C, Madeo AC, Manichaikul A,Pryor SP, et al. SLC26A4 genotype, but not cochlear radio-logic structure, is correlated with hearing loss in ears with anenlarged vestibular aqueduct. Laryngoscope 2009;120:384–9.
[4] Boston M, Halsted M, Meinzen-Derr J, Bean J,Vijayasekaran S, Arjmand E, et al. The large vestibularaqueduct: a new definition based on audiologic and com-puted tomography correlation. Otolaryngol Head Neck Surg2007;136:972–7.
[5] Pryor SP, Madeo AC, Reynolds JC, Sarlis NJ, Arnos KS,Nance WE, et al. SLC26A4/PDS genotype-phenotype cor-relation in hearing loss with enlargement of the vestibularaqueduct (EVA): evidence that Pendred syndrome and non--syndromic EVA are distinct clinical and genetic entities.J Med Genet 2005;42:159–65.
[6] Delange F. Iodine deficiency in Europe and its conse-quences: an update. Eur J Nucl Med Mol Imaging 2002;29:S404–16.
[7] Madden C, Halsted M, Meinzen-Derr J, Bardo D,Boston M, Arjmand E, et al. The influence of mutationsin the SLC26A4 gene on the temporal bone in a populationwith enlarged vestibular aqueduct. Arch Otolaryngol HeadNeck Surg 2007;133:162–8.
[8] WangQJ, Zhao YL, Rao SQ,Guo YF, YuanH, Zong L, et al.A distinct spectrum of SLC26A4 mutations in patients withenlarged vestibular aqueduct in China. Clin Genet 2007;72:245–54.
[9] Wu CC, Lin SY, Su YN, Fang MY, Chen SU, Hsu CJ.Preimplantation genetic diagnosis (embryo screening) forenlarged vestibular aqueduct due to SLC26A4 mutation.Audiol Neurootol 2010;15:311–17.
[10] Pera A, Villamar M, Vinuela A, Gandia M, Meda C,Moreno F, et al. A mutational analysis of theSLC26A4 gene in Spanish hearing-impaired families pro-vides new insights into the genetic causes of Pendred syn-drome and DFNB4 hearing loss. Eur J Hum Genet 2008;16:888–96.
[11] Campbell C, Cucci RA, Prasad S, Green GE, Edeal JB,Galer CE, et al. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possi-ble genotype-phenotype correlations. Hum Mutat 2001;17:403–11.
[12] Usami S, Abe S, Weston MD, Shinkawa H, Van Camp G,Kimberling WJ. Non-syndromic hearing loss associated with
1248 F.-F. Zhao et al.
Act
a O
tola
ryng
ol D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y U
nive
rsity
of
Tex
as a
t Aus
tin o
n 11
/25/
14Fo
r pe
rson
al u
se o
nly.

enlarged vestibular aqueduct is caused by PDS mutations.Hum Genet 1999;104:188–92.
[13] Van Hauwe P, Everett LA, Coucke P, Scott DA, Kraft ML,Ris-Stalpers C, et al. Two frequent missense mutations inPendred syndrome. Hum Mol Genet 1998;7:1099–104.
[14] Choi BY, Stewart AK, Madeo AC, Pryor SP, Lenhard S,Kittles R, et al. Hypo-functional SLC26A4 variants associ-ated with nonsyndromic hearing loss and enlargement of thevestibular aqueduct: genotype-phenotype correlation or coin-cidental polymorphisms? Hum Mutat 2009;30:599–608.
[15] Tsukamoto K, Suzuki H, Harada D, Namba A, Abe S,Usami S. Distribution and frequencies of PDS (SLC26A4)mutations in Pendred syndrome and nonsyndromic hearingloss associated with enlarged vestibular aqueduct: a uniquespectrum of mutations in Japanese. Eur J Hum Genet 2003;11:916–22.
[16] Wu CC, Lu YC, Chen PJ, Yeh PL, Su YN, Hwu WL, et al.Phenotypic analyses and mutation screening of the
SLC26A4 and FOXI1 genes in 101 Taiwanese familieswith bilateral nonsyndromic enlarged vestibular aqueduct(DFNB4) or Pendred syndrome. Audiol Neurootol 2009;15:57–66.
[17] Azaiez H, Yang T, Prasad S, Sorensen JL, Nishimura CJ,Kimberling WJ, et al. Genotype-phenotype correlations forSLC26A4-related deafness. Hum Genet 2007;122:451–7.
[18] Gopen Q, Zhou G, Whittemore K, Kenna M. Enlargedvestibular aqueduct: review of controversial aspects. Laryn-goscope 2011;121:1971–8.
[19] Iwasaki S, Tsukamoto K, Usami S, Misawa K, Mizuta K,Mineta H. Association of SLC26A4 mutations with clinicalfeatures and thyroid function in deaf infants with enlargedvestibular aqueduct. J Hum Genet 2006;51:805–10.
[20] Merchant SN, Nakajima HH, Halpin C, Nadol JB Jr,Lee DJ, Innis WP, et al. Clinical investigation and mecha-nism of air-bone gaps in large vestibular aqueduct syndrome.Ann Otol Rhinol Laryngol 2007;116:532–41.
Genotype and phenotype correlations in children with EVAS 1249
Act
a O
tola
ryng
ol D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y U
nive
rsity
of
Tex
as a
t Aus
tin o
n 11
/25/
14Fo
r pe
rson
al u
se o
nly.





























![Aqueduct en[1]](https://img.pdfslide.us/doc/110x75/557e9f8ed8b42a1d048b535e/aqueduct-en1.jpg)