Embed Size (px)
Citation preview

U. S. Department of CommerceNational Bureau of Standards
Research Paper RP1937Volume 41, November 1948
Part of the Journal of Research of the National Bureau of Standards
Copolymerization*By Robert Simha and Leo A. Wall
A critical discussion of the mechanism of formation of copolymers by addition polymeri-
zation is presented. It deals mainly with the following three fundamental aspects: First,
the quantitative treatment of the reaction starting with a scheme consisting of initiation,
growth, and termination mechanisms. Expressions for the instantaneous and total copolymer
composition and for the over-all rate of reaction as function of monomer composition and
of conversion are derived in terms of quantities characteristic of the reaction. Graphical
and numerical methods for the determination of these parameters from experimental data
are given in detail. The statistical distribution of molecular weights and compositions in
the product is considered in relation to the constants of the reaction and to the analogous
case of simple polymers.
Second, methods for the analysis of copolymer compositions are discussed and experi-
mental results are summarized. Reactivity ratios describing the behavior in growth of a
given radical toward a pair of monomers are tabulated for a series of systems.
Third, these results are interpreted on the basis of resonance and of electrostatic and
steric effects as encountered in the study of certain organic reactions.
In addition, degradation of copolymers is briefly considered in the light of the possible
types of sequences in the chain. A numerical relation between yield and copolymer com-
position is derived.
The p r o b l e m s remaining are principally the following: Experimental methods of
copolymer analysis, determination of over-all rates of reaction and of individual rate con-
stants, and a more fundamental correlation between structure of monomers and behavior
in copolymerization. Also, systematic data on the thermodynamic and rate properties of
copolymer solutions should be of great interest, and studies of the bulk properties and their
relation to copolymer structure represent a field where research has only recently been
initiated.
I. Introduction
Following the extensive experimental and theo-retical attack on the general problem of chainpolymerization reactions, recent years havebrought a series of fundamental investigationsregarding copolymerization reactions. A frame-work for the analysis of over-all monomer con-sumption and resulting change in average polymercomposition has been created [1, 18, 26, 31]2 andtested experimentally [5, 18]. Equations for thesize and composition distributions have also beendeveloped [26, 28]. However, no systematic ex-perimental results on such distributions are avail-
1 This article is scheduled to appear as a chapter in a forthcoming AmericanChemical Society Monograph, published by the Eeinhold Publishing Co.,New York, N. Y.
2 Figures in brackets indicate the literature references at the end of thispaper.
able at present. Finally, interpretations of thedifferences in relative reactivities have been madeon the basis of the electronic structure of theindividual monomers [15, 24]. It is the purposeof this article to review the main points of thesetheoretical and experimental investigations. Inrespect to the former, stress will be laid on thoseaspects which also have been examined experi-mentally.
II. List of Important Symbols
A, B = respective numbers of monomer molecules ofcopolymerizing species. Also used to indi-cate species.
Ao, Bo • = initial values of A and B.z = ratio A/B.Zo = initial value of z.C = catalyst concentration.
Copolymerization 521

nra(A) = number of radical chains with an active A-end containing r ^4-units and s fi-units.
nr8(B) = number of radical chains with an active fi-end, containing r ^4-units and s fi-units.
A* = total number, 2 J nrs(A) of radical chainsr,s
with an active ^L-end.B* = total number, ]>^ nrs(B) of radical chains
r,s
with an active B-end.Nrs = number of stable polymer chains contain r A-
units and s fi-units.IA = first order rate constant for initiation of mono-
mer species A.IB = first order rate constant for initiation of mono-
mer species B.I =rate constant for production of radicals from
catalyst.kgA{A) =rate constant for propagation by addition of
^L-monomer to a radical with an active A-end.
kgB(A) =rate constant for propagation by addition of5-monomer to a radical with an active A-end.
kgA(B) =rate constant for propagation by addition ofi-monomer to a radical with an active fi-end.
kgB(B) =rate constant for propagation by addition offi-monomer to a radical with an active fi-end.
kgA(r,s,^4)=rate constant for propagation defined in thesame manner as kgA{A) above, but depend-ent on composition r,s of growing radical.
kt(A, A) =rate constant for mutual termination of tworadicals with active ^4-ends.
kt(A, B) =rate constant for mutual termination of tworadicals with active A- and fi-ends, respect-ively.
kt(B, B) =rate constant for mutual termination of tworadicals with active fi-ends.
a = reactivity ratio kgA{A)lkgB(A)n = reactivity ratio kgB{B)lkgA{B)VA -\-VB = over-all rate of copolymerization per unit
number of radicals.wv = total weight of polymer at a given instant.wo = initial weight of monomer.I = degree of polymerization of radical or stable
polymer.y = composition deviation of individual chain
from the mean.w(l,y) = distribution function of composition and chain
length expressed as weight fraction in termsof above quantities I and y.
X = number of average degree of polymerization ofradical chains.
Pi(A) = probability of occurence of i ^.-units in suc-cession in a copolymer chain.
Pi(B) = probability of occurence of i fi-units in suc-cession in a copolymer chain.
co (A) = probability of formation of an A-A linkage bypropagation.
co(fi) = probability of formation of B-B linkage bypropagation.
III. Quantitative Treatment
1. General Remarks on Chain PolymerizationReactions
It is well established today that initiation,growth, and termination are the principal, al-though not necessarily the only mechanisms thatdetermine the kinetics of chain polymerizationreactions. The rates of these individual stepsvary widely. The growth reaction is the fastest.The initiation, which produces, by one means oranother, out of a stable monomer an activatedradical is by far the slowest step, whenever longchains are formed. Otherwise, the supply ofactive monomer would be too large comparedwith the demand of the growth reaction for stablemonomer.
The crucial step then, to begin with, is the pro-duction of a certain number of radicals able togrow before they are terminated. Their tota]concentration is determined by the initiation andtermination only, since the growth merely changestheir molecular weight. If they are terminatedmuch faster than they are produced, an equilibriumis established. The exact condition for this tobe true requires the mean life time of the activeradicals to be small in comparison with that ofstable monomer. This defines a quasistationarystate and allows the expression of the "steadystate" concentration of free radicals by means ofan "equilibrium" constant given by the ratio
Equilibrium _ rate of production of free radicalsconstant ~~rate of destruction of free radicals
This additional, and in most cases of interest,valid assumption simplifies considerably thequantitative treatment. It is then possible todevelop completely the kinetics of the polymeriza-tion reaction and the resulting molecular sizedistribution [12] on the basis of a postulated reac-tion mechanism.
It will be shown here how to carry this programthrough when two or more competing monomerspecies are present. Additional problems thenarise. One of the most important questions isconcerned with the change in average polymercomposition with changing composition ofmonomer residue. The mean composition de-pends on the relative rate with which the different
522 Journal of Research

species enter the growing chain and hence, uponthe relative growth rates, if we exclude theinsignificant number of dimers, trimers, and othervery short chains. In addition to the inhomo-geneity in respect to chain length necessarilyoccuring in polymerizing systems, there will nowexist also fluctuations of the composition fromchain to chain. These fluctuations depend uponthose in the long chain radicals. All three stepsare essential for the determination of the distribu-tion curves.
2. Basic Reaction Scheme and EquationsAfter these general remarks, we turn to the
treatment of the copolymerization of a binarysystem, assuming the simplest possible reactionscheme, as indicated above. No kinetic studiesexist at present that would necessitate the consid-eration of additional elementary acts, as is thecase for one-component systems. In view ofwhat was said previously this would not effectthe calculation of average polymer compositions.In what follows, let nrs(A) be the number ofgrowing radical chains, each of which containsaltogether r units of component A and s units ofcomponent B, while having an activated endconsisting of an ^L-type monomer. nrs(B) is thencorrespondingly defined. Nrs represents the num-ber of stabilized chains of specified composition.C is a catalyst molecule and R a radical producedby its decomposition. The following scheme maythen be considered.Initiation:
>nlo(A); B mOiCB)
Growth:C
kgA(A)nrs(A)+A >nri.hs(A)
kgB{A)nrs(A)+B mr,s+1(B)
kgA{B)nrs(B) +A >nr+1>s(A)
kgB(B)nrs(B)+B >nr>sH(B)
Termination:kt{AyA)
nrs(A)+nik(A) > Nr+itS+k or Nrs+Nik
HA,B)nrs(A)+nik(B) > Nr+i.8+* or Nrs+Nik
kt(B,B)nrs(B)+nik(B) > Nr+Wk or Nrs+Nik J
(1)
In writing the initiation equation we have con-sidered two types of activation. In the catalyzedactivation the elementary act consists in a de-composition of the catalyst C. In the equationsfor growth and termination, we have differentiatedbetween A-A, A-B, B-A, and B-B addition ofmonomer to radical and radical to radical, respec-tively. The rates of consumption of monomerare given according to eq 1 by
nrs(A)+klA(B) S nt.(B)
TfS (2)
The terms IA and IB are omitted for a catalyzedpolymerization. Otherwise, they are small andcan be neglected in comparison with the growthterms in eq 2. The summations are carried outover all values of r and s. The concentrations offree radicals 2nr8(A) and 2nrs(B) in a steadystate obey the following relations if the initiationterm is omitted:
kt(A,
0. (3)
An analogous equation results for 2nrs(B).Equation 3 expresses the fact that ^1-type radicalsare produced by addition of ^L-type monomer toJ?-type radicals, and they are destroyed by addingB-type monomer and by termination with an A—or 5-type radical. The terms contributed by thechain-breaking reaction are small compared withthe growth terms in eq 3, if long chains are to beformed. Hence, they may be neglected, and weobtain the simple relation:
(3a)
It expresses the fact that free radicals with anactive end A are produced as rapidly by additionof monomer A to free radicals with an active endB as they are destroyed by addition of monomerB to free radicals with an active end A.
3. Compositional Relationships: Average Com-position, Relation to Conversion
Insertion of eq 3a into eq 3 leads to the following
Copolymerization 523

relation for the change in composition of monomerresidue :
dA=A ku(B)_ kgA(A)A+kgB(A)BdB B kgB(A) kgA(B)A+kgB(B)B (4)
It has been assumed in eq 1 that the rate con-stants are essentially independent of chain length,an assumption commonly made in chain polymer-ization reactions and shown to be true in polycon-densation reactions [II].3 Furthermore, they areindependent of chain composition r,s in eq 1.Herington and Robertson [12] have establishedequations that allow in principle a deduction ofsuch a dependence from molecular-weight dis-tributions. Analogous relations for copolymeriz-ing systems have been developed by Simha andBranson [26]. However, the equations are toocomplex and experimental results nonexistent tomerit further discussion here. It may be notedonly that eq 4 is unaffected by any such assump-tion. If the growth rates depend upon the com-position, then the constants kgL(M) in eq 4 repre-sent mean values averaged over the radicals nrs{M)with the particular end M, that is, for instance:
7 / A\—kgA(A)-
Equation 4 gives the relative rates of consump-tion of the two monomer species A and B underthe approximations stated previously. It alsorepresents the instantaneous average compositionof copolymer formed at an instant in which themonomer residue consisted of A moles of speciesone and B moles of species two. This compositioncan not depend on the absolute magnitude of thegrowth rates kg but only upon the relative ratesof addition of each monomer species. Hence,the following parameters are defined [18:]
+ B1 JceB{B)
(5)
The symbols in the brackets indicate the modes ofaddition to which the constants a and n refer.4
3 H. W. Melville (lecture presented at the National Bureau of Standards onApril 11, 1947) finds that growth and termination rates decrease but slightlywith increasing chain length in the polymerization of vinyl acetate in theliquid phase.
4 Various other symbols have been used for the ratios denoted here by <rand n, namely, I/a, p [1], <r, 9 [3], a, 0[28], n, r2 (T. Alfrey, F . R. Mayo F., T.Wall, J. Polymer Sci. 1, 581 (1946).
Both parameters express the growth rate bymeans of addition of monomer of the same kindto an activated end relative to the addition of amonomer of the other kind. Defining further-more the mole ratio A/B as z, eq 4 is transformedinto
dA ((72+1)dB (z+n) dA+dB~
dA r i (g+jQT1
L(4a)
Figures 1, 2, and 3 show plots of eq 4a as afunction of the mole fraction A/A+B in themonomer residue for a set of various values ofthe parameters a and JJL. The straight line corres-ponds to a situation in which monomer composi-tion and instantaneous polymer composition are
.2 .8 .9 10.3 .4 .5 .6 .7
A/A+B MONOMER
FIGURE 1. Plot of instantaneous mole fraction of com-ponent A in polymer as function of mole fraction of A inmonomer mixture.
A, n=l, <r=l; B , M = 2 , <r=l; C, n=5, <r=l; D , n = 5, <r=H; E , n = 5 , <r=Vo\
F , M = 5 , <r=0; G, M = 1 0 , o-=0.
always equal. That is, a and /JL must both equalunity. In some instances, (figs. 2 and 3) it will benoted that the curves intersect the diagonal,indicating that for a particular monomer chargethe above-mentioned equality between the twocompositions holds true. The meaning of thisspecial case is discussed below. It is easy tovisualize the limiting trends of these curves. Forinstance, large /J, and small a, that is, preferentialB-B and A-B addition must have the effect of
524 Journal of Research
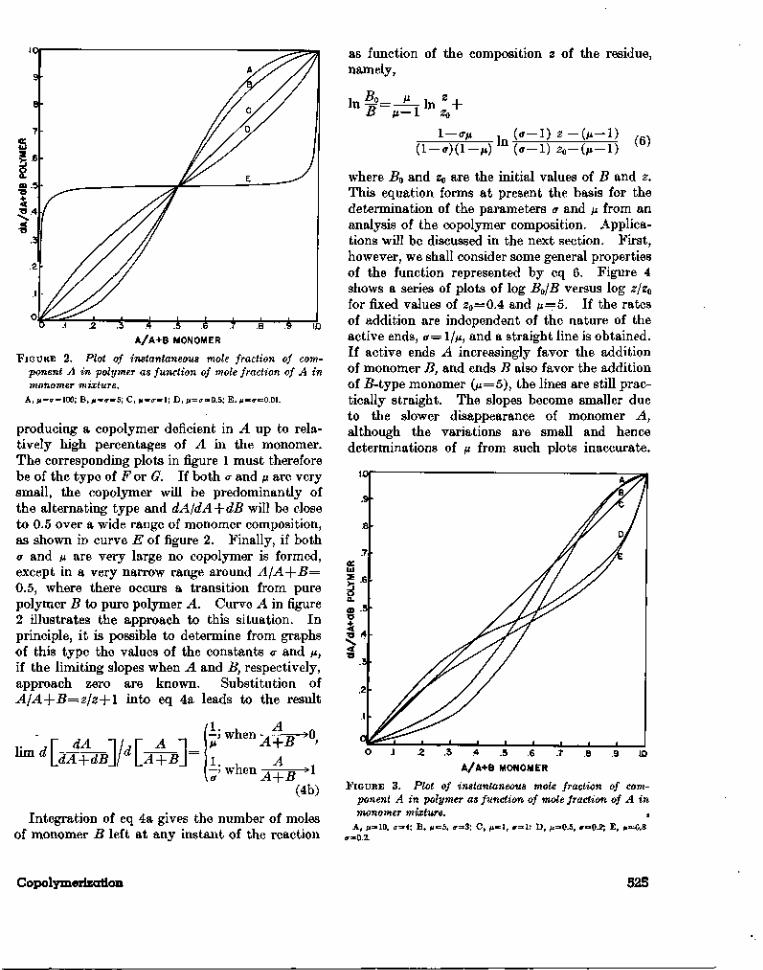
.7 .8 .9 1.0.5 .6A/A+B MONOMER
FIGURE 2. Plot of instantaneous mole fraction of com-ponent A in polymer as function of mole fraction of A inmonomer mixture.
A, M=<r=l00; B, AI=O-=5; C, ix=a=l; D, M=<r=0.5; E? M=<r=O.OL
producing a copolymer deficient in A up to rela-tively high percentages of A in the monomer.The corresponding plots in figure 1 must thereforebe of the type of F or G. If both a and /x are verysmall, the copolymer will be predominantly ofthe alternating type and dA/dA-{-dB will be closeto 0.5 over a wide range of monomer composition,as shown in curve E of figure 2. Finally, if both<r and fx are very large no copolymer is formed,except in a very narrow range around A/A-{-B=0.5, where there occurs a transition from purepolymer B to pure polymer A. Curve A in figure2 illustrates the approach to this situation. Inprinciple, it is possible to determine from graphsof this type the values of the constants a and /z,if the limiting slopes when A and B, respectively,approach zero are known. Substitution ofA/A+B=z/z+l into eq 4a leads to the result
Integration of eq 4a gives the number of molesof monomer B left at any instant of the reaction
as function of the composition z of the residue,namely,
— CTfJL-in
where i?0 and z0 are the initial values of B and 2.This equation forms at present the basis for thedetermination of the parameters a and /x from ananalysis of the copolymer composition. Applica-tions will be discussed in the next section. First,however, we shall consider some general propertiesof the function represented by eq 6. Figure 4shows a series of plots of log Bo/B versus log z/z0
for fixed values of 20=0.4 and /*=5. If the ratesof addition are independent of the nature of theactive ends, a= l//x, and a straight line is obtained.If active ends A increasingly favor the additionof monomer B, and ends B also favor the additionof 5-type monomer (/x=5), the lines are still prac-tically straight. The slopes become smaller dueto the slower disappearance of monomer A,although the variations are small and hencedeterminations of \x from such plots inaccurate.
-2 .8 .9 1.0.3 4 .5 .6 .7
A/A+B MONOMER
FIGURE 3. Plot of instantaneous mole fraction of com-ponent A in polymer as function of mole fraction of A inmonomer mixture. t
A, M=10, <r=4; B, M=5, c=3; C, n=l, <r=l: D, n=Q.5, <r=0.2; E, u—G.8o-=0.2.
Copolymerization 525

100
100
FIGURE 4. Plots of log BQ/B VS log z/z0.o=O.4; <r=5. A, <r=0; B , < /z; C, o-=2; D , <r=
The final polymer formed consists, in these exam-ples, entirely of species A, z~ oo y for 5 = 0 .When AI>1 and o\>l, A-A and B-B linkages aremore probable than cross-overs. The correspond-ing curves exhibit an upward curvature and possessa vertical asymptote shifting to the left as a in-creases. In other words, the final polymer formedis a copolymer of fixed composition. Under suchconditions the ratios between the rates balancethe concentration ratios in the monomer residuein such a way that the polymer compositionequals the composition of the monomer mixture or
dA_AdB~B
Relation 4a then gives for the critical composi-tion zc
(7)~C ( 7 - 1
Thus a and /x must both be either smaller or largerthan unity.
The fact that there exists a mixture of definitecomposition that copolymerizes without changingits composition, suggests an analogy with thefamiliar azeotropic mixtures often encountered indistillation processes [31]. One can constructcurves analogous to distillation curves by con-sidering the sum of the rates of consumption of
,i ,2 .6 .9 1.0.4 .5
A/AtB
FIGURE 5. Plot of over-all rate for unit concentration, ofradical V\ + VB on an arbitrary scale vs mole fraction ofA in monomer (M) and in polymer (P).
kgA(A)=2kgB(B); M=1.5; <7=0.5.
each monomer species as a function of the com-position. Specifically, one may plot the over-allrate of polymerization VA+ VB for unit concentra-tion of free radicals [31]:
1
By combining eq 2 and 3a the following result isobtained:
1
+Figures 5 and 6 show such plots, assuming A-Aaddition to be twice as fast as B-B growth. Thecurves for the polymer (P) are constructed from eq
i i i i i i I i i i I i i I i l i l I.8 1.0.5
A/A+B
FIGURE 6. Plot of VA+VB on an arbitrary scale vs molefraction of A in monomer (M) and in polymer (P), show-ing "azeotropic" composition X.
kgA(A)=2kgB(B); M=(r=0.5.
526 Journal of Research

4a. The curves for the monomer (M) representVA+VB as a function of A/A+B=z/z+ly ac-cording to the expression above. The abcissas ofconjugate points on the two lines P and M in-dicate the composition of the copolymer and thecomposition of the corresponding monomer resi-due, respectively. Figure 6 depicts an "azeo-trope." It will be noted that here the composi-tion X does not correspond to an extremum in thecurves, which represents a necessary thermodyna-mic condition for azeotropic boiling mixtures.Clearly the second intersection in figure 6 does notindicate a common composition for polymer andmonomer. The discussion of these curves followsotherwise familiar lines. If n and a are bothsmaller than unity, that is, if cross-combinationsare preferred, one derives from eq 6 that z neverreaches the critical value zc but approaches zeroor infinity, depending on whether z0 is respectivelysmaller or greater than zc. If both reactivity ra-tios exceed unity, then the azeotropic compositionis gradually approached.
It is of interest to follow the changes in com-position taking place in the course of the copoly-merization. Figure 7 depicts the relation betweenthe instantaneous and total copolymer com-position and the percentage conversion. Suchcurves are computed in the following manner.The extent of reaction, that is, the ratio between
0.2 -
O.I -
O.I 0.2 0.8 0.9 1.00.3 0.4 0.5 0.6 0.7
EXTENT OF CONVERSION
FIGURE 7. Instantaneous {full lines) and total composition{dashed lines) as function of the extent of conversion.
A, A', n=o, <r=l/n,zo=OA; B, B', n=5, a=2. ZQ=0.4; C, C , /z=5, <T=H, ZQ=0.7.
the weight of polymer at a given instant and theinitial weight of the monomer mixture is given bythe equation:
WQ Mxza+M2(8)
and Z/ZQ have the same meaning as in eq 6.Mi and M2 are the molecular weights of the twomonomers. With the aid of eq 6, B/Bo can beeliminated and wp/w0 obtained as function ofz/z0. The instantaneous copolymer compositionis directly given by eq 4a. The total compositionresulting up to a given instant equals:
Jo dA+dBdw*= B (9)
and is again obtained with the aid of eq 6. As isobvious, the difference between instantaneous andtotal values increases with time and is more pro-nounced for systems with widely different re-activity ratios. In case an azeotrope exists, andthe initial composition has been suitably chosen,the changes are slight as shown in curves (7 and C .On the whole, the total copolymer compositiondoes not change very much over a wide range ofconversion. These variations can be eliminatedby compensatory addition of the more activespecies during the course of the polymerization.
4. Over-all rate of reaction
It is evident that the calculation of the averagecopolymer composition does not require a knowl-edge of the total concentration of free radicals butmerely the ratio of A- and 5-type radicals. For theover-all reaction rate, however, this information isrequired and obtained in the following manner.In a steady state and for the simplest caseof a catalyzed reaction, we can write
i(A*+B*) =IC-kt{A,A)A*2-2kt(A,B)A*B*-kt(B,B)B*2=0,
(3b)
Copolymerization
dt
where
Combination of eq 3a, 3b, and 2 then leads to the
527

FIGURE 8. Plots of copolymerization rate on an arbitraryscale vs composition of monomer mixture according to eq2a.
A, 0-=7 = 3, fi=tr=0.5, scale multiplied by 4; B, 0=T=--2, fi=<r=l; C, 0=1,7 = M6,/i=0.5,<r=2.
following expression for the over-all rate ofcopolymerization [26]:
where: (2a)
r IC 7,kgB(B) kt(A,AY
y~a2 a2LkgB(B)_\ kt(A,A)
It should be noted again that in this derivation theinitiation is described by a single constant / . Thisrestricts the generality of this equation. For it ispossible that the rate of production of the initialradicals varies with the composition of themixture. In the most general case of the reactionscheme described, nine constants altogether wouldbe needed to describe the process completely.As is to be expected under the assumptions made,the rate is proportional to the square root of thecatalyst concentration, a familiar result in poly-merization kinetics. Equation 2a contains besideso- and /z, three constants. A knowledge of theseallows a determination of the product
kt(A;A)kt(B,B)/kt(A,B).
No use has as yet been made of these relations.Rate studies on the system styrene-methyl metha-crylate have been presented by Norrish and Brook-man [21]. However, they have been interpretedon the basis of an equation that assumes the con-centrations A* and S* to be independent of themonomer ratio 2. Plots of the rate (eq 2a) as func-tion of the monomer composition A are shown infigure 8 for a few extreme cases.
5. Composition, Size, and IntramolecularSequence Distributions
For the considerations hitherto presented, it isnecessary only to know the total concentrationsof radicals Xnrs(A) and 2nrs(B), regardless of sizeand composition. In order to obtain the distribu-tion of polymer sizes and compositions, we mustconsider the mode of production of individualradical chains of specified chain length and com-position. From the postulated mechanism (eq 1),we find for the rate of production of these radicals:
kgA(A)Anrs(A) -kgB(A)Bnrs(A)~
kt{A,B)nrs(A)Vnik{B)(3c)
kgB(B)Bnrs(B)-kgA(B)Anrs(B) -
kt(B,B)nrs(B)2nik(B)-
kt(A,B)nr8(B)2nik(A).
Equation 3c may be compared with the corre-sponding eq 3 for the total concentrations of A-and B-radicals and interpreted in the same man-ner. The positive terms refer to the growth ofsmaller chains to the desired size by monomeraddition, and the negative ones measure the rateof disappearance of the radicals in question byeither further growth or termination. Equation3c holds for r and s both larger than unity. Therate of production of species n10 (A) and nOi (B)is governed by the rate of initiation such that inthe first set of eq 3c the positive terms are replacedby IAA and in the second one by IBB or corre-sponding expressions involving the catalyst con-centration. In a steady state the left-hand sides
528 Journal of Research

of eq 3c vanish. The following quantities maybe defined:
fA. kgA(A)Aw U U kgA(A)A+kgB(A)B+kt(A,A)Xnik{A)+kt(A,B)Xnik(B)
/ D N _ kgB(B)B
kgB(A)kgA(B)_ 1x~kgA{A)kgB(B) <nx'
It will be noted that <a(A) represents the prob-ability of formation of an A-A linkage by propaga-tion, u(B) that of a B-B bond, x measures theprobability of occurrence of B-A and A-B linkagesrelative to that of A-A and B-B bonds. Thesolution of eq 3c has been shown by Simha andBranson [26] to be
nrs{Ji) =
nrs{B) =
Equation 3d may be interpreted in the followingmanner [26]: The terms multiplied by IA give thetotal number of possibilities of producing a chainof specified composition by initiation through an^[-radical. For each combinatory factor in thesum is a measure of the number of ways in whichone possible internal arrangement characterizedby a fixed number of A-A, B-B, A-B and B-Aconfigurations may be realized by permutations.The exponential factors indicate the probabilityof occurrence of these configurations. The sum-mation is then carried out over all possible internalarrangements compatible with the condition ofhaving r ^4-units and s B-units. It is taken be-tween the extremes of having one long sequenceof ^t-units followed by one sequence of 5-units,(j= 1), and the opposite extreme of a checker boardarrangement of these two monomers. The IB
terms then stand for chains initiated by a B-radical. The meaning of the last equation maybe seen also by specializing to the case <r/z=l, inwhich propagation is independent of the nature ofthe growing end. Equation 3d then reduces to [10]
IB (r+s-\kgB{B) \ r (3e)
Clearly the first term enumerates all ways ofobtaining the polymer nrs from a nucleus n10; thesecond term from a nucleus nOi.
From the known radical distribution, eq 3d, weobtain the distribution of stable polymer by meansof the relations:
Combination:
»r-«. s-k(A)nn{A) +
2kt{A,
Disproportionation:
(10)
Equation 10 determines the instantaneous distri-bution of sizes and compositions in a copolymerformed from a monomer mixture of a givencomposition A/A + B, which, in turn, determinesthe values of u(A) and <a(B). If the rate of thereaction has been measured, integration then yieldsthe total distribution obtained up to a giveninstant or degree of conversion.
For the practically important case of long chainseq 3d has been simplified by Stockmayer [28] ina manner analogous to that for simple polymers[12]. It will be noted that for the latter case(s—0), eq 3e reduces to
/ I~~ke
A
the result obtained by Herington and Robertson[12] for the distribution of radical lengths. Itsphysical significance is obvious. Since co deviatesfrom unity only slightly because of the small con-centration of radicals present, cor can, in a goodapproximation, be replaced for large r by 6"(1
6" ( 1 " c o ) r
Copolymerization 529

The final result is best expressed in terms of thenumber average chain length X of radicals, whichequals the ratio between the rate of reaction andthe rate of production (or destruction) of radicals.Inserting this value and noting that the total con-centration of radicals is obtained from an equa-tion analogous to eq 3b, one finds for the fractionof radicals of specified size [12]:
^n~\ e
To obtain the corresponding expressions for copoly-mers we denote the total chain length r-\-s by I.Deviations in the composition of a chain from theaverage value as given by eq 4a will be measuredby the quantity
r dA rV
As we are concerned with large values of Z, thesums in eq 3d may be approximated by integralsand the individual terms expressed by means ofStirling's formula. Considering that the devia-tion from the average composition will not bevery large in long chains, one can furthermoreexpand the relevant expressions in terms of y.The final result may be expressed as a function of /and y [28]
, y)dldy=exp ( - £ ) 2̂ dl
which gives the weight fraction of radicals withpolymerization degrees between I and l\dl andcomposition deviations between y and y-\-dy, ir-respective of whether they terminate in A- or B-units. K is defined by the relation
(13)
It is a measure of the spread of the compositiondistribution. X is again the number averagedegree of polymerization of radicals
\
employing the notation of eq 3b and noting thatthe factor in the bracket represents the total rateof destruction of radicals which equals the rateof production in the steady state. The first factor,the over-all rate of the reaction can be expressedas in eq 2a or by corresponding expressions forother reaction mechanisms. It is assumed in eq12 that monomers A and B have equal molecularweights.
The instantaneous size distribution of the stablepolymer follows from eq 10. If termination occursby disproportionation, the weight fraction ofcopolymer in the specified range of I and y isdirectly given by eq 12. If a fraction p of theradical chains is terminated by combination, theweight fraction is obtained by multiplying eq 12by a factor
l1 - P + 2X
Thus in the approximations used to simplify eq 3dand granted the validity of the reaction scheme 1and of eq 4, the distribution function consists oftwo factors. One characterizes the distributionof molecular weights and the other the distribu-tion of molecular composition for a fixed molecularweight. The specific nature of the terminationprocess affects the former factor, but not thecomposition distribution.
Finally, one derives from eq 12 the chain-lengthdistribution irrespective of composition by inte-gration over all compositions,
w(l)dl= f°Jo
w(l,y)dy-dl=e ^dL (12a)
Equation 12a has the same form as the resultobtained for the instantaneous distribution in purepolymers produced by disproportionation [12].The distribution of composition fluctuations is
w
with
(2
l / 2
2kt(A,B)A*B*+kt(B,B)B*2]-\ (14)
Plots of the expression (eq 12) as function of thereduced variables l/\ and rj are shown in figures 9and 10, respectively, as presented by Stockmayer[28]. It will be noted,that large values of rj, i. e.,large deviations from the average * composition,occur primarily in shorter chains, the longer chains
530 Journal of Research

0.4 .*>
FIGURE 9. Full lines: Chain length distribution, eq 12,(weight fractions), as function of reduced chain length Z/Xfor fixed values of the reduced composition deviation rj.Dotted line* Chain length distribution, eq 12a, irrespective of chain com-
position.
having mainly the average composition. Finally,the composition distribution (eq 12b) is shown infigure 11 for different values of the parameter K.With increasing value of K, that is, increasing valuesof a M for a fixed p0, the fluctuations in compositionbecome larger. This is understandable, sincein this instance self addition of A- and 5-unitsbecomes increasingly preferred. However, it willbe seen that the deviations from the mean valuey=0 are not large. Actually it can be derivedfrom eq 12b that 88 percent by weight of the copol-ymer is found in ' the range \y\<C[2po(l—po)K/\]1/2,which is small for large values of X. Itshould be noted again that application of theseresults to a complete copolymer product requiresa knowledge of the complete reaction mechanismwhich determines the variation of the parameters Xand K with average composition and conversion.No quantitative data seem to be available.Fractionation of various copolymer systems hasactually indicated the existence of a dispersion inrespect to composition [13, 17, 27].
The discussion of eq 3d has shown what isobvious, that even for a fixed composition in agiven chain, there are a variety of possible in-ternal arrangements of the species A and B inthe chain corresponding to sequences of identical
FIGURE 10. Composition distribution, eq 12, as function ofthe reduced composition deviation t\ for fixed values of thereduced chain length l/\.
units of varying lengths. The frequency of oc-currence of such sequences can be calculated andis of interest in connection with questions of struc-ture [1,25,31] in copolymers. For sufficiently longchains the probabilities Pi(A) and Pt(B) of se-quences of i A- or i?-units produced by propaga-tion are given by
i> 1(15)
The co's have the same meaning as in eq 3d, namely,that of propagation probabilities A-A and B-B.The validity of eq 15 is then immediately evident.Here the P/s are normalized so as to representfractions of the total number of A- or 5-sequences,respectively. If based on the average concentra-tion of A, Pt(A) is multiplied by c/B/dA+dB.
7K = 0.25/
-
/1
w(y)
r- 30
\
\
20
\
- \ 10
- . 0 5 + .05
FTGURE 11. Composition distribution, eq 12b, as functionof the composition deviation y from the mean for fixed
values of K, eq 13, and assuming „—-p. c = 400.£p\\ p)
Copolymerization 531

If termination is effected by combination, the dis-tribution (eq 15) of sequences in the radical chainsis not strictly identical with that in the stablecopolymer. Neglecting the effect of terminationaltogether, we simplify the equation for co(A) and«(JB)to:
If the polymer in question has been obtained forinstance, by copolymerizing mono and divinylunits, eq 15 gives the distribution of chain lengthsbetween cross links and a number average chain
length for a copolymer prepared from a monomermixture containing B moles of cross-linking agent.
IV. Experimental Studies1. Determination of Reactivity Ratios
The first thorough experimental investigation ofcopolymerization reactions has been made byLewis, Mayo, and Hulse [15, 18], in which theparameters a and /x were found for several pairsof monomers. As eq 6 cannot be solved readilyfor o~ and /x, the following procedure was adopted:Equation 6 can be transformed into
/ * = - (6a)
where
One experimental run gives a set of z,z0 and B,B0
values. Arbitrary values of p are then chosen,and the corresponding /* is computed using eq 6a.The value for <r is obtained from the expressionfor p. A plot of <r versus fi gives practically astraight line. A second run is utilized to getanother a-p line. The intersection of two or moresuch curves then gives the unique values for aand M, satisfying theoretically all runs. In prac-tice the intersection of three lines form a triangle,the area of which is a measure of the experimentalerrors. On this basis [18] table 1 and figure 12represent, as an example, the best data obtainedby Mayo and Lewis on the copolymerization of
6 o.sh-
0.8
FIGURE 12. Determination of n and a values from data intable 1.
styrene and methyl methacrylate. The difficultiesinvolved in the analysis of a partially polymerizedsystem have been emphasized [14]. Separation ofmonomers from the polymer has been in mostcases open to improvement. The usual techniqueshave been the precipitation of the polymer bysuitable combinations of solvents and vacuumdistillation. In general these methods do notadequately separate the polymers, as shown bythe fact that the results obtained by the aboveworkers [18] differed considerably, depending onthe procedure used for the isolation of the polymer.The technique finally developed utilizes the rela-tively high vapor pressure of frozen benzene attemperatures near 0° C. This method, known asthe frozen benzene technique, involves severalprecipitations, after which the polymer is dissolvedin benzene and then the solution quickly frozen.Subsequently the benzene is sublimed off undervacuum. The polymer is then in the form of avery fine powder, which is easily handled. Byusing such means for the isolation of polymer,results [15] were produced that are very accuratefor work of this nature.
Almost all of the monomer pairs so far studied
532 Journal of Research

TABLE 1. Styrene-methyl methacrylate copolymerization
Experiment
A
BCDEF
Concentration of unreacted monomer
Original
Ao
0. 7980
.5010
.2021
.8064
.5020
.1980
Bo
0. 2020
.4990
.7979
.1996
.4980
.8020
Final
A
0. 7435
.4571
.1828
.7450
.4552
.1623
B
0.1813
.4556
.7468
.1796
.4520
.7058
Reactiontime
hr5
52.68
898968.5
Polymer
wt%7.57
8.747.057.539.28
13, 23
Carbon %83.66
76.5269.1383.9876.7068.90
Carbon %|83. 77[83.9476.6169.1184.0076.5369.07
<r
0.48 to 0.50
0.48 to 0.52
*
0.48 to 0.50
0.48 to 0.52
have been those in which one monomer containeda different and easily analyzed element or group.In view of the availability and the high develop-ment of spectrometric and other physical methodsof analysis [9, 16, 19], there is room for techniquesusing these methods for the study of copolymeri-zation. Instead of analyzing the polymer, itshould be possible to determine directly the com-position of the monomer residue. One could placea sample of a polymerizing mixture in a highvacuum system and remove monomers from poly-mer by pumping off volatiles into a large residualvolume or condensing in a liquid air trap. Thenthe volatiles could be analyzed by either mass,infrared, or ultraviolet spectrometry, dependingon the nature of the system. An analysis of arelatively large bulk of residual monomers shouldbe less subject to errors due to small amounts ofmonomers trapped in the polymer than an analy-sis of the polymer itself at low conversion. Suchprocedures should be particularly useful in copoly-merization studies of isomers or compounds havingsmall differences in their elemental analyses.
2. Summary of Reactivity Ratios
Table 2 summarizes the published results onmonomer reactivities found by copolymerizingvarious pairs of monomers.5 It is seen, for in-stance, that in the reaction of styrene andmethyl methacrylate, the addition of styrenemonomer to styrene radical occurs half as fast asthe addition of methyl methacrylate monomer tostyrene radical. Also methyl methacrylate mono-mer adds half as fast to a methyl methacrylate
5 We are indebted to K. R. Henery-Logan and R. V. V. Nicholls for placingtheir results at our disposal prior to publication. The work was sponsoredby the Office of Rubber Reserve.
Copolymerization807127—48 12
radical as does styrene monomer. Furthermore,styrene monomer adds to styrene radical twice asfast as vinylidene chloride monomer to styreneradical. On the other hand, vinylidene chloridemonomer adds to vinylidene chloride radical ap-proximately one seventh as fast as styrene mono-mer. The estimated degrees of precision are in-dicated whenever given by the authors. In thecase of references [3, 5, 7, 38, 39], the reactivityratios were obtained from plots according to eq 4aby fitting the "best" curve. As an example, thesystem styrene-dichlorostyrene investigated byAlfrey, Merz, and Mark [ 5] is shown in figure 13.
A special case investigated is that wherein oneof the monomers does not polymerize with itself
1.0
0.8
0.6ecLJ
2
oCL
0.4
0.2
\/ 1O.2
/
I0.4
10.6
/
10.8 1.0
MONOMER
FIGURE 13. Plot of instantaneous concentration of dichloro-styrene in polymer vs concentration of dichlorostyrene inmonomer according to eq 4a and data in [5].
533
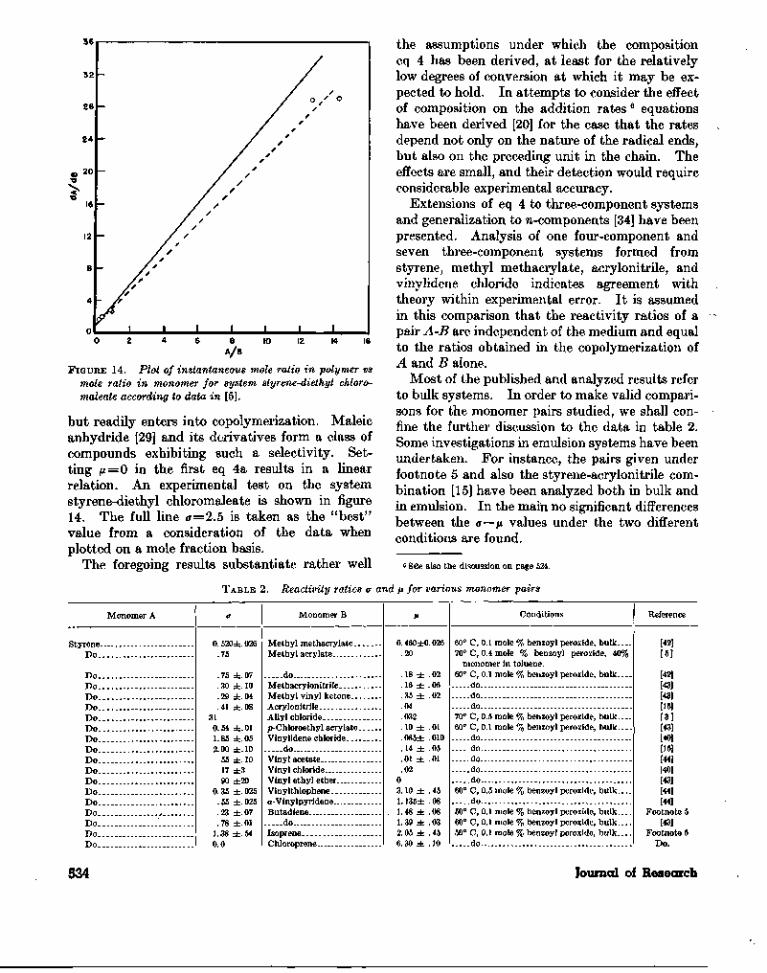
FIGURE 14. Plot of instantaneous mole ratio in polymer vsmole ratio in monomer for system styrene-diethyl chloro-maleate according to data in [5].
but readily enters into copolymerization. Maleicanhydride [29] and its derivatives form a class ofcompounds exhibiting such a selectivity. Set-ting /x = 0 in the first eq 4a results in a linearrelation. An experimental test on the systemstyrene-diethyl chloromaleate is shown in figure14. The full line (7=2.5 is taken as the "best"value from a consideration of the data whenplotted on a mole fraction basis.
The foregoing results substantiate rather well
the assumptions under which the compositioneq 4 has been derived, at least for the relativelylow degrees of conversion at which it may be ex-pected to hold. In attempts to consider the effectof composition on the addition rates 6 equationshave been derived [20] for the case that the ratesdepend not only on the nature of the radical ends,but also on the preceding unit in the chain. Theeffects are small, and their detection would requireconsiderable experimental accuracy.
Extensions of eq 4 to three-component systemsand generalization to ^-components [34] have beenpresented. Analysis of one four-component andseven three-component systems formed fromstyrene, methyl methacrylate, acrylonitrile, andvinylidene chloride indicates agreement withtheory within experimental error. It is assumedin this comparison that the reactivity ratios of apair A-B are independent of the medium and equalto the ratios obtained in the copolymerization ofA and B alone.
Most of the published and analyzed results referto bulk systems. In order to make valid compari-sons for the monomer pairs studied, we shall con-fine the further discussion to the data in table 2.Some investigations in emulsion systems have beenundertaken. For instance, the pairs given underfootnote 5 and also the styrene-acrylonitrile com-bination [15] have been analyzed both in bulk andin emulsion. In the main no significant differencesbetween the o—/x values under the two differentconditions are found.
° See also the discussion on page 524.
TABLE 2. Reactivity ratios a and n for various monomer pairs
Monomer A
Styrene..Do_.
Do.Do_Do.Do.Do.Do.Do.Do.Do-Do.Do-Do.Do.Do-Do.Do-Do.
0. 520±. 026.75
.75
.29
.41310.541.852.00
551790
0.35.55.23.78
1.380.0
±.07±.10±.04±.08
±.01±.05±.10±.10±3±20±.025±.025±.07±.01±.54
Monomer B
Methyl methacrylate.Methyl aery late
doMethacrylonitrileMethyl vinyl ketone_._AcrylonitrileAllyl chloridep-Chloroethyl aery late .Vinylidene chloride
doVinyl acetateVinyl chlorideVinyl ethyl ether..Vinylthiophene. _.a - Viny lpyridene -..Butadiene
doIsoprene..--.Chloroprene..
0. 460±0.026.20
.18 ± . 02
.16 ± . 06.02.35 ±
.04
.032
.10 ± .01
.085± .010
.14 ± . 05
.01 ± .01
.0203.10 ± . 451.135± .081.48 ± .081.39 ± .032.05 ± .456.30 ± .10
Conditions
60° C, 0.1 mole % benzoyl peroxide, bulk70° C, 0.4 mole % benzoyl peroxide, 40%
monomer in toluene.60° C, 0.1 mole % benzoyl peroxide, bulk.. . .
dododo
70° C, 0.5 mole %60° C, 0.1 mole %
do
benzoyl peroxide, bulk.benzoyl peroxide, bulk.
.do-._do.._do_.do
60° C, 0.5 mole %do
benzoyl peroxide, bulk
50° C, 0.1 mole %60° C, 0.1 mole %50° C, 0.1 mole %_—do
benzoyl peroxide, bulk,benzoyl peroxide, bulk,benzoyl peroxide, bulk..
Reference
[42][ 5 ]
[42][43][43][15][3][43][40][15][44][40][43][44][44]
Footnote 5[43]
Footnote 5Do.
534 Journal of Research

TABLE 2. Reactivity ratios a and /x for various monomer pairs—Continued
Monomer A
StyreneDoDoDo _._.DoDoDoDoDoDoDoDoDoDoDoDoDoDoDoDoDoDoDoDoDoDoDoDo
Methyl methacrylate..DoDoDoDo
DoDoDoDoDoDoDoDoDoDoDoDoDoDoDo
Vinyl acetate _Do
Do.Do.
DoDo
DoDoDoDoDoDoDoDo
Do.Do.Do.
. 043±. 092.50.13 ± . 018.5 ±.205.06. 52 ± . 500.19 db. 01. 18 d=. 10.21 ±.02.30 ±.02.19 ±.03
±2±20±3±15
1618537
2101050-1001.16 ± . 091.015±.060. 74 ± . 03. 695±. 02.62 ±.05.64 ±.05. 55 ± . 03.28 ±.025. 19 ± . 02.20. 56 ± . 03. 67 ± . 10
1.2 ±.142. 53 ± . 1
20 ± 310
0. 395±. 025. 25 ± . 03.29 ±.03. 205±. 02. 405±. 025.53 ±.025. 415±. 02. 395±. 02. 36 ± . 03.47 ±.075. 48 ± . 02. 22 ± . 02.50 ±.03.50 ± .03.41
0.1 ±.1
. 60 ±. 15
.1
.0
.3±.03
.353.0 ± .1. 23 ± . 02
0.17 ± . 01.011±.001.67. 66 ± . 04
5
6.8 ±.50.852.8
Monomer B
Maleic anhydrideDiethy] chloromaleate..Monoethyl maleateDimethyl maleateDiethyl maleate..—doMaleonitrileMonoethyl fumarateDimethyl fumarateDiethyl f umarateFumaronitrileTrichloroethyleneTetrachloroethylenetrans-Dichloroethylenecis-Dichloroethylenetrans-Dichloroethylenecis-Dichloroethylenep-Methoxystyrenep-Dimethylamino-styrene..p-Chlorostyrenep-Bromostyrenep-Iodostyrenew-Chlorostyrenew-Bromostyrenep-Cyanostryrenep-Nitrostyrene2,5 Dichlorostyreneo-ChlorostyreneMethacrylonitrileAcrylonitrileVinylidene chlorideVinyl-acetateVinyl chloride
a-Vinyl pyridineButadiene 'p-Methoxystyrenep-Dimethylaminostyrene..p-Methylstyrenera-Methylstyrenep-Chlorostyrenep-Bromostyrenep-Iodostyrenew-Chlorostyrenew-Bromostyrenep-Cyanostyreneo-Chlorostyrenea-Methyl styrene2,5 Dichlorostyrene
Methyl acrylate.Allyl chloride
Allyl acetateVinylidene chloride.
. . . .doVinyl chloride-
Vinyl bromideVinyl ethyl etherVinyl chlorideDiethyl maleateDiethyl fumarateTrichloroethylene
Tetrachloroethylene _.
trans-Dichloroethylene_.m-Dichloroethylene
00.035± .01.03 ± .01
0.005± .01
00. 25 ±0.10.25 ± . 015.070± .007
0.01 ± .01
000000.82 ± .07.84 ± .05
1.025± .050. 99 ± . 071. 25 ± . 301.09 ± .231.05 ± . 211.16 ± . 131.15 ± . 200.801. 64 ± . 070.65 ± .060.15 ± . 07.24 ± .03.015± .015.1
0.86 ±.75 ±.32 ±.11 ±.44 ±.49 ±.89 ±
1.10 ±0.95 ±.91 ±
1.17 ±1.41 ±1.37 ±0.14 ±2.55
9 ±2.50.67
. 45 ± .15
3.6 ± .52.1
4.5 ±1.201. 68 ± . 08.043± .005.444± .003
0.01 ± .01
0
000
Conditions
80° C, benzoyl peroxide, benzene solvent...70° C, 0.4 mole % benzoyl peroxide, bulk—60° C, 0.2 mole % benzoyl peroxide, bulk—. . . d o70° C, 0.4 mole % benzoyl peroxide, bulk...60° C, 0.1 mole % benzoyl peroxide, bulk...60° C, 0.2 mole % benzoyl peroxide, bulk...
do_do_.
60° C, 0.1 mole % benozoyl peroxide, bulk...60° C, 0.2 mole % benzoyl peroxide, bulk60° C, 0.1 mole % benzoyl peroxide, bulk..... . .do
60° C, 0.2 mole % benzoyl peroxide, bulk—...do
68° C, 0.1 weight % benzoyl peroxide, bulk.do
60° C, 0.1 mole % benzoyl peroxide, bulkdo
. . . .dodo
.do.,
.do.,
.do.— d o
do70° C, 0.4 mole % benzoyl peroxide, bulk—60° C, 0.5 mole % benzoyl peroxide, bulk60° C, 0.1 mole % benzoyl peroxide, bulk—60° C, 0.1 mole % benzoyl peroxide, bulk....
do— d o68° C, 0.2-0.4 weight % benzoyl peroxide
bulk.60° C, 0.5 mole % benzoyl peroxide, bulk60° C, 0.1 mole % benzoyl peroxide, bulk....— d o
dodo
— d o.. . .do
— d ododo
— d odo
60° C, 0.5 mole % benzoyl peroxide, bulk—do
68° C, 0.2-0.4 weight % benzoyl peroxide,bulk.
60° C, 0.1 mole % benzoyl peroxide, bulk...68° C, 0.2-0.4 weight % benzoyl peroxide,
bulk.60° C, 0.1 mole % benzoyl peroxide, bulk68° C, 0.2-0.4 weight % benzoyl peroxide,
bulk.60° C, 0.1 mole % benzoyl peroxide, bulk68° C, 0.2-0.4 weight % benzoyl peroxide,
bulk.60° C, 0.1 mole % benzoyl peroxide, bulk_-_do60° C, 0.1 mole % benzoyl peroxide, bulk60° C, 0.2 mole % benzoyl peroxide, bulk
do68° C, 0.1 weight % benzoyl peroxide, bulk..60° C, 0.5 mole % benzoyl peroxide, bulk68° C, 0.2-0.4 weight % benzoyl peroxide,
bulk.60° C, 0.1 mole % benzoyl peroxide, bulk.__.68° C, 0.1 weight % benzoyl peroxide, bulk..
do
Reference
[4][3]
[41][41][ 5 ][42][41][41][41][42][41][40][40][41][41][39][39][35][35][35][35][35][35][35][35][35][3][46][43][15][15][44][38]
[46][43][35][35][35][35][35][35][35][35][35][35][46][46][38]
[44][38]
[43][38]
[40][38]
[44][44][44][41][41][39][44][38]
[40][39][39]
Copolymerization 535

TABLE 2. Reactivity ratios a and n for various monomer pairs—Continued
Monomer A Monomer B Conditions Reference
Vinyl Acetate.Do
Acrylonitrile---DoDo
DoDoDoDoDo
Vinyl chloride.
Do.Do_
Do.Do.
DoVinylidene chloride.
Do.Do.Do.
Maleic AnhydrideDo
0 Chloroethyl acrytate.DoDo
p-ChlorostyreneDoDo
ChloropreneDo
0.99±0.026.3 ± .2.61± .04.91± .1
4.05± .3
3.28± .060.0 ± . 04.03± .03.01± .01
4700.14
.77
.42
-12± .015.0
2.05± .30.35
.3512. 2 ±2.03.8
. 03±. 03
.13 up5. 5 ± 14 ± 10.9 ± . 11.15± .050.86± .08
.70± .083.41d= .073. 65± . 11
£r(ms-Dichloroethylenecis-T> ichloroethyleneMethyl vinyl ketoneVinylidene chlorideVinyl acetate
Vinyl chlorideButadieneIsopreneChloropreneTetr achloroethylene..Vinylidene chloride..
Diethyl maleate-.Dioctyl maleate..
Diethyl fumarate.Pentene 1
IsobutyleneEthyl methacrylate.
Butyl methacrylate.Diethyl fumarate- __Ally 1 chloride
StilbeneAUyl acetate -
doMethallyl acetate. .Methyl aery latep-Methylstyrene. _ _p-Methoxystyrene _p-NitrostyreneButadieneIsoprene
0.086±0.01• 018=h .003
1. 78 ± . 220.37 ± . 1.061± .013
. 02 =t .02
.35 d= .08
.45 ± .056.07 ± . 53
60° C, 0.2 mole % benzoyl peroxide, bulkdo
60° C, 0.1 mole % benzoyl peroxide, bulk...do
60° C, 0.1 mole % benzoyl peroxide, 45 mole% monomer in acetonitrile.
60° C, 0.1 mole % benzoyl peroxide, bulk50° C, 0.1 mole % benzoyl peroxide, b u l k . . .
dodo.
0Large
.009± .003
.47 ± . 05
.2
. 08 ± . 102.2
2.20.046± .015.26
.03 d= .03
. 0075 up00.9 ± .1
60° C, 0.1 mole % benzoyl peroxide, bulk. . .68° C, 0.2-0.4 weight % benzoyl peroxide,
bulk.60° C, 0.2 mole % benzoyl peroxide, bulk_._68° C, 0.2-0.4 weight % benzoyl peroxide,
bulk.60° C, 0.2 mole % benzoyl peroxide, bulk.. .68° C, 0.2-0.4 weight % benzoyl peroxide,
bulk.60° C, 0.1 mole % benzoyl peroxide, bulk.. .68° C, 0.2-0.4 weight % benzoyl peroxide,
bulk.do
60° C, 0.1 mole % benzoyl peroxide, bulk68° C, 0.2-0.4 weight % benzoyl peroxide,
bulk.60° C, 0.1 mole % benzoyl peroxide, bulk30° and 38.5° C, approximately 0.5 mole %..60° C, 0.1 mole % benzoyl peroxide, bulk
do
.61 ± .03
. 58 ± . 03
.91 ± .37
..do..
..do.._do_._do.
.059± .015
.133± .02550° C, 0.1 mole % benzoyl peroxide, bulk..
do
[41][41][43][15][44]
[43]Footnote 5.
Do.Do.
[40][38]
[41][38]
[41][38]
]43][38]
[38][40][38]
[41][7]
[43][43][43][35][35][35]
Footnote 5.Do .
V. Discussion
1. Remarks on Effect of Substituents in OrganicReactions
Tables 3 and 4 summarize the experimentalinformation in a slightly different way by givingthe relative reactivities of a series of differentmonomers vs a given radical, e. g., styrene. Thescale is arbitrarily fixed by setting the reactivityof a monomer toward a radical of its own kindequal to unity. Values above unity then signifya higher activity than that exhibited by the radicalin question toward the identical monomer, whereasvalues below unity signify a lower activity. Someof the values may be affected by large errors, ascan be judged by the limits given for the a and \xvalues from which they were calculated.
The interpretation of these results in terms ofthe electronic structure and internal geometry ofradical and monomer is no easy task. However,certain qualitative attacks, at least, can be made
TABLE 3. Relative reactivities of monomers with styreneradical shown in comparison with Hammett's a-valuesfor the aromatics
Monomer
Vinyl acetateAllyl chlorideDiethyl maleateDiethyl chloromaleateVinylidene chlorideIsoprenep-Methoxystyrenep-D imethylaminostyreneStyreneMethyl acrylatep-Chlorostyrenep-Bromostyrenew-Chlorostyrenep-Iodostyrenew-BromostyreneMethyl methacrylateAcrylonitrilep-CyanostyreneButadiene....Dichlorostyrenep-NitrostyreneMaleic anhydrideChloroprene
Reactivity
0.02
Hammett's a
.0322
.4
. 5
.73
.86
.981.001.331.351.441.561.611.822.002.503.574.455.005.2624.0
- 0 . 2 6 8- . 2 0 5
.000
+.227.232.373.276.391
1.000
1.27
536 Journal of Research

along the lines established in the study of organicreactions of small molecules. That we are dealinghere with large radicals is no objection, since wehave established within the limits of the systemstudied, that the growth rates are independentof molecular size.
TABLE 4. Relative reactivities of various monomers withvarious radicals
Monomer
Styrene .Methyl methacrylate-.AcrylonitrileVinylidene chlorideIsopreneButadieneChloroprene
Eadical type
1.02.02.50.5
.74.5
2.01.00.8.4
4.0
2571.01.1
33.0CO
200
74.12.71.0
0.5
2.2
1.0
7.5
0.71.33.1
1.017.0
0.2
.3
.31.0
It is perhaps worthwhile to precede the dis-cussion of the subject proper with a cursory andnecessarily simplified summary of certain resultsand concepts regarding organic reactions. Weneed to consider the influence certain substituentgroups such as CH3, Cl, NO2, etc., in vinyl-typemonomers exert on the electronic configuration inthe adjacent double bond. The presence of suchgroups in a benzene ring leads to a change, ascompared with benzene, in the rates of furthersubstitution and affects also the locus of substi-tution. These facts have been known for sometime in classical organic chemistry.7 They arecaused by the tendency of an electrophilic groupto attack the ring at the region of highest (relative)electron density. Furthermore, if the over-alldensity is reduced compared to that in benzene,the rate of reaction is accordingly reduced.
In considering the mechanisms of such distor-tions of the charge distribution, we shall some-what arbitrarily separate two factors, inductiveeffects in the sense of Ingold8 and resonanceeffects. The former lead to an increase or de-crease of the over-all availability of electrons inthe ring, the substituent acting as an electronsource or sink. For instance, a methyl group
7 See for instance, G. E. K. Branch and M. Calvin, The theory of organicchemistry, (Prentice Hall, Inc., New York, N. Y., 1941).
8 See, for instance, L. P. Hammett, Physical organic chemistry (McGraw-Hill Publishing Co., Inc., New York, N. Y., 1940).
increases the electronic density in the ring andthus should increase the rate of substitution byan electrophilic agent while a chlorine atom, withits strong electron affinity, has the opposite tend-ency. Similarly, substituents such as NO2, CN,or COOR tend to diminish the electron density.Analogous effects by such groups are observed inaddition reactions to double bonds in simpleolefins. Resonance effects can determine thelocus of attack. For instance, groups with anunshared pair of electrons such as Cl, OH, NH2,C6H5, contribute structures to the activated com-plex of the substitution reaction (see footnote 7)which make the ortho- and para-positions morenegative
>e x=.
while no structure making the meta-positionnegative can be written. The resonance effect ofNO2, CN, COOR, and similar groups tends toleave the meta-position relatively more negative.Hence they are meta-directing. These directiveeffects persist in addition reactions to olefins.In general, ortho-para-directing substituents causeaddition to proceed in accordance with Markowni-koff's rule, meta-directing ones in opposition to it.These directive effects have also been explainedwithout introducing explicitly the notion ofresonance [23].
A quantitative measure of changes in electrondensity produced by substituents is given byHammett's a (see footnote 8), constants. Theyare defined as the logarithm of the ratio of theionization constant of the meta or para substitutedbenzoic acid to that in the unsubstituted acid.A high positive value of Hammett's a indicates adecrease in electron density. The fundamentalcorrectness of these concepts may be judged fromthe results of certain calculations regarding thecharge distribution in substituted benzenes [37].
2. Induction and Polarization Effects and Rela-tive Reactivities
Reverting now to the problem posed in the be-ginning, it may be seen that several factors oughtto be considered in attempting to account for therelative reactivities obtained and to predict thecomparative behavior of monomers toward thesame radical. The first ones are the over-all
Copolymerization 537

availability of electrons and, perhaps to a lesserextent, the direction of polarization of the doublebond. Let us take first the styrene radical as abasis for comparison. In the stable monomer,the double bond should have a relatively highelectron density because of the character of thephenyl group as an electron donor as evidencedby the direction of the dipole moment, which isopposite to that in toluene [45]. It should beremarked, however, that Hammett's a for thephenyl groups is positive. Thus
One would similarly expect propylene to showlow activity with styrene.
It is reasonable to assume that this over-allnegativity carries over to the radical end of agrowing vinyl chain [24]:
. . . . — CH2— CH—I
One can then expect that, under otherwise equalconditions (see below) addition to styrene of amonomer in which the inductive effect decreasesthe over-all availability of electrons in the pertinentdouble bond is favored in comparison with amonomer in which it is increased. It will benoticed, for instance, in table 3 that out of thethree dienes, chloroprene shows the highestactivity, isoprene the least and butadiene isintermediate.
CH2=C-CH=CH2I I
Cl=CC
Also from tables 3 and 4 it can be seen thatacrylonitrile monomer adds preferentially tostyrene radical and styrene monomer adds pref-erentially to acrylonitrile radical because of theopposing effects of the substituent groups. Thecyanide group decreases the electronic density.
N
Even allyl chloride has a low reactivity withstyrene, as seen in table 3. Contrary to what onemight expect by analogous reasoning, methylmethacrylate is slightly more active than methylacrylate. The difference however, may not besignificant.
The data on the substituted styrenes [35] in table3 provide further support for the viewpoint ex-pressed above. They indicate the existence of acorrelation between Hammett's a values for therespective substituent group and the correspond-ing relative reactivities of the substituted styrenemonomers with styrene radical. A high <r-value,which is characteristic, as said before, for a de-creased electron availability on other groupsattached to the ring is accompanied by greaterreactivity. A similar arrangement can be madein respect to the methyl methacrylate radical.Exceptions are encountered with respect to2>-OCH3, and ^-N(CH3)2-styrene. The reactiv-ities are higher than would correspond to theposition of the substituent on the c-scale. Theauthors [35] point out that these compounds areparticularly effective in forming complexes withconjugated carbonyl systems. Generally suchcomplexes can be formed by electron transferbetween constituents of the complex [36]. Inthis connection the high selectivity (<r and /x verysmall) of allyl acetate and maleic anhydride is ofinterest [7]. Here there exists a possibility ofresonance structures between radical and monomerwhich correspond to charge transfer within theactivated complex. This possibility appears likelyin view of the colors formed by maleic anhydridein mixtures with electron donor-type moleculessuch as stilbene, indene, and styrene. Thedifference between maleic anhydride and a qui-none-type inhibitor would be a matter of degreeand depend on the extent of resonance stabiliza-tion of the new radical formed. Inhibition isthen effected by the removal of the stable radicalthrough some further side reaction.
The direction and extent of polarization of thedouble bond can be of importance in favoring
538 Journal of Research

a high reactivitybelow
A monomer such as the one
Pen _CH2=CH,
should tend to react with a styrene radical byadding head to head, which would bring into playinterference by the group X, resulting in decreasedactivity as compared to the case:
®CH2
eCH-NP
Consideration of the resonance structures inbiphenyl (see footnote 8) suggests that the phenylgroup should be o-p-directing and thereforepolarize the double bond in the direction indicatedby the first of the above formulas.
The relative stabilities of the radical ends are asecond factor governing the propagation rates.The styrene radical is relatively stable because ofresonance through the benzene ring. In consider-ing two reactions, the one producing the morestable radical end will be favored. Thus in com-paring vinyl acetate with methyl acrylate mono-mers, the latter will exhibit the greater activitybecause of conjugation. The inductive effectsshould be approximately equal in magnitude forthese two isomers. Resonance in the styreneradical would be more important than in theacrylate radical. The value for the acrylate mono-mer is nevertheless slightly larger than unity, prob-ably, because of the direction of the inductiveeffect away from the double bond. One might ex-pect the formation of acrylonitrile radical to besomewhat favored by the resonance effect. Fromthis point of view all dienes should representfavorable cases.
We have been using for the purpose of illustra-tion almost exclusively the styrene radical becauseof the wealth of data available. The conceptsdeveloped are in fair agreement with experimentalresults obtained on other radicals. In the caseof vinylidene chloride, the radical resonancestabilization can play no significant role. Thechloride groups make the monomer positive in thesense previously discussed. Therefore, it shows ahigh activity, with styrene monomer and a lesserone with methyl methacrylate, which would not beextensively polarized because of the opposingeffects of the methyl and methyl substitutedcarboxyl groups. With the more positively polar-
ized acrylonitrile, the activity is still furtherreduced. For the same reasons, a similar order ofactivities is obtained, at least in a qualitative way,for acrylonitrile radical and methyl methacrylateradical with the same monomers. The electro-static effects of groups on diene radicals should beless important than in their monomers, becausethe effects would be most important on the doublebond, and the group is always one or more bonddistances away from the activated end in thecase of 1,4-addition. The double bond itselfshould act as an electron source. Actually thefew data available could be interpreted by assum-ing that the diene radicals are somewhat negativeregardless of the character of the substituent.However, the values for acrylonitrile relative tothe dienes do not appear to fit this picture.
The mass of data given can be summarized interms of constants that refer to pairs of radicalsand monomers (see table 2). It would be desir-able indeed to obtain constants characteristic foreach monomer as such. This would in principleallow the prediction of reactivity ratios. Thecomplete realization of such a program seemsremote in view of the many factors involved andthe probable existence of coupling effects.
3. Semiempirical relationships
The preceding discussion makes it evident thatthe polarity of radical and monomer and therelative stabilities of the radicals are the most im-portant factors to be considered. An attempt tofind a set of characteristic numbers in terms ofthese two effects has been recently made byAlfrey and Price [6]. It is suggested that thevarious influences are separable and can be repre-sented in the following way:
kgB(A)=PAQBexp( — eAeB),
where kgB(A) is the rate of addition of monomer Bto radical A. PA is characteristic for the radical.The e's are a measure of the effective charge on theend of the radical taken to be identical with thecharge on the double bond of monomer A and onthe double bond of monomer B, respectively.QB represents a mean reactivity of monomer Bobtained by forming the geometric mean of thereactivities of B with a series of radicals A, B,. . . and then assigning to one monomer a refer-
ence value of unity. This equation implies thatthe free energy of activation for the propagation
Copolymerization 539

step is additive in respect to the above-namedeffects. In comparing the behavior of monomersA and B toward radical A, PA cancels out and oneobtains for the ratios:
(17)
(18)<rn=exp[—(eA—eB)2].
By the use of these equations, a set of relative Q-and e-values can be derived from the experimentalresults. They are presented in table 5 for sometypical vinyl monomers. The result for thepolarities is in general agreement with the pointof view discussed before, styrene being the mostnegative and acrylonitrile the most positive of thefour. The order of the Q's show a parallelism tothe stabilities expected on the basis of the possibleresonance structures for the radicals previouslymentioned. Furthermore, the set of values isself-consistent in as much as they give c's and fi'sthat are in agreement with the experimental dataon all combinations of these four monomers.
TABLE 5 Q- and e-values for vinyl compounds
Monomer
Styrene _ _.Methyl methacrylateAcrylonitrileVinylidene chloride.
Q
1.00.64.34.16
e
- 10
+10
Some difficulties arise if one attempts to fit theother olefins studied into the framework repre-sented by table 5. The Q-values are not alwaysconsistent with the interpretation given to themand neither are the polarities. No satisfactoryresults are obtained by applying eq 17 and 18 tothe data on dienes obtained by Henery-Loganand Nicholls (see footnote 5). In deriving theseresults the equality of effective charges for mono-mer and radical is assumed. As mentioned previ-ously, it seems to us that in the case of dienes adifferentiation ought to be made particularly whenthe inductive effect of a substituent acts to de-crease the electron availability [32]. On thisbasis one can evaluate the pertinent data in con-junction with the values for the vinyl compounds
given in table 5. We modify the two eq 17 and18 by replacing the first factors eA and eB by e*A
and e*B and one factor (eA — eB) in eq 18 by{V*A — &*B), respectively. The star refers to theradical. The result of these calculations is sum-marized in table 6. The differences in sign be-tween the 6-values are in the expected direction.All 6*-numbers are negative, the one for chloro-prene being the least. The few data available foran independent check are rather well reproducedby using table 6. It can be seen from eq 17 thata large value of the ratio QA/QB for a set of e'scorresponds to large values of a and small valuesof fx. Thus monomers with great disparity in theQ's, will copolymerize poorly. Large differencesin the e's, of course, lead to good copolymeriza-tion.
TABLE 6. Q, e, and e* for dienes
Monomer
Chloroprene .Butadiene.. ._Isoprene
Q
2.22.53.5
e
0.9- 1 . 4- 1 . 4
e*
- 0 . 6- 1 . 2- 1 . 3
4. Steric Effects
In considering the copolymerization of com-pounds such as stilbene, indene, maleic anhydride,the maleates, and the fumarates, another factorbecomes important, namely steric hindrance.For example, the symmetrical substitution ofanother phenyl group in styrene leads to a com-pound, stilbene, which does not polymerize.Maleic anhydride and other symmetrical disub-stituted ethylene derivatives polymerize withdifficulty, if at all. However, stilbene and maleicanhydride form copolymers. The hindrance insuch a case should still be great but is apparentlyovercome by the influence of polarity effects sincethe pertinent double bond can be expected to bepositive in maleic anhydride and negative instilbene.
A striking example of steric hindrance is pro-vided by the comparison of maleic anhydride,diethyl chloromaleate, and diethyl maleate "[4, 5].The reactivities with styrene radical are respec-tively 24, 0.4, and 0.2. There differences havebeen ascribed to the opening of the anhydridering [24]. Diethyl fumarate has a reactivity of2.5 toward styrene monomer [24]. The increaseover that of its cis-isomer can be understood on the
540 Journal of Research

basis of the geometry of the two molecules, if therespective resonance structures of maleates andfumarates are considered.
The preceding discussion dealt entirely withrelative rates of propagation in copolymerization,which are the ones determining average composi-tion. Differences between monomers are, ofcourse, to be expected in respect to the other stepsof the chain reaction. In considering for instance,the rate of peroxide induced initiation, the polari-ties of monomer and catalyst radical and thestabilities of the radicals formed are of importance.We would expect the phenyl and the benzoylradicals to be negative. It is not possible tocompare directly the rates of initiation in twobinary systems, since the rate of decompositionof the peroxide depends markedly on the medium[22]. This can be minimized by using dilutesolutions of the monomers in an identical solvent.It serves no purpose to discuss in any greaterdetail possible effects of monomer structure on therelative rates of elementary acts other than propa-gation, until a complete kinetic analysis of thecopolymerization of at least some typical pairshas been obtained.
5. Effect of Intramolecular Arrangement onDegradation
The whole discussion, has hitherto referred to thebuilding up of copolymer chains. It is of interestto consider also the reverse process. It is not ourpurpose here to discuss in detail thermal decom-position of polymers. We merely wish to point outbriefly the relationship between the structure ofthe copolymer as considered previously and theresults to be expected in its degradation. Studiesof the thermal decomposition of various copolymershave shown that in many cases the yield of mon-omers are much lower than what would beexpected from the number of monomer units knownto be in the polymer and the behavior of the simplepolymer [33]. For example, the yield of styrenefrom GR-S is much less than the correspondingyield of styrene from polystyrene.
Assuming that the effect of side reactions on theyield of a given monomer remains constant ingoing from simple polymer to copolymer depoly-merizations, the pyrolysis yield of certain types ofmonomers obtained from a copolymer of givencomposition may be calculated. Let A representmonomers of the mono or asvmmetrical disubsti-
tuted ethylene type (CH2CXY) with head-to-tailarrangement in the simple chain and in thecopolymer, and B monomers of the diene of sym-metrically disubstituted ethylene types. In com-paring the expected yield of A from a copolymerwith the corresponding one from a pure polymerA, we proceed in the following manner. Con-sider a sequence of i ^4-units, which in the caseof the copolymer is bounded by B units. Thereare 2% possibilities of producing a split, 2%—1in the interior and 2XK at the boundaries, wherethe factor % is included to avoid twofold count-ing of the bonds joining the sequence to the restof the chain. 2i—1 of these splits producemonomer A. If the probability of occurrenceof a sequence of length i is denoted by Pi(A) (eq15), the yield Yc of A from the copolymer becomesequal to:
(19)2%
where Yo denotes the expected yield of monomerfrom the pure polymer A. In deriving eq 19, it hasbeen assumed that splitting occurs at random andindependent of the nature of the adjacent units inthe chain. Also recombination is excluded. Onewould expect large positive deviations from thecalculated yields to be an indication of head-to-head and tail-to-tail structures in the polymer.For in such a case, the effect of the sequenceboundaries considered in eq 19 is absent. Inpractice, the number of such configurations isusually small and obscured by other factors.Using the expressions (eq 15 and 16), we finallyobtain from eq 19:
Yc B 1<TA+B_\
(19a)
Since the composition of the copolymer can bedetermined from eq 4a, the thermal decomposi-tion yield can be plotted against the instan-taneous polymer composition as shown in fig-ure 15 for the styrene-butadiene system usingthe pertinent values of a and /x. For certain co-polymers, such as the polybutenes, where themonomers are isomers or otherwise similar, thismay be a useful tool for the development of apyrolytic analytical technique, particularly sincemost monomers can be determined spectrometri-cally, while the copolymers cannot always beanalyzed. The full application of this technique
Copolymerization 541

35
STYRENE IN MONOMER, MOLE FRACTIONO.I 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
O.I 0.2 0.3 0.4 0.5 0.6 0.7 0.8
STYRENE IN POLYMER, MOLE FRACTION0.9 LO
FIGURE 15. Theoretical yield of styrene from butadiene-styrene copolymer as function of instantaneous polymercomposition.
A, Experimental for GR-S.
will require precise control of pyrolysis conditionsand highly refined analytical methods. At present,the above concepts will account for some of theresults found in the depolymerization of co-polymers.
6. Conclusion
In conclusion, the obvious main lines for furtherwork may be sketched. Kinetic studies on copoly-mers are few and will have to be extended to includeover-all rates, and at least in some typical cases,determinations of the individual rate constants ofthe chain reaction. It will be further interestingto see whether the observed reactivity ratios canbe related to the structure of the monomer throughother constants characteristic of the same. Con-sideration of dipole moments, polarizabilities, andionization potentials may offer some clues. Indi-cations as to steric influences in the copolymerchain may be gained by a comparison of the heatsof reaction of the pure and mixed species.9 Thepreceding discussion deal b primarily with the mecha-nism of formation and the resulting structure of thecopolymer. Apart from the work on GR-S, nosystematic studies of the relation between thesefactors and the thermodynamic [26] and rateproperties of copolymer solutions seem to havebeen undertaken. Some physical properties of
9 See in this connection a remark made by M. G. Evans, J. Chem. Soc,1947, 264. L. K. J. Tong and W. O. Kenyon, 113th meeting of the AmericanChemical Society, Chicago, 111., April 19 to 23, 1948
certain copolymers besides synthetic rubbers havebeen systematically investigated.10
VI. References
[1] T. Alfrey and G. Goldfinger, J. Chem. Phys. 12, 205(1944).
[2] T. Alfrey and G. Goldfinger, J. Chem. Phys. 13, 322(1944); 14, 115 (1946).
[3] T. Alfrey and J. G. Harrison, J. Am. Chem. Soc. 68,299 (1946).
[4] T. Alfrey and E. Lavin, J. Am. Chem. Soc. 67, 2044(1945).
[5] T. Alfrey, E. Merz, and H. Mark, J. Polymer Sci. 1,37 (1946).
[6] T. Alfrey and C. C. Price, J. Polymer Sci. 2, 101(1947).
[7] P. D. Bartlett and K. Nozaki, J. Am. Chem. Soc. 68,1495 (1946).
[8] R. F. Boyer, J. Phys. and Colloid Chem. 51, 80 (1947).[9] R. F. Boyer, R. S. Spencer, and R. M. Wiley, J.
Polymer Sci. 1, 249 (1946).[10] H. Branson and R. Simha, J. Chem. Phys. 11, 297
(1943).[11] P. J. Flory, J. Am. Chem. Soc. 61, 3334 (1939).[12] E. F. G. Herington and A. Robertson, Trans. Faraday
Soc. 38, 490 (1942).[13] E. Jenckel, Zeits. f. physik. Chem. 190A, 24 (1942).[14] F. M. Lewis and F. R. Mayo, Ind. Eng. Chem.,
Anal. Ed., 17, 134 (1945).
[15] F. M. Lewis, F. R. Mayo, and W. F. Hulse, J. Am.Chem. Soc. 67, 1701 (1945).
[16] I. Madorsky and L. A. Wood, work sponsored by theOffice of Rubber Reserve.
[17] C. S. Marvel, G. D. Jones, T. W. Mastin, and G. L.Schertz, J. Am. Chem. Soc. 64, 2356 (1942).
[18] F. R. Mayo and F. M. Lewis, J. Am. Chem. Soc.66, 1594 (1944).
[19] E. J. Meehan, J. Polymer Sci. 1, 175 (1946).[20] E. Merz, T. Alfrey, and G. Goldfinger, J. Polymer
Sci. 1, 75 (1946).[21] R. G. W. Norrish and E. F. Brookman, Proc. Roy.
Soc. London [A] 171, 147 (1939).[22] K. Nozaki and P. D. Bartlett, J. Am. Chem. Soc. 68,
1686 (1946).[23] C. C. Price, Chem. Rev. 29, 37 (1941); Reactions at
carbon-carbon double bonds (Interscience Pub-lishers, Inc., New York, N. Y., 1946).
[24] C. C. Price, J. Polymer Sci. 1, 83 (1946).[25] R. Simha, J. Am. Chem. Soc. 63, 1479 (1941).[26] R. Simha and H. Branson, J. Chem. Phys. 13, 253
(1944).[27] H. Staudinger and J. Schneiders, J. Ann. 541, 151
(1939).[28] W. H. Stockmayer, J. Chem. Phys. 13, 199 (1945).[29] T. Wagner-Jauregg, Ber. 63, 3213 (1930).
10 See, for instance, W. O. Baker, in Advancing fronts in chemistry I,High polymers (Reinhold Publishing Corp., New York, N. Y., 1945).
542 Journal of Research

[30] F. T. Wall, J. Am. Chem. Soc. 62, 803 (1940); 63,821 (1941).
[31] F. T. Wall, J. Am. Chem. Soc. 66, 2050 (1944).[32] L. A. Wall, J. Polymer Sci. 2, 542 (1947).[33] L. A. Wall, J. Research NBS 41, 315 (1948) RP 1928.[34] C. Walling and E. R. Briggs, J. Am. Chem. Soc. 67,
1774 (1945).[35] C. Walling, E. R. Briggs, K. B. Wolfstirn, and F. R.
Mayo, J. Am. Chem. Soc. 70, 1537 (1948).[36] J. Weiss, J. Chem. Soc. (London), 245 (1942).[37] T. Ri and H. Eyring, J. Chem. Phys. 8, 433 (1940).[38] P. Agron, T. Alfrey, J. Bohner, H. Haas, and H.
Wechsler, J. Polymer Sci. 3, 157 (1948).[39] T. Alfrey and S. Greenberg, J. Polymer Sci. 3, 297
(1948).[40] K. W. Doak, J. Am. Chem. Soc. 70, 1525 (1948).
[41] F. M. Lewis and F. R. Mayo, J. Am. Chem. Soc. 70,1533 (1948).
[42] F. M. Lewis, C. Walling, W. Cummings, E. R. Briggs,and F. R. Mayo, J. Am. Chem. Soc. 70, 1519 (1948).
[43] F. M. Lewis, C. Walling, W. Cummings, E. R. Briggs,and E. J. WTenisch, J. Am. Chem. Soc. 70, 1527(1948).
[44] F. R. Mayo, C. Walling, F. M. Lewis, and W. F.Hulse, J. Am. Chem. Soc. 70, 1523 (1948).
[45] M. M. Otto and H. H. Wenzke, J. Am. Chem. Soc.57, 294 (1935).
[46] C. Walling, E. R. Briggs, K. B. Wolfstirn, J. Am.Chem. Soc. 70, 1543 (1948).
WASHINGTON, November 25, 1947.
Copolymerization 543
![Oligomerization of Indole Derivatives with Incorporation ... · dimerization of the expected products with addition of 1,2-ethanedithiol. Molecules 2008, 13 1848 Dimers [5-10], trimers](https://img.pdfslide.us/doc/110x75/5ea22301e7d6870d1e338962/oligomerization-of-indole-derivatives-with-incorporation-dimerization-of-the.jpg)




























