Embed Size (px)
Citation preview

Electroanalytical Chemistry and InterJacial Electrochemistry, 52 (1974) 403-412 403 ~) Elsevier Sequoia S.A., Lausanne Printed in The Netherlands
C O N V O L U T I O N POTENTIAL S W E E P VOLTAMMETRY
III. EFFECT OF SWEEP RATE CYCLIC VOLTAMMETRY
L. NADJO, J. M. SAVI~ANT and D. TESSIER
Laboratoire d'Electrochimie de l'UniversitO de Paris VII, 2 place Jussieu-75221 Paris Cedex 05 (France)
(Received 2nd January 1974)
The essential feature of convolution potential sweep voltammetry (CPSV) is that quantities proportional to the concentrations of the diffusing species at the electrode surface are immediately derivable from the convoluted current:
, = ' , t' i/o/ J0 (1)
which is recorded as a function of the electrode potential z. In other words the convolution procedure appears as a mathematical means for eliminating the mass- transfer polarization from the current-potential data.
Since the early presentation of the method I (note that "semi-integral electro- analysis 2'' is basically founded on the same principles as CPSV) its potentialities and particularly its advantages over conventional linear sweep voltammetry (LSV) have become apparent through the analysis of the formal kinetics pertaining to the main basic reaction schemes: linear diffusion control 3, accompanying homogeneous chemical reaction 3, slow and "quasi-reversible" charge transfer 3, nernstian double waves 3'4. Procedures for correcting the I E plots from sphericity effects and iR drop have also been defined and tested 3. The practical applicability of the method has been demonstrated until now mainly on reversible single-wave z 3 ~ 6 and double- wave 3-6 systems and for a few irreversible and quasi-reversible systems 6. However all these experiments were performed in narrow ranges of low sweep rates, less than 1 0 V s -1.
For very slow scans the sphericity effect, using a mercury drop as a working electrode, becomes exceedingly large and natural convection may also interfere. Both effects lead to a bent limiting convoluted current. If one does not wish to have recourse to empirical procedures, similar to those used in polarography and chrono- potentiometry, for determining the actual limiting I value 6, 0.1 V s -1 appears as a reasonable lower limit for the sweep rate in order to avoid natural convection and to maintain the sphericity effect under a value where a correction procedure 3 is applicable.
Raising the sweep rate the double layer charging current increases and may interfere, together with the faradaic current, in the iR drop effect as already shown in L S V 7. It can be estimated that this effect may become significant depending on the depolarizer concentration when the sweep rate reaches several hundred V s-1.

404 L. NADJO, J. M. SAVI~ANT, D. TESSIER
As concerns mechanism analysis the role of the sweep rate is twofold: (i) For an irreversible accompanying chemical reaction an increase of the sweep rate tends to lower the kinetic control by the chemical reaction leading to diffusion or charge- transfer control (see ref. 8 and references therein). For reversible homogeneous reac- tions and/or "quasi-reversible" charge transfers an increase of the sweep rate leads to an enhanced kinetic control by either the chemical reaction or the charge transfer 9,10. (ii) Inside a zone of complete kinetic control by either a chemical reaction or the charge transfer the peak potential in LSV shifts linearly with the logarithm of the sweep rate, the shift rate being a characteristic of the mechanism. In CPSV the logarithmic plot corresponding to the mechanism gives rise to the same straight line whatever the sweep rate. This means that the effect of the sweep rate is in fact included in the form of the logarithmic analysis featuring a given mechanism: It follows that, if a better accuracy in mechanism determination is looked for using CPSV instead of LSV, this implies that the logarithmic plot must be linear and give the right slope not only at a given sweep rate but also in the largest possible range of sweep rates, i.e., of the same order as that used in LSV.
The sweep rate thus appears as an experimental parameter as important as in LSV. One goal of the present communication is therefore to test the validity of the convolution procedure over a large range of sweep rates. This has been done on the f luorenone~uorenone anion couple which is a simple nernstian diffusion controlled system already used as an example for iR drop studies in LSV 7'11. The initial concentration is another important experimental parameter. Its variation mainly concerns the mechanism diagnosis of second-order versus first-order mechanisms. Some results showing the effect of concentration on the same system will therefore be presented.
Th effect of iR drop and- double-layer charging as well as the usefulness of the convolution procedure in cyclic voltammetry will be discussed.
EXPERIMENTAL
The experiments were performed in acetonitrile with 0.1 mol 1-1 tetraethyl- ammonium perchlorate as supporting electrolyte. The cell, electrodes, LSV ap- paratus, digitalization and convolution procedures were essentially the same as already described 3, a 2. Better results were obtained with a droplet of mercury hanging on a mercury plated gold disk than with a conventional long drop capillary. This electrode was therefore used throughout the present work. The corrections for sphericity and for iR drop effect were made using procedures previously described 3, the sphericity factor D½/r ° being evaluated by extrapolation at zero sweep rate.
The value 6f the largest usable sweep rate was determined by the shortest sampling time (30 #s) for an accuracy of 8 bits on the acquisition system and by the number of samples taken on each voltammogram. This last number was generally 200 except for the highest sweep rates where it was reduced to 50.
VARIATION OF SWEEP RATE AND INITIAL CONCENTRATION
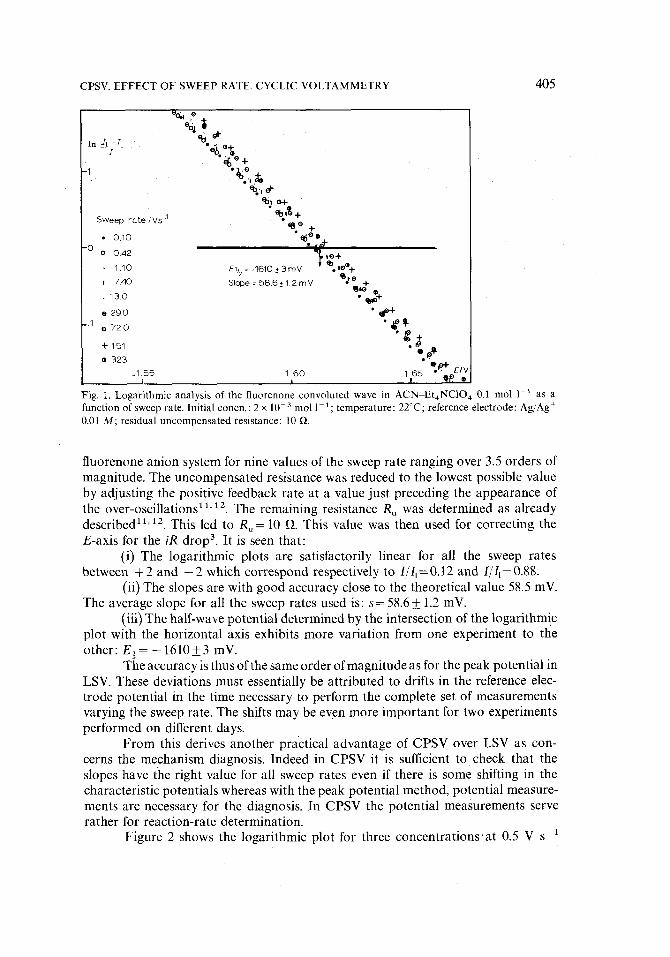
Figure 1 shows the logarithmic plots In [ ( I i - I ) / I] vs. E (11 =limiting con- voluted current, E=working electrode potential) obtained with the fluorenone-
CPSV. EFFECT OF SWEEP RATE. CYCLIC VOLTAMMETRY 405
In Jl --T ~ ~"
: "%'~o + "%'~o + ~l~o. 0 ~"
• 1 e-I-
Sweel3 rate Vs J ° e8"1~ + ~ e +
0 1 0
" 0 o 0 4 2 l e +
" 10 E1,2 = -1610 Z 3 m V ~ t e ° +
, s,opo: 8. .,2 v
• 1 3 0 * ~ +
e 29.0 " ~ + _1 " ~-
e 7 2 0 " ~ +
• 323 ~'?+ El' _1 55 1 60 _1 65 O~ I
1 I I - -
F i g . 1. Logarithmic analysis of the fluorenone convoluted wave in ACN Et,,NCIO 4 0.1 mol 1 I as a function of sweep rate. Initial concn.: 2 x 10-a mol 1 ~; temperature: 22°C; reference electrode: Ag/Ag + 0.01 M; residual uncompensated resistance: 10 f~.
fluorenone anion system for nine values of the sweep rate ranging over 3.5 orders of magnitude. The uncompensated resistance was reduced to the lowest possible value by adjusting the positive feedback rate at a value just preceding the appearance of the over-oscillations 11't2. The remaining resistance R. was determined as already described 11'12. This led to R~= 10 ft. This value was then used for correcting the E-axis for the iR drop 3. It is seen that:
(i) The logarithmic plots are satisfactorily linear for all the sweep rates between + 2 and - 2 which correspond respectively to I/I]=0.12 and 1/1]=0.88.
(ii) The slopes are with good accuracy close to the theoretical value 58.5 mV. The average slope for all the sweep rates used is: s= 58.6-t-1.2 mV.
(iii) The half-wave potential determined by the intersection of the logarithmic plot with the horizontal axis exhibits more variation from one experiment to the other: E{= - 1610+3 mV.
The accuracy is thus of the same order of magnitude as for the peak potential in LSV. These deviations must essentially be attributed to drifts in the reference elec- trode potential in the time necessary to perform the complete set of measurements varying the sweep rate. The shifts may be even more important for two experiments performed on different days.
From this derives another practical advantage of CPSV over LSV as con- cerns the mechanism diagnosis. Indeed in CPSV it is sufficient to check that the slopes have (he right value for all sweep rates even if there is some shifting in the characteristic potentials whereas with the peak potential method, potential measure- ments are necessary for the diagnosis. In CPSV the potential measurements serve rather for reaction-rate determination.
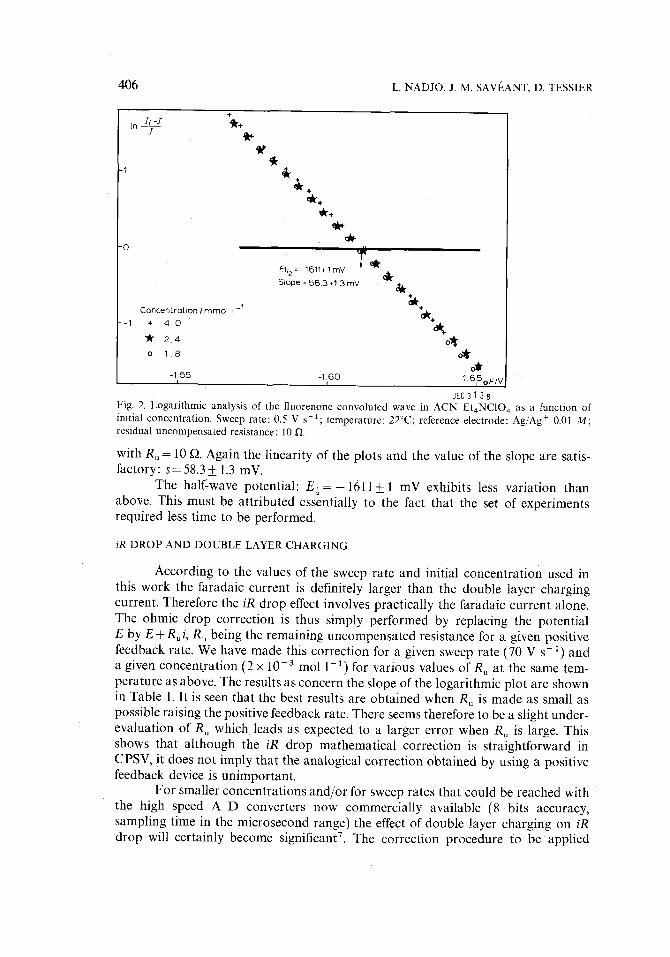
Figure 2 shows the logarithmic plot for three concentrations 'at 0.5 V s 1
406 L. NADJO, J. M. SAVI2ANT, D. TESSIER
In I I - I l
-1
- 0
C o n c e n t r a t i o n / m m o l I - I
- - 1 ÷ 4 . 0
2 . 4
o 1 8
- 1 5 5 I
.1.
&-+
El; 2 : - 1611t 1 mV
S lope = 5 8 3 ± 1 3 m V
- 1 . 60 I
4~
&.
o~ o~
1 6 1 5 o E I V
JEC31 38
Fig. 2. Logarithmic analysis of the fluorenone convoluted wave in ACN-Et4NC10 4 as a function of initial concentration. Sweep rate: 0.5 V s-1; temperature: 22°C; reference electrode: Ag/Ag + 0.01 M; residual uncompensated resistance: 10 ~.
with Ru = 10 ~. Again the linearity of the plots and the value of the slope are satis- factory: s=58.3_+ 1.3 mV.
The half-wave potential: E ~ = - 1611+ 1 mV exhibits less variation than above. This must be attributed essentially to the fact that the set of experiments required less time to be performed.
iR DROP AND DOUBLE LAYER CHARGING
According to the values of the sweep rate and initial concentration used in this work the faradaic current is definitely larger than the double layer charging current. Therefore the iR drop effect involves practically the faradaic current alone. The ohmic drop correction is thus simply performed by replacing the potential E by E + R u i, R, being the remaining uncompensated resistance for a given positive feedback rate. We have made this correction for a given sweep rate (70 V s -1) and a given concentration (2 × 10 -3 tool 1-1) for various values of R u at the same tem- perature as above. The results as concern the slope of the logarithmic plot are shown in Table 1. It is seen that the best results are obtained when R. is made as small as possible raising the positive feedback rate. There seems therefore to be a slight under- evaluation of R. which leads as expected to a larger error when R. is large. This shows that although the iR drop mathematical correction is straightforward in CPSV, !t does not imply that the analogical correction obtained by using a positive feedback device is unimportant.
For smaller concentrations and/or for sweep rates that could be reached with the high speed A-D converters now commercially available (8 bits accuracy, sampling time in the microsecond range) the effect of double layer charging on iR drop will certainly become significant v. The correction procedure to beappl ied
CPSV. EFFECT OF SWEEP RATE. CYCLIC VOLTAMMETRY
TABLE 1
LOGARITHMIC ANALYSIS FOR VARIOUS VALUES OF THE UNCOMPENSATED RESISTANCE
407
R u/~ 474 343 ! 53 60 12 Slope/mV decade- 1 63.0 62.1 60.8 60.0 58.6
tt~ ~ RRe~e~'ence e)ectrode
........ ----'-~ Working electrode
E >
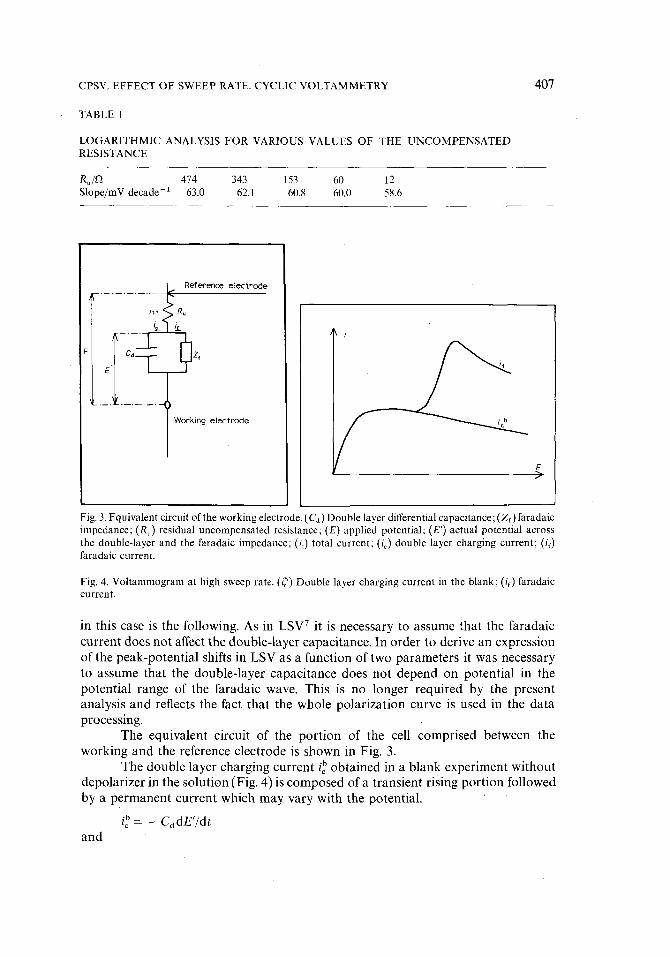
Fig. 3. Equivalent circuit of the working electrode. (Ca) Double layer differential capacitance; (Zf) faradaic impedance; (Ru) residual uncompensated resistance; (E) applied potential; (E') actual potential across the double-layer and the faradaic impedance; (i,) total current; (ic) double layer charging current; (i~) faradaic current.
Fig. 4. Voltammogram at high sweep rate. (i~) Double layer charging current in the blank; (if) faradaic current.
in this case is the following. As in LSV 7 it is necessary to assume that the faradaic current does not affect the double-layer capacitance. In order to derive an expression of the peak-potential shifts in LSV as a function of two parameters it was necessary to assume that the double-layer capacitance does not depend on potential in the potential range of the faradaic wave. This is no longer required by the present analysis and reflects the fact that the whole polarization curve is used in the data processing.
The equivalent circuit of the portion of the cell comprised between the working and the reference electrode is shown in Fig. 3.
The double layer charging current i b obtained in a blank experiment without depolarizer in the solution (Fig. 4) is composed of a transient rising portion followed by a permanent current which may vary with the potential.
• b Cd d E ' / d t
and

408 L. NADJO, J. M. SAVI~ANT, D. TESSIER
"b E ' = E i - v t + R u tc
Thus: Ca ( E ' ) = ib/( V-- R , d ib /d t )
• b and dib /d t in the blank exper iment Knowing Ro 1'. the measurement of ~¢ thus provides Cd as a function of time or potential in the potential region of the faradaic wave.
The depolarizer is now introduced in the solution giving rise to a faradaic current if. The total current is:
it = i~ -+- if
where the double layer charging current i~ may be different from i b due to the occurrence of the faradaic current.
Now:
T h u s :
ic - - C a d E ' d t
E ' = E - v t + Ru i t
ic = Cd(v -- Ru d i , /d t )
It follows that:
if = it - Cd v + R u Cd d i t /d t
Ca is known as a function of potential from the blank experiment. The measurement of it and d i , / d t along the faradaic wave thus provides if to which the usual con- volution procedure can be applied. This series of successive operations that would be tedious to perform manually becomes easy when done in a computer from the analog-to-digital converted data.
CYCLIC VOLTAMMETRY
As noted before 2. 3, the curve convoluted current vs. potential is not dependent upon the exact form of the potential scanning from the foot to the top of the wave for diffusion-controlled systems. It is therefore interesting to discuss what would be the interest of applying the convolution procedure to the "return" wave in cyclic voltammetry.
It will be still assumed that diffusion is linear and semi-infinite and that adsorption of the reactants does not occur at the electrode surface.
As long as the electrochemical system under examination involves only electron-transfer steps and homogeneous chemical reactions that remain at equi- librium in the considered time range, i.e., as long as diffusion remains the only rate- limiting factor, the I - E curves can be expressed in a closed form. This means that the I - E wave does not depend on the current itself since the concentrations at the electrode are only functions of the electrode potential. In such conditions the I E curves are independent of the way and the time taken to reach each particular value of the electrode potential. It follows that the "return" I E curve is exactly super- imposable on the forward curve.
The situation becomes different as soon as the charge transfer and/or

CPSV. EFFECT OF SWEEP RATE. CYCLIC ¥OLTAMMETRY 409
secondary chemical reactions interfere in the kinetic control of the overall process: This is clearly seen in the various wave equations previously derived 3 for slow and "quasi-reversible" charge transfers and for homogeneous chemical reactions under pure kinetic conditions where the convoluted current is not only expressed as a function of the electrode potential but also of the current i itself. Since the current changes from the forward to the reverse scan this is also true for I. In other words the I-E wave equation is actually, as the i E wave equation, an integral equation involving the imposed potential-time relationship. Indeed, the reverse of eqn. (1) is:
1, f l dI(~ ) do i = 7c ~ do ( t-o) ~
Taking as examples the cases of: (i) A "quasi-reversible" charge transfer, the I-E wave equation (see ref. 3, p.
181) becomes:
~ f t dI(v) dv k(E){It-I[I+exp(nF/RT)(E-E~)]}D~A = ~2 o do (t(E)-v) ~
(ii) A Nernstian charge transfer followed by a fast and irreversible dimeriza- tion, the I-E wave equation (see ref. 3, p. 177) becomes:
( l l f l E ) dI do ) ~ nF {E ( RT ~k ~ dv (t(E)- v) = (I~- I) exp - R-~ - E ° + ~ In nFSD-=W~Tj]
It is thus seen that the I E curve does depend on the form and speed of the potential scanning and that the reverse curve is no more superimposable on the forward one.
Provided the charge transfer is not preceded by any chemical reaction and that there is no chemical regeneration of the starting material (catalytic process) the limiting convoluted current does not depend on the charge-transfer rate or on the nature and rate of the chemical reactions since the very existence of the plateau is related to the condition (CA)0 = 0 at the electrode surface.
It follows that contrarily to what has been incorrectly expressed in ref. 3 the observation in CV of a reverse I-E wave superimposed on the forward one does provide a criterion of reversibility. What was actually meant is that in many other cases than diffusion control, i.e. for non-reversible charge transfer and for homogeneous chemical reaction under pure kinetic conditions, the same logarithmic plot of the wave involving both I and i is traced out in the reverse as well as in the forward direction. This is clearly seen on the wave expressions given in ref. 3 (Table 1 p. 179, and p. 181).
In the case of, e.g. a ,'quasi-reversible" charge transfer ihe whole cyclic I-E pattern exhibits an hysteresis which is the more pronounced the higher the sweep rate and the slower the charge transfer. Measurement of the potential separation between the forward and the reverse curves may provide a mean for determining the charge transfer rate, similarly to the peak separation in CV. It must be emphasized however that the working curves relating this potential separation to the charge transfer imply the a priori knowledge of the potential dependence of:the
410 L. NADJO, J. M. SAVI~ANT, D. TESSIER
rate law'in order to be calculated as it is the case in CV. If one does not wish to state the rate law a priori but rather to derive it from the experimental data it is therefore more advisable to use both the I and i data using the following wave equation3:
k (E) = D A i /l -- I [1 + exp (nF/R T)(E - E{)]
which leads to the same curve in the reverse and forward scans. In the case of a fol low-up chemical reaction, the reverse curve is the more
I /A s-l/2
5'10 - 6
10 _6
- 1 4 - 1 6 _18 E/V
2
o
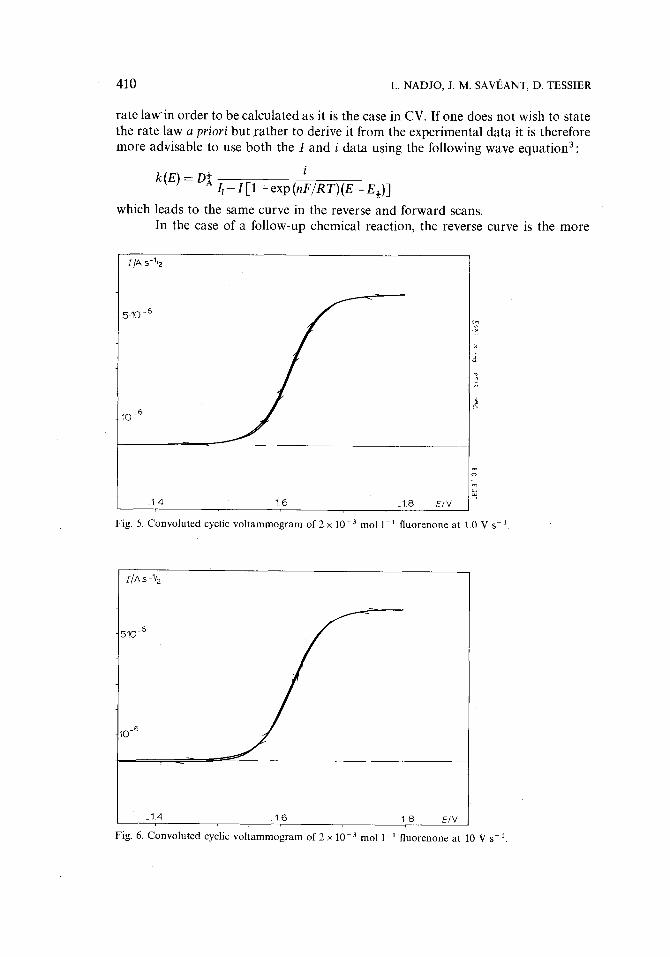
Fig. 5. Convoluted cyclic voltammogram of 2 x l 0 - 3 mol 1-1 fluorenone at 1.0 V s-1.
] /A s -1/2
510 _6
10 -6
_14 _1.6 _ 1 8 E/V
Fig. 6. Convoluted cyclic voltammogram of 2 x 10 3 mol 1-1 fluorenone at 10 V s - 2.
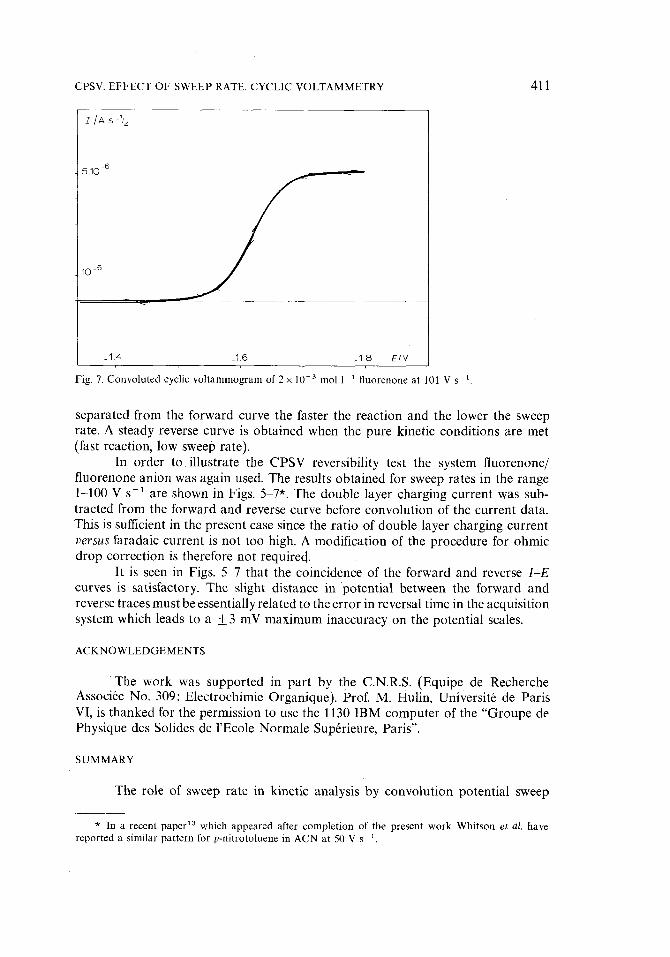
CPS¥. EFFECT OF SWEEP RATE. CYCLIC VOLTAMMETRY 411
T / A S -1/2
5 1 0 _6
10 -6
_1 4 1 6 _1 8 E /V
Fig. 7. Convoluted cyclic voltammogram of 2 x 10 -3 mol 1 1 fluorenone at 101 V s i
separated from the forward curve the faster the reaction and the lower the sweep rate. A steady reverse curve is obtained when the pure kinetic conditions are met (fast reaction, low sweep rate).
In order t o illustrate the CPSV reversibility test the system fluorenone/ fluorenone anion was again used. The results obtained for sweep rates in the range 1-100 V s-1 are shown in Figs. 5 7*. The double layer charging current was sub- tracted from the forward and reverse curve before convolution of the current data. This is sufficient in the present case since the ratio of double layer charging current v e r s u s faradaic current is not too high. A modification of the procedure for ohmic drop correction is therefore not required.
It is seen in Figs. 5-7 that the coincidence of the forward and reverse I - E
curves is satisfactory. The slight distance in potential between the forward and reverse traces must be essentially related to the error in reversal time in the acquisition system which leads to a _+ 3 mV maximum inaccuracy on the potential scales.
ACKNOWLEDGEMENTS
'The work was supported in part by the C.N.R.S. (Equipe de Recherche Associ6e No. 309: Electrochimie Organique). Prof. M. Hulin, Universit6 de Paris VI, is thanked for the permission to use the 1130 IBM computer of the "Groupe de Physique des Solides de l'Ecole Normale Sup6rieure, Paris".
SUMMARY
The role of sweep rate in kinetic analysis by convolution potential sweep
* In a recent paper 13 which appeared after completion of the present work Whitson et M. have reported a similar pattern for p-nitrotoluene in ACN at 50 V s 1.

412 L, NADJO, J. M. SAVI~ANT, D. TESSIER
voltammetry is underlined. The method is evaluated over 3.5 orders of magnitude of sweep rate using the fluorenone-fluorenone anion couple in acetonitrile. The applica- tion of convolution procedures to cyclic voltammetry is discussed.
REFERENCES
1 C. P. Andrieux, L. Nadjo and J. M. Sav6ant, J. Electroanal. Chem., 26 (1970) 147. 2 K. B. Oldham, Anal. Chem., 44 (1972) 196. ° 3 J. C. Imbeaux and J. M. Say,ant, J. Electroanal. Chem., 44 (1973) 169. 4 F. Ammar and J. M. Sav6ant, J. Electroanal. Chem., 47 (1973) 115. 5 M. Grenness and K. B. Oldham, Anal. Chem., 44 (1972) 1121. 6 M. Goto and K. B. Oldham, Anal. Chem., 45 (1973) 2043. 7 J. C. lmbeaux and J. M. Sav6ant, J. Electroanal. Chem., 28 (1970) 325. 8 L. Nadjo and J. M. Sav6ant, J. Electroanal. Chem., 48 (1973) 113. 9 H. Matsuda and Y. Ayabe, Z. Elektrochem., 59 (1955) 494.
10 J. M. Sav6ant and E. Vianello, Electrochim. Acta, 12 (1967) 629. 11 D. Garreau and J. M. Saveant, J. Electroanal. Chem., 35 (1972) 309. 12 D. Garreau and J. M. Sav6ant, J. Electroanal. Chem., 50 (1974) 1. 13 P. E. Whitson, H. W. Vandenborn and D. H. Evans, Anal. Chem., 45 (1973) 1298.





























