Embed Size (px)
Citation preview

Proc. Natl. Acad. Sci. USAVol. 90, pp. 3182-3186, April 1993Medical Sciences
Conversion of truncated and elongated prion proteins into thescrapie isoform in cultured cells
(glycosylinositol phospholipid/Creutzfeldt-Jakob disease/posttranslational modification)
MARK ROGERS*t, FRUMA YEHIELY*, MICHAEL SCOrr*, AND STANLEY B. PRUSINER*t§Departments of *Neurology and tBiochemistry and Biophysics, University of California, San Francisco, CA 94143
Contributed by Stanley B. Prusiner, December 10, 1992
ABSTRACT The only known component of the infectiousprion is a posttranslationafly modified protein known as thescrapie isoform of the prion protein, PrP&. Upon limitedproteolysis, a protease-resistant fragment designated PrP 27-30is formed. Using in vitro mutagenesis, we examined the role ofthe N and C termini in the formation of prP& in persistentlyinfected, mouse neuroblastoma (ScN2a) cells. Neither deletionof amino acids 23-88, which are also removed by proteinase Kin the formation of PrP 27-30, nor deletion of the five octapep-tide repeats within this region altered synthesis of PrPSc.Elongation of PrP with one, two, four, or six octapeptiderepeats in addition to the five found in wild-type PrP did notalter the synthesis of PrPsc. Truncation of the C terminus wasaccomplished by substituting a translation stop codon for thepredicted glycosylinositol phospholipid (GPI) anchor-attachment signal corresponding to amino acids 231-254.Expression of this C-terminal PrP mutant in ScN2a cellsproduced PrPsc that appeared to lack a GPI anchor. Weconclude that neither the GPI anchor nor the N-terminal 66amino acids are required for the synthesis ofPrPsc as measuredby the acquisition of limited resistance to proteinase K diges-tion. Whether these truncated or elongated PrP molecules arecompetent to participate in the formation of infectious prionsremains to be established.
Many lines of evidence indicate that prions are composedlargely, if not entirely, of the scrapie isoform of the prionprotein (PrPSC) (1). Since conversion of the cellular isoform,PrPC, into PrPSc is a posttranslational process (2-4), wedetermined whether unglycosylated PrP synthesized in thepresence of tunicamycin or from an expression vector withnonfunctional Asn-linked glycosylation sites could be con-verted into PrPSc (5, 6). Using scrapie-infected mouse neu-roblastoma (ScN2a) cells, we found that PrPSC synthesisproceeded in the absence of Asn-linked glycosylation. Thedevelopment of an efficient expression vector for use inScN2a cells (7) and the discovery that a chimeric PrP mole-cule designated MHM2 could be converted into PrPSc (8)made the investigations possible. To extend those studies, weremoved a 24-amino-acid segment containing three octapep-tide repeats (ORs) within the N terminus, all five of the ORs,or the N-terminal amino acids 23-88. The conversion ofPrPSc into the 27- to 30-kDa proteinase K-resistant core ofPrPSc (PrP 27-30) occurs when the N-terminal amino acids23-88 of PrPSc are hydrolyzed (9-13). The formation of PrPScin ScN2a cells required neither the five ORs nor the N-ter-minal 66 amino acids. Since the addition of the glycosyli-nositol phospholipid (GPI) anchor to Syrian hamster PrP isaccompanied by removal of the C-terminal peptide of 24amino acids (14-16), we constructed a mouse PrP genelacking the corresponding C-terminal 24 codons. Although
the synthesis ofrecombinant PrPSc was diminished comparedwith wild-type (wt) PrPsc, sufficient amounts were producedfor detection by immunoblotting.We also investigated the synthesis of PrPFs from recom-
binant PrP molecules with additional ORs. The formation ofPrPsr from PrPC or a precursor with one, two, four, or sixadditional ORs in ScN2a cells was similar to that observed forwt PrP. In families with the inherited Creutzfeldt-Jakobdisease, affected members have four to nine ORs in additionto the five ORs found in wt PrP (17-24).We conclude that the regions of PrP that feature in the
conversion ofPrPC or a precursor into PrPsc lie within regionsof the molecule unaltered by these mutations. This conclu-sion is consistent with studies on four synthetic PrP peptideswhich lie between residues 109 and 218 (25). Each of thesepeptides, 'z15 amino acids in length, was predicted to be ana-helix (26), but three of the four have high 3-sheet contentand form amyloid fibrils, suggesting that the transition fromPrPC to PrPSc might be accompanied by a transformation ofputative a-helices into (-sheets (25).
MATERIALS AND METHODSCells. ScN2a cells were grown in Dulbecco's modified
Eagle's medium H16 (GIBCO) supplemented with 10%'o fetalbovine serum, penicillin at 100 units/ml, and streptomycin at100 pLg/ml. OptiMem was obtained from GIBCO/BRL.Recombinant PrP Genes. Generally, manipulation ofDNA
used protocols described by Sambrook et al. (27). Restrictionenzymes were obtained from New England Biolabs and BRL.Modified PrP genes were constructed as follows. The MHM2PrP gene, a chimeric mouse-Syrian hamster construct, wasused for all modifications (8, 28). To generate MHM2 PrP(del23-88), an oligonucleotide (5'-GGCCTCTGTGGCCAAG-GAGG-3') was synthesized which had homology to the 3' endof the signal sequence and to the sequence directly aftercodon 88. The oligonucleotide was used to delete the inter-vening sequences in the MHM2 PrP gene by the strategy ofKunkel (29). This mutagenesis resulted in the in-frame fusionof codon 22 to codon 89. The nature of the mutation wasconfirmed by restriction analysis and DNA sequencingacross the junction. The MHM2 PrP(del 231-254) gene wasconstructed by using two complementary oligonucleotides(5'-CCTATTACGACGGCCGACGATCGTGAC-3' and 5'-TCGAGTCACGATCGTCGGCCGTCGTAATAGG-3') toreplace the sequences between the unique Stu I restrictionsite at codon 223 (93 bases in from the 3' end of the PrP open
Abbreviations: PrP, prion protein; PrPsc, scapie isoform of PrP; PrP27-30, proteinase K-resistant core of PrPsc; PrPC, cellular isoform ofPrP; wt, wild type; GPI, glycosylinositol phospholipid; OR, octapep-tide repeat; mAb, monoclonal antibody.tPresent address: Department of Zoology, University College, Bel-field, Dublin 4, Ireland.1To whom reprint requests should be addressed at: Department ofNeurology, HSE-781, University of California, San Francisco, CA94143-0518.
3182
The publication costs of this article were. defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.

Proc. Natl. Acad. Sci. USA 90 (1993) 3183
reading frame) and the Xho I site located at the 3' end of theMHM2 PrP gene construct. This substitution replacedcodons 231-254 in the MHM2 PrP gene with a translation stopcodon effectively removing the GPI anchor-attachment sig-nal sequence.We constructed variants of the MHM2PrP gene (8) con-
taining 0, 2, 5, 6, 7, 9, and 11 copies of the OR sequencePro-(Gln/His)-Gly-Gly-(Gly/Ser)-Trp-Gly-Gln (9, 13). Firstwe constructed a variant of MHM2 with a single copy of theOR sequence. The wt MHM2 PrP molecule contains anOxaNI-Kpn I fragment of 129 bp which encompasses the ORregion. A double-stranded OxaNI-Kpn I fragment of 33 bpwas synthesized by annealing two oligonucleotides: 5'-TCAGGGTGGTACCTGGTGGGGCCAAGGAGGTGGTG-GTAC-3' and 5'-CACCTCCTTGGCCCCAGGTACCACCC-3'. This synthetic fragment was inserted between the OxaNIand Kpn I sites of the MHM2 PrP gene (28), creating a variantwith a single OR designated MHM2 PrP(OR -4). The syn-thetic OxaNI-Kpn I fragment in MHM2 PrP(OR -4) DNAcontains an additional Kpn I site. Cleavage of this constructwith Kpn I and religation removes the 24-bp Kpn I fragmentcomprising the single remaining OR, yielding a variant withno ORs, termed MHM2 PrP(OR -5). The wt PrP DNAcontains two restriction cleavage sites for BstYI within theOR region. Following cleavage and religation a variant ofMHM2 with only two copies of the OR, MHM2 PrP(OR -3)was produced. Next, a 24-bp fragment corresponding to asingle Pro-His-Gly3-Trp-Gly-Gln OR unit was created withBstYI compatible sticky ends by annealing two oligonucle-otides: 5'-ATGAGGCTGTCCCCAGCCACCACC-3' and 5'-GTGGCTGGGGACAGCCTCATGGTG-3'. This was in-serted into the single BstYI site of MHM2 PrP(OR -3) DNA.By varying the ratio of OR fragment to target DNA, wecreated variants with different numbers of ORs. These wereidentified by fine-structure restriction mapping and DNAsequencing, yielding variants with 6, 7, 9, and 11 copies of theOR, denoted MHM2 PrP(OR + 1), -(OR +2), -(OR +4), and-(OR +6), respectively (Fig. 1).
Lipofectin (BRL)-mediated transfection was used to intro-duce recombinant plasmids into ScN2a cells according to themanufacturer's instructions. One to two micrograms of plas-mid DNA was used for the generation of stable transfectants.Transfected cells were grown in nonselective medium for 24hr, then split and plated onto 100-mm Petri dishes and grownin the presence of Geneticin (GIBCO) at 1 mg/ml, to selectfor cells stably expressing the neomycin-resistance gene.Individual clones were picked with cloning rings, expanded,and examined for their ability to express recombinant PrPdetected on immunoblots. Suitable clones were maintainedfor further analysis.
Immunoblots. After SDS/PAGE, proteins were trans-ferred to Immobilon-P membrane (Millipore) and probed asdescribed (28). R073 is a rabbit polyclonal antiserum raisedagainst electrophoretically purified PrP 27-30. The 3F4mouse monoclonal antibody (mAb) was kindly provided byR. Kascsak (Institute for Basic Research, Staten Island, NY).
Metabolic Labeling and Immunoprecipitation. Sampleswere prepared and analyzed as described (7, 28).
In the experiment involving treatment with aqueous HF,cells were labeled as described above, in the presence oftunicamycin (30 ,ug/ml) and 0.5 mCi (18.5 MBq) of [35S]me-thionine for 1 hr. Cells grown in 90-mm plates were lysed in2 ml of 0.5% (vol/vol) Nonidet P-40 in phosphate-bufferedsaline. Cell extracts were incubated for 18 hr at 4°C in thepresence of 0.1% (wt/vol) N-lauroylsarcosine, 1% bovineserum albumin, 10 AM phenylmethanesulfonyl fluoride, and3F4 at a dilution of 1:1000. Then, 160 ,ul of 10o (vol/vol)protein A-Sepharose in 150 mM NaCI/10 mM Tris-HCl, pH7.8/1% N-lauroylsarcosine was added and the mixture wasincubated at 4°C for 3 hr. The protein A-Sepharose-antibody
M M CHO CHO
* S-S
M M CHO rHO
S-S
M M CHO CHO
* anE.: ::' . . .S-S
M M CHO CHO
~~~88
M M CHO C-O
I U
ScS
M M C'HO CHO
* lm }
SS
Q M M CHO CHO/ I
M- CHOmM M CHO CHO
/ U
S-Ss
iJM M CHO CHO
.'' ... ''!-88!
MHM2 PrP
MHM2 PrP (del 23-88)
MHM12 PrP (del 231-254)
MHM2 PrP (OR -5)
MHM2 PrP (OR -3)
MHM2 PrP OR +1)
MHM2 PrP (OR +2)
MHM2 PrP (OR +4)
MHM2 PrP (OR +6)
FIG. 1. Structures of truncated and elongated MHM2 PrP mol-ecules. The PrP gene is shown as a horizontal box. Deletions areshown as gaps, sites for the addition of Asn-linked oligosaccharidesas CHO, the disulfide bond as S-S, the GPI attachment signal as theC-terminal black box, and the signal peptide as the N-terminal blackbox. The shaded boxes contain amino acids 23-88, which remainsensitive to digestion by proteinase K after PrPSc synthesis. Thehatched boxes depict the ORs. The methionines in MHM2 PrP thatallow this protein to be recognized by the anti-Syrian hamster PrPmAb 3F4 (30) are designated as M. In MHM2 PrP these methioninesare at positions 108 and 111.
complexes were washed six times in the same buffer andboiled in 200 ,ul of 10 mM Tris HCl, pH 7.8/1 mM EDTA/0.1% SDS. The proteins released from the beads wereincubated with phosphatidylinositol-specific phospholipaseC (PIPLC, 0.1 unit/ml) for 6 hr at 37°C in the presence of 10,uM phenylmethanesulfonyl fluoride, then precipitated in thepresence of7 volumes ofmethanol and bovine serum albumin(6 ,g/ml) for 18 hr at -20°C. Dry pellets were suspended in25 ,ul of aqueous 50% HF, incubated for 36 hr at 4°C, andneutralized with 75 ,ul of cold water, and proteins wereconcentrated by drying (Speed Vac, Savant). Control sam-ples were suspended in HF, immediately neutralized, anddried. Pellets were suspended in gel loading buffer for SDS/PAGE.
RESULTS
The ability to convert recombinant PrP into PrPSc wasassessed by measuring protease-resistant PrP on immuno-blots. To distinguish the recombinant from the endogenousPrP, a chimeric mouse-Syrian hamster PrP sequence desig-nated MHM2 PrP was used in which Leul' and Val"1' werereplaced with methionines (8). These amino acid substitu-tions allow the MHM2 PrP to be recognized by the anti-Syrian hamster PrP mAb 3F4 but do not hinder its conversionto PrPSc in ScN2a cells (8, 28, 31). 3F4 does not recognizemouse PrP (28, 30).
Truncation ofMHM2 PrP at the N Terminus. The deletionintroduced into the MHM2 PrP(del 23-88) construct should
Medical Sciences: Rogers et al.
3184 Medical Sciences: Rogers et al.
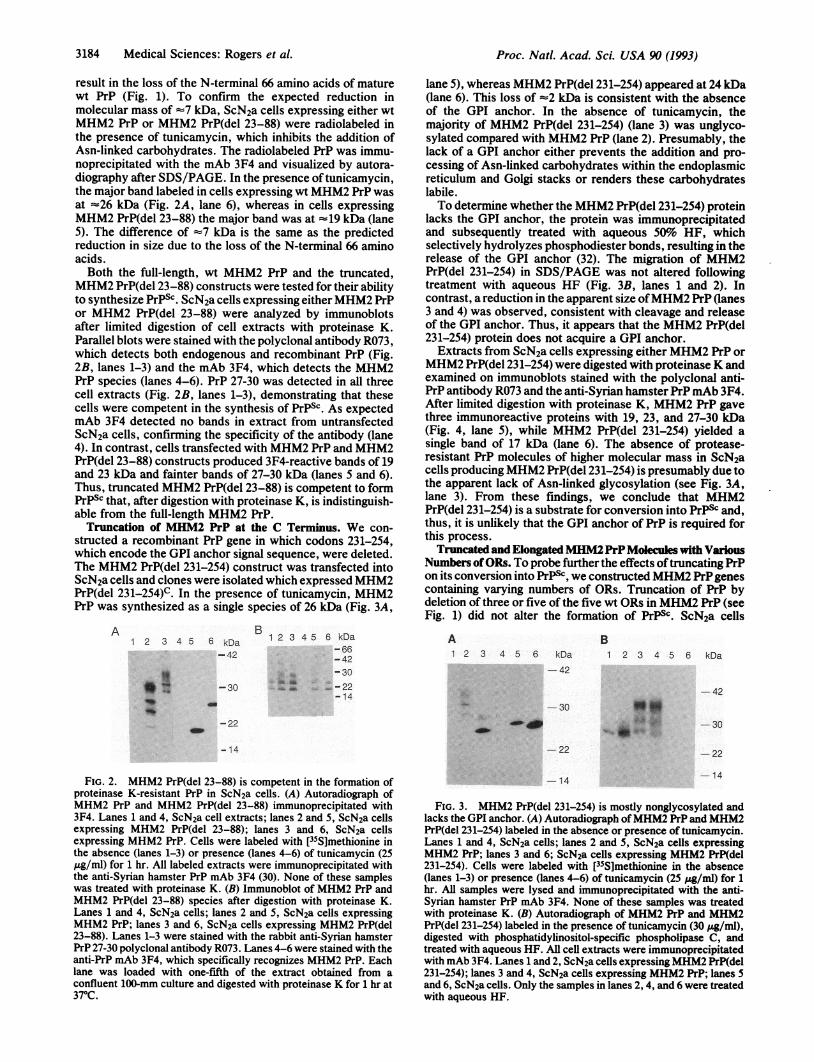
result in the loss of the N-terminal 66 amino acids of maturewt PrP (Fig. 1). To confirm the expected reduction inmolecular mass of -7 kDa, ScN2a cells expressing either wtMHM2 PrP or MHM2 PrP(del 23-88) were radiolabeled inthe presence of tunicamycin, which inhibits the addition ofAsn-linked carbohydrates. The radiolabeled PrP was immu-noprecipitated with the mAb 3F4 and visualized by autora-diography after SDS/PAGE. In the presence oftunicamycin,the major band labeled in cells expressing wt MHM2 PrP wasat -26 kDa (Fig. 2A, lane 6), whereas in cells expressingMHM2 PrP(del 23-88) the major band was at -19 kDa (lane5). The difference of -7 kDa is the same as the predictedreduction in size due to the loss of the N-terminal 66 aminoacids.Both the full-length, wt MHM2 PrP and the truncated,
MHM2 PrP(del 23-88) constructs were tested for their abilityto synthesize PrPSC. ScN2a cells expressing eitherMHM2 PrPor MHM2 PrP(del 23-88) were analyzed by immunoblotsafter limited digestion of cell extracts with proteinase K.Parallel blots were stained with the polyclonal antibody R073,which detects both endogenous and recombinant PrP (Fig.2B, lanes 1-3) and the mAb 3F4, which detects the MHM2PrP species (lanes 4-6). PrP 27-30 was detected in all threecell extracts (Fig. 2B, lanes 1-3), demonstrating that thesecells were competent in the synthesis of PrPSc. As expectedmAb 3F4 detected no bands in extract from untransfectedScN2a cells, confirming the specificity of the antibody (lane4). In contrast, cells transfected with MHM2 PrP and MHM2PrP(del 23-88) constructs produced 3F4-reactive bands of 19and 23 kDa and fainter bands of 27-30 kDa (lanes 5 and 6).Thus, truncated MHM2 PrP(del 23-88) is competent to formPrPSc that, after digestion with proteinase K, is indistinguish-able from the full-length MHM2 PrP.
Truncation of MHM2 PrP at the C Terminus. We con-structed a recombinant PrP gene in which codons 231-254,which encode the GPI anchor signal sequence, were deleted.The MHM2 PrP(del 231-254) construct was transfected intoScN2a cells and clones were isolated which expressed MHM2PrP(del 231-254)C, In the presence of tunicamycin, MHM2PrP was synthesized as a single species of 26 kDa (Fig. 3A,
lane 5), whereas MHM2 PrP(del 231-254) appeared at 24 kDa(lane 6). This loss of =2 kDa is consistent with the absenceof the GPI anchor. In the absence of tunicamycin, themajority of MHM2 PrP(del 231-254) (lane 3) was unglyco-sylated compared with MHM2 PrP (lane 2). Presumably, thelack of a GPI anchor either prevents the addition and pro-cessing of Asn-linked carbohydrates within the endoplasmicreticulum and Golgi stacks or renders these carbohydrateslabile.To determine whether the MHM2 PrP(del 231-254) protein
lacks the GPI anchor, the protein was immunoprecipitatedand subsequently treated with aqueous 50o HF, whichselectively hydrolyzes phosphodiester bonds, resulting in therelease of the GPI anchor (32). The migration of MHM2PrP(del 231-254) in SDS/PAGE was not altered followingtreatment with aqueous HF (Fig. 3B, lanes 1 and 2). Incontrast, a reduction in the apparent size ofMHM2 PrP (lanes3 and 4) was observed, consistent with cleavage and releaseof the GPI anchor. Thus, it appears that the MHM2 PrP(del231-254) protein does not acquire a GPI anchor.
Extracts from ScN2a cells expressing either MHM2 PrP orMHM2 PrP(del 231-254) were digested with proteinase K andexamined on immunoblots stained with the polyclonal anti-PrP antibody R073 and the anti-Syrian hamster PrPmAb 3F4.After limited digestion with proteinase K, MHM2 PrP gavethree immunoreactive proteins with 19, 23, and 27-30 kDa(Fig. 4, lane 5), while MHM2 PrP(del 231-254) yielded asingle band of 17 kDa (lane 6). The absence of protease-resistant PrP molecules of higher molecular mass in ScN2acells producing MHM2 PrP(del 231-254) is presumably due tothe apparent lack of Asn-linked glycosylation (see Fig. 3A,lane 3). From these findings, we conclude that MHM2PrP(del 231-254) is a substrate for conversion into PrPSc and,thus, it is unlikely that the GPI anchor of PrP is required forthis process.
Truncated and ElongatedMHM2 PrP Moecules with VarousNumbers of ORs. To probe further the effects oftruncating PrPon its conversion into PrPSc, we constructedMHM2 PrPgenescontaining varying numbers of ORs. Truncation of PrP bydeletion of three or five of the five wt ORs in MHM2 PrP (seeFig. 1) did not alter the formation of PrPSc. ScN2a cells
1 2 3 4 5 6 kDa -2 3 4 5 6 kDa
-42 -42
t#gg- -30-30 -
- 22-14
A1 2 3 4 5 6 kDa
-42
-30-22
-14 -22
B1 2 3 4 5 6 kDa
- 42
- 30
-22
FIG. 2. MHM2 PrP(del 23-88) is competent in the formation ofproteinase K-resistant PrP in ScN2a cells. (A) Autoradiograph ofMHM2 PrP and MHM2 PrP(del 23-88) immunoprecipitated with3F4. Lanes 1 and 4, ScN2a cell extracts; lanes 2 and 5, ScN2a cellsexpressing MHM2 PrP(del 23-88); lanes 3 and 6, ScN2a cellsexpressing MHM2 PrP. Cells were labeled with [35S]methionine inthe absence (lanes 1-3) or presence (lanes 4-6) of tunicamycin (25pug/ml) for 1 hr. All labeled extracts were immunoprecipitated withthe anti-Syrian hamster PrP mAb 3F4 (30). None of these sampleswas treated with proteinase K. (B) Immunoblot of MHM2 PrP andMHM2 PrP(del 23-88) species after digestion with proteinase K.Lanes 1 and 4, ScN2a cells; lanes 2 and 5, ScN2a cells expressingMHM2 PrP; lanes 3 and 6, ScN2a cells expressing MHM2 PrP(del23-88). Lanes 1-3 were stained with the rabbit anti-Syrian hamsterPrP 27-30 polyclonal antibody R073. Lanes 4-6 were stained with theanti-PrP mAb 3F4, which specifically recognizes MHM2 PrP. Eachlane was loaded with one-fifth of the extract obtained from aconfluent 100-mm culture and digested with proteinase K for 1 hr at370C.
-14-14
FIG. 3. MHM2 PrP(del 231-254) is mostly nonglycosylated andlacks the GPI anchor. (A) Autoradiograph ofMHM2 PrP and MHM2PrP(del 231-254) labeled in the absence or presence of tunicamycin.Lanes 1 and 4, ScN2a cells; lanes 2 and 5, ScN2a cells expressingMHM2 PrP; lanes 3 and 6; ScN2a cells expressing MHM2 PrP(del231-254). Cells were labeled with [35S]methionine in the absence(lanes 1-3) or presence (lanes 4-6) of tunicamycin (25 Ag/ml) for 1hr. All samples were lysed and immunoprecipitated with the anti-Syrian hamster PrP mAb 3F4. None of these samples was treatedwith proteinase K. (B) Autoradiograph of MHM2 PrP and MHM2PrP(del 231-254) labeled in the presence of tunicamycin (30 Ag/ml),digested with phosphatidylinositol-specific phospholipase C, andtreated with aqueous HF. All cell extracts were immunoprecipitatedwith mAb 3F4. Lanes 1 and 2, ScN2a cells expressing MHM2 PrP(del231-254); lanes 3 and 4, ScN2a cells expressing MHM2 PrP; lanes 5and 6, ScN2a cells. Only the samples in lanes 2, 4, and 6 were treatedwith aqueous HF.
A
. I*_0_ Z
Proc. Natl. Acad Sci. USA 90 (1993)
-9.,
O.4* i,-..,z;.11-- u6

Proc. Natl. Acad. Sci. USA 90 (1993) 3185
1 2 3 4 5wsv* jx
$.4
...
I 6 kDa
-42
-30
-22
-14
FIG. 4. MHM2 PrP(del 231-254) produces a 17-kDa protein afterdigestion with proteinase K in ScN2a cells. Immunoblot of extractsof ScN2a cells expressing MHM2 PrP and MHM2 PrP(del 231-254)after digestion with proteinase K (20 ,ug/ml at 37°C for 1 hr). Lanes1 and 4, ScN2a cells; lanes 2 and 5, ScN2a cells expressing MHM2PrP; lanes 3 and 6, ScN2a cells expressing MHM2 PrP(del 231-254).Lanes 1-3 were stained with the rabbit anti-Syrian hamster PrP 27-30polyclonal antibody R073. Lanes 4-6 were stained with the anti-PrPmAb 3F4, which specifically recognizes MHM2 PrP. Each lane wasloaded with one-fifth (lanes 1-3) or four-fifths (lanes 4-6) of theextract obtained from a confluent 100-mm culture.
transfected with MHM2 PrP (Fig. 5, lane 3), MHM2 PrP(OR-5) (lane 1), or MHM2 PrP(OR -3) (lane 2) all producedrecombinant PrPSc with equal efficiency. Elongated forms ofthe MHM2 PrP molecule were constructed by adding one,two, four, or six additional ORs (see Fig. 1). ScN2a cellstransfected with MHM2 PrP, MHM2 PrP(OR +1), MHM2PrP(OR +2), MHM2 PrP(OR +4), or MHM2 PrP(OR +6) allproduced recombinant PrPSC with similar efficiency (Fig. 5,lanes 3-7). These data suggest that varying the number of ORsdoes not affect the steady-state levels of PrPSc.
DISCUSSIONMany lines of evidence argue that the conversion of prPC or
a precursor into PrPSc is essential for prion multiplication. Awealth of data now provides convincing evidence that the
1 2 3 4 5 6 7
- PK
t PK
FIG. 5. MHM2 PrP with 0, 2, 5, 6, 7, 9, or 11 ORs. RecombinantPrP genes with various numbers of ORs were expressed by theSPOX.Ilneo vector in ScN2a cells. Lane 1, MHM2 PrP(OR -5); lane2, MHM2 PrP(OR -3); lane 3, MHM2 PrP; lane 4, MHM2 PrP(OR+1); lane 5, MHM2 PrP(OR +2); lane 6, MHM2 PrP(OR +4); lane7, MHM2 PrP(OR +6). (Upper) Lanes were loaded with extractscontaining '50 ,ug of protein and analyzed by immunoblotting afterSDS/PAGE. None of the samples was digested with proteinase K (-PK). (Lower) Samples containing -250 jg of protein were digestedwith proteinase K (20 Ag/ml) for 30 min at 37°C (+ PK). All laneswere stained with the anti-PrP mAb 3F4, which recognizes MHM2PrP. Positions of prestained molecular weight markers are shown atthe left of each panel. The apparent molecular sizes of the markerproteins in kilodaltons are, from top to bottom, 49.5, 32.5, 27.5, and18.5.
formation of PrPSc is a posttranslational process (2-5, 13, 31,33, 34). In the study reported here, we extended earlier workshowing that PrPSc synthesis occurs in the absence of Asn-linked glycosylation (5) by investigating PrPSC synthesis inScN2a cells transfected with a recombinant PrP gene lackingthe C-terminal 24 codons. Deletion of these codons appearsto prevent the addition of the GPI anchor to PrP (Fig. 3). LikeC-terminal truncation, deletion of as many as 66 amino acidsfrom the N-terminus of PrP did not prevent PrPsc synthesis.Thus, more than one-third (112 of 254) of the PrP residues arenot required for a conformational change which occurs duringthe conversion of PrPC or a precursor into PrPSc.The low levels of PrPSc observed with the MHM2 PrP(del
231-254) construct present three problems with respect toconclusions about the role of the GPI anchor in PrPScformation. First, the small amounts of the MHM2 PrP(del231-254) scrapie isoform produced in ScN2a cells makeextended chemical analysis impractical (Fig. 4). Second, asmall proportion ofMHM2 PrP(del 231-254) molecules mighttransiently carry GPI anchors and be preferentially convertedinto PrPSc. Third, we cannot eliminate the remote possibilitythat a GPI is added to MHM2 PrP(del 231-254) at a site otherthan the C terminus through a chemical bond which isinsensitive to aqueous HF cleavage.
Studies of wt PrP argue that PrPC or a subset of PrPmolecules currently indistinguishable from PrPC transit to thecell surface, where they are bound by their GPI anchors priorto reentry and conversion into PrPsc (3, 4, 34, 35). While thetransit of PrP molecules to the cell surface appears to precedePrPSc synthesis, the data presented here support the scenarioof an intracellular, less efficient pathway for the formation ofPrPSc. Thus, the mechanism by which MHM2 PrP(del 231-254)sc is synthesized remains unknown. The apparent lowlevels of MHM2 PrP(del 231-254)sc may be due, at least inpart, to the poor binding ofMHM2 PrP(del 231-254)Sc, whichlacks a GPI anchor, to the poly(vinylidene difluoride) mem-branes used for immunoblotting. Nevertheless, it should benoted that the -z17-kDa MHM2 PrP(del 231-254)sc is indeedPrPSc as defined by two criteria: resistance to proteinase Kand insolubility in detergents.
Structural studies of the GPI anchor attached to PrPScindicate that its glycan may exist in at least six forms, two ofwhich are sialylated (16). Two different "strains" or distinctisolates of scrapie prions passaged in Syrian hamsters had thesame set of GPI glycans. Although only limited amounts ofPrPc were available for structural studies (36), sufficientquantities were purified to allow determination that the GPIglycans of PrPC also contained sialic acid.That the deletion of the N-terminal 66 residues of PrP,
which include five ORs, did not alter the synthesis of PrPScis of considerable interest, since an insert of 144 bp at codon53 containing an additional six ORs has been described inpatients with Creutzfeldt-Jakob disease from southern En-gland (20). The logarithm-of-odds score for this extendedpedigree exceeds 11 (23, 24). While our investigations indi-cate that the addition of one, two, four, or six ORs did notchange the efficiency of PrPSc formation in ScN2a cells (Fig.5), clinical studies have demonstrated that four to nine ORsin addition to the normal five ORs segregate with affectedindividuals in families with inherited Creutzfeldt-Jakob dis-ease (17-20, 22, 37), whereas deletion of one of the fivenormal ORs has been found to be unassociated with neuro-logic disease (38, 39).Although much information has been gained from studies
assessing the formation of recombinant PrPsc in scrapie-infected cultured cells transfected with recombinant PrPgenes, studies of prion infectivity generated from recombi-nant PrP molecules have been confined to transgenic miceexpressing foreign or mutant PrP genes (8, 40-43). Unfortu-nately, the efficiency of infection of cultured cells is rather
Medical Sciences: Rogers et al.

3186 Medical Sciences: Rogers et al.
low, which makes infecting many different cell lines express-ing a variety of recombinant PrP genes impractical. Tocircumvent this problem, transgenic mice expressing thetruncated and elongated PrP molecules can be constructed byusing mice with both murine PrP alleles ablated to eliminateany background contribution from the host (44). From theresults reported here, it seems likely that all the truncated andelongated PrP constructions will support the synthesis ofprion infectivity. Examining the effects ofchanging the lengthof PrP on the propagation of distinct prion isolates will be ofconsiderable interest with respect to elucidating how infor-mation is carried within the infectious prion particle (45).
We thank A. Taraboulos, D. Serban, and D. Borchelt for helpfuldiscussions. This work was supported by research grants from theNational Institutes of Health (AG02132, NS14069, AG08%7, andNS22786) and by gifts from the Sherman Fairchild Foundation andfrom National Medical Enterprises.
1. Prusiner, S. B. (1991) Science 252, 1515-1522.2. Borchelt, D. R., Scott, M., Taraboulos, A., Stahl, N. &
Prusiner, S. B. (1990) J. Cell Biol. 110, 743-752.3. Borchelt, D. R., Taraboulos, A. & Prusiner, S. B. (1992) J.
Biol. Chem. 267, 6188-6199.4. Caughey, B. & Raymond, G. J. (1991) J. Biol. Chem. 266,
18217-18223.5. Taraboulos, A., Rogers, M., Borchelt, D. R., McKinley,
M. P., Scott, M., Serban, D. & Prusiner, S. B. (1990) Proc.Natl. Acad. Sci. USA 87, 8262-8266.
6. Rogers, M., Taraboulos, A., Scott, M., Groth, D. & Prusiner,S. B. (1990) Glycobiology 1, 101-109.
7. Scott, M., Butler, D., Bredesen, D., Walchli, M., Hsiao, K. &Prusiner, S. B. (1988) Protein Eng. 2, 69-76.
8. Scott, M. R., Kohler, R., Foster, D. & Prusiner, S. B. (1992)Protein Sci. 1, 986-997.
9. Oesch, B., Westaway, D., Walchli, M., McKinley, M. P.,Kent, S. B. H., Aebersold, R., Barry, R. A., Tempst, P.,Teplow, D. B., Hood, L. E., Prusiner, S. B. & Weissmann, C.(1985) Cell 40, 735-746.
10. McKinley, M. P., Meyer, R., Kenaga, L., Rahbar, F., Cotter,R., Serban, A. & Prusiner, S. B. (1991) J. Virol. 65, 1440-1449.
11. Meyer, R. K., McKinley, M. P., Bowman, K. A., Braunfeld,M. B., Barry, R. A. & Prusiner, S. B. (1986) Proc. Natl. Acad.Sci. USA 83, 2310-2314.
12. Prusiner, S. B., Groth, D. F., Bolton, D. C., Kent, S. B. &Hood, L. E. (1984) Cell 38, 127-134.
13. Basler, K., Oesch, B., Scott, M., Westaway, D., Wilchli, M.,Groth, D. F., McKinley, M. P., Prusiner, S. B. & Weissmann,C. (1986) Cell 46, 417-428.
14. Stahl, N., Borchelt, D. R., Hsiao, K. & Prusiner, S. B. (1987)Cell 51, 229-240.
15. Stahl, N., Borchelt, D. R. & Prusiner, S. B. (1990) in Molecularand Cell Biology of Membrane Proteins, ed. Turner, A. J.(Horwood, Chichester, England), pp. 189-216.
16. Stahl, N., Baldwin, M. A., Hecker, R., Pan, K.-M., Burlin-game, A. L. & Prusiner, S. B. (1992) Biochemistry 31, 5043-5053.
17. Goldfarb, L. G., Brown, P., McCombie, W. R., Goldgaber, D.,Swergold, G. D., Wills, P. R., Cervenakova, L., Baron, H.,Gibbs, C. J., Jr., & Gajdusek, D. C. (1991) Proc. Natl. Acad.Sci. USA 88, 10926-10930.
18. Owen, F., Poulter, M., Collinge, J., Leach, M., Lofthouse, R.,Crow, T. J. & Harding, A. E. (1992) Mol. Brain Res. 13,155-157.
19. Owen, F., Poulter, M., Shah, T., Collinge, J., Lofthouse, R.,Baker, H., Ridley, R., McVey, J. & Crow, T. (1990) Mol. BrainRes. 7, 273-276.
20. Owen, F., Poulter, M., Lofthouse, R., Collinge, J., Crow, T. J.,
Risby, D., Baker, H. F., Ridley, R. M., Hsiao, K. & Prusiner,S. B. (1989) Lancet i, 51-52.
21. Owen, F., Poulter, M., Collinge, J., Leach, M., Shah, T.,Lofthouse, R., Chen, Y. F., Crow, T. J., Harding, A. E. &Hardy, J. (1991) Exp. Neurol. 112, 240-242.
22. Collinge, J., Harding, A. E., Owen, F., Poulter, M., Lofthouse,R., Boughey, A. M., Shah, T. & Crow, T. J. (1989) Lancet ii,15-17.
23. Collinge, J., Brown, J., Hardy, J., Mullan, M., Rossor, M. N.,Baker, H., Crow, T. J., Lofthouse, R., Poulter, M., Ridley, R.,Owen, F., Bennett, C., Dunn, G., Harding, A. E., Quinn, N.,Doshi, B., Roberts, G. W., Honavar, M., Janota, I. & Lantos,P. L. (1992) Brain 115, 687-710.
24. Poulter, M., Baker, H. F., Frith, C. D., Leach, M., Lofthouse,R., Ridley, R. M., Shah, T., Owen, F., Collinge, J., Brown, G.,Hardy, J., Mullan, M. J., Harding, A. E., Bennett, C., Doshi,R. & Crow, T. J. (1992) Brain 115, 675-685.
25. Gasset, M., Baldwin, M. A., Lloyd, D., Gabriel, J.-M., Holtz-man, D. M., Cohen, F., Fletterick, R. & Prusiner, S. B. (1992)Proc. Natl. Acad. Sci. USA 89, 10940-10944.
26. Cohen, F. E., Abarbanel, R. M., Kuntz, I. D. & Fletterick,R. J. (1986) Biochemistry 25, 266-275.
27. Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989) MolecularCloning: A Laboratory Manual (Cold Spring Harbor Lab.,Plainview, NY), 2nd Ed.
28. Rogers, M., Serban, D., Gyuris, T., Scott, M., Torchia, T. &Prusiner, S. B. (1991) J. Immunol. 147, 3568-3574.
29. Kunkel, T. A. (1985) Methods Enzymol. 154, 367-382.30. Taraboulos, A., Serban, D. & Prusiner, S. B. (1990) J. Cell
Biol. 110, 2117-2132.31. Kascsak, R. J., Rubenstein, R., Merz, P. A., Tonna-DeMasi,
M., Fersko, R., Carp, R. I., Wisniewski, H. M. & Diringer, H.(1987) J. Virol. 61, 3688-3693.
32. Ferguson, M. A. J., Homans, S. W., Dwek, R. A. & Radema-cher, T. W. (1988) Science 239, 753-759.
33. Taraboulos, A., Raeber, A., Borchelt, D., McKinley, M. P. &Prusiner, S. B. (1991) FASEB J. 5, A1177 (abstr.).
34. Caughey, B., Raymond, G. J., Ernst, D. & Race, R. E. (1991)J. Virol. 65, 6597-6603.
35. Taraboulos, A., Raeber, A. J., Borchelt, D. R., Serban, D. &Prusiner, S. B. (1992) Mol. Biol. Cell 3, 851-863.
36. Pan, K.-M., Stahl, N. & Prusiner, S. B. (1992) Protein Sci. 1,1343-1352.
37. Collinge, J., Owen, F., Poulter, H., Leach, M., Crow, T.,Rosser, M., Hardy, J., Mullan, H., Janota, I. & Lantos, P.(1990) Lancet 336, 7-9.
38. Laplanche, J.-L., Chatelain, J., Launay, J.-M., Gazengel, C. &Vidaud, M. (1990) Nucleic Acids Res. 18, 6745.
39. Vnencak-Jones, C. L. & Phillips, J. A. (1992) Am. J. Hum.Genet. 50, 871-872.
40. Westaway, D., Mirenda, C. A., Foster, D., Zebaijadian, Y.,Scott, M., Torchia, M., Yang, S.-L., Serban, H., DeArmond,S. J., Ebeling, C., Prusiner, S. B. & Carlson, G. A. (1991)Neuron 7, 59-68.
41. Scott, M., Foster, D., Mirenda, C., Serban, D., Coufal, F.,Walchli, M., Torchia, M., Groth, D., Carlson, G., DeArmond,S. J., Westaway, D. & Prusiner, S. B. (1989) Cell 59, 847-857.
42. Prusiner, S. B., Scott, M., Foster, D., Pan, K.-M., Groth, D.,Mirenda, C., Torchia, M., Yang, S.-L., Serban, D., Carlson,G. A., Hoppe, P. C., Westaway, D. & DeArmond, S. J. (1990)Cell 63, 673-686.
43. Hsiao, K. K., Scott, M., Foster, D., Groth, D. F., DeArmond,S. J. & Prusiner, S. B. (1990) Science 250, 1587-1590.
44. Bueler, H., Fischer, M., Lang, Y., Bluthmann, H., Lipp,H.-L., DeArmond, S. J., Prusiner, S. B., Aguet, M. & Weiss-mann, C. (1992) Nature (London) 356, 577-582.
45. Hecker, R., Taraboulos, A., Scott, M., Pan, K.-M., Torchia,M., Jendroska, K., DeArmond, S. J. & Prusiner, S. B. (1992)Genes Dev. 6, 1213-1228.
Proc. Natl. Acad. Sci. USA 90 (1993)





























