Embed Size (px)
Citation preview

THE JOURNAL OF BIOUXICAL CHEMISTRY Q 1993 by The American Society for Biochemistry and Molecular Biology, Inc
Vol. 268, No. 32, Issue of November 15, pp. 23799-23805, 1993 Printed in U.S.A.
Control of Yeast Glycogen Synthase-2 by COOH-terminal Phosphorylation*
(Received for publication, May 28, 1993, and in revised form, July 21, 1993)
Thomas A. Hardy and Peter J. Roach From the DeDartment of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, Indiana 46202-5122
The budding yeast Saccharomyces cerevisiae ex- presses two isoforms of glycogen synthase, of which gly- cogen synthase-2 (GS-2) appears to be the most impor- tant determinant of glycogen accumulation (Farkas, I., Hardy, T. A, Goebl, M. G., and Roach, P. J. (1991) J. Biol. Chem 266, 15602-15607). Partial proteolysis of purified yeast glycogen synthase activated the enzyme, mimick- ing the effects of dephosphorylation. The cleavage was localized to the COOH terminus of the molecule and trypsin treatment released 32P from enzyme labeled in vivo with 32P or in vitro by cyclic AMP-dependent pro- tein kinase. Similarly, when cells were labeled with 32P, no radioactivity was incorporated into a mutant form of (38-2 truncated at residue 643 while the wild type en- zyme was phosphorylated at both Ser and Thr residues. The 9 Ser and Thr residues COOH-terminal to position 643 were mutated individually to Ala, and the GS-2 mu- tants were expressed from a low copy plasmid in yeast that lacked functional chromosomal copies of the two glycogen synthase genes. Mutations at Ser-650, Ser-654, and Thr-667 resulted in significant activation of yeast glycogen synthase and elevation in the level of accumu- lated glycogen as compared with wild type. Likewise, expression of the truncated GS-2 resulted in hyperactive enzyme and the overaccumulation of glycogen. None of the other Ser or Thr mutations substantially affected glycogen synthase activity and glycogen storage. We conclude that Ser-650, Ser-654, and Thr-667 are regula- tory phosphorylation sites in vivo. However, in vitro, cy- clic AMP-dependent protein kinase modified Ser resi- due(s) COOH-terminal to position 659, and so the identity of the physiological 08-2 kinases is unclear. Yeast strains bearing glc7 and gacl mutations are defec- tive in genes encoding type 1 protein phosphatase com- ponents and are impaired in their ability to accumulate glycogen. Expression of the truncated GS-2 in these strains restored glycogen accumulation, as did the pres- ence of (38-2 mutated at ser-650, ser-654, or Thr-667. These data are consistent with the hypothesis that type 1 phosphatase regulates GS-2 by controlling its phos- phorylation state.
Glycogen is produced in organisms as diverse as bacteria and man as a deposit of reserve carbohydrate. In the budding yeast Saccharomyces cerevisiae, this branched polymer of glucose can represent more than 20% of the cell's dry weight (l), although the extent to which yeast cells synthesize and store glycogen
Grant DK 42576 (to P. J. R.) and a Medical Scientist Scholar fellowship * This work was supported in part by National Institutes of Health
from the Indiana University School of Medicine (to T. A. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "aduertise- rnent" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
depends on environmental conditions. For instance, glycogen deposition is stimulated in response to starvation for carbon, nitrogen, sulfur, or phosphorus (1). Exposure of anaerobically maintained cells to oxygen (2, 3) or of haploid cells to mating pheromone (4) opposes glycogen accumulation. The molecular mechanisms by which yeast cells respond to these various en- vironmental conditions are largely unknown, but ultimately there must be regulation of the key glycogen-metabolizing en- zymes.
Glycogen synthase (EC 2.4.1.11) is of central importance in the biosynthesis of glycogen (5, 6). S. cerevisiae expresses two isoforms of glycogen synthase, designated GS-1 and GS-2,' en- coded by the GSYl and GSY2 genes, respectively (7, 8). GS-2 represents the predominant activity in vegetatively growing yeast cells and expression of GSY2, but not GSYl, appears to be linked to nutrient limitation (8). The sequences of the yeast glycogen synthases are about 50% identical to those of the corresponding mammalian enzymes (7, 8), which have been more extensively studied (Fig. 1). Mammalian glycogen syn- thase, especially the rabbit muscle isoform, has served as a valuable model for studies of protein phosphorylation and en- zyme regulation (5, 6). The enzyme is believed to be regulated by multisite phosphorylation at sites concentrated near the NH2- and COOH-termini of the molecule. The greatest diver- gence in sequence as compared with the yeast enzymes is pre- cisely in these regulatory regions. The yeast enzymes both lack the mammalian NH2-terminal phosphorylation sites. An im- portant cluster of five phosphorylation sites (designated sites 3a, 3b, 3c, 4 and 5) in mammalian glycogen synthase is located toward the COOH terminus. Sites 3a and 3b are conserved in the yeast sequence, but in the region of the other three sites, there is no recognizable sequence similarity (Fig. 1). There is considerable biochemical evidence that yeast glycogen syn- thase also undergoes reversible phosphorylation (9-13). As is true for the mammalian protein, phosphorylation converts the enzyme to a less active form that requires the presence of the allosteric effector glucose-6-P to realize full activity (10, 11). Knowledge of the molecular mechanisms involved in control- ling yeast glycogen synthase is still limited, and neither the phosphorylation sites nor the relevant protein kinases have been fully identified.
Genetic analysis has uncovered many cellular factors that influence glycogen accumulation in yeast and some of these are potentially involved in the regulation of glycogen synthase. Examples are genes encoding components of a type I protein phosphatase (14, 15) and CAMP PK (16, 17), both of which are
synthase-2; CAMP PK, cyclic AMP-dependent protein kinase; PAGE, The abbreviations used are: GS-1, glycogen synthase-1; glycogen
polyacrylamide gel electrophoresis; PCR, polymerase chain reaction; PVDF, polyvinylidene difluoride; TLCK, N"-p-L-lysine chloromethyl ke- tone; kb, kilobase paifis); HPLC, high performance liquid chromatog- raphy.
23799

23800 Control of East Glycogen Synthase-2
MUSCLE 11n1 II
05-2 II I \
"""""
""". \ \ "-
""
\ \
' 6 4 3 6 5 0 654 6 6 7 I 05-2: K K L K V A R P L Q V P G Q P R D L R Y ~ G D L G ~ L Q E ~ ~ D Y F S L G ~ P ~ D D D D G P Y ~ D S
U : Q G Y R Y P R P A S V P P S P S L S R H S S P H Q S E D E E E P R D G L P E E D G E R Y D E D E E ~ D ~ I ~ E W ~ ~
MRASCTSSSGGSKRSNSVDTSSLSTPSEPLSPASSLGEERN
FIG. 1. Primary structures of rabbit skeletal muscle glycogen synthase and yeast GS-2. In the upper portion of the figure, line representations of the two proteins are shown, to scale, with phospho- rylation sites indicated by vertical bars. In the lower part of the figure, the amino acid sequences of the indicated COOH-terminal regions of GS-2 and muscle ( M ) glycogen synthase are given. Residues in GS-2 which were mutated in this study are underlined. Boldface residues correspond to phosphorylation sites determined in this and other stud- ies. The sites are identified by amino acid position number for GS-2 and by a commonly used nomenclature for the muscle enzyme (5,6). Amino acids 630-734 of the muscle enzyme are shown.
3a 3b 3c 4 5
la lb
thought to be involved in the post-translational control of mam- malian glycogen synthase (5, 6, 18). The GACl and GLC7I DZS2Sl genes are thought to encode regulatory and catalytic subunits, respectively, of a yeast type I protein phosphatase (14, 151, and mutations in either gene can lead to diminished glycogen accumulation. The glycogen synthase in these mutant strains is predominantly in a glucose-6-P-dependent form, con- sistent with its dephosphorylation being impaired (13-15). There is an inverse correlation between glycogen accumulation and the activity of the RASIcAMP pathway in yeast (16, 17, 19-22). This finding, together with the observation that CAMP PK can phosphorylate glycogen synthase in vitro (7,13), has led to the expectation that CAMP PK directly phosphorylates yeast glycogen synthase in vivo (7, 16, 23, 24). Although the genetic evidence clearly supports the importance of both CAMP PK and type I phosphatase in controlling glycogen accumulation, it does not establish whether or not these enzymes act directly on glycogen synthase.
The studies reported here focus on the post-translational control of yeast GS-2. Using site-directed mutagenesis, we have identified three regulatory phosphorylation sites near the COOH terminus of the enzyme whose mutation causes glyco- gen hyperaccumulation or, ingacl orglc7 hosts, a restoration of glycogen accumulation. The results support the proposal that the GACl and GLC7 gene products function as a physiological glycogen synthase phosphatase.
EXPERIMENTAL PROCEDURES
Strains and Vectors-The glycogen synthase-deficient S. cereuisiae strains IF3 (MAT a ura3 lys2 ade2 his3 trpl gsylAl::URA3 gsyZAl::HIS3) and IF1 (MAT a ura3 lys2 ade2 his3 trpl gsylA1::URAS) were derived from strain YPH52 as described previously (7, 8). Strain E8-llC (a cir" pep4 leu21 was originally from E. Young, University of Seattle. Strains EG327-1D (glc7-1; Ref. 15) and EG328-2D (gaclA::LEU2; Ref. 15) were isogenic to the wild type strain EG328-1A (MAT a his4 leu2 ura3 t r p l ) and were kindly provided by K. Tatchell, North Carolina State University. Escherichia coli strain TG-1 was used as host for DNAmanipulations involving M13 vectors. E. coli DH5a was used in all other cases. The yeast shuttle vector pRS314 (25) was ob- tained from P. Hieter, Johns Hopkins University. This plasmid contains the CENl, ARSl, and TRPl sequences and was used for expression of GSY2 in yeast. Other E. coli vectors included pUCHE (26) and M13 mp19 (27).
Phosphorylation and Partial Proteolysis of Yeast Glycogen Synthase "Purified glycogen synthase (7) was incubated for 2 h with the cata- lytic subunit of yeast CAMP PK (TPK1 provided by M. Zoller, ARIAD Pharmaceuticals, Cambridge, MA) in a reaction containing 50 mM Tris- C1, pH 7.5,20 mM Mg(CH3COOH)2, 175 PM [y32PlATP(1500 cpdpmol), 100 pg glycogen synthasdml, and 5 pg TPKl/ml. For studies of the effect of proteolysis on enzyme activity, purified glycogen synthase was incubated with trypsin (Sigma; 5:l glycogen synthase to trypsin mass
ratio) at 30 "C in a reaction containing 130 pg glycogen synthasdml, 50 mM Tris-C1, pH 7.5,1.75 mM EDTA, 0.65 mM EGTA, 10% glycerol, 50 mM 6-mercaptoethanol, and 250 mM NaCl. At various times, aliquots were removed and added to 5 x sample buffer (1 x sample buffer is 10% (v/v) glycerol, 12.5 mM Tris phosphate, pH 6.8, 0.46% (w/v) SDS, and 1% P-mercaptoethanol) for analysis by SDS-PAGE (281, or diluted 10-fold in glycogen synthase reaction buffer for activity assays.
Isolation and Sequencing of GS-2 Phosphopeptides-To locate CAMP PK phosphorylation sites in yeast glycogen synthase, 400 pg of purified glycogen synthase was phosphorylated with TPKl as described above. An equal mass of trypsin was then added directly to the phosphoryla- tion reaction, and incubation was continued for 2 h. One volume of cold acetone was then added, and after 1 h at -20 "C, the sample was centrifuged for 30 min at 13,000 x g . Preliminary studies had shown that the phosphopeptides generated by trypsin action were soluble in 50% acetone. The supernatant was dried completely and resuspended in 200 pl of 0.1% trifluoroacetic acid before loading onto a C8 reverse phase microbore HPLC column (RP-300 cartridge, Applied Biosystems). Pep- tides were eluted from the C8 column with a linear gradient of aceto- nitrile in 0.1% trifluoroacetic acid (%loo% acetonitrile in 45 rnin). Fractions (75 pl) were collected and analyzed by counting Cerenkov radiation. Two peaks of radioactivity were resolved by C8 chromatog- raphy. The major peak (peak 1) contained 75% of the radioactivity and eluted at approximately 43% acetonitrile. The remainder of the radio- activity, peak 2, eluted a t approximately 39% acetonitrile. The radioac- tivity recovered from the column represented 80% of the 32P originally incorporated into the glycogen synthase. Fractions corresponding to these peaks of radioactivity were separately pooled for chromatography on a C18 reverse phase microbore column (OD-300, Applied Biosys- tems). Elution from the C18 column was achieved by a linear gradient from 37 to 47% acetonitrile in 0.1% trifluoroacetic acid over a 15-min period. Purified peptides were sequenced on a Porton Instruments (Tar- zana, CA) model 2090 integrated microsequencing system.
Purification of GS-2 Labeled in Viuo-Radiolabeled GS-2 was puri- fied from IF1 cells grown in the presence of 32Pi. A 12-liter YPD culture was grown to early stationary phase. Unlabeled glycogen synthase was extracted from 11.5 liters of this culture. Cells were harvested from the remaining 0.5 liters by centrifugation for 5 min at 8000 x g and resus- pended in 0.5 liters of MIN-P media. This is a synthetic medium which contains no phosphate (29). 15 mCi of 32Pi (phosphoric acid form; 10 mCi/ml; DuPont-NEN) was added, and the cells were incubated for 3 h a t 23 "C with stimng. Glycogen synthase was purified as described previously (7) except that all buffers included 50 mM sodium fluoride. Samples from radioactive and unlabeled cells were processed in parallel and pooled prior to concanavalin Achromatography to provide sufficient glycogen synthase protein for subsequent analysis.
Immunoprecipitation and Phosphoamino Acid Analysis-Polyclonal GS-2 antisera were raised in guinea pigs. Animals were immunized with the synthetic peptide RELVGEELNDSNMDA, which was coupled to keyhole limpet hemocyanin (30). This peptide corresponds to amino acids 622-636 of GS-2. In vivo labeling and immunoprecipitations uti- lized strain IF3 expressing wild type GS-2 or the truncation mutant, GS-2A643, from plasmid pRS314. Cells were grown at 30 "C in 10 ml of phosphate-depleted media (YPD-Pi) prepared according to Rubin (31). Cultures were split in early stationary phase (approximately 1.5 X 10' cells/ml) and inorganic 32Pi was added to half of the cells a t a final concentration of 100 pCi/ml. After 1 h at 30 "C, cells were collected by centrifugation, the media was removed, and the cell pellet was frozen on dry ice. Cells were resuspended in 300 pl of ice-cold buffer containing 50 mM TriS-Cl, pH 7.5, 1 mM EDTA, 0.1% Triton X-100, 100 mM sodium fluoride, 0.1 mM sodium vanadate, 1 mM sodium pyrophosphate, 0.1 mM TLCK, 5 nm benzamidine, 0.25 pg leupeptidml, and 0.5 pg aprotinid ml. Cell extracts were prepared as for glycogen synthase assays. Im- munoprecipitation was accomplished by adding 4 p1 of GS-2 antiserum to 250 pl of cell extract. After 1 h on ice, antigen-antibody complexes were adsorbed with protein A-agarose (Pierce Chemical Co.) for 1 h a t 4 "C. Proteins were separated from the agarose beads by boiling for 5 min in 2 x Laemmli sample buffer, separated by 10% SDS-PAGE (28L and transferred to a PVDF membrane (Immobilon-P, Millipore). Immu- noblotting was performed by incubation with a 1:200 dilution of anti- GS-2 antiserum, followed by addition of lZ5I-protein A and visualization by autoradiography. For phosphoamino acid analysis, labeled GS-2 was excised from PVDF, and the membrane fragment was incubated 2 h a t 110 "C in 300 pl of constant boiling HCI (Pierce). The hydrolysate was analyzed by thin-layer electrophoresis (32).
Site-directed Mutagenesis and Plasmid Construction-To facilitate mutagenesis of the GSY2 COOH terminus, a SmaI site was generated in the 3"flanking sequence of the cloned GSY2 gene (at nucleotide 2178
Control of Yeast Glycogen Synthase-2 23801 of the published sequence, Ref. 8) by a two-step PCR (33). The primer pairs for the initial PCR were 5'GATCAGTTCAGAGAGAGCTC3'
(GSY2-PCR2), and 5'CATACGACA"TCCCGGGGTITITGC?TTTI
where GSY2-PCR2 and GSY2-PCR3 are mutagenic. GSY2-PCR1 and GSY2-PCR4 were used as primers in the secondary amplification reac- tion. GSY2-PCR1 spans the internal SacI site in GSY2. The GSY2- PCR4 sequence is derived from the pBluescript polylinker sequence that flanked the 3' end of our subcloned GSYZ genomic fragment (8). The 1.1-kb SacI-Sal1 fragment of the secondary PCR product, contain- ing the newly introduced SmaI site, was ligated to a 2.7-kb SalI-Sac1 fragment of GSY2, and the 3.8-kb product was inserted into the Sal1 site of pUCHE. From this construct, a 0.3-kb SacI-SmaI fragment span- ning the COOH terminus of GSYZ was transferred into M13 mp19 for site-directed mutagenesis using the oligonucleotide-directed in vitro mutagenesis system version 2.1 as suggested by the manufacturer (Am- ersham International, United Kingdom). Mutations included a stop codon introduced a t codon 644 of GSY2 and a series of serine to alanine or threonine to alanine substitutions. The mutagenic oligonucleotides used were as follows, with the mutations underlined and the name of the mutant product as indicated: 5'AAGAAA'ITGTAAG'ITGCATGAC-
(GSY2-PCR1) PIUS 5'AAAAGCAAAAACCCCGGGAAATGTCGTATG3'
(GSY2-PCR3) PIUS 5'CTTATCGATACCGTCGTCGAC3' (GSY2-PCR4),
CGCTTAGT3' (GS-2A643); 5'TGCAAGACCGC'ITmTGTACCTGGCT- CACC3' (GS-2S650A); 5'TAGTGTACCTGGCGCACCAAGAGATT- TGAG3' (GS-2S654A); 5'CAAGAGATTTGAGAGCAAACAGCA- CAGTCT3' (GS-2S660Ak 5'TTTGAGATCAAACaCACAGTCTACAT- GAC3' (GS-2S662A); 5'GAGATCAAACAGCGCAGTCTACATG- ACCCC3' (GS-2T663A); 5'CACAGTCTACATGGCCCCTGGTGA- TJTGGG3' (GS-2T667A); 5'TGGTGA"TGGGTGCTCTGCAGGAGGT- TAA3' (GS-2T673A); 5'GGACGA'ITATTlTWA'ITGGGAGTGAATCC3' (GS-2S685A); 5'CATATGCTGATGACETTAAATCCTATGAG3' (GS- 2S704A); and 5 ' T m T G T A C C T G G C W A C C A A G A G A G 3 ' (GS- 2S650AlS654A). To generate the double mutant, GS-2S650AlS654A, single-stranded DNA from mutant GS-2S650A was used as template.
Mutant 0.3-kb cassettes were transferred back into GSY2 in pUCHE, and the sequence of the entire cassette was confirmed using the dideoxy chain termination method of Sanger et al. (34). For expression in yeast, a 3.2-kb EcoRV-EcoRV fragment was subcloned into the SmaI site of the vector pRS314. This fragment includes 503 bp of 5"noncoding sequence from the GSYZ gene. The constructs used for expression were all ori- ented with the GSY2 NH2 terminus toward the SacI side of the pRS314 multiple cloning site. Plasmids were transformed into yeast cells by the lithium acetate method of Ito et al. (35).
Growth of Yeast and Measurement of Glycogen and Glycogen Syn- thase Activity-For qualitative analysis of glycogen accumulation, cells were grown to stationary phase in selective media (SD-TRP; Ref. 36) and 5-10 pl was spotted onto YPD or SD-TRP plates (36). Patches were typically grown for 24 h a t 30 "C before detection of glycogen by expos- ing plates to iodine vapor.
For biochemical analyses, 10-ml SD-TRP cultures were grown to early stationary phase. Cells were collected by centrifugation for 2 min a t 3000 x g, the medium was removed by aspiration and the cells rapidly frozen on dry ice. This process was carried out a t room tem- perature and required less than 5 min. Cell breakage was accomplished by resuspending cells in 300 pl ice-cold homogenization buffer (50 mM Tris-C1, pH 7.5,l mM EDTA, 4 mM Mg(CH,COOH),, 3 mM dithiothreitol, 0.1 mM TLCK, 5 mM benzamidine, 0.25 pg leupeptin/ml, 0.5 pg aprotinidml) and vortexing with an equal volume of cold glass beads in four 30-s pulses. The homogenate was recovered from the beads by centrifugation for 90 s a t 3000 x g, and the supernatant was used for measurement of glycogen and glycogen synthase activity.
Glycogen synthase was assayed as described by Thomas et al. (37). Routine assays were of 10-min duration. A unit of activity is defined as the amount of enzyme that catalyzes the transfer of 1 pmol of glucose from UDP-glucose to glycogen in 1 min under conditions of the standard assay. The -/+ glucose-6-P activity ratio is the ratio of glycogen synthase activity in the absence of glucose-6-P to that measured in the presence of 7.2 mM sugar phosphate. Glycogen levels were determined by a modi- fication of the method used previously (8). Trichloroacetic acid was added to 100 pl of extract until 10% (w/v). After centrifugation for 15 min a t 10.000 x g, glycogen was precipitated from the supernatant by the addition of 4 volumes methanol and centrifugation as above. The dried pellet was digested 4 h at 37 "C with 30 pg amyloglucosidasdml (Sigma) in 150 mM sodium acetate, pH 4.6. Glycogen was estimated from the amount of glucose released (38). Protein was quantitated by the method of Bradford (39), using bovine serum albumin as standard.
8 M = 0.7
0.6
+trypsin
0.3
0.2
0.1
0.0 + 1 0 0 200 300
time (min)
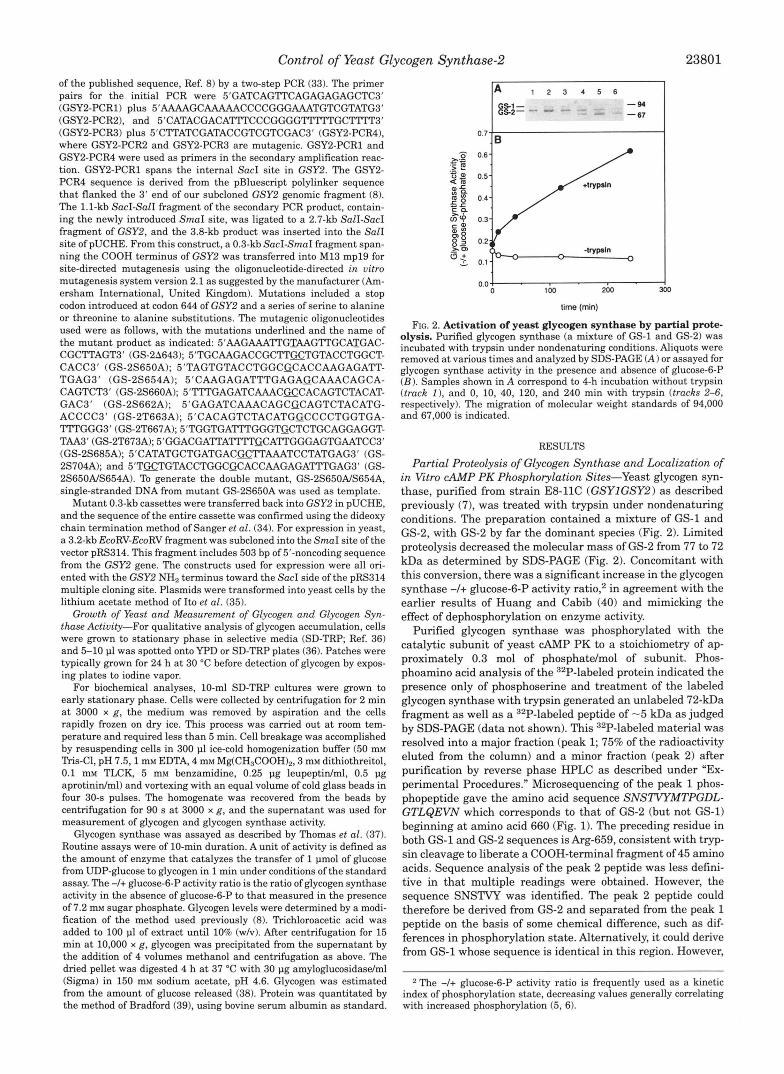
FIG. 2. Activation of yeast glycogen synthase by partial prote- olysis. Purified glycogen synthase (a mixture of GS-1 and GS-2) was incubated with trypsin under nondenaturing conditions. Aliquots were removed a t various times and analyzed by SDS-PAGE (A) or assayed for glycogen synthase activity in the presence and absence of glucose-6-P (B) . Samples shown in A correspond to 4-h incubation without trypsin (track I ) , and 0, 10, 40, 120, and 240 min with trypsin (tracks 2-6, respectively). The migration of molecular weight standards of 94,000 and 67,000 is indicated.
RESULTS
Partial Proteolysis of Glycogen Synthase and Localization of in Vitro CAMP PK Phosphorylation Sites-Yeast glycogen syn- thase, purified from strain E8-llC (GSYIGSY2) as described previously (71, was treated with trypsin under nondenaturing conditions. The preparation contained a mixture of GS-1 and GS-2, with GS-2 by far the dominant species (Fig. 2). Limited proteolysis decreased the molecular mass of GS-2 from 77 to 72 kDa as determined by SDS-PAGE (Fig. 2). Concomitant with this conversion, there was a significant increase in the glycogen synthase -I+ glucose-6-P activity ratio: in agreement with the earlier results of Huang and Cabib (40) and mimicking the effect of dephosphorylation on enzyme activity.
Purified glycogen synthase was phosphorylated with the catalytic subunit of yeast CAMP PK to a stoichiometry of ap- proximately 0.3 mol of phosphatdmol of subunit. Phos- phoamino acid analysis of the 32P-labeled protein indicated the presence only of phosphoserine and treatment of the labeled glycogen synthase with trypsin generated an unlabeled 72-kDa fragment as well as a 32P-labeled peptide of -5 kDa as judged by SDS-PAGE (data not shown). This 32P-labeled material was resolved into a major fraction (peak 1; 75% of the radioactivity eluted from the column) and a minor fraction (peak 2) after purification by reverse phase HPLC as described under "Ex- perimental Procedures." Microsequencing of the peak 1 phos- phopeptide gave the amino acid sequence SNSTVYMTPGDL- GTLQEVN which corresponds to that of GS-2 (but not GS-1) beginning at amino acid 660 (Fig. 1). The preceding residue in both GS-1 and GS-2 sequences is Arg-659, consistent with tryp- sin cleavage to liberate a COOH-terminal fragment of 45 amino acids. Sequence analysis of the peak 2 peptide was less defini- tive in that multiple readings were obtained. However, the sequence SNSTVY was identified. The peak 2 peptide could therefore be derived from GS-2 and separated from the peak 1 peptide on the basis of some chemical difference, such as dif- ferences in phosphorylation state. Alternatively, it could derive from GS-1 whose sequence is identical in this region. However,
* The -/+ glucose-6-P activity ratio is frequently used as a kinetic index of phosphorylation state, decreasing values generally correlating with increased phosphorylation (5, 6).
23802 Control of Yeast Glycogen Synthase-2
neither interpretation changes the assignment of a proteolytic cleavage site corresponding to Arg-659 in GS-2.
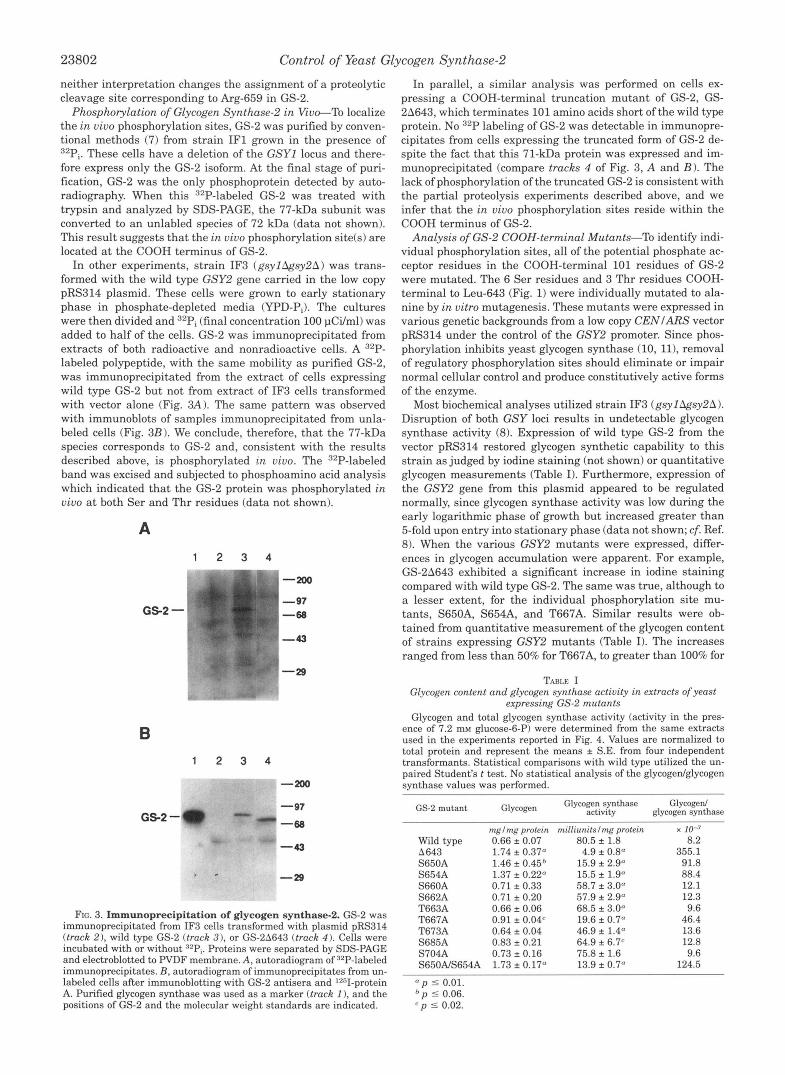
Phosphorylation of Glycogen Synthase2 in Vivo-To localize the in vivo phosphorylation sites, GS-2 was purified by conven- tional methods (7) from strain IF1 grown in the presence of 32Pi. These cells have a deletion of the GSYl locus and there- fore express only the GS-2 isoform. At the final stage of puri- fication, GS-2 was the only phosphoprotein detected by auto- radiography. When this 32P-labeled GS-2 was treated with trypsin and analyzed by SDS-PAGE, the 77-kDa subunit was converted to an unlabled species of 72 kDa (data not shown). This result suggests that the in vivo phosphorylation site(s) are located at the COOH terminus of GS-2.
In other experiments, strain IF3 (gsyl&sy2A) was trans- formed with the wild type GSY2 gene carried in the low copy pRS314 plasmid. These cells were grown to early stationary phase in phosphate-depleted media (YPD-Pi). The cultures were then divided and 32Pi (final concentration 100 pCi/ml) was added to half of the cells. GS-2 was immunoprecipitated from extracts of both radioactive and nonradioactive cells. A "P- labeled polypeptide, with the same mobility as purified GS-2, was immunoprecipitated from the extract of cells expressing wild type GS-2 but not from extract of IF3 cells transformed with vector alone (Fig. 3A). The same pattern was observed with immunoblots of samples immunoprecipitated from unla- beled cells (Fig. 3B). We conclude, therefore, that the 77-kDa species corresponds to GS-2 and, consistent with the results described above, is phosphorylated in vivo. The 32P-labeled band was excised and subjected to phosphoamino acid analysis which indicated that the GS-2 protein was phosphorylated in vivo a t both Ser and Thr residues (data not shown).
A 1 2 3 4
G S 2 - -43
B 1 2 3 4
G S 2 - ' -97
' -68 -43
-29
immunoprecipitated from IF3 cells transformed with plasmid pRS314 FIG. 3. Immunoprecipitation of glycogen synthase-2. GS-2 was
(track 2 ) , wild type GS-2 (track 3 ) , or GS-211643 (track 4 ) . Cells were incubated with or without azPi. Proteins were separated by SDS-PAGE and electroblotted to PVDF membrane. A, autoradiogram of "P-labeled immunoprecipitates. B, autoradiogram of immunoprecipitates from un- labeled cells after immunoblotting with GS-2 antisera and '2"I-protein A. Purified glycogen synthase was used as a marker (track I ) , and the positions of GS-2 and the molecular weight standards are indicated.
In parallel, a similar analysis was performed on cells ex- pressing a COOH-terminal truncation mutant of GS-2, GS- 2A643, which terminates 101 amino acids short of the wild type protein. No 32P labeling of GS-2 was detectable in immunopre- cipitates from cells expressing the truncated form of GS-2 de- spite the fact that this 71-kDa protein was expressed and im- munoprecipitated (compare tracks 4 of Fig. 3, A and B) . The lack of phosphorylation of the truncated GS-2 is consistent with the partial proteolysis experiments described above, and we infer that the in vivo phosphorylation sites reside within the COOH terminus of GS-2.
Analysis of GS-2 COOH-terminal Mutants-To identify indi- vidual phosphorylation sites, all of the potential phosphate ac- ceptor residues in the COOH-terminal 101 residues of GS-2 were mutated. The 6 Ser residues and 3 Thr residues COOH- terminal to Leu-643 (Fig. 1) were individually mutated to ala- nine by in vitro mutagenesis. These mutants were expressed in various genetic backgrounds from a low copy CENIARS vector pRS314 under the control of the GSY2 promoter. Since phos- phorylation inhibits yeast glycogen synthase (10, ll), removal of regulatory phosphorylation sites should eliminate or impair normal cellular control and produce constitutively active forms of the enzyme.
Most biochemical analyses utilized strain IF3 (gsylAgsy2A). Disruption of both GSY loci results in undetectable glycogen synthase activity (8). Expression of wild type GS-2 from the vector pRS314 restored glycogen synthetic capability to this strain as judged by iodine staining (not shown) or quantitative glycogen measurements (Table I). Furthermore, expression of the GSY2 gene from this plasmid appeared to be regulated normally, since glycogen synthase activity was low during the early logarithmic phase of growth but increased greater than 5-fold upon entry into stationary phase (data not shown; cf. Ref. 8). When the various GSY2 mutants were expressed, differ- ences in glycogen accumulation were apparent. For example, GS-2A643 exhibited a significant increase in iodine staining compared with wild type GS-2. The same was true, although to a lesser extent, for the individual phosphorylation site mu- tants, S650A, S654A, and T667A. Similar results were ob- tained from quantitative measurement of the glycogen content of strains expressing GSY2 mutants (Table I). The increases ranged from less than 50% for T667A, to greater than 100% for
TARLE I Glycogen content and glycogen synthase activity in extracts of yeast
expressing GS-2 mutants Glycogen and total glycogen synthase activity (activity in the pres-
ence of 7.2 mM glucose-6-P) were determined from the same extracts used in the experiments reported in Fig. 4. Values are normalized to total protein and represent the means f S.E. from four independent transformants. Statistical comparisons with wild type utilized the un- paired Student's t test. No statistical analysis of the glycogedglycogen synthase values was performed.
GS-2 mutant Glycogen Glycogen synthase Glycogen/ activity glycogen synthase
Wild type 0.66 f 0.07 mglmgprotein milliunitslmg protein x IO-.'
80.5 f 1.8 8.2 A643 1.74 f 0.37" 4.9 f 0.8" 355.1 S650A 1.46 f 0.45h 15.9 f 2.9" 91.8 S654A 1.37 f 0.22" 15.5 f 1.9" 88.4 S660A 0.71 f 0.33 58.7 f 3.0" 12.1 S662A 0.71 f 0.20 57.9 f 2.9" 12.3 T663A 0.66 f 0.06 68.5 f 3.0" 9.6 T667A 0.91 f 0.04' 19.6 f 0.7" 46.4 T673A 0.64 f 0.04 46.9 f 1.4" 13.6 S685A 0.83 f 0.21 64.9 f 6.7' 12.8 S704A 0.73 f 0.16 75.8 f 1.6 9.6 S650NS654A 1.73 f 0.17" 13.9 f 0.7" 124.5
" p c 0.01.
C p c 0.02. h p 5 0.06.
Control of Yeast Glycogen Synthase-2 23803
S650A and S654A. From Table I it is also apparent that most mutants, with the exception of S704A, were expressed a t lower levels of total activity compared with wild type, perhaps due to reduced stability of the mutant proteins. However, those mu- tants that correlated with overaccumulation of glycogen were expressed a t markedly reduced levels compared with wild type GS-2 so that the accumulation of glycogen was achieved with much less total glycogen synthase activity. This point can be appreciated better by normalizing glycogen levels to the spe- cific activity of glycogen synthase (Table I). For example, GS- 28643 was able to synthesize 40-fold more glycogen per glyco- gen synthase molecule than its wild type counterpart (assuming a constant specific activity determined in the pres- ence of glucose-6-P for the different glycogen synthase mu- tants). Similarly, the mutants S650A, S654A, and T667A showed significant increases in this parameter. No other mu- tant was significantly different from wild type in terms of its ability to accumulate glycogen, whether judged by iodine stain- ing or quantitative measurememt.
Measurements of glycogen synthase activity in cell extracts indicated alterations in enzyme kinetic properties for this same set of mutations. The truncation mutant, and mutants S650A, S654A, and T667A, all had significantly elevated -I+ glucose- 6-P activity ratios compared with wild type (Fig. 4). Under conditions in which wilU type GS-2 had an activity ratio of 0.05, the activity ratio of GS-28643 was greater than 0.8. Mutation of Ser-650, Ser-654, or Thr-667 each caused a significant in- crease in activity ratio, to -0.34, although none as large as that associated with GS-2A643. A Ser-6501Ser-654 double mutant resulted in a larger change, to an activity ratio of 0.63. None of the other COOH-terminal mutations altered the activity ratio except T673A which correlated with a smaller increase to an activity ratio of 0.15.
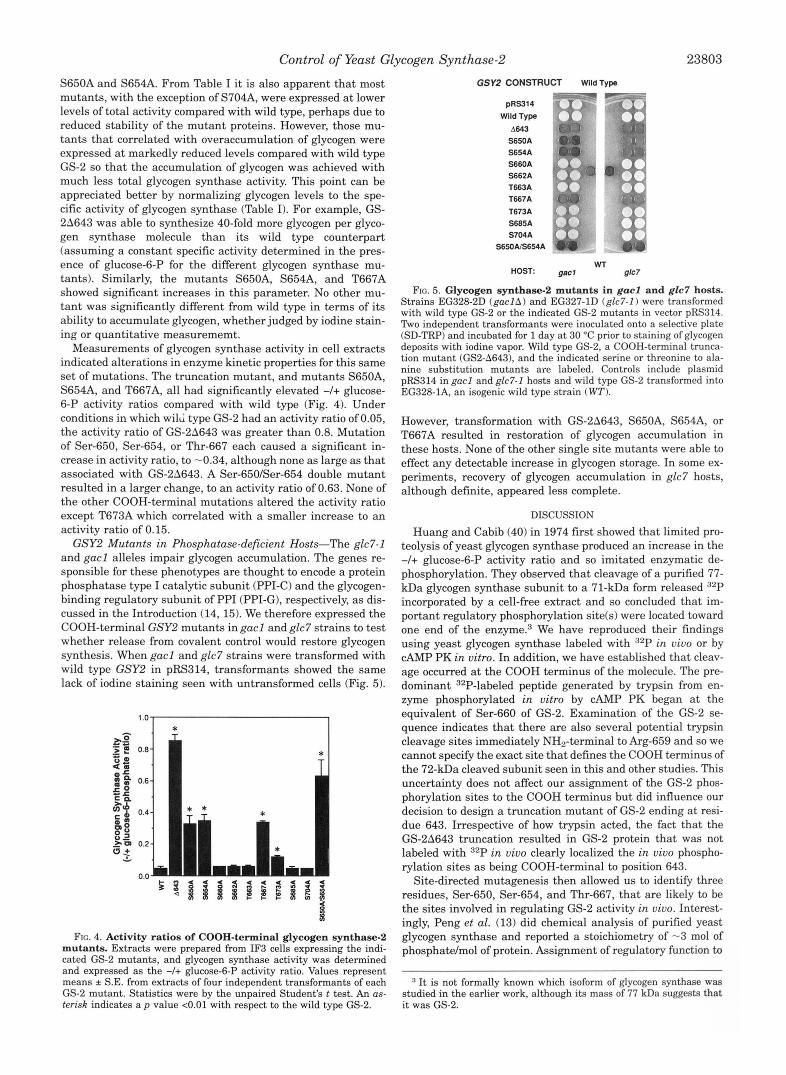
GSY2 Mutants in Phosphatase-deficient Hosts-The glc7-1 and gacl alleles impair glycogen accumulation. The genes re- sponsible for these phenotypes are thought to encode a protein phosphatase type I catalytic subunit (PPI-C) and the glycogen- binding regulatory subunit of PPI (PPI-G), respectively, as dis- cussed in the Introduction (14,15). We therefore expressed the COOH-terminal GSY2 mutants in gacl and glc7 strains to test whether release from covalent control would restore glycogen synthesis. When gacl and glc7 strains were transformed with wild type GSYZ in pRS314, transformants showed the same lack of iodine staining seen with untransformed cells (Fig. 5).
FIG. 4. Activity ratios of COOH-terminal glycogen synthase-2 mutants. Extracts were prepared from IF3 cells expressing the indi- cated GS-2 mutants, and glycogen synthase activity was determined and expressed as the -/+ glucose-6-P activity ratio. Values represent means * S.E. from extracts of four independent transformants of each GS-2 mutant. Statistics were by the unpaired Student's t test. An as- terisk indicates a p value <0.01 with respect to the wild type GS-2.
GSY2 CONSTRUCT Wlld Type - k- pRS314
Wild Type A643
S650A S654A S660A S662A T663A T667A T673A S685A S704A
SSOAIS654A
-
HOST: gacl WT
g/c7
FIG. 5. Glycogen synthase-2 mutants in gacl and glc7 hosts. Strains EG328-2D (gac lA) and EG327-1D (glc7-I) were transformed with wild type GS-2 or the indicated GS-2 mutants in vector pRS314. Two independent transformants were inoculated onto a selective plate (SD-TRP) and incubated for 1 day a t 30 "C prior to staining of glycogen deposits with iodine vapor. Wild type GS-2, a COOH-terminal trunca- tion mutant (GS2-A643), and the indicated serine or threonine to ala- nine substitution mutants are labeled. Controls include plasmid pRS314 in gacl and glc7-I hosts and wild type GS-2 transformed into EG328-1A, an isogenic wild type strain (WT).
However, transformation with GS-28643, S650A, S654A, or T667A resulted in restoration of glycogen accumulation in these hosts. None of the other single site mutants were able to effect any detectable increase in glycogen storage. In some ex- periments, recovery of glycogen accumulation in glc7 hosts, although definite, appeared less complete.
DISCUSSION Huang and Cabib (40) in 1974 first showed that limited pro-
teolysis of yeast glycogen synthase produced an increase in the -I+ glucose-6-P activity ratio and so imitated enzymatic de- phosphorylation. They observed that cleavage of a purified 77- kDa glycogen synthase subunit to a 71-kDa form released 32P incorporated by a cell-free extract and so concluded that im- portant regulatory phosphorylation site(s) were located toward one end of the e n ~ y m e . ~ We have reproduced their findings using yeast glycogen synthase labeled with 32P in vivo or by CAMP PK in vitro. In addition, we have established that cleav- age occurred at the COOH terminus of the molecule. The pre- dominant 32P-labeled peptide generated by trypsin from en- zyme phosphorylated in vitro by CAMP PK began at the equivalent of Ser-660 of GS-2. Examination of the GS-2 se- quence indicates that there are also several potential trypsin cleavage sites immediately NH2-terminal to Arg-659 and so we cannot specify the exact site that defines the COOH terminus of the 72-kDa cleaved subunit seen in this and other studies. This uncertainty does not affect our assignment of the GS-2 phos- phorylation sites to the COOH terminus but did influence our decision to design a truncation mutant of GS-2 ending at resi- due 643. Irrespective of how trypsin acted, the fact that the GS-2A643 truncation resulted in GS-2 protein that was not labeled with 32P in vivo clearly localized the in vivo phospho- rylation sites as being COOH-terminal to position 643.
Site-directed mutagenesis then allowed us to identify three residues, Ser-650, Ser-654, and Thr-667, that are likely to be the sites involved in regulating GS-2 activity in vivo. Interest- ingly, Peng et al. (13) did chemical analysis of purified yeast glycogen synthase and reported a stoichiometry of -3 mol of phosphatdmol of protein. Assignment of regulatory function to
It is not formally known which isoform of glycogen synthase was studied in the earlier work, although its mass of 77 kDa suggests that it was GS-2.

23804 Control of Yeast Glycogen Synthase-2 Ser-650, Ser-654, and Thr-667 is based on several consider- ations. First, extracts of cells expressing the S65OA, S654A, and T667A mutants had elevated glycogen synthase activity ratios compared with wild type enzyme. Second, these muta- tions correlated with elevated glycogen storage compared with wild type. Third, when glycogen synthase bearing these muta- tions was expressed in yeast with impaired phosphatase activ- ity, the ability to accumulate glycogen was restored. Through all of these tests, none of the other mutations differed signifi- cantly from wild type, except the mutant T673A, which corre- lated with a modest but statistically significant increase in activity ratio. However, this change did not correlate with ei- ther increased glycogen accumulation or an override of defec- tive phosphatase, and we consider Thr-673 a less likely candi- date for physiologically significant control of GS-2 function. The truncation mutant GS-2A643 consistently gave a pheno- type similar to the S650A, S654A, and T667A mutants but more extreme in degree. For example, the activity ratio was 0.85 for GS-2A643 but only -0.34 for the single site mutants. A possible explanation is that the individual phosphorylations are additive for the inactivation of GS-2. Indeed, the double mutant, with both Ser-650 and Ser-654 mutated, had a higher activity ratio, 0.63, than either single mutant. All of the data are consistent with the hypothesis that the COOH-terminal region of GS-2 is regulatory and that regulation involves phos- phorylation at Ser-650, Ser-654, and Thr-667. Deletion of the COOH terminus or mutation of these residues by-passes, to- tally or partially, the control mechanisms.
Those mutations that affected glycogen synthase activity and glycogen accumulation correlated with lower levels of GS-2 expression (Table I). In fact, the level of expression of the GS-2 forms analyzed in this study could be inversely correlated with their activity ratio. Comparison of glycogen contents may therefore underestimate the biosynthetic impact of those mu- tant enzymes expressed at low levels. The reason for the de- creased expression is not clear. One possibility is that there is a selective force against expression of unregulated glycogen synthase. Consistent with this idea, we have seen that cells harboring these mutants grow slower than cells carrying the wild type construct (data not shown). Alternatively, there may be other factors limiting glycogen deposits. The ability of cells to sense the amount of glycogen and react by suppressing GS-2 levels could represent a separate regulatory mechanism, pre- sumably independent of the phosphorylation control.
Previous studies have implicated several protein phosphata- ses in the control of yeast glycogen metabolism. The PPG (41) and SIT4 (42) genes encode homologs of mammalian type 2A protein phosphatase. Disruption of the PPG gene impaired gly- cogen accumulation, but the glycogen synthase activity ratio was unchanged in these mutants (41). In contrast, disruption of SIT4 caused a decrease in the -I+ glucose-6-P activity ratio (23), but the impact that SIT4 has on glycogen metabolism is unclear, since no abberrant glycogen accumulation has been reported for sit4 mutants. From the previous work, the best candidate for the post-translational control of glycogen SF- thase is a yeast type I protein phosphatase encoded by the GLCIIDZS2Sl gene (15). The glc7-1 mutation is associated with a nearly complete impairment of glycogen accumulation, and the glycogen synthase in glc7-1 strains was shown to be locked in the glucose-6-P-dependent state, consistent with the enzyme being in the phosphorylated form (13, 15). LOSS of func- tion of a putative type 1 phosphatase regulatory subunit, en- coded by the GACl gene, produced similar effects (15). These findings suggest that the glycogen synthase phosphatase has been conserved between yeast and mammals, where the en- zyme thought to activate glycogen synthase is PPlG, composed of the type 1 catalytic subunit and the mammalian counterpart
of GACl (18). Our data are consistent with the earlier reports (13-15) on glycogen accumulation in gacl and glc7 strains. We found that substitutions at residues 650, 654, and 667, or COOH-terminal truncation of GS-2, resulted in glycogen accu- mulation in these hosts consistent with the removal of inhibi- tory phosphorylation sites. These results provide further evi- dence that the products of the GLC7 and GACl genes comprise a physiological glycogen synthase phosphatase. Since single amino acid changes in the putative target protein can rescue glycogen storage in glc7-1 and gacl strains, it is possible that PPI acts directly as a GS-2 phosphatase.
At the outset of this study, we had expectations that physi- ological phosphorylation of GS-2 would involve, at least in part, the direct action of CAMP PK. We and others (7, 13) had dem- onstrated the in vitro phosphorylation of yeast glycogen syn- thase by this enzyme and Peng et al. (13) had shown inactiva- tion. However, from the present study, cAMP PK phosphorylated serine(s) in our GS-2 preparation located COOH-terminal to residue 660. The high recovery of 32P-la- beled peptides makes it unlikely that major phosphorylated peptides were missed in our analysis. From the mutagenesis experiments, the 2 serines whose alteration affected glycogen synthase activity in vivo were Ser-650 and Ser-654. Since both are NH,-terminal to residue 660, we have no evidence that cAMP PK modifies the physiologically relevant residues. How- ever, we cannot rigorously exclude the possibility that the fail- ure of CAMP PK to modify the physiological sites in vitro was due to their already being fully phosphorylated. I t is relevant that Peng et al. (13) were able to inactivate only the glycogen synthase “I-form,” that is, enzyme isolated to be in a minimally phosphorylated state. Of the in vivo sites identified in the pre- sent study, only Ser-650 is in a local sequence context at all compatible with the recognition requirements of CAMP PK. The surrounding sequence, -R-X-X-S-, does not define the canonical cAMP PK site (-R-R-X-S-; Ref. 431, but there are other NH2- terminal basic residues characteristic of legitimate sites (44, 45). Although we have no direct evidence that CAMP PK phos- phorylates Ser-650, we cannot formally exclude this possibility.
Whether or not CAMP PK phosphorylates Ser-650, other pro- tein kinase(s) must be involved in modifying GS-2. Inspection of the local sequences around Ser-650, Ser-654, and Thr-667 reveals that Ser-654 conforms to the consensus recognition site reported for the mammalian MAP kinases (-P-X-S-P-; Refs. 46 and 47). FranGois et al. (4) found that the mating pheromone causes an inhibition of glycogen synthase that is dependent on the yeast M A P kinase homolog, FUSS. However, fus3 null mu- tants accumulated glycogen normally in the absence of phero- m ~ n e . ~ Likewise, attempts to phosphorylate purified GS-2 with mammalian MAP kinases or recombinant FUS3 have been un- suc~ess fu l .~~ However, yeast appears to contain a family of MAP kinase homologs involved in intracellular signaling cas- cades (48, 49), and one of these enzymes could be a glycogen synthase kinase. With knowledge of the GS-2 sites modified, an immediate goal will be to try to identify the corresponding yeast glycogen synthase kinasek).
Another point of interest, regardless of the protein kinases responsible, is that Ser-650 and Ser-654 align with the phos- phorylation sites 3a and 3b of the mammalian enzymes (Fig. 1). Although the muscle and liver isoforms display greatest vari- ation in their COOH-terminal segments, there is still a signifi- cant level of identity in the region corresponding to sites 3, 4, and 5 (50-52). Recent work has shown that site 3a, and to a lesser degree site 3b, are critical in the control of mammalian
T. A. Hardy and P. J. Roach, unpublished observations. A. Gartner and G. Ammerer, personal communication.

Control of Yeast Glycogen Synthase-2 23805
glycogen synthase activity.6 From an evolutionary perspective, it will be of interest to know whether the conservation seen in the mechanism of glycogen synthase control is limited to the location of the phosphorylation sites in the substrate or whether other elements of the signal transducing systems have also been conserved.
Acknowledgments-We are appreciative of the Biochemistry Biotech- nology Facility for synthesis of oligonucleotides and for peptide sequenc- ing. Besides supplying some yeast strains used in this work, Dr. Kelly Tatchell has been invaluable through many discussions and the sharing of unpublished data. We are also indebted to Drs. Anna DePaoli-Roach, Mark Goebl, and Ron Wek who offered criticisms and advice during the course of this work.
REFERENCES 1. Lillie, S. E., and Pringle, J. R. (1980) J. Bacteriol. 143, 1384-1394
3. Quain, D. E. (1988) J. Inst. Brew. 95, 315423 2. Chester, V. E. (1963) Biochem. J . 86, 153-160
4. FranGois, J., Higgins, D. L., Chang, F., and Tatchell, K. (1991) J. Biol. Chem.
5. Roach, P. J. (1990) FASEB J. 4, 2961-2968 6. Cohen, P. (1986) The Enzymes (Boyer, P. D., ed) Vol. XVII, pp. 462497, Aca-
7. Farkas, I., Hardy, T. A,, DePaoli-Roach, A. A., and Roach, P. J. (1990) J. Biol.
8. Farkas, I., Hardy, T. A,, Goebl, M. G., and Roach, I? J. (1991) J. Biol. Chem.
9. Rothman-Denes, L. B., and Cabib, E. (1970) Proc. Natl. Acad. SCL. U. S. A. 66,
266,6174-6180
demic Press, New York
Chem. 285,20879-20886
266,15602-15607
967-974 10. Rothman-Denes, L. B., and Cabib, E. (1971) Biochemistry 10, 12361242 11. Frantois, J., Villanueva, M. E., and Hers, H.-G. (1988) Eur. J . Biochem. 174,
12. Francois, J., and Hers, H.G. (1988) Eur. J . Biochem. 174, 561567 13. Peng, Z.-Y., Trumbly, R. J., and Reimann, E. M. (1990) J. Biol. Chem. 265,
14. Feng, Z., Wilson, S. E., Peng, Z.-Y., Schlender, K. K., Reimann, E. M., and
15. Francois, J. M., Thompson-Jaeger, S., Skroch, J., Zellenka, U., Spevak, W., and
16. Cannon, J. F., and Tatchell, K. (1987) Mol. Cell. Bid . 7, 2653-2663 17. Cameron, S., Levin, L., Zoller, M., and Wigler, M. (1988) Cell 53, 555-566 18. Cohen, P., and Hardie, D. G. (1991) Biochim. Biophys. Acta 1094, 292-299 19. Toda, T., Uno, I., Ishikawa, T., Powers, S., Kataoka, T., Broek, D., Cameron, S.,
20. Tatchell, K., Robinson, L. C., and Brietenbach, M. (1985) Proc. Natl. Acad. Sci. Broach, J., Matsumoto, K., and Wigler, M. (1985) Cell 40, 27-36
(I. S. A. 82,3785-3789
551-559
13871-13877
Trumbly, R. J. (1991) J. Biol. Chem. 266,2379623801
Tatchell, K. (1992) EMBO J. 11, 87-96
A. Skurat, Y. Wang, and P. J. Roach, unpublished results. ~~ ~
21. 22.
23. 24. 25.
27. 26.
28. 29.
30. 31. 32.
33.
34.
35.
36. 37.
38.
39. 40. 41.
42. 43.
44.
45.
46.
47.
48.
49. 50.
51.
52.
Cannon, J . F., Gibbs, J . B., and Tatchell, K. (1986) Genetics 113, 247-264 Tanaka, K., Nakafuku, M., Tamanoi, F., Kaziro, Y., Matsumoto, K., and lbh-e,
Posas, F., Clotet, J., and Arino, J. (1991) FEBS Lett. 279,341-345 Rowen, D. W., Meinke, M., and LaPorte, D. C. (1992) Mol. Cell. Biol. 12,22-29 Sikorski, R. S., and Hieter, P. (1989) Genetics 122, 19-27 Edenberg, H. J. , Moss, L. G., and Rutter, W. J. (1987) Gene (Amst. 158,297-298 Yaniseh-Perron, C., Vieira, J., and Messing. J. (1985) Gene (Amst.) 33, 103-119 Laemmli, U. K. (1970) Nature 227, 680-685 Johnston, G. C. , F'ringle, J. R., and Hartwell, L. H. (1977) Exp. Cell Res. 105,
Bauminger, S., and Wilchek, M. (1980) Methods Enzymol. 70, 151-159 Rubin, G . M. (1975) Mol. Cell. Biol. 12, 45-64 Zioncheck, T. F., Hamson, M. L., and Geahlen, R. L. (1986) J. Biol. Chem. 261,
15637-15643 Higuchi, R. (1990) in PCR Protocols: A Guide to Methods and Applications
(Innis, M. A., Gelfand, D. H., Sninsky, J. J., and White, T. J., eds) pp. 177-183, Academic Press, San Diego, CA
Sanger, F., Nicklen, S., and Coulson, A. R. (19771 Proc. Natl. Acad. Sci. U. S. A. 74,5463-5467
Ito, H., Fukuda, Y., Muruta, K., and Kimura, A. (1983) J. Bacteriol. 153, 163-168
Sherman, F., (1991) Methods Enzymol. 194, 3-21 Thomas, J. A., Schlender, K. K., and Lamer, J. (1968) Anal. Biochem. 25, 486-499
Bergmeyer, H. U., Bernt, E., Schmidt, F., and Stork, H. (1974) in Methods of Enzymatic Analysis (Bergmeyer, H. U., ed) 2nd Ed., Vol. 3, pp. 11961201, Academic Press, New York
A. (1990) Mol. Cell. Biol. 10, 43034313
79-98
Bradford, M. M. (1976) Anal. Biochem. 72,248-254 Huang, K.-P., and Cabib, E. (1974) J. Biol. Chem. 249, 3858-3861 Posas, F., Clotet, J., Muns, M. T., Corominas, J . , Casamayor, A,, and Arifio, J.
Amdt, K. T., Styles, C. A,, and Fink, G. R. (1989) Cell 56, 527-537 Kemp, B. E., Graves, D. J. , Benjamini, E., and Krebs, E. G. (1977) J. Biol.
Denis, C. L., Kemp, B. E., and Zoller, M. J. (1991) J. Biol. Chem. 266, 17932-
Prorok, M., and Lawrence, D. S. (1989) Biochem. Biophys. Res. Commun. 165,
Clark-Lewis, I., Sanghera, J. S., and Pelech, S. L. (1991) J. Biol. Chem. 266,
Gonzalez, F. A., Raden, D. L., and Davis, R. J. (1991) J. Biol. Chem. 266,
Brewster, J. L., de Valoir, T., Dwyer, N. D., Winter, E., and Gustin, M. C. (1993)
Nishida, E., and Gotoh, Y. (1993) 'lFends Biochem. Sci. 18, 128-131 Browner, M. F., Nakano, K., Bang, A. G., and Fletterick, R. J. (1989) Prac. Natl.
Zhang, W., Browner, M. F., Fletterick, R. J., DePaoli-Roach, A. A,, and Roach,
Bai, G., Zhang, Z., Werner, R., Nuttall, F. Q., Tan, A. W. H., and Lee, E. Y. C.
(1993) J. Biol. Chem. 268, 134S1354
Chem. 252,48884894
17935
3 6 a m
15180-15184
22159-22163
Science 259, 1760-1763
Acad. Sci. ti. S. A. 86, 1443-1447
J. R. (1989) FASEB J. 3, 2532-2536
(1990) J. B i d . Chem. 265,7843-7848





























