Embed Size (px)
Citation preview

JOURNAL OF VIROLOGY, Jan. 2009, p. 167–180 Vol. 83, No. 10022-538X/09/$08.00�0 doi:10.1128/JVI.01719-08Copyright © 2009, American Society for Microbiology. All Rights Reserved.
Control of the Papillomavirus Early-to-Late Switch by DifferentiallyExpressed SRp20�†
Rong Jia,1 Xuefeng Liu,1 Mingfang Tao,1 Michael Kruhlak,2 Ming Guo,3 Craig Meyers,4Carl C. Baker,5 and Zhi-Ming Zheng1*
HIV and AIDS Malignancy Branch,1 Experimental Immunology Branch,2 and Laboratory of Cellular Oncology,5 Center forCancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892; Department of Pathology,
The University of Texas M. D. Anderson Cancer Center, Houston, Texas 770303; and Department of Microbiology andImmunology, Pennsylvania State University College of Medicine, Hershey, Pennsylvania 170334
Received 7 August 2008/Accepted 9 October 2008
The viral early-to-late switch of papillomavirus infection is tightly linked to keratinocyte differentiation andis mediated in part by alternative mRNA splicing. Here, we report that SRp20, a cellular splicing factor,controls the early-to-late switch via interactions with A/C-rich RNA elements. An A/C-rich SE4 elementregulates the selection of a bovine papillomavirus type 1 (BPV-1) late-specific splice site, and binding of SRp20to SE4 suppresses this selection. Expression of late BPV-1 L1 or human papillomavirus (HPV) L1, the majorcapsid protein, inversely correlates with SRp20 levels in the terminally differentiated keratinocytes. In HPVtype 16, a similar SRp20-interacting element also controls the viral early-to-late switch. Keratinocytes in raftcultures, which support L1 expression, make considerably less SRp20 than keratinocytes in monolayer cul-tures, which do not support L1 expression. Conversely, abundant SRp20 in cancer cells or undifferentiatedkeratinocytes is important for the expression of the viral early E6 and E7 by promoting the expression ofcellular transcription factor SP1 for transactivation of viral early promoters.
Papillomaviruses are small DNA tumor viruses that infectcutaneous or mucosal epithelial cells and cause benign tumorsand sometimes malignant neoplasms, including cervical cancerin women (36). Papillomavirus infections are transmittedmainly by close skin-to-skin or mucosa-to-mucosa contact. In-fecting viral particles reach the keratinocytes in the basal layerof the squamous epithelium via microwounds that expose thebasal keratinocytes to incoming virus. After infection of thebasal keratinocytes, viral-gene expression and replication pro-ceed in a tightly controlled fashion regulated by keratinocytedifferentiation (25, 34).
Although we do not fully understand how keratinocyte dif-ferentiation regulates papillomavirus gene expression and vi-rus production, different parts of the viral life cycle occur atdifferent stages of keratinocyte differentiation. The early stageof virus infection takes place in undifferentiated or intermedi-ately differentiated keratinocytes in basal or parabasal layers;at this stage, the viral early genes (E1, E2, E5, E6, and E7) areexpressed from the early region of the viral genome and en-code all five viral regulatory nonstructural proteins. In con-trast, the expression of two structural viral capsid proteins (L1and L2) from the late region of the virus genome at the latestage of viral infection occurs only in keratinocytes undergoingterminal differentiation in the granular and cornified layers ofthe epithelium (34, 41). Although the early-to-late switch ofviral-gene expression involves a switch in the use of viral pro-
moters during the viral life cycle (21, 48, 49), strict regulationof viral-RNA processing, including alternative RNA splicingand polyadenylation, is absolutely necessary for expression ofthe viral genes at the appropriate times (42, 57).
Alternative RNA splicing and polyadenylation occur duringRNA processing in most eukaryotic and viral genes, usuallywhen the RNA bears weak splice sites or multiple poly(A)signals (38, 56). Because it depends on the local availability ofthe correct forms of splicing factors, alternative splicing of aparticular RNA can be found in different cell types and atdifferent stages of cell differentiation. Although the exactmechanism by which alternative RNA splicing is regulatedremains largely unknown, it is the general consensus that oneor more cis-acting elements in the regulated exons or intronsinteract with one or more locally available cellular splicingfactors to select an alternative splice site (24, 54). This alter-native RNA splicing provides an alternative poly(A) site, whichmay or may not be used for RNA polyadenylation (7, 57). Inpapillomaviruses, both viral early and late transcripts are inbicistronic or polycistronic forms that contain two or moreopen reading frames (ORFs) and multiple introns and exons.These bicistronic or polycistronic transcripts overlap with eachother and feature suboptimal splicing signals. Thus, their ex-pression undergoes extensive alternative RNA splicing andpolyadenylation at either an early or a late poly(A) site (57). Atthe early stage of papillomavirus infection, only the earlypromoters are activated, and viral early transcripts utilizeearly splice sites and the early poly(A) signal; this occurs inundifferentiated or intermediately differentiated keratino-cytes, where the late splice sites and the late poly(A) site areblocked. This strategy allows the virus to avoid any expressionof the viral late proteins from the early transcripts and enablesthe viral early proteins to take control of various cellular path-
* Corresponding author. Mailing address: HIV and AIDS Malig-nancy Branch, National Cancer Institute, National Institutes of Health,10 Center Dr. 6N106, Bethesda, MD 20892-1868. Phone: (301) 594-1382. Fax: (301) 480-8250. E-mail: [email protected].
† Supplemental material for this article may be found at http://jvi.asm.org/.
� Published ahead of print on 22 October 2008.
167
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

ways before an infectious virus can be produced. In contrast, atthe late stage of papillomavirus infection, when the infectedcell differentiates and transcription from the late promoterbecomes active, the late-specific splice sites and the latepoly(A) signal are activated for the expression of viral lategenes. However, during this time, the usage of the viral earlysplice sites and the early poly(A) signal continues, suggestingthat some cellular differentiation factors are crucial for thisregulation.
To investigate this process in more detail, our laboratoriesand others have attempted to elucidate the underlying mech-anisms that control the viral early-to-late switch. In particular,we have focused on the viral-RNA processing that occurs dur-ing late-gene expression of bovine papillomavirus type 1 (BPV-1), which has served as a model system for studies of themolecular biology of papillomaviruses. In the late stage ofBPV-1 infection in differentiated cells, the late leader 5� splicesite (5� ss) of bicistronic L1L2 transcripts alternatively splicesto a proximal 3� splice site (3� ss) (nucleotide [nt] 3225 3� ss) toexpress L2 or to a distal 3� ss (nt 3605 3� ss) to express L1. Thisalternative splicing of late transcripts involves the active par-ticipation of three exonic splicing enhancers (ESEs), SE1, SE2,and SE4, and two exonic splicing suppressors (ESS), ESS1 andESS2, in exon 2 (56, 57). SE1, SE2, and ESS1 regulate theselection of the proximal 3� ss, which is also active in theexpression of viral early genes. SE4 is an A/C-rich ESE andfunctions along with ESS2 in vitro to select the distal 3� ss,which is essential for defining a downstream 5� ss (nt 3764 5�ss) to remove intron 2 during splicing of the L1 pre-mRNAs(61); retention of intron 2 results in production of the viralminor capsid protein L2 instead of the viral major capsid pro-tein L1. Notably, selection of the distal 3� ss to remove intron2 takes place only in the upper layers of the epidermis, whereasselection of the proximal 3� ss, which also occurs during splic-ing of viral early transcripts, appears in both the basal andupper layers of the epidermis (4). However, how SE4 functionsremains unknown.
Here, we verified the role of SE4 in the selection of the 3� ssand further investigated its function by searching for cellularproteins that bind to it. We identified SRp20, a cellular splicingfactor (8) and RNA export mediator (27), as a specific SE4-interacting protein. Subsequently, we examined the effect ofSRp20 on the splicing of BPV-1 and human papillomavirustype 16 (HPV16) late transcripts. We found that low levels ofcellular SRp20 permit the expression of viral late genes andcontrol the early-to-late switch in viral-RNA processing. Incontrast, a high level of SRp20 was found to promote theexpression of the HPV early genes E6 and E7.
MATERIALS AND METHODS
Plasmids. Plasmids SE4m1 (pXFL102), SE4m2 (pXFL103), and SE4m3(pXFL106) were all derived from plasmid p3033 (SE2m) (59) and contain pointmutations in both the SE2 and SE4 elements. Plasmid pTMF25-8 was derivedfrom plasmid p3231(59) and has no point mutations in SE2 but has the samepoint mutations in SE4 elements as SE4m2. Plasmid pJR1, derived frompCBG1(33), has a simian virus 40 promoter-driven BPV-1 late minigene with amutant (mt) SE2 and wild-type (wt) SE4. Plasmid pXFL101 has a simian virus 40promoter-driven BPV-1 late minigene similar to that of plasmid p3231(59). Bothplasmids pJR1 and pXFL101 contain a blasticidin resistance gene for selection ofstable transfection.
Plasmid pJR5 contains a subgenomic HPV16 DNA from nt 686 to 7471 under
the control of a cytomegalovirus (CMV) immediate-early (IE) promoter. Plas-mid pJR9, derived from plasmid pJR5, contains point mutations in the HPV16ESE sequence (see Fig. 8B).
Raft cultures. Raft (organotypic) cultures were derived from HPV16-immor-talized primary human vaginal keratinocytes and were grown as previously de-scribed (14, 40).
Transient and stable transfections. Both U2OS cells and 293 cells were usedfor transfection with mammalian expression vectors in the presence of Lipo-fectamine 2000 (Invitrogen). Total cell RNA was prepared 48 h after transfectionand analyzed for viral-late-gene expression by reverse transcription (RT)-PCR orNorthern blotting. For stable transfection, U2OS cells transfected with 2 �g ofEcoRI-linearized plasmid pJR1 or pXFL101 were maintained in Dulbecco’smodified Eagle’s medium containing 10% fetal bovine serum with the addition of10 �g/ml of blasticidin (Invitrogen).
Synthetic siRNAs and RNAi. All small interfering RNAs (siRNAs) were syn-thesized from Dharmacon, Inc. (Lafayette, CO), siRNA 393 (5�-UGACACCAAGGAAGAUGUATT-3�), siRNA 394 (5�-GCAGCCGAUCCACCAGCUGTT-3�), and siRNA oJR1 (5�-GGUCAUCGCAACGAAGGUU-3�) for humanYB-1. SRp20 siRNA was purchased as a siGenome SMARTpool (SFRS3; cat-alog no. M-030081-00), and a nonspecific (NS) siRNA with 52% GC content(catalog no. D-001206-08-20) was used as a negative control. For RNA interfer-ence (RNAi), U2OS cells transfected with BPV-1 or HPV16 and CaSki andHeLa cells were transfected with each gene-specific siRNA in two separatetransfections at an interval of 48 h in the presence of Lipofectamine 2000 (52,53). The siRNAs used for transfection were SRp20 siRNA (40 nM), YB-1 siRNA(30 nM; 10 nM each of siRNAs 393, 394, and oJR1), and NS siRNA (30 nM forU2OS cells and 40 nM for CaSki or HeLa cells). Protein samples and total cellRNA were prepared 24 or 48 h after the second siRNA transfection.
Western blotting. Protein samples were blotted separately with the followingmouse monoclonal antibodies (MAbs): anti-SRp20 (7B4; ATCC, Manassas,VA), anti-pan-SR protein MAb104 (ATCC), anti-hnRNP K (D-6), anti-SP1(1C6), and anti-HPV16 E7 (ED17), all from Santa Cruz Biotechnology (SantaCruz, CA); anti-�-tubulin (5H1), anti-SC35 (�SC35), anti-p21 (6B6), and anti-pRb (G3-245), all from BD Pharmingen (San Diego, CA); anti-ASF/SF2 anti-body (clone 96; Invitrogen); anti-HPV L1 (K1H8; Lab Vision Corporation,Fremont, CA); anti-hnRNP L (4D11; Abcam, Cambridge, MA); and anti-p53(Ab-6; Oncogene, Cambridge, MA). Goat anti-HPV18 E7 (SC-1590) was fromSanta Cruz Biotechnology, and rabbit polyclonal anti-YB-1 was from Abcam.
RNA preparation and RT-PCR. Following DNase I treatment, 1 �g of RNAwas reverse transcribed at 42°C using random hexamers and then amplified for35 cycles using the primer pair Pr7345 (5�-CAATGGGACGCGTGCAAAGC-3�) and Pr3738 (5�-CAGTATTTGTGCTTGTCCTT-3�) or Pr3715 (5�-TTTCAGCACCGTTGTCAGCAACTGTG-3�) for BPV-1 L1 and L2 detection. BPV-1L2 and L1 cDNAs spliced using the nt 3225 3� ss and nt 3605 3� ss, respectively,were used as positive controls. The primer pair oSB23 (nt 3385 to 3402; 5�-TATTAGGCAGCACTTGGC-3�) and oXHW41 (nt 5723 to 5699; 5�-CAACATATTCATCCGTGCTTACAAC-3�) was also used for HPV16 L1 detection.
RNA pull-down assays. Biotin-labeled BPV-1 RNA oligonucleotides oJR4(5�-biotin-CUGCACCACCACCUGGUUCTT-3�; wt1 SE4), oJR5 (5�-biotin-CUGUGUCACUGUCUGGUUCTT-3� [underlining indicates mutations]; mt1SE4), oJR6 (5�-biotin-GGCAGGAAGAAGAGGAGCATT-3�; wt SE2), oJR7(5�-biotin-CUGCACCACCACCUAUCUATT-3�; wt2 SE4), and oJR8 (5�-biotin-CUGUGUCACUGUCUAUCUATT-3�; mt2 SE4), and biotin-labeledHPV16 RNA oligonucleotides oJR9 (5�-biotin-CCAGACACCGGAAACCCCUGCCACACCAC-3�; wt ESE) and oJR10 (5�-biotin-CCAGAUGUCGGAAACCUCUGCUGUGCUGU-3�; mt ESE) were synthesized by Integrated DNATechnologies (Coralville, IA). We obtained the 293 nuclear extract (7.5 mg/ml)from ProteinOne (Bethesda, MD). The U2OS cell extract was prepared fromactively growing cells in exponential phase. After washes, the cell pellets wereresuspended in radioimmunoprecipitation assay (RIPA) buffer (Boston Bio-Products, Ashland, MA) with the addition of Complete Mini EDTA-free Pro-tease Inhibitor Mixture (Roche, Indianapolis, IN), repeat pipetted, and soni-cated (10 s) on ice. After centrifugation, the supernatant was collected as U2OStotal cell extract. Each biotin-labeled RNA oligonucleotide (10 �l at 40 �M) wasfirst immobilized onto 100 �l of NeutrAvidin beads (50% slurry; Pierce, Rock-ford, IL) in a final volume of 300 �l of 1� binding buffer (20 mM Tris, 200 mMNaCl, 6 mM EDTA, 5 mM potassium fluoride, 5 mM �-glycerophosphate, 2�g/ml aprotinin, pH 7.5) at 4°C for 2 h, followed by incubation with 10 �l of 293nuclear extract or 100 �l of U2OS total cell extract in 1� binding buffer in a finalvolume of 400 �l at 4°C for 2 h. The beads were washed three times with bindingbuffer, resuspended in 40 �l of 2� sodium dodecyl sulfate sample buffer, andboiled for 5 min. Proteins in the pull-down assays were analyzed by Westernblotting.
168 JIA ET AL. J. VIROL.
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

In vitro splicing assay and spliceosome assembly. In vitro splicing of 4 ng of32P-labeled BPV-1 late pre-mRNAs was carried out using 40% of the HeLanuclear extract (ProteinOne) in a volume of 25 �l in splicing buffer by incubationat 30°C for 2 h. The spliced products were analyzed by electrophoresis in adenaturing 8% polyacrylamide-8 M urea gel. Spliceosomal complex formationwas performed under the conditions described previously (60).
Northern blotting. Equal amounts of the poly(A)-selected RNAs from U2OScells transiently transfected with a papillomavirus late-minigene vector were runin 1% agarose-formaldehyde gels and blotted onto a GeneScreen Plus mem-brane (Perkin Elmer, Waltham, MA). To detect CaSki and HeLa E6E7 RNAs,approximately 10 �g of the cytoplasmic and nuclear fractions of total RNA with(CaSki) or without (HeLa) poly(A) selection was prepared 24 h after the secondsiRNA transfection. Northern blot analyses were performed with a 32P-labeledantisense HPV16 E6E7 probe (nt 442 to 816) transcribed in vitro or with anoligonucleotide probe end labeled with 32P. The following oligonucleotideprobes were used: oXHW41 for HPV16 L1, oZMZ433 (5�-CACTGAGGTAC/CTGCTGGGATGCACACCAC-3�) for HPV18 E6E7, Pr3738 for BPV-1 latetranscripts, and oZMZ270 (5�-TGAGTCCTTCCACGATACCAAA-3�) for cel-lular GAPDH (glyceraldehyde-3-phosphate dehydrogenase).
Immunohistochemical and immunofluorescence staining. Bovine fibropapil-loma tissues and cervical tissues with cervical intraepithelial neoplasia I (CIN I)lesions were from previous studies (4, 5, 22). Normal cervical sections werepurchased from US Biomax (Rockville, MD). Immunohistochemistry was per-formed with the Vectastain ABC kit (Vector Laboratories, Burlingame, CA).Sections were incubated with MAb 7B4 (anti-SRp20; ATCC), anti-BPV-1 L1(Chemicon) (60), or anti-HPV L1 K1H8 overnight at 4°C, followed by secondaryantibody for 30 min and ABC reagent for 30 min. The specific signal wasdeveloped with a DAB substrate kit (Vector). Immunofluorescence double stain-ing was performed with the Vector M.O.M. Immunodetection Kit. Briefly, tissuesections were processed as described above without endogenous peroxidasequenching and incubated with anti-SRp20 7B4 overnight at 4°C, followed bybiotin-labeled secondary antibody for 30 min. The specific signal was developedby using Alexa Fluor 488 streptavidin for 5 min. The same section was thentreated with the reagents in the Streptavidin/Biotin Blocking Kit (Vector), fol-lowed by Image-iT FX Signal Enhancer (Invitrogen) for 30 min and M.O.MMouse Ig Blocking Reagent for 1 h. Subsequently, the sections were incubatedwith anti-BPV-1 L1 or anti-HPV L1 K1H8 antibody overnight at 4°C and withbiotin-labeled secondary antibody for 30 min. The specific signal was developedusing Alexa Fluor 546 streptavidin for 5 min and was imaged by epifluorescenceor confocal microscopy. The background signals captured from control slidesstained with no primary antibody, anti-SRp20 only, anti-BPV-1 L1 only, oranti-HPV L1 only were used to subtract the background signal (if any) fromdouble-stained images.
RESULTS
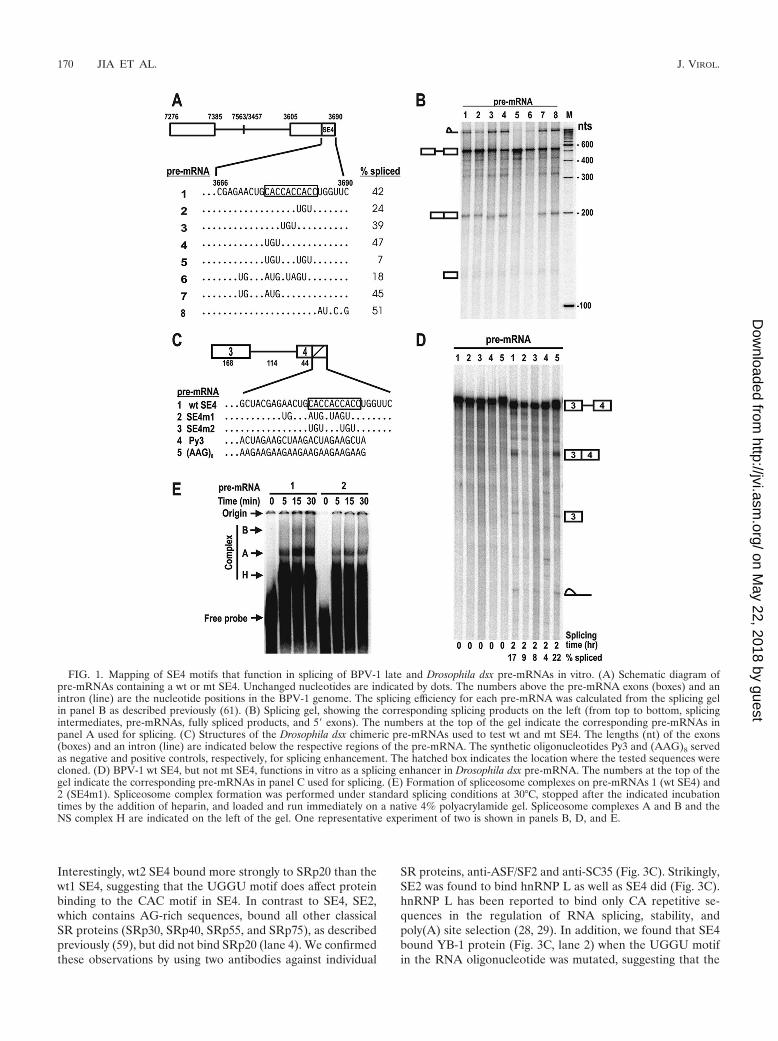
Selection of a viral late-specific splice site is facilitated by anexonic A/C-rich element. SE4, an A/C-rich element reported tobe an ESE in the BPV-1 late transcript (61), has three consec-utive tandem repeats of CAC sequences followed by a sup-pressor (ESS2) core, UGGU. We analyzed the structure andfunction of SE4 by introducing point mutations. As shown inFig. 1A and B, point mutations in the third CAC (from 5� to 3�)reduced the ability of SE4 to promote RNA splicing in vitro(compare pre-mRNAs 2, 5, and 6 to pre-mRNA 1). In contrast,introduction of point mutations into the first CAC had noeffect on the function of SE4 (compare pre-mRNA 4 to pre-mRNA 1); however, mutation of both the first and third CACtogether (pre-mRNA 5) was more suppressive than mutationof the third CAC alone (pre-mRNA 2). Mutations in the sec-ond CAC had little effect (pre-mRNA 3), and changes ofsequences outside of the CAC repeats (pre-mRNA 8) alone orin combination with mt CACs (pre-mRNAs 6 and 7) did nothave any additional effect. We confirmed this observation in aheterologous pre-mRNA by using a Drosophila double-sex(dsx) pre-mRNA (58). The Drosophila dsx pre-mRNA containsa suboptimal intron and cannot be spliced in vitro in the ab-sence of an ESE. When the dsx exon 4 was attached to a wt SE4
or a purine-rich enhancer (AAG)8, the dsx pre-mRNAs werespliced efficiently (Fig. 1C and D, pre-mRNAs 1 and 5). Aspredicted, an mt SE4 (SE4m1 or SE4m2) or a negative controlsequence, Py3, lacked this activity when placed in the sameposition (Fig. 1C and D, pre-mRNAs 2, 3, and 4). Mechanis-tically, SE4, but not its mutant, more efficiently enhances theformation of spliceosomal complexes A and B (Fig. 1E). To-gether, these data suggest that the A/C-rich SE4 functions asan ESE mainly through its A/C-rich motif.
To understand how the A/C-rich SE4 functions in vivo inalternative RNA splicing, we utilized a BPV-1 late-minigeneexpression vector driven by a CMV IE promoter for in vivoassays. This minigene has a large deletion in its intron 1 toremove all viral early promoters and contains two alternative 3�ss in its exon 2 (Fig. 2a). A proximal 3� ss at nt 3225 in the virusgenome is a major 3� ss for all early transcripts, but the samesplice site is also used for splicing of viral late transcripts toexpress the viral minor capsid protein L2. Thus, selection ofthis 3� ss takes place in both undifferentiated and highly dif-ferentiated keratinocytes and in both the early and late stagesof virus infection (4). It is controlled by three downstreamRNA cis elements, SE1, SE2, and ESS1(57). In contrast, adistal 3� ss at nt 3605 in the virus genome is a late-specific 3� ssand is controlled by two other downstream cis elements, SE4and ESS2 (61). However, selection of this distal 3� ss is activeonly in highly differentiated keratinocytes at the late stage ofvirus infection, and its selection is essential for activating adownstream 5� ss at nt 3764 for splicing to remove intron 2,where the L2 ORF resides (4), to produce the viral majorcapsid protein L1.
The introduction of point mutations into SE2 (Fig. 2b) led toa switch of 3� ss usage from nt 3225 (proximal) to nt 3605(distal) (Fig. 2c, lane 3), showing the importance of SE2 in theselection of the proximal 3� ss. As predicted, the introductionof additional point mutations into SE4 to disrupt its CAC motifpartially restored the usage of the proximal 3� ss (Fig. 2c, lanes4 and 5), indicating that SE4 plays a key role in selection of thedistal 3� ss in vivo. Consistent with the dominant function ofSE4, introduction of point mutations into the UGGU se-quence immediately downstream of the CAC motif in SE4 didnot affect the choice of splice sites (Fig. 2c, lane 6). This wasfurther confirmed by RT-PCR analysis when SE4 was mutatedon its own in the context of wt SE2. The choice of the nt 36053� ss was not detectable in vivo when the late transcript con-tained an mt SE4 (SE4m2) alone (data not shown).
Interaction of SRp20 with the CAC motif in SE4 blocksselection of the viral late-specific splice site. To understandwhich cellular factors are involved in the SE4-mediated selec-tion of splice sites, we carried out RNA pull-down assays usingbiotin-labeled SE4 RNA with or without the presence of mu-tations. The UGGU sequence immediately downstream of theCAC motif was also mutated (Fig. 3A, wt2 and mt2) to avoidpossible base pairing with the overlapped ACCACCAs in SE4that might affect the binding of cellular proteins to SE4. Inpull-down assays using nuclear extracts from 293 cells, asshown in Fig. 3B, the CAC motif in SE4 selectively interactedwith SRp20, the smallest member of the SR protein family(55), with high affinity (lanes 1 and 2). When point mutationswere introduced into the first and third CAC in the SE4 CACmotif, the mt SE4 bound weakly to SRp30 and SRp75 (lane 3).
VOL. 83, 2009 SRp20 IN VIRAL EARLY-TO-LATE SWITCH 169
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Interestingly, wt2 SE4 bound more strongly to SRp20 than thewt1 SE4, suggesting that the UGGU motif does affect proteinbinding to the CAC motif in SE4. In contrast to SE4, SE2,which contains AG-rich sequences, bound all other classicalSR proteins (SRp30, SRp40, SRp55, and SRp75), as describedpreviously (59), but did not bind SRp20 (lane 4). We confirmedthese observations by using two antibodies against individual
SR proteins, anti-ASF/SF2 and anti-SC35 (Fig. 3C). Strikingly,SE2 was found to bind hnRNP L as well as SE4 did (Fig. 3C).hnRNP L has been reported to bind only CA repetitive se-quences in the regulation of RNA splicing, stability, andpoly(A) site selection (28, 29). In addition, we found that SE4bound YB-1 protein (Fig. 3C, lane 2) when the UGGU motifin the RNA oligonucleotide was mutated, suggesting that the
FIG. 1. Mapping of SE4 motifs that function in splicing of BPV-1 late and Drosophila dsx pre-mRNAs in vitro. (A) Schematic diagram ofpre-mRNAs containing a wt or mt SE4. Unchanged nucleotides are indicated by dots. The numbers above the pre-mRNA exons (boxes) and anintron (line) are the nucleotide positions in the BPV-1 genome. The splicing efficiency for each pre-mRNA was calculated from the splicing gelin panel B as described previously (61). (B) Splicing gel, showing the corresponding splicing products on the left (from top to bottom, splicingintermediates, pre-mRNAs, fully spliced products, and 5� exons). The numbers at the top of the gel indicate the corresponding pre-mRNAs inpanel A used for splicing. (C) Structures of the Drosophila dsx chimeric pre-mRNAs used to test wt and mt SE4. The lengths (nt) of the exons(boxes) and an intron (line) are indicated below the respective regions of the pre-mRNA. The synthetic oligonucleotides Py3 and (AAG)8 servedas negative and positive controls, respectively, for splicing enhancement. The hatched box indicates the location where the tested sequences werecloned. (D) BPV-1 wt SE4, but not mt SE4, functions in vitro as a splicing enhancer in Drosophila dsx pre-mRNA. The numbers at the top of thegel indicate the corresponding pre-mRNAs in panel C used for splicing. (E) Formation of spliceosome complexes on pre-mRNAs 1 (wt SE4) and2 (SE4m1). Spliceosome complex formation was performed under standard splicing conditions at 30°C, stopped after the indicated incubationtimes by the addition of heparin, and loaded and run immediately on a native 4% polyacrylamide gel. Spliceosome complexes A and B and theNS complex H are indicated on the left of the gel. One representative experiment of two is shown in panels B, D, and E.
170 JIA ET AL. J. VIROL.
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
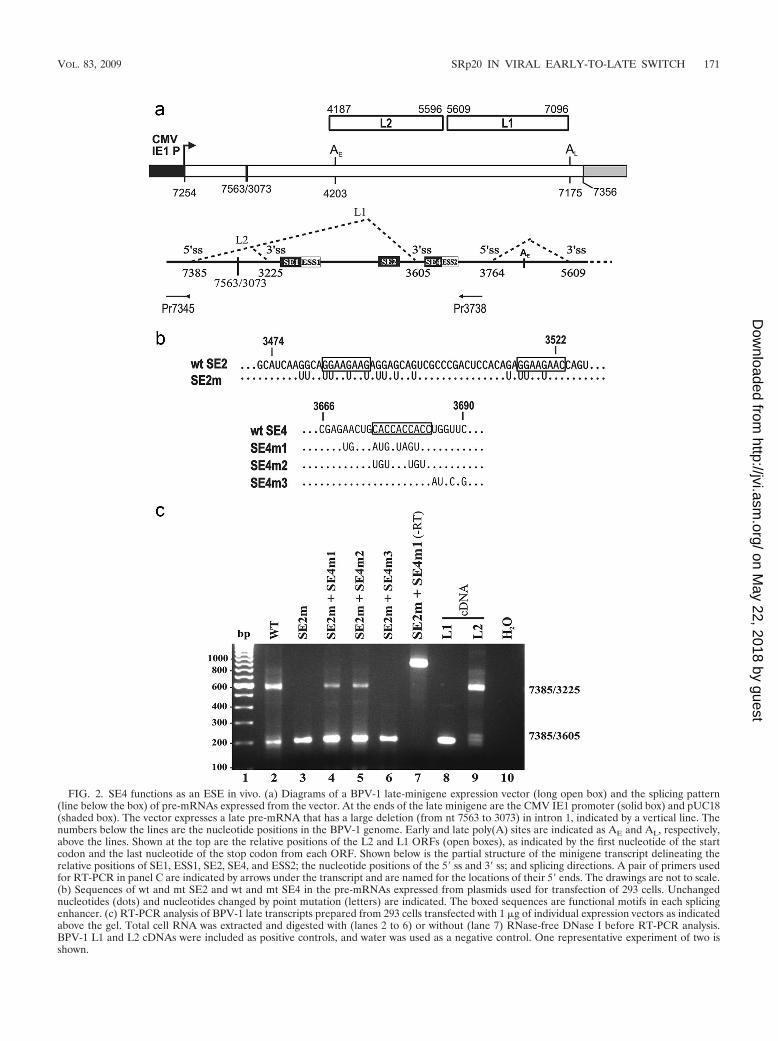
FIG. 2. SE4 functions as an ESE in vivo. (a) Diagrams of a BPV-1 late-minigene expression vector (long open box) and the splicing pattern(line below the box) of pre-mRNAs expressed from the vector. At the ends of the late minigene are the CMV IE1 promoter (solid box) and pUC18(shaded box). The vector expresses a late pre-mRNA that has a large deletion (from nt 7563 to 3073) in intron 1, indicated by a vertical line. Thenumbers below the lines are the nucleotide positions in the BPV-1 genome. Early and late poly(A) sites are indicated as AE and AL, respectively,above the lines. Shown at the top are the relative positions of the L2 and L1 ORFs (open boxes), as indicated by the first nucleotide of the startcodon and the last nucleotide of the stop codon from each ORF. Shown below is the partial structure of the minigene transcript delineating therelative positions of SE1, ESS1, SE2, SE4, and ESS2; the nucleotide positions of the 5� ss and 3� ss; and splicing directions. A pair of primers usedfor RT-PCR in panel C are indicated by arrows under the transcript and are named for the locations of their 5� ends. The drawings are not to scale.(b) Sequences of wt and mt SE2 and wt and mt SE4 in the pre-mRNAs expressed from plasmids used for transfection of 293 cells. Unchangednucleotides (dots) and nucleotides changed by point mutation (letters) are indicated. The boxed sequences are functional motifs in each splicingenhancer. (c) RT-PCR analysis of BPV-1 late transcripts prepared from 293 cells transfected with 1 �g of individual expression vectors as indicatedabove the gel. Total cell RNA was extracted and digested with (lanes 2 to 6) or without (lane 7) RNase-free DNase I before RT-PCR analysis.BPV-1 L1 and L2 cDNAs were included as positive controls, and water was used as a negative control. One representative experiment of two isshown.
VOL. 83, 2009 SRp20 IN VIRAL EARLY-TO-LATE SWITCH 171
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
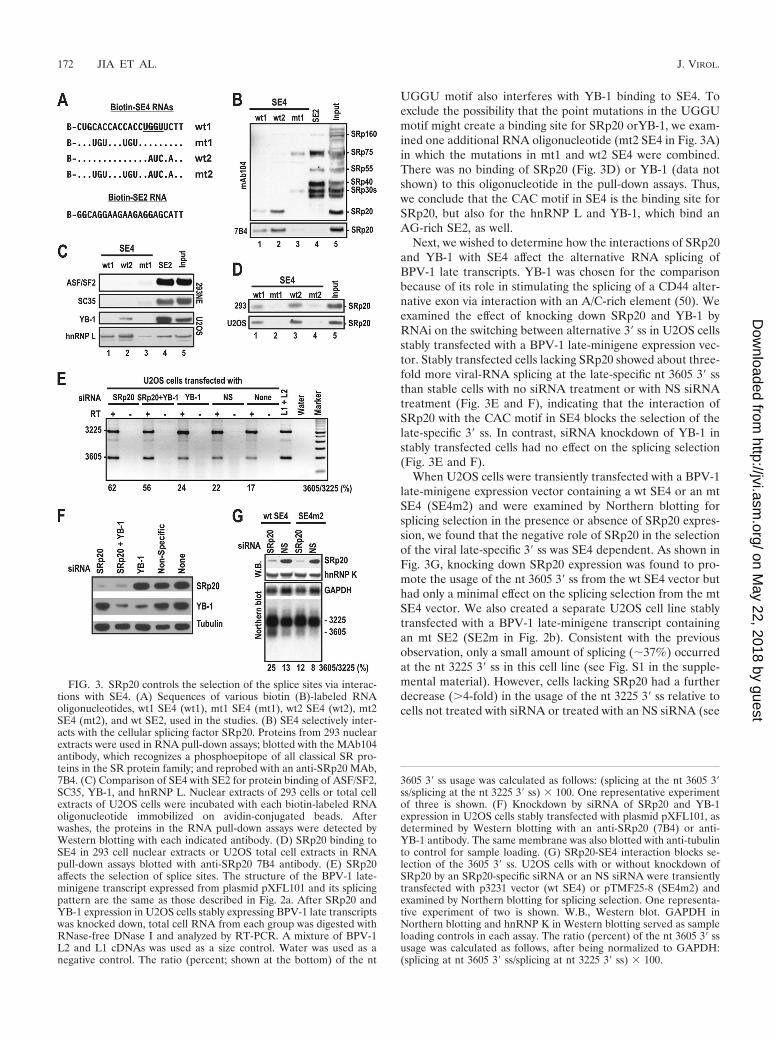
UGGU motif also interferes with YB-1 binding to SE4. Toexclude the possibility that the point mutations in the UGGUmotif might create a binding site for SRp20 orYB-1, we exam-ined one additional RNA oligonucleotide (mt2 SE4 in Fig. 3A)in which the mutations in mt1 and wt2 SE4 were combined.There was no binding of SRp20 (Fig. 3D) or YB-1 (data notshown) to this oligonucleotide in the pull-down assays. Thus,we conclude that the CAC motif in SE4 is the binding site forSRp20, but also for the hnRNP L and YB-1, which bind anAG-rich SE2, as well.
Next, we wished to determine how the interactions of SRp20and YB-1 with SE4 affect the alternative RNA splicing ofBPV-1 late transcripts. YB-1 was chosen for the comparisonbecause of its role in stimulating the splicing of a CD44 alter-native exon via interaction with an A/C-rich element (50). Weexamined the effect of knocking down SRp20 and YB-1 byRNAi on the switching between alternative 3� ss in U2OS cellsstably transfected with a BPV-1 late-minigene expression vec-tor. Stably transfected cells lacking SRp20 showed about three-fold more viral-RNA splicing at the late-specific nt 3605 3� ssthan stable cells with no siRNA treatment or with NS siRNAtreatment (Fig. 3E and F), indicating that the interaction ofSRp20 with the CAC motif in SE4 blocks the selection of thelate-specific 3� ss. In contrast, siRNA knockdown of YB-1 instably transfected cells had no effect on the splicing selection(Fig. 3E and F).
When U2OS cells were transiently transfected with a BPV-1late-minigene expression vector containing a wt SE4 or an mtSE4 (SE4m2) and were examined by Northern blotting forsplicing selection in the presence or absence of SRp20 expres-sion, we found that the negative role of SRp20 in the selectionof the viral late-specific 3� ss was SE4 dependent. As shown inFig. 3G, knocking down SRp20 expression was found to pro-mote the usage of the nt 3605 3� ss from the wt SE4 vector buthad only a minimal effect on the splicing selection from the mtSE4 vector. We also created a separate U2OS cell line stablytransfected with a BPV-1 late-minigene transcript containingan mt SE2 (SE2m in Fig. 2b). Consistent with the previousobservation, only a small amount of splicing (�37%) occurredat the nt 3225 3� ss in this cell line (see Fig. S1 in the supple-mental material). However, cells lacking SRp20 had a furtherdecrease (�4-fold) in the usage of the nt 3225 3� ss relative tocells not treated with siRNA or treated with an NS siRNA (see
FIG. 3. SRp20 controls the selection of the splice sites via interac-tions with SE4. (A) Sequences of various biotin (B)-labeled RNAoligonucleotides, wt1 SE4 (wt1), mt1 SE4 (mt1), wt2 SE4 (wt2), mt2SE4 (mt2), and wt SE2, used in the studies. (B) SE4 selectively inter-acts with the cellular splicing factor SRp20. Proteins from 293 nuclearextracts were used in RNA pull-down assays; blotted with the MAb104antibody, which recognizes a phosphoepitope of all classical SR pro-teins in the SR protein family; and reprobed with an anti-SRp20 MAb,7B4. (C) Comparison of SE4 with SE2 for protein binding of ASF/SF2,SC35, YB-1, and hnRNP L. Nuclear extracts of 293 cells or total cellextracts of U2OS cells were incubated with each biotin-labeled RNAoligonucleotide immobilized on avidin-conjugated beads. Afterwashes, the proteins in the RNA pull-down assays were detected byWestern blotting with each indicated antibody. (D) SRp20 binding toSE4 in 293 cell nuclear extracts or U2OS total cell extracts in RNApull-down assays blotted with anti-SRp20 7B4 antibody. (E) SRp20affects the selection of splice sites. The structure of the BPV-1 late-minigene transcript expressed from plasmid pXFL101 and its splicingpattern are the same as those described in Fig. 2a. After SRp20 andYB-1 expression in U2OS cells stably expressing BPV-1 late transcriptswas knocked down, total cell RNA from each group was digested withRNase-free DNase I and analyzed by RT-PCR. A mixture of BPV-1L2 and L1 cDNAs was used as a size control. Water was used as anegative control. The ratio (percent; shown at the bottom) of the nt
3605 3� ss usage was calculated as follows: (splicing at the nt 3605 3�ss/splicing at the nt 3225 3� ss) � 100. One representative experimentof three is shown. (F) Knockdown by siRNA of SRp20 and YB-1expression in U2OS cells stably transfected with plasmid pXFL101, asdetermined by Western blotting with an anti-SRp20 (7B4) or anti-YB-1 antibody. The same membrane was also blotted with anti-tubulinto control for sample loading. (G) SRp20-SE4 interaction blocks se-lection of the 3605 3� ss. U2OS cells with or without knockdown ofSRp20 by an SRp20-specific siRNA or an NS siRNA were transientlytransfected with p3231 vector (wt SE4) or pTMF25-8 (SE4m2) andexamined by Northern blotting for splicing selection. One representa-tive experiment of two is shown. W.B., Western blot. GAPDH inNorthern blotting and hnRNP K in Western blotting served as sampleloading controls in each assay. The ratio (percent) of the nt 3605 3� ssusage was calculated as follows, after being normalized to GAPDH:(splicing at nt 3605 3� ss/splicing at nt 3225 3� ss) � 100.
172 JIA ET AL. J. VIROL.
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
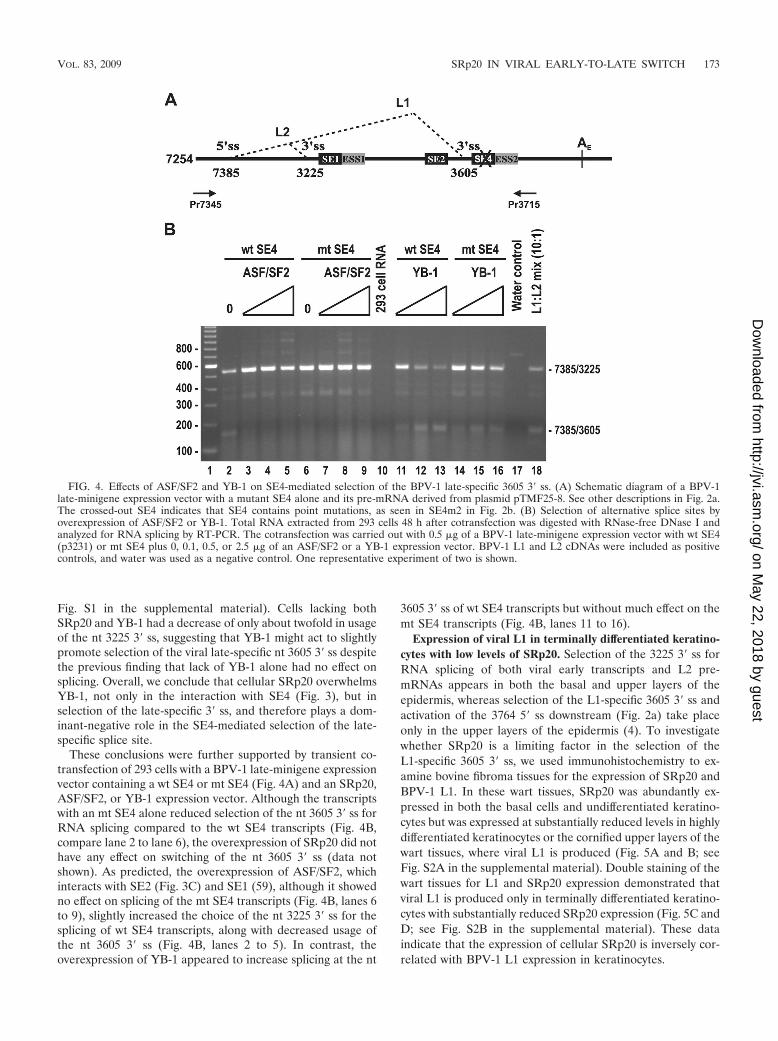
Fig. S1 in the supplemental material). Cells lacking bothSRp20 and YB-1 had a decrease of only about twofold in usageof the nt 3225 3� ss, suggesting that YB-1 might act to slightlypromote selection of the viral late-specific nt 3605 3� ss despitethe previous finding that lack of YB-1 alone had no effect onsplicing. Overall, we conclude that cellular SRp20 overwhelmsYB-1, not only in the interaction with SE4 (Fig. 3), but inselection of the late-specific 3� ss, and therefore plays a dom-inant-negative role in the SE4-mediated selection of the late-specific splice site.
These conclusions were further supported by transient co-transfection of 293 cells with a BPV-1 late-minigene expressionvector containing a wt SE4 or mt SE4 (Fig. 4A) and an SRp20,ASF/SF2, or YB-1 expression vector. Although the transcriptswith an mt SE4 alone reduced selection of the nt 3605 3� ss forRNA splicing compared to the wt SE4 transcripts (Fig. 4B,compare lane 2 to lane 6), the overexpression of SRp20 did nothave any effect on switching of the nt 3605 3� ss (data notshown). As predicted, the overexpression of ASF/SF2, whichinteracts with SE2 (Fig. 3C) and SE1 (59), although it showedno effect on splicing of the mt SE4 transcripts (Fig. 4B, lanes 6to 9), slightly increased the choice of the nt 3225 3� ss for thesplicing of wt SE4 transcripts, along with decreased usage ofthe nt 3605 3� ss (Fig. 4B, lanes 2 to 5). In contrast, theoverexpression of YB-1 appeared to increase splicing at the nt
3605 3� ss of wt SE4 transcripts but without much effect on themt SE4 transcripts (Fig. 4B, lanes 11 to 16).
Expression of viral L1 in terminally differentiated keratino-cytes with low levels of SRp20. Selection of the 3225 3� ss forRNA splicing of both viral early transcripts and L2 pre-mRNAs appears in both the basal and upper layers of theepidermis, whereas selection of the L1-specific 3605 3� ss andactivation of the 3764 5� ss downstream (Fig. 2a) take placeonly in the upper layers of the epidermis (4). To investigatewhether SRp20 is a limiting factor in the selection of theL1-specific 3605 3� ss, we used immunohistochemistry to ex-amine bovine fibroma tissues for the expression of SRp20 andBPV-1 L1. In these wart tissues, SRp20 was abundantly ex-pressed in both the basal cells and undifferentiated keratino-cytes but was expressed at substantially reduced levels in highlydifferentiated keratinocytes or the cornified upper layers of thewart tissues, where viral L1 is produced (Fig. 5A and B; seeFig. S2A in the supplemental material). Double staining of thewart tissues for L1 and SRp20 expression demonstrated thatviral L1 is produced only in terminally differentiated keratino-cytes with substantially reduced SRp20 expression (Fig. 5C andD; see Fig. S2B in the supplemental material). These dataindicate that the expression of cellular SRp20 is inversely cor-related with BPV-1 L1 expression in keratinocytes.
FIG. 4. Effects of ASF/SF2 and YB-1 on SE4-mediated selection of the BPV-1 late-specific 3605 3� ss. (A) Schematic diagram of a BPV-1late-minigene expression vector with a mutant SE4 alone and its pre-mRNA derived from plasmid pTMF25-8. See other descriptions in Fig. 2a.The crossed-out SE4 indicates that SE4 contains point mutations, as seen in SE4m2 in Fig. 2b. (B) Selection of alternative splice sites byoverexpression of ASF/SF2 or YB-1. Total RNA extracted from 293 cells 48 h after cotransfection was digested with RNase-free DNase I andanalyzed for RNA splicing by RT-PCR. The cotransfection was carried out with 0.5 �g of a BPV-1 late-minigene expression vector with wt SE4(p3231) or mt SE4 plus 0, 0.1, 0.5, or 2.5 �g of an ASF/SF2 or a YB-1 expression vector. BPV-1 L1 and L2 cDNAs were included as positivecontrols, and water was used as a negative control. One representative experiment of two is shown.
VOL. 83, 2009 SRp20 IN VIRAL EARLY-TO-LATE SWITCH 173
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

This inverse correlation of BPV-1 L1 and SRp20 expressionwas also verified in the HPV-infected cervix (Fig. 6). When cer-vical tissue infected with HPV18, -52, and -66 with active viral-DNA replication (22) was used for double staining with viral L1and SRp20 antibodies, we demonstrated that viral-L1 expressiontook place only in the upper layers of the infected cervix, wherethe infected keratinocytes exhibited remarkable deficiency ofSRp20 (Fig. 6A). In contrast, SRp20 was found to be expressedabundantly in the undifferentiated or intermediately differenti-ated keratinocytes in the basal and parabasal layers of the cervix.This was further confirmed by immunohistochemical staining of
the cervical tissues with the two antibodies (data not shown). Indouble-staining and confocal-imaging analyses, a few cellsshowed high levels of SRp20 but low levels of L1 (Fig. 6Aand B) or vice versa, indicating an inverse correlation of theexpression of SRp20 and viral L1 in these cervical cells, asshown in bovine wart tissues.
In raft cultures derived from human vaginal keratinocytesimmortalized by HPV16, we also demonstrated that SRp20appeared mostly in basal cells but only weakly in spinous cells,with none in granular or cornified cells (Fig. 7), indicating itsdifferentiation-dependent expression.
FIG. 5. Inverse expression of SRp20 and L1 in bovine warts caused by BPV-1 infection. (A and B) Immunohistochemical staining of wart tissueswith anti-SRp20 antibody 7B4 (A) or an anti-BPV-1 L1 antibody (B), showing that BPV-1 L1 is expressed in superficial layers of the wart tissues,where SRp20 expression is low. (C) Expression of BPV-1 L1 in keratinocytes with terminal differentiation and substantially reduced SRp20. Theconfocal images were taken separately from the superficial layers of wart tissues double-stained for BPV-1 L1 (red) and SRp20 (green) and thenmerged to show the colocalization of the two proteins. DAPI (4�,6�-diamidino-2-phenylindole) shows nuclear staining of the cells. Scale bar, 50 �m.(D) (Left) Enlarged, merged image from panel C showing the colocalization and expression of BPV-1 L1 (red) and SRp20 (green) in individualcells. (Right) L1 and SRp20 staining in individual cells.
174 JIA ET AL. J. VIROL.
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

SRp20 suppresses the expression of HPV late genes butpromotes the expression of the HPV early genes E6 and E7.Recent studies have shown that HPV16 late transcripts alsocontain an A/C-rich, SE4-like element (45) in a correspondingexon, as illustrated in Fig. 8A. Interestingly, this ESE elementalso interacts with SRp20, YB-1, and hnRNP L (Fig. 8B to D)through its CAC motifs. However, unlike BPV-1 SE4, whichdoes not interact at all with SRp30, SRp55, and SRp75 (Fig. 8Cand 3B), HPV16 ESE binds to all three (Fig. 8C). Since YB-1–SE4 interaction had only a minimal effect on BPV-1 alternativesplicing and hnRNP L also bound to AG-rich SE2 (Fig. 3C)
and a UAUCUA motif (data not shown), we focused onSRp20 binding in the regulation of the expression of HPV16late genes. U2OS cells were cotransfected with SRp20 siRNAor an NS siRNA (Fig. 8E), along with a subgenomic HPV16construct, pJR5 (wt ESE), or were transfected only with pJR5or pJR9 (an HPV16 construct with an mt ESE) (Fig. 8F).Because the viral late promoter has no activity in cell cultures,transcription of viral late transcripts from pJR5 and pJR9 wasdriven by a CMV IE promoter. We found an increase inHPV16 late transcripts in the cells either with reduced SRp20but transfected with pJR5 (wt ESE) (Fig. 8F, left) or with a
FIG. 6. Inverse expression of SRp20 and L1 in HPV-infected cervical tissues. The cervical tissue had mixed infections with HPV18, HPV52,and HPV56 and displayed CIN I lesions with active viral-DNA replication (22). (A) Expression of HPV L1 is inversely correlated with the SRp20level in cervical keratinocytes. Confocal images of cervical tissues double stained with anti-SRp20 7B4 antibody (green) and anti-HPV L1 K1H8antibody (red) were taken separately and merged to show the colocalization of the two proteins. The arrows and arrowheads indicate, respectively,the cells inversely coexpressing SRp20 and L1. (B) Colocalization and expression of HPV L1 (red) and SRp20 (green) in individual cells enlargedfrom a merged image in panel A (magnification, �20).
VOL. 83, 2009 SRp20 IN VIRAL EARLY-TO-LATE SWITCH 175
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

normal level of SRp20 but transfected with pJR9 (mt ESE)(Fig. 8F, right), by RT-PCR analysis. The former was furtherverified by Northern blot analysis (Fig. 8G), showing a sub-stantial increase in both L1 and L2 in the cells with knockingdown of SRp20 expression. These data suggest that SRp20suppresses the expression of HPV16 late genes through anSRp20-ESE interaction. To test this hypothesis, we comparedthe expression levels of SRp20 in human vaginal keratinocytes inraft cultures, which support L1 production, and in monolayercultures, which do not support L1 production (Fig. 8H) but dosupport the expression of viral early genes. We found that kera-tinocytes in raft cultures make considerably less SRp20 than thecorresponding cells in monolayer cultures or HPV16� CaSki cells(Fig. 8H), indicating that a natural reduction of SRp20 in raftcultures is at least partially responsible for the early-to-late switchand the resulting expression of viral late genes.
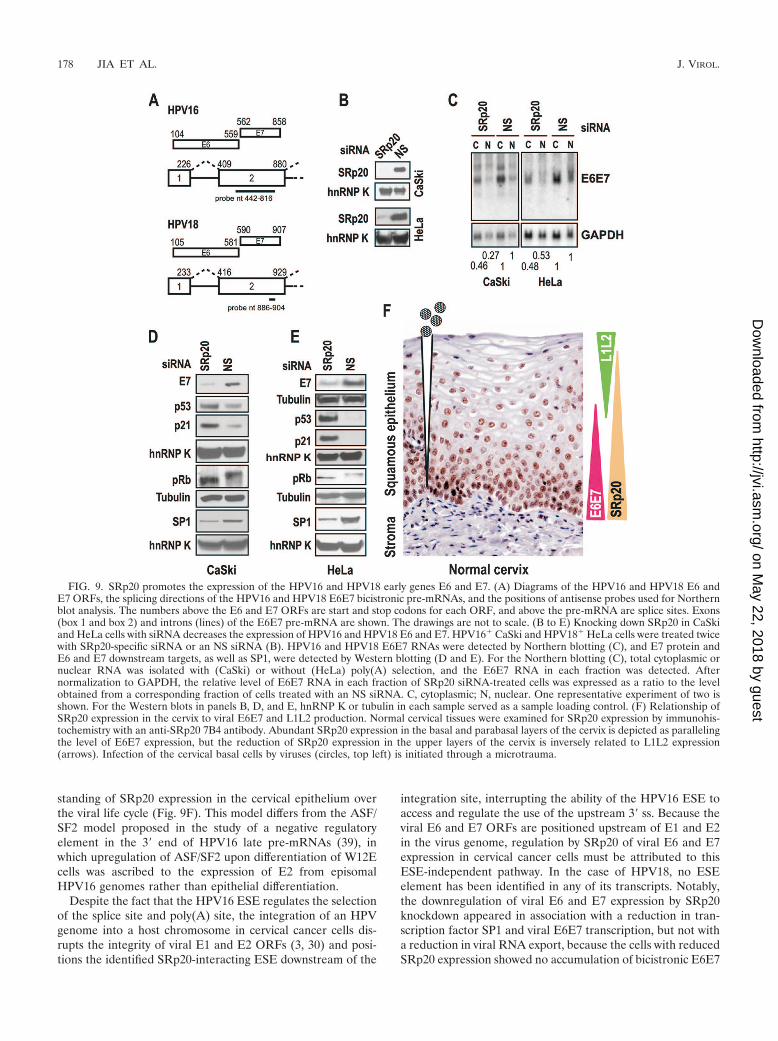
Subsequently, we asked whether the presence of SRp20 at ahigh level is required for expression of viral early genes. We usedan siRNA to knock down the production of SRp20 in HPV-positive cervical cancer cells, which produce large amounts of theE6 and E7 oncoproteins but do not express viral late genes. Insharp contrast to the effect of SRp20 on the expression of virallate genes, the expression of viral early E6 and E7 depended ona high level of SRp20 in the cells (Fig. 9). Reduction of cellularSRp20 by siRNA led to an approximately twofold reduction ofbicistronic viral E6E7 transcripts (Fig. 9A to C) and a substantialreduction of the viral E7 oncoprotein in both HPV16� CaSki cells(Fig. 9D) and HPV18� HeLa cells (Fig. 9E). The accumulation ofp53 and activation of a p53 downstream target, p21, in these cellswere indications that viral E6 was downregulated, because viralE6 targets cellular p53 for degradation (47). Downregulation ofE7 expression in both cell lines also resulted in the accumulationof hypophosphorylated p105Rb, an E7 target (20). Strikingly, SP1,an essential transcription factor for E6 and E7 expression (2, 13,15, 51), was also greatly reduced in both cells with SRp20 knock-down (Fig. 9D and E). Altogether, these data indicate that cel-lular SRp20 promotes the expression of viral early genes by main-taining SP1 at a physiological level in the cells.
DISCUSSION
SRp20 is the smallest member of the SR protein family (55),with a single N-terminal RNA recognition motif (RRM) im-
mediately followed by an RS-rich domain. In addition to itsregulation of RNA splicing via interactions with ESEs (17, 18),SRp20 has been found to play an important role in alternativeRNA polyadenylation (35, 37), RNA export (26, 27), andtranslation (6). Furthermore, blastocyst formation is blocked inmouse embryos lacking SRp20 (31). Although SRp20 wasinitially shown to bind a pyrimidine-rich sequence with aconsensus (A/U)C(A/U)(A/U)C sequence (8), a recentstudy demonstrated that the SRp20 RRM binds the RNAsequence 5�-CAUC-3� in a semi-sequence-specific manner,with only the 5� cytosine recognized specifically by the RRM(23). In this report, we have described SRp20 as an importantfactor controlling the papillomaviral early-to-late switchthrough its interactions with an A/C-rich element in viral tran-scripts, SE4, that controls the selection of a BPV-1 late-specificsplice site. We found that binding of SRp20 to the three con-secutive CAC motifs in SE4 suppresses this selection, whereasknocking down SRp20 expression in U2OS cells promotesselection of the L1-specific splice site. We also found that theterminally differentiating keratinocytes in bovine wart tissuesthat express BPV-1 L1 or in human cervical tissues with HPVinfection-induced CIN I lesions that express HPV L1 havemuch reduced expression of SRp20, suggesting an inverse cor-relation between SRp20 expression and L1 production duringdifferentiation. This observation is consistent with the conceptthat viral early genes are expressed in the basal and undiffer-entiated cells of the cervix but that the expression of papillo-mavirus late genes and production of the viral major capsidprotein L1 take place only in the most terminally differentiatedkeratinocytes in the most superficial layers of the cervix (25).Finding of a suppressive, rather than enhancing, activity ofSRp20 on the selection of a viral late-specific splice site isconsistent with the report that SRp20 inhibits fibronectin EDI(extra domain I) splicing (12).
In HPV16, a similar SRp20-interacting element, ESE, con-trols the viral early-to-late switch of gene expression. An earlystudy of this ESE by deletion mutations showed that it pro-motes the use of a common 3� ss at nt 3358, an important 3� ssin E6, E7, E4, and L1 pre-mRNAs, and the use of polyadenyl-ation at an early poly(A) site. Thus, ESE indirectly blocks theexpression of HPV16 late genes (45). In this report, we foundthat this ESE interacts with SRp20, YB-1, and hnRNP Lthrough its CAC motifs. It also interacts with SRp30s, SRp55,
FIG. 7. SRp20 expression in 10-day-old raft cultures derived from HPV16-immortalized vaginal keratinocytes. Immunohistochemical stainingof the raft tissue section was performed with an anti-SRp20 antibody (7B4), showing strong nuclear SRp20 staining mainly in the basal-layer cellsand weak nuclear staining in the spinous layer cells, but no staining in granular and cornified cells.
176 JIA ET AL. J. VIROL.
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

and SRp75, deviating somewhat from BPV-1 SE4, which doesnot bind any of the SR proteins other than SRp20. Knockingdown SRp20 expression in U2OS cells with an siRNA or dis-ruption of the SRp20 binding sites in the ESE by point muta-tion promoted the expression of viral late genes from a sub-genomic HPV16 expression vector, confirming that the ESEsuppresses the expression of viral late genes (45), presumablyby interacting with SRp20. The lack of conventional cell cul-tures for HPV multiplication has largely restrained our under-standing of the HPV life cycle. However, raft cultures derivedfrom human keratinocytes have been successfully utilized toproduce infectious HPVs (14, 40). We found that human ke-ratinocytes in raft cultures, which support viral-L1 expression,
make much less SRp20 than the corresponding monolayercultures, which do not support viral-L1 expression. Conversely,cervical cancer cell lines produce a large amount of SRp20 butdo not express any late genes. Instead, the abundant SRp20 incervical cancer cell lines is important for the substantial ex-pression of the viral early genes E6 and E7. The finding thatSRp20 is produced primarily in basal and parabasal cells of thecervix, where only viral early genes (including viral E6 and E7)are expressed (41), further supports an important role forSRp20 in the regulation of the viral life cycle. Altogether,along with the observation of viral-L1 expression inverselyrelated to reduced SRp20 in terminally differentiated kerati-nocytes, we propose a model to illustrate our current under-
FIG. 8. SRp20 suppresses the expression of HPV16 late genes. (A) Diagram of an HPV16 late transcript, the splicing pattern, and the positionof an identified ESE. The numbers below the diagram are the nucleotide positions in the HPV16 genome. The arrows indicate a pair of primersused for the RT-PCR assays shown in panel F. (B) Sequences of biotin-labeled wt ESE, mt ESE, and wt2 SE4; underlining indicates mutations.(C) Interactions of HPV16 ESE with cellular splicing factors from U2OS total cell extracts. BPV-1 SE4 was used for comparison in the bindingassays. The proteins in the biotin-labeled RNA pull-down assays were examined by Western blotting with anti-SRp20 7B4, anti-YB-1, or MAb104antibody. The cell extracts (15% of the input in the binding assays) were blotted as positive controls. (D) The CAC motifs in HPV16 ESE are thebinding sites for SRp20 and hnRNP L (hnR L). Wt ESE and SE4 were compared with mt ESE for hnRNP L and SRp20 binding in the presenceof U2OS total cell extracts or 293 cell nuclear extracts. The cell extracts (10% for U2OS or 40% for 293 of the input in the binding assays) werealso blotted as positive controls. (E) Knockdown of SRp20 expression in U2OS cells. U2OS cells transfected once with an SRp20 siRNA or anNS siRNA were cotransfected with a subgenomic (nt 686 to 7471) HPV16 late-gene expression vector, pJR5, along with the second siRNAtreatment. Protein samples collected 24 h after the cotransfection were examined by Western blotting with the indicated antibodies. hnRNP K ineach sample served as a loading control. (F) Interaction of SRp20 and HPV16 ESE plays a negative role in the expression of HPV16 L1. Knockingdown SRp20 in U2OS cells (left) or disruption of HPV16 ESE by point mutation (right) increases the expression of HPV16 late genes. HPV16vector (pJR5)-transfected U2OS cells with or without knockdown of SRp20 or U2OS cells transfected only with an HPV16 vector containing a wtESE (pJR5) or mt ESE (pJR9), as shown in panel B, were examined by RT-PCR for L1 expression 24 h after transfection. One representativeexperiment of three is shown. (G) Northern blot analysis for the expression of HPV16 late genes from HPV16 vector containing a wt ESE (pJR5)in U2OS cells with or without knocking down SRp20. Cellular GAPDH RNA served as a loading control. L1 and L2 expression levels werenormalized to GAPDH before the ratio to corresponding NS siRNA controls was calculated. (H) Expression of HPV16 L1 and SRp20 in humanvaginal keratinocytes in raft and monolayer cultures. HPV16� CaSki cells were used as a cancer cell control for Western blotting. Tubulin servedas a loading control.
VOL. 83, 2009 SRp20 IN VIRAL EARLY-TO-LATE SWITCH 177
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
standing of SRp20 expression in the cervical epithelium overthe viral life cycle (Fig. 9F). This model differs from the ASF/SF2 model proposed in the study of a negative regulatoryelement in the 3� end of HPV16 late pre-mRNAs (39), inwhich upregulation of ASF/SF2 upon differentiation of W12Ecells was ascribed to the expression of E2 from episomalHPV16 genomes rather than epithelial differentiation.
Despite the fact that the HPV16 ESE regulates the selectionof the splice site and poly(A) site, the integration of an HPVgenome into a host chromosome in cervical cancer cells dis-rupts the integrity of viral E1 and E2 ORFs (3, 30) and posi-tions the identified SRp20-interacting ESE downstream of the
integration site, interrupting the ability of the HPV16 ESE toaccess and regulate the use of the upstream 3� ss. Because theviral E6 and E7 ORFs are positioned upstream of E1 and E2in the virus genome, regulation by SRp20 of viral E6 and E7expression in cervical cancer cells must be attributed to thisESE-independent pathway. In the case of HPV18, no ESEelement has been identified in any of its transcripts. Notably,the downregulation of viral E6 and E7 expression by SRp20knockdown appeared in association with a reduction in tran-scription factor SP1 and viral E6E7 transcription, but not witha reduction in viral RNA export, because the cells with reducedSRp20 expression showed no accumulation of bicistronic E6E7
FIG. 9. SRp20 promotes the expression of the HPV16 and HPV18 early genes E6 and E7. (A) Diagrams of the HPV16 and HPV18 E6 andE7 ORFs, the splicing directions of the HPV16 and HPV18 E6E7 bicistronic pre-mRNAs, and the positions of antisense probes used for Northernblot analysis. The numbers above the E6 and E7 ORFs are start and stop codons for each ORF, and above the pre-mRNA are splice sites. Exons(box 1 and box 2) and introns (lines) of the E6E7 pre-mRNA are shown. The drawings are not to scale. (B to E) Knocking down SRp20 in CaSkiand HeLa cells with siRNA decreases the expression of HPV16 and HPV18 E6 and E7. HPV16� CaSki and HPV18� HeLa cells were treated twicewith SRp20-specific siRNA or an NS siRNA (B). HPV16 and HPV18 E6E7 RNAs were detected by Northern blotting (C), and E7 protein andE6 and E7 downstream targets, as well as SP1, were detected by Western blotting (D and E). For the Northern blotting (C), total cytoplasmic ornuclear RNA was isolated with (CaSki) or without (HeLa) poly(A) selection, and the E6E7 RNA in each fraction was detected. Afternormalization to GAPDH, the relative level of E6E7 RNA in each fraction of SRp20 siRNA-treated cells was expressed as a ratio to the levelobtained from a corresponding fraction of cells treated with an NS siRNA. C, cytoplasmic; N, nuclear. One representative experiment of two isshown. For the Western blots in panels B, D, and E, hnRNP K or tubulin in each sample served as a sample loading control. (F) Relationship ofSRp20 expression in the cervix to viral E6E7 and L1L2 production. Normal cervical tissues were examined for SRp20 expression by immunohis-tochemistry with an anti-SRp20 7B4 antibody. Abundant SRp20 expression in the basal and parabasal layers of the cervix is depicted as parallelingthe level of E6E7 expression, but the reduction of SRp20 expression in the upper layers of the cervix is inversely related to L1L2 expression(arrows). Infection of the cervical basal cells by viruses (circles, top left) is initiated through a microtrauma.
178 JIA ET AL. J. VIROL.
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

RNAs in the nuclear fraction. Instead, the number of bicis-tronic E6E7 transcripts was reduced two- to fourfold in boththe cytoplasmic and nuclear fractions (Fig. 9C). Thus, reducedviral E6 and E7 transcription might be a result of the reducedexpression of SP1 in the SRp20 knockdown cells, since SP1 isessential for transcription activation of both HPV16 andHPV18 E6E7 promoters (2, 13, 15, 51).
Initially, we thought that YB-1 could be a major player inSE4-mediated regulation of BPV-1 L1 expression. YB-1 is aDNA/RNA-binding nucleocytoplasmic shuttling protein that isinvolved in regulation of DNA repair (19), mRNA transcrip-tion (46), splicing (1, 50), translation (10, 43), and stability (9,16). YB-1 has been identified as a spliceosome-associated fac-tor (11) and interacts with splicing factors SRp30c (44),SRrp86 (32), and phosphatase PP2C (1) to regulate alterna-tive RNA splicing. In human CD44 alternative RNA splicing,YB-1 binding to an A/C-rich exon enhancer controls the in-clusion of variable exon 4 (50). However, in our study, YB-1bound the A/C-rich SE4 only with low affinity compared to itsbinding of the purine-rich SE2 and had little positive effect onthe selection of a viral-L1-specific 3� ss. Although YB-1 is anabundant cellular protein, the observed minimal effect of YB-1on the function of A/C-rich SE4 in selection of the L1-specific3� ss could be a consequence of the overwhelming suppressiveeffect of the SRp20-SE4 interaction on splice site selection.Altogether, our data support a compelling model of the pap-illomavirus early-to-late switch that is closely tied to SRp20function and its differential expression in infected keratino-cytes.
ACKNOWLEDGMENTS
We thank Tom Cooper (Baylor College of Medicine) for YB-1plasmids, James Manley (Columbia University) for the ASF/SF2 plas-mid pCGNF1, and Javier Caceres (MRC Human Genetics Unit, Ed-inburgh, United Kingdom) for the SRp20 plasmid. We also thankJennice Gullett for preparation of the raft culture tissues.
This study was supported by the Intramural Research Program ofthe Center for Cancer Research, National Cancer Institute, NIH, andNIH grant R01 AI057988-01 to C.M.
REFERENCES
1. Allemand, E., M. L. Hastings, M. V. Murray, M. P. Myers, and A. R.Krainer. 2007. Alternative splicing regulation by interaction of phosphatasePP2C with nucleic acid-binding protein YB-1. Nat. Struct. Mol. Biol. 14:630–638.
2. Apt, D., R. M. Watts, G. Suske, and H. U. Bernard. 1996. High Sp1/Sp3ratios in epithelial cells during epithelial differentiation and cellular trans-formation correlate with the activation of the HPV-16 promoter. Virology224:281–291.
3. Baker, C. C., W. C. Phelps, V. Lindgren, M. J. Braun, M. A. Gonda, andP. M. Howley. 1987. Structural and transcriptional analysis of human papil-lomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 61:962–971.
4. Barksdale, S., and C. C. Baker. 1995. Differentiation-specific alternativesplicing of bovine papillomavirus late mRNAs. J. Virol. 69:6553–6556.
5. Barksdale, S. K., and C. C. Baker. 1993. Differentiation-specific expressionfrom the bovine papillomavirus type 1 P2443 and late promoters. J. Virol.67:5605–5616.
6. Bedard, K. M., S. Daijogo, and B. L. Semler. 2007. A nucleo-cytoplasmic SRprotein functions in viral IRES-mediated translation initiation. EMBO J.26:459–467.
7. Bruce, S. R., R. W. Dingle, and M. L. Peterson. 2003. B-cell and plasma-cellsplicing differences: a potential role in regulated immunoglobulin RNAprocessing. RNA 9:1264–1273.
8. Cavaloc, Y., C. F. Bourgeois, L. Kister, and J. Stevenin. 1999. The splicingfactors 9G8 and SRp20 transactivate splicing through different and specificenhancers. RNA 5:468–483.
9. Chen, C. Y., R. Gherzi, J. S. Andersen, G. Gaietta, K. Jurchott, H. D. Royer,
M. Mann, and M. Karin. 2000. Nucleolin and YB-1 are required for JNK-mediated interleukin-2 mRNA stabilization during T-cell activation. GenesDev. 14:1236–1248.
10. Cobbold, L. C., K. A. Spriggs, S. J. Haines, H. C. Dobbyn, C. Hayes, C. H.de Moor, K. S. Lilley, M. Bushell, and A. E. Willis. 2008. Identification ofinternal ribosome entry segment (IRES)-trans-acting factors for the Mycfamily of IRESs. Mol. Cell. Biol. 28:40–49.
11. Deckert, J., K. Hartmuth, D. Boehringer, N. Behzadnia, C. L. Will, B.Kastner, H. Stark, H. Urlaub, and R. Luhrmann. 2006. Protein compositionand electron microscopy structure of affinity-purified human spliceosomal Bcomplexes isolated under physiological conditions. Mol. Cell. Biol. 26:5528–5543.
12. de la Mata, M., and A. R. Kornblihtt. 2006. RNA polymerase II C-terminaldomain mediates regulation of alternative splicing by SRp20. Nat. Struct.Mol. Biol. 13:973–980.
13. Demeret, C., M. Yaniv, and F. Thierry. 1994. The E2 transcriptional repres-sor can compensate for Sp1 activation of the human papillomavirus type 18early promoter. J. Virol. 68:7075–7082.
14. Dollard, S. C., J. L. Wilson, L. M. Demeter, W. Bonnez, R. C. Reichman,T. R. Broker, and L. T. Chow. 1992. Production of human papillomavirus andmodulation of the infectious program in epithelial raft cultures. Genes Dev.6:1131–1142.
15. Dong, X. P., and H. Pfister. 1999. Overlapping YY1- and aberrant SP1-binding sites proximal to the early promoter of human papillomavirus type16. J. Gen. Virol. 80:2097–2101.
16. Evdokimova, V., P. Ruzanov, H. Imataka, B. Raught, Y. Svitkin, L. P.Ovchinnikov, and N. Sonenberg. 2001. The major mRNA-associated proteinYB-1 is a potent 5� cap-dependent mRNA stabilizer. EMBO J. 20:5491–5502.
17. Fairbrother, W. G., R. F. Yeh, P. A. Sharp, and C. B. Burge. 2002. Predictiveidentification of exonic splicing enhancers in human genes. Science 297:1007–1013.
18. Galiana-Arnoux, D., F. Lejeune, M. C. Gesnel, J. Stevenin, R. Breathnach,and F. Gatto-Konczak. 2003. The CD44 alternative v9 exon contains asplicing enhancer responsive to the SR proteins 9G8, ASF/SF2, and SRp20.J. Biol. Chem. 278:32943–32953.
19. Gaudreault, I., D. Guay, and M. Lebel. 2004. YB-1 promotes strand sepa-ration in vitro of duplex DNA containing either mispaired bases or cisplatinmodifications, exhibits endonucleolytic activities and binds several DNArepair proteins. Nucleic Acids Res. 32:316–327.
20. Gonzalez, S. L., M. Stremlau, X. He, J. R. Basile, and K. Munger. 2001.Degradation of the retinoblastoma tumor suppressor by the human papillo-mavirus type 16 E7 oncoprotein is important for functional inactivation andis separable from proteasomal degradation of E7. J. Virol. 75:7583–7591.
21. Grassmann, K., B. Rapp, H. Maschek, K. U. Petry, and T. Iftner. 1996.Identification of a differentiation-inducible promoter in the E7 open readingframe of human papillomavirus type 16 (HPV-16) in raft cultures of a newcell line containing high copy numbers of episomal HPV-16 DNA. J. Virol.70:2339–2349.
22. Guo, M., Y. Gong, M. Deavers, E. G. Silva, Y. J. Jan, D. E. Cogdell, R.Luthra, E. Lin, H. C. Lai, W. Zhang, and N. Sneige. 2008. Evaluation of acommercialized in situ hybridization assay for detecting human papilloma-virus DNA in tissue specimens from patients with cervical intraepithelialneoplasia and cervical carcinoma. J. Clin. Microbiol. 46:274–280.
23. Hargous, Y., G. M. Hautbergue, A. M. Tintaru, L. Skrisovska, A. P.Golovanov, J. Stevenin, L. Y. Lian, S. A. Wilson, and F. H. Allain. 2006.Molecular basis of RNA recognition and TAP binding by the SR proteinsSRp20 and 9G8. EMBO J. 25:5126–5137.
24. Hastings, M. L., and A. R. Krainer. 2001. Pre-mRNA splicing in the newmillennium. Curr. Opin. Cell Biol. 13:302–309.
25. Howley, P. M., and D. R. Lowy. 2001. Papillomaviruses and their replication,p. 2197–2229. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb,M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology. LippincottWilliams & Wilkins, Philadelphia, PA.
26. Huang, Y., R. Gattoni, J. Stevenin, and J. A. Steitz. 2003. SR splicing factorsserve as adapter proteins for TAP-dependent mRNA export. Mol. Cell11:837–843.
27. Huang, Y., and J. A. Steitz. 2001. Splicing factors SRp20 and 9G8 promotethe nucleocytoplasmic export of mRNA. Mol. Cell 7:899–905.
28. Hui, J., L. H. Hung, M. Heiner, S. Schreiner, N. Neumuller, G. Reither, S. A.Haas, and A. Bindereif. 2005. Intronic CA-repeat and CA-rich elements: anew class of regulators of mammalian alternative splicing. EMBO J. 24:1988–1998.
29. Hung, L. H., M. Heiner, J. Hui, S. Schreiner, V. Benes, and A. Bindereif.2008. Diverse roles of hnRNP L in mammalian mRNA processing: a com-bined microarray and RNAi analysis. RNA 14:284–296.
30. Jeon, S., B. L. Allen-Hoffmann, and P. F. Lambert. 1995. Integration ofhuman papillomavirus type 16 into the human genome correlates with aselective growth advantage of cells. J. Virol. 69:2989–2997.
31. Jumaa, H., G. Wei, and P. J. Nielsen. 1999. Blastocyst formation is blockedin mouse embryos lacking the splicing factor SRp20. Curr. Biol. 9:899–902.
32. Li, J., I. C. Hawkins, C. D. Harvey, J. L. Jennings, A. J. Link, and J. G.
VOL. 83, 2009 SRp20 IN VIRAL EARLY-TO-LATE SWITCH 179
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

Patton. 2003. Regulation of alternative splicing by SRrp86 and its interactingproteins. Mol. Cell. Biol. 23:7437–7447.
33. Liu, X., A. Mayeda, M. Tao, and Z. M. Zheng. 2003. Exonic splicing enhanc-er-dependent selection of the bovine papillomavirus type 1 nucleotide 32253� splice site can be rescued in a cell lacking splicing factor ASF/SF2 throughactivation of the phosphatidylinositol 3-kinase/Akt pathway. J. Virol. 77:2105–2115.
34. Longworth, M. S., and L. A. Laimins. 2004. Pathogenesis of human papil-lomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 68:362–372.
35. Lou, H., K. M. Neugebauer, R. F. Gagel, and S. M. Berget. 1998. Regulationof alternative polyadenylation by U1 snRNPs and SRp20. Mol. Cell. Biol.18:4977–4985.
36. Lowy, D. R., and P. M. Howley. 2001. Papillomaviruses, p. 2231–2264. InD. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B.Roizman, and S. E. Straus (ed.), Fields virology, vol. 2. Lippincott Williams& Wilkins, Philadelphia, PA.
37. Maciolek, N. L., and M. T. McNally. 2007. Serine/arginine-rich proteinscontribute to negative regulator of splicing element-stimulated polyadenyl-ation in Rous sarcoma virus. J. Virol. 81:11208–11217.
38. Maniatis, T., and B. Tasic. 2002. Alternative pre-mRNA splicing and pro-teome expansion in metazoans. Nature 418:236–243.
39. McPhillips, M. G., T. Veerapraditsin, S. A. Cumming, D. Karali, S. G.Milligan, W. Boner, I. M. Morgan, and S. V. Graham. 2004. SF2/ASF bindsthe human papillomavirus type 16 late RNA control element and is regulatedduring differentiation of virus-infected epithelial cells. J. Virol. 78:10598–10605.
40. Meyers, C., M. G. Frattini, J. B. Hudson, and L. A. Laimins. 1992. Biosyn-thesis of human papillomavirus from a continuous cell line upon epithelialdifferentiation. Science 257:971–973.
41. Middleton, K., W. Peh, S. Southern, H. Griffin, K. Sotlar, T. Nakahara, A. ElSherif, L. Morris, R. Seth, M. Hibma, D. Jenkins, P. Lambert, N. Coleman,and J. Doorbar. 2003. Organization of human papillomavirus productivecycle during neoplastic progression provides a basis for selection of diagnos-tic markers. J. Virol. 77:10186–10201.
42. Milligan, S. G., T. Veerapraditsin, B. Ahamet, S. Mole, and S. V. Graham.2007. Analysis of novel human papillomavirus type 16 late mRNAs in dif-ferentiated W12 cervical epithelial cells. Virology 360:172–181.
43. Nekrasov, M. P., M. P. Ivshina, K. G. Chernov, E. A. Kovrigina, V. M.Evdokimova, A. A. Thomas, J. W. Hershey, and L. P. Ovchinnikov. 2003. ThemRNA-binding protein YB-1 (p50) prevents association of the eukaryoticinitiation factor eIF4G with mRNA and inhibits protein synthesis at theinitiation stage. J. Biol. Chem. 278:13936–13943.
44. Raffetseder, U., B. Frye, T. Rauen, K. Jurchott, H. D. Royer, P. L. Jansen,and P. R. Mertens. 2003. Splicing factor SRp30c interaction with Y-boxprotein-1 confers nuclear YB-1 shuttling and alternative splice site selection.J. Biol. Chem. 278:18241–18248.
45. Rush, M., X. Zhao, and S. Schwartz. 2005. A splicing enhancer in the E4coding region of human papillomavirus type 16 is required for early mRNAsplicing and polyadenylation as well as inhibition of premature late geneexpression. J. Virol. 79:12002–12015.
46. Samuel, S., J. C. Twizere, and L. R. Bernstein. 2005. YB-1 represses AP1-dependent gene transactivation and interacts with an AP-1 DNA sequence.Biochem. J. 388:921–928.
47. Scheffner, M., B. A. Werness, J. M. Huibregtse, A. J. Levine, and P. M.Howley. 1990. The E6 oncoprotein encoded by human papillomavirus types16 and 18 promotes the degradation of p53. Cell 63:1129–1136.
48. Sen, E., S. Alam, and C. Meyers. 2004. Genetic and biochemical analysis ofcis regulatory elements within the keratinocyte enhancer region of the hu-man papillomavirus type 31 upstream regulatory region during differentstages of the viral life cycle. J. Virol. 78:612–629.
49. Spink, K. M., and L. A. Laimins. 2005. Induction of the human papilloma-virus type 31 late promoter requires differentiation but not DNA amplifica-tion. J. Virol. 79:4918–4926.
50. Stickeler, E., S. D. Fraser, A. Honig, A. L. Chen, S. M. Berget, and T. A.Cooper. 2001. The RNA binding protein YB-1 binds A/C-rich exon enhanc-ers and stimulates splicing of the CD44 alternative exon v4. EMBO J.20:3821–3830.
51. Stunkel, W., and H. U. Bernard. 1999. The chromatin structure of the longcontrol region of human papillomavirus type 16 represses viral oncoproteinexpression. J. Virol. 73:1918–1930.
52. Tang, S., M. Tao, J. P. McCoy, and Z. M. Zheng. 2006. Short-term inductionand long-term suppression of HPV16 oncogene silencing by RNA interfer-ence in cervical cancer cells. Oncogene 25:2094–2104.
53. Tang, S., M. Tao, J. P. McCoy, Jr., and Z. M. Zheng. 2006. The E7 onco-protein is translated from spliced E6*I transcripts in high-risk humanpapillomavirus type 16- or type 18-positive cervical cancer cell lines viatranslation reinitiation. J. Virol. 80:4249–4263.
54. Ule, J., G. Stefani, A. Mele, M. Ruggiu, X. Wang, B. Taneri, T. Gaasterland,B. J. Blencowe, and R. B. Darnell. 2006. An RNA map predicting Nova-dependent splicing regulation. Nature 444:580–586.
55. Zahler, A. M., W. S. Lane, J. A. Stolk, and M. B. Roth. 1992. SR proteins: aconserved family of pre-mRNA splicing factors. Genes Dev. 6:837–847.
56. Zheng, Z. M. 2004. Regulation of alternative RNA splicing by exon defini-tion and exon sequences in viral and mammalian gene expression. J. Biomed.Sci. 11:278–294.
57. Zheng, Z. M., and C. C. Baker. 2006. Papillomavirus genome structure,expression, and post-transcriptional regulation. Front. Biosci. 11:2286–2302.
58. Zheng, Z. M., P. He, and C. C. Baker. 1996. Selection of the bovine papil-lomavirus type 1 nucleotide 3225 3� splice site is regulated through an exonicsplicing enhancer and its juxtaposed exonic splicing suppressor. J. Virol.70:4691–4699.
59. Zheng, Z. M., P. J. He, and C. C. Baker. 1997. Structural, functional, andprotein binding analyses of bovine papillomavirus type 1 exonic splicingenhancers. J. Virol. 71:9096–9107.
60. Zheng, Z. M., M. Huynen, and C. C. Baker. 1998. A pyrimidine-rich exonicsplicing suppressor binds multiple RNA splicing factors and inhibits spliceo-some assembly. Proc. Natl. Acad. Sci. USA 95:14088–14093.
61. Zheng, Z. M., E. S. Reid, and C. C. Baker. 2000. Utilization of the bovinepapillomavirus type 1 late-stage-specific nucleotide 3605 3� splice site ismodulated by a novel exonic bipartite regulator but not by an intronicpurine-rich element. J. Virol. 74:10612–10622.
180 JIA ET AL. J. VIROL.
on May 22, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from





























