Embed Size (px)
Citation preview

Vol. 169, No. 7JOURNAL OF BACTERIUOLOGY, July 1987, P. 3321-33280021-9193/87/073321-08$02.00/0Copyright © 1987, American Society for Microbiology
Construction and Use of Signal Sequence Selection Vectors inEscherichia coli and Bacillus subtilis
HILDE SMITH,'* SIERD BRON,1 JAN VAN EE,2 AND GERARD VENEMA'Department of Genetics, Center ofBiological Sciences, 9751 NN Haren (Gn),' and Gist-Brocades N.V., 2600 MA Delft,2
The Netherlands
Received 10 February 1987/Accepted 17 April 1987
To study the diversity and efficiency of signal peptides for secreted proteins in gram-positive bacteria, twoplasmid vectors were constructed which were used to probe for export signal-coding regions in Bacilus subtilis.The vectors contained genes coding for extracellular proteins (the aL-amylase gene from Bacilus licheniformisand the j3-lactamase gene from Escherichia colt) which lacked a functional signal sequence. By shotgun cloningof restriction fragments from B. subtilis chromosomal DNA, a great variety of different export-coding regionswere selected. These regions were functional both in B. subtlis and in E. coli. In a number of cases whereprotein export had been restored, intracellular precursor proteins of increased size could be detected, whichupon translocation across the cellular membrane were processed to mature products. The high frequency withwhich export signal-coding regions were obtained suggests that, in addition to natural signal sequences, manyrandomly cloned sequences can function as export signal.
The ability of bacilli to secrete large amounts ofexoenzymes into the culture medium (17) makes theseorganisms interesting for the synthesis of foreign gene prod-ucts on an industrial scale. This is one of the reasons why,from an applicational point of view, studies on proteinsecretion in these organisms are of interest. Research on themechanism of protein secretion in gram-positive bacteria isalso important from a scientific point of view. In bothgram-positive and gram-negative bacteria, secreted proteinsare initially synthesized with N:terminal signal sequences,which are removed during translocation across the cellularmembrane. Signal sequences of gram-positives are usuallylonger than those of gram-negatives and frequently containmore charged amino acid residues in the N terminus (26). Inaddition, several gram-positive exoproteins, such as severalproteases, are synthesized as even larger precursor forms,called prepropolypeptides (20, 25, 28).To optimize the efficiency of protein secretion by bacilli, a
better understanding of the parameters affecting the translo-cation mechanism(s) is desirable. So far, the approach mostfrequently used has been to introduce mutations in thevarious regions of the signal sequence (for review, seereference 19). Particularly in Escherichia coli, this hasresulted in valuable information about functional propertiesof the different regions of the signal peptides. An alternativeapproach would be to select a considerable number of signalsequences naturally occurring in the chromosome and tocompare the properties of the corresponding signal peptides.We have chosen for this approach to study the properties ofsignal sequences in Bacillus subtilis. The present paperdescribes an efficient system for the selection of exportsignal sequences. The approach was to construct two plas-mid vectors containing genes coding for extracellular pro-teins which lacked part of or the entire signal sequence.Since signal sequences are usually functionally interchange-able (3, 15, 23), we expected that cloning in the correctreading frame of DNA fragments coding for a promoter,ribosomal binding site, start codon, and signal sequencewould result in the synthesis and export of the protein. Two
* Corresponding author.
different genes coding for easily assayable extracellularproteins were chosen as signal sequence selection probes: (i)the a-amylase gene from Bacillus licheniformis and (ii) theP-lactamase gene from E. coli. These genes have alreadybeen cloned (16; Gist-Brocades N.V., European patentapplication 8320106.9, 1983) and sequenced (21, 30).The results showed that, by shotgun cloning of restriction
fragments from B. subtilis chromosomal DNA in the signalsequence selection vectors, a large variety of different ex-port-coding regions could be selected. These regions ap-peared to function both in B. subtilis and in E. coli.
MATERIALS AND METHODSBacteria and plasmids. Table 1 lists the bacterial strains
and plasmids used. B. subtilis 8G-5 (amy) was obtained bycongression after transformation of competent 8G-5 cellswith DNA from B. subtilis 1-85 and selection for his'recombinants.Media and plates. TY medium contained (per liter) 10 g of
tryptone, 5 g of yeast extract, and 10 g of NaCl (pH 7.4).Minimal media used in the competence regimen for B.subtilis were as described before (2). Media were supple-mented with antibiotics as follows: for B. subtilis, chloram-phenicol at 5 ,ug/ml and erythromycin at 1 ,ug/ml; and for E.coli, ampicillin at 25 ,ug/ml and erythromycin at 100 ,ug/ml(unless stated otherwise).
(Bio)chemicals. Restriction enzymes, DNA polymerase I,T4 DNA polymerase, and T4 DNA ligase, obtained fromBoehringer (Mannheim, Federal Republic of Germany) orfrom New England Biolabs (Beverly, Mass.), were used asindicated by the suppliers.DNA preparations. Chromosomal DNA was extracted
from B. subtilis as described by Bron and Venema (2).Preparative amounts of plasmid DNA were obtained basi-cally according to the alkaline lysis procedure described byManiatis et al. (11). Selected restriction fragments wereobtained by separation on 0.8 or 2.0% agarose gels, followedby extraction and purification on DEAE NA-45 membranes(Schleicher & Schuell, Dassel, Federal Republic of Ger-many) as indicated by the manufacturer. The analytical"miniprep" procedure described by Ish-Horowicz and
3321
on June 3, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

3322 SMITH ET AL.
TABLE 1. Bacterial strains and plasmids
Plasmid or strain Properties and genotype reference
PlasmidspKTH74 E. coli plasmid; 5.8 kb; Apr, I. Palva
TcrpGK13 Streptococcus cremoris plas- Laboratory col-
mid, derivative of pGK12; lection, Kok4.8 kb; Cmr, Emr et al. (9)
pGK13-amy+ pGK13, carrying the Laboratory col-ox-amylase gene of B. lectionlicheniformis; 7.8 kb; Cmr,Emr; a-amylase+
pRW101 Bifunctional replicon, carrying Mezes et al. (13)the penicillinase gene of B.licheniformis; 7.0 kb; Kmr(B. subtilis); Apr (E. coli)
pUC13 E. coli plasmid; 2.7 kb; Apr Messing et al.(12)
pPL608 B. subtilis plasmid; 5.0 kb; Williams et al.Kmr, Cmr (27)
pTA1060 Cryptic B. subtilis plasmid; Uozumi et al.8.6 kb (24)
pHP14 Bifunctional replicon; 4.2 kb; This paperEmr; carrying the MCS ofm13 mpll
pGPB11 Export signal selection vector; This paper5.7 kb; Emr
pGPB14 As pGPB11 This paperpGPA11 Export signal selection vector; This paper
5.8 kb; EmrpGPA14 As pGPA11 This paper
StrainsB. subtilis
8G-5 trpC2 tyr his nic ura rib met Bron et al. (2)ade
8G-5 amy a-Amylase-negative derivative Laboratory col-of 8G-5 lection
DB104 his nprR2 nprEJ8 aprA3 Kawamura et al.(8)
1-85 trpC2 amy Yuki (29)E. coliBHB2600 803 supE supF rk- mk- met K. Murray
Burke (5) was used to extract plasmids from 2.5-ml culturesof E. coli. Minipreps from B. subtilis were prepared basicallyaccording to the same procedure with the modification thatcells from 5-ml cultures were washed once by centrifugationwith 50 mM Tris hydrochloride (pH 7.4)-10 mM EDTA-200mM NaCl before being lysed. Miniprep DNAs were incu-bated with 100 ,ug of pancreatic RNase (BDH, Poole, En-gland) per ml for 30 min at 37°C.
Molecular cloning procedures. Vector molecules and re-
stricted target DNAs were mixed in approximately a 1:4weight ratio at a total concentration of 100 jig/ml. Ligationwas carried out as described by Maniatis et al. (11). Ligatedsamples were used to transform competent B. subtilis or E.coli cells.
Transformations. Transformation of competent B. subtilisand E. coli cells was as described by Bron and Luxen (1).Transformants were selected on TY plates supplementedwith selective antibiotics as follows: for B. subtilis, erythro-mycin at 1 ,ug/ml and kanamycin at 5 p.g/ml; and for E. coli,erythromycin at 200 ,ug/ml or erythromycin at 50 ,ug/ml plusampicillin at 2 ,ug/ml.Probing for chromosomal export signals. B. subtilis chro-
mosomal DNA was digested with Sau3A, AluI, HaeIII, orRsaI and subsequently ligated with BamHI- or SmaI-
digested vector molecules (pGPB14 or pGPA14) in a 4:1weight ratio. The construction of these vectors is describedin Results. Ligated samples were used to transform compe-tent E. coli cells. Transformants were selected by plating onTY plates containing erythromycin (200 [ig/ml) plus 1%soluble starch (for pGPA14) or erythromycin (50 jig/ml) plusampicillin (2 p.g/ml) (for pGPB14). Plasmid DNAs extractedfrom the transformants were subsequently used to transformcompetent B. subtilis cells to erythromycin resistance.Transformants were analyzed for their capacity to secreteeither amylase or TEM 3-lactamase into the culture mediumas described below.
Agarose gel electrophoresis. Gel electrophoresis was car-ried out on 0.8 to 2.0% agarose (Bio-Rad, Richmond, Calif.).Electrophoresis buffer consisted of 89 mM Tris, 89 mM boricacid, 10 mM EDTA (pH 8.3), and 1 ,ug of ethidium bromideper ml.PAA gel electrophoresis. Sodium dodecyl sulfate (SDS)-
polyacrylamide (PAA) gel electrophoresis was performed onslab gels by the method of Laemmli (10). Electrophoresiswas conducted at room temperature for about 4 h at aconstant current of 20 mA. Samples were heated for 5 min at100°C before being applied to the gels.
Assays for a-amylase activity. (i) In solutions. Assay mix-tures contained 2 ml of substrate solution (0.1% solublestarch in 100 mM potassium phosphate, pH 7.0) and 2 ml ofvarious dilutions of the enzyme solutions. The mixtureswere incubated for 1 h at 60°C, and the reactions werestopped by the addition of 0.2 ml of iodine reagent (0.3% 12+ 0.6% KI). One unit of (x-amylase was defined as theamount of enzyme which hydrolyzed 0.1 mg of solublestarch in 1 min under the conditions used.
(ii) In colonies. E. coli colonies were grown on TY agarsupplemented with 1% soluble starch. B. subtilis colonieswere grown on minimal agar without glucose, supplementedwith 1% soluble starch. After the addition of iodine reagent(0.3% 12 + 0.6% KI), clear halos appeared around ot-amylase-producing colonies.
(iii) In SDS-PAA gels. Stacking and separating gels con-tained 0.25% soluble starch. After electrophoresis of theot-amylase-containing samples, the gels were washed (fourtimes for 30 min) in 10 mM Tris hydrochloride (pH 6.8) plus0.25% starch to remove the SDS and to allow renaturation ofthe proteins. Subsequently, the gels were incubated for 2 h at65°C (or overnight at 37°C) in the same buffer. ot-Amylaseactivity was visualized as clear zones after the starch in thegels was stained with '2 (0.5%) + KI (1.5%).
Assay of -lactamase activity. P-Lactamase activity in B.subtilis culture supernatants was determined spectrophoto-metrically using nitrocefin (Becton Dickinson B.V.,Amersfoort, The Netherlands). Nitrocefin (5 mg) was dis-solved in 0.5 ml dimethyl sulfoxide and diluted to 10 ml with100 mM potassium phosphate (pH 7.0). Assay mixturescontaining 0.3 ml of the nitrocefin substrate solution, 2.7 mlof potassium phosphate buffer (pH 7.0), and appropriateamounts (25 to 500 ,ul) of the enzyme solutions were incu-bated for 5 to 10 min at room temperature. One unit ofP-lactamase was defined as the amount of enzyme thatincreased absorbance at 486 nm by 0.001 in 1 min at roomtemperature.ICDH assay. Isocitrate dehydrogenase (ICDH), an enzyme
involved in the tricarboxylic acid pathway, is exclusivelylocalized in the cytoplasm. Therefore, the amount of ICDHfound in B. subtilis culture supernatants is a measure of thedegree of cell lysis. Assay mixtures containing 500 ,ul of 100mM potassium phosphate (pH 8.0), 20 ,ul of 250 mM MgCl2,
J. BACTERIOL.
on June 3, 2018 by guesthttp://jb.asm
.org/D
ownloaded from
BACTERIAL SIGNAL SEQUENCE SELECTION VECTORS
M bo I
Bcl I i . *. ori,>// *--. pW VO I
pGK13moumylose BmHI
7.8 k b
EcoRi EcoR IBomHI Pst I
Hindm - laISal I
1,6kb Pst I-Hind m fragmentcarrying the aiomylase gene
locking most of Its signol sequence
Inserted inpHP14 digestedwith Hind IIIand Pst I
EcoR I Hind ImPst I
?- -0 Pst I
pKTH74 %5.8kb
Hind m
\ ' BamHI
Sal I
AvaI
1,5kb Hind III fragment carryingthe 19 lactamose gene lacking
Its signal sequence
*\ wN Bcl I
Oi/ HP14PTAlO0O. P4.2 kb / or/s*44fflJ pBR322
NaI loc CS (mp 13)
Inserted inHind III siteof pHP14
= - Bcl I
/<
o
pTA16 oril pGPA11 (14) pBR 322; 5,8kb
amyase Hind m
mp'3) CIa l I
,H\ B Bcl I
pTA186\ oril p GPB11(14) pBR 322
\ 5,7kb~.lactamase Hind m
MCSlmp13) PstIl
Key: -npC194 pBR322pE194 pTA1060
............. pWVO1cloned fragments (ocamylase, R lactomase )
- fusionpoints
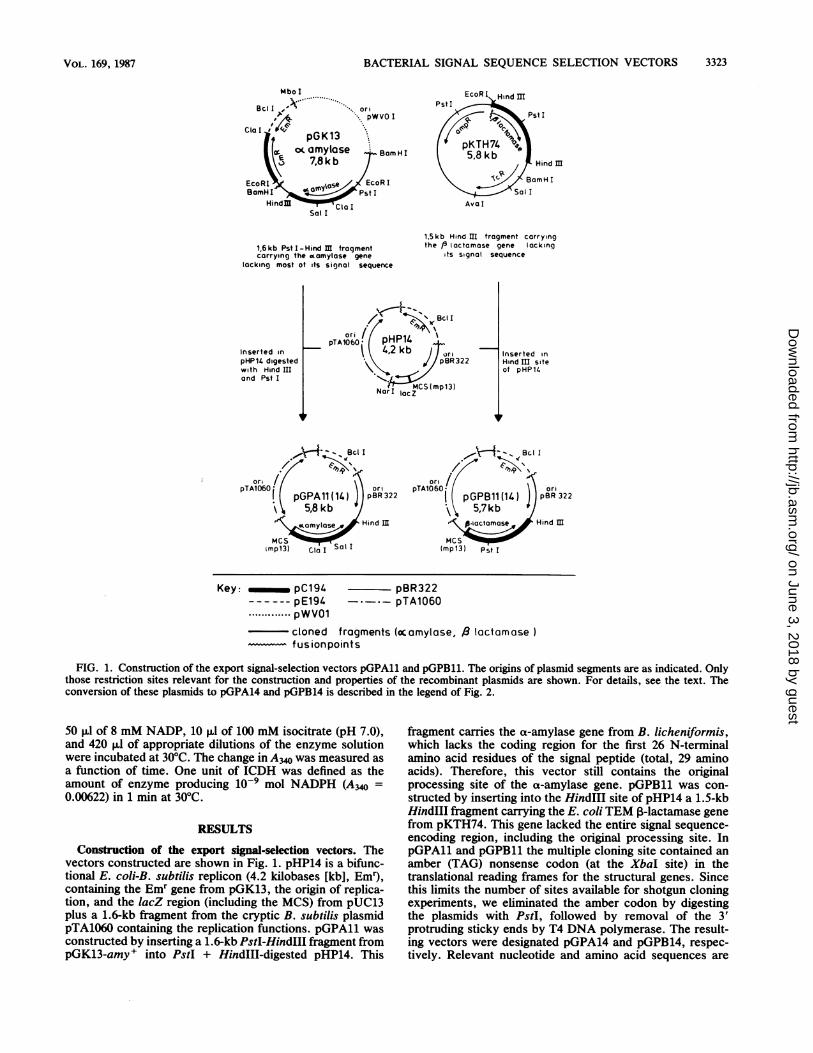
FIG. 1. Construction of the export signal-selection vectors pGPAll and pGPB11. The origins of plasmid segments are as indicated. Onlythose restriction sites relevant for the construction and properties of the recombinant plasmids are shown. For details, see the text. Theconversion of these plasmids to pGPA14 and pGPB14 is described in the legend of Fig. 2.
50 IL1 of 8 mM NADP, 10 p.1 of 100 mM isocitrate (pH 7.0),and 420 ,ul of appropriate dilutions of the enzyme solutionwere incubated at 30°C. The change in A3 was measured asa function of time. One unit of ICDH was defined as theamount of enzyme producing 10-9 mol NADPH (A3 =0.00622) in 1 min at 30°C.
RESULTS
Construction of the export signal-selection vectors. Thevectors constructed are shown in Fig. 1. pHP14 is a bifunc-tional E. coli-B. subtilis replicon (4.2 kilobases [kb], Em9,containing the Em" gene from pGK13, the origin of replica-tion, and the lacZ region (including the MCS) from pUC13plus a 1.6-kb fragment from the cryptic B. subtilis plasmidpTA1060 containing the replication functions. pGPA11 wasconstructed by inserting a 1.6-kb PstI-HindIII fragment frompGK13-amy+ into PstI + HindIII-digested pHP14. This
fragment carries the ax-amylase gene from B. licheniformis,which lacks the coding region for the first 26 N-terminalamino acid residues of the signal peptide (total, 29 aminoacids). Therefore, this vector still contains the originalprocessing site of the a-amylase gene. pGPBll was con-structed by inserting into the HindIII site of pHP14 a 1.5-kbHindIII fragment carrying the E. coli TEM P-lactamase genefrom pKTH74. This gene lacked the entire signal sequence-encoding region, including the original processing site. InpGPAll and pGPBll the multiple cloning site contained anamber (TAG) nonsense codon (at the XbaI site) in thetranslational reading frames for the structural genes. Sincethis limits the number of sites available for shotgun cloningexperiments, we eliminated the amber codon by digestingthe plasmids with PstI, followed by removal of the 3'protruding sticky ends by T4 DNA polymerase. The result-ing vectors were designated pGPA14 and pGPB14, respec-tively. Relevant nucleotide and amino acid sequences are
3323VOL. 169, 1987
on June 3, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

3324 SMITH ET AL.
MCS mp 13 uamylpose Hind rIT
/ ~ - - CIa! Sal I
,onmylase -
-3 -2 -1*jX ~~~~~~~~~~~stop?ser Lrg pro Gl aLa ala qaiaW,l
GAATTCGAGCTCGCCCGGGGATCCTCTAGAGTCGACCTGCAGCAGUGGCG GC
EcoRI Sac! Smal BamHI Xbol Sail PstI
|oamylase
p9y asp pro Leu glu ser thr ala ala alai,010GAATTCGAGCTCGCCCGGGGATCCTCTAGA(,TC ACCGCAGCGGCGGCAEcoRI Sacl SImaI BamHl Xbol Sal!
®E) MCSmp13 f3.iactamose
- - Pst I
Hind III
stop ser org p ala ala ginff ala cysGAATTCGAGCTCGCCCGGGGATCCTCTAGAGTCGACCTGCAGCCCAAGCTTGEcoRI Sacl Smal BamHI Xbal Soil Psi I Hind M
g!y asp pro lugla ser thr ala gLn ala scyPpoGAATrCGAGCTCGCCCGGGGATCCTCTAGAGTCGACCGCCCAAGCTTGCCCCEcoR! Sac! Smal BamH1 Xbol Sal! Hind IU
FIG. 2. Construction of pGPA14 and pGPB14. The Dquences at the fusion points of the multiple cloning site (MCthe structural genes are shown. The vertical arrows indicoriginal cleavage sites for leader peptidase. Several codonsthe processing sites are numbered, and the correspondingacids are indicated. The pGPA14 and pGPB14 derivativeconstructed by digesting pGPA11 and pGPB11 with PstI, fiby removal of the four-base 3' protruding sticky ends with Tpolymerase.
indicated in Fig. 2. An additional vector was construcinserting a 280-base-pair (bp) EcoRI fragment from p]containing a phage SPO2 promoter, into the uniquesite of pGPA14. These vectors were constructed waim of detecting additional promoterless, exportcoding fragments, which would remain undetected woriginal vector.
Applicability of the export signal selection systeiexamine the general applicability for the selection ofsignals, the truncated P-lactamase gene was first futwo known signal sequences (Fig. 3). pGK13-amydigested with BamHI "nd PstI. The 940-bp BamIfragment, containing the entire promoter and sigiquence of a-amylase except for the last three aminowas purified and ligated to BamHI + partially PstI-dipGPB11. Since Apr transformants cannot be selecrectly in B. subtilis, the ligation mixture was used toform E. coli BHB2600 cells. The resulting plasipGPB15 (Fig. 3), yielded Apr E. coli transformants103/,g of DNA), indicating that with the truncalactamase gene probe in pGPB11, the export siga-amylase could easily be detected. We also awhether an incomplete signal sequence would reprotein secretion. To that aim a 300-bp BamHI-Psiment from pRW101, containing the promoter andsequence for the first 13 amino acids of the penPpeptide, was fused to BamHI-plus-PstI-cleaved pGP]this case, the hybrid signal sequence lacked a hydrocore. With the resulting plasmid (pGPB16), E. coli cobe transformed to Apr. This indicates that with pGPB
complete or nearly complete signal sequences will be pickedup in E. coli. On the basis of these findings, we tried to select
pOPAll export signal sequences from the B. subtilis chromosome- ~ with both pGPB14 and pGPA14. These experiments arela described next.,A. Selection of export-coding funcos from B. subtilis DNA by
shotgun cloning in E. coli. To minimize the risk of missingpGPA14 potential export signals by cloning the inserts in incorrect
translational reading frames relative to the target genes, anumber of different restriction enzymes (Sau3A, AluI,HaeIII, RsaI) were used to cleave the B. subtilis chromo-somal DNA. After the B. subtilis DNA fragments wereligated into both vectors, clones were selected in E. coli onthe basis either of their ability to form halos on starch plates(pSPA series of plasmids) or their resistance to ampicillin(pSPB series of plasmids). Out of thousands of transform-
pGPB 11 ants tested, a-amylase positive clones were obtained withefficiencies varying from 0.1 to 0.5%, and Apr clones were
.1 *2 obtained with about a fivefold lower efficiency (0.02 toCCCC-CCA 0.1%). A possible explanation for this difference is that, in
contrast to pGPB14, the vector used to select a-amylase-pGPB14 positive transformants contained the SPO2 promoter. A
random selection of 35 potential export-proficient clones isP.T2 presented in Table 2. Restriction analysis showed that in-CCA serts of various length had been cloned. Moreover, the
various inserts rendered the E. coli cells resistant to differentNA se- concentrations of ampicillin (Table 2; pSPB series) or gave'S) with rise to different levels of a-amylase production (results not,ate the shown; pSPA series). These results indicate that a largearound variety of potential export signal coding sequences had been
g amino picked up.*s were a-Amylase activities produced by the recombinant plasmidsollowed in B. subtilis. To study whether the inserts initially selectedr4 DNA in E. coli were also effective in protein secretion in B.
subtilis, a number of recombinant plasmids were used totransform competent B. subtilis cells. The transformants
:ted by were analyzed for the production of ,B-lactamase and a-PL608, amylase in the culture supernatants. The assays of theEcoRI a-amylase activities (Fig. 4 and Table 3) showed that con-,ith the siderable amounts of a-amylase activity were present in thesignal- culture supematants of B. subtilis cells containing theiith the recombinant plasmids. These results strongly supported the
view that export coding regions, effective in both E. coli andm. To B. subtilis, had been cloned. The amounts of a-amylaseexport produced by the various clones differed considerably. It isised to interesting that B. subtilis strains bearing pSPA2, pSPA8,+ was and pSPA12 exhibited a-amylase activities which were sub-HI-PstI stantially higher than those produced by the control strain,nal se- containing the wild-type a-amylase on pGK13. Since allacids,
igestedted di-) trans-mid of(abouttted I-gnal ofLssayedsult intI frag-codingsignal
Bl1. Inphobiculd not11 only
-29 -4 /3-lactamase
Pst I
-34 -21 /3-lactomosePst I
pGPB15
pGPB16
FIG. 3. Construction of pGPB15 and pGPB16. Schematic pre-sentation of the construction of fusions between penP and the genefor 3-lactamase (pGPB16) and the a-amylase and P-lactamase genes(pGPB15). The numbers refer to the amino acid residues in thesignal sequence. Rectangles, Fragment derived from pGK13-amy+;dot-dash line, fragment derived from pRW101; solid line, truncated,B-lactamase gene from pGPBll; +++, charged amino acid resi-dues. For details, see the text.
J. BACTERIOL.
on June 3, 2018 by guesthttp://jb.asm
.org/D
ownloaded from
BACTERIAL SIGNAL SEQUENCE SELECTION VECTORS
TABLE 2. Properties of potential export-proficient clonesobtained in E. coli
Size of insertb Ampicilln d Size of insertPlasmida resistancec Plasmid (b )
pSPB4' 710 10 pSPA2 730pSPB5' 650 20 pSPA8 340pSPB6' 780 10 pSPA9 1,400pSPB8' 600 300 pSPA12 700pSPB9' 360 50 pSPA13 1,200pSPB11' 270 5 pSPA26 260pSPB12' 830 10 pSPA27 <100pSPB13' 650 10 pSPA28 880pSPB16' 780 50 pSPA29 940pSPB19 420 300 pSPA31 50pSPB20' 1,500 300 pSPA32 <100pSPB21' 740 50 pSPA33 100pSPB23 490 300 pSPA34 100pSPB28 280 100 pSPA41 <100pSPB29 300 5 pSPA42 400pSPB30 260 5pSPB31 420 5pSPB32 1,080 300pSPB34 370 10pSPB38 660 20
a The pSPB clones shown lacked the SP02 promoter.b All pSPB clones contain Sau3A inserts. The size of the inserts was
determined by restriction analysis.c The level of resistance to ampicillin was defined as the maximal concen-
tration of ampicillin to which 100%o of the viable cells were resistant.d All pSPA clones shown contain the SP02 promoter.pSPA2, -26, -27, and -33 contain an AluI insert; pSPA8 contains an HaelIl
insert; and pSPA9, -12, -13, -28, -29, -31, -32, -34, -41, and 42 contain an RsaIinsert.
a-amylase-positive transformants were selected with a vec-tor containing the SPO2 promoter, the inserts do not neces-sarily contain promoter activity of their own. Deletion of theSPO2 promoter in pSPA8, pSPA9, pSPA12, pSPA41, andpSPA42 revealed (Table 3) that, indeed, some inserts did
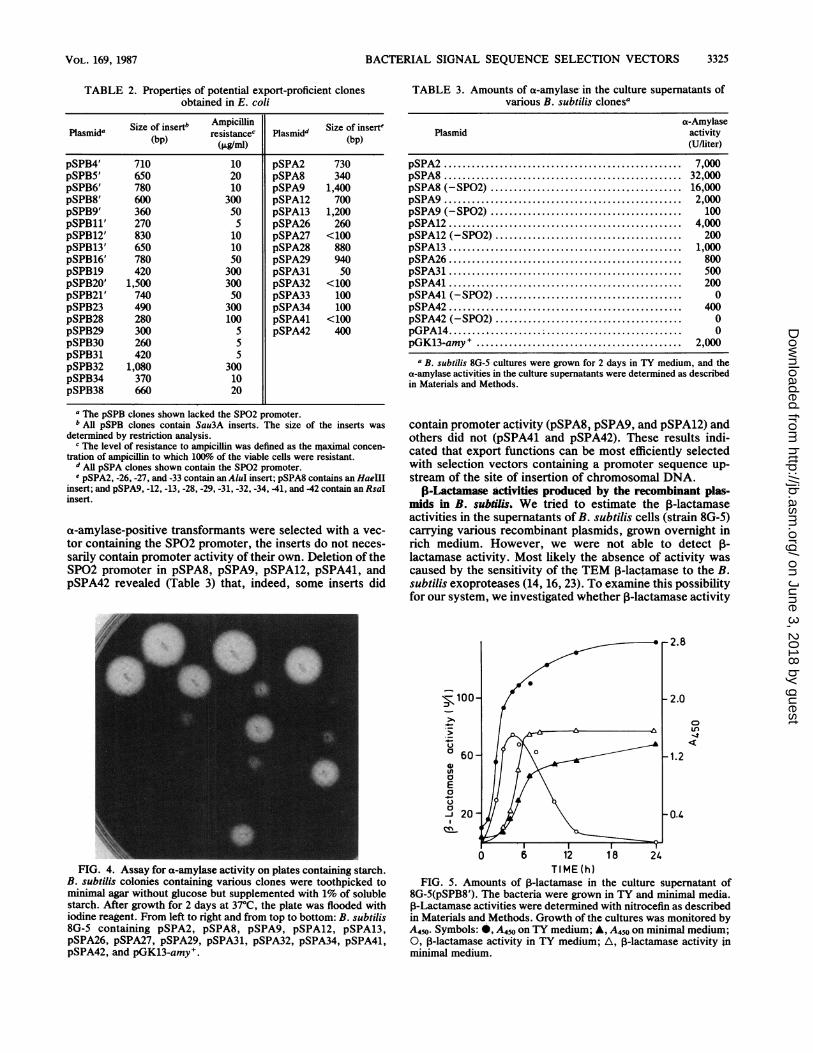
FIG. 4. Assay for a-amylase activity on plates containing starch.B. subtilis colonies containing various clones were toothpicked tominimal agar without glucose but supplemented with 1% of solublestarch. After growth for 2 days at 37°C, the plate was flooded withiodine reagent. From left to right and from top to bottom: B. subtilis8G-5 containing pSPA2, pSPA8, pSPA9, pSPA12, pSPA13,pSPA26, pSPA27, pSPA29, pSPA31, pSPA32, pSPA34, pSPA41,pSPA42, and pGK13-amy+.
TABLE 3. Amounts of ca-amylase in the culture supernatants ofvarious B. subtilis clonesa
a-AmylasePlasmid activity
(U/liter)
pSPA2 ........................................... 7,000pSPA8 ........................................... 32,000pSPA8 (-SP02) ........................................ 16,000pSPA9...... ... ............ 2,000pSPA9 (-SP02) ......................................... 100pSPA12... 4,000pSPA12 (-SP02) ....................................... 200pSPA13 .......................................... 1,000pSPA26... 800pSPA31 .......................................... 500pSPA41... 200pSPA41 (-SP02) ........................................ 0pSPA42 .......................................... 400pSPA42 (-SP02) ........................................ 0pGPA14 .. 0pGK13-amy . .......................................... 2,000
a B. subtilis 8G-5 cultures were grown for 2 days in TY medium, and thea-amylase activities in the culture supernatants were determined as describedin Materials and Methods.
contain promoter activity (pSPA8, pSPA9, and pSPA12) andothers did not (pSPA41 and pSPA42). These results indi-cated that export functions can be most efficiently selectedwith selection vectors containing a promoter sequence up-stream of the site of insertion of chromosomal DNA.
P-Lactamase activities produced by the recombinant plas-mids in B. sublils. We tried to estimate the P-lactamaseactivities in the supernatants of B. subtilis cells (strain 8G-5)carrying various recombinant plasmids, grown overnight inrich medium. However, we were not able to detect 1-lactamase activity. Most likely the absence of activity wascaused by the sensitivity of the TEM 1-lactamase to the B.subtilis exoproteases (14, 16, 23). To examine this possibilityfor our system, we investigated whether ,B-lactamase activity
2.8
-100- / -2.0
U.)u <
a 0 1.28 24
0
a2004
TIME (h)FIG. 5. Amounts of 13-lactamase in the culture supernatant of
8G-5(pSPB8'). The bacteria were grown in TY and minimal media.,B-Lactamase activities were determined with nitrocefin as describedin Materials and Methods. Growth of the cultures was monitored byA450. Symbols: 0, A450 on TY medium; A, A450 on minimal medium;0, 3-lactamase activity in TY medium; A, ,-lactamase activity inminimal medium.
VOL. 169, 1987 3325
on June 3, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

3326 SMITH ET AL.
TABLE 4. a-Amylase and ICDH activities in cell pellets andculture supernatants of two B. subtilis clonesa
cx-Amylase activity ICDH activityPlasmid (U/liter) (U/ml)
Supematant Pellet Supernatant Pellet
pSPA2 100 1 3.9 287pSPA8 83 8 23 122
a Cultures were grown overnight in minimal medium without glucose.Portions (1 ml) were centrifuged for 3 min in an Eppendorf centrifuge. Thecells were washed twice with 100 mM potassium phosphate (pH 8.0). Thepellets were suspended in 0.5 ml of 100 mM potassium phosphate (pH 8.0)plus 200 ,ug of lysozyme per ml and incubated for 15 min at 37°C. The culturesupernatants were filtered (0.45.um pore size) to remove cells and debris.a-Amylase and ICDH activities were determined as described in Materialsand Methods.
would be detectable in a protease-deficient mutant, DB104(8), and whether P-lactamase would be more stable incultures grown in minimnal compared to rich medium. Theresults, obtained with pSPB8', are shown in Fig. 5. In TYmedium, ,-lactamase was only detectable during the loga-rithmic and early stationary phases of growth. However, theamount of ,-lactamase started to decrease after prolongedperiods of growth and was completely lost in overnightcultures. In contrast, the amount of P-lactamase remainedconstant for up to 24 h in minimal medium. These resultssupported the idea that, at least in TY medium, P-lactamasewas unstable. Qualitatively similar results were obtainedwith the protease-deficient strain DB104 (results not shown).The inability to improve the stability of ,B-lactamase isprobably due to the residual levels of protease in this strain(2.6%) (8).
Secretion versus lysis. The results presented in the previ-ous sections strongly suggest that pGPB14 and pGPA14 areefficient vectors to select export-coding regions which func-tion both in E. coli and in B. subtilis. To exclude thepossibility that the enzymatic activities measured in B.subtilis were the result of cell lysis rather than of secretion,we compared the cellular locations of a-amylase, producedby pSPA2 and pSPA8, and a cytoplasmic marker protein, forwhich ICDH was chosenl. First, we tested whether ICDHitself was stable in various media. After 24 h of incubation at37°C in minimal medium and in the supernatants of B.subtilis overnight cultures in minimal medium, 80% of theoriginal ICDH activity was still present. However, in TYmedium ICDH activity was completely lost after 3 to 5 h at37°C. Similar results were obtained with supernatants ofcultures of B. subtilis 8G-5 and DB104. These results indi-cated that ICDH can be used as a cytoplasmic markerprotein in minimal medium. The cellular locations of a-amylase and ICDH in B. subtilis cultures grown in minimalmedium are shown in Table 4. Whereas the a-amylaseactivity was predominantly found in the culture superna-tants, most of the ICDH activity was associated with the cellpellets. These results strongly favor the idea that the a-amylase activities measured in the supernatants were theresult of secretion rather than of lysis. In addition, it isworthwhile to mention that in strain 8G-5(pSPA8) 10 to 20%P/of the total a-anlylase activity was reproducibly found to beassociated with the cell pellet.
Analysis of a-amylase activities in SDS-PAA gels. As shownin Table 4, in strain 8G-5(pSPA8) substantial amounts ofca-amylase activity were associated with the cell pellet. Thisis surprising, since the wild-type precursor is very rapidly
processed to the mature form. To examine the nature of thepellet-associated activity, we analyzed the a-amylase activ-ities in the culture supernatants and cell pellets of strain8G-5(pSPA8) and a number of other clones on SDS-PAAgels (Fig. 6). Purified a-amylase from B. licheniformis wasused as a control to determine the molecular weight positionof the mature a-amylase. The molecular weight positions ofa-amylases isolated from the culture supernatants of strain8G-5(pGK13-amy') and strain 8G-5 carrying either pSPA2,pSPA8, or pSPA9 seemed to be identical to that of thecontrol, suggesting that in all constructs cleavage of theexport signal function had occurred at, or very close to, theoriginal processing site of a-amylase. Interestingly, extractsof cell pellets from strain 8G-5(pSPA2) and 8G-5(pSPA8)showed a-amylase bands at higher molecular-weight posi-tions than that found in the supernatants. Presumably thesebands correspond to the precursor forms of a-amylasesynthesized in the various clones. The precursors in 8G-5(pSPA2) are of higher molecular weight than those in8G-5(pSPA8). Analogous to the wild-type situation, no pre-cursor forms could be detected in the cell pellets of strain8G-5 carrying pSPA9 (Fig. 6, lane 2). This applied also tostrain 8G-5(pSPA12) and strain 8G-5(p$PA13) (results notshown).
DISCUSSION
The present report describes a system that can be used inan efficient way to probe for export signal-coding regionswhich function in E. coli and B. subtilis. Two plasmidvectors containing genes for extracellular proteins, whichlacked the promoter and a functional signal sequence, wereconstructed. Therefore, expression of the genes and secre-tion of their products requires the fusion in the correctreading frame and correct orientation to DNA fragmentscontaining a promoter, a ribosomal binding site, a transla-tional start site, and a signal sequence-coding region.
2 3 4 6 7 8 9 10
FIG. 6. SDS-PAA gel analysis ofthe a-amylase patterns. Culturesupernatants and cell pellets of various B. subtilis clones wereanalyzed. Portions (10 ml) of overnight cultures in TY medium were
centrifuged for 10 min at 6,000 x g. The cells were washed twice bycentrifugation with starvation medium. The pellets were suspendedin 5 ml of starvation medium + 200 gg of lysozyme per ml andincubated for 15 min at 37°C. The culture supernatants were filteredthrough 0.45-p.m-pore-size ifiters (Schleicher and Schuell). Both thepellet and the supernatant fractions were concentrated about 10times by dialysis against dry sucrose. Samples (30 p1) were appliedto 7.5% PAA gels. a-Amylase patterns were, visualized in starch-containing SDS-PAA gels as described in Materials and Methods.Lanes: 1, control, purified a-amylase of B. licheniformis; 2, pSPA9,pellet; 3, pSPA9, supernatant; 4, pSPA8, pellet; 5, pSPA8, super-natant; 6, pSPA2, pellet; 7, pSPA2, supernatant; 8, pGK13-amy+,pellet; 9, pGK13-amy+, supernatant; 10, purified a-amylase. Thevertical arrow indicates the direction of electrophoresis; horizontalarrows indicate the positions of the mature and precursor forms ofa-amylase.
J. BACTERIOL.
on June 3, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

BACTERIAL SIGNAL SEQUENCE SELECTION VECTORS
Using this system, we have selected a great variety ofdifferent export signal-coding regions from chromosomal B.subtilis DNA. Clones in which the export of a-amylase orP-lactamase had been restored were selected in E. coli. Asignificant advantage of using this organism as initial hostwas that, in contrast to B. subtilis, E. coli cells producingand exporting f-lactamase are resistant to ampicillin and,therefore, can be positively selected. Since cytoplasmicP-lactamase cannot protect a cell against ampicillin, andsince the catalytic activity of pre-p-lactainase is very low (6,18, 22), the selection of Apr transformants in E. coli willrequire both the correct translocation and processing ofpre-i-lactamase.The bifunctional B. subtilis-E. coli character of the present
system allowed us to examine whether the export-restoringclones selected in E. coli also restored protein export in B.subtilis. The results indicated that this was the case with allclones selected with both the a-amylase and the 3-lactamaseprobe vector. Recently, an analogous system for the selec-tion of export signals has been described (4) which, how-ever, can only be used in E. coli.
Restriction analysis of approximately 35 randomly chosenexport-proficient recombinant clones indicated that thesecontained inserts of different size. Moreover, the variousclones showed different levels of a-amylase or 1-lactamaseactivities. This indicates that a considerable variety of dif-ferent export-restoring DNA inserts had been picked up.Recent DNA sequence data (Smnith et al., manuscript inpreparation) have provided further proof for this idea. Aninteresting question was whether the selected inserts resem-bled typical signal sequences for exported proteins. Onecriterion for this is that the initial translation products of thefusions should be of increased size. The results showed thatthis was indeed the case for two clones: in cell pellets ofstrains 8G-5(pSPA2) and 8G-5(pSPA8), a-amylase activitiescould be detected at considerably higher molecular-weightpositions than in the supernatants. Since these activitieswere found at the molecular-weight position of maturewild-type a-amylase, the results also indicate that processingof fused precursor proteins had occurred at or close to theoriginal cleavage site. Also, in this respect the properties ofthe cloned sequences in pSPA2 and pSPA8 are similar tothose of typical signal sequences.The pSPA2 and pSPA8 clones revealed several additional
properties of interest. First, strains 8G-5(pSPA8) and 8G-5(pSPA2) produced substantially higher a-amylase activitiesthan the control strain containing the plasmid with thewild-type a-amylase gene. At present it is not clear whetherthis increase is caused by differences at the transcriptional ortranslational level or by altered efficiencies of secretion ofthe enzymes.
Second, the precursor bands could be detected immuno-logically (data not shown) as well as enzymatically with thesame intensities as the mature products. This shows thatthese precursor forms of the B. licheniformis a-amylase areenzymatically active and that the specific activities of theprecursors and mature products are similar. In fact, twoprecursor bands were observed with pSPA2 and pSPA8.One possible explanation for this is that two alternativetranscription/translation start points were present on theinserts.
Third, the observation that, in strains 8G-5(pSPA2) and8G-5(pSPA8) under steady-state conditions, considerableamounts of precursor could be detected is noteworthy, sinceprecursor forms could not be detected with wild-type a-amylase. This suggests that the rate of secretion or process-
ing of a-amylases in these fusions was delayed compared tothe wild type.The large number of different export-proficient clones
obtained in these studies deserves further consideration.Expression of the a-amylase and ,-lactamase genes andsecretion of their products required fusion to DNA frag-ments containing a promoter, ribosomal binding site, trans-lational start site, and a signal sequence-coding region,which we assume to correspond to approximately 30 aminoacids. Altogether we estimate this region to amount to about175 bp. Since DNA inserts were obtained with restrictionenzymes recognizing 4-bp nucleotide sequences, and theaverage gene can be estimated to consist of about 1,000 bp,the probability that an inserted sequence would contain astretch of at least 175 bp of DNA containing the transcrip-tion/translation signals and a coding region for 30 aminoacids would be 256/1,000 x (256-175)/256, which amountsto 0.081. Moreover, only one third of the inserted sequencesare expected to be in the correct reading frame, and one halfare expected to be in the correct orientation. Furthermore,since religation of the vector was not prevented, we esti-mated that about one third of the transformants contained aninsert. Therefore, about 0.081 x 1/3 x 1/2 x 1/3, or 0.45% ofall transformants will be expected to carry a fusion withappropriate transcription/translation signals and a 30-amino-acid coding region in the correct reading frame and orienta-tion. Based on the frequencies of export signal-positiveclones among the total number of transformants which weselected with the P-lactamase probe vector (0.02 to 0.1%),this would mean that approximately 4 to 15% of the clonedsequences, expected to be in frame and to carry the requiredstretch of 175 bp for transcription/translation, would func-tion as an export signal. This seems an exceptionally highfrequency, particularly since in B. subtilis only about 20 to25 different exported proteins seem to be present (17). Onthe assumption that the total number of B. subtilis genes willbe approximately 3,000, this would mean that only about0.83% of the B. subtilis genes code for secreted proteins.Therefore, our results seem to indicate that, in addition tonatural signal sequences, we have selected a remarkablyhigh frequency of more or less random sequences which canfunction as export signals. Very interestingly, similar con-clusions were recently drawn by Kaiser et al. (7) fromstudies on a eucaryotic system. These authors showed thatin Saccharomyces cerevisiae 20% of random DNA se-quences cloned in frame were functional as export signal. Itthus seems likely that the specificity with which signalsequences are recognized by both eucaryotic and procary-otic systems is low.Sequence analysis of the various export signals selected in
the present studies (specifying high as well as low levels ofenzyme production and secretion) may provide valuableinformation with respect to features essential for efficientprotein export in gram-positive bacteria. Future experimentswill be focused on this purpose.
ACKNOWLEDGMENTS
We thank Anne de Jong for excellent technical assistance andHenk Mulder for preparing the photographs.Funding for the project of which this work is a part was provided
by Gist Brocades N.V., Delft, the Netherlands.
LITERATURE CITED
1. Bron, S., and E. Luxen. 1985. Segregational instability ofpUB1l1-derived recombinant plasmids in Bacillus subtilis. Plas-
3327V6L. 169, 1087
on June 3, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

3328 SMITH ET AL.
mid 14:235-244.2. Bron, S., and G. Venema. 1972. Ultraviolet inactivation and
excision repair in Bacillus subtilis. I. Construction and charac-terization of a transformable eightfold-auxotrophic strain andtwo ultraviolet-sensitive derivatives. Mutat. Res. 15:1-10.
3. Gbrayeb, J., H. Kimura, M. Takahara, H. Hsiung, Y. Masui,and M. Inouye. 1984. Secretion cloning vectors in Escherichiacoli. EMBO J. 3:2437-2442.
4. Hoffman, C. S., and A. Wright. 1985. Fusions of secretedproteins to alkaline phosphatase: an approach for studyingprotein secretion. Proc. Natl. Acad. Sci. USA 82:5107-5111.
5. Ish-Horowicz, D., and J. F. Burke. 1981. Rapid and efficientcosmid cloning. Nucleic Acids Res. 9:2989-2998.
6. Kadonaga, J. T., A. E. Gautier, D. R. Straus, A. D. Charles,M. D. Edge, and J. R. Knowles. 1984. The role of the ,B-lactamase signal sequence in the secretion of proteins byEscherichia coli. J. Biol. Chem. 259:2149-2154.
7. Kaiser, C. A., D. Preuss, P. Grisafl, and D. Botstein. 1987. Manyrandom sequences functionally replace the secretion signalsequence of yeast invertase. Science 235:312-317.
8. Kawamura, F., and R. H. Doi. 1984. Construction of a Bacillussubtilis double mutant deficient in extracellular alkaline andneutral proteases. J. $acteriol. 160:442-444.
9. Kok, J., J. M. B. M. van der Vossen, and G. Venema. 1984.Construction of plasmid cloning vectors for lactic streptococciwhich also replicate in Bacillus subtilis and Escherichia coli.Appl. Environ. Microbiol. 48:726-731.
10. Laemmli, U. K. 1970. Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature (London)227:680-685.
11. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
12. Messing, J. 1983. New M13 vectors for cloning. MethodsEnzymol. 101:20-78.
13. Mezes, P. S. F., W. Wang, E. C. H. Yeh, and J. 0. Lampen.1983. Construction of penPAI, Bacillus licheniformis 749/CP-lactamase lacking site for lipoprotein modification. J. Biol.Chem. 258:11211-11218.
14. Nakamura, K., T. Furusato, T. Shiroza, and K. Yamane. 1985.Stable hyper-production of Escherichia coli P-lactamase byBacillus subtilis grown on a 0.5M succinate-medium using a B.subtilis ax-amylase secretion vector. Biochim. Biophys. Res.Commun. 128:601-06.
15. Ohmura, K., T. Shiroza, K. Nakamura, A. Nakayama, K.Yamane, K. Yoda, M. Yamasaki, and G. Tamura. 1984. ABacillus subtilis secretion vector system derived from the B.subtilis a-amylase promoter and signal sequence region, andsecretion of Escherichia coli P-lactamase by the vector system.J. Biochem. 95:87-93.
16. Palva, I., M. Sarvas, P. Lehtovaara, M. Sibakov, and L.Kaaririinen. 1982. Secretion of Escherichia coli ,B-lactamasefrom Bacillus subtilis by the aid of a-amylase signal sequence.Proc. Natl. Acad. Sci. USA 79:5582-5586.
17. Priest, F. G. 1977. Extracellular enzyme synthesis in the genusBacillus. Bacteriol. Rev. 41:711-753.
18. Roggenkamp, R., H. Dargatz, and C. P. Hollenberg. 1985.Precursor of p-lactamase is enzymatically inactive. J. Biol.Chem. 260:1508-1512.
19. Silhavy, T. J., S. A. Benson, and S. D. Emr. 1983. Mechanismsof protein localization. Microbiol. Rev. 47:313-344.
20. Stahl, M. L., and E. Ferrari. 1984. Replacement of the Bacillussubtilis subtilisin structural gene with an in vitro-derived dele-tion mutation. J. Bacteriol. 158:411-418.
21. Sutcliff, J. G. 1978. Complete nucleotide sequence of theEscherichia coli plasmid pBR322. Cold Spring Harbor Symp.Quant. Biol. 43:77-90.
22. Tai, P. C., N. Zyk, and N. Citri. 1985. In situ detection ofP-lactamase activity in sodium dodecyl sulfate-polyacrylamidegels. Anal. Biochem. 144:199-203.
23. Ulmanen, I., K. Lundstrom, P. Lehtovaara, M. Sarvas, M.Ruohonen, and I. Palva. 1985. Transcription and translation offoreign genes in Bacillus subtilis by the aid of a secretion vector.J. Bacteriol. 162:176-182.
24. Uozumi, T., A. Ozaki, T. Beppu, and K. Arima. 1980. Newcryptic plasmid of Bacillus subtilis and restriction analysis ofother plasmids found by general screening. J. Bacteriol.142:315-318.
25. Vasantha, N., and L. D. Thompson. 1986. Secretion of aheterologous protein from Bacillus subtilis with the aid ofprotease signal sequences. J. Bacteriol. 165:837-842.
26. Watson, M. E. E. 1984. Compilation of published signal se-quences. Nucleic Acids Res. 12:5145-5164.
27. Williams, D. M., E. J. Duvall, and P.S. Lovett. 1981. Cloningrestriction fragments that promote expression of a gene inBacillus subtilis. J. Bacteriol. 146:1162-1165.
28. Yang, M. Y., E. Ferrari, and D. J. Henner. 1984. Cloning of theneutral protease gene of Bacillus subtilis and the use of thecloned gene to create an in vitro-derived deletion mutation. J.Bacteriol. 160:15-21.
29. Yuki, S. 1967. Genetic studies on amylase of different electro-phoretic mobility produced by strains of Bacillus subtilis. J.Genet. 42:251-261.
30. Yuuki, T., T. Nomura, H. Tezuka, A. Tsuboi, H. Yamagatal N.Tsukagoshi, and S. Udaka. 1985. Complete nucleotide sequenceof a gene coding for heat- and pH-stable a-amylase of Bacilluslicheniformis: comparison of the amino acid sequence of threebacterial liquefying a-amylases deduced from the DNA se-quence. J. Biochem. 98:1147-1156.
J. BACTERIOL.
on June 3, 2018 by guesthttp://jb.asm
.org/D
ownloaded from





























