Embed Size (px)
Citation preview

Congenital myasthenic syndromesDaniel Hantaı a, Pascale Richardb, Jeanine Koeniga and Bruno Eymarda
Purpose of review
Congenital myasthenic syndromes are a heterogeneous group
of diseases caused by genetic defects affecting neuromuscular
transmission. In this article, a strategy that leads to the
diagnosis of congenital myasthenic syndromes is presented,
and recent advances in the clinical, genetic and molecular
aspects of congenital myasthenic syndrome are outlined.
Recent findings
Besides the identification of new mutations in genes already
known to be implicated in congenital myasthenic syndromes
(genes for the acetylcholine receptor subunits and the collagen
tail of acetylcholinesterase), mutations in other genes have more
recently been discovered and characterized (genes for choline
acetyltransferase, rapsyn, and the muscle sodium channel
SCN4A). Fluoxetine has recently been proposed as an
alternative treatment for ‘slow channel’ congenital myasthenic
syndrome.
Summary
The characterization of congenital myasthenic syndromes
comprises two complementary steps: establishing the diagnosis
and identifying the pathophysiological type of congenital
myasthenic syndrome. Characterization of the type of congenital
myasthenic syndrome has allowed it to be classified as caused
by presynaptic, synaptic and postsynaptic defects. A clinically
and muscle histopathologically oriented genetic study has
identified several genes in which mutations cause the disease.
Despite comprehensive characterization, the phenotypic
expression of one given gene involved is variable, and the
aetiology of many congenital myasthenic syndromes remains to
be discovered.
Keywords
electromyography, genetic diagnosis, microelectrophysiology,
neuromuscular junction molecules, neuromuscular transmission,
treatment
Curr Opin Neurol 17:539–551. # 2004 Lippincott Williams & Wilkins.
aInserm U582 and Unite Clinique de Pathologie Neuromusculaire, Institut deMyologie, and bUnite Fonctionnelle de Cardiogenetique et Myogenetique, Hopital dela Salpetriere, Paris, France
Correspondence to Daniel Hantaı, Inserm U582, Institut de Myologie, Hopital de laSalpetriere, 47 Boulevard de l’Hopital, 75651 Paris Cedex 13, FranceTel: +33 1 42165706; fax: +33 1 42165700;e-mail: [email protected]
Current Opinion in Neurology 2004, 17:539–551
Abbreviations
ChAT choline acetyltransferaseCMAP compound muscle action potentialCMS congenital myasthenic syndrome
# 2004 Lippincott Williams & Wilkins1350-7540
IntroductionCongenital myasthenic syndromes (CMSs) form a
heterogeneous group of genetic diseases characterized
by a dysfunction of neuromuscular transmission. This
dysfunction causes muscle weakness, which is increased
by exertion and usually starts during childhood. The
prevalence of CMS is estimated at one in 500 000 in
Europe, and CMSs are much more uncommon than
autoimmune myasthenia [1].
Knowledge of the mechanisms underlying CMS has
increased considerably in the past 25 years, because of
the pioneering work undertaken by the group of Engel etal. [2]. Acetylcholinesterase deficiency was the first CMS
identified, based on the lack of the enzyme at
neuromuscular junctions [2]. Progressively, the patho-
physiological heterogeneity of CMS was demonstrated:
besides synaptic CMS caused by acetylcholinesterase
deficiency, pre- and postsynaptic CMS were described,
the latter including quantitative deficiency or kinetic
anomalies of the acetylcholine receptor. In the past 15
years, many gene mutations responsible for CMS were
identified, affecting the different acetylcholine receptor
subunits and the collagenic tail of acetylcholinesterase
[3,4 ..]. Mutations in the genes for choline acetyltrans-
ferase (ChAT) [5], rapsyn [6], and more recently the
sodium channel SCN4A have been reported to cause
CMS [7.].
Several reviews have been devoted to CMS, one of the
more recent being that of Engel et al. [8 ..]. The
objectives of the present review are to highlight the
principal phenotypical and pathophysiological character-
istics of CMS, to pinpoint the more recent advances in
the field, and to propose a strategy for the accurate
characterization of these disorders.
Classification of congenital myasthenicsyndromes and recent findingsThe current classification of CMS is based on patho-
physiology, i.e. on the precise identification of the
neuromuscular transmission anomaly. The location of
the dysfunction of neuromuscular transmission (Fig. 1)
[9], which is specific to the different CMSs, is either
presynaptic (generally caused by an anomaly of ChAT),
synaptic (corresponding to an anomaly of the acetylcho-
linesterase collagen tail), or postsynaptic (secondary to an
anomaly of acetylcholine receptor or rapsyn). In the
experience of Engel’s group, postsynaptic CMSs are
three times more frequent than acetylcholinesterase
deficiency and 10 times more frequent than presynaptic
539
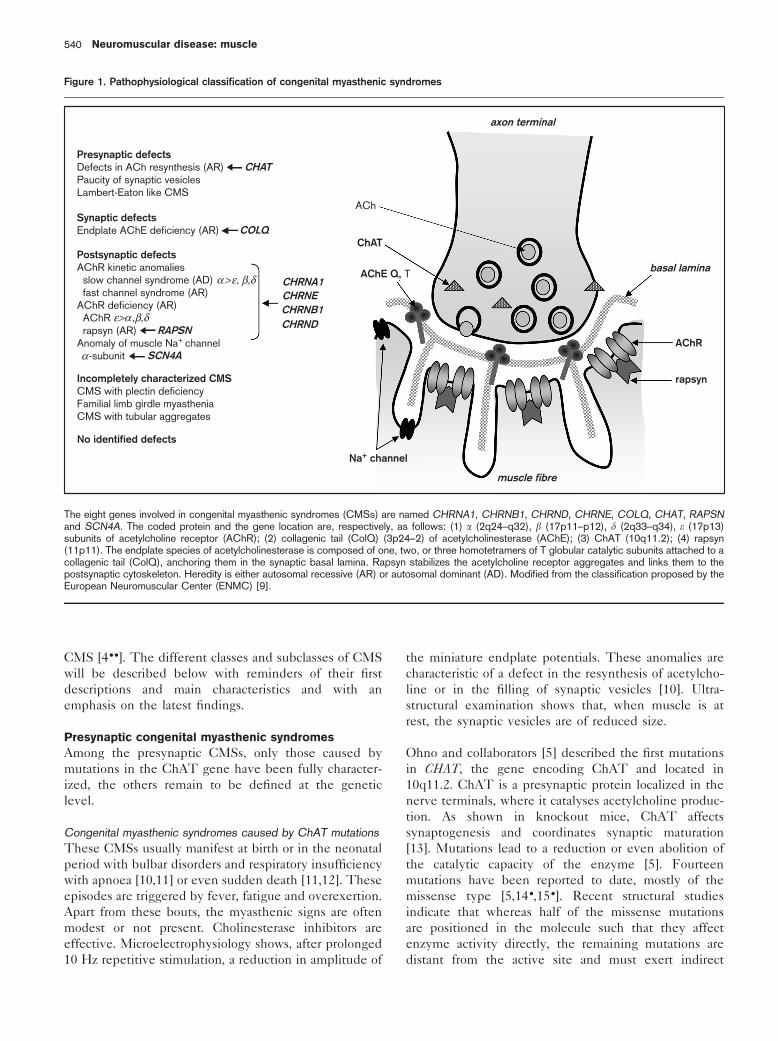
CMS [4..]. The different classes and subclasses of CMS
will be described below with reminders of their first
descriptions and main characteristics and with an
emphasis on the latest findings.
Presynaptic congenital myasthenic syndromes
Among the presynaptic CMSs, only those caused by
mutations in the ChAT gene have been fully character-
ized, the others remain to be defined at the genetic
level.
Congenital myasthenic syndromes caused by ChAT mutations
These CMSs usually manifest at birth or in the neonatal
period with bulbar disorders and respiratory insufficiency
with apnoea [10,11] or even sudden death [11,12]. These
episodes are triggered by fever, fatigue and overexertion.
Apart from these bouts, the myasthenic signs are often
modest or not present. Cholinesterase inhibitors are
effective. Microelectrophysiology shows, after prolonged
10 Hz repetitive stimulation, a reduction in amplitude of
the miniature endplate potentials. These anomalies are
characteristic of a defect in the resynthesis of acetylcho-
line or in the filling of synaptic vesicles [10]. Ultra-
structural examination shows that, when muscle is at
rest, the synaptic vesicles are of reduced size.
Ohno and collaborators [5] described the first mutations
in CHAT, the gene encoding ChAT and located in
10q11.2. ChAT is a presynaptic protein localized in the
nerve terminals, where it catalyses acetylcholine produc-
tion. As shown in knockout mice, ChAT affects
synaptogenesis and coordinates synaptic maturation
[13]. Mutations lead to a reduction or even abolition of
the catalytic capacity of the enzyme [5]. Fourteen
mutations have been reported to date, mostly of the
missense type [5,14 .,15.]. Recent structural studies
indicate that whereas half of the missense mutations
are positioned in the molecule such that they affect
enzyme activity directly, the remaining mutations are
distant from the active site and must exert indirect
Figure 1. Pathophysiological classification of congenital myasthenic syndromes
axon terminal
basal lamina
AChR
rapsyn
muscle fibre
Na+ channel
ACh
ChAT
AChE Q, T
Presynaptic defectsDefects in ACh resynthesis (AR) CHATPaucity of synaptic vesiclesLambert-Eaton like CMS
Synaptic defectsEndplate AChE deficiency (AR) COLQCOLQ
CHAT
Postsynaptic defectsAChR kinetic anomalies slow channel syndrome (AD) α>ε, β.δ fast channel syndrome (AR)AChR deficiency (AR) AChR α>ε, β.δ rapsyn (AR) RAPSNAnomaly of muscle Na+ channel α-subunit SCN4A
RAPSN
SCN4A
α > ε, β,δ
ε>α ,β,δ
Incompletely characterized CMSCMS with plectin deficiencyFamilial limb girdle myastheniaCMS with tubular aggregates
No identified defects
CHRNB1CHRND
CHRNA1CHRNE
α
The eight genes involved in congenital myasthenic syndromes (CMSs) are named CHRNA1, CHRNB1, CHRND, CHRNE, COLQ, CHAT, RAPSNand SCN4A. The coded protein and the gene location are, respectively, as follows: (1) a (2q24–q32), b (17p11–p12), d (2q33–q34), e (17p13)subunits of acetylcholine receptor (AChR); (2) collagenic tail (ColQ) (3p24–2) of acetylcholinesterase (AChE); (3) ChAT (10q11.2); (4) rapsyn(11p11). The endplate species of acetylcholinesterase is composed of one, two, or three homotetramers of T globular catalytic subunits attached to acollagenic tail (ColQ), anchoring them in the synaptic basal lamina. Rapsyn stabilizes the acetylcholine receptor aggregates and links them to thepostsynaptic cytoskeleton. Heredity is either autosomal recessive (AR) or autosomal dominant (AD). Modified from the classification proposed by theEuropean Neuromuscular Center (ENMC) [9].
Neuromuscular disease: muscle540

effects [16 ..]. The possibility that the I336T ChAT
mutation found in three consanguineous Turkish
families was a founder was postulated [14 .].
Other presynaptic myasthenic syndromes still incompletely
characterized
The paucity of synaptic vesicles was described in one
patient with early-onset CMS. The density of acetylcho-
line synaptic vesicles was reduced by 80% and the
number of quanta released was drastically reduced [17].
The exact cause of this CMS is still unknown.
The first case of Lambert–Eaton-like CMS was first
reported in a child [18]. His myasthenic syndrome was
characterized by a good response to guanidine and by
electrophysiological anomalies identical to those of a
Lambert–Eaton syndrome, i.e. diminished action poten-
tials markedly potentiated by tetanic stimulation. A
second case presented with severe hypotonia and
respiratory distress at birth [4..]. No mutation was found
in the gene coding for the presynaptic calcium channel.
Three patients were reported with a sporadic myasthenic
syndrome with associated signs of attack of the central
nervous system (cerebellar ataxia or nystagmus) [19].
None presented, as in the Lambert–Eaton syndrome,
with a reduction of the action potentials or with
potentiation after high frequency stimulation. Microelec-
trophysiology revealed a marked reduction in the
spontaneous or nerve stimulation-induced release of
acetylcholine quanta.
Synaptic congenital myasthenic syndrome:
acetylcholinesterase deficiency
CMSs caused by acetylcholinesterase deficiency were
first described in 1977 [2]. Since then, many cases of
partial or complete deficiency of the enzyme located in
the synaptic basal lamina have been reported [20]. The
first symptoms usually arise in the neonatal period, and
the symptoms are severe with a significant lethal risk.
However, the disease may start later, during infancy, and
is not so severe. Several observations point to the
diagnosis of acetylcholinesterase deficiency: autosomal
recessive heredity, repetitive compound muscle action
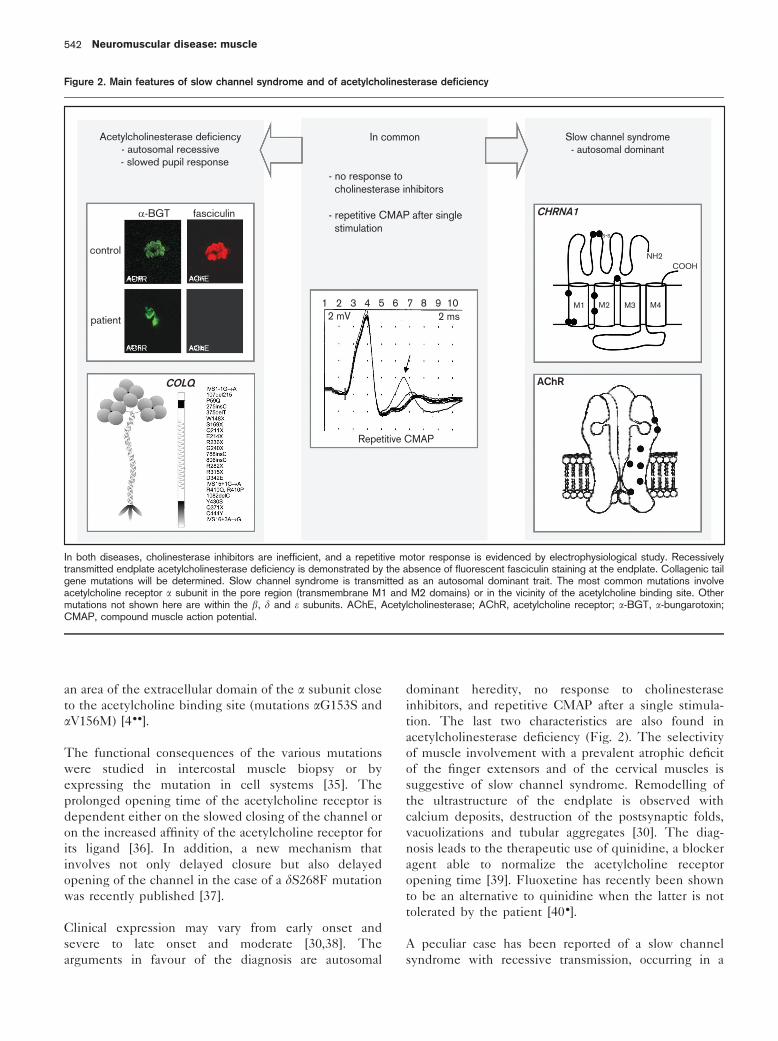
potential (CMAP) after single stimulation (Fig. 2), the
absence of response to cholinesterase inhibitors, and
slowed [20] but inconstant [21] pupil responses to light.
Diagnosis using muscle biopsies is indicated by no or
poor visualization of acetylcholinesterase at the neuro-
muscular junction. Acetylcholinesterase deficiency is
related to mutations in the COLQ gene coding for the
collagenic tail of acetylcholinesterase [22–24]. At the
neuromuscular synapse, acetylcholinesterase is present
as asymmetric acetylcholinesterase, which is made up of
three homotetramers each comprising four globular
catalytic subunits linked together by a collagenic tail
(ColQ; Q for ‘queue’ in French, which means ‘tail’) of
trimeric helicoidal structure. The collagenic tail concen-
trates and anchors the enzyme within the synaptic basal
lamina.
Twenty-four recessive mutations have been described to
date (Fig. 2). They are more often homozygous than
heterozygous, and nonsense than missense [4 ..]. The
fact that the same homozygous G240X mutation was
detected in several Arab families and in one Iraqi Jewish
patient suggests that it is not uncommon in Near and
Middle East countries [25]. Depending on their localiza-
tion, COLQ mutations have different consequences: in
the N-terminal proline-rich attachment domain, they
prevent attachment of the acetylcholinesterase catalytic
subunits to the collagenic tail; in the mid-part they
prevent the trimerization of the collagenic tail; in the C-
terminal domain they most often impair anchoring of the
enzyme within the synaptic basal lamina [24,26,27.,28.].
This impaired attachment of the collagenic tail to the
synaptic basal lamina has recently been elegantly and
comprehensively tested using purified C-terminal do-
main mutant ColQ applied to frog neuromuscular
junctions [29 ..].
To date there is no effective treatment for this type of
CMS.
Postsynaptic congenital myasthenic syndromes
Among these postsynaptic CMSs, two categories are
described: CMS in connection with a kinetic anomaly of
the acetylcholine receptor; and CMS with a decreased
number of acetylcholine receptors at the neuromuscular
junction. In the latter category, besides the CMSs with
acetylcholine receptor deficiency as a result of numerous
mutations in the different acetylcholine receptor subunit
genes, those caused by mutations in the rapsyn gene
were identified recently [6] and are far from infrequent.
Congenital myasthenic syndrome caused by acetylcholine
receptor kinetic anomalies
Slow channel syndrome is the most frequent kinetic
anomaly of the acetylcholine receptor. This entity, of
autosomal dominant inheritance, is characterized by a
prolonged opening time of the acetylcholine receptor
[30]. Fifteen autosomal dominant missense point muta-
tions causing a gain of function of the acetylcholine
receptor were identified [4..]. Although most of the
mutations were found in the acetylcholine receptor asubunit [31], other subunits are also concerned [32]. The
mutations are located in two transmembrane domains
taking part in the formation of the acetylcholine receptor
pore through which passes the sodium flux [33], M1 for
mutations of the a and b subunits and M2 for those,
more frequent, affecting the a, b, d and e subunits [34];
Congenital myasthenic syndromes Hantaı et al. 541
an area of the extracellular domain of the a subunit close
to the acetylcholine binding site (mutations aG153S and
aV156M) [4 ..].
The functional consequences of the various mutations
were studied in intercostal muscle biopsy or by
expressing the mutation in cell systems [35]. The
prolonged opening time of the acetylcholine receptor is
dependent either on the slowed closing of the channel or
on the increased affinity of the acetylcholine receptor for
its ligand [36]. In addition, a new mechanism that
involves not only delayed closure but also delayed
opening of the channel in the case of a dS268F mutation
was recently published [37].
Clinical expression may vary from early onset and
severe to late onset and moderate [30,38]. The
arguments in favour of the diagnosis are autosomal
dominant heredity, no response to cholinesterase
inhibitors, and repetitive CMAP after a single stimula-
tion. The last two characteristics are also found in
acetylcholinesterase deficiency (Fig. 2). The selectivity
of muscle involvement with a prevalent atrophic deficit
of the finger extensors and of the cervical muscles is
suggestive of slow channel syndrome. Remodelling of
the ultrastructure of the endplate is observed with
calcium deposits, destruction of the postsynaptic folds,
vacuolizations and tubular aggregates [30]. The diag-
nosis leads to the therapeutic use of quinidine, a blocker
agent able to normalize the acetylcholine receptor
opening time [39]. Fluoxetine has recently been shown
to be an alternative to quinidine when the latter is not
tolerated by the patient [40 .].
A peculiar case has been reported of a slow channel
syndrome with recessive transmission, occurring in a
Figure 2. Main features of slow channel syndrome and of acetylcholinesterase deficiency
Acetylcholinesterase deficiency- autosomal recessive
- slowed pupil response
α-BGT fasciculin
control
patient
AChR AChE
AChR AChE
COLQ
In common
- no response to cholinesterase inhibitors
- repetitive CMAP after single stimulation
1 2 3 4 5 6 7 8 9 102 mV 2 ms
Repetitive CMAP
Slow channel syndrome- autosomal dominant
CHRNA1
AChR
M1 M2 M3 M4
s-s
NH2COOH
In both diseases, cholinesterase inhibitors are inefficient, and a repetitive motor response is evidenced by electrophysiological study. Recessivelytransmitted endplate acetylcholinesterase deficiency is demonstrated by the absence of fluorescent fasciculin staining at the endplate. Collagenic tailgene mutations will be determined. Slow channel syndrome is transmitted as an autosomal dominant trait. The most common mutations involveacetylcholine receptor a subunit in the pore region (transmembrane M1 and M2 domains) or in the vicinity of the acetylcholine binding site. Othermutations not shown here are within the b, d and e subunits. AChE, Acetylcholinesterase; AChR, acetylcholine receptor; a-BGT, a-bungarotoxin;CMAP, compound muscle action potential.
Neuromuscular disease: muscle542

consanguineous family in connection with a homozygous
mutation of the e subunit (eL78P) located in the
extramembrane region. This mutation was pathogenic
only if present on two alleles [41]. In addition, a slow
channel syndrome associated with a chromosomal
translocation 2q31–9p27 was described [42].
Fast channel syndromes are of autosomal recessive
transmission, although a case of autosomal dominant
transmission was reported recently [43.]. The diagnosis
is made by microelectrophysiology showing a shortening
of the acetylcholine receptor opening time [44]. Clinical
severity is variable. Arthrogryposis was reported in one
case [45]. The patients are responsive to the combina-
tion of 3,4-diaminopyridine and cholinesterase inhibitors.
Eight mutations were identified affecting a, d and esubunits and are located either in the extracellular
domain, in the M3 transmembrane domain (mutation
aV285I), or in the cytoplasmic loop between the M3 and
M4 domains (e mutations only) [8 ..]. Of the two
mutations present in the patient, one is a nonsense
mutation whereas the other is responsible for the kinetic
anomalies. This second mutation can modify the kinetics
of the receptor by various mechanisms that can be
determined on intercostal biopsy or after in-vitro
expression in cell models. A recent article thus details
the detrimental effects of a V132L mutation located in
the acetylcholine receptor a subunit within the signature
cystine loop on acetylcholine binding and channel gating
[46 ..]. The different mechanisms underlying fast chan-
nel syndromes are the topic of a recent review [47.].
Congenital myasthenic syndromes with predominant
acetylcholine receptor deficiency (with absent or only slight
kinetic anomalies)
These account for approximately half of CMS patients
[4..]. The majority are related to mutations of the
acetylcholine receptor. No peculiar clinical findings point
to this type of autosomal recessive CMS, whose severity
is variable. Nevertheless a founder effect in the Gypsy
population of the e1267delG mutation has been pro-
posed [48]. An extensive study on five disease loci in the
different Gypsy groups has demonstrated a strong
founder effect and a carrier rate of 3.74% for this
mutation [49].
Cholinesterase inhibitors are most often active and 3,4-
diaminopyridine can provide additional benefit.
The described mutations are numerous (60 or more),
either homozygous or heterozygous [4 ..]. They are of all
types: missense mutations, chromosomal deletions,
insertions, deletions. The mutations are located on the
whole gene encoding the acetylcholine receptor esubunit, most being located in the extracellular domain
and in the cytoplasmic loop between the M3 and M4
transmembrane domains [4 ..]. Recently, a chromosomal
microdeletion was identified for the first time in CHRNE[50], showing that this type of mutation may be missed
by standard screening techniques. Lately a frameshift
mutation in exon 7 of CHRNE (e553del7) was shown to
provoke skipping of the preceding exon both in muscle
tissue and when expressed in COS cells [51 .].
Mutations in the promoter were also described [52,53].
Interestingly, the injection of the corresponding recom-
binant in the rat allowed the authors to demonstrate that
a mutation in an N-box of the CHRNE promoter leads to
less acetylcholine receptor synaptic expression [50].
Another experimental approach, namely the cell expres-
sion of green fluorescent tagged acetylcholine receptor,
allowed others to show that mutations affecting cysteine
470 of the e subunit prevent acetylcholine receptor
surface expression [54].
More rarely, other subunits of acetylcholine receptor a, band d subunits are implicated [4..]. The preponderance
of mutations of the e subunit may be caused by the
possibility of the re-expression of the g fetal acetylcho-
line receptor isoform in the case of null mutations of
CHRNE [55,56].
Curiously, CMS not only affects humans but also South
African Red Brahman calves. These calves suspected of
myasthenia have been shown to bear a homozygous 20
basepair deletion mutation in bovine CHRNE. This
mutation leads to a non-functional allele and a severe
phenotype [57,58.].
Congenital myasthenic syndromes with mutations of the rapsyn
gene (RAPSN)
These were first identified in 2002 [6]. Rapsyn is a
43 000 Mr postsynaptic cytoplasmic protein, which
participates in acetylcholine receptor assembly at the
neuromuscular junction [59] and allows its anchoring to
the cytoskeleton by b-dystroglycan among other mole-
cules [60]. Most mutations of this gene located in 11p11
were identified in the tetratricopeptide repeat domain,
and cell expression studies revealed that the co-
expression of mutant rapsyn and acetylcholine receptor
subunits impair the recruitment of acetylcholine recep-
tors to rapsyn clusters, an essential step for the anchoring
of the acetylcholine receptor to the cytoskeleton [6].
These mutations are responsible for a reduction of
rapsyn and consequently of the acetylcholine receptor
itself at the neuromuscular junction. The reduction in
rapsyn expression is not specific, because it is also
observed in primary acetylcholine receptor deficiencies.
The inheritance of this CMS is autosomal recessive.
Since the first four cases were published, nearly 50 other
cases have been reported [61–63,64.,65,66 .,67.,68]. Half
Congenital myasthenic syndromes Hantaı et al. 543

of them bear the homozygous N88K. The other half
bears N88K on one allele and a second mutation on the
other allele. This second mutation is localized all along
the rapsyn molecule, and nearly 20 different mutations
have been identified to date (Fig. 3a) [6]. Missense
mutations predominate (approximately two-thirds of
cases). When the second mutation is not identified by
direct sequencing, the search for a chromosomal micro-
deletion of RAPSN is recommended [69].
Two E-box mutations were identified in the rapsyn
promoter [64.] (Fig. 3b). Seven of the eight patients
reported originated from the Jewish population of Iraq
and Iran and had already been described for their
peculiar clinical phenotype: benign CMS with facial
malformations (mandibular prognathism, elongated face)
[70].
A founder effect of the frequently identified N88K
mutation is likely at least in the European or Indo-
European population [61,62,71..], although the exis-
tence of other founders has been proposed [72]. The
high frequency of the N88K mutation may lead to cases
of pseudo-dominant inheritance.
Genotype–phenotype correlation is not easy. Analysis
of the corpus of clinical observations confirms the
existence of two phenotypes: (1) a neonatal form,
even antenatal (with arthrogryposis multiplex congeni-
ta), with major respiratory disorders and severe
progression of the disease; and (2) mild forms
beginning during childhood or in adulthood. On the
basis of 16 cases, a distinction between early and late
onset was proposed [66.]. The importance of the
identification of the late-onset cases is to avoid
improper immunotherapy. Patients with the rapsyn
mutation responded well to cholinesterase inhibitors
[66 .] or to the combination of cholinesterase inhibitors
and 3,4-diaminopyridine [67.].
In summary, mutations in RAPSN and the resulting
rapsyn deficiency appear to be an important cause of
Figure 3. Diagram depicting the main domains of rapsyn and the localization of the identified mutations
Tetratricopeptide repeats (TPR) necessaryto rapsyn self-association
AChR
TPR1 TPR2 TPR3 TPR4 TPR5 TPR6 TPR7 coiledcoil RING
Q3K
L14P
46in
sCA
25V
F81
LY
86X
N88
KR
91L
C97
X
Q12
4X
A14
2DR
151P
V16
5M
553i
ns5
IVS
4-2A
→G
A24
6V
Y26
9X
G29
1D
E33
3X
1083
_108
4dup
CT
1177
delA
A
(a)
(b)
–38A→G –27C→G
E box E box
β-d
ystr
ogly
can
(a) Seven tetratricopeptide repeat domains (TPR1–7) are necessary for rapsyn to self-associate. The coiled-coil domain binds to the large cytoplasmicloop of the acetylcholine receptor (RACh) subunits. The RING domain binds rapsyn to b-dystroglycan. A serine phosphorylation site is located atcodon 406. Modified from Ohno et al. [6]. (b) Two E-box (CANNTG-type sequence, on grey background), to which myogenic factors can bind arelocated upstream of the transcription initiation site in the rapsyn gene promoter region. Localization of the two different rapsyn promoter mutations.Adapted from Ohno et al. [64.].
Neuromuscular disease: muscle544

CMS associated with endplate acetylcholine receptor
deficiency.
Congenital myasthenic syndrome with plectin deficiency
Plectin is a highly preserved structural protein of the
cytoskeleton expressed in several cell types, including
skeletal muscle and the postsynaptic membrane. Plectin
deficiency was described in a patient presenting with
progressive myopathy, associated with myasthenic syn-
drome (involving facial, limb and oculomotor muscles),
and epidermolysis bullosa [73]. The pathophysiology of
this CMS is poorly understood.
Congenital myasthenic syndrome caused by a mutation in the
sodium channel SCN4A
The case was recently reported of a 20-year-old patient
presenting since birth with very short bouts (3–30 min)
of respiratory distress and bulbar paralysis [7 .]. The
diagnosis was made by electrophysiology of the inter-
costal muscle, which revealed the impossibility of
evoking an action potential after nerve stimulation.
Two mutations of SCN4A were identified, including
only one (V1442E) located in the S3/S4 extracellular
domain, which was found to be pathogenic when
expressed in HEK cells. The clinical aspect is quite
different from that usually associated with a SCN4Amutation (dyskalemic paralysis, congenital paramyot-
ony).
Incompletely characterized congenital myasthenic
syndromes
These CMSs are described on clinical or histological
grounds, but their molecular origin and more generally
their pathophysiology remain unknown in the absence of
an exhaustive exploration.
Familial limb girdle myasthenia
Several families have been reported [74,75]. This
previously named ‘myasthenic myopathy’ is of recessive
inheritance. Clinically, the absence of oculobulbar signs
was remarkable. The weakness and fatigability involved
the girdles. The peculiarity of this not yet understood
entity was recently stressed with the publication of five
cases, who all presented with tubular aggregates in their
muscle biopsy and who all responded favourably to
cholinesterase inhibitors [76].
Congenital myasthenic syndrome with tubular aggregates
This CMS is associated with tubular aggregates at the
histological muscle examination. The case of three
sisters presenting with a slowly progressive myopathy
beginning in early childhood associated with cardio-
myopathy was reported. A favourable response to
cholinesterase inhibitors was noted [77]. Similar char-
acteristics were described in another family [75]. A
sporadic case was reported recently [78]. In the absence
of thorough investigations of neuromuscular transmis-
sion, the classification of these cases remains delicate,
more especially as the presence of tubular aggregates is
not specific and can be associated with isolated
myopathy, painful cramps [79] and with slow channel
syndromes [31].
Approach to the diagnosis of congenitalmyasthenic syndromesOn the basis of these historical advances in the knowl-
edge of CMS, the diagnostic strategy includes roughly
two successive steps: (1) the association of a clinical-
electrophysiological picture of a myasthenic syndrome,
and data in favour of a congenital origin; and (2) the
recognition of the pathophysiological type, which is
based on clinical data, the type of hereditary transmis-
sion, the response to cholinesterase inhibitors, the results
of electromyography, and finally the muscle biopsy and
molecular genetics. The sequential order of these two
last investigations depends upon the initial clinical-
electromyographical data.
Clinical presentation
The various CMSs share a common clinical presenta-
tion. The onset is in general early. Late appearance of
the symptoms during adolescence, or even in the adult,
is more rarely reported. Some clinical signs suggest an
anomaly of neuromuscular transmission: ophthalmople-
gia and ptosis, dysphonia and swallowing disturbance,
facial paresis, and muscle fatigability. In the young child,
the ptosis is not easy to recognize because hypotonia,
poor mimicry, suction disorders, and weakness of the cry
are in the foreground. The occurrence of bouts and
worsening by exertion are characteristics of the disease.
The favourable effect of cholinesterase inhibitors is a
significant argument in favour of a myasthenic syn-
drome. However, two types of CMS are worsened by
cholinesterase inhibitors: slow channel syndrome and
acetylcholinesterase deficiency. With the proper
myasthenic signs, myopathic signs are often associated:
amyotrophy, tendinous retractions, facial malformation
and scoliosis. The severity of the CMS is variable,
depending upon the severity of the walking deficit, the
bulbar disorders and the respiratory difficulties. Acute
respiratory failure may occur, triggered by infectious
episodes, and is frequent in the first months of life. In
the absence of respiratory assistance, the risk of death is
high [12].
A family history of the disease is an essential argument in
favour of the genetic origin of myasthenic syndrome.
Most CMSs are of autosomal recessive inheritance. Slow
channel syndrome is the only autosomal dominant CMS
characterized hitherto. The progression patterns of
CMSs are highly variable, including in a given patient,
at various periods of life. Myasthenic bouts are fre-
Congenital myasthenic syndromes Hantaı et al. 545

quently triggered by infectious episodes, pregnancy and
even periods. Progressive aggravation of the disease may
sometimes occur late in adulthood, with the appearance
of respiratory insufficiency [27 .]. A favourable progres-
sion is possible after a severe neonatal onset [4..].
Titration of anti-acetylcholine receptor antibodies in the
serum
The absence of antibodies against acetylcholine receptor
and muscle-specific receptor tyrosine kinase [80,81.] is a
prerequisite for the diagnosis of CMS, although an
exception was reported [82 .].
Electromyography
The electrophysiology of neuromuscular transmission is
the determinant for the diagnosis of CMS. This includes
searching for neuromuscular block, repetitive motor
responses and increments [83,84]. The observation of a
neuromuscular transmission block affirms the myasthe-
nic syndrome. The decrement can be absent in CMS,
particularly in patients who are not highly symptomatic,
and in cases of CMS caused by mutations in ChAT [5] or
rapsyn [6]. In the case of a ChAT deficiency, the
decrement may appear only after an initial 5-min 10 Hz
stimulation [14 .]. Repetitive CMAPs are pathognomonic
of two varieties of CMS: slow channel syndrome and
acetylcholinesterase deficiency (Fig. 2). The search for
an increment is imperative. An increment greater than
100% in amplitude and in area is suggestive of a
presynaptic origin.
Muscle biopsy
A first role of the muscle biopsy is to eliminate the
diagnosis of myopathy (congenital myopathy or mito-
chondrial cytopathy). Although non-specific, the pre-
dominance of type I fibres and the marked atrophy of
type II fibres is suggestive of CMS. The presence of
tubular aggregates is frequent, but poses the problem of
the group of CMSs with tubular aggregates. The
neuromuscular junctions are visualized for (acetyl)choli-
nesterase by the histochemical technique of Koelle,
fasciculin or specific antibodies, and for acetylcholine
receptor by fluorescent a-bungarotoxin, which binds to
it. The neuromuscular junctions frequently exhibit
variable anomalies: reduced size, the disappearance of
synaptic folds, all modifications are not specific, however,
to a given CMS.
Two types of information are decisive: (1) the absence of
acetylcholinesterase at the neuromuscular junction
establishes the diagnosis of acetylcholinesterase defi-
ciency; a study by ultracentrifugation on sucrose gradient
will generally reveal the absence of asymmetrical
(synaptic) forms of the enzyme; and (2) a significant
reduction in the number of acetylcholine receptors,
further quantified by binding with iodinated a-bungar-
otoxin, points to a primary anomaly of acetylcholine
receptor or rapsyn.
The expression of the fetal g subunit of the acetylcho-
line receptor argues in favour of a primary anomaly of
the acetylcholine receptor e subunit [55,56]. Immuno-
cytochemical study of the expression of other markers of
the neuromuscular synapse can also be performed: agrin,
muscle-specific receptor tyrosine kinase, rapsyn, neur-
egulin, a-dystrobrevin or utrophin [85]. It is aetiologi-
cally suggestive if there is a major and selective
reduction of the expression of a protein, but the primary
nature of the deficit is, however, not established (a
deficit in rapsyn is found in CMS with mutations in both
the rapsyn gene and the acetylcholine receptor subunit
genes).
Molecular genetics
The diagnosis of CMS can be confirmed by molecular
analyses in the eight genes whose mutations are so far
known to cause CMS: four genes encoding the various
acetylcholine receptor subunits (CHRNE, CHRNA1,CHRNB1, CHRND), the genes encoding rapsyn
(RAPSN), the collagen tail of acetylcholinesterase
(COLQ), choline acetyltransferase (CHAT), and the
sodium channel (SCN4A). With the exception of the
Gypsy e1267del mutation [48] and the RAPSN N88K
mutation [6,71 ..], a search has been made to identify
‘private’ mutations. Analysis of the coding sequences
and flanking intronic regions by direct sequencing after
polymerase chain reaction amplification of each fragment
on genomic DNA is required. Many mutations have
been described to date, and the two predominant genes
in postsynaptic CMS appear to be CHRNE and RAPSN.In approximately half of the cases, the analysis of these
genes does not identify a mutation causing the disease,
suggesting that other genes could be involved. When a
new mutation is identified, its pathogenic character can
be demonstrated by expression studies of this mutation
in HEK cells, COS cells or oocytes, but other experi-
mental models can also be used.
Microelectrophysiology of neuromuscular transmission
Microelectrophysiology of the intercostal muscle can be
used to specify the pre- or postsynaptic location of the
dysfunction of neuromuscular transmission, and in
postsynaptic CMS, to find kinetic anomalies of the
acetylcholine receptor [4..]. The complexity of these
techniques (patch clamp) and the risks of general
anaesthesia in a myasthenic patient limit the indications
for this exploration, more especially because the expres-
sion of the mutations in experimental cell systems by
itself allows the pathophysiological characterization of
CMS. In addition, the study of other muscles under local
anaesthesia was proposed: quadriceps [86] and ancone
[15].
Neuromuscular disease: muscle546

Difficulties of diagnosisThe diagnosis of CMS is not always easy. Faced with
sporadic CMS beginning after the neonatal period, the
diagnosis of seronegative autoimmune myasthenia may
be difficult to eliminate, more especially as long periods
of remission are possible in both afflictions and bouts can
occur in the adult CMS patient during pregnancy [38].
Muscle-specific receptor tyrosine kinase (MuSK) anti-
bodies have been detected in more than
half of the patients presenting with seronegative
(no acetylcholine receptor antibodies) auto-immune
myasthenia [80,81 .]. In case of uncertainty, the absence
of MuSK antibodies must be verified before establishing
a diagnosis of CMS.
Three recent observations have stressed that it is
sometimes difficult to draw clear boundaries between
autoimmune myasthenia and CMS [82,87,88.]. The first
reported two sisters carrying heteroallelic mutations of
the acetylcholine receptor a subunit, both presenting
with neonatal myasthenic syndrome, but one developed
autoimmune myasthenia as an adult, attested by the
transitory presence of anti-acetylcholine receptor anti-
bodies and a favourable response to plasmaphereses and
corticotherapy [82]. The authors suggested that the
genetic anomaly of the acetylcholine receptor could
constitute the factor triggering autoimmune myasthenia.
The second observation concerned a patient presenting
with acquired slow channel syndrome beginning at 30
years of age. The passive transfer of the serum of this
patient to a mouse reproduced kinetic anomalies of the
acetylcholine receptor, which demonstrated its autoim-
mune origin and excluded a congenital affliction [87].
The third observation reported an acetylcholine recep-
tor-seronegative, MuSK-seropositive myasthenia gravis
patient, in whom no acetylcholine receptor or MuSK
deficiency was found in muscle biopsies despite the
electrophysiological impairment. Mutation analysis of
MUSK did not reveal mutations but polymorphisms. The
authors concluded that circulating anti-MuSK antibodies
may not have caused the myasthenic syndrome in this
patient [88.].
Phenotype–genotype correlations andprognosisThe genotype and clinical phenotype are not correlated
in CMS. Mutations in different synaptic proteins give
similar clinical pictures: the occurrence of apnoeic
episodes in early childhood was reported in CMSs with
a deficit in ChAT, in those caused by primary anomalies
of the acetylcholine receptor, of the acetylcholinesterase
or of rapsyn. Arthrogryposis has been described in CMS
caused by mutations in the gene encoding rapsyn [66.]
and the acetylcholine receptor d subunit [45]. The same
mutation could lead to very different clinical phenotypes:
for example, the homozygous N88K rapsyn mutation
leads either to a very severe neonatal form or to a late-
onset and benign form [6,66 .]. Finally, variability within a
family was noted in certain cases of CMS.
Prognosis is difficult to assess. A favourable outcome is
possible in cases of CMS initially thought to be severe
because of respiratory or bulbar bouts. In contrast, motor
and respiratory degradation occurring late in adulthood
has been reported in patients initially only slightly
affected [27.]. As indicated above, knowledge of the
primary molecular anomaly of CMS does not enable the
prediction of disease progression. The response to
treatments known to ameliorate neuromuscular transmis-
sion is a significant prognostic factor: thus in acetylcho-
linesterase deficiency, the absence of amelioration by
cholinesterase inhibitors or any other drug may be
alarming.
TreatmentNon-specific measures are essential: immediate treat-
ment of respiratory distress, the prevention of infections
and of malnutrition as a result of swallowing disorders,
and orthopaedic surveillance of spinal complications and
retractions. Drug contraindications must be respected as
for any other myasthenic syndrome. In the case of CMS,
there is no reason to apply the immunosuppressive
therapy used for myasthenia gravis. Cholinesterase
inhibitors are efficient in all CMSs, with the exception
of slow channel syndrome and acetylcholinesterase
deficiency, which they can even worsen. They exert a
preventative effect on the respiratory decompensations
of CMS caused by ChAT mutations [4 ..]. 3,4-Diamino-
pyridine, whose mode of action is presynaptic, is
sometimes effective in pre- or postsynaptic CMSs [39].
Patients suffering from slow channel syndrome benefit
from the regulatory action of acetylcholine receptor
blockers: quinidine is effective by correcting the
prolonged opening of the acetylcholine receptor [89],
but is formally contraindicated in all the other forms of
CMS. A favourable effect of fluoxetine was recently
demonstrated in some patients, and is of interest despite
the large amount needed [40 .]. At present, there is no
specific treatment for acetylcholinesterase deficiency.
ConclusionAlthough the epidemiology of the CMSs is poorly
understood, these disorders constitute the major cause
of the myasthenic syndrome in the young child and are a
minor cause of adult myasthenic syndrome. The
diagnosis is often difficult to ascertain because of the
frequent absence of a family history of the disease, and
because of the pre-eminence of the myopathic signs
compared with myasthenic signs. The early onset of the
first symptoms, the presence of fluctuations, the
demonstration of a neuromuscular block, repetitive
CMAP after single stimulation, and the cholinesterase
Congenital myasthenic syndromes Hantaı et al. 547
inhibitor test all enable the rectification of the diagnosis
and the proposal of an effective treatment and genetic
counseling. The numerous studies devoted to CMS over
more than 20 years have demonstrated the patho-
physiological heterogeneity of CMS. Characterization
of the CMS is based on the mode of transmission, the
search for a CMAP, the positive or negative response to
cholinesterase inhibitors, the study of motor endplates,
which is easily done on a deltoid muscle biopsy, and
molecular genetics. It will thus be possible to identify
the majority of CMSs: a primary anomaly of one of the
various acetylcholine receptor subunits, of rapsyn,
acetylcholinesterase, ChAT or even SCN4A. However,
the origin of a significant fraction of CMSs remains
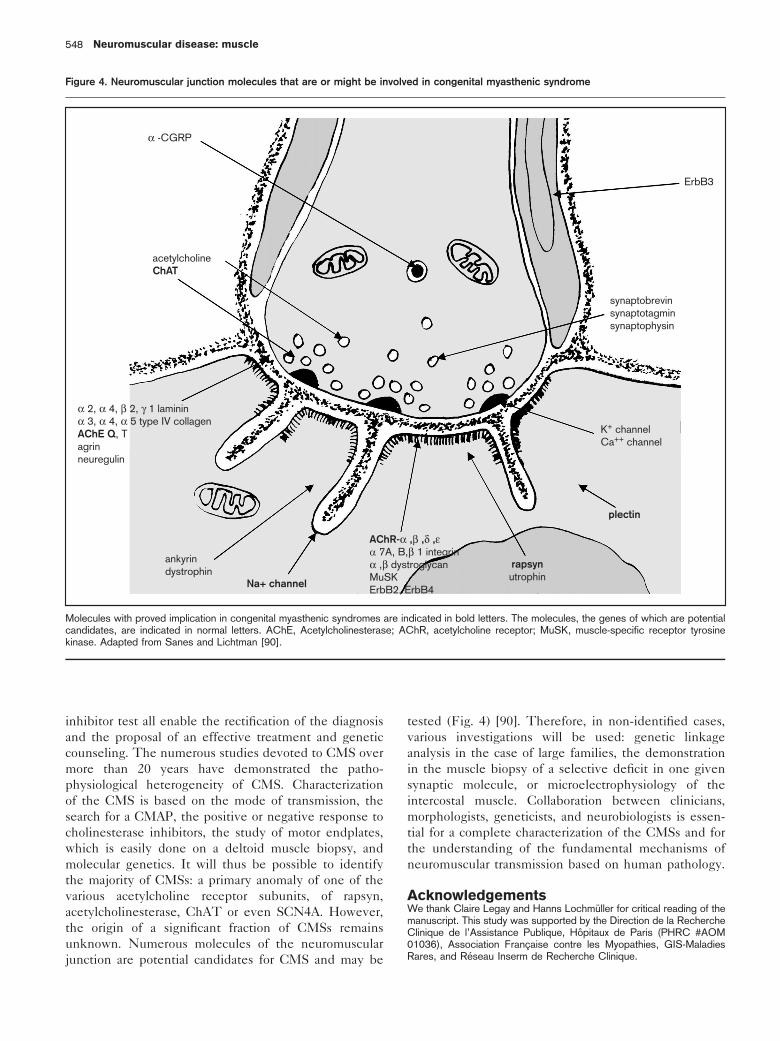
unknown. Numerous molecules of the neuromuscular
junction are potential candidates for CMS and may be
tested (Fig. 4) [90]. Therefore, in non-identified cases,
various investigations will be used: genetic linkage
analysis in the case of large families, the demonstration
in the muscle biopsy of a selective deficit in one given
synaptic molecule, or microelectrophysiology of the
intercostal muscle. Collaboration between clinicians,
morphologists, geneticists, and neurobiologists is essen-
tial for a complete characterization of the CMSs and for
the understanding of the fundamental mechanisms of
neuromuscular transmission based on human pathology.
AcknowledgementsWe thank Claire Legay and Hanns Lochmuller for critical reading of themanuscript. This study was supported by the Direction de la RechercheClinique de l’Assistance Publique, Hopitaux de Paris (PHRC #AOM01036), Association Francaise contre les Myopathies, GIS-MaladiesRares, and Reseau Inserm de Recherche Clinique.
Figure 4. Neuromuscular junction molecules that are or might be involved in congenital myasthenic syndrome
α -CGRP
acetylcholineChAT
α 2, α 4, β 2, γ 1 lamininα 3, α 4, α 5 type IV collagenAChE Q, Tagrinneuregulin
ankyrindystrophin
Na+ channel
AChR-α ,β ,δ ,εα 7A, B,β 1 integrinα ,β dystroglycanMuSKErbB2, ErbB4
rapsynutrophin
plectin
K+ channelCa++ channel
ErbB3
synaptobrevinsynaptotagminsynaptophysin
Molecules with proved implication in congenital myasthenic syndromes are indicated in bold letters. The molecules, the genes of which are potentialcandidates, are indicated in normal letters. AChE, Acetylcholinesterase; AChR, acetylcholine receptor; MuSK, muscle-specific receptor tyrosinekinase. Adapted from Sanes and Lichtman [90].
Neuromuscular disease: muscle548

References and recommended readingPapers of particular interest, published within the annual period of review, havebeen highlighted as:. of special interest.. of outstanding interest
1 Millichap JG, Dodge PR. Diagnosis and treatment of myasthenia gravis ininfancy, childhood, and adolescence. Neurology 1960; 10:1007–1014.
2 Engel AG, Lambert EH, Gomez MR. A new myasthenic syndrome withendplate acetylcholinesterase deficiency, small nerve terminals, and reducedacetylcholine release. Ann Neurol 1977; 1:315–330.
3 Shillito P, Vincent A, Newsom-Davis J. Congenital myasthenic syndromes.Neuromusc Disord 1993; 3:183–190.
4. .
Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes: progressover the past decade. Muscle Nerve 2003; 27:4–25.
An excellent review on CMS (see also Ref. [8..]).
5 Ohno K, Tsujino A, Shen XM, et al. Choline acetyltransferase mutationscause myasthenic syndrome associated with episodic apnea in humans. ProcNatl Acad Sci U S A 2002; 98:2017–2022.
6 Ohno K, Engel AG, Shen XM, et al. Rapsyn mutations in humans causeendplate acetylcholine-receptor deficiency and myasthenic syndrome. Am JHum Genet 2002; 70:875–885.
7.
Tsujino A, Maertens C, Ohno K, et al. Myasthenic syndrome caused bymutation of the SCN4A sodium channel. Proc Natl Acad Sci U S A 2003;100:7377–7382.
The first described SCN4A mutations in CMS: the inability of a normal evokedendplate potential to activate the sodium channel SCN4A is the result of thesemutations and the cause of this CMS.
8. .
Engel AG, Ohno K, Sine SM. Sleuthing molecular targets for neurologicaldiseases at the neuromuscular junction. Nature Rev Neurosci 2003; 4:339–352.
An important and comprehensive review on CMS that covers the symptomatologyof CMS in the light of the underlying molecular pathogenic mechanisms.
9 Engel AG. Congenital myasthenic syndromes. In: 73rd European Neuromus-cular Center (ENMC) International Workshop. Naarden, the Netherlands,22–23 October 1999. Neuromusc Disord 2001; 11:315–321.
10 Mora M, Lambert EH, Engel AG. Synaptic vesicle abnormality in familialinfantile myasthenia. Neurology 1987; 37:206–214.
11 Conomy JP, Levisohn M, Fanaroff A. Familial infantile myasthenia gravis: acause of sudden death in young children. J Pediatr 1975; 87:428–429.
12 Byring RF, Pihko H, Shen XM, et al. Congenital myasthenic syndromeassociated with episodic apnea and sudden infant death. Neuromusc Disord2002; 12:548–553.
13 Misgeld T, Burgess RW, Cunningham JM, et al. Roles of neurotransmitter insynapse formation: development of neuromuscular junctions lacking cholineacetyltransferase. Neuron 2002; 36: 635–648.
14.
Schmidt C, Abicht A, Krampfl K, et al. Congenital myasthenic syndrome duea novel missense mutation in the gene encoding choline acetyltransferase.Neuromusc Disord 2003; 13:245–251.
Three Turkish families presented with a homozygous I336T in CHAT. A foundereffect may be considered.
15.
Maselli RA, Chen D, Mo D, et al. Choline acetyltransferase mutations inmyasthenic syndrome due to deficient acetylcholine resynthesis. MuscleNerve 2003; 27:180–187.
This study reports the pathophysiological characterization of patients sufferingfrom CMSs caused by CHAT mutations. A cold bath triggered the myasthenicsyndrome instead of improving it. A thorough microelectrophysiological study ofthe ancone muscle was performed.
16. .
Cai Y, Cronin CN, Engel AG, et al. Choline acetyltransferase structurereveals distribution of mutations that cause motor disorders. EMBO J 2004;23:2047–2058.
An original study showing the positioning of the 12 missense mutations identifiedto date in the crystal structure of ChAT and providing clues as to how they canaffect the enzyme activity.
17 Walls TJ, Engel AG, Nagel AS, et al. Congenital myasthenic syndromeassociated with paucity of synaptic vesicles and reduced quantal release.Ann NY Acad Sci 1993; 681:461–468.
18 Bady B, Chauplannaz G, Carrier H. Congenital Lambert–Eaton myasthenicsyndrome. J Neurol Neurosurg Psychiatry 1987; 50:476–478.
19 Maselli R, Kong DZ, Bowe CM, et al. Presynaptic congenital myasthenicsyndrome due to quantal release deficiency. Neurology 2001; 57:279–289.
20 Hutchinson DO, Walls TJ, Nakano S, et al. Congenital endplate acetylcho-linesterase deficiency. Brain 1993; 116:633–653.
21 Kohara N, Lin TS, Fukudome T, et al. Pathophysiology of weakness in apatient with congenital end-plate acetylcholinesterase deficiency. MuscleNerve 2002; 25:585–592.
22 Ohno K, Brengman JM, Tsujino A, Engel AG. Human endplate acetylcho-linesterase deficiency caused by mutations in the collagen-like tail subunit(ColQ) of the asymmetric enzyme. Proc Natl Acad Sci U S A 1998; 95:9654–9659.
23 Donger C, Krejci E, Serradell P, et al. Mutation in the human acetylcholi-nesterase-associated gene, COLQ, is responsible for congenital myasthenicsyndrome with end-plate acetylcholinesterase deficiency. Am J Hum Genet1998; 63:967–975.
24 Ohno K, Engel AG, Brengman JM, et al. The spectrum of mutations causingendplate acetylcholinesterase deficiency. Ann Neurol 2000; 47:162–170.
25 Shapira YA, Sadeh ME, Bergtraum MP, et al. The novel COLQ mutationsand variation of phenotypic expressivity due to G240X. Neurology 2002;58:603–609.
26 Ohno K, Brengman JM, Felice KJ, et al. Congenital endplate acetylcholines-terase deficiency caused by a nonsense mutation and an A-to-G splice sitemutation at position 3 of the collagen-like tail subunit gene (COLQ): Howdoes G at position 3 result in aberrant splicing? Am J Hum Genet 1999;65:635–644.
27.
Ishigaki K, Nicolle D, Krejci E, et al. Two novel mutations in the ColQ genecause endplate acetylcholinesterase deficiency. Neuromusc Disord 2003;13:236–244.
A common splicing mutation associated with another truncating mutation causedissimilar phenotypes in two patients.
28.
Muller JS, Petrova S, Kiefer R, et al. Synaptic congenital myasthenicsyndrome in three patients due to a novel missense mutation (T441A) of theCOLQ gene. Neuropediatrics 2004; 35:183–189.
The same homozygous COLQ mutation results here again in various phenotypicexpressions.
29. .
Kimbell LM, Ohno K, Engel AG, Rotundo RL. C-terminal and heparin-bindingdomains of collagenic tail subunit are both essential for anchoringacetylcholinesterase at the synapse. J Biol Chem 2004; 279:10997–11005.
By testing the binding of the purified collagenic tail on frog neuromuscularjunctions, the authors demonstrate, among other things, how mutations in the C-terminal domain of the collagenic tail impair attachment of the collagenic tail to thesynaptic basal lamina.
30 Engel AG, Lambert EH, Mulder DM, et al. A newly recognized congenitalmyasthenic syndrome attributed to a prolonged open time of the acetylcho-line-induced ion channel. Ann Neurol 1982; 11:553–569.
31 Sine SM, Ohno K, Bouzat C, et al. Mutation of the acetylcholine receptoralpha subunit causes a slow-channel myasthenic syndrome by enhancingagonist binding affinity. Neuron 1995; 15:229–239.
32 Ohno K, Hutchinson DO, Milone M, et al. Congenital myasthenic syndromecaused by prolonged acetylcholine receptor channel openings due to amutation in the M2 domain of the epsilon subunit. Proc Natl Acad Sci U S A1995; 92:758–762.
33 Wang HL, Auerbach A, Bren N, et al. Mutation in the M1 domain of theacetylcholine receptor alpha subunit decreases the rate of agonist dissocia-tion. J Gen Physiol 1997; 109:757–766.
34 Milone M, Wang HL, Ohno K, et al. Slow-channel syndrome caused byenhanced activation, desensitization, and agonist binding affinity due tomutation in the M2 domain of the acetylcholine receptor alpha subunit. JNeurosci 1997; 17:5651–5665.
35 Croxen R, Newland C, Beeson D, et al. Mutations in different functionaldomains of the human muscle acetylcholine receptor alpha subunit in patientswith the slow-channel congenital myasthenic syndrome. Hum Mol Genet1997; 6:767–774.
36 Engel AG, Ohno K, Milone M, et al. New mutations in acetylcholine receptorsubunit genes reveal heterogeneity in the slow-channel congenital myasthenicsyndrome. Hum Mol Genet 1996; 5:1217–1227.
37 Gomez CM, Maselli R, Vohra BPS, et al. Novel delta subunit mutation inslow-channel syndrome causes severe weakness by novel mechanisms. AnnNeurol 2002; 51:102–112.
38 Oosterhuis H, Newsom-Davis J, Wokke J et al. The slow channel syndrome.Two new cases. Brain 1987; 110:1061–1079.
39 Harper CM, Engel AG. Quinidine sulfate therapy for the slow-channelcongenital myasthenic syndrome. Ann Neurol 1998; 43:480–484.
40.
Harper CM, Engel AG, Fukudome T, et al. Treatment of slow channelcongenital myasthenic syndrome with fluoxetine. Neurology 2003; 60:1710–1713.
The authors show that a high dosage of fluoxetine has a favourable effect on slowchannel patients and may be an alternative to quinidine.
Congenital myasthenic syndromes Hantaı et al. 549

41 Croxen R, Hatton C, Shelley C, et al. Recessive inheritance and variablepenetrance of slow-channel congenital myasthenic syndromes. Neurology2002; 59:162–168.
42 Zeevaert B, Hansen I, Crielaard JM, Wang FC. Slow channel syndrome dueto an autosomal translocation at 2q31–9p27 [in French]. Rev Neurol (Paris)2002; 158:606–609.
43.
Webster R, Brydson M, Croxen R, et al. Mutation in the AChR ion channelgate underlies a fast channel congenital syndrome. Neurology 2004;62:1090–1096.
A case of dominant inheritance in fast channel syndrome.
44 Uchitel O, Engel AG, Walls TJ, et al. Congenital myasthenic syndromes: II. Asyndrome attributed to abnormal interaction of acetylcholine with its receptor.Muscle Nerve 1993; 16:1293–1301.
45 Brownlow S, Webster R, Croxen R, et al. Acetylcholine receptor deltasubunit mutations underlie a fast-channel myasthenic syndrome andarthrogryposis multiplex congenita. J Clin Invest 2001; 108:125–130.
46. .
Shen XM, Ohno K, Tsujino A, et al. Mutation causing severe myastheniareveals functional asymmetry of AChR signature cystine loops in agonistbinding and gating. J Clin Invest 2003; 111:497–505.
The a V132L mutation located in the signature cystine loops reduces both theacetylcholine binding affinity of the closed channel and the total acetylcholinecurrent. Corresponding amino acid substitution in the signature cystine loops ofthe other acetylcholine receptor subunits leads to different if not oppositephenotypes.
47.
Sine SM, Wang HL, Ohno K, et al. Mechanistic diversity underlying fastchannel congenital myasthenic syndromes. Ann NY Acad Sci 2003;998:128–137.
A comprehensive review of fast channel syndromes.
48 Abicht A, Stucka R, Karcagi V, et al. A common mutation (e1267delG) incongenital myasthenic patients of Gypsy ethnic origin. Neurology 1999;53:1564–1569.
49 Morar B, Gresham D, Angelicheva D, et al. Mutation history of the Roma/Gypsies. Am J Hum Genet 2004; in press.
50 Abicht A, Stucka R, Schmidt C, et al. A newly identified chromosomalmicrodeletion and an N-box mutation of the AChRe gene cause a congenitalmyasthenic syndrome. Brain 2002; 125:1005–1013.
51.
Ohno K, Milone M, Shen XM, Engel AG. A frameshifting mutation in CHRNEunmasks skipping of the preceding exon. Hum Mol Genet 2003; 12:3055–3066.
An interesting study showing that the frameshift e553del7 mutation in exon 7enhances the expression of an aberrrantly spliced transcript that skips exon 6.
52 Nichols P, Croxen R, Vincent A, et al. Mutation of the acetylcholine receptorepsilon-subunit promoter in congenital myasthenic syndrome. Ann Neurol1999; 45:439–443.
53 Ohno K, Anlar B, Engel AG. Congenital myasthenic syndrome caused by amutation in the Ets-binding site of the promoter region of the acetylcholinereceptor subunit gene. Neuromusc Disord 1999; 9:131–135.
54 Ealing J, Webster R, Brownlow S, et al. Mutations in congenital myasthenicsyndromes reveal an e subunit C-terminal cysteine, C470, crucial formaturation and surface expression of adult AChR. Hum Mol Genet 2002;11:3087–3096.
55 Engel AG, Ohno K, Bouzat C, et al. End-plate acetylcholine receptordeficiency due to nonsense mutations in the epsilon subunit. Ann Neurol1996; 40:810–817.
56 Croxen R, Young C, Slater C, et al. Endplate gamma and epsilon-subunitmRNA levels in AChR deficiency syndrome due to epsilon subunit nullmutations. Brain 2001; 124:1362–1372.
57 Kraner S, Sieb JP, Thompson PN, Steinlein OK. Congenital myasthenia inBrahman calves caused by homozygosity for a CHRNE truncating mutation.Neurogenetics 2002; 4:87–91.
58.
Thompson PN, Steinlein OK, Harper CK, et al. Congenital myasthenicsyndrome of Brahman cattle in South Africa. Veterin Rec 2003; 153:779–781.
When calves are affected by CMS too . . . See also Ref. [57].
59 Ramarao MK, Bianchetta MJ, Lauken J, Cohen JB. Role of rapsyntetratricopeptide repeat and coiled-coil domains in self association andnicotinic acetylcholine receptor clustering. J Biol Chem 2001; 9:7475–7483.
60 Cartaud A, Coutant S, Petrucci TC, Cartaud J. Evidence for in situ and invitro association between b-dystroglycan and the subsynaptic 43 K rapsynprotein. Consequence for acetylcholine receptor clustering at the synapse. JBiol Chem 1998; 273:11321–11326.
61 Richard P, Gaudon K, Andreux F, et al. Possible founder effect of rapsynN88K mutation and identification of novel rapsyn mutations in congenitalmyasthenic syndromes. J Med Genet 2003; 40:e81–e85.
62 Muller JS, Mildner G, Muller-Felber W, et al. Rapsyn N88K is a frequentcause of congenital myasthenic syndromes in European patients. Neurology2003; 60:1805–1810.
63 Dunne V, Maselli R. Identification of pathogenic mutations in the humanrapsyn gene. J Hum Genet 2003; 48:204–207.
64.
Ohno K, Sadeh M, Blatt I, et al. E-box mutations in the RAPSN promoterregion in eight cases with congenital myasthenic syndrome. Hum Mol Genet2003; 12:739–748.
Two E-box mutations were identified in the rapsyn promoter in eight CMS patients,of whom seven were Jews originating from Iraq or Iran, and had already beendescribed for their peculiar clinical phenotype (elongated face and prognathism).
65 Maselli RA, Dunne V, Pascual-Pascual SI, et al. Rapsyn mutations inmyasthenic syndrome due to impaired receptor clustering. Muscle Nerve2003; 28:293–301.
66.
Burke G, Cossins J, Maxwell S, et al. Rapsyn mutations in hereditarymyasthenia: distinct early- and late-onset phenotypes. Neurology 2003;61:826–828.
This extensive study of 16 unrelated CMS patients with rapsyn mutationsdistinguishes early- and late-onset phenotypes whose consequences in terms ofdiagnosis and therapy are important.
67.
Banwell BL, Ohno K, Sieb JP, Engel AG. Novel truncating RAPSN mutationscausing congenital myasthenic syndrome responsive to 3,4-diaminopyridine.Muscle Nerve 2004; 14:202–207.
This study reported that a combination of 3,4-diaminopyridine and cholinesteraseinhibitors is of interest in patients with either severe or mild CMS as a result ofrapsyn deficiency.
68 Yasaki E, Prioleau C, Barbier J, et al. Electrophysiological and morphologicalcharacterization of a case of autosomal recessive congenital myasthenicsyndrome with acetylcholine receptor deficiency due to N88K rapsynhomozygous mutation. Neuromusc Disord 2004; 14:24–32.
69 Muller JS, Abicht A, Christen HJ, et al. A newly identified chromosomalmicrodeletion of the rapsyn gene causes a congenital myasthenic syndrome.Neuromusc Disord 2004; in press.
70 Goldhammer Y, Blatt I, Sadeh M, Goodman RM. Congenital myastheniaassociated with facial malformations in Iraqui and Iranian Jews. A new geneticsyndrome. Brain 1990; 113:1291–1306.
71. .
Muller JS, Abicht A, Burke G, et al. The congenital myasthenic syndrome(CMS) mutation RAPSN N88K derives from an ancient Indo-Europeanfounder. J Med Genet 2004; 41:e104–e106.
The reality of a founder effect for the RAPSN N88K missense mutation wasdisputed (see Refs [61,62,72]). The goal of the present paper was to gain enoughstatistical power to decide by increasing the number of single nucleotidepolymorphisms and of alleles analysed in a large number of patients.
72 Ohno K, Engel AG. Lack of founder haplotype for the rapsyn N88K mutation:N88K is an ancient founder mutation or arises from multiple founders. J MedGenet 2004; 41:e8–e10.
73 Banwell BL, Russel J, Fukudome T, et al. Myopathy, myasthenic syndrome,and epidermolysis bullosa simplex due to plectin deficiency. J NeuropatholExp Neurol 1999; 58:832–846.
74 McQuillen MP. Familial limb–girdle myasthenia. Brain 1966; 89:121–132.
75 Johnes TR, Campa SF, Adelman IS. Familial myasthenia gravis with tubularaggregates treated with prednisone. Neurology 1973; 28:426–433.
76 Rodolico C, Toscano A, Autunno M, et al. Limb–girdle myasthenia: clinical,electrophysiological and morphological features in familial and autoimmunecases. Neuromusc Disord 2002; 12:964–969.
77 Dobkin BH, Verity MA. Familial neuromuscular disease with type I fiberhypoplasia, tubular aggregates, cardiomyopathy and myasthenic features.Neurology 1978; 28:1135–1140.
78 Zephir H, Stojkovic T, Maurage C, et al. Tubular aggregate congenitalmyopathy associated with neuromuscular block [in French]. Rev Neurol(Paris) 2001; 157:1293–1296.
79 Fardeau M, Tome FMS. Congenital myopathies. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill; 1994. pp. 1487–1532.
80 Hoch W, McConville J, Helms S, et al. Auto-antibodies to the receptortyrosine kinase MuSK in patients with myasthenia gravis without acetylcholinereceptor antibodies. Nat Med 2001; 7:365–368.
81.
Vincent A, McConville J, Farrugia ME, Newsom-Davis J. Seronegativemyasthenia gravis. Semin Neurol 2004; 24:125–133.
An up-to-date review on ‘seronegative’ autoimmune myasthenia.
82 Croxen R, Vincent A, Newsom-Davis J, Beeson D. Myasthenia gravis in awoman with congenital AChR deficiency due to epsilon-subunit mutations.Neurology 2002; 58:1563–1565.
Neuromuscular disease: muscle550

83 Fournier E. Study of neuromuscular transmission and muscle excitability. In:Fournier E, editor. Electromyographical examination and nerve conductionstudy, electrophysiological semiology. Cachan: Editions Medicales Inter-nationales; 1998. pp. 254–283.
84 Harper CM. Electrodiagnosis of endplate disease. In: Engel AG, editor.Myasthenia gravis and myasthenic disorders. New York: Oxford UniversityPress; 2002. pp. 65–84.
85 Sieb JP, Kraner S, Rauch M, Steinlein OK. Immature end-plates and utrophindeficiency in congenital myasthenic syndrome caused by epsilon-AChRsubunit truncating mutations. Hum Genet 2000; 107:160–464.
86 Slater CR, Lyons PR, Walls TJ, et al. Structure and function ofneuromuscular junctions in the vastus lateralis of man. Brain 1992;115:451–478.
87 Wintzen AR, Plomp JJ, Molenaar PC, et al. Acquired slow-channel syndrome:a form of myasthenia gravis with prolonged open time of the acetylcholinereceptor channel. Ann Neurol 1998; 44:657–664.
88.
Selcen D, Fukuda T, Shen XM, Engel AG. Are MuSK antibodies the primarycause of myasthenic symptoms? Neurology 2004; 62:1945–1950.
A debated pathogenic role for MuSK antibodies in MuSK-seropositive autoimmunemyasthenia.
89 Fukudome T, Ohno K, Brengman JM, Engel AG. Quinidine normalizes theopen duration of slow-channel mutants of the acetylcholine receptor.Neuroreport 1998; 9:1907–1911.
90 Sanes JR, Lichtman JW. Development of the vertebrate neuromuscularjunction. Annu Rev Neurosci 1999; 22:389–442.
Congenital myasthenic syndromes Hantaı et al. 551





























