Embed Size (px)
Citation preview

Control and Prediction of the Organic Solid State
A Basic Technology project of the Research Councils UK
Computational Crystal Energy Landscapes
as an aid to polymorph screening
Sarah (Sally) L Price Department of Chemistry, UCL Overview: for details see references on www.cposs.org.uk

What is the computed crystal energy landscape? Price SL 2009. Accounts Chem Res 42:117
v The crystal structures which are sufficiently low in energy to be thermodynamically feasible polymorphs v Within ~ 10 kJ mol-1 (?) of most stable
v Global minimum predicts THE (thermodynamically most stable) crystal structure
v Are local minima possible practically important polymorphs?
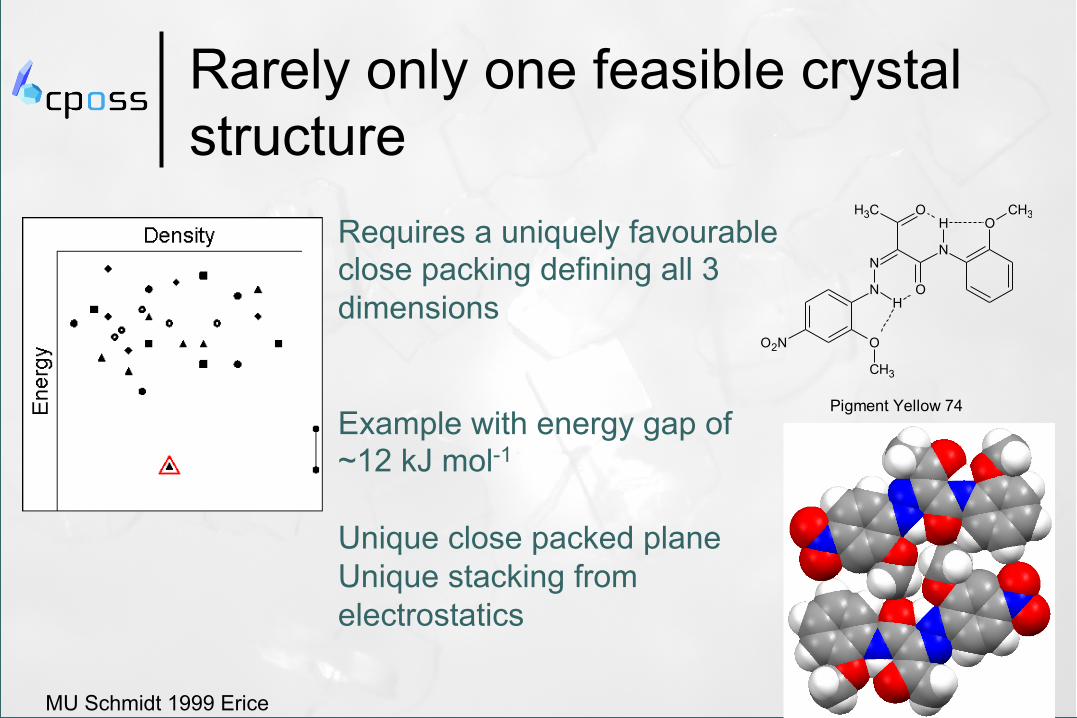
Rarely only one feasible crystal structure
O
CH3
N
O2N
N
OH
CH3 O
N
H OCH3
Pigment Yellow 74
Requires a uniquely favourable close packing defining all 3 dimensions Example with energy gap of ~12 kJ mol-1 Unique close packed plane Unique stacking from electrostatics
MU Schmidt 1999 Erice

More typical: small differences for 3 chiral molecules that spontaneously resolve E. D’Oria, PG Karamertzanis & SLP, 2010 CG&D 10, 1749.
v Energy differences all small
<1 kcal mol-1
v Racemic structures qualitatively similar to chiral
v Even if have a chiral 1D or 2D motif, having packing in 3rd dimension strongly favouring spontaneous resolution will be unusual
Observed enantiopure P212121 at global min Elatt = -106.3 kJ mol-1
Lowest racemic P21/n Elatt = -106.0 kJ mol-1

Benefits & limitations
v Gives range of packing types that are thermodynamically plausible
v needs packing analysis v Hydrogen-bond graph
sets v XPAC v Compack similarity
RMSDn
v Within search performed v extent, space groups, Z’
v Accuracy of energies of static perfect lattice v Missing temperature v Depends on method (Manolis’ poster)

-104
-102
-100
-98
-96
-94
1,55 1,6 1,65 1,7 1,75 1,8
Latti
ce E
nerg
y / k
J m
ol-1
Density / g cm-3
C2/c
P-1
P2/c
P21
P21/c
P212121
Pbcn
Pc
Pca21
Pna21
Form I
Form II
E.g. 5-fluorouracil
Form II found experimental search from dry nitromethane
Form I Z’=4
Form II & solvate
In two solvates
75% of these structures are free energy minima at 310 K
Hulme AT, Tocher DA, SLP, 2005 J. Am Chem Soc, 127, 1116 Karamertzanis PG, Raiteri P, Parrinello M, Leslie M, SLP 2007 J Phys Chem B 112:4298.
Form I more stable than form II ~ 550 K with no polymorphic transitions

Why calculate crystal energy landscapes? v to design new molecular materials prior to synthesis v to understand crystallization behaviour of a molecule
as a complement to polymorph screening and “Quality by Design” crystallization processes v target finding new polymorphs (THD or metastable) v to help solve structures from PXRD or other
experimental evidence v to see whether disorder may be affecting crystallisation
v to understand contrasting crystallisation behaviour of related molecules (see Ogaga’s poster)

Interpretation: when low energy structures are similar v if no barrier to rearrangement in nucleating/
growing cluster to most stable form, then only this form will be obtained
v if enantiotropic may get polymorphs v if solid state transformations facile may get
dynamically “averaged” structures plastic phases, higher symmetry, methyl rotation, ...
v may get intrinsic static disorder giving crystal growth reproducibility problems

Parallel ribbons
-114.9 kJ mol-1
Anti-parallel ribbons
-116.1 kJ mol-1
PXRD_2?
-115.5 kJ mol-1
PXRD_1
-116.4 kJ mol-1
From Eniluracil Crystal Energy Landscape
Non-polar ribbons
Also little energy discrimination for the stacking variations for C4O C6H interchange
Stacking & interdigitation errors hard to avoid & barrier to correction
Polar ribbons N
N
H
H
OO
HH

Experimental: variable disorder in single XRD on 4 crystals
P21/n disordered anti-parallel non-polar 0.742(3) 0.705(3) 0.738(3) 0.841(3)
Crystal 4 better R1 P21 Z’=2 minor polar
Single crystal analysis could be interpreted as polymorphism.
Powder patterns are very similar
Variable disorder challenging for devising robust production process
Simulated PXRD
Copley RCB, Barnett SA, Karamertzanis PG, Harris KDM, Kariuki BM, Xu MC, Nickels EA, Lancaster RW, Price SL 2008. Cryst Growth Des 8:3474

Site Occupancy Disorder calculations on eniluracil M Habgood R Grau-Crespo & SLP2011 Phys Chem Chem Phys 13, 9590
v In 16 molecule supercell, generate all possible configurations
v Apply thermodynamics, including configurational entropy, excluding structures unlikely to attach during crystal growth
-118.2
-118
-117.8
-117.6
-117.4
-117.2
-117
0 0.1 0.2 0.3 0.4 0.5
Minor component proportion (=τ)
Ener
gy [k
J m
ol-1
]
-0.9
-0.8
-0.7
-0.6
-0.5
-0.4
-0.3
-0.2
-0.1
0
Entro
pic
ener
gy (TS
) [kJ
mol
-1]
AETS
Disorder favoured ~ over observed range See Doris’ poster for
intergrowths

Interpretation: when low energy structures are different v Once crystallised, difficulties of solid state
transformation may mean that you have practically useful polymorphs
v How to experimentally find them? v Are they stable relative to other forms? v May it be impossible to find appropriate
conditions?

Finding catemeric CBZ V required seeded sublimation suggested by related landscapes
Pbca a/Å b/Å c/Å Expt 9.1245(5) 10.4518(5) 24.8224(11)
Rigid calc 2006
9.3124 10.5979 24.8819
Flexible calc 2011
9.4816 10.3426 24.7227
DHC form II (seed)
CBZ form V CBZ form V
Arlin J-B, Price LS, Price SL, Florence AJ, Chem. Commun., 2011, 47, 7074

Related molecules all have dimer & catemer structures competitive
dimers
form II 2008
form I 2008
chains
form IV 2002
form III 1981
form II 1987
form I 2003
form V 2011
form IV 2010
form III 2007
form II 2006
form I 1992
form II 2008
form I 2007
1:1 CBZ:DHC solid solution 2006
N
O NH2
CBZ
N
O NH2
DHC O NH2
CYH O NH2
CYT
isostructural relationships
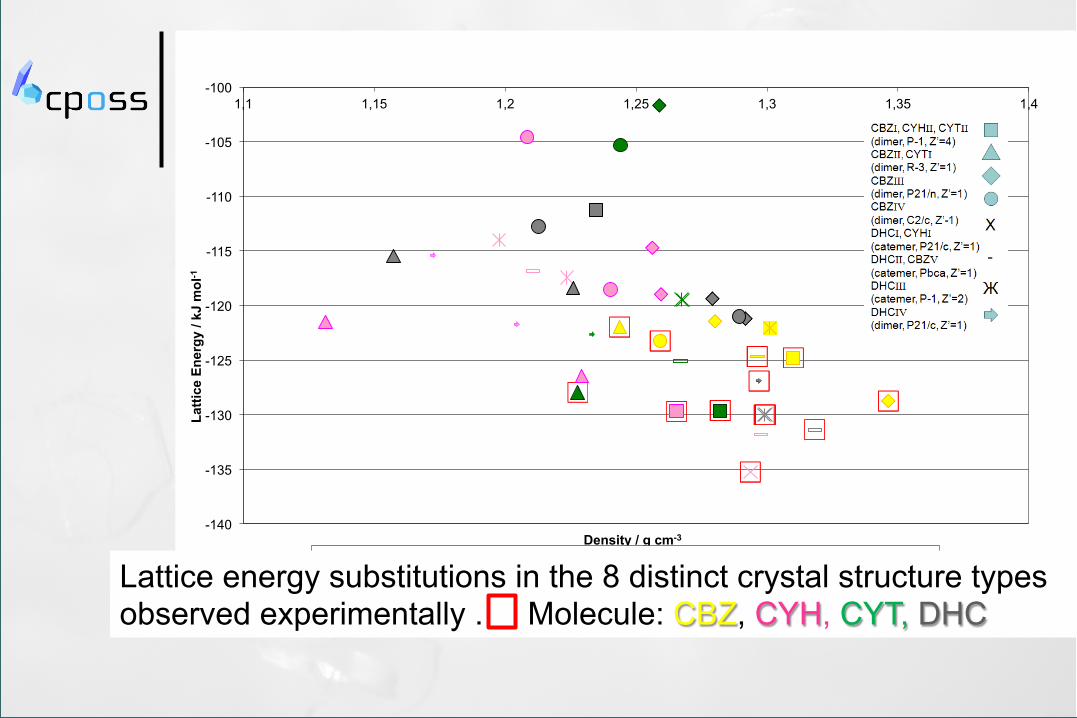
-140
-135
-130
-125
-120
-115
-110
-105
-100 1,1 1,15 1,2 1,25 1,3 1,35 1,4
Latti
ce E
nerg
y / k
J m
ol-1
Density / g cm-3
Observed Structures Carbamazepine Cyheptamide Cytenamide Dihydrocarbamazepine
Lattice energy substitutions in the 8 distinct crystal structure types observed experimentally . Molecule: CBZ, CYH, CYT, DHC

Finding the right crystallization conditions may be even harder
Racemic crystal could not be formed without racemization
Lancaster, RW; Karamertzanis, PG; Hulme, AT; Tocher, DA; Covey, DF; Price, SL, Chem.Commun., 2006, 47, 4921
- or obliging synthetic chemist
Other molecules may have less impossible barrier to conformational change

Disappearing polymorph of progesterone
v Attempts to reproduce form 2 failed v Found concomitantly with form 1 and
pregnenolone:progesterone
v Form II moderately unstable v Level of pregnenolone too high for impurity
stabilization mechanism v 1 mol% ethamindosulphathiazole stabilizes form I sulphathiazole
Lancaster RW, Karamertzanis PG, Hulme AT, Tocher DA, Lewis TC, Price SL 2007. J Pharm Sci 96:3419-3431. N. Blagden, R. J. Davey, R. Rowe and R. Roberts, Int. J. Pharm., 1998, 172, 169-177.

50 year old samples from Innsbruck Special thanks to Ulrich Griesser
Liquid Chromatography-Mass Spec Form 2 11 impurities total 4.85% Form 1 3 impurities total ~1.5%, Aldrich 1.3% different impurities irreproducible cocktail of impurities needed for long-lived form 2 Lancaster RW, Harris LD, Pearson D 2011 CrystEngComm, 13, 1775

The right crystallization experiment cannot be performed because
v crystal may be unstable relative to other products, inherent in all possible crystallization experiments
v Solvates versus polymorphs v Which, if any, hydrates will crystallise? Predicting hydrate formation of
two dihydroxybenzoic acids. Braun DE, Karamertzanis PG, Price SL 2011. Chem Commun 47: 5443 v Cocrystals versus components v Salt versus cocrystal ΔpKa <3

Cocrystal not found N Issa SA Barnett, S Mohamed, DE Braun, RCB Copley, DA Tocher, SL Price CrystEngComm ASAP
N
N
All low energy structures are segregated
O
OO
OH
H
H O
O
O
O H
C CN N
3 lowest, next 3
No cocrystal hydrogen bonds!
Could this pack densely?

Proton positions matter S Mohammed, DA Tocher & SLP, 2011 Int J Pharm, 418 187
Experimental salt
Hypothetical Cocrystal
Disordered proton structure was unique in salt & cocrystal version both low

Solid-State Forms of β-Resorcylic Acid Braun DE, Karamertzanis PG, Arlin J-B, Florence AJ, Kahlenberg V, Tocher DA, Griesser UJ, Price SL 2010. Cryst Growth Des 11, 210
New polymorph I CSP added confidence to PXRD solution and evidence for proton disorder
Similar structures,
unlikely to be distinguishable
polymorphs

Characterising racemic naproxen DE Braun M Ardid-Candel E D’Oria,PG Karamertzanis J-B Arlin AJ Florence, AG Jones, SLP Cryst. Growth Des., 2011, 11 5659
Racemic structure had to be solved from powder data - confidence from match to global minima + ssNMR for Zʹ′=1 - other stackings of these layers

Energy difference between forms - more challenging than structures
ΔHR+S→RScry
v −1.5 ± 0.3 kJ·mol–1
at T~ 156 °C by DSC ΔHfus
v −2.4 ± 1.0 kJ·mol–1 T = 10–40 °C from solubilities
v -6.1 to -9.2 kJ·mol–1 at T= 0 K from ΔElatt

Interpreting the crystal energy landscape v Polymorphs only occur when barrier to
rearrangement to most stable form at nucleation/growth stage
v Solid state transformations usually difficult, so kinetically-favoured metastable polymorphs may be trapped.
v Need to determine which structures are sufficiently different to be feasible as polymorphs – through collaboration with simultaneous experimental screening

Where are we now?
v Crystal energy landscapes complement crystal engineering and solid form screening, answering
“What are the alternatives?” v likely motifs in solid forms v possible types of disorder v range of possible target structures
v Can be calculated with “good enough” accuracy for increasing range of molecules & multi-component systems.

Where do we need to go?
v To bigger molecules v increased difficulty of changing
conformation and packing during crystallisation
v To more accurate relevant energies v Plenty of impetus in computational
chemistry and from desired applications v Need for more validation data
O
O
NH
N
S
CH3
SO2
CH3
DA Bardwell et 40 al. 2011 Acta Cryst B67, 535. GM Day, CS Adjiman, SLP et 5 al. 2011 Int J Pharm 418,168

Grateful Thanks to
v Doris Braun, Matthew Habgood, Ogaga Uzoh, Louise Price v Nizar Issa, Gareth Welch, Sharmarke Mohamed v Derek Tocher, Maurice Leslie (ex-CCLRC) Bob Lancaster (ex-GSK) v Manolis Vasileiadis, Andrei Kazantsev, Panos Karamertzanis,
Costas Pantelides, Claire Adijman (Imperial College) v Alastair Florence, Rajni Miglani, Andrea Johnston, Jean-Baptiste
Arlin, Phillipe Fernandes (Strathclyde University)
v Other coworkers in CPOSS and many collaborators v Other Programs: AJ Stone (Cambridge), H Ammon (Maryland), CCDC
v Funding EPSRC (including E-Science), CCDC v Basic Technology Program of RC UK for funding Control and Prediction
of the Organic Solid State www.cposs.org.uk, including “Translation” funding for Knowledge Transfer in CPOSS Industrial Alliance.

Database of computed crystal structures >150 molecules
O
OOHH
N
O
NH2O
N+
H
H
HO-
O
N
NH
SCH3
N
N
CH3

Basic method for crystal energy landscapes ~ thermodynamically feasible crystal structures
v Use quantum mechanics to predict molecular structure and represent the charge distribution within the molecule (repeat with multiple conformers for flexible molecules, using intramolecular energy penalty ΔEintra)
v Use search method to generate plausible crystal structures (~3000 MOLPAK or ~105 Crystal Predictor for each rigid conformation, or >106 for flexible CrystalPredictor) for Z’=1,...
v Use advanced models of the intermolecular forces (distributed multipoles to represent lone pair & π electron density) to minimize the intermolecular lattice energy Uinter of each crystal structure.
v Refine conformation within crystal to minimize Elatt= Uinter + ΔEintra > Basic Crystal (Lattice) Energy Landscape
v Estimate lattice modes, elastic tensor & harmonic free energies for rigid molecules and confidence in relative stabilities.
v Calculate other properties: PXRD, morphologies Karamertzanis PG, Kazantsev AV, Issa N, Welch GWA, Adjiman CS, Pantelides CC, Price SL 2009. J Chem
Theory Comput 5, 1432

Rearrangement more likely in nucelation Hulme AT, Johnston A, Florence AJ, Fernandes P, Shankland K, Bedford C, Welch GWA, Sadiq G, Haynes DA, Motherwell WDS, Tocher DA, SLP J.Am.Chem.Soc. 2007, 129, 3649
2001 blindtest predicted dimers ~ chains invited search for dimer
polymorph Screen found plastic phase
+ chain polymorph (Z’=2 arrested crystallisation) & chain solvates
Imide can readily rearrange hydrogen bonds to most stable form
chain within computational error
Electrostatic potential on the water-accessible surface Scale ±60 kJ/mol
Predicted Observed





























