Embed Size (px)
Citation preview

Computational Chemistry Approach for the Early
Detection of Drug-Induced Idiosyncratic Liver Toxicity
MAYKEL CRUZ-MONTEAGUDO,1,2 M. NATALIA D. S. CORDEIRO,3 FERNANDA BORGES1
1Physico-Chemical Molecular Research Unit, Department of Organic Chemistry, Faculty ofPharmacy, University of Porto, 4150-047 Porto, Portugal
2CEQA & CBQ, Central University of ‘‘Las Villas’’, Santa Clara 54830, Cuba3REQUIMTE, Department of Chemistry, Faculty of Sciences, University of Porto,
4169-007 Porto, Portugal
Received 2 May 2007; Revised 21 June 2007; Accepted 5 July 2007DOI 10.1002/jcc.20812
Published online 17 August 2007 in Wiley InterScience (www.interscience.wiley.com).
Abstract: Idiosyncratic drug toxicity (IDT), considered as a toxic host-dependent event, with an apparent lack of
dose response relationship, is usually not predictable from early phases of clinical trials, representing a particularly
confounding complication in drug development. Albeit a rare event (usually \1/5000), IDT is often life threatening
and is one of the major reasons new drugs never reach the market or are withdrawn post marketing. Computational
methodologies, like the computer-based approach proposed in the present study, can play an important role in
addressing IDT in early drug discovery. We report for the first time a systematic evaluation of classification models
to predict idiosyncratic hepatotoxicity based on linear discriminant analysis (LDA), artificial neural networks (ANN),
and machine learning algorithms (OneR) in conjunction with a 3D molecular structure representation and feature
selection methods. These modeling techniques (LDA, feature selection to prevent over-fitting and multicollinearity,
ANN to capture nonlinear relationships in the data, as well as the simple OneR classifier) were found to produce
QSTR models with satisfactory internal cross-validation statistics and predictivity on an external subset of chemicals.
More specifically, the models reached values of accuracy/sensitivity/specificity over 84%/78%/90%, respectively in
the training series along with predictivity values ranging from ca. 78 to 86% of correctly classified drugs. An LDA-
based desirability analysis was carried out in order to select the levels of the predictor variables needed to trigger the
more desirable drug, i.e. the drug with lower potential for idiosyncratic hepatotoxicity. Finally, two external test sets
were used to evaluate the ability of the models in discriminating toxic from nontoxic structurally and pharmacologi-
cally related drugs and the ability of the best model (LDA) in detecting potential idiosyncratic hepatotoxic drugs,
respectively. The computational approach proposed here can be considered as a useful tool in early IDT prognosis.
q 2007 Wiley Periodicals, Inc. J Comput Chem 29: 533–549, 2008
Key words: chemoinformatics; computational prediction; drug development; early detection; idiosyncratic hepato-
toxicity; quantitative structure–toxicity relationships
Introduction
Adverse drug reactions (ADRs) are major clinical concern as
being between the fourth and sixth leading causes of death in
hospitals.1 However, the true incidence of adverse drug reactions
is largely unknown since only 10–15% of recognized reactions
are ever reported.2 In particular, drug idiosyncrasy is an ADR of
unknown etiology observed in only a small fraction of the
human population on certain drug regimens, with an apparent
lack of a dose-response or temporal relationship, and displaying
a variable time to onset in relation to the start of therapy. These
particularities make its occurrence by large unpredictable. ADRs
are also commonly thought to arise either from drug metabolism
polymorphisms or from allergic response to a drug or its metab-
olite(s). However, supporting evidence for either hypothesis is
lacking for majority of drugs.3
Idiosyncratic drug toxicity (IDT), often not detected during
preclinical trials due to its rather low incidence, dose-independ-
ence, and species sensitivity, is a particular barrier to drug de-
velopment.4 In fact, IDT is usually not observed until the drug
Contract/grant sponsor: Portuguese Fundacao para a Ciencia e a Tecno-
logia (FCT); contract/grant number: SFRH/BD/30698/2006
Contract/grant sponsor: Physico-Chemical Molecular Research Unit
(Department of Organic Chemistry, Faculty of Pharmacy, University of
Porto, Portugal)
Correspondence to: F. Borges; e-mail: [email protected]
q 2007 Wiley Periodicals, Inc.

has been on the market for some time and large populations are
exposed to it. Nevertheless, its results can be devastating, and
represent an unacceptable health risk to patient populations as
well as an undesirable financial risk for the pharmaceutical
industry.5–7 Idiosyncratic drug hepatotoxicity in particular, which
can lead in many cases to fatal liver failures,8–14 is amongst the
most frequent causes of post-marketing drugs withdrawals.15
Very recently, Li16 proposed an interesting hypothesis aimed
at explaining the low incidence of this type of toxic event.
According to this author, factors such as genetic differences,
age, sex, exposure, environmental factors, and chemical proper-
ties play key roles in IDT. This multifactorial approach is more
likely to yield useful results for a drug already known to induce
idiosyncratic toxicity, but may not be useful for the prediction
of sensitivity towards new drugs with unknown idiosyncratic
toxic potential.16
As soon as the relevant role of chemical properties in the
occurrence of an IDT event become apparent, one can recognize
a unique opportunity to attempt to develop alternative computa-
tional (in silico-based) models that could be used to predict this
toxicological feature.
The use of in silico-based models, particularly quantitative
structure-property/activity/toxicity relationships (QSPR/QSAR/
QSTR) date back to much longer than the first popularity of
computers.17,18 These studies are nowadays central in the toxi-
cology field, particularly in drug development being this
research facilitated by the availability of powerful computers
and high quality molecular databases. These models have proved
to be useful tools for predicting a vast number of chemical prop-
erties19,20 and biological features21–23 with many applications in
the chemical and life sciences. In particular, the efficacy of
QSTR in predicting toxicological features has been widely
reported in the literature.5,6,24–26
In this work we have examined the ability of a structurally
and pharmacologically diverse dataset of 74 drugs (from which
33 drugs are known to be associated with idiosyncratic hepato-
toxic events and 41 are not associated) to provide robust and
predictive QSTR models of idiosyncratic hepatotoxicity, based
on several classification modeling techniques (parametric and
nonparametric) along with a 3D molecular structure representa-
tion and feature selection algorithms. Additionally, the discrimi-
native power of all the developed models was evaluated by
using an external set comprising three pairs of drugs, chemically
and pharmacologically related, but having an observed opposite
toxicological profile (troglitazone vs. pioglitazone; tolcapone vs.
entacapone; clozapine vs. olanzapine). Finally, the ability of the
best model found (LDA model) in detecting potential idiosyn-
cratic hepatotoxic drugs was definitively checked by using an
external set of 13 new idiosyncratic hepatotoxic drugs never
used for training.
Methods
Radial Distribution Function Approach
The so-called Radial Distribution Function (RDF) descriptors27
form a set of consistent 3D-chemical descriptors, which are
becoming increasingly used in QSARs, in both drug design and
environmental sciences.28,29 The RDF of a group of N atoms
can be interpreted as the probability distribution of finding an
atom in a spherical volume of radius r. These descriptors are
then based on the distance distribution in the geometrical repre-
sentation of a molecule30 and defined, accordingly, as follows:
RDFðrÞ ¼ f �XN�1
i¼1
XN
j>i
AiAje�Bðr�rijÞ2 (1)
where f is a scaling factor.
By including characteristic atomic properties A of the atoms iand j, the RDF codes30 can be used in different tasks to fit the
requirements of the information to be modeled. These atomic
properties enable the discrimination of the atoms of a molecule
for almost any property that can be attributed to an atom. The
exponential term contains the distance rij between the atoms iand j and the smoothing parameter B, which defines the proba-
bility distribution of the individual distances; B can be inter-
preted as a temperature factor that defines the movement of the
atoms.
The radial distribution function in this form meets all the
requirements for being considered a 3D structural descriptor: it
is independent of the number of atoms, i.e. of the size of a mol-
ecule, it is unique regarding the three-dimensional arrangement
of the atoms, and it is resistant towards translation and rotation
of the entire molecule. Additionally, the RDF descriptors can be
restricted to specific atom types or distance ranges to represent
specific information in a certain three-dimensional structure
space (e.g. to describe steric hindrance or structure/activity prop-
erties within a molecule).
Finally, the RDF chemical description is reversible, since the
RDF codes are interpretable by using simple rules sets, they can
easily be converted back into the corresponding 3D structure.
Besides information about interatomic distances in the entire
molecule, the RDF descriptors provide further valuable informa-
tion, for instance, about bond distances, ring types, planar and
nonplanar systems, and atom types.
Data Set
The ideal training set for any predictive drug toxicity model is
large, diverse and internally consistent; i.e. composed of drugs
able to represent the structural and pharmacological universe
related to the toxicological problem intended to be modelled.
Accordingly, our training set comprises a subset of 33 drugs
known to be associated with idiosyncratic hepatotoxicity (IH),
which have been reported previously by Li,16 along with another
subset of 41 drugs with no idiosyncratic hepatotoxicity (NIH)
reported (see Table 1 for details). The latter subset was selected
from the Index of Pharmaceutical Specialties INTERCOM’94,31
by selecting one drug per pharmacological class which was not
associated with any type of idiosyncratic hepatotoxicity. There-
fore, most known pharmacological groups are included.
The molecular structures of all drugs were represented on
ChemDraw Ultra 9.0.32 Compounds were initially sketched into
HyperChem (version 7.5).33 Hyperchem provided reasonable
starting geometries for each compound, by resorting to the
534 Cruz-Monteagudo, Cordeiro, and Borges • Vol. 29, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc
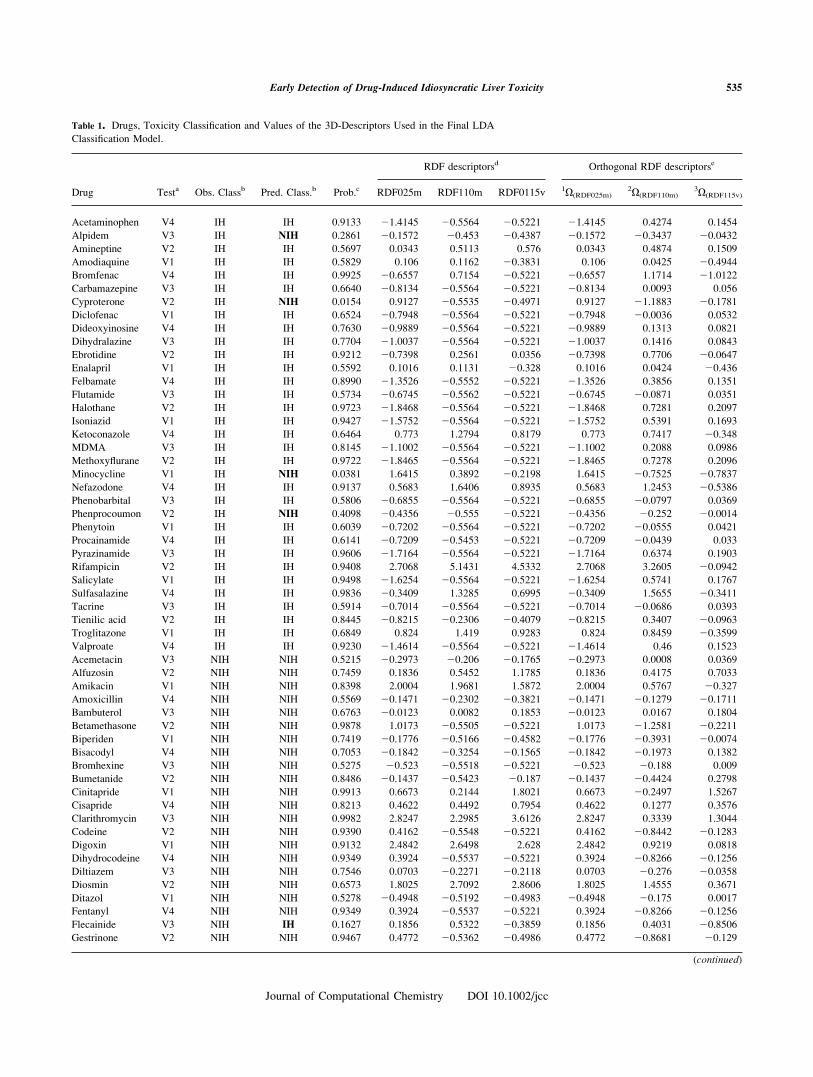
Table 1. Drugs, Toxicity Classification and Values of the 3D-Descriptors Used in the Final LDA
Classification Model.
Drug Testa Obs. Classb Pred. Class.b Prob.c
RDF descriptorsd Orthogonal RDF descriptorse
RDF025m RDF110m RDF0115v 1X(RDF025m)2X(RDF110m)
3X(RDF115v)
Acetaminophen V4 IH IH 0.9133 21.4145 20.5564 20.5221 21.4145 0.4274 0.1454
Alpidem V3 IH NIH 0.2861 20.1572 20.453 20.4387 20.1572 20.3437 20.0432
Amineptine V2 IH IH 0.5697 0.0343 0.5113 0.576 0.0343 0.4874 0.1509
Amodiaquine V1 IH IH 0.5829 0.106 0.1162 20.3831 0.106 0.0425 20.4944
Bromfenac V4 IH IH 0.9925 20.6557 0.7154 20.5221 20.6557 1.1714 21.0122
Carbamazepine V3 IH IH 0.6640 20.8134 20.5564 20.5221 20.8134 0.0093 0.056
Cyproterone V2 IH NIH 0.0154 0.9127 20.5535 20.4971 0.9127 21.1883 20.1781
Diclofenac V1 IH IH 0.6524 20.7948 20.5564 20.5221 20.7948 20.0036 0.0532
Dideoxyinosine V4 IH IH 0.7630 20.9889 20.5564 20.5221 20.9889 0.1313 0.0821
Dihydralazine V3 IH IH 0.7704 21.0037 20.5564 20.5221 21.0037 0.1416 0.0843
Ebrotidine V2 IH IH 0.9212 20.7398 0.2561 0.0356 20.7398 0.7706 20.0647
Enalapril V1 IH IH 0.5592 0.1016 0.1131 20.328 0.1016 0.0424 20.436
Felbamate V4 IH IH 0.8990 21.3526 20.5552 20.5221 21.3526 0.3856 0.1351
Flutamide V3 IH IH 0.5734 20.6745 20.5562 20.5221 20.6745 20.0871 0.0351
Halothane V2 IH IH 0.9723 21.8468 20.5564 20.5221 21.8468 0.7281 0.2097
Isoniazid V1 IH IH 0.9427 21.5752 20.5564 20.5221 21.5752 0.5391 0.1693
Ketoconazole V4 IH IH 0.6464 0.773 1.2794 0.8179 0.773 0.7417 20.348
MDMA V3 IH IH 0.8145 21.1002 20.5564 20.5221 21.1002 0.2088 0.0986
Methoxyflurane V2 IH IH 0.9722 21.8465 20.5564 20.5221 21.8465 0.7278 0.2096
Minocycline V1 IH NIH 0.0381 1.6415 0.3892 20.2198 1.6415 20.7525 20.7837
Nefazodone V4 IH IH 0.9137 0.5683 1.6406 0.8935 0.5683 1.2453 20.5386
Phenobarbital V3 IH IH 0.5806 20.6855 20.5564 20.5221 20.6855 20.0797 0.0369
Phenprocoumon V2 IH NIH 0.4098 20.4356 20.555 20.5221 20.4356 20.252 20.0014
Phenytoin V1 IH IH 0.6039 20.7202 20.5564 20.5221 20.7202 20.0555 0.0421
Procainamide V4 IH IH 0.6141 20.7209 20.5453 20.5221 20.7209 20.0439 0.033
Pyrazinamide V3 IH IH 0.9606 21.7164 20.5564 20.5221 21.7164 0.6374 0.1903
Rifampicin V2 IH IH 0.9408 2.7068 5.1431 4.5332 2.7068 3.2605 20.0942
Salicylate V1 IH IH 0.9498 21.6254 20.5564 20.5221 21.6254 0.5741 0.1767
Sulfasalazine V4 IH IH 0.9836 20.3409 1.3285 0.6995 20.3409 1.5655 20.3411
Tacrine V3 IH IH 0.5914 20.7014 20.5564 20.5221 20.7014 20.0686 0.0393
Tienilic acid V2 IH IH 0.8445 20.8215 20.2306 20.4079 20.8215 0.3407 20.0963
Troglitazone V1 IH IH 0.6849 0.824 1.419 0.9283 0.824 0.8459 20.3599
Valproate V4 IH IH 0.9230 21.4614 20.5564 20.5221 21.4614 0.46 0.1523
Acemetacin V3 NIH NIH 0.5215 20.2973 20.206 20.1765 20.2973 0.0008 0.0369
Alfuzosin V2 NIH NIH 0.7459 0.1836 0.5452 1.1785 0.1836 0.4175 0.7033
Amikacin V1 NIH NIH 0.8398 2.0004 1.9681 1.5872 2.0004 0.5767 20.327
Amoxicillin V4 NIH NIH 0.5569 20.1471 20.2302 20.3821 20.1471 20.1279 20.1711
Bambuterol V3 NIH NIH 0.6763 20.0123 0.0082 0.1853 20.0123 0.0167 0.1804
Betamethasone V2 NIH NIH 0.9878 1.0173 20.5505 20.5221 1.0173 21.2581 20.2211
Biperiden V1 NIH NIH 0.7419 20.1776 20.5166 20.4582 20.1776 20.3931 20.0074
Bisacodyl V4 NIH NIH 0.7053 20.1842 20.3254 20.1565 20.1842 20.1973 0.1382
Bromhexine V3 NIH NIH 0.5275 20.523 20.5518 20.5221 20.523 20.188 0.009
Bumetanide V2 NIH NIH 0.8486 20.1437 20.5423 20.187 20.1437 20.4424 0.2798
Cinitapride V1 NIH NIH 0.9913 0.6673 0.2144 1.8021 0.6673 20.2497 1.5267
Cisapride V4 NIH NIH 0.8213 0.4622 0.4492 0.7954 0.4622 0.1277 0.3576
Clarithromycin V3 NIH NIH 0.9982 2.8247 2.2985 3.6126 2.8247 0.3339 1.3044
Codeine V2 NIH NIH 0.9390 0.4162 20.5548 20.5221 0.4162 20.8442 20.1283
Digoxin V1 NIH NIH 0.9132 2.4842 2.6498 2.628 2.4842 0.9219 0.0818
Dihydrocodeine V4 NIH NIH 0.9349 0.3924 20.5537 20.5221 0.3924 20.8266 20.1256
Diltiazem V3 NIH NIH 0.7546 0.0703 20.2271 20.2118 0.0703 20.276 20.0358
Diosmin V2 NIH NIH 0.6573 1.8025 2.7092 2.8606 1.8025 1.4555 0.3671
Ditazol V1 NIH NIH 0.5278 20.4948 20.5192 20.4983 20.4948 20.175 0.0017
Fentanyl V4 NIH NIH 0.9349 0.3924 20.5537 20.5221 0.3924 20.8266 20.1256
Flecainide V3 NIH IH 0.1627 0.1856 0.5322 20.3859 0.1856 0.4031 20.8506
Gestrinone V2 NIH NIH 0.9467 0.4772 20.5362 20.4986 0.4772 20.8681 20.129
(continued)
535Early Detection of Drug-Induced Idiosyncratic Liver Toxicity
Journal of Computational Chemistry DOI 10.1002/jcc
MM1 molecular mechanics force field. Structures were then
further optimized by the semiempirical molecular orbital pack-
age MOPAC 6.0.34 The PM3 Hamiltonian35 was used to obtain
minimal-energy geometry optimized structures, which in turn
were also checked by frequency calculations. RDF molecular
descriptors were then calculated at radii ranging from 1.0 to
15.5 A, with increments of 0.5 A, using the Dragon software
package.36 As weighting properties, we tried all the atomic prop-
erties available in the Dragon software (masses, van der Waals
volumes, Sanderson electronegativities and polarizabilities),
finally computing a total of 150 descriptors.30
LDA Classification Model
The task here is to find a classification function [eq. (2)] that
best describes the idiosyncratic hepatotoxicity IH, as a linear
combination of the predictor X-variables (the RDF descriptors)
weighed by the coefficients bk:
IH ¼ b0 þ b1 3 RDFx1 þ b2 3 RDFx2 þ � � � þ bk 3 RDFxk (2)
In developing this classification function, IH values of 11 and
21 were assigned to toxic and nontoxic drugs, respectively, and
the equation coefficients were obtained by the least squares
method as implemented in the general discriminant analysis
(GDA) module of the STATISTICA 6.0 software package.37 In
addition, all the predictor variables were standardized38 after
LDA modeling and the forward stepwise (FS) method was
selected as the strategy for variable selection.39
The quality of the final LDA model was established by
examining its discriminatory power—Wilk’s U statistic and the
square of Mahalanobis’s distance (D2), and statistical signifi-
cance—Fisher ratio (F) and p-level (p). The ratio between cases
and predictor variables (q value) was also inspected to avoid
over-fitting or chance correlation problems.38
Specifically, the Wilk’s U statistic for the overall discrimina-
tion is computed as the ratio of the determinant of the within-
groups variance/covariance matrix (W) over the determinant of
the total variance/covariance matrix (T)40:
U ¼ Det:ðWÞDet:ðTÞ (3)
The Mahalanobis distance is the distance of a case from the
centroid in the multidimensional space, defined by the correlated
independent variables. It is computed as follows40:
Table 1. (Continued)
Drug Testa Obs. Classb Pred. Class.b Prob.c
RDF descriptorsd Orthogonal RDF descriptorse
RDF025m RDF110m RDF0115v 1X(RDF025m)2X(RDF110m)
3X(RDF115v)
Gliclazide V1 NIH NIH 0.7733 20.2385 20.3543 0.0696 20.2385 20.1884 0.3961
Indapamide V4 NIH NIH 0.5774 20.3289 20.4541 20.5113 20.3289 20.2253 20.0894
Ketoprofen V3 NIH IH 0.4441 20.6493 20.5564 20.5221 20.6493 20.1048 0.0316
Ketotifen V2 NIH NIH 0.8720 0.1207 20.5564 20.5221 0.1207 20.6404 20.083
Loratadine V1 NIH NIH 0.8908 0.3206 20.3336 20.2744 0.3206 20.5566 20.0481
Mesterolone V4 NIH NIH 0.9486 0.479 20.5564 20.5221 0.479 20.8896 20.1363
Methylergometrine V3 NIH IH 0.4919 0.36 0.2405 20.3551 0.36 20.0099 20.6062
Nimodipine V2 NIH NIH 0.9176 0.3806 20.4408 20.4142 0.3806 20.7055 20.1087
Nitrendipine V1 NIH NIH 0.8862 0.1688 20.5564 20.5221 0.1688 20.6738 20.0901
Oxazepam V4 NIH NIH 0.5564 20.4872 20.5564 20.5221 20.4872 20.2176 0.0074
Pefloxacine V3 NIH NIH 0.9244 0.4004 20.4526 20.4169 0.4004 20.7311 20.1047
Penicillin V2 NIH NIH 0.5533 20.4688 20.4994 20.4402 20.4688 20.1733 0.0397
Quazepam V1 NIH NIH 0.7975 20.0587 20.5432 20.5221 20.0587 20.5024 20.0672
Quinapril V4 NIH NIH 0.9742 0.7621 20.4408 20.3245 0.7621 20.9709 20.0758
Retinol V3 NIH NIH 0.8121 0.0152 20.322 20.1292 0.0152 20.3326 0.1331
Tetracycline V2 NIH NIH 0.9934 1.2267 20.4368 20.2622 1.2267 21.29 20.0858
Tetrazepam V1 NIH NIH 0.7439 20.1854 20.5564 20.5221 20.1854 20.4275 20.0374
Ursodeoxycholic acid V4 NIH NIH 0.9237 1.0383 0.3361 0.2034 1.0383 20.3861 20.2271
Veralipride V3 NIH NIH 0.7652 0.0008 20.3041 20.2291 0.0008 20.3047 0.0206
aV1-V4 denote drugs used for prediction in the respective validation experiment; on the other three experiments each
drug was used for training.bThe observed/predicted classification for the selected drugs; the symbols indicate idiosyncratic hepatotoxic (IH) and
nonidiosyncratic hepatotoxic (NIH) drugs.cCalculated posterior probability of the drugs to be toxic (IH) or not (NIH) according to the final LDA model.dRDF025m, RDF110m and RDF115v represent, respectively, the radial distribution function weighted by atomic
masses at 2.5 A, at 11.0 A and by van der Waals volumes at 11.5 A.e1X(RDF025m),
2X(RDF110m), and3X(RDF115v) are the orthogonal complements of descriptors RDF025m, RDF110m and
RDF115v, respectively, determined by Randic’s approach.
536 Cruz-Monteagudo, Cordeiro, and Borges • Vol. 29, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc
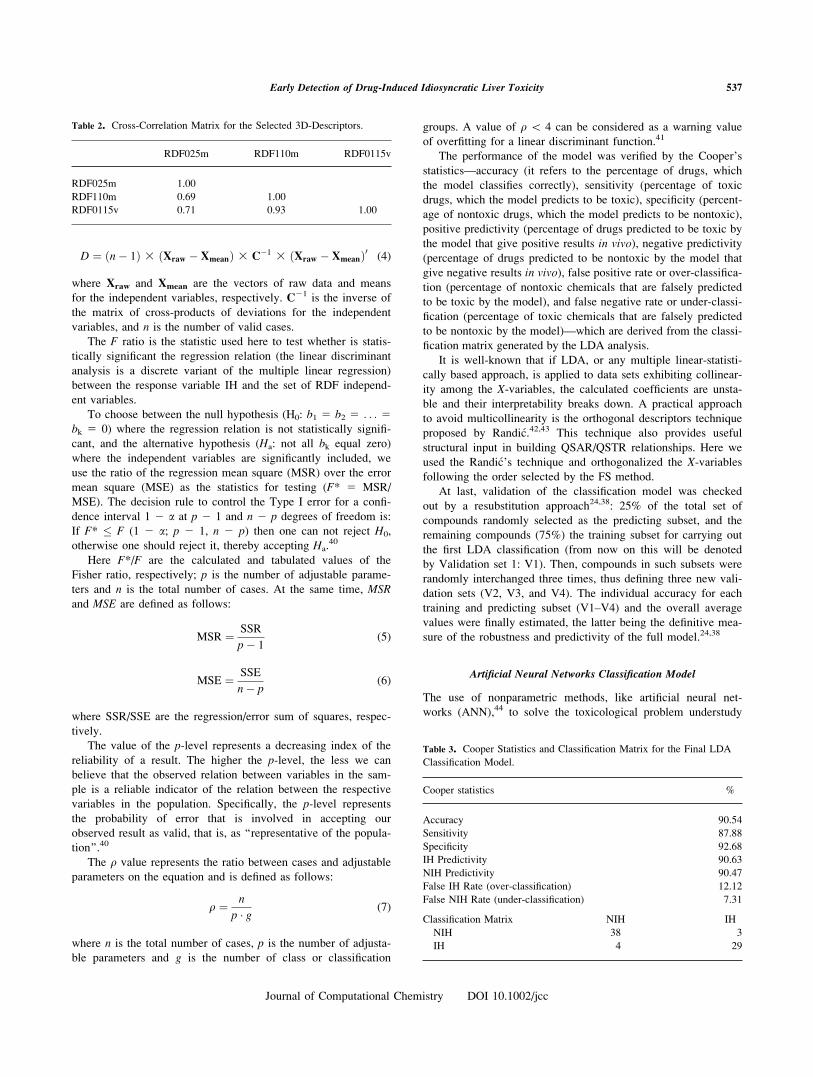
D ¼ ðn� 1Þ 3 ðXraw � XmeanÞ 3 C�1 3 ðXraw � XmeanÞ0 (4)
where Xraw and Xmean are the vectors of raw data and means
for the independent variables, respectively. C21 is the inverse of
the matrix of cross-products of deviations for the independent
variables, and n is the number of valid cases.
The F ratio is the statistic used here to test whether is statis-
tically significant the regression relation (the linear discriminant
analysis is a discrete variant of the multiple linear regression)
between the response variable IH and the set of RDF independ-
ent variables.
To choose between the null hypothesis (H0: b1 5 b2 5 . . . 5bk 5 0) where the regression relation is not statistically signifi-
cant, and the alternative hypothesis (Ha: not all bk equal zero)
where the independent variables are significantly included, we
use the ratio of the regression mean square (MSR) over the error
mean square (MSE) as the statistics for testing (F* 5 MSR/
MSE). The decision rule to control the Type I error for a confi-
dence interval 1 2 a at p 2 1 and n 2 p degrees of freedom is:
If F* � F (1 2 a; p 2 1, n 2 p) then one can not reject H0,
otherwise one should reject it, thereby accepting Ha.40
Here F*/F are the calculated and tabulated values of the
Fisher ratio, respectively; p is the number of adjustable parame-
ters and n is the total number of cases. At the same time, MSRand MSE are defined as follows:
MSR ¼ SSR
p� 1(5)
MSE ¼ SSE
n� p(6)
where SSR/SSE are the regression/error sum of squares, respec-
tively.
The value of the p-level represents a decreasing index of the
reliability of a result. The higher the p-level, the less we can
believe that the observed relation between variables in the sam-
ple is a reliable indicator of the relation between the respective
variables in the population. Specifically, the p-level represents
the probability of error that is involved in accepting our
observed result as valid, that is, as ‘‘representative of the popula-
tion’’.40
The q value represents the ratio between cases and adjustable
parameters on the equation and is defined as follows:
q ¼ n
p � g (7)
where n is the total number of cases, p is the number of adjusta-
ble parameters and g is the number of class or classification
groups. A value of q \ 4 can be considered as a warning value
of overfitting for a linear discriminant function.41
The performance of the model was verified by the Cooper’s
statistics—accuracy (it refers to the percentage of drugs, which
the model classifies correctly), sensitivity (percentage of toxic
drugs, which the model predicts to be toxic), specificity (percent-
age of nontoxic drugs, which the model predicts to be nontoxic),
positive predictivity (percentage of drugs predicted to be toxic by
the model that give positive results in vivo), negative predictivity
(percentage of drugs predicted to be nontoxic by the model that
give negative results in vivo), false positive rate or over-classifica-tion (percentage of nontoxic chemicals that are falsely predicted
to be toxic by the model), and false negative rate or under-classi-
fication (percentage of toxic chemicals that are falsely predicted
to be nontoxic by the model)—which are derived from the classi-
fication matrix generated by the LDA analysis.
It is well-known that if LDA, or any multiple linear-statisti-
cally based approach, is applied to data sets exhibiting collinear-
ity among the X-variables, the calculated coefficients are unsta-
ble and their interpretability breaks down. A practical approach
to avoid multicollinearity is the orthogonal descriptors technique
proposed by Randic.42,43 This technique also provides useful
structural input in building QSAR/QSTR relationships. Here we
used the Randic’s technique and orthogonalized the X-variablesfollowing the order selected by the FS method.
At last, validation of the classification model was checked
out by a resubstitution approach24,38: 25% of the total set of
compounds randomly selected as the predicting subset, and the
remaining compounds (75%) the training subset for carrying out
the first LDA classification (from now on this will be denoted
by Validation set 1: V1). Then, compounds in such subsets were
randomly interchanged three times, thus defining three new vali-
dation sets (V2, V3, and V4). The individual accuracy for each
training and predicting subset (V1–V4) and the overall average
values were finally estimated, the latter being the definitive mea-
sure of the robustness and predictivity of the full model.24,38
Artificial Neural Networks Classification Model
The use of nonparametric methods, like artificial neural net-
works (ANN),44 to solve the toxicological problem understudy
Table 2. Cross-Correlation Matrix for the Selected 3D-Descriptors.
RDF025m RDF110m RDF0115v
RDF025m 1.00
RDF110m 0.69 1.00
RDF0115v 0.71 0.93 1.00
Table 3. Cooper Statistics and Classification Matrix for the Final LDA
Classification Model.
Cooper statistics %
Accuracy 90.54
Sensitivity 87.88
Specificity 92.68
IH Predictivity 90.63
NIH Predictivity 90.47
False IH Rate (over-classification) 12.12
False NIH Rate (under-classification) 7.31
Classification Matrix NIH IH
NIH 38 3
IH 4 29
537Early Detection of Drug-Induced Idiosyncratic Liver Toxicity
Journal of Computational Chemistry DOI 10.1002/jcc
was investigated. Specifically, the ability of a classification
ANN model derived from the descriptors identified by FS-LDA
was evaluated. The model was built with the aid of STATIS-
TICA 6.037 using a Radial Basis Function (RBF) architecture.45
RBF networks combine a single radial hidden layer with a dot
product output layer. The hidden layer neurons act as clustering
centers, grouping similar training cases, and the output layer
forms a discriminant function. Since the clustering transforma-
tion is nonlinear, a linear output layer is sufficient to perform an
overall nonlinear function. These networks follow a two stage
training process, namely: (i) assignment of the radial centers and
their deviations and (ii) optimization of the output layer. The
quality of the RBF-ANN model was assessed by examining its
performance, errors and percentages of correctly classified IH/
NIH drugs in training and testing series, based on the distribu-
tion of compounds in the V1 set above referred to. Network
training was conducted by using 75% of the drugs (labeled with
V2, V3, or V4 on Table 1) and the remaining 25% of the drugs
(labeled with V1 on Table 1) were used for model testing.
Machine Learning Classification Algorithm
Here, the ability of the OneR classification algorithm,46 as
implemented in software WEKA (Waikato Environment for
Knowledge Analysis),46,47 to flag potential idiosyncratic hepato-
toxicity was tested. OneR, short for ‘‘One Rule’’, is a simple
machine learning classification algorithm that generates a one-
level decision tree. OneR is able to infer typically simple classi-
fication rules from a set of instances that are straightforward for
humans to interpret. This algorithm creates one rule for each at-
tribute in the training data, and then selects the best rule as its
‘‘one rule’’. To create a rule for a specific attribute, the most fre-
quent class for each attribute value must be determined. A rule
is thus simply a set of attribute values bound to their majority
class; the rule is based on such binding for each value of the at-
tribute. One can also define the error rate of a rule as the num-
ber of misclassified training data instances by using the rule.46
Typically machine learning algorithms select the rule with
the lowest error rate as the best one. If two or more rules
have the same error rate, the rule is chosen at random. In oppo-
sition, the OneR algorithm in WEKA picks the rule with the
highest number of correct instances, not the lowest error rate,
and does not randomly select a rule when error rates are identi-
cal. The best rule obtained was then validated by means of four-
fold cross internal validation (an iterative process of rederiving
the model leaving the 25% of the data out each time).
Desirability Analysis
Response/desirability profiling allows tracing the response sur-
face produced by fitting the observed responses using an equa-
tion based on levels of the predictor variables. The predicted
values for the dependent variable at different combinations of
Figure 1. Receiver operating characteristic curve (ROC curve) for
the final LDA model. [Color figure can be viewed in the online
issue, which is available at www.interscience.wiley.com.]
Table 4. LDA Model Validation Experiment by Using a Resubstitution of Cases in Four External
Predicting Series.
Validation set
Robustness
Accuracy Sensitivity Specificity (IH) Predictivity (NIH) Predictivity (IH) False rate (NIH) False rate
V1 89.29 84.00 93.55 91.30 87.88 16.00 6.45
V2 89.29 84.00 93.55 91.30 87.88 16.00 6.45
V3 92.73 88.00 96.67 95.65 90.63 12.00 3.33
V4 81.82 83.33 80.65 76.92 86.21 16.67 19.35
Average 88.28 84.83 91.10 88.79 88.15 15.17 8.90
Predictivity
V1 83.33 75.00 90.00 85.71 81.82 25.00 10.00
V2 83.33 62.50 100.00 100.00 76.92 37.50 0.00
V3 78.94 87.50 72.73 70.00 88.89 12.50 27.27
V4 100.00 100.00 100.00 100.00 100.00 0.00 0.00
Average 86.40 81.25 84.09 86.91 86.91 18.75 9.32
538 Cruz-Monteagudo, Cordeiro, and Borges • Vol. 29, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc
levels of the X-variables can be studied, specify desirability
functions for the dependent variable, and search for the levels of
the X-variables that produce the most desirable responses on the
dependent variable.48 In the present work, the desirability analy-
sis was conducted with STATISTICA 6.0, by setting the current
level of each predictor variable to the respective mean. Curva-
ture s and t parameters were fixed at 1.00 considering the linear
form of the function used to perform the discriminant analysis.
Least squares method was selected for fitting the desirability
function and surface/contours maps.
The desirability of responses need not to decrease (or
increase) linearly between inflection points in the desirability
function. Perhaps there is a ‘‘critical region’’ close to a desired,
intermediate response on a dependent variable beyond which the
desirability of the response at first drops off very quickly, but
drops off less quickly as the departure from the ‘‘targeted’’ value
becomes greater. Modeling this type of desirability function
requires ‘‘curvature’’ parameters to take into account the nonli-
nearity in the ‘‘falloff’’ of desirability between inflection points.
The s and t parameters can be specified as the value for the
exponent of the desirability function representing the curvature
in the desirability function between the low and medium inflec-
tion points of the function, and between the medium and high
inflection points of the function, respectively. Assuming that an
intermediate response is most desirable, values greater than 1.0
for the s and t parameters represent initial quicker ‘‘falloff’’ in
desirability but subsequent slower ‘‘falloff’’ in desirability as the
departure from the ‘‘targeted’’ value becomes greater. Values
less than 1.0 for the s and t parameters represent initial slower
‘‘falloff’’ in desirability but subsequent quicker ‘‘falloff’’ in
desirability as the departure from the ‘‘targeted’’ value becomes
greater. The default specifications for both these parameters are
values of 1.0, representing linear ‘‘falloff’’ in desirability
between the medium and low inflection points as well as
between the medium and high inflection points.48
Results and Discussion
LDA Classification Model
Having designed a structurally and pharmacologically represen-
tative data set of drugs, we carried out FS-LDA to find the best
linear discriminant model detailed below:
IH ¼ 4:484� 0:235 RDF025m
þ 0:770 RDF110m� 0:443 RDF115v (8)
In this equation, RDF025m, RDF110m, and RDF115v represent
RDF molecular descriptors; the first two derived from the RDF
computed at a radius of 2.5 and 11.0 A, respectively, weighed
by atomic masses, while the latter from the RDF at 11.5 A
weighted by van der Waals volumes.
The collinearity among the different variables of this model
can be checked by analyzing the cross-correlation matrix (see
Table 2). It was observed that all variables are highly correlated
with each other (R 5 0.69–0.93), suggesting that multicollinear-
ity may be a problem. As previously mentioned, the primary
concern with multicollinearity is that uncertainty is introduced
into the estimated coefficients. Thus, to obtain more efficient
coefficients of the predictor variables, orthogonal complements
were determined for all variables in eq. (8) by the Randic’s tech-
nique, deriving the equation given below:
IH ¼ �0:473� 1:516 1XðRDF025mÞþ 1:581 2XðRDF110mÞ � 0:646 3XðRDF115vÞ (9)
F 5 17.64, p\ 0.00000001, D2 5 2.98, U 5 0.57, q 5 9.25
Here, the symbol IX(RDFx) indicates that the variable RDFx is
selected with order of importance (I) after former orthogonaliza-
tion (X).This model is able to correctly classify 90.54% of the drugs
in the training set using only three predictive variables, and also
shows a good accuracy (large F index and small p value) and
discriminatory power, see Table 3 (satisfactory U and D2 value),
as well as parsimony (q 5 9.25). It also displays, as shown in
Table 3, satisfactory values of sensitivity, specificity, positive/
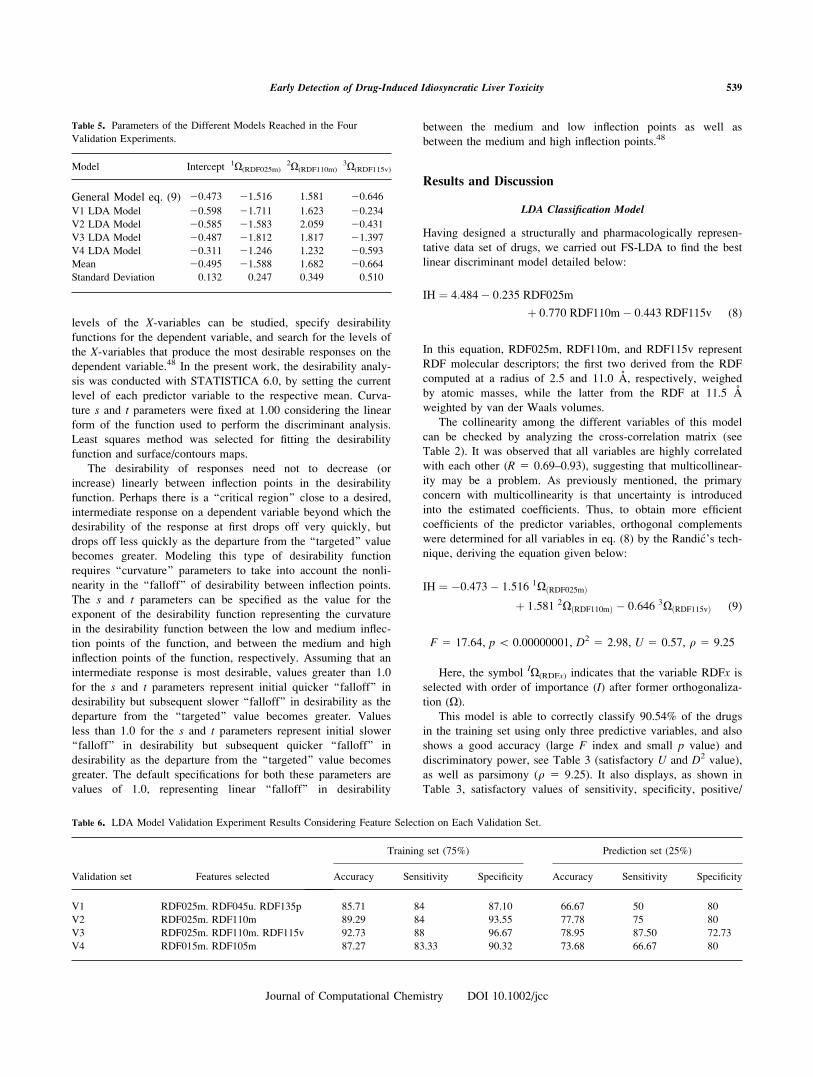
Table 5. Parameters of the Different Models Reached in the Four
Validation Experiments.
Model Intercept 1X(RDF025m)2X(RDF110m)
3X(RDF115v)
General Model eq. (9) 20.473 21.516 1.581 20.646
V1 LDA Model 20.598 21.711 1.623 20.234
V2 LDA Model 20.585 21.583 2.059 20.431
V3 LDA Model 20.487 21.812 1.817 21.397
V4 LDA Model 20.311 21.246 1.232 20.593
Mean 20.495 21.588 1.682 20.664
Standard Deviation 0.132 0.247 0.349 0.510
Table 6. LDA Model Validation Experiment Results Considering Feature Selection on Each Validation Set.
Validation set Features selected
Training set (75%) Prediction set (25%)
Accuracy Sensitivity Specificity Accuracy Sensitivity Specificity
V1 RDF025m. RDF045u. RDF135p 85.71 84 87.10 66.67 50 80
V2 RDF025m. RDF110m 89.29 84 93.55 77.78 75 80
V3 RDF025m. RDF110m. RDF115v 92.73 88 96.67 78.95 87.50 72.73
V4 RDF015m. RDF105m 87.27 83.33 90.32 73.68 66.67 80
539Early Detection of Drug-Induced Idiosyncratic Liver Toxicity
Journal of Computational Chemistry DOI 10.1002/jcc
negative predictivity, and false positive/negative rates (over/
under-classification). In addition, the receiver operating charac-
teristic curve (ROC curve) obtained, indicate that the model is
not a random, but a statistically significant, classifier. A ROC
curve49,50 plots the sensitivity versus one minus the specificity.
An ideal classifier hugs the left side and top side of the graph,
and the area under the curve is 1.0. A random classifier should
achieve �0.5 (Fig. 1).
Validation of the model was undertaken by the re-substitution
technique24,51 and the results derived for four different valida-
tion subsets are summarized in Table 4. Judging from the aver-
aged values of accuracy in the training/test subsets (88.28%/
86.40%), as well as of sensitivity, specificity, etc., the model is
robust, exhibits little dependence on the composition of the
training and test subsets, and has high predictive ability.
Since selection of test compounds from the full set is ran-
dom, we replicate four times the selection by including other
compounds than the previously selected in order to access the
influence of the test set variation over the performance of the
model. As shown in Table 4, the performance in the different
validation experiments showed slight variations. For this reason,
we report averaged values which are a more representative mea-
sure of the performance of the model. In Table 5, one can notice
the stability of model parameters for each validation experiment
contrasted to the general model.
Additionally, the robustness of the model proposed is verified
by re-doing the descriptor selection and line fitting with 75%
compounds without touching the remaining 25% compounds.
After the model is developed, the 25% compounds were used to
test the predictive ability of the model. This procedure is carried
out on the four validation sets. By looking at Table 6, one can
see that the descriptors selected each time are similar and pre-
diction results are in the similar range, which confirms the
robustness of the model.
Anyhow, the definitive measure of the predictive ability of
any model can only be assessed by using an independent set of
drugs never used for training, as carried out afterwards.
The next step is to find out if the basic assumptions of LDA
are fulfilled.38,52 As the names implies, LDA establishes a linear,
additive relationship between the molecular descriptors and the
underlying bioactivity and indeed, this is the simplest functional
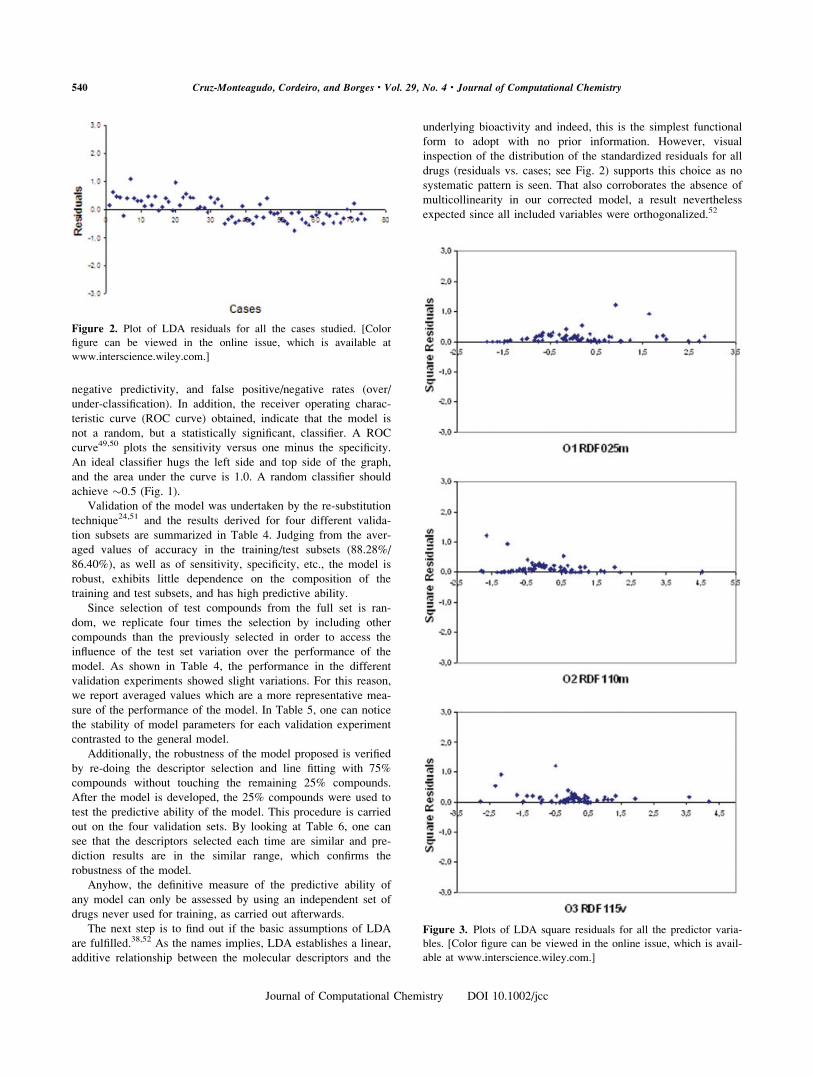
form to adopt with no prior information. However, visual
inspection of the distribution of the standardized residuals for all
drugs (residuals vs. cases; see Fig. 2) supports this choice as no
systematic pattern is seen. That also corroborates the absence of
multicollinearity in our corrected model, a result nevertheless
expected since all included variables were orthogonalized.52
Figure 2. Plot of LDA residuals for all the cases studied. [Color
figure can be viewed in the online issue, which is available at
www.interscience.wiley.com.]
Figure 3. Plots of LDA square residuals for all the predictor varia-
bles. [Color figure can be viewed in the online issue, which is avail-
able at www.interscience.wiley.com.]
540 Cruz-Monteagudo, Cordeiro, and Borges • Vol. 29, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc
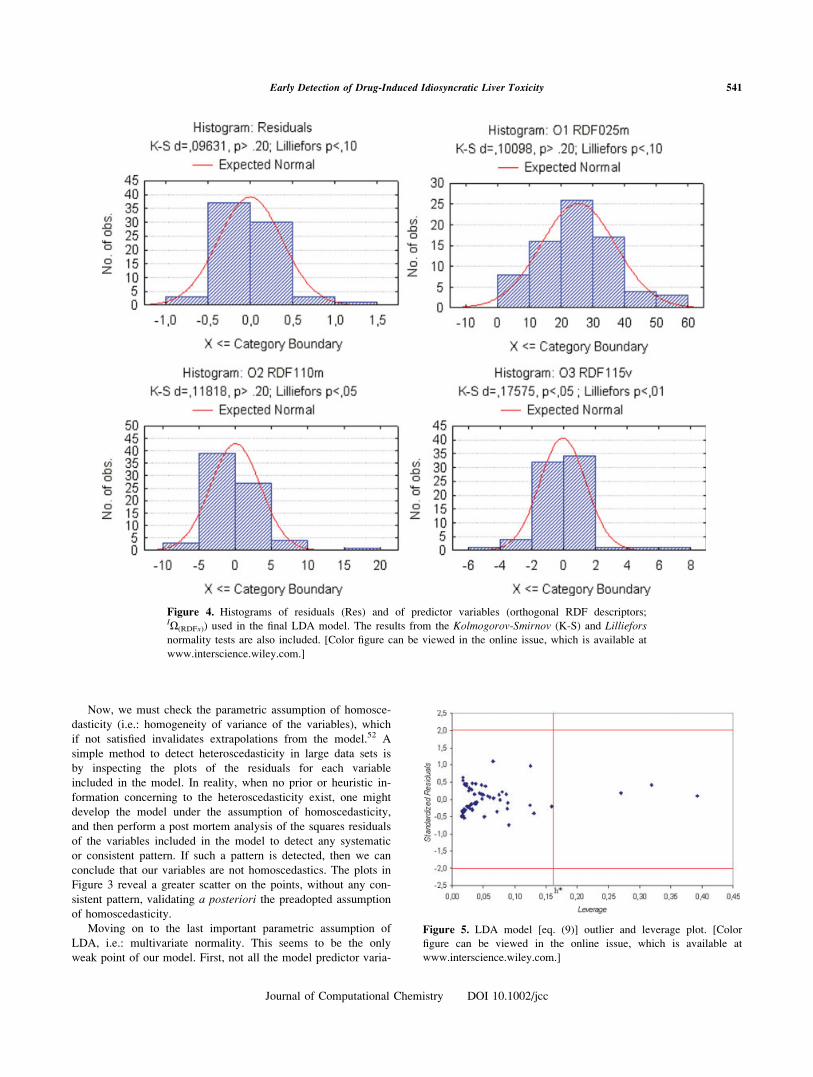
Now, we must check the parametric assumption of homosce-
dasticity (i.e.: homogeneity of variance of the variables), which
if not satisfied invalidates extrapolations from the model.52 A
simple method to detect heteroscedasticity in large data sets is
by inspecting the plots of the residuals for each variable
included in the model. In reality, when no prior or heuristic in-
formation concerning to the heteroscedasticity exist, one might
develop the model under the assumption of homoscedasticity,
and then perform a post mortem analysis of the squares residuals
of the variables included in the model to detect any systematic
or consistent pattern. If such a pattern is detected, then we can
conclude that our variables are not homoscedastics. The plots in
Figure 3 reveal a greater scatter on the points, without any con-
sistent pattern, validating a posteriori the preadopted assumption
of homoscedasticity.
Moving on to the last important parametric assumption of
LDA, i.e.: multivariate normality. This seems to be the only
weak point of our model. First, not all the model predictor varia-
Figure 4. Histograms of residuals (Res) and of predictor variables (orthogonal RDF descriptors;IX(RDFx)) used in the final LDA model. The results from the Kolmogorov-Smirnov (K-S) and Lillieforsnormality tests are also included. [Color figure can be viewed in the online issue, which is available at
www.interscience.wiley.com.]
Figure 5. LDA model [eq. (9)] outlier and leverage plot. [Color
figure can be viewed in the online issue, which is available at
www.interscience.wiley.com.]
541Early Detection of Drug-Induced Idiosyncratic Liver Toxicity
Journal of Computational Chemistry DOI 10.1002/jcc
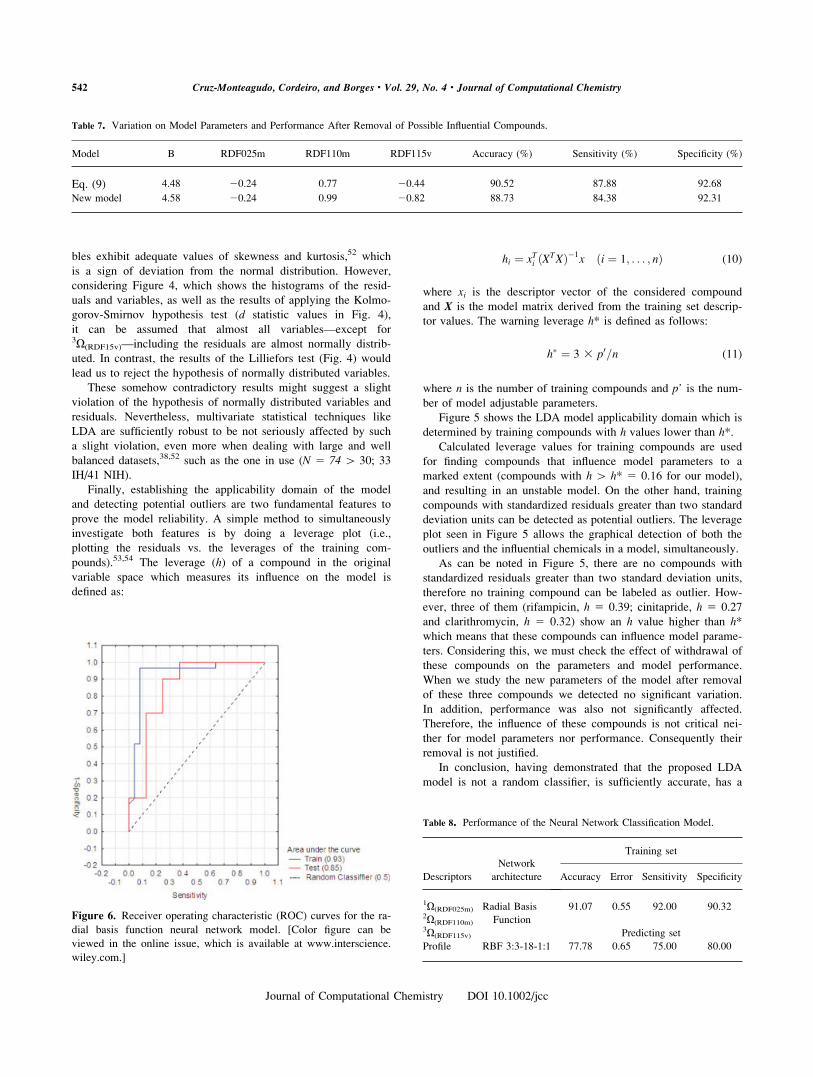
bles exhibit adequate values of skewness and kurtosis,52 which
is a sign of deviation from the normal distribution. However,
considering Figure 4, which shows the histograms of the resid-
uals and variables, as well as the results of applying the Kolmo-
gorov-Smirnov hypothesis test (d statistic values in Fig. 4),
it can be assumed that almost all variables—except for3X(RDF15v)—including the residuals are almost normally distrib-
uted. In contrast, the results of the Lilliefors test (Fig. 4) would
lead us to reject the hypothesis of normally distributed variables.
These somehow contradictory results might suggest a slight
violation of the hypothesis of normally distributed variables and
residuals. Nevertheless, multivariate statistical techniques like
LDA are sufficiently robust to be not seriously affected by such
a slight violation, even more when dealing with large and well
balanced datasets,38,52 such as the one in use (N 5 74 [ 30; 33
IH/41 NIH).
Finally, establishing the applicability domain of the model
and detecting potential outliers are two fundamental features to
prove the model reliability. A simple method to simultaneously
investigate both features is by doing a leverage plot (i.e.,
plotting the residuals vs. the leverages of the training com-
pounds).53,54 The leverage (h) of a compound in the original
variable space which measures its influence on the model is
defined as:
hi ¼ xTi ðXTXÞ�1x ði ¼ 1; . . . ; nÞ (10)
where xi is the descriptor vector of the considered compound
and X is the model matrix derived from the training set descrip-
tor values. The warning leverage h* is defined as follows:
h� ¼ 3 3 p0=n (11)
where n is the number of training compounds and p’ is the num-
ber of model adjustable parameters.
Figure 5 shows the LDA model applicability domain which is
determined by training compounds with h values lower than h*.Calculated leverage values for training compounds are used
for finding compounds that influence model parameters to a
marked extent (compounds with h [ h* 5 0.16 for our model),
and resulting in an unstable model. On the other hand, training
compounds with standardized residuals greater than two standard
deviation units can be detected as potential outliers. The leverage
plot seen in Figure 5 allows the graphical detection of both the
outliers and the influential chemicals in a model, simultaneously.
As can be noted in Figure 5, there are no compounds with
standardized residuals greater than two standard deviation units,
therefore no training compound can be labeled as outlier. How-
ever, three of them (rifampicin, h 5 0.39; cinitapride, h 5 0.27
and clarithromycin, h 5 0.32) show an h value higher than h*which means that these compounds can influence model parame-
ters. Considering this, we must check the effect of withdrawal of
these compounds on the parameters and model performance.
When we study the new parameters of the model after removal
of these three compounds we detected no significant variation.
In addition, performance was also not significantly affected.
Therefore, the influence of these compounds is not critical nei-
ther for model parameters nor performance. Consequently their
removal is not justified.
In conclusion, having demonstrated that the proposed LDA
model is not a random classifier, is sufficiently accurate, has a
Table 7. Variation on Model Parameters and Performance After Removal of Possible Influential Compounds.
Model B RDF025m RDF110m RDF115v Accuracy (%) Sensitivity (%) Specificity (%)
Eq. (9) 4.48 20.24 0.77 20.44 90.52 87.88 92.68
New model 4.58 20.24 0.99 20.82 88.73 84.38 92.31
Figure 6. Receiver operating characteristic (ROC) curves for the ra-
dial basis function neural network model. [Color figure can be
viewed in the online issue, which is available at www.interscience.
wiley.com.]
Table 8. Performance of the Neural Network Classification Model.
Descriptors
Network
architecture
Training set
Accuracy Error Sensitivity Specificity
1X(RDF025m)2X(RDF110m)
Radial Basis
Function
91.07 0.55 92.00 90.32
3X(RDF115v) Predicting set
Profile RBF 3:3-18-1:1 77.78 0.65 75.00 80.00
542 Cruz-Monteagudo, Cordeiro, and Borges • Vol. 29, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc

satisfactory parsimony, displays good robustness and predictivity
and nearly completely fulfill the main LDA assumptions, we can
state that it can be efficiently used for flag potential idiosyncratic
hepatotoxicity. However, due to the slight deviations from nor-
mality, the model is not as efficient as possible, justifying the
application of nonparametrical methods to seek for improved
models.
ANN Classification Model
As discussed above, LDA-based QSTRs can only capture linear
relationships between molecular characteristics and activities. In
contrast, neural networks can recognize highly nonlinear rela-
tionships between structural or physicochemical descriptors and
biological endpoints or any other properties. This inherent fea-
ture makes neural networks particularly well suited for tackling
generally nonlinear structure–toxicity relationships.
To build a NN classification scheme able to overcome the
deficiencies of the LDA model, the same set of descriptors of
the LDA model were used. In addition, we selected a RBF
architecture since this type of NN has been successfully used to
solve a variety of problems in many modeling and classification
studies, leading in most cases to superior results over standard
statistical models.
As expected, for the training series, the RBFNN model is a
better classifier than the LDA model, with a global accuracy of
91.07%, a sensitivity of 92.00% and a specificity of 90.32% (see
Table 7). In addition, as seen in Figure 6, the area under the
ROC curves pertaining to the training and test series are 0.92
and 0.85, respectively, suggesting that its good results are by no
means due to chance correlation.
However, for the testing series, the performance of the neural
networks (NN) model is inferior compared to that of the LDA
model (accuracy: 77.78% vs. 86.4%; sensitivity: 75% vs. 81.25%;
specificity: 80% vs. 84.09%, respectively; see Tables 4 and 8).
Although quite impressive, given the complexity of the bio-
logic response being modeled, it does not justify replacing the
LDA model taking into account specifically its lower sensitivity
(75%). However, if we compare the predictive performance of
the ANN model considering the results showed by the LDA
model in the same validation set (V1) rather than considering
the averaged results, both models exhibit identical sensitivity
values. Further, the significance of such slight improvement
remains unclear or, at least, does not offer any reason to reject a
linear relationship between the IH endpoint and the present 3D-
descriptors.
OneR Classification Algorithm
Even though an accurate and predictive model has been
obtained, it is worthwhile to search for a simple rule to classify
drugs according to their toxicological characteristics. To accom-
plish this goal we examined the use of another nonparametrical
method, namely the OneR.Classifier from software WEKA, with
a long history in chemical and life sciences applications.55,56
OneR is a machine learning algorithm aimed at providing onesimple rule for two-way categorical classifications based on a
Table 9. Classification Results Obtained with the OneR
Classifier Algorithm.
Training set % 4-fold cross validation %
Accuracy 83.78 Accuracy 81.08
Sensitivity 77.40 Sensitivity 75.50
Specificity 100.00 Specificity 97.60
IH predictivity 63.60 IH predictivity 60.60
NIH predictivity 100.00 NIH predictivity 97.60
False IH Rate 36.40 False IH Rate 39.40
False NIH Rate 0.00 False NIH Rate 2.40
Kappa statistic 0.6598 Kappa statistic 0.6031
Table 10. Other Machine Learning Classifications Algorithms.
Classification scheme Rules/Tree/Function Performance
weka.classifiers.rules.JRip (1X(RDF025m) � 20.655689) 5[ Class 5 IH Accuracy: 89.2%
(3X(RDF115v) � 20.9538) 5[ Class 5 IH Sensitivity: 81.8%
5[ Class 5 NIH Specificity: 95.1%
weka.classifiers.rules.part.PART 1X(RDF025m) [20.655689 AND Accuracy: 90.54%3X(RDF115v) [20.93486: NIH Sensitivity: 84.8%
Specificity: 95.1%
weka.classifiers.trees.j48 1X(RDF025m) � 20.655689: IH Accuracy: 91.9%1X(RDF025m) [20.655689 Sensitivity: 81.8%
| 3X(RDF115v) � 20.93486 Specificity: 100%
| | 3X(RDF115v) � 21.66124: NIH
| | 3X(RDF115v) [21.66124: IH
| 3X(RDF115v) [20.93486: NIH
Variable Coefficient
weka.classifiers.functions.Logistic 1X(RDF025m) 21.8553 Accuracy: 89.2%2X(RDF110m) 1.6733 Sensitivity: 84.8%3X(RDF115v) 21.2576 Specificity: 92.7%
Intercept 20.5765
543Early Detection of Drug-Induced Idiosyncratic Liver Toxicity
Journal of Computational Chemistry DOI 10.1002/jcc
single parameter or molecular descriptor. The descriptor used
here was 1X(RDF025m), which is the more significant variable
included in the LDA model.
The One Rule for classifying the current drugs as IH or NIH
was found to be:
IF1X(RDF025m) \20.652517515, THEN CLASS 5 IH
OTHERWISE, THEN CLASS 5 NIH
It allows for a correct classification of 62 out of the 74 drugs
(83.78% of accuracy) from the training sample. To be more spe-
cific, 77.4% of the IH drugs and all the NIH drugs were classi-
fied correctly. Moreover, good overall predictability (81.08%)
and sensitivity/specificity (75.5%/97.6%, respectively) is found,
as judged by fourfold internal cross validation (see Table 9).
In conclusion, this one rule displays a good overall perform-
ance but has a rather low sensitivity. This last is the most impor-
tant feature when aiming at detecting potential IH drugs in early
drug development, since the consequences of misclassify an IH
drug (classify an IH drug as NIH) can be more devastating (the
false safe drug can reach the market with a potential risk to
human life) than the opposite case (classify an NIH drug as IH).
So, a potential drug is unnecessarily eliminated and will never
reach the market; it is regrettable but instead no life is compro-
mised.
Other rules or algorithms can be developed by using the
other two variables included in the LDA model. However, no
significant improvement is reached through more complex
schemes (see Table 10).
Desirability Analysis
A typical problem in drug development is to find a set of condi-
tions, or levels of the predictor variables, that generate the most
desirable product in terms of characteristics, or responses on the
predictor variables. In the development of any drug whose char-
acteristics are known to depend on its properties or molecular
descriptors, generating the greatest potential product requires
determining the effects of those, and then finding the balance
that optimizes the overall desirability of the final product. Typi-
cally, this involves two main steps: (1) predicting responses on
the property under study (in this case, the idiosyncratic hepato-
toxicity), by fitting the observed responses using an equation
based on the levels of the predictor variables (in this case, the
RDF descriptors), and (2) finding the levels of the X-variablesthat simultaneously produce the most desirable predicted
responses of the studied property.
Considering the LDA model as the best choice for predicting
idiosyncratic hepatotoxicity, we decided to perform a desirability
analysis57 based on the levels of the predictor variables used in
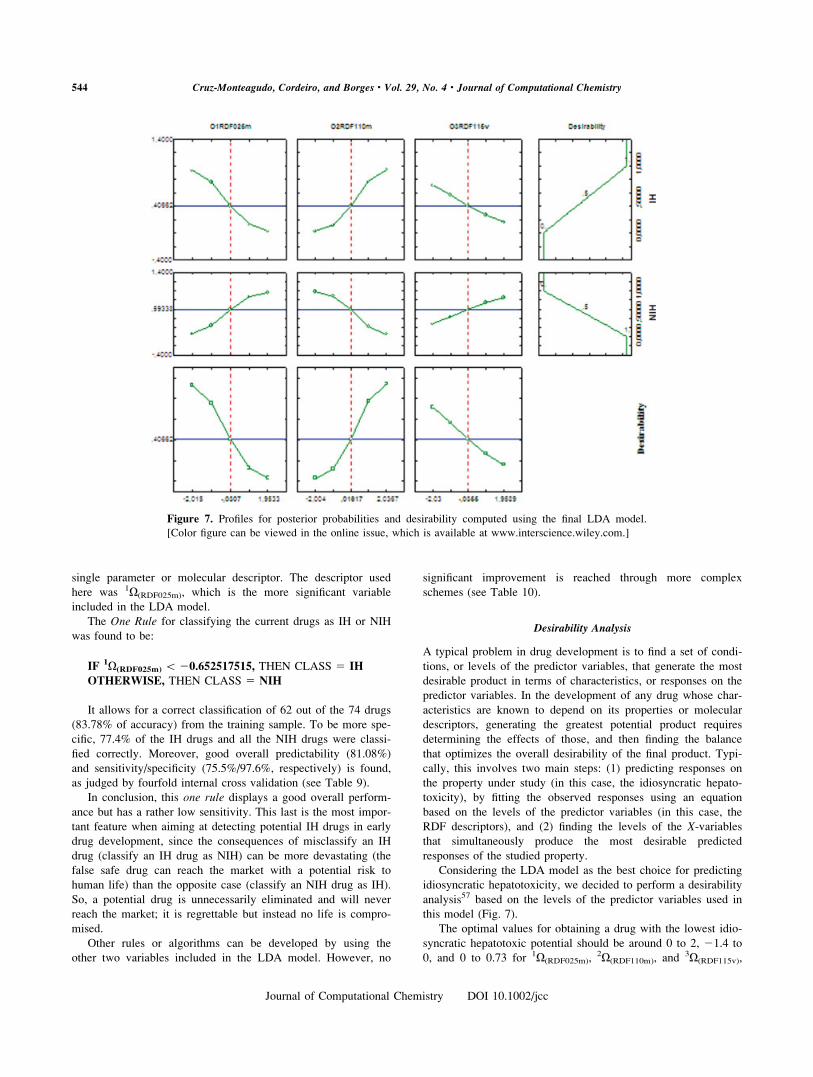
this model (Fig. 7).
The optimal values for obtaining a drug with the lowest idio-
syncratic hepatotoxic potential should be around 0 to 2, 21.4 to
0, and 0 to 0.73 for 1X(RDF025m),2X(RDF110m), and
3X(RDF115v),
Figure 7. Profiles for posterior probabilities and desirability computed using the final LDA model.
[Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
544 Cruz-Monteagudo, Cordeiro, and Borges • Vol. 29, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc
respectively, fixing the other two variables at their present mean
values (see Fig. 6). However, if the three variables are used at
their current values, it is possible to obtain a drug with a low
desirability value of 0.374 for the propensity to induce IH. On
the contrary, if the same current (mean) values are used it is
possible to obtain a drug with a desirability value of 0.626 for
the propensity to not induce IH.
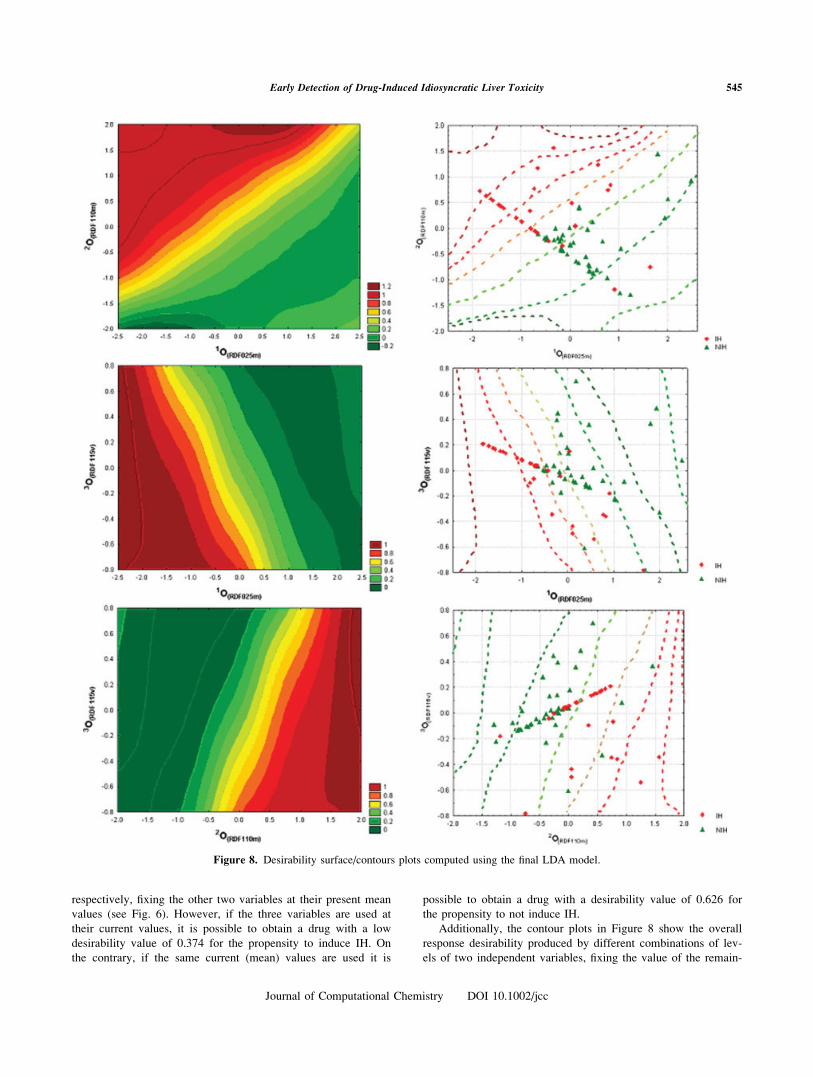
Additionally, the contour plots in Figure 8 show the overall
response desirability produced by different combinations of lev-
els of two independent variables, fixing the value of the remain-
Figure 8. Desirability surface/contours plots computed using the final LDA model.
545Early Detection of Drug-Induced Idiosyncratic Liver Toxicity
Journal of Computational Chemistry DOI 10.1002/jcc
ing third variable at its mean value. They were built by previ-
ously transforming scores of each of the three variables into
desirability scores that could range from 0.0 (undesirable; repre-
sented in green) to 1.0 (very desirable; represented in red). This
means that the green zone in the contour plots represents the
zone of higher probability to obtain a drug with lower potential
to induce an idiosyncratic hepatotoxic event, and the opposite
for the red zone. As can be noticed, most of the cases are
located in the proper zone of the plane, i.e.: most of the IH
drugs lay in the red (toxic) zone where the desirability values
are clearly over 0.75 while the NIH drugs lay in the green (safe)
zone where the desirability values are clearly under 0.3. The
maximum overlapping zones (between IH and NIH drugs) are
the zones with medium desirability values (ca. 0.5). The consis-
tence between the results from the desirability analysis and the
location of cases in the contour plots further demonstrates the
advantages of using this kind of analysis in drug development.
In Silico Virtual Screening: A Case Study
In an effort to illustrate the applicability of the method proposed
to the drug development process and at the same time to test its
predictive ability an external set of three pairs of chemically and
pharmacological related drugs having an observed opposite toxi-
Table 11. Chemical Structures and Calculated Posterior Probability of the Drugs to be Toxic (IH) or Not
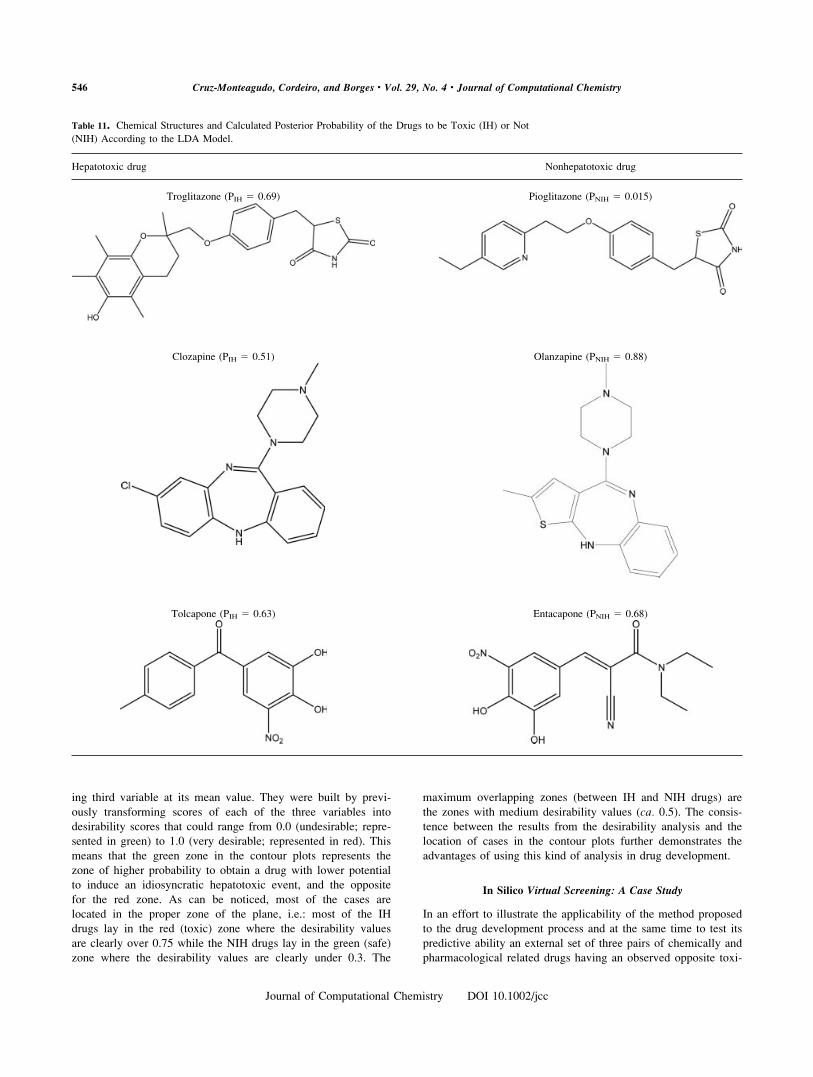
(NIH) According to the LDA Model.
Hepatotoxic drug Nonhepatotoxic drug
Troglitazone (PIH 5 0.69) Pioglitazone (PNIH 5 0.015)
Clozapine (PIH 5 0.51) Olanzapine (PNIH 5 0.88)
Tolcapone (PIH 5 0.63) Entacapone (PNIH 5 0.68)
546 Cruz-Monteagudo, Cordeiro, and Borges • Vol. 29, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc
cological profile (not used here for training), namely the pairs
troglitazone vs. pioglitazone, tolcapone vs. entacapone, and clo-
zapine vs. olanzapine, were used.
Thiazolidinediones or glitazones specifically target insulin re-
sistance. However, troglitazone, the first compound approved by
the FDA in the United States, proved to be hepatotoxic and was
withdrawn from the market after the report of several dozens of
deaths or cases of severe hepatic failure requiring liver trans-
plantation.58–61 In contrast, just a few cases of severe hepatotox-
icity have been reported with pioglitazone,62–64 consequently
this drug has not yet been recognized as hepatotoxic and is con-
sidered safe in the treatment of type 2 diabets.
On the other hand, two psychotropic drugs (clozapine and
olanzapine) provide another example. Although the main side
effects reported to clozapine is the agranulocytosis, its liver tox-
icity is also significant and life threatening.65,66 In opposition,
olanzapine is considered as a safe drug.67
Another case of similar drugs with different toxic profiles is
tolcapone and entacapone. Both are catechol-O-methyltransfer-
ase (COMT) inhibitors and were developed during the 1990’s to
be used as adjuncts to levodopa (LD)—dopa decarboxylase
(DDC) inhibitors in the treatment of Parkinson’s disease. Enta-
capone is currently in wide clinical use, while tolcapone can be
used in restricted indications only, due to its hepatotoxicity.68
The bioactivation and hepatotoxicity of this type of nitroaro-
matics drugs is currently under study.69 The chemical structures
among the calculated posterior probability of the drugs to be
toxic (IH) or not (NIH) according to the LDA model are shown
in Table 11.
Table 12 details the performance of each of the three models
developed for predicting the hepatotoxicity of all selected drugs
among their respective descriptors values. In agreement with our
previous proposal in using the LDA model as the best option,
Table 12. Results for the External Validation Experiment.
Drug Obs. Class LDA Pred. Class ANN Pred. Class OneR Pred. Class LDA IH Post. Prob. LDA NIH Post. Prob.
Clozapine IH IH NIH NIH 0.507 0.493
Olanzapine NIH NIH NIH NIH 0.121 0.879
Tolcapone IH IH IH IH 0.625 0.375
Entacapone NIH NIH NIH NIH 0.317 0.683
Pioglitazone NIH IH IH NIH 0.985 0.015
Troglitazone IH IH IH IH 0.685 0.315
Orthogonal RDF Descriptors RDF Descriptors
1X RDF025m 2X RDF110m 3X RDF115v RDF025m RDF110m RDF115v
Clozapine 0.210 20.119 20.541 0.210 0.023 20.492
Olanzapine 0.214 20.663 20.061 0.214 20.517 20.453
Tolcapone 20.693 20.026 0.066 20.693 20.495 20.442
Entacapone 20.356 20.261 0.228 20.356 20.501 20.235
Pioglitazone 20.408 1.360 20.564 20.408 1.084 0.264
Troglitazone 0.824 0.846 20.360 0.824 1.419 0.9283
Models Performance
Model Accuracy Sensitivity Specificity
LDA 83.33% 100% 66.67%
ANN 66.67% 66.67% 66.67%
OneR 83.33% 66.67% 100%
LDA/ANN/OneR Pred. Class: Predicted class (IH or NIH) according to the LDA/ANN/OneR model.
LDA IH/NIH Post. Prob.: Calculated posterior probability of the drugs to be toxic (IH) or not (NIH) according to
the LDA model.
Table 13. Results of the External Validation Experiment for the
LDA Model.
No. Drug
Obs.
Class.
Pred.
Class
Post.
Prob.
Std.
Res. Leverage
1 Benoxaprofen IH IH 0.5995 1.1435 0.0171
2 Depakote IH IH 0.9198 0.3799 0.0463
3 Iproniazid IH IH 0.8692 0.5847 0.0366
4 Leflunomide IH IH 0.9925 20.5374 0.1025
5 Nevirapine IH NIH 0.3848 1.4707 0.0173
6 Pemoline IH IH 0.9111 0.4221 0.0441
7 Penicillamine IH IH 0.9274 0.3395 0.0484
8 Ranitidine IH IH 0.8442 0.6611 0.0262
9 Serzone IH NIH 0.2002 1.8141 0.0204
10 Temafloxacina IH NIH 0.0135 2.9050 0.0535
11 Ticrynafen IH IH 0.6753 1.0201 0.0215
12 Trovafloxazinea IH NIH 0.0297 2.6026 0.0431
13 Zileuton IH IH 0.7518 0.8791 0.0259
Predictive Accuracy 5 Predictive Sensitivity 5 81.82%/69.23%b
aOutlier drugs not considered on the calculation of the predictive accu-
racy.bPredictive accuracy without to consider the model’s applicability do-
main.
547Early Detection of Drug-Induced Idiosyncratic Liver Toxicity
Journal of Computational Chemistry DOI 10.1002/jcc

one can see that this model again shows the best performance of
the three evaluated models. Notice that the ANN model and
OneR classification algorithm were able to predict more than 60
and 80% of the data used, respectively. These results further
confirm the reliability of the three classification methods devel-
oped in the present work.
Is interesting to note that the only misclassified drug by the
LDA model was pioglitazone which is reasonable since until
today remains unclear whether or not its hepatotoxicity is a class
effect or it is related to the unique tocopherol side chain of tro-
glitazone.58 In addition, though not significant in number, some
reports of hepatotoxicity have been made for this drug.62,64 So,
we cannot be certain about the ‘‘misclassification’’ of the model
for this drug.
Another fact in support of the suitability of the LDA model
is that the leverage value of all drugs do not exceed the h* criti-
cal value of 0.16 (htolcapone 5 0.02; hentacapone 5 0.02; holanzapine5 0.02; hclozapine 5 0.04; hpioglitazone 5 0.09; htroglitazone 50.05), which means that these drugs fall within the applicability
domain of the model and so the predictions are reliable.
Since the data previously analyzed is insufficient for external
model evaluation, we decided to use a collection of 13 published
drugs causing idiosyncratic liver toxicity not included in the
training list. Such drugs can be found in the Physicians’ Desk
Reference and on the FDA website.70 Table 13 lists the names
of the idiosyncratic hepatotoxic drugs used as well as the respec-
tive predicted LDA classifications.
As can be noted in Table 13, 9 out of 13 drugs were cor-
rectly classified, evidencing a predictive ability near to 70%
(69.23%). However, as mentioned previously the applicability
domain of the model has to be checked for the new drugs to be
tested. From the leverage plot for the testing compounds shown
in Figure 9, we can note the outlier nature of two structurally
and pharmacologically related drugs used for testing the predic-
tive ability of the model, i.e.: temafloxacin and trovafloxacine.
In fact, temafloxacin and trovafloxacine show standardized resid-
uals greater than two standard deviation units (see Table 13) and
consequently fall out of the model’s applicability domain. Con-
sequently, predictions of such drugs are very questionable. So, if
these two drugs are not used for testing the model (as advised
for such situations) a higher and more realistic model predictive
ability is attained (9 out of 11 drugs classified correctly, leading
to a predictive accuracy of 81.82%). Is important to note that
the other two miss predicted drugs (nevirapine and sarzone) pre-
sented values of standardized residuals (1.5 and 1.8, respec-
tively) near to 2.
Even though the drugs used previously are known, the same
approach can be applied in similar situations but for novel drugs
candidates in the drug development process.
Conclusions
In summary, the present work provides a discussion about the
viability of addressing idiosyncratic hepatotoxicity in early drug
discovery by means of computational QSTR-based approaches.
For that purpose, a structurally and pharmacologically diverse
set of 74 drugs (from which 33 had reported IH) was assembled
from the literature, and used to create several QSTR classifica-
tion models for probing potential IH: we have thoroughly eval-
uated the performance of LDA, RBFNN and OneR.Classifiermodels derived from 3D-RDF molecular descriptors and feature
selection algorithms.
All QSTR classification models developed exhibited satisfac-
tory internal accuracy and predictivity. However, the LDA
model is suggested to be the best model, as judged by extensive
validation tests, accompanied by its simplicity and wide inter-
pretability compared to the other nonlinear models. Further,
desirability analysis based on such LDA model allows asserting
the characteristics or descriptor values that a drug candidate
should have for ensuring a lower IH potential. Nevertheless, the
most important conclusion concerns the ability of the alternative
QSTR models proposed here in accurately predicting the idio-
syncratic hepatotoxic potential of drugs. We believe that in a
near future, this kind of computational approaches could be
applied in early drug discovery to minimize the selection of
chemicals with IDT for further development.
Acknowledgment
Prof. Paula Fresco is specially acknowledged for her valuable
help during the correction of the manuscript.
References
1. Lazarou, J.; Pomeranz, B. H.; Corey, P. N. JAMA 1998, 279, 1200.
2. Galati, G.; Tafazoli, S.; Sabzevari, O.; Chan, T. S.; O’Brien, P. J.
Chem Biol Interact 2002, 142, 25.
3. Ulrich, R. G. Annu Rev Med 2007, 58, 17.
4. Ulrich, R. G.; Bacon, J. A.; Brass, E. P.; Cramer, C. T.; Petrella, D.
K.; Sun, E. L. Chem Biol Interact 2001, 134, 251.
5. Rietjens, I. M.; Alink, G. M. Chem Res Toxicol 2006, 19, 977.
Figure 9. Outlier and leverage plot for thirteen idiosyncratic hepa-
totoxic drugs never used for training.
548 Cruz-Monteagudo, Cordeiro, and Borges • Vol. 29, No. 4 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc

6. Richard, A. M. Chem Res Toxicol 2006, 19, 1257.
7. Federsel, H. J. Drug Discov Today 2006, 11, 966.
8. Baty, V.; Denis, B.; Goudot, C.; Bas, V.; Renkes, P.; Bigard, M. A.;
Boissel, P.; Gaucher, P. Gastroenterol Clin Biol 1994, 18, 1129.
9. Bernuau, J.; Larrey, D.; Campillo, B.; Degott, C.; Verdier, F.; Rueff,
B.; Pessayre, D.; Benhamou, J. P. J Hepatol 1988, 6, 109.
10. Hunter, E. B.; Johnston, P. E.; Tanner, G.; Pinson, C. W.; Awad, J. A.
Am J Gastroenterol 1999, 94, 2299.
11. Fontana, R. J.; McCashland, T. M.; Benner, K. G.; Appelman, H.
D.; Gunartanam, N. T.; Wisecarver, J. L.; Rabkin, J. M.; Lee, W.
M. Liver Transpl Surg 1999, 5, 480.
12. Rabkin, J. M.; Smith, M. J.; Orloff, S. L.; Corless, C. L.; Stenzel,
P.; Olyaei, A. J. Ann Pharmacother 1999, 33, 945.
13. Min, D. I.; Burke, P. A.; Lewis, W. D.; Jenkins, R. L. Pharmaco-
therapy 1992, 12, 68.
14. Aranda-Michel, J.; Koehler, A.; Bejarano, P. A.; Poulos, J. E.;
Luxon, B. A.; Khan, C. M.; Ee, L. C.; Balistreri, W. F.; Weber, F.
L., Jr. Ann Intern Med 1999, 130, 285.
15. Gao, J.; Ann Garulacan, L.; Storm, S. M.; Hefta, S. A.; Opiteck, G.
J.; Lin, J. H.; Moulin, F.; Dambach, D. M. Toxicol In Vitro 2004,
18, 533.
16. Li, A. P. Chem Biol Interact 2002, 142, 7.
17. Hansch, C.; Muir, R. M.; Fujita, T.; Maloney, P. P.; Geiger, F.;
Streich, M. J Am Chem Soc 1963, 85, 2817.
18. Hansch, C.; Fujita, T. J Am Chem Soc 1964, 86, 1616.
19. Estrada, E.; Gonzalez-Diaz, H. J Chem Inf Comput Sci 2003, 43,
75.
20. Cabrera Perez, M. A.; Gonzalez-Diaz, H.; Jimenez-Lopez, E.; Perez,
D.; Gomez-Quintero, E.; Castanedo Cancio, N. Eur Bull Drug Res
2001, 9, 1.
21. Gonzalez-Diaz, H.; Cruz-Monteagudo, M.; Vina, D.; Santana, L.;
Uriarte, E.; De Clercq, E. Bioorg Med Chem Lett 2005, 15,
1651.
22. Gonzalez-Diaz, H.; Gia, O.; Uriarte, E.; Hernadez, I.; Ramos, R.;
Chaviano, M.; Seijo, S.; Castillo, J. A.; Morales, L.; Santana, L.;
Akpaloo, D.; Molina, E.; Cruz, M.; Torres, L. A.; Cabrera, M. A.
J Mol Model (Online) 2003, 9, 395.
23. Gonzalez-Diaz, H.; Torres-Gomez, L. A.; Guevara, Y.; Almeida, M.
S.; Molina, R.; Castanedo, N.; Santana, L.; Uriarte, E. J Mol Model
(Online) 2005, 11, 116.
24. Cruz-Monteagudo, M.; Gonzalez-Diaz, H. Eur J Med Chem 2005,
40, 1030.
25. Cruz-Monteagudo, M.; Gonzalez-Diaz, H.; Borges, F.; Gonzalez-
Diaz, Y. Bull Math Biol 2006, 68, 1555.
26. Rothfuss, A.; Steger-Hartmann, T.; Heinrich, N.; Wichard, J. Chem
Res Toxicol 2006, 19, 1313.
27. Hemmer, M. C.; Steinhauer, V.; Gasteiger, J. Vib Spectrosc 1999,
19, 151.
28. Helguera, A. M.; Cabrera Perez, M. A.; Gonzalez, M. P. J Mol
Model (Online) 2006, 12, 769.
29. Gonzalez, M. P.; Puente, M.; Fall, Y.; Gomez, G. Steroids 2006, 71,
510.
30. Gasteiger, J.; Sadowski, J.; Schuur, J.; Selzer, P.; Steinhauer, L.;
Steinhauer, V. J Chem Inf Comput Sci 1996, 36, 1030.
31. Garcia, A. G.; Horga de la Parte, J. F. Indice de especialidades farm-
aceuticas. Prescripcion racional de farmacos. INTERCON; Editores
Medicos S. A.; EDIMSA: Madrid, 1994.
32. ChemDraw Ultra 9.0; CambridgeSoft, 2004.
33. HyperChem 7.5; Hypercube, Inc., 2002.
34. MOPAC 6.0; Frank J. Seiler Research Laboratory, US Air Force
Academy: Colorado Springs, CO, 1993.
35. Stewart, J. J. P. J Comput Chem 1989, 10, 209.
36. Todeschini, R.; Consonni, V.; Pavan, M. Dragon Software; Milano
Chemometrics: Milano, 2002.
37. STATISTICA 6.0; Statsoft Inc., 2001.
38. Van Waterbeemd, H. Chemometric Methods in Molecular Design;
Wiley-VCH: New York, 1995.
39. Van de Waterbeemd, H.; Testa, B.; Carrupt, P. A.; el Tayar, N. Prog
Clin Biol Res 1989, 291, 123.
40. Kutner, M. H.; Nachtsheim, C. J.; Neter, J.; Li, W. Applied Linear
Statistical Models; McGraw Hill: New York, 2005.
41. Van Waterbeemd, H. In Chemometric Methods in Molecular Design;
Van Waterbeemd, H., Ed.; Wiley-VCH: New York, 1995; pp. 265–
282.
42. Randic, M. New J Chem 1991, 15, 517.
43. Klein, D. J.; Randic, M.; Babic, D.; Lucic, B.; Nikolic, S.; Trinajs-
tic, N. Int J Quantum Chem 1997, 63, 215.
44. Zupan, J.; Gasteiger, J. Neural Networks in Chemistry and Drug
Design; Wiley-VCH: Weinheim, 1999.
45. Wan, C.; Harrington, P. B. J Chem Inf Comput Sci 1999, 39, 1049.
46. Witten, I. H.; Frank, E. Data Mining: Practical Machine Learning
Tools and Techniques with Java Implementations; Morgan Kauf-
mann: San Francisco, 2000.
47. WEKA Software; University of Waikato: New Zealand, 2002.
48. Derringer, G.; Suich, R. J Quality Technol 1980, 12, 214.
49. Zweig, M. H. Arch Pathol Lab Med 1994, 118, 141.
50. Zweig, M. H.; Broste, S. K.; Reinhart, R. A. Clin Chem 1992, 38,
1425.
51. Gonzalez-Diaz, H.; Prado-Prado, F. J.; Santana, L.; Uriarte, E. Bio-
org Med Chem 2006, 14, 5973.
52. Stewart, J.; Gill, L. Econometrics; Prentice Hall: London, 1998.
53. Atkinson, A. C. Plots, Transformations and Regression; Oxford:
Clarendon Press, 1985.
54. Eriksson, L.; Jaworska, J.; Worth, A. P.; Cronin, M. T.; McDowell,
R. M.; Gramatica, P. Environ Health Perspect 2003, 111, 1361.
55. Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I. H. Bioinfor-
matics 2004, 20, 2479.
56. Nascimento, D. G.; Rates, B.; Santos, D. M.; Verano-Braga, T.; Bar-
bosa-Silva, A.; Dutra, A. A.; Biondi, I.; Martin-Eauclaire, M. F.;
Lima, M. E.; Pimenta, A. M. Toxicon 2006, 47, 628.
57. Aguero-Chapin, G.; Gonzalez-Diaz, H.; Molina, R.; Varona-Santos,
J.; Uriarte, E.; Gonzalez-Diaz, Y. FEBS Lett 2006, 580, 723.
58. Scheen, A. J. Diabetes Metab 2001, 27, 305.
59. Menon, K. V. N.; Angulo, P.; Lindor, K. D. Am J Gastroenterol
2001, 96, 1631.
60. Rothwell, C.; McGuire, E. J.; Altrogge, D. M.; Masuda, H.; de la
Iglesia, F. A. J Toxicol Sci 2002, 27, 35.
61. Masubuchi, Y. Drug Metab Pharmacokinet 2006, 21, 347.
62. May, L. D.; Lefkowitch, J. H.; Kram, M. T.; Rubin, D. E. Ann In-
tern Med 2002, 136, 449.
63. Farley-Hills, E.; Sivasankar, R.; Martin, M. BMJ 2004, 329, 429.
64. Chase, M. P.; Yarze, J. C. Am J Gastroenterol 2002, 97, 502.
65. Macfarlane, B.; Davies, S.; Mannan, K.; Sarsam, R.; Pariente, D.;
Dooley, J. Gastroenterology 1997, 112, 1707.
66. Panagiotis, B. J Neuropsychiatry Clin Neurosci 1999, 11, 117.
67. Boelsterli, U. A.; Bouis, P.; Donatsch, P. Cell Biol Toxicol 1987, 3,
231.
68. Gordin, A.; Kaakkola, S.; Teravainen, H. J Neural Transm 2004,
111, 1343.
69. Boelsterli, U. A.; Ho, H. K.; Zhou, S.; Leow, K. Y. Curr Drug
Metab 2006, 7, 715.
70. www.fda.gov/cder/livertox/clinical.pdf.
549Early Detection of Drug-Induced Idiosyncratic Liver Toxicity
Journal of Computational Chemistry DOI 10.1002/jcc





























