Embed Size (px)
Citation preview

Computational Biology, Part 5
Multiple Sequence Alignment
Computational Biology, Part 5
Multiple Sequence Alignment
Robert F. MurphyRobert F. Murphy
Copyright Copyright 1996-2006. 1996-2006.
All rights reserved.All rights reserved.

Multiple Sequence AlignmentMultiple Sequence Alignment Goal: Goal: Create best possible Create best possible “overall” alignment of a family “overall” alignment of a family of sequences (more than two)of sequences (more than two)
Ideal approach: Ideal approach: Compare all Compare all sequences “simultaneously”sequences “simultaneously”
Short-cut approach: Short-cut approach: Align all Align all of the members of the members pairwise pairwise with with oneone of the members of the members

Pairwise Multiple Sequence Alignment Example - MacVector
Pairwise Multiple Sequence Alignment Example - MacVector ““Align to FolderAlign to Folder””
InputsInputs An open sequence fileAn open sequence file A folder containing a set of A folder containing a set of sequencessequences
Settings for sequence comparisonSettings for sequence comparison OutputsOutputs
Aligned sequence map (graphical)Aligned sequence map (graphical) Aligned sequence listing (text)Aligned sequence listing (text)

Pairwise Multiple Sequence Alignment Example 1
Pairwise Multiple Sequence Alignment Example 1 Input: Input: Folder containing a Folder containing a set of protein sequences for set of protein sequences for various various and and tubulin tubulin chainschains
Task: Task: Compare first sequence Compare first sequence to all others and display mapto all others and display map

Pairwise Multiple Sequence Alignment Example 1
Pairwise Multiple Sequence Alignment Example 1 Open first tubulin sequence Open first tubulin sequence (A23035)(A23035)

Pairwise Multiple Sequence Alignment Example 1
Pairwise Multiple Sequence Alignment Example 1
Under Under DatabaseDatabase, pull , pull down to down to Align to Align to FolderFolder
Click on Click on Folder Folder to to SearchSearch
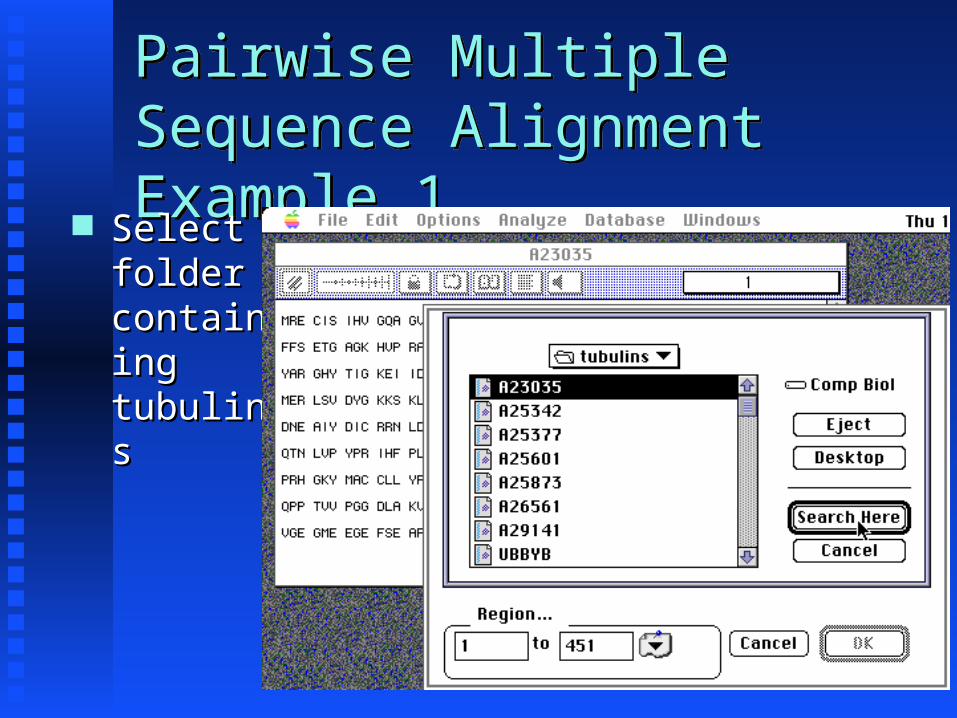
Pairwise Multiple Sequence Alignment Example 1
Pairwise Multiple Sequence Alignment Example 1 Select Select folder folder containcontaining ing tubulintubulinss

Pairwise Multiple Sequence Alignment Example 1
Pairwise Multiple Sequence Alignment Example 1 Use Use defauldefaults for ts for search search settinsettings and gs and click click OKOK

Pairwise Multiple Sequence Alignment Example 1
Pairwise Multiple Sequence Alignment Example 1 Click all boxes for Click all boxes for Display Display OptionsOptions and and OKOK
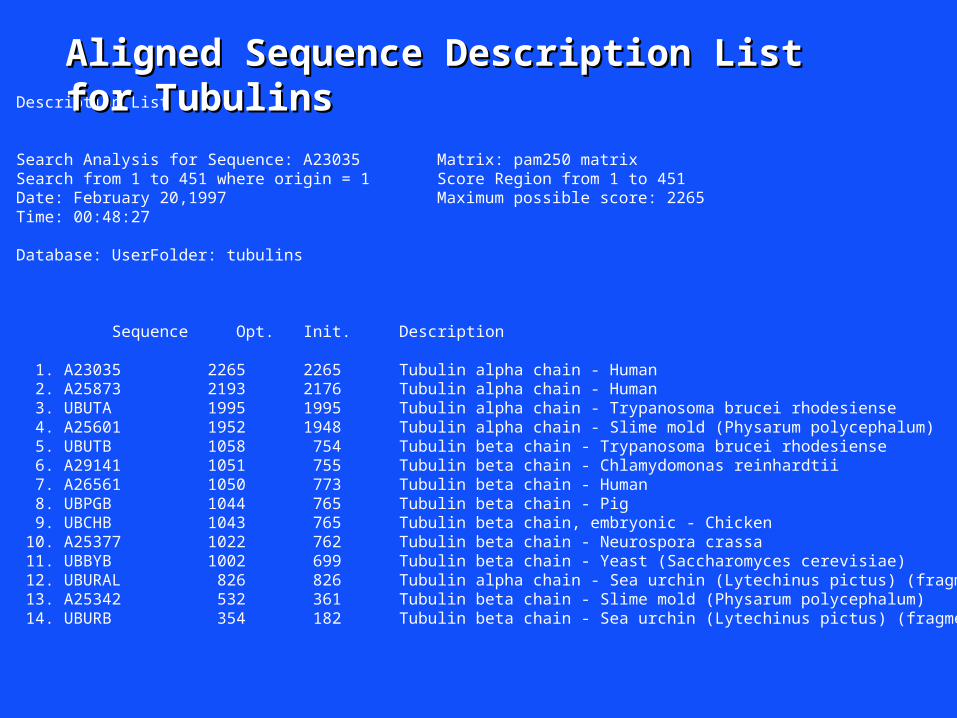
Description List
Search Analysis for Sequence: A23035 Matrix: pam250 matrixSearch from 1 to 451 where origin = 1 Score Region from 1 to 451Date: February 20,1997 Maximum possible score: 2265Time: 00:48:27
Database: UserFolder: tubulins
Sequence Opt. Init. Description
1. A23035 2265 2265 Tubulin alpha chain - Human 2. A25873 2193 2176 Tubulin alpha chain - Human 3. UBUTA 1995 1995 Tubulin alpha chain - Trypanosoma brucei rhodesiense 4. A25601 1952 1948 Tubulin alpha chain - Slime mold (Physarum polycephalum) 5. UBUTB 1058 754 Tubulin beta chain - Trypanosoma brucei rhodesiense 6. A29141 1051 755 Tubulin beta chain - Chlamydomonas reinhardtii 7. A26561 1050 773 Tubulin beta chain - Human 8. UBPGB 1044 765 Tubulin beta chain - Pig 9. UBCHB 1043 765 Tubulin beta chain, embryonic - Chicken 10. A25377 1022 762 Tubulin beta chain - Neurospora crassa 11. UBBYB 1002 699 Tubulin beta chain - Yeast (Saccharomyces cerevisiae) 12. UBURAL 826 826 Tubulin alpha chain - Sea urchin (Lytechinus pictus) (fragment) 13. A25342 532 361 Tubulin beta chain - Slime mold (Physarum polycephalum) 14. UBURB 354 182 Tubulin beta chain - Sea urchin (Lytechinus pictus) (fragment)
Aligned Sequence Description List Aligned Sequence Description List for Tubulinsfor Tubulins

Search Analysis for Sequence: A23035 Matrix: pam250 matrixSearch from 1 to 451 where origin = 1 Score Region from 1 to 451Date: February 20,1997 Maximum possible score: 2265Time: 00:48:27
Database: UserFolder: tubulins
1. A23035 2265 2. A25873 2193 3. UBUTA 1995 4. A25601 1952 5. UBUTB 1058 6. A29141 1051 7. A26561 1050 8. UBPGB 1044 9. UBCHB 1043 10. A25377 1022 11. UBBYB 1002 12. UBURAL 826 13. A25342 532 14. UBURB 354
50
50
100
100
150
150
200
200
250
250
300
300
350
350
400
400
450
450
Aligned Sequence Map for Aligned Sequence Map for TubulinsTubulins

Pairwise Multiple Sequence Alignment Example 2
Pairwise Multiple Sequence Alignment Example 2 Input: Input: Folder containing Folder containing sequence files for just three sequence files for just three tubulinstubulins
Task: Task: Compare using different Compare using different query sequences and examine query sequences and examine aligned sequence listing for aligned sequence listing for differencesdifferences

Pairwise Multiple Sequence Alignment Example 2
Pairwise Multiple Sequence Alignment Example 2 Open first of three sequencesOpen first of three sequences

Pairwise Multiple Sequence Alignment Example 2
Pairwise Multiple Sequence Alignment Example 2
Align Align to to Folder Folder with with itself itself and and two two other other tubulitubulinsns
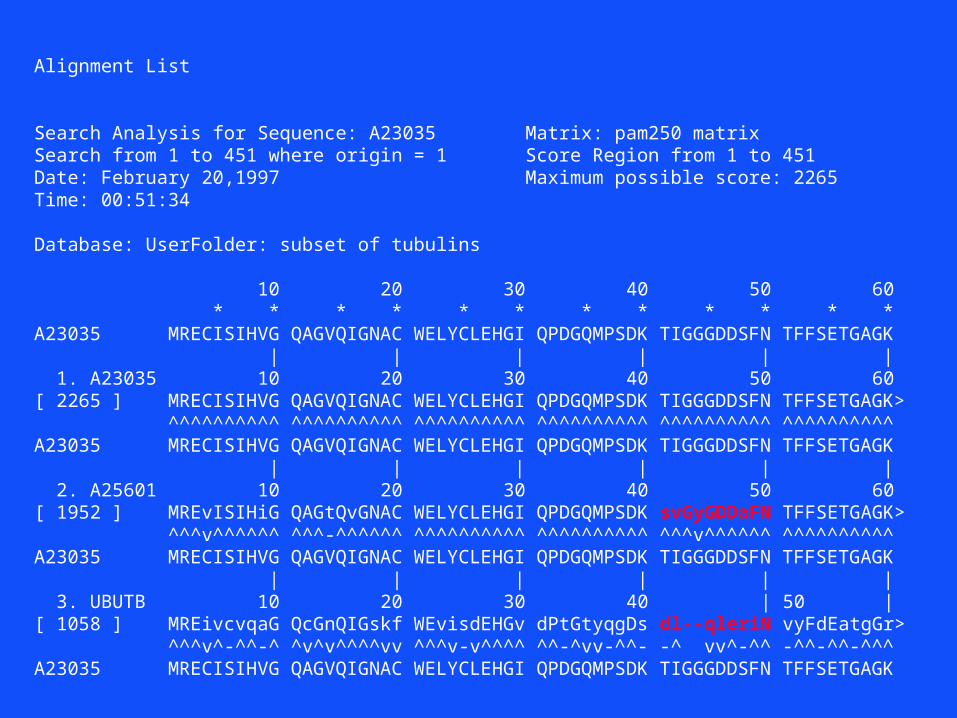
Alignment List
Search Analysis for Sequence: A23035 Matrix: pam250 matrixSearch from 1 to 451 where origin = 1 Score Region from 1 to 451Date: February 20,1997 Maximum possible score: 2265Time: 00:51:34
Database: UserFolder: subset of tubulins
10 20 30 40 50 60 * * * * * * * * * * * *A23035 MRECISIHVG QAGVQIGNAC WELYCLEHGI QPDGQMPSDK TIGGGDDSFN TFFSETGAGK | | | | | | 1. A23035 10 20 30 40 50 60[ 2265 ] MRECISIHVG QAGVQIGNAC WELYCLEHGI QPDGQMPSDK TIGGGDDSFN TFFSETGAGK> ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^A23035 MRECISIHVG QAGVQIGNAC WELYCLEHGI QPDGQMPSDK TIGGGDDSFN TFFSETGAGK | | | | | | 2. A25601 10 20 30 40 50 60[ 1952 ] MREvISIHiG QAGtQvGNAC WELYCLEHGI QPDGQMPSDK svGyGDDaFN TFFSETGAGK> ^^^v^^^^^^ ^^^-^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^v^^^^^^ ^^^^^^^^^^A23035 MRECISIHVG QAGVQIGNAC WELYCLEHGI QPDGQMPSDK TIGGGDDSFN TFFSETGAGK | | | | | | 3. UBUTB 10 20 30 40 | 50 |[ 1058 ] MREivcvqaG QcGnQIGskf WEvisdEHGv dPtGtyqgDs dl--qleriN vyFdEatgGr> ^^^v^-^^-^ ^v^v^^^^vv ^^^v-v^^^^ ^^-^vv-^^- -^ vv^-^^ -^^-^^-^^^A23035 MRECISIHVG QAGVQIGNAC WELYCLEHGI QPDGQMPSDK TIGGGDDSFN TFFSETGAGK

Pairwise Multiple Sequence Alignment Example 2
Pairwise Multiple Sequence Alignment Example 2
Close Close first, first, open open second second sequensequence and ce and repeat repeat Align Align to to FolderFolder

Alignment List
Search Analysis for Sequence: A25601 Matrix: pam250 matrixSearch from 1 to 450 where origin = 1 Score Region from 1 to 450Date: February 20,1997 Maximum possible score: 2161Time: 00:52:46
Database: UserFolder: subset of tubulins
10 20 30 40 50 60 * * * * * * * * * * * *A25601 MREVISIHIG QAGTQVGNAC WELYCLEHGI QPDGQMPSDK SVGYGDDAFN TFFSETGAGK | | | | | | 1. A25601 10 20 30 40 50 60[ 2161 ] MREVISIHIG QAGTQVGNAC WELYCLEHGI QPDGQMPSDK SVGYGDDAFN TFFSETGAGK> ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^A25601 MREVISIHIG QAGTQVGNAC WELYCLEHGI QPDGQMPSDK SVGYGDDAFN TFFSETGAGK | | | | | | 2. A23035 10 20 30 40 50 60[ 1948 ] MREcISIHvG QAGvQiGNAC WELYCLEHGI QPDGQMPSDK tiGgGDDsFN TFFSETGAGK> ^^^v^^^^^^ ^^^-^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^v^^^^^^ ^^^^^^^^^^A25601 MREVISIHIG QAGTQVGNAC WELYCLEHGI QPDGQMPSDK SVGYGDDAFN TFFSETGAGK | | | | | | 3. UBUTB 10 20 30 40 | 50 |[ 1014 ] MREivcvqaG QcGnQiGskf WEvisdEHGv dPtGtyqgDs dlql-er-iN vyFdEatgGr> ^^^^^-^^v^ ^v^-^^^^vv ^^^v-v^^^^ ^^-^vv-^^- -^vv ^v ^^ -^^-^^-^^^A25601 MREVISIHIG QAGTQVGNAC WELYCLEHGI QPDGQMPSDK SVGYGDDAFN TFFSETGAGK

Alignment List
Search Analysis for Sequence: A23035 Matrix: pam250 matrixSearch from 1 to 451 where origin = 1 Score Region from 1 to 451Date: February 20,1997 Maximum possible score: 2265Time: 00:51:34
Database: UserFolder: subset of tubulins
10 20 30 40 50 60 * * * * * * * * * * * *A23035 MRECISIHVG QAGVQIGNAC WELYCLEHGI QPDGQMPSDK TIGGGDDSFN TFFSETGAGK | | | | | | 1. A23035 10 20 30 40 50 60[ 2265 ] MRECISIHVG QAGVQIGNAC WELYCLEHGI QPDGQMPSDK TIGGGDDSFN TFFSETGAGK> ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^A23035 MRECISIHVG QAGVQIGNAC WELYCLEHGI QPDGQMPSDK TIGGGDDSFN TFFSETGAGK | | | | | | 2. A25601 10 20 30 40 50 60[ 1952 ] MREvISIHiG QAGtQvGNAC WELYCLEHGI QPDGQMPSDK svGyGDDaFN TFFSETGAGK> ^^^v^^^^^^ ^^^-^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^ ^^^v^^^^^^ ^^^^^^^^^^A23035 MRECISIHVG QAGVQIGNAC WELYCLEHGI QPDGQMPSDK TIGGGDDSFN TFFSETGAGK | | | | | | 3. UBUTB 10 20 30 40 | 50 |[ 1058 ] MREivcvqaG QcGnQIGskf WEvisdEHGv dPtGtyqgDs dl--qleriN vyFdEatgGr> ^^^v^-^^-^ ^v^v^^^^vv ^^^v-v^^^^ ^^-^vv-^^- -^ vv^-^^ -^^-^^-^^^A23035 MRECISIHVG QAGVQIGNAC WELYCLEHGI QPDGQMPSDK TIGGGDDSFN TFFSETGAGK

Pairwise Multiple Sequence Alignment Example 2
Pairwise Multiple Sequence Alignment Example 2
1st alignment A23035 TIGGGDDSFN A25601 svGyGDDaFN UBUTB dl--qleriN2nd alignment A23035 tiGgGDDsFN A25601 SVGYGDDAFN UBUTB dlql-er-iN
Note that a different (better) alignment Note that a different (better) alignment of of A25601A25601 and and UBUTBUBUTB is obtained from is obtained from direct direct comparison to each other (2nd comparison to each other (2nd alignment) than when each is compared alignment) than when each is compared indirectlyindirectly via comparison to A23035 (1st via comparison to A23035 (1st alignment)alignment)

“True” Multiple Sequence Alignment“True” Multiple Sequence Alignment Find optimal alignment of Find optimal alignment of multiple sequences by considering multiple sequences by considering all possible alignments of each all possible alignments of each position of position of kk sequences - NP hard sequences - NP hard
Approximation: Approximation: First do First do allall k(k-1)/2 k(k-1)/2 pairwise pairwise alignments (not just one sequence alignments (not just one sequence with all others)with all others)
Then combine pairwise alignments to Then combine pairwise alignments to generate overall alignmentgenerate overall alignment

Global vs. LocalGlobal vs. Local
Just as for pairwise Just as for pairwise alignments, we can choose to alignments, we can choose to find either a find either a globalglobal multiple multiple alignment (in which the final alignment (in which the final alignment is full length) or alignment is full length) or a a locallocal alignment that may alignment that may match only blocks of match only blocks of conserved sequence shared conserved sequence shared among the sequencesamong the sequences

How do we score a multiple-sequence alignment?
How do we score a multiple-sequence alignment? What score should we assign to What score should we assign to each position in a multiple each position in a multiple alignment?alignment?
Each position corresponds to Each position corresponds to aligning three (or more) amino aligning three (or more) amino acidsacids
How good is matching L with S How good is matching L with S with T?with T?
Solution: Solution: Carrillo-Lipman sum of Carrillo-Lipman sum of pairs methodpairs method

How do we score a multiple-sequence alignment?
How do we score a multiple-sequence alignment?
Any position Any position in an MSA can in an MSA can be projected be projected onto onto individual individual sequence sequence pairs and the pairs and the scores of scores of those pairs those pairs summed to summed to give overall give overall score for score for that positionthat position

Scoring Global Multiple Sequence Alignments
Scoring Global Multiple Sequence Alignments SP methodSP method For a set of sequences For a set of sequences x x and and a position a position kk, sum scores , sum scores using scoring function using scoring function S S for for all pairs of sequences at all pairs of sequences at that positionthat position
€
g(k) = S(x(k)i,x(k) j )j≠ i
∑i
∑

Problem with SP methodProblem with SP method
Using SP method, what is the Using SP method, what is the effect of adding one (or a effect of adding one (or a small number) of sequences small number) of sequences that don’t match at a given that don’t match at a given position to a set that position to a set that matches perfectly?matches perfectly?
€
gnmatch= n = Smatchn(n −1) /2
gnmatch= n−1 = Smatch n(n −1) /2 − (n −1)( ) + Smismatch (n −1)
Δg = (Smatch − Smismatch )(n −1)
Δg /gnmatch= n = (2 /n)(Smatch − Smismatch ) /Smatch

Problem with SP methodProblem with SP method
Thus the larger the set of Thus the larger the set of sequences, the more the sequences, the more the relative effect of having one relative effect of having one mismatch. This is counter to mismatch. This is counter to our expectation for a our expectation for a reasonable method.reasonable method.
€
Δg /gnmatch= n = 2(Smatch − Smismatch ) /(Smatchn)

Alternative scoring approachesAlternative scoring approaches Use a phylogenetic tree to Use a phylogenetic tree to order the sequences and only order the sequences and only sum scores for all nodes of sum scores for all nodes of the treethe tree

Multiple Sequence Alignment programsMultiple Sequence Alignment programs MSAMSA
attempts to find optimal alignment attempts to find optimal alignment by simultaneous dynamic programmingby simultaneous dynamic programming
available via web serveravailable via web server ClustalWClustalW
progressive pairwise alignment of progressive pairwise alignment of sequencessequences
available via web serveravailable via web server included within MacVectorincluded within MacVector

MSAMSA
Use pairwise alignments to Use pairwise alignments to build a tree showing how build a tree showing how sequences could have divergedsequences could have diverged
Use it to define region Use it to define region examined for multiple examined for multiple alignmentalignment

MSAMSA
Then use dynamic programming Then use dynamic programming in that regionin that region

Weighting scores to de-emphasis distant relatives
Weighting scores to de-emphasis distant relatives When scoring a position, MSA When scoring a position, MSA calculates weights for each calculates weights for each pair of sequences using the pair of sequences using the treetree

MSAMSA
Only possible for small Only possible for small number of short sequencesnumber of short sequences
U. Washington server can U. Washington server can handle 8 sequences of 500 handle 8 sequences of 500 amino acidsamino acids
PSC supercomputer can handle PSC supercomputer can handle 10 sequences of 1000 amino 10 sequences of 1000 amino acidsacids

CLUSTALWCLUSTALW
Group sequences by similarity and Group sequences by similarity and then align them then align them progressivelyprogressively from most to least similarfrom most to least similar
The grouping is done by building The grouping is done by building a phylogenetic tree (called a a phylogenetic tree (called a guide tree)guide tree)
Can be done for large number of Can be done for large number of long sequences even on desktop long sequences even on desktop computerscomputers


ClustalW within MacVectorClustalW within MacVector Use same set of tubulins from Use same set of tubulins from the MacVector Sample Files the MacVector Sample Files folderfolder
Open each sequenceOpen each sequence Under Under AnalyzeAnalyze, select , select ClustalW AlignmentClustalW Alignment

ClustalW within MacVectorClustalW within MacVector

0.073 UBUTB
0.073 UBUTA
0.050 UBURAL
A230350.038
0.013A258730.068
0.003
0.089 A25601
0.215
0.396 FVVFBA
0.402 CVJB0.225
0.180
0.134 UBBYB
0.015
0.089 A25377
0.029
0.080 A25342
0.023
0.083 UBURB
UBCHB0.018
UBPGB0.027
0.016A26561
0.046
0.043 A29141
0.007
ClustalW
Guide Tree
ClustalW
Guide Tree
From pairwise alignments, build tree that links similar sequences

ClustalW within MacVectorClustalW within MacVector

ClustalW within MacVectorClustalW within MacVector Three ways to save resultsThree ways to save results
Click Save button on initial “Aligned Click Save button on initial “Aligned Sequences Formatted Alignments” screen Sequences Formatted Alignments” screen (or do Save As... on “Aligned Sequences”)(or do Save As... on “Aligned Sequences”)
get BW PICT fileget BW PICT file Use Save As... on “Aligned Sequences Use Save As... on “Aligned Sequences Multiple Alignments”Multiple Alignments”
get interleaved text file useful for printing get interleaved text file useful for printing but requires significant editing for input to but requires significant editing for input to other programsother programs
Use Save As... on “Aligned Sequences” Use Save As... on “Aligned Sequences” displaydisplay
Select FAST formatSelect FAST format get sequential text file useful for input to get sequential text file useful for input to otherother

ClustalW graphical display (partial)ClustalW graphical display (partial)
Color (or shade of gray) shows type of amino acid

ClustalW interleaved fileClustalW interleaved fileClustal W(1.4) multiple sequence alignment
16 Sequences Aligned. Alignment Score = 145639Gaps Inserted = 114 Conserved Identities = 1
Pairwise Alignment Mode: SlowPairwise Alignment Parameters: Open Gap Penalty = 10.0 Extend Gap Penalty = 0.1 Similarity Matrix: blosum
Multiple Alignment Parameters: Open Gap Penalty = 10.0 Extend Gap Penalty = 0.1 Delay Divergent = 40% Gap Distance = 8 Similarity Matrix: blosum
Processing time: 21.2 seconds
UBUTB MREIVCVQAGQCGNQIGSKFWEVISDEHGVDPTGTYQGDSDLQL--ERINVYFDEATGGRYVPRSVLIDLEPGTMDSVRAGPYGQIFRPDNFIFGQSGAGUBUTA MREAICIHIGQAGCQVGNACWELFCLEHGIQPDGAMPSDKTIGVEDDAFNTFFSETGAGKHVPRAVFLDLEPTVVDEVRTGTYRQLFHPEQLISGKEDAAUBURB UBURAL UBPGB MREIVHIQAGQCGNQIGAKFWEVISDEHGIDPTGSYHGDSDLQL--ERINVYYNEAAGNKYVPRAILVDLEPGTMDSVRSGPFGQIFRPDNFVFGQSGAGUBCHB MREIVHIQAGQCGNQIGAKFWEVISDEHGIDPTGSYHGDSDLQL--ERINVYYNEATGNKYVPRAILVDLEPGTMDSVRSGPFGQIFRPDNFVFGQSGAGUBBYB MREIIHISTGQCGNQIGAAFWETICGEHGLDFNGTYHGHDDIQK--ERLNVYFNEASSGKWVPRSINVDLEPGTIDAVRNSAIGNLFRPDNYIFGQSSAGFVVFBA TDE--I--------------------TSFS-------IP--KFR---P---D--------Q---P-NLIF-Q---GCVJB ADT--I------------------VAVELDT------YPNTDIGD--PS--------------YP-----------A29141 MREIVHIQGGQCGNQIGAKFWEVVSDEHGIDPTGTYHGDSDLQL--ERINVYFNEATGGRYVPRAILMDLEPGTMDSVRSGPYGQIFRPDNFVFGQTGAGA26561 MREIVHIQAGQCGNQIGAKFWEVISDEHGIDPTGTYHGDSDLQL--DRISVYYNEATGGKYVPRAILVDLEPGTMDSVRSGPFGQIFRPDNFVFGQSGAGA25873 MRECISVHVGQAGVQMGNACWELYCLEHGIQPDGQMPSDKTIGGGDDSFTTFFCETGAGKHVPRAVFVDLEPTVIDEIRNGPYRQLFHPEQLITGKEDAAA25601 MREVISIHIGQAGTQVGNACWELYCLEHGIQPDGQMPSDKSVGYGDDAFNTFFSETGAGKXXXXAVFLDLEPTVIDEVRTGTYRQLFHPEQLITGKEDAAA25377 MREIVHLQTGQCGNQIGAAFWQTISGEHGLDASGVYNGTSELQL--ERMNVYFNEASGNKYVPRAVLVDLEPGTMDAVRAGPFGQLFRPDNFVFGQSGAGA25342 MREIVHIQAGQCGNQIGAKFWEVISDEHGIDPTGSYHGDSDLQL--ERINVYYNEATGGKYVPRAVLVDLEPGTMDSVRAGPFGQIFRPDNFVFGQTGAGA23035 MRECISIHVGQAGVQIGNACWELYCLEHGIQPDGQMPSDKTIGGGDDSFNTFFSETGAGKHVPRAVFVDLEPTVIDEVRTGTYRQLFHPEQLITGKEDAA
UBUTB NNWAKGHYTEGAELIDSVLDVCCKEAESCDCLQGFQICHSLGGGTGSGMGTLLISKLREQYPDRIMMTFSIIPSPKVSDTVVEPYNTTLSVHQLVENSDEUBUTA NNYARGHYTIGKEIVDLCLDRIRKLADNCTGLQGFLVYHAVGGGTGSGLGALLLERLSVDYGKKSKLGYTVYPSPQVSTAVVEPYNSVLSTHSLLEHTDVUBURB UBURAL UBPGB NNWAKGHYTEGAELVDSVLDVVRKESESCDCLQGFQLTHSLGGGTGSGMGTLLISKIREEYPDRIMNTFSVVPSPKVSDTVVEPYNATLSVHQLVENTDEUBCHB NNWAKGHYTEGAELVDSVLDVVRKESESCDCLQGFQLTHSLGGGTGSGMGTLLISKIREEYPDRIMNTFSVMPSPKVSDTVVEPYNATLSVHQLVENTDE

ClustalW sequential fileClustalW sequential fileMREIVCVQAGQCGNQIGSKFWEVISDEHGVDPTGTYQGDSDLQL--ERINVYFDEATGGRYVPRSVLIDLEPGTMDSVRAGPYGQIFRPDNFIFGQSGAGNNWAKGHYTEGAELIDSVLDVCCKEAESCDCLQGFQICHSLGGGTGSGMGTLLISKLREQYPDRIMMTFSIIPSPKVSDTVVEPYNTTLSVHQLVENSDESMCIDNEALYDICFRTLKLTTPTFGDLNHLVSAVVSGVTCCLRFPGQLNSDLRKLAVNLVPFPRLHFFMMGFAPLTSRGSQQYRGLSVPELTQQMFDAKNMMQAADPRHGRYLTASALFRGRMSTKEVDEQMLNVQNKNSSYFIEWIPNNIKSSVCDIP----PKG----LKMAVTFIGNNTCIQEMFRRV-GEQFTLMFRRKAFLHWYTGEGMDEMEFTEAESNMNDLVSEYQQYQDATIEEEGE------FDEEEQY---------MREAICIHIGQAGCQVGNACWELFCLEHGIQPDGAMPSDKTIGVEDDAFNTFFSETGAGKHVPRAVFLDLEPTVVDEVRTGTYRQLFHPEQLISGKEDAANNYARGHYTIGKEIVDLCLDRIRKLADNCTGLQGFLVYHAVGGGTGSGLGALLLERLSVDYGKKSKLGYTVYPSPQVSTAVVEPYNSVLSTHSLLEHTDVAAMLDNEAIYDLTRRNLDIERPTYTNLNRLIGQVVSSLTASLRFDGALNVDLTEFQTNLVPYPRIHFVLTSYAPVISAEKAYHEQLSVSEISNAVFEPASMMTKCDPRHGKYMACCLMYRGDVVPKDVNAAVATIKTKRTIQFVDWSPTGFKCGINYQPPTVVPGGDLAKVQRAVCMIANSTAIAEVFARI-DHKFDLMYSKRAFVHWYVGEGMEEGEFSEAREDLAALEKDYEEVGAESADMDGE--------EDVEEY---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------FAPLTSRGSQQYRALTVSELTQQMFDAKNMMAACDPRHGRYLTVAAIFRGRMSMKEVDEQMLNVQNKNSSYFVEWIPNNVKTAVCDIP----PRG----LKMSATFIGNSTAIQELFKRI-SEQFTAMFRRKAFLHWYTGEGMDEMEFTEAESNMNDLVSEYQQYQDATAEEEGE------FDEEEGDEEAA-----

Multiple Alignment ServersMultiple Alignment Servers
CLUSTALWCLUSTALW http://www.bioinformatics.nl/http://www.bioinformatics.nl/tools/clustalw.htmltools/clustalw.html
BCM Multiple Sequence BCM Multiple Sequence Alignment LauncherAlignment Launcher http://searchlauncher.bcm.tmc.edhttp://searchlauncher.bcm.tmc.edu/multi-align/multi-align.htmlu/multi-align/multi-align.html
ExPASyExPASy http://www.expasy.org/tools/http://www.expasy.org/tools/#align#align

Reading for next classReading for next class
Mount, Chapter 6 through p. Mount, Chapter 6 through p. 259259
Altschul paperAltschul paper