Embed Size (px)
Citation preview

SURFACE AND INTERFACE ANALYSISSurf. Interface Anal. 26, 498È511 (1998)
Compositional Changes in Plasma-depositedFluorocarbon Films during Ageing
Thomas R. Gengenbach* and Hans J. GriesserCSIRO Molecular Science, Private Bag 10, Clayton South MDC, Clayton 3169, Australia
Plasma polymer coatings were deposited from perÑuoro-1,3-dimethylcyclohexane (PFDMCH) and their composi-tion and surface properties studied by XPS, grazing-angle Fourier transform infrared spectroscopy and contactangle measurements as a function of time after fabrication as they were stored under ambient conditions for morethan 2 years. The spontaneous ambient oxidation of PFDMCH plasma polymers was found to be a multi-stepprocess. The rapid initial oxygen uptake, assigned to reaction between carbon-centred radicals incorporated into thecoating during deposition and in-di†using atmospheric was similar to that of plasma polymers deposited fromO
2,
hydrocarbon-based monomers (alkanes, alkylamines, alcohols), suggesting that the density of radicals incorporatedduring deposition was similar. Subsequently, however, the extent of oxidation was much lower for PFDMCHcoatings. This can be attributed to the lack of availability of hydrogen abstraction reactions, which are importantfor radical propagation in hydrocarbon-based plasma polymers. While XPS recorded a continuous incorporation ofoxygen for more than 2 years, the air/water contact angles decreased only during the Ðrst 2 months and on furtherstorage remained stable. There appeared to be only a small extent of surface restructuring as assessed from thesmall depth variations of compositions. The surface was enriched in groups at all times. 1998 John Wiley &CF
3(
Sons, Ltd.
KEYWORDS: plasma polymers ; Ñuorocarbons ; oxidation ; ageing ; surface restructuring ; XPS; FTIR
INTRODUCTION
Polymeric thin Ðlms deposited from Ñuorocarbonradiofrequency plasmas are of applied interest as aresult of their advantageous chemical and surfaceproperties. They o†er the prospect of combining favour-able characteristics, such as chemical inertness and verylow surface energy, of conventional Ñuoropolymers suchas polytetraÑuoroethylene (PTFE) with the unique fea-tures of plasma-deposited polymeric coatings. Thesefeatures comprise a dense, amorphous, cross-linked andpinhole-free structure, high uniformity of coating thick-ness, excellent adhesion to substrates and versatility.1,2Plasma-deposited Ñuorocarbon coatings thus have beenstudied for use in the fabrication of optical devices andmicroelectronics, as protective coatings, di†usion bar-riers in controlled drug delivery systems, selective bar-riers on gas separation membranes, andnon-thrombogenic coatings for vascular implantdevices.2,3
Ultimately, however, the usefulness of plasma-deposited coatings depends on whether their propertiesare maintained through the lifetime of the device. Earlyreports have already suggested that plasma polymersmay undergo oxidation, after fabrication, on storage inair.1 However, the kinetics, mechanisms and products ofoxidation have only recently been studied using modern
* Correspondence to : T. R. Gengenbach, CSIRO MolecularScience, Private Bag 10, Clayton South MDC, Clayton 3169, Aus-tralia. E-mail : Thomas.Gengenbach=molsci.csiro.au.
spectroscopic methods. Plasma-deposited polymericcoatings from hydrocarbon,4 alkylamine5h7 and methylmethacrylate monomers8,9 have thus been shown toundergo spontaneous oxidative compositional changes,without exposure to UV radiation, from the momentthey are exposed to air.
While conventional Ñuoropolymers possess excellentresistance to chemical attack and oxidative degrada-tion,10 analogous properties have not been demon-strated for plasma-polymerized Ñuoropolymer coatings.Various aspects of the deposition and applications ofÑuorocarbon plasma polymers have beendescribed ;2,3,11h26 however, the question of the long-term stability of these coatings is rarely discussed. A fewreports have mentioned the detection of oxygen byx-ray photoelectron spectroscopy (XPS) or Fouriertransform infrared spectroscopy (FTIR)16,17,19 but donot discuss its origin. It would be of interest to assignthe origin of oxygen either to the presence of an impu-rity gas in the plasma phase or to post-deposition oxi-dation. Other studies have presented evidence thatÑuorocarbon plasma polymers are susceptible to oxida-tive and/or hydrolytic degradation, which might beassociated with free radicals in the freshly preparedcoatings.11,13,15,21h23,26
Post-deposition formation of polar chemical groupsupon oxidation could be disadvantageous for applica-tions that rely on the very low surface free energy ofÑuoropolymeric coatings. We have shown previouslythat an initially hydrophobic hydrocarbon plasmapolymer became more wettable as oxidation generatedpolar chemical groups. However, physical processessuch as rearrangements of polymer chain segments(“surface restructuringÏ) also contributed to the Ðnal
CCC 0142È2421/98/070498È14 $17.50 Received 20 October 1997( 1998 John Wiley & Sons, Ltd. Accepted 27 February 1998

AGEING OF PLASMA-DEPOSITED FLUOROCARBON FILMS 499
surface composition. By removing carbonÈoxygen func-tional groups from the very surface, these processespartly counteracted the e†ects of oxidation on thesurface structure.27 The resulting surface propertiestherefore were determined by the overall balance ofthese concurrent, competing processes.
To anticipate possible adverse e†ects of oxidation inpotential applications of Ñuorocarbon plasma polymers,it is desirable to improve characterization of the rates,products and mechanism(s) of oxidative ageing, and ofthe surface mobility of these materials. Abstraction ofhydrogen plays an important role in the oxidation reac-tion sequences of hydrocarbon and alkylamine plasmapolymers.4h7 However, except by in-di†usion of watervapour, Ñuorocarbon polymers do not contain hydro-gen and thus their oxidative reactions may di†er con-siderably from those operating in other plasmapolymers. In this study, the composition and surfacewettability of coatings from perÑuoro-1,3-dimethyl-cyclohexane were monitored over a period in excess of 2years in order to characterize long-term consequencesof oxidative ageing and surface restructuring.
EXPERIMENTAL
Sample preparation
The substrate for plasma deposition was a polyimideweb (Kapton 100HN, 25 lm thick, 12.7 mm wide) thathad been coated with D0.1 lm of aluminium by elec-tron beam evaporation. The highly reÑective metallizedsurface not only provided a good substrate for FTIRanalysis in the specular reÑectance mode but also facili-tated estimation of the thickness of the plasma polymerÐlms via their interference fringes.
Plasma polymer Ðlms were deposited using perÑuoro-1,3-dimethylcyclohexane (PFDMCH) as the “monomerÏliquid (Aldrich, Castle Hill, Australia). The monomerwas used as received. A fresh batch of the monomerliquid was placed in a round-bottom Ñask and con-nected to the reactor chamber by a stainlees-steel lineand a manual Ñow control valve. Volatile impuritieswhich may have been present were removed bypumping on the liquid for a few minutes prior to igni-tion of the plasma.
Plasma depositions were carried out in a reactor ofconventional design comprising two capacitivelycoupled, parallel, disc-shaped electrodes in a horizontalconÐguration within a cylindrical glass vessel (7.4 lvolume). Samples were placed on the base electrode(155 mm diameter, earthed). The upper (active) elec-trode was separated from the lower electrode by D150mm. The oscillator used in this study was a commercialplasma generator (ENI HPG-2) equipped with a match-ing network and operating in the range 125È375 kHz.The conditions used for the deposition of the samplesstudied in detail were : initial monomer pressure \ 0.13mbar ; maximum monomer pressure \ 0.17 mbar ;plasma load power \ 5 W; plasma frequency\ 215kHz; deposition time\ 10 min.
In the initial experiments, plasma conditions werevaried until visual examination of the interference
fringes exhibited by samples coated while placed atvarious locations on the electrode showed a high degreeof uniformity. The interference fringes of samples coatedunder the above conditions indicated a thickness in therange 200È250 nm. The chemical uniformity of thedepositions was assessed by XPS analysis of freshlycoated specimens from di†erent locations on the elec-trode. A large number of identical specimens werestored in clean glass Petri dishes at room temperature(20 ^ 1 ¡C) ; during storage the relative humidity variedbetween 30% and 70%, and samples were not exposedto sources of UV radiation. They were analysed period-ically, with the analysis of the Ðrst specimen takingplace within 20 min of venting the plasma reactor with
and the subsequent exposure to air.N2
Analysis by XPS
Generally, the surface analysis procedures employedwere analogous to those used earlier for studying theoxidation of other plasma polymers.4,5 BrieÑy, XPSanalysis was performed in a Vacuum GeneratorsEscalab V unit with a non-monochromatic Al Kasource at a power of 200 W (10 kV, 20 mA) and a hemi-spherical analyser operating in the Ðxed analyser trans-mission mode. Typically the pressure in the mainvacuum chamber during analysis was 2] 10~9 mbar.Calibration procedures have been detailed previously.4A value of 285.0 eV was used for the binding energy(BE) of the “neutralÏ C 1s component to correct forcharging of the specimens under irradiation.28 Elementspresent were identiÐed from survey spectra, and high-resolution spectra recorded of individual peaks at 30 eVpass energy. The elemental composition of the surfacewas determined as described in a previous report.4 Therandom error associated with quantiÐcation was deter-mined to be between 5% and 10% following the methodsuggested by Harrison and Hazell.29 Peak positionswere determined according to the procedure of Wagneret al.30 Possible errors introduced by this method andby calibration procedures of the energy scale of thespectrometer and correction of peak positions forsample charging have been discussed earlier.6
Each specimen was analysed at the two di†erentemission angles (#) of 0¡ and 75¡ as measured from thesurface normal, to obtain qualitative information aboutthe variation with depth of the chemical composition.Quantitative interpretation requires knowledge of theelectron attenuation length j of photoelectrons inplasma polymers, which may di†er from that in conven-tional polymers due to the di†erent structure (usuallyhigher density) of the former. The magnitude of jappears to have been determined experimentally foronly one plasma polymer.31 Assuming that di†erentplasma polymers have similar relevant physical proper-ties, such as density, we calculate the j values for C 1s,O 1s and F 1s photoelectrons to be in the range 1.5È2.0nm, which implies that the depth from which 95% ofthe detected XPS signals originate is D4.5È6 nm fornormal emission and D1È1.5 nm for 75¡ emission.
The e†ects of sample decomposition under x-radiation, particularly with non-monochromatized radi-ation, were considered. Specimens were exposed tox-radiation for \1 min before starting data acquisition.
( 1998 John Wiley & Sons, Ltd. Surf. Interface Anal. 26, 498È511 (1998)

500 T. R. GENGENBACH AND H. J. GRIESSER
The total exposure time never exceeded 40 min ; changesto the elemental composition due to sample degrada-tion were thus kept below 5%, as veriÐed in separatecontrol experiments. To avoid accumulation of radi-ation damage due to repeated XPS analysis, a newspecimen was used for each analysis. While thisrequired the preparation of a large number of identicalspecimens, this procedure was essential for undertakingreliable monitoring of long-term compositional changes.
Curve Ðtting of XPS spectra
Individual components of the C 1s signal were assumedto have a Gaussian/Lorentzian lineshape and weredeconvoluted using damped non-linear least-squaresregression.32 We used the following curve-Ðtting proto-col : Ðve spectral components, representing the Ðvemajor types of chemical environments of a carbon atomin a Ñuorocarbon structure, were used : CÈC/CH
x(“neutralÏ or hydrocarbon), CF, andCÈCFx, CF2 CF3 .
All components were assigned the same (adjustable)width (full width at half-maximum, FWHM). Theirrespective positions were not Ðxed. Assuming the BE ofCÈC to be 285.0 eV, the mean BEs obtained for thedi†erent C 1s components were 287.02 eV^ 0.08 eV
289.14 eV^ 0.06 eV (CF), 291.44 eV^ 0.08(CÈCFx),
eV and 293.63 eV^ 0.04 eV These values(CF2) (CF3).(and the corresponding assignments) agree well withdata published in the literature (Ref. 2 and referencestherein). The C 1s spectra recorded at various timesafter fabrication were thus curve Ðtted to determine therelative amounts of the various components as a func-tion of time of storage. Later in the study, a spectro-meter with monochromated Al Ka x-radiation (KratosAXIS-HSi) became available, which enabled conÐrma-tion that these positions had been reasonable estimatesby recording selected spectra of the same coatings withsubstantially higher resolution (see Results section).Although di†erential charging e†ects and the presenceof a range of binding environments should be con-sidered, with two di†erent heteroatoms inducing vari-able secondary binding energy shifts, this is inherentlydifficult for multifunctional, heterogeneous materialssuch as plasma polymers. Four of these Ðve signals (theexception being the component) may contain con-CF3tributions from oxygen-containing species ; although itmay be chemically meaningful to add correspondingcomponents at appropriate positions, it is not war-ranted given the resolution of the photoelectron signaland it would not contribute signiÐcant information tothe Ðnal curve-Ðt result. The simpliÐed protocol usedinstead appears reasonable in light of the low oxygencontent of the plasma polymer even after extendedstorage.
Uncertainties associated with this curve-Ðtting proto-col were estimated in the same way as described pre-viously for another material,5 assuming a worst-casescenario where the error in the intensity is the sum of allpossible errors due to the uncertainties associated withthe three peak parameters (position, width, height). Theerror bars included in the Ðgures are therefore regardedas the upper limits for the possible uncertainties.
The weak O 1s signal was broad and clearly asym-metric. Because the signal-to-noise (S/N) ratio was low,
an iterative curve-Ðt protocol was employed. Initially,two components with the same, adjustable peak width(FWHM) were used to Ðt the experimental spectra. Inorder to achieve internal consistency,5 all curve Ðts werethen recalculated a second time with the separation ofthe two components Ðxed at the average value obtainedafter the Ðrst round of curve Ðtting (see Results section).The error associated with O 1s BE values was estimatedas reported previously.6 Taking into account uncer-tainties due to calibration of the spectrometer, chargereferencing and O 1s curve Ðtting, the standard devi-ation was determined to be 0.41 eV.
Infrared spectroscopy
Grazing-angle infrared spectra of the plasma polymerÐlms were measured on a BOMEM MB-100 FTIRspectrometer Ðtted with a SpectraTech FT80 specularreÑectance attachment using a Ðxed incident angle of80¡. Spectra were scanned 128 times with a resolution of8 cm~1 to increase the S/N ratio. Because the instru-mental contribution to peak broadening in the FTIRspectra of plasma polymers appears generally less sig-niÐcant than the intrinsic broadening of spectral linesdue to the structural heterogeneity of plasma polymers,use of a higher instrumental resolution only reduced theS/N ratio without improving spectral resolution. Priorto each analysis a spectrum of a clean mirror wasrecorded to be able to account for the inevitable pres-ence of vibrational bands due to water vapour and
However, often these bands could not be fullyCO2 .compensated for on account of atmospheric variabilitywithin the IR spectrometer.
In order to visually enhance small spectral changesover time, some FTIR spectra will be presented as dif-ference spectra : the spectrum of the freshly depositedplasma polymer Ðlm, used as a reference, was sub-tracted from the spectra recorded at various times ofstorage. For experimental reasons the Ðrst FTIR spec-trum was acquired after 1 day of storage in air.
Assignments of vibrational bands are based on refer-ence infrared frequencies tabulated by Bellamy33 anddata listed in a review.2 QuantiÐcation of chemicalcompositions from FTIR spectra was not attemptedbecause it is considered unreliable on account of uncer-tainties in polarization e†ects and absorption coeffi-cients.
Contact angle measurements
Contact angles were measured with triply distilleddeionized water using a modiÐed Kernco Model G-IIgoniometer within 20 min of exposing the freshly pre-pared samples to the ambient atmosphere, and then atvarious times of storage. The goniometer was equippedwith a reversible, micrometer-driven plunger in thesyringe to enable determination of the advancing (hA),sessile and receding contact angles. Readings(hS) (hR)were taken from four drops and the mean and standarddeviation calculated from these values. Clean referencesurfaces of Ñuorinated ethyleneÈpropylene co-polymer(FEP) were used to ascertain reproducibility of mea-surements and purity of the water. Separate washing
Surf. Interface Anal. 26, 498È511 (1998) ( 1998 John Wiley & Sons, Ltd.
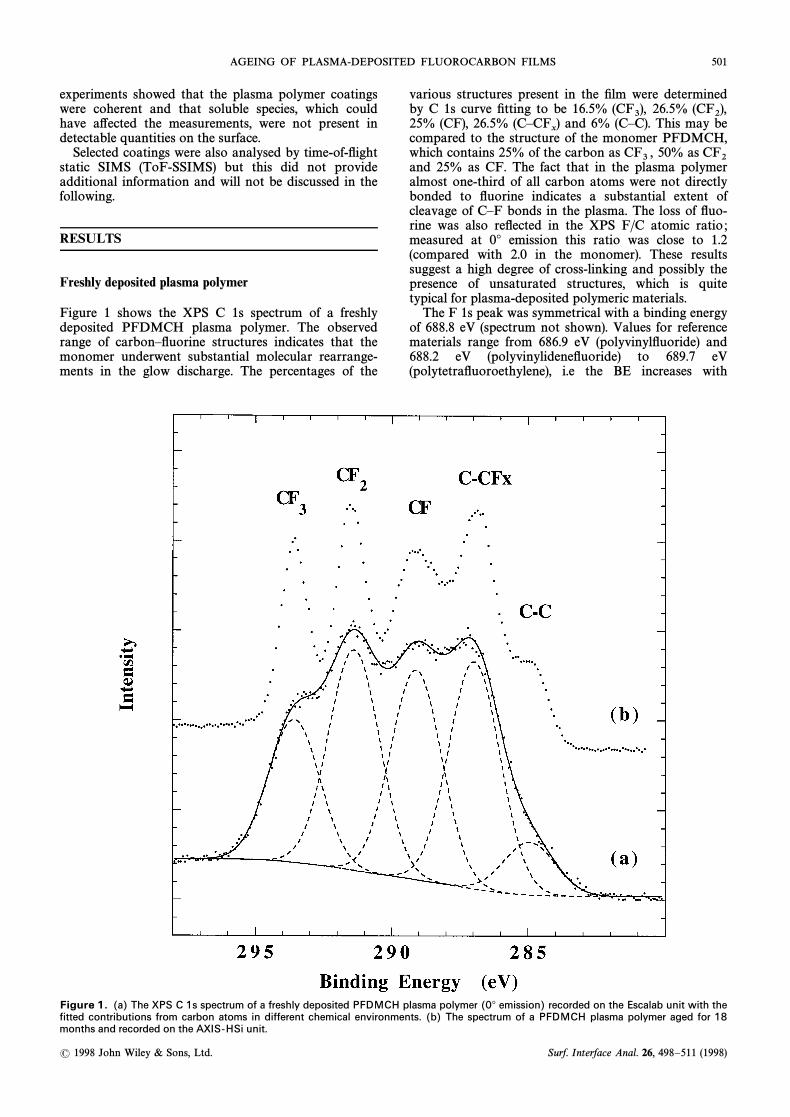
AGEING OF PLASMA-DEPOSITED FLUOROCARBON FILMS 501
experiments showed that the plasma polymer coatingswere coherent and that soluble species, which couldhave a†ected the measurements, were not present indetectable quantities on the surface.
Selected coatings were also analysed by time-of-Ñightstatic SIMS (ToF-SSIMS) but this did not provideadditional information and will not be discussed in thefollowing.
RESULTS
Freshly deposited plasma polymer
Figure 1 shows the XPS C 1s spectrum of a freshlydeposited PFDMCH plasma polymer. The observedrange of carbonÈÑuorine structures indicates that themonomer underwent substantial molecular rearrange-ments in the glow discharge. The percentages of the
various structures present in the Ðlm were determinedby C 1s curve Ðtting to be 16.5% 26.5%(CF3), (CF2),25% (CF), 26.5% and 6% (CÈC). This may be(CÈCF
x)
compared to the structure of the monomer PFDMCH,which contains 25% of the carbon as 50% asCF3 , CF2and 25% as CF. The fact that in the plasma polymeralmost one-third of all carbon atoms were not directlybonded to Ñuorine indicates a substantial extent ofcleavage of CÈF bonds in the plasma. The loss of Ñuo-rine was also reÑected in the XPS F/C atomic ratio ;measured at 0¡ emission this ratio was close to 1.2(compared with 2.0 in the monomer). These resultssuggest a high degree of cross-linking and possibly thepresence of unsaturated structures, which is quitetypical for plasma-deposited polymeric materials.
The F 1s peak was symmetrical with a binding energyof 688.8 eV (spectrum not shown). Values for referencematerials range from 686.9 eV (polyvinylÑuoride) and688.2 eV (polyvinylideneÑuoride) to 689.7 eV(polytetraÑuoroethylene), i.e the BE increases with
Figure 1. (a) The XPS C 1s spectrum of a freshly deposited PFDMCH plasma polymer (0¡ emission) recorded on the Escalab unit with thefitted contributions from carbon atoms in different chemical environments. (b) The spectrum of a PFDMCH plasma polymer aged for 18months and recorded on the AXIS-HSi unit.
( 1998 John Wiley & Sons, Ltd. Surf. Interface Anal. 26, 498È511 (1998)
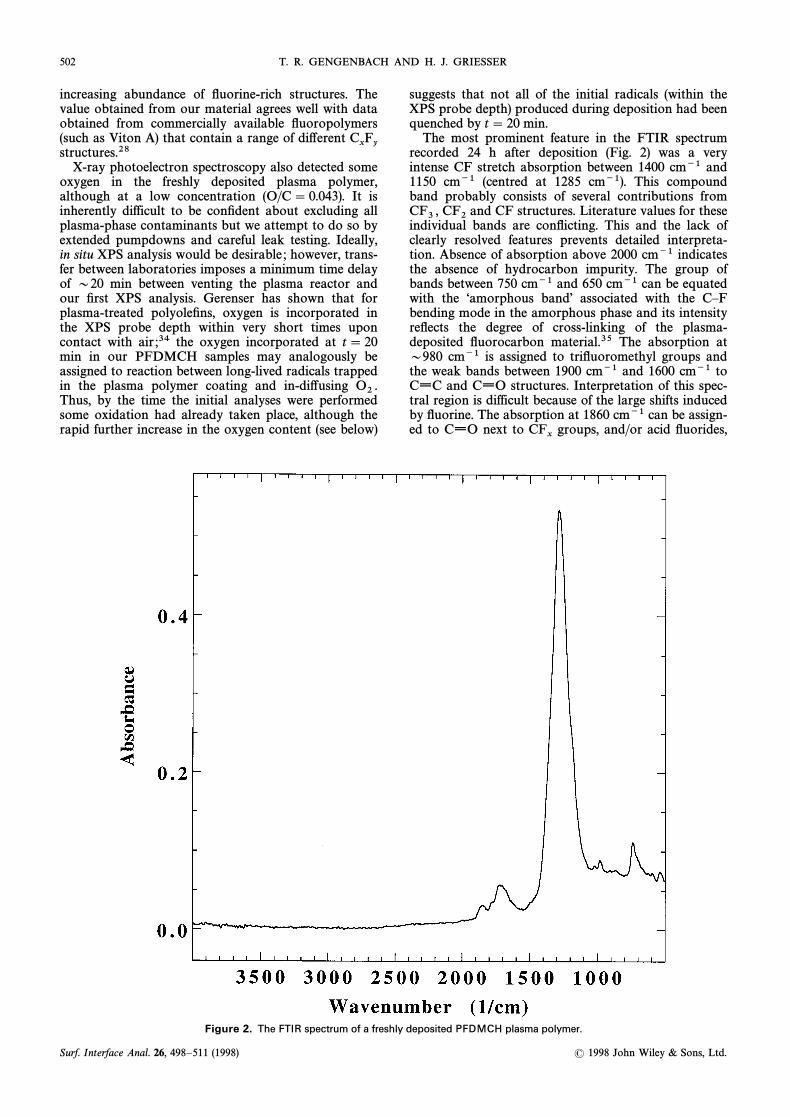
502 T. R. GENGENBACH AND H. J. GRIESSER
increasing abundance of Ñuorine-rich structures. Thevalue obtained from our material agrees well with dataobtained from commercially available Ñuoropolymers(such as Viton A) that contain a range of di†erent C
xFystructures.28
X-ray photoelectron spectroscopy also detected someoxygen in the freshly deposited plasma polymer,although at a low concentration (O/C \ 0.043). It isinherently difficult to be conÐdent about excluding allplasma-phase contaminants but we attempt to do so byextended pumpdowns and careful leak testing. Ideally,in situ XPS analysis would be desirable ; however, trans-fer between laboratories imposes a minimum time delayof D20 min between venting the plasma reactor andour Ðrst XPS analysis. Gerenser has shown that forplasma-treated polyoleÐns, oxygen is incorporated inthe XPS probe depth within very short times uponcontact with air ;34 the oxygen incorporated at t \ 20min in our PFDMCH samples may analogously beassigned to reaction between long-lived radicals trappedin the plasma polymer coating and in-di†using O2 .Thus, by the time the initial analyses were performedsome oxidation had already taken place, although therapid further increase in the oxygen content (see below)
suggests that not all of the initial radicals (within theXPS probe depth) produced during deposition had beenquenched by t \ 20 min.
The most prominent feature in the FTIR spectrumrecorded 24 h after deposition (Fig. 2) was a veryintense CF stretch absorption between 1400 cm~1 and1150 cm~1 (centred at 1285 cm~1). This compoundband probably consists of several contributions from
and CF structures. Literature values for theseCF3 , CF2individual bands are conÑicting. This and the lack ofclearly resolved features prevents detailed interpreta-tion. Absence of absorption above 2000 cm~1 indicatesthe absence of hydrocarbon impurity. The group ofbands between 750 cm~1 and 650 cm~1 can be equatedwith the “amorphous bandÏ associated with the CÈFbending mode in the amorphous phase and its intensityreÑects the degree of cross-linking of the plasma-deposited Ñuorocarbon material.35 The absorption atD980 cm~1 is assigned to triÑuoromethyl groups andthe weak bands between 1900 cm~1 and 1600 cm~1 toCxC and CxO structures. Interpretation of this spec-tral region is difficult because of the large shifts inducedby Ñuorine. The absorption at 1860 cm~1 can be assign-ed to CxO next to groups, and/or acid Ñuorides,CF
x
Figure 2. The FTIR spectrum of a freshly deposited PFDMCH plasma polymer.
Surf. Interface Anal. 26, 498È511 (1998) ( 1998 John Wiley & Sons, Ltd.

AGEING OF PLASMA-DEPOSITED FLUOROCARBON FILMS 503
Figure 3. The XPS atomic ratios measured on a PFDMCH plasma polymer, at 0¡ emission angle, as a function of storage time: F/C (=)and O/C Inset : O/C ratio during the first 2 months. Vertical bars indicate, for selected data points, the standard deviation.(…).
although the latter are usually observed at slightlyhigher frequencies (1870È1880 cm~1). The band at 1780cm~1 can be assigned to structures.CFxCF2Absorption between 1700 cm~1 and 1650 cm~1 isusually indicative of unsaturated structures, althoughÈCFxCFÈ moieties absorb near 1730 cm~1. The broadabsorption between 1730 cm~1 and 1600 cm~1 may,however, also partly equate with absorption between1735 cm~1 and 1710 cm~1 observed in PTFE after irra-diation in vacuo, assigned to branching and cross-linking bonding conÐgurations.36 The CxO groups notlocated next to Ñuorine-containing structures alsoabsorb near 1730 cm~1 but the low oxygen contentdetected by XPS argues against them being important.As FTIR probes a larger thickness region of the coatingthan XPS, and our previous studies suggest that it takesseveral days for the oxidation front to move into thebulk of the plasma polymer, oxygen-containing groupsshould be of low intensity in the FTIR spectra of freshPFDMCH coatings.
The air/water contact angles (h) measured on thefreshly deposited material and(hA \ 115¡, hS\ 104¡
were comparable to those recorded on the ref-hR\ 76¡)erence material FEP and(hA \ 117¡, hS\ 107¡ hR\
except for which was markedly lower. The98¡), hRresulting contact angle hysteresis of 38¡ (cf. 19¡ for FEP)is assigned to chemical heterogeneity caused by thepresence on the surface of a wide variety of structuresranging from polar oxygen-containing groups (albeit alow amount) to extremely non-polar triÑuoromethylgroups. To exclude a contribution from surface rough-ness to the hysteresis, we measured the root meansquare (RMS) roughness of PFDMCH plasma polymerwith an AFM (Nanoscope III, Digital Instruments) ;several scans (10 lm ] 10 lm) yielded RMS values ofbetween 3 nm and 4 nm. Surface roughness of this mag-
nitude probably does not add signiÐcantly to contactangle hysteresis.
Changes to the chemical structure during ageing
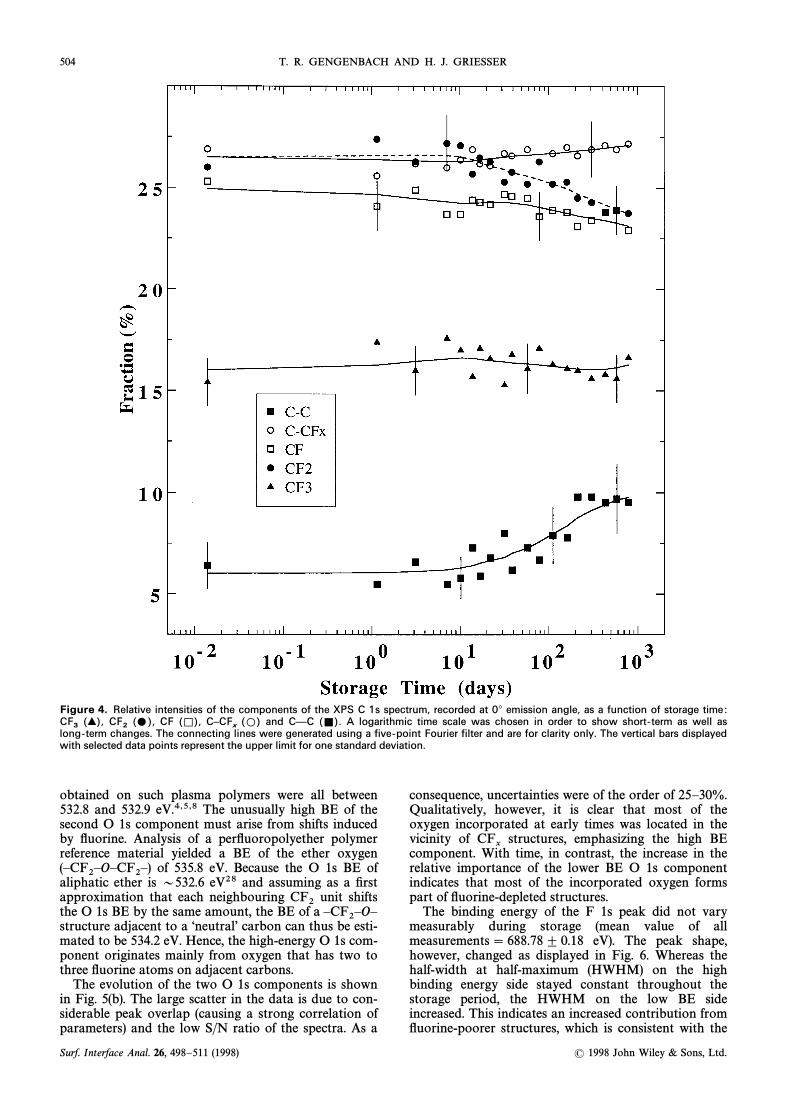
Figures 3È7 illustrate the chemical changes that wereobserved in the course of 18 months of storage in air.The F/C ratio was observed to decrease slowly from aninitial value of 1.23 to 1.1 (Fig. 3). We note that themagnitude of the decrease in the F/C ratio was abouttwice that of the increase of the O/C ratio (Fig. 3), sug-gesting that conversion of elements to carbonylÈCF2Èmay be an important process. Figures 4 and 5 displaythe results of curve Ðtting the C 1s and O 1s spectra.After 7È14 days the fractions of and CF decreasedCF2slightly, whereas the percentages of carbon atoms notdirectly bonded to Ñuorine and CÈC) increased.(CÈCF
xThe overall abundance of species stayed constantCF3within experimental error. With the exception of CF3groups, it appeared that the more Ñuorine-rich thestructural element, the greater the observed loss withtime.
The oxygen content measured by XPS (Fig. 3)increased rapidly during the Ðrst day ; then oxygenincorporation slowed down markedly. The ratedecreased further after 1È2 months but the amount ofoxygen continued to increase for many more months.Curve Ðtting revealed more detail. The range of BEvalues determined for the two O 1s components was532.7È532.9 eV for component O1 and 534.5È534.7 eVfor O2 [Fig. 5(a)]. The former value is typical of O 1ssignals measured on oxidizing hydrocarbon-basedplasma polymers where a range of di†erent carbonÈoxygen functionalities (e.g. hydroxyls, ethers, carbonyls)contribute to the peak : experimental BE values
( 1998 John Wiley & Sons, Ltd. Surf. Interface Anal. 26, 498È511 (1998)
504 T. R. GENGENBACH AND H. J. GRIESSER
Figure 4. Relative intensities of the components of the XPS C 1s spectrum, recorded at 0¡ emission angle, as a function of storage time:CF and C—C A logarithmic time scale was chosen in order to show short-term as well asCF
3(>), CF
2(…), (K), C–CF
x(L) (=).
long-term changes. The connecting lines were generated using a five-point Fourier filter and are for clarity only. The vertical bars displayedwith selected data points represent the upper limit for one standard deviation.
obtained on such plasma polymers were all between532.8 and 532.9 eV.4,5,8 The unusually high BE of thesecond O 1s component must arise from shifts inducedby Ñuorine. Analysis of a perÑuoropolyether polymerreference material yielded a BE of the ether oxygen
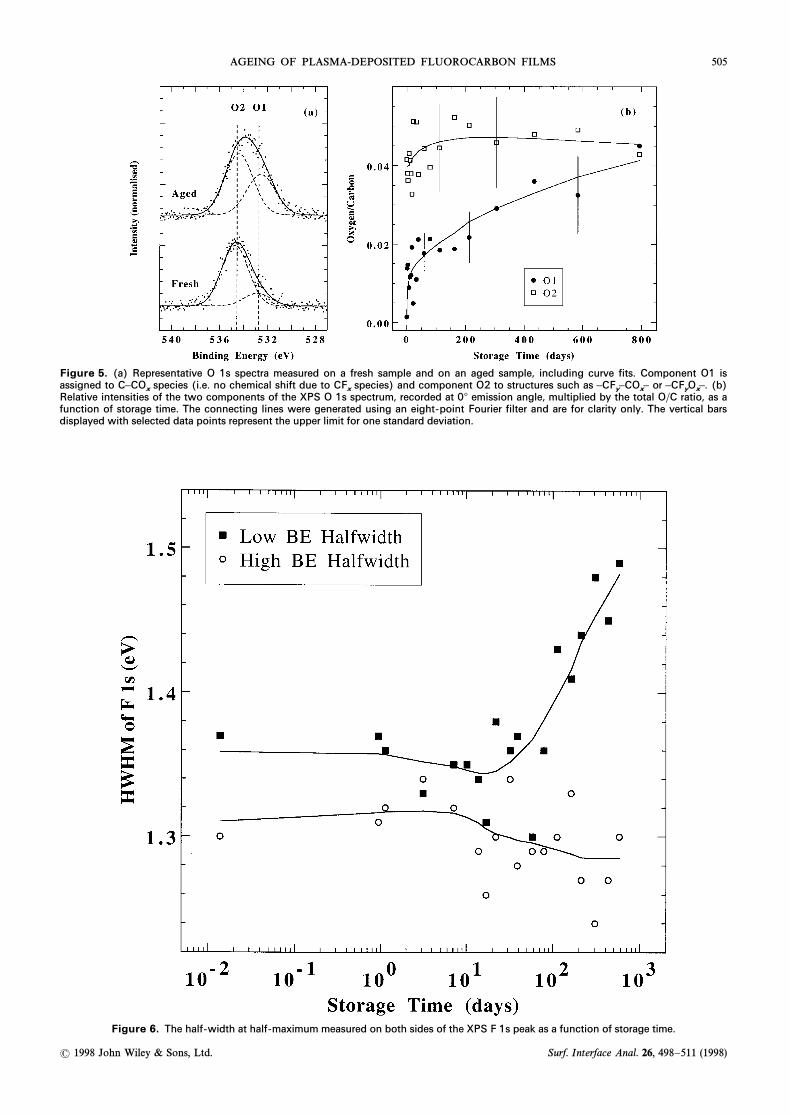
of 535.8 eV. Because the O 1s BE of(ÈCF2ÈOÈCF2È)aliphatic ether is D532.6 eV28 and assuming as a Ðrstapproximation that each neighbouring unit shiftsCF2the O 1s BE by the same amount, the BE of a ÈCF2ÈOÈstructure adjacent to a “neutralÏ carbon can thus be esti-mated to be 534.2 eV. Hence, the high-energy O 1s com-ponent originates mainly from oxygen that has two tothree Ñuorine atoms on adjacent carbons.
The evolution of the two O 1s components is shownin Fig. 5(b). The large scatter in the data is due to con-siderable peak overlap (causing a strong correlation ofparameters) and the low S/N ratio of the spectra. As a
consequence, uncertainties were of the order of 25È30%.Qualitatively, however, it is clear that most of theoxygen incorporated at early times was located in thevicinity of structures, emphasizing the high BECF
xcomponent. With time, in contrast, the increase in therelative importance of the lower BE O 1s componentindicates that most of the incorporated oxygen formspart of Ñuorine-depleted structures.
The binding energy of the F 1s peak did not varymeasurably during storage (mean value of allmeasurements \ 688.78^ 0.18 eV). The peak shape,however, changed as displayed in Fig. 6. Whereas thehalf-width at half-maximum (HWHM) on the highbinding energy side stayed constant throughout thestorage period, the HWHM on the low BE sideincreased. This indicates an increased contribution fromÑuorine-poorer structures, which is consistent with the
Surf. Interface Anal. 26, 498È511 (1998) ( 1998 John Wiley & Sons, Ltd.
AGEING OF PLASMA-DEPOSITED FLUOROCARBON FILMS 505
Figure 5. (a) Representative O 1s spectra measured on a fresh sample and on an aged sample, including curve fits. Component O1 isassigned to species (i.e. no chemical shift due to species) and component O2 to structures such as or (b)C–CO
xCF
x–CF
y–CO
x– –CF
yO
x–.
Relative intensities of the two components of the XPS O 1s spectrum, recorded at 0¡ emission angle, multiplied by the total O/C ratio, as afunction of storage time. The connecting lines were generated using an eight-point Fourier filter and are for clarity only. The vertical barsdisplayed with selected data points represent the upper limit for one standard deviation.
Figure 6. The half-width at half-maximum measured on both sides of the XPS F 1s peak as a function of storage time.
( 1998 John Wiley & Sons, Ltd. Surf. Interface Anal. 26, 498È511 (1998)
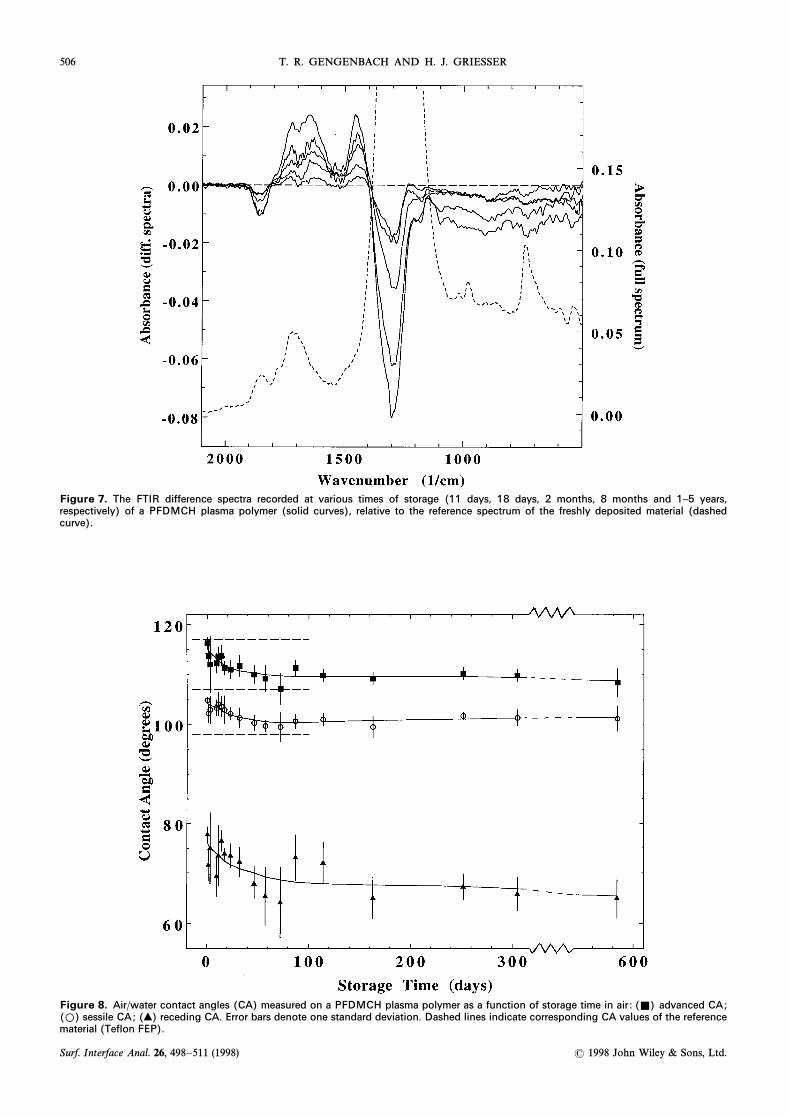
506 T. R. GENGENBACH AND H. J. GRIESSER
Figure 7. The FTIR difference spectra recorded at various times of storage (11 days, 18 days, 2 months, 8 months and 1–5 years,respectively) of a PFDMCH plasma polymer (solid curves), relative to the reference spectrum of the freshly deposited material (dashedcurve).
Figure 8. Air/water contact angles (CA) measured on a PFDMCH plasma polymer as a function of storage time in air : advanced CA;(=)sessile CA; receding CA. Error bars denote one standard deviation. Dashed lines indicate corresponding CA values of the reference(L) (>)
material (Teflon FEP).
Surf. Interface Anal. 26, 498È511 (1998) ( 1998 John Wiley & Sons, Ltd.
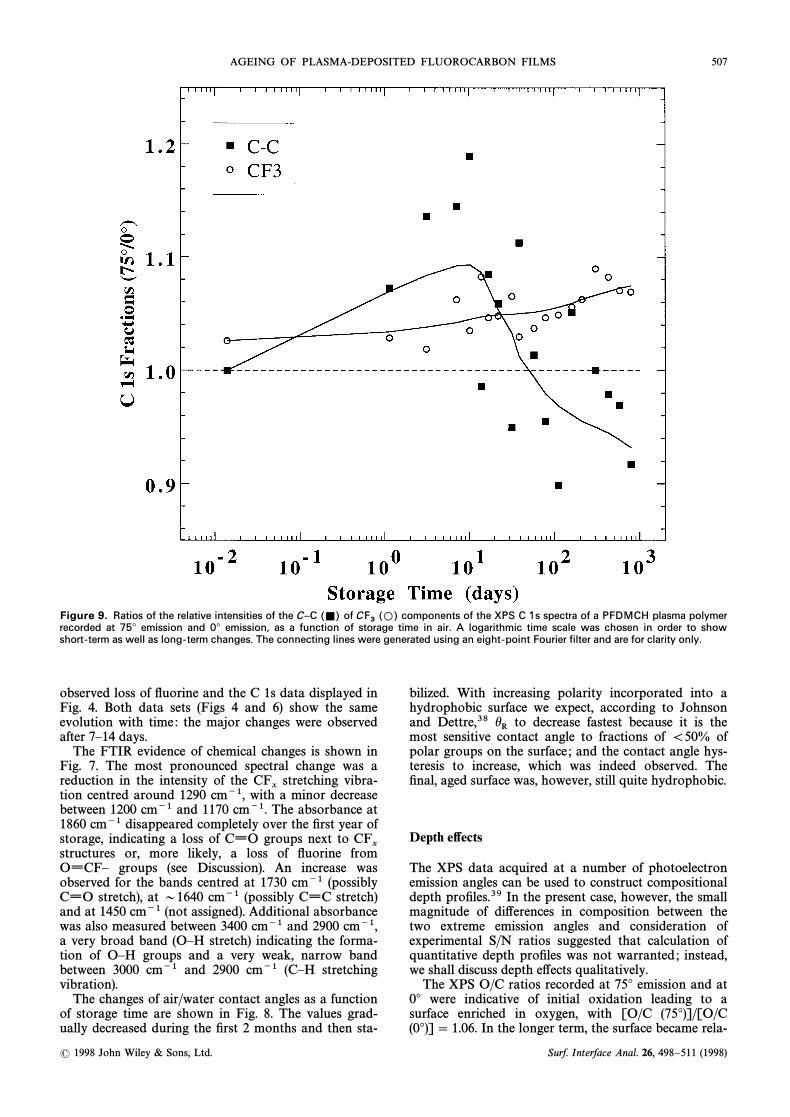
AGEING OF PLASMA-DEPOSITED FLUOROCARBON FILMS 507
Figure 9. Ratios of the relative intensities of the C–C of components of the XPS C 1s spectra of a PFDMCH plasma polymer(=) CF3
(L)recorded at 75¡ emission and 0¡ emission, as a function of storage time in air. A logarithmic time scale was chosen in order to showshort-term as well as long-term changes. The connecting lines were generated using an eight-point Fourier filter and are for clarity only.
observed loss of Ñuorine and the C 1s data displayed inFig. 4. Both data sets (Figs 4 and 6) show the sameevolution with time : the major changes were observedafter 7È14 days.
The FTIR evidence of chemical changes is shown inFig. 7. The most pronounced spectral change was areduction in the intensity of the stretching vibra-CF
xtion centred around 1290 cm~1, with a minor decreasebetween 1200 cm~1 and 1170 cm~1. The absorbance at1860 cm~1 disappeared completely over the Ðrst year ofstorage, indicating a loss of CxO groups next to CF
xstructures or, more likely, a loss of Ñuorine fromOxCFÈ groups (see Discussion). An increase wasobserved for the bands centred at 1730 cm~1 (possiblyCxO stretch), at D1640 cm~1 (possibly CxC stretch)and at 1450 cm~1 (not assigned). Additional absorbancewas also measured between 3400 cm~1 and 2900 cm~1,a very broad band (OÈH stretch) indicating the forma-tion of OÈH groups and a very weak, narrow bandbetween 3000 cm~1 and 2900 cm~1 (CÈH stretchingvibration).
The changes of air/water contact angles as a functionof storage time are shown in Fig. 8. The values grad-ually decreased during the Ðrst 2 months and then sta-
bilized. With increasing polarity incorporated into ahydrophobic surface we expect, according to Johnsonand Dettre,38 to decrease fastest because it is thehRmost sensitive contact angle to fractions of \50% ofpolar groups on the surface ; and the contact angle hys-teresis to increase, which was indeed observed. TheÐnal, aged surface was, however, still quite hydrophobic.
Depth e†ects
The XPS data acquired at a number of photoelectronemission angles can be used to construct compositionaldepth proÐles.39 In the present case, however, the smallmagnitude of di†erences in composition between thetwo extreme emission angles and consideration ofexperimental S/N ratios suggested that calculation ofquantitative depth proÐles was not warranted ; instead,we shall discuss depth e†ects qualitatively.
The XPS O/C ratios recorded at 75¡ emission and at0¡ were indicative of initial oxidation leading to asurface enriched in oxygen, with [O/C (75¡)]/[O/C(0¡)]\ 1.06. In the longer term, the surface became rela-
( 1998 John Wiley & Sons, Ltd. Surf. Interface Anal. 26, 498È511 (1998)

508 T. R. GENGENBACH AND H. J. GRIESSER
tively depleted in oxygen (compared to deeper levels)even though the oxygen levels continued to increase inabsolute terms. In other words, the oxygen levels deter-mined at 0¡ emission increased at a faster rate than thecorresponding data measured at 75¡, reversing the gra-dient in the depth distribution over time. Althoughthere is a fair degree of scatter in the data, there is aconsistent downward trend (data not shown), reaching avalue of [O/C(75¡)]/[O/C (0¡)]\ 0.98 after 1 year.Fluorine was enriched in the uppermost surface layer atall times. The ratio of 75¡ F/C data vs. 0¡ data initiallywas 1.07 and showed no systematic changes with time(data not shown). After 1 year the value was 1.06 ; thesevalues are identical within experimental accuracy.
Fitting of the C 1s components to signals recorded atvarious times of storage also enabled assessment oftime-dependent depth variations in composition. Themost pronounced e†ect was seen in the “neutralÏ C 1scomponent, which showed a behaviour analogous tothat of oxygen : overall, the fraction of this componentincreased at all times ; however, the relative intensityratio of this component recorded at 75¡ to that record-ed at 0¡ increased at Ðrst and then dropped steadilyover a period of 2 years (Fig. 9). This indicates that, inrelative terms, the uppermost surface layer becameenriched in this component during the Ðrst few weeksbut eventually depleted. The highest BE component
groups) was always enriched at the surface ; the(CF3ratios (75¡/0¡) increased signiÐcantly on storage (Fig. 9).The depth distribution of the three remaining species
CF and remained essentially unchanged(CÈCFx, CF2)during storage. The latter two were uniformly distrib-
uted over the probe depth (75¡/0¡ D 1.0) whereasstructures were slightly depleted within theCÈCF
xuppermost surface layer (75¡/0¡ D 0.97).
DISCUSSION
Changes to the chemical structure during ageing
The spontaneous ambient oxidation of hydrocarbon-based plasma polymers has been documented in earlierreports.4,27 It is initiated during the deposition processas radicals are incorporated into the growing Ðlm.1 Pre-sumably due to steric constraints in the cross-linkedmaterial, some of these radicals are unable to satisfytheir bonding requirements during Ðlm formation. Onexposure to air these remaining radicals react with in-di†using oxygen. The peroxy radicals thus producedrapidly convert to peroxides and, by abstraction ofhydrogen, to hydroperoxides. These two types of groupsare metastable and decay in turn, forming stable endproducts as well as regenerating radicals that continuethe oxidative reaction cycles.4 Whereas the initial stepof radical quenching with oxygen is very fast, the sub-sequent reactions happen on a much slower time scale.Apart from the plasma-induced initiation step, the reac-tion sequences are basically the same as those operatingin the oxidative degradation of conventional hydrocar-
bon polymers, but the plasma polymer environment sig-niÐcantly alters the importance and relative yields of thevarious reactions. Most importantly, the oxidation ofthe plasma polymers studied to date4h7 slows downwith time and eventually becomes negligible, in contrastto the autocatalytically accelerated degradation of poly-oleÐns.
The chemical changes upon ageing observed forPFDMCH plasma polymer are much less pronouncedthan those taking place in the hydrocarbon-basedplasma polymers studied previously. The O/C ratioafter extended storage was D0.09 for PFDMCHplasma polymer compared with 0.25È0.3 for plasma-deposited materials from n-hexane4 and n-heptylamine.5Consistent with the relatively small amount of oxygenincorporation, the F/C ratio decreased by only D10%over the same period of time. These observationssuggest that Ñuoropolymer coatings have a much lowerpropensity for oxidative ageing.
Yet, inspection of the kinetics of oxygen incorpor-ation reveals that while the extents of long-term oxida-tion di†er markedly between PFDMCH andhydrocarbon-based plasma polymers, there is a sur-prising similarity in the oxidative behaviour at shorttime intervals. The amount of oxygen incorporated inthe initial, very rapid phase of oxidation di†ered little,with values in the range 0.04È0.07 for the variousplasma polymers studied so far.4h7 This stage (i.e. theÐrst day after exposure to air) is dominated by the reac-tion between radicals and in-di†using oxygen. Hence, itappears reasonable to conclude that the density of rad-icals is not substantially lower in freshly depositedPFDMCH plasma polymer (prepared under the presentset of conditions) than in freshly deposited alkane andalkylamine plasma polymers deposited under similarplasma conditions.
The subsequent increase in the oxygen content after 1day was, however, 5È10 times less for PFDMCHplasma polymer samples compared with hydrocarbon-based plasma polymers. Evidently the e†ectiveness ofsecondary oxidation reactions was substantially reducedfor the Ñuorocarbon plasma polymer. Hydrogenabstraction is a major requirement for auto-oxidation,being involved in a number of key reaction sequences.With no hydrogen present in the freshly deposited Ðlm,the conversion of peroxy radicals to hydroperoxides isnot possible, and thus a major oxidative reactionpathway is inoperative in the PFDMCH plasma poly-mers. Other subsequent reactions known to operate inpolyoleÐn oxidation also are not feasible in the oxida-tion of PFDMCH plasma polymers. In the absence ofhydrogen-associated reactions, radical dissipation mustoccur by alternative pathways ; studies of the e†ects ofionizing radiation on PTFE36 provide guidance for pos-tulating possible reactions in the ambient oxidation ofPFDMCH plasma polymers.
The initially generated, carbon-centred radicals arebelieved to react rapidly with in-di†using oxygen toform peroxy radicals, which are evident in the high BEO 1s XPS signal but not detectable by FTIR becausethe CÈO stretch band absorbs in the region dominatedby the intense CÈF stretch band. Peroxy radicals cancombine with carbon-centred radicals to form peroxi-des, which can subsequently dissociate to formperÑuoro-alkoxy radicals. These can convert to acid
Surf. Interface Anal. 26, 498È511 (1998) ( 1998 John Wiley & Sons, Ltd.

AGEING OF PLASMA-DEPOSITED FLUOROCARBON FILMS 509
Ñuorides and a secondary radical. The acid Ñuoride isexpected to possess an XPS O 1s (OCÈF) BE similar tothat of peroxy and peroxide oxygens (ÈOÈCF2È),thereby preventing direct detection of this reactionpathway. The FTIR absorption at 1860 cm~1 (Fig. 2),however, is consistent with acid Ñuorides.
Acid Ñuorides tend to hydrolyse, leading to carbox-ylic acid groups and the release of hydrogen Ñuoride.The decrease in absorbance at 1860 cm~1 over time andthe increase in intensity of bands expected for carbox-ylic acids (OÈH stretch between 3400 and 2900 cm~1and CO stretch between 1800 and 1700 cm~1) (Fig. 7)indicates that this process occurs during the ageing ofPFDMCH plasma polymers. The postulated release ofHF is consistent with the reduction in the Ñuorinecontent observed by XPS and the reduced intensity ofIR CÈF bands.
The perÑuoro-alkoxy radicalsCF2-based (ÈCF2ÈO~)can decay by releasing carbonyl Ñuoride in the(CF2O),process generating a secondary carbon-centred radical.This reaction also leads to a loss of Ñuorine, in particu-lar groups, as observed by XPS (Fig. 4). TheCF2increase in IR absorbance around 1640 cm~1 and theincrease in the XPS fraction of carbon atoms notbonded to F or indicates the formation of cross-CF
xlinks and unsaturated structures as a result of ageing.Steric e†ects may a†ect reaction yields signiÐcantly.
The high thermal stability of linear perÑuoropolyetherpolymers is substantially reduced when branching isintroduced ; this reduced stability is mainly due to stericstrain caused by the large van der Waals radius of Ñuo-rine (1.47 compared to 1.2 for H).10 The XPS F/C ratioindicates that our PFDMCH plasma polymers arehighly branched. In conjunction with limited segmentalmobility on account of cross-links, this probably resultsin considerable micro-strain in the freshly depositedcoatings and thus increased susceptibility for reactionsat strained sites.
Most of the properties of freshly deposited plasmapolymers that inÑuence the ageing characteristics ofthese materials are determined by the deposition condi-tions. In particular, the plasma power input permonomer mass unit (W/FM) plays a substantial role, asshown in a recent study of the long-term evolution ofthe chemical structure of methylmethacrylate (MMA)plasma deposited polymeric coatings :8 A low value ofW/FM led to a high degree of retention of the originalmonomer structure whereas a high value of W/FMresulted in substantial monomer fragmentation and theformation of a partially unsaturated material consider-ably di†erent to conventional poly(methyl methacrylate)(PMMA). As the MMA plasma coatings were stored inambient air after fabrication, all showed spontaneousoxidative changes to their composition. Deposition atlow W/FM led to moderate oxidative changes, whereashigh power led to a pronounced increase in the oxygencontent over time and resulted in a wide range ofcarbonÈoxygen functionalities in the aged material. Asthe initial plasma deposition conditions thus inÑuencedthe oxidative post-deposition reactions, MMA plasmapolymers deposited under di†erent conditions not onlyvaried in their initial composition but became evenmore diverse as they aged. We hypothesize that thisapplies to plasma-deposited materials in general ;however, it is beyond the scope of this report to verify
the hypothesis for the speciÐc case of PFDMCH plasmapolymers.
Depth e†ects
Three possible reasons might cause the composition ofPFDMCH plasma polymers to vary with depth and onageing :(1) The outermost layer of the freshly deposited plasma
polymer may di†er from the deeper layers becauseradical reactions may proceed for a short while afterthe plasma is extinguished. The distribution andtypes of active species present after switching o† thedriving power may di†er from that in the activeplasma, leading to a surface layer with a di†erentmix of chemical groups or possibly even containinggroups not present deeper down.
(2) Oxidation results from a combination of carbon-centred radicals with in-di†using atmospheric O2 ;4the initial reaction thus is di†usion-terminated andinitially concentrated in the surface layers.
(3) Although plasma polymers are usually considered tobe highly cross-linked and to have limited segmentalmobility, surface restructuring can occur leading tochanges in the depth distributions of polar and non-polar groups with time.7,27
Accordingly, compositional di†erences with depth infreshly deposited plasma polymers would be indicativeof post-glow e†ects on the surface layers, whilst onageing either oxygen-terminated di†usion e†ects orsurface restructuring might be revealed. Di†usion ofoxygen and surface restructuring37 are fast processes inthe case of conventional Ñuoropolymers (due to theirhigh permeability and high chain mobility) ; forO2plasma polymers, in contrast, these processes are sub-stantially slower. We attribute this di†erence to the highcross-link density of the latter materials.
Uncertainties in quantiÐcation of grazing-anglereÑectance IR data prevent direct quantitative compari-son of the experimental XPS and IR data for eluci-dation of compositional depth information ;qualitatively, however, the spectral data appear to be inagreement. More informative is the comparison of XPSdata recorded at two emission angles. The initial surfaceenrichment of oxygen suggests, as for other plasmapolymers,7,27 a reaction-terminated di†usion boundaryon oxidation. As the oxidation front moves inwards, thesurface enrichment disappears. The similarity in thekinetics and depth distribution of oxygen and carbonatoms that do not carry Ñuorine atoms suggests thatoxygen incorporation and Ñuorine loss are linked.
The surface enrichment of Ñuorine in freshly depos-ited PFDMCH plasma polymers (Fig. 9) is of interest.The observation that groups were more abundantCF3at the surface accords with the greater F/C ratio at 75¡emission. We speculate that either the groups areCF3surface-enriched because, as in other polymers, chainends preferentially orient towards the surface for entro-pic reasons or that as the plasma is extinguished someadditional material is deposited from monomer frag-ments that are less fragmented and thus richer in Ñuo-rine. As oxidation proceeds, the groups appear notCF3
( 1998 John Wiley & Sons, Ltd. Surf. Interface Anal. 26, 498È511 (1998)

510 T. R. GENGENBACH AND H. J. GRIESSER
to be attacked ; Fig. 4 shows that oxidation favourscarbon atoms carrying less than three Ñuorine atoms
and CF groups).(CF2The contact angle data reach Ðnal values muchearlier than the XPS and FTIR data sets. Contactangles are thought to probe polar groups to a depth ofD0.5 nm,40 whereas XPS analyses a layer of D1.5È2nm at 75¡ (glancing) emission and D6 nm at 0¡ (normal)emission. Over the course of the Ðrst 2 months afterdeposition, the polarity of the surface layers as mea-sured by contact angles changed gradually. After thattime, contact angles no longer were a†ected by ageing,indicating that the surface layers had stabilized ; thefurther changes evident in XPS and IR spectra tookplace in deeper regions.
Surface restructuring
Many polymer surfaces become more hydrophobic asthey are stored in air ; this phenomenon is commonlycalled “surface restructuringÏ.41 We have found surfacerestructuring to operate in a number of other plasmapolymers, with a depletion in oxygen and nitrogen inthe surface layers relative to deeper regions.7,27 ForPFDMCH plasma polymers a “hydrophobicrecoveryÏÈincrease in the contact angles as polargroups move away from the air interfaceÈwas notobserved on further storage after the 2 month point atwhich oxidation had apparently terminated in the out-ermost layers. On the other hand, as for other plasmapolymers, XPS provided evidence of changes with timeto the depth distribution of oxygen, and the componentto the C 1s signal of carbon atoms not carrying Ñuorinealso showed a time-dependent depth distribution (Fig.9) paralleling that of the O/C ratios. The oxygencontent at the surface relative to deeper regionsdecreases gradually with time over 1 year, but the mag-nitude of the total decrease was small, indicating verylimited surface restructuring.
The extent of surface restructuring might depend onseveral factors, one of them being the inherent di†usion-al mobility of polymer segments. The outermost layersof plasma polymers might be more mobile as a result ofdeposition of less cross-linked material in the post-plasma atmosphere. However, there is also a need for adriving force such as interfacial enthalpy or a concen-tration gradient (translational entropy). The low-densityair environment probably has little interfacial e†ect onthe surface layers, and thus surface restructuring incontact with air, or any gaseous medium, may be domi-nated by translational entropy forces. Although it is notlarge, the reactiveÈdi†usive surface enrichment of polargroups during the initial stages of oxidation provides anentropic driving force for surface restructuring in thePFDMCH plasma polymer.
The gradual surface enrichment of groups onCF3storage also is assignable to surface restructuring. Thesegroups move to the air interface, compensating for theinward migration of some of the polar structures. The
groups preferentially migrate to the air interfaceCF3because they a†ord the lowest interfacial energy contri-bution.
CONCLUSIONS
In summary, spontaneous post-deposition oxidation ofPFDMCH plasma polymers was observed when theywere stored under ambient conditions. This propensityfor slow oxidation after fabrication is shared with mostother plasma polymers (deposited in our apparatusunder similar conditions), but the extent of oxidation ismuch less for PFDMCH coatings than when usinghydrocarbon-based monomers (alkanes, alkylamines,alcohols). As for those plasma polymers, incorporationof oxygen into PFDMCH coatings shows multi-stepkinetics. The Ðrst, rapid step, within 1 day after fabrica-tion, led to an oxygen amount similar to that recordedfor other plasma polymers. This suggests that thedensity of radicals incorporated during deposition andcapable of reacting with in-di†using is similar. TheO2subsequent oxidation steps are, however, far less effi-cient in PFDMCH plasma polymer compared withhydrocarbon-based plasma polymers, markedlyreducing the oxygen uptake rate on extended storage.
There also appears to be only a small extent ofsurface restructuring as assessed from the small depthvariations of compositions. The surface is enriched in
groups at all times. While XPS recorded contin-CF3uing incorporation of oxygen for more than 2 years, theair/water contact angles decreased only during the Ðrst2 months and on further storage remained stable. Thefurther oxidation shown by XPS and FTIR after 2months therefore must occur exclusively in deeper([0.5 nm) regions of the polymer. The gradual declinein oxidation allows the coatings to retain their structur-al integrity. The PFDMCH plasma polymers are there-fore well suited for applications that require long-termstability and predictable, stable interfacial properties ; ifnecessary, incubation for 2 months would stabilizesurface properties prior to end use.
Acknowledgement
The aluminized Kapton (Al-Kapton) tape was a generous gift fromMr R. Spahn, Eastman Kodak Research Laboratories, Rochester,NY, USA.
REFERENCES
1. H. von Boenig, Fundamentals of Plasma Chemistry and Tech-nology, Technomic, Lancaster, PA (1988).
2. R. d’Agostino, F. Cramarossa, F. Fracassi and F. Illuzi, inPlasma Deposition, Treatment and Etching of Polymers , 1stEdn, ed. by R. d’Agostino, p. 95. Academic Press, San Diego(1990).
3. Y. Haque and B. D. Ratner, J . Polym.Sci . B 26, 1237 (1988).4. T. R. Gengenbach, Z. R. Vasic, R. C. Chatelier and H. J.
Griesser, J . Polym.Sci . A 32, 1399 (1994).5. T. R. Gengenbach, R. C. Chatelier and H. J. Griesser, Surf .
Interface Anal . 24, 271 (1996).6. T. R. Gengenbach, R. C. Chatelier and H. J. Griesser, Surf .
Surf. Interface Anal. 26, 498È511 (1998) ( 1998 John Wiley & Sons, Ltd.

AGEING OF PLASMA-DEPOSITED FLUOROCARBON FILMS 511
Interface Anal . 24, 611 (1996).7. T. R. Gengenbach, Z. R. Vasic, S. Li, R. C. Chatelier and H. J.
Griesser, Plasmas Polym. 00, 000 (1997).8. T. R. Gengenbach and H. J. Griesser, J . Polym. Sci . A 36, 985
(1998).9. W. Potter, A. J. Ward and R. D. Short, Polym. Degrad. Stab.
43, 385 (1994).10. B. E. Smart, in Organofluorine Chemistry : Principles and Com-
mercial Applications , 1st Edn, ed. by R. E. Banks, B. E. Smartand J. C. Tatlow, Chapt. 3, Plenum Press, New York (1994).
11. U. Hetzler and E. Kay, J.Appl . Phys. 49, 5617 (1978).12. S. Kaplan and A. Dilks, J . Appl . Polym. Sci .: Appl . Polym.
Symp. 38, 105 (1984).13. M. Friedrich, D. Hinze and W. Ebert, Thin Solid Films 112, 61
(1984).14. N. Inagaki, S. Tasaka and T. Murata, J. Appl . Polym. Sci . 38,
1869 (1989).15. D. Wang and J. Chen, J.Appl . Polym.Sci . 42, 233 (1991).16. A. G. Shard, H. S. Munro and J. P. S. Badyal, Polym.
Commun. 32, 152 (1991).17. M. A. Golub, T. Wydeven and R. T. Cormia, J. Polym. Sci . A
30, 2683 (1992).18. C. R. Savage, R. B. Timmons and J. W. Lin, Adv. Chem. Ser .
236, 745 (1993).19. H. S. Munro, R. J. Ward, M. C. Davies and R. D. Short,
Polymer 34, 2250 (1993).20. M. J. O’Keefe and J. M. Rigsbee, J. Appl . Polym. Sci . 53,
1631 (1994).21. E. Krentsel, H. Yasuda, M. Miyama and T. Yasuda, J. Polym.
Sci . 33, 2887 (1995).22. M. Horie, J . Vac. Sci . Technol . A 13, 2490 (1995).23. H. H. Chen and M. D. Ries, J . Adhes. Sci . Technol . 10, 495
(1996).24. A. Tasaka, A. Komura, Y. Uchimoto, M. Inaba and Z. Ogumi,
J. Polym.Sci . A 34, 193 (1996).25. A. M. Hynes, M. J. Shenton and J. P. S. Badyal, Macro-
molecules 29, 18 (1996).
26. R. Chen and M. S. Silverstein, J. Polym. Sci . A 34, 207(1996).
27. T. R. Gengenbach, Z. R. Vasic, R. C. Chatelier and H. J.Griesser, Plasmas Polym. 1, 207 (1996).
28. G. Beamson and D. Briggs, High Resolution XPS of OrganicPolymers . The Scienta ESCA300 Database , 1st Edn. Wiley,Chichester (1992).
29. K. Harrison and L. B. Hazell, Surf . Interface Anal . 18, 368(1992).
30. C. D. Wagner, W. M. Riggs, L. E. Davis, J. F. Moulder and G.E. Muilenberg (eds), Handbook of X-Ray PhotoelectronSpectroscopy. Perkin-Elmer, Physical Electronics Division,Eden Prairie, MN (1979).
31. H. W. StJohn, T. C. Vaithianathan and H. J. Griesser, Surf .Interface Anal ., in preparation.
32. A. E. Hughes and B. A. Sexton, J. Electron Spectrosc . Relat .Phenom. 46, 31 (1988).
33. L. J. Bellamy, The Infrared Spectra of Complex Molecules ,Vol. 1, 3rd Edn. Halsted Press, Wiley, New York (1975).
34. L. J. Gerenser, J . Adhes. Sci . Technol . 1, 303 (1987).35. R. D’Agostino, L. Martinu and V. Pische, Proc. 9th Int . Symp.
Plasma Chem. ISPC-9, Pugnochiuso, Italy, 1989, pp. 1201and 1642.
36. W. K. Fisher and J. C. Corelli, J . Polym. Sci : Polym. Chem. Ed.19, 2465 (1981).
37. T. R. Gengenbach, X. Xie, R. C. Chatelier and H. J. Griesser, J .Adhes. Sci . Technol ., 8, 305 (1994).
38. R. E. Johnson and R. H. Dettre, Surf . Colloid Sci . 2, 85(1969).
39. B. J. Tyler, D. G. Castner and B. D. Ratner, Surf . InterfaceAnal . 14, 443 (1989).
40. C. D. Bain and G. M. Whitesides, J. Am. Chem. Soc. 110,5897 (1988).
41. F. Garbassi, M. Morra and E. Occhiello, Polymer Surfaces—From Physics to Technology, Chapt. 2. Wiley, Chichester(1994).
( 1998 John Wiley & Sons, Ltd. Surf. Interface Anal. 26, 498È511 (1998)





























