Embed Size (px)
Citation preview

Comparing phylogenetic and statistical classification methods
for DNA barcoding
Frederic Austerlitz, Olivier David, Brigitte Schaeffer, Sisi Ye, Michel
Veuille & Catherine Laredo

Testing the assignation methods
• Individual to be tested:– X1 : ATATGTACCTAGTA
– X2 : TTATCTACCTAGAA
• Reference sample :– 1_1: ATATGTACGTAGTA
– 1_2: ATATCTACGAAGTA
– 1_3: ATATCTACTAAGTA
– 2_1: ATATGTACGTAGTT
– 2_2: ATATGTACGAAGTT
– 2_3: ATTTCTACTAAGTT
Species 1
Species 2

Phylogenetic methods
1_1 1_3 2_1 1_2 2_2 2_3X1
• Unanimity rule: X1 classified as ambiguous, X2 classified as species 1
• Majority rule: X1 and X2 classified as belonging to species 1
• Two methods tested: neighbor joining and maximum likelihood (PhyML, Guindon and Gascuel 2003)
X2
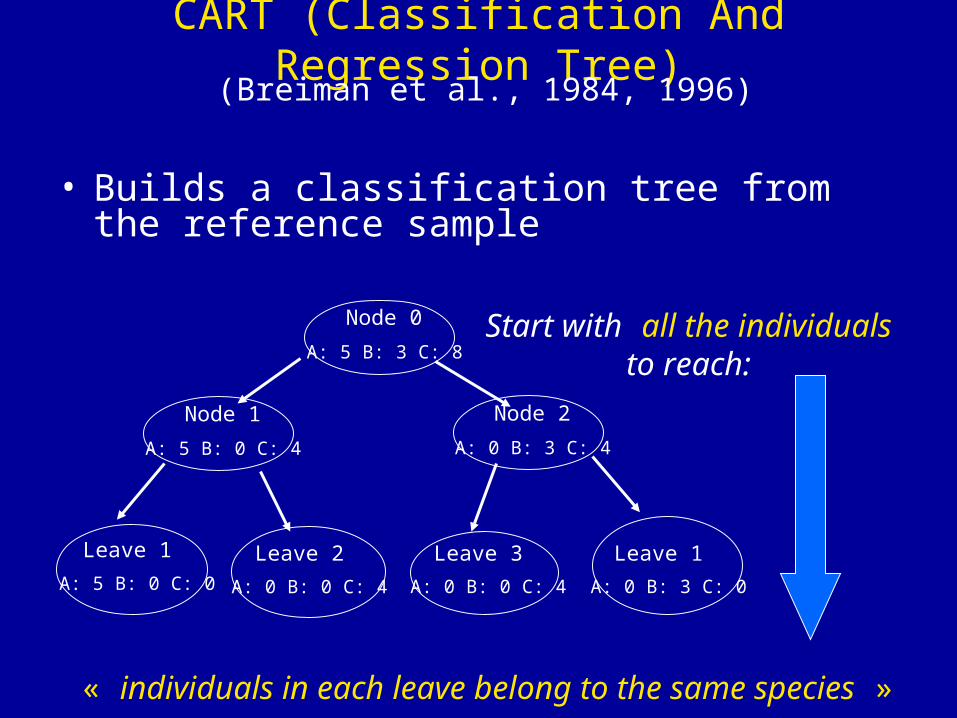
Node 0 Start with all the individualsto reach:
« individuals in each leave belong to the same species »
A: 5 B: 3 C: 8
CART (Classification And Regression Tree)
• Builds a classification tree from the reference sample
(Breiman et al., 1984, 1996)
Node 1
A: 5 B: 0 C: 4
Node 2
A: 0 B: 3 C: 4
Leave 1
A: 5 B: 0 C: 0
Leave 2
A: 0 B: 0 C: 4
Leave 3
A: 0 B: 0 C: 4
Leave 1
A: 0 B: 3 C: 0
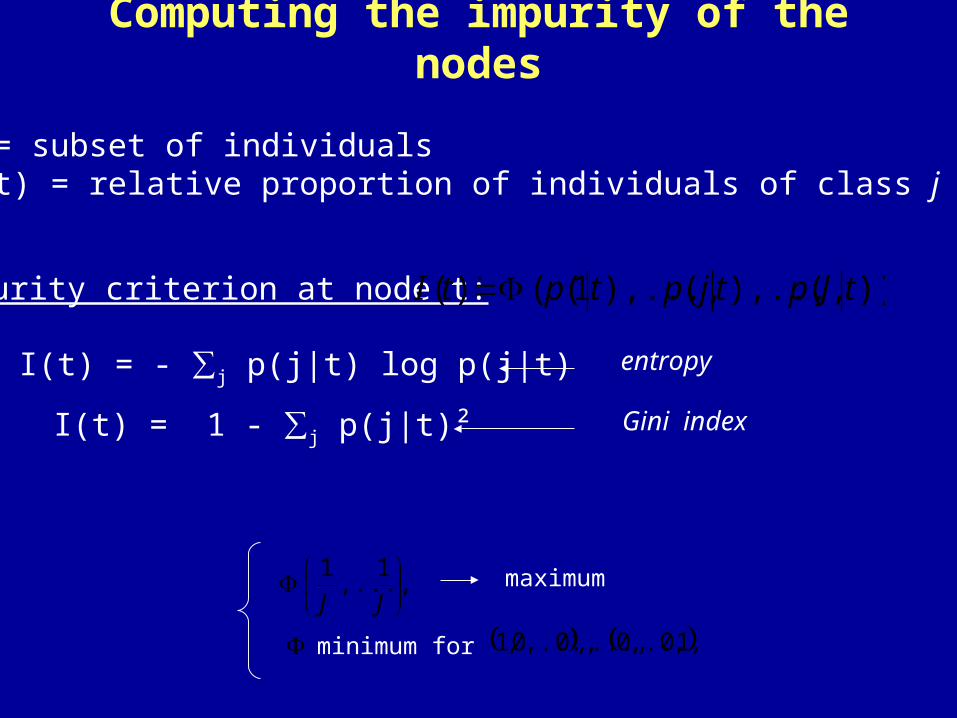
node t = subset of individualsp (j | t) = relative proportion of individuals of class j in node t
JJ
1,...,
1 maximum
1,0,...,0,...,0,...,0,1 minimum for
Computing the impurity of the nodes
Impurity criterion at node t:
I(t) = - ∑j p(j|t) log p(j|t) entropy
I(t) = 1 - ∑j p(j|t)² Gini index
))(),...,(),...,1(()( tJptjptptI

PL + PR = 1
t
tLtR
s PRPL s SS: set of variables
t: set of individuals
Finding the best split
ΔI(s*,t) = max { ΔI(s,t), s S }
ΔI(s,t) = I(t) - pL I(tL) - pR I(tR)
Rule to select a splitting candidate:
Decrease in impurity:
s* selected as
Stop splitting rule: e.g. threshold β
max { ΔI(s,t), s S } < β
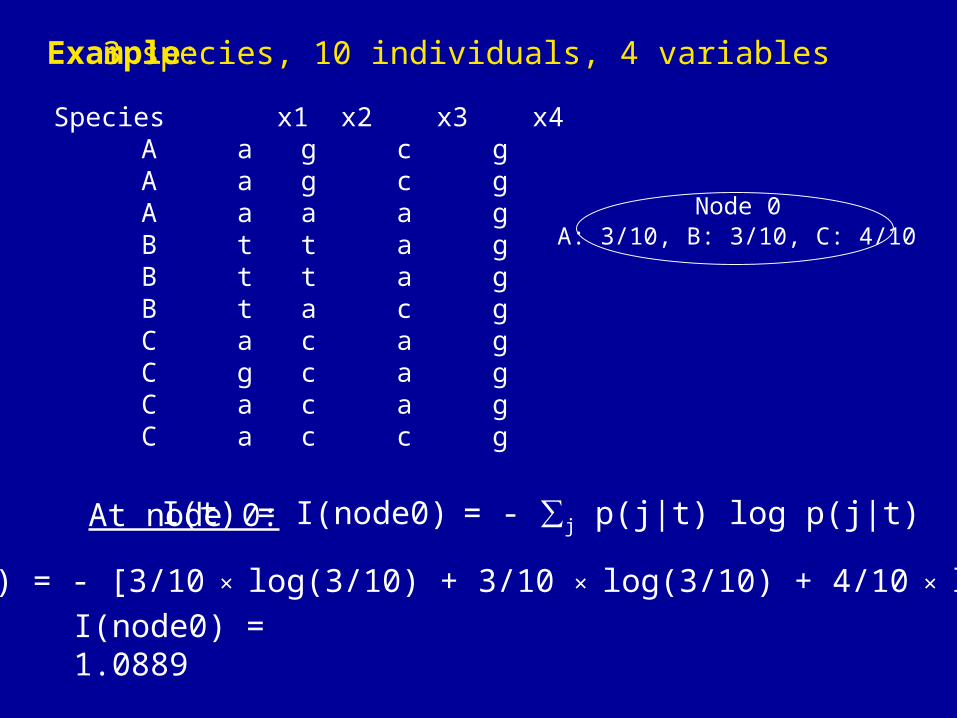
Example:
I(t) = I(node0) = - ∑j p(j|t) log p(j|t)At node 0:
I(node0) = - [3/10 × log(3/10) + 3/10 × log(3/10) + 4/10 × log(4/10)]
3 species, 10 individuals, 4 variables
I(node0) = 1.0889
Species x1 x2 x3 x4 A a g c g A a g c g A a a a g B t t a g B t t a g B t a c g C a c a g C g c a g C a c a g C a c c g
Node 0A: 3/10, B: 3/10, C: 4/10
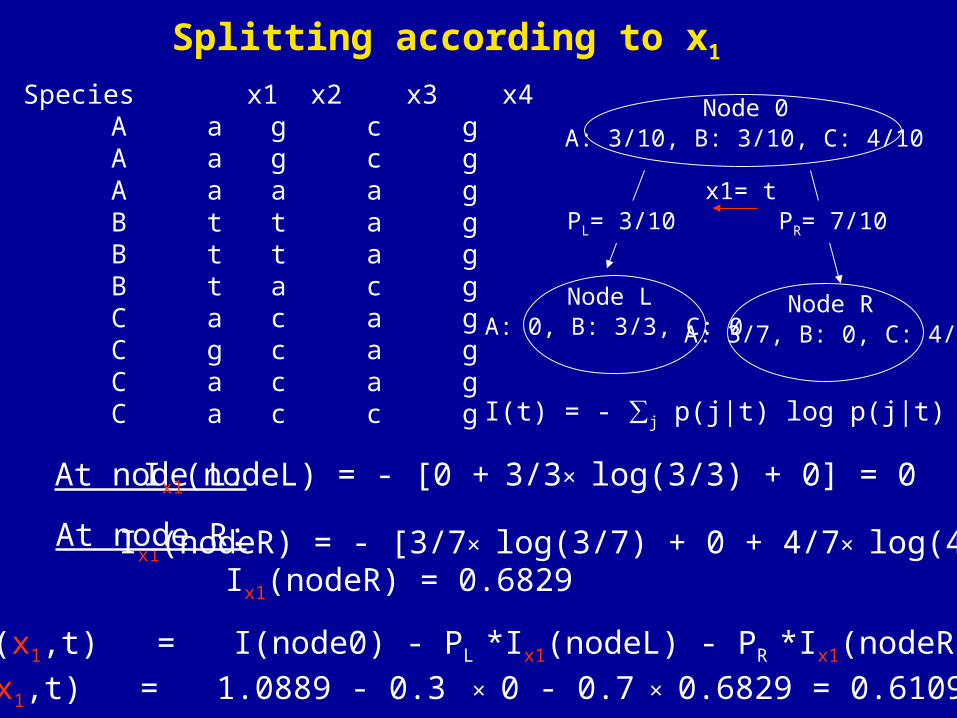
I(t) = - ∑j p(j|t) log p(j|t)
Splitting according to x1
Species x1 x2 x3 x4 A a g c g A a g c g A a a a g B t t a g B t t a g B t a c g C a c a g C g c a g C a c a g C a c c g
At node L: Ix1(nodeL) = - [0 + 3/3× log(3/3) + 0] = 0
Ix1(nodeR) = - [3/7× log(3/7) + 0 + 4/7× log(4/7)]At node R:
Ix1(nodeR) = 0.6829
A: 0, B: 3/3, C: 0 A: 3/7, B: 0, C: 4/7
Node 0A: 3/10, B: 3/10, C: 4/10
x1= t
Node L Node R
PR= 7/10PL= 3/10
ΔI(x1,t) = I(node0) - PL *Ix1(nodeL) - PR *Ix1(nodeR)
ΔI(x1,t) = 1.0889 - 0.3 × 0 - 0.7 × 0.6829 = 0.6109
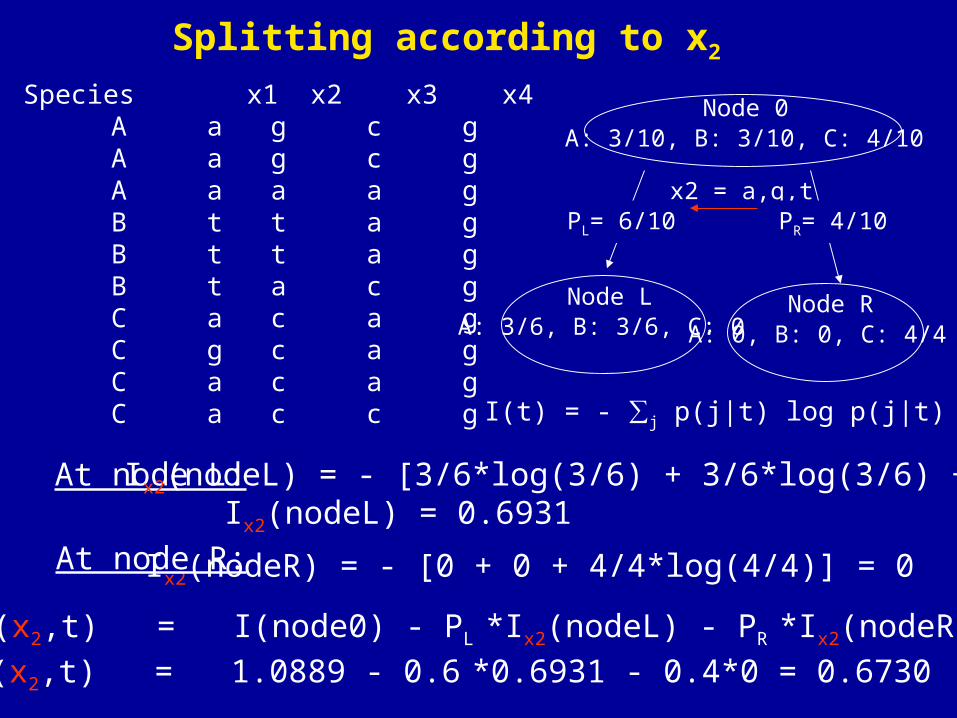
I(t) = - ∑j p(j|t) log p(j|t)
At node L:
Ix2(nodeR) = - [0 + 0 + 4/4*log(4/4)] = 0
Species x1 x2 x3 x4 A a g c g A a g c g A a a a g B t t a g B t t a g B t a c g C a c a g C g c a g C a c a g C a c c g
Ix2(nodeL) = - [3/6*log(3/6) + 3/6*log(3/6) +0]
At node R:
Node 0A: 3/10, B: 3/10, C: 4/10
x2 = a,g,t
Node L Node RA: 3/6, B: 3/6, C: 0 A: 0, B: 0, C: 4/4
PR= 4/10PL= 6/10
ΔI(x2,t) = I(node0) - PL *Ix2(nodeL) - PR *Ix2(nodeR)
ΔI(x2,t) = 1.0889 - 0.6 *0.6931 - 0.4*0 = 0.6730
Ix2(nodeL) = 0.6931
Splitting according to x2
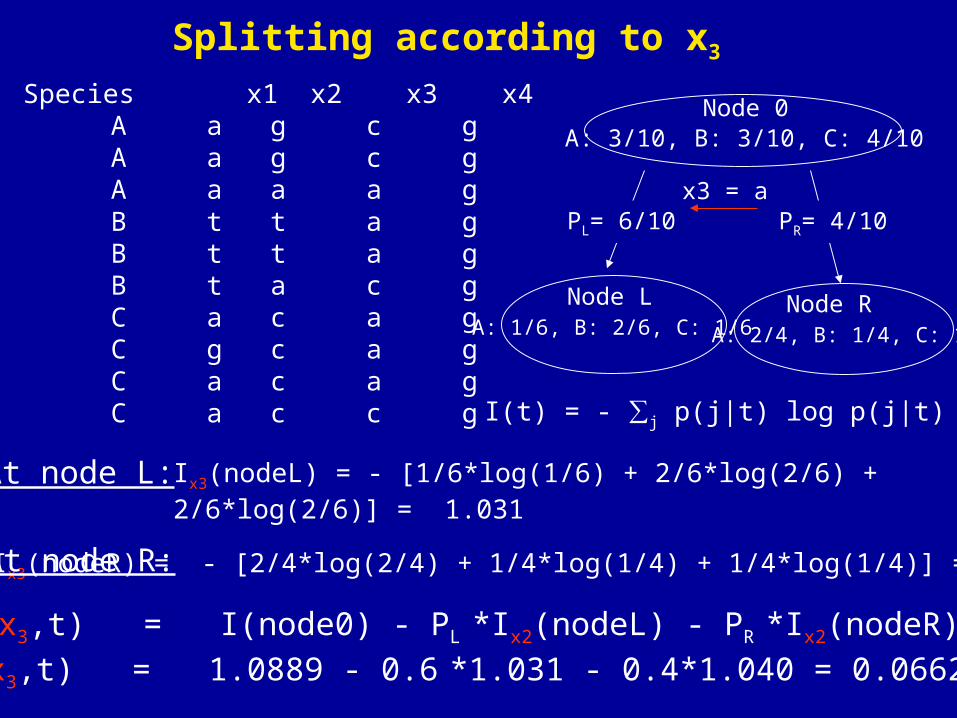
I(t) = - ∑j p(j|t) log p(j|t)
Ix3(nodeR) = - [2/4*log(2/4) + 1/4*log(1/4) + 1/4*log(1/4)] = 1.040
Species x1 x2 x3 x4 A a g c g A a g c g A a a a g B t t a g B t t a g B t a c g C a c a g C g c a g C a c a g C a c c g
At node L: Ix3(nodeL) = - [1/6*log(1/6) + 2/6*log(2/6) + 2/6*log(2/6)] = 1.031
At node R:
Node 0A: 3/10, B: 3/10, C: 4/10
x3 = a
Node L Node RA: 1/6, B: 2/6, C: 1/6 A: 2/4, B: 1/4, C: 1/4
PR= 4/10PL= 6/10
ΔI(x3,t) = I(node0) - PL *Ix2(nodeL) - PR *Ix2(nodeR)
ΔI(x3,t) = 1.0889 - 0.6 *1.031 - 0.4*1.040 = 0.0662
Splitting according to x3
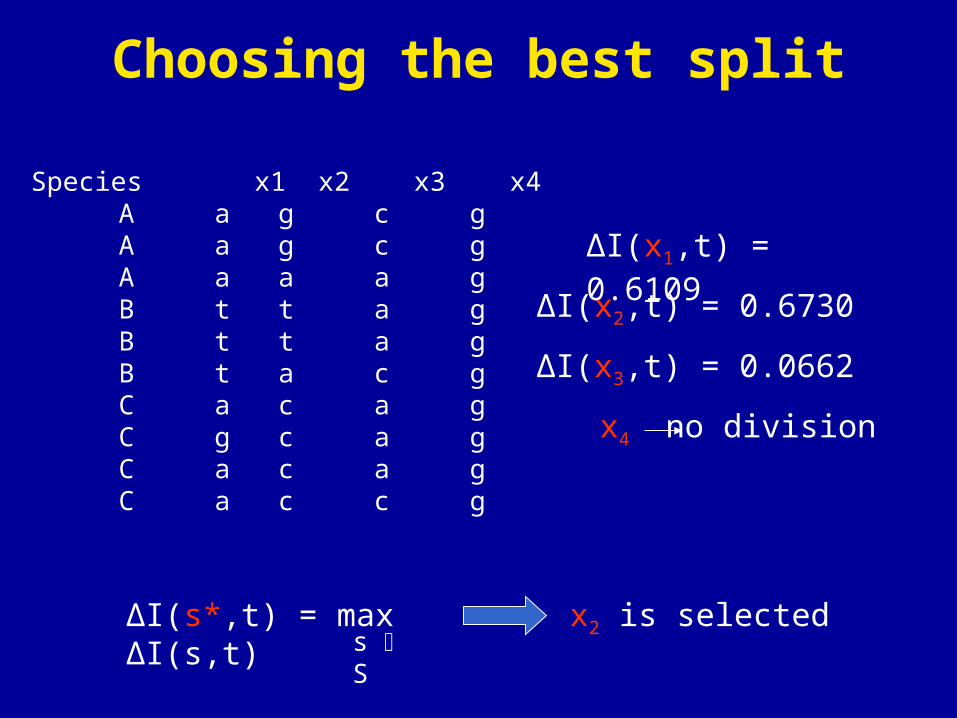
Species x1 x2 x3 x4 A a g c g A a g c g A a a a g B t t a g B t t a g B t a c g C a c a g C g c a g C a c a g C a c c g
ΔI(x2,t) = 0.6730
ΔI(s*,t) = max ΔI(s,t)s S
ΔI(x1,t) = 0.6109
ΔI(x3,t) = 0.0662
x4 no division
x2 is selected
Choosing the best split
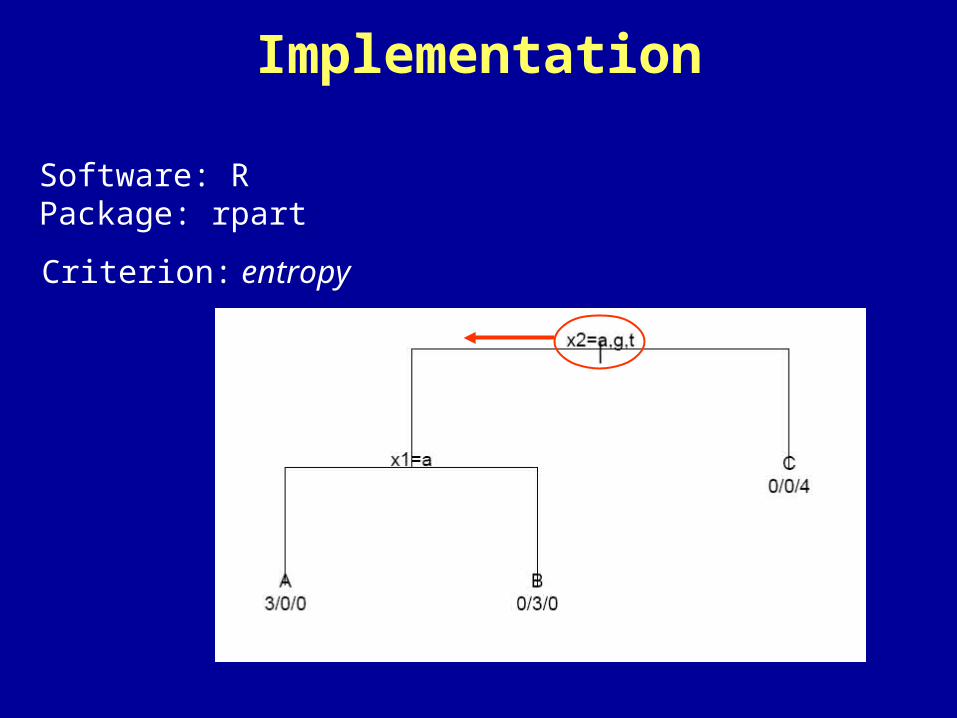
Criterion: entropy
Software: RPackage: rpart
Implementation

Bagging or bootstrap aggregation
N bootstrap samples from original data
N classification trees
N assignements for a new individual
Majority rule → class of the new individual

Simulation method
• one haploid population that splits into two (or more), T generations in the past.
• The ancestral population and the two new populations of constant size N.
• Sequences with mutation rate parameter of interest = 2N• Simulations performed with simcoal 2.1.2 (Excoffier et al)
T

Evaluation of the different classification methods
• We simulate n +1 individuals in each species.
• n individuals are considered as the reference samples, and the last one as the individual to test.
• Using repeated simulations, we compute the proportion of cases in which each test individual is correctly assigned to its species.
T

Parameters assumed for the simulation study
• = 3 (e.g. Litoria) or 30 (e.g. Astraptes)
• Reference sample size n = 3, 5, 10, 25
• Effective population size: N = 1000
• Separation time T = N/10, N/2, N, 5N or 10N
• Number of newly founded populations: from 2 to 5 In all cases, all populations assumed to be founded
simultaneously.
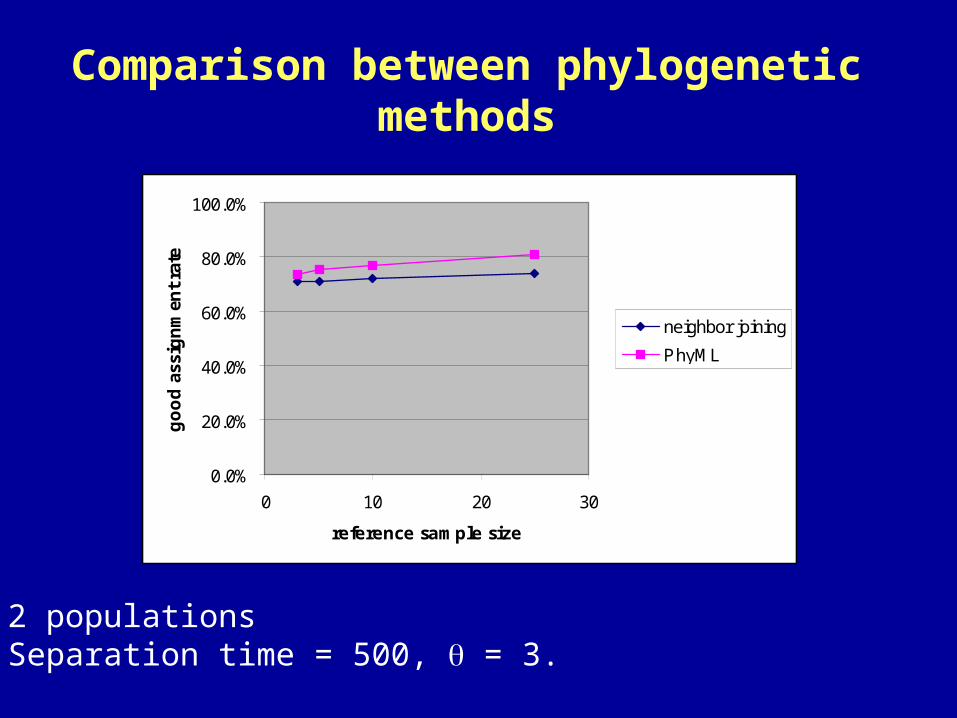
Comparison between phylogenetic methods
0.0%
20.0%
40.0%
60.0%
80.0%
100.0%
0 10 20 30
reference sample size
go
od
ass
ign
men
t ra
te
neighbor joining
PhyML
• 2 populations • Separation time = 500, = 3.
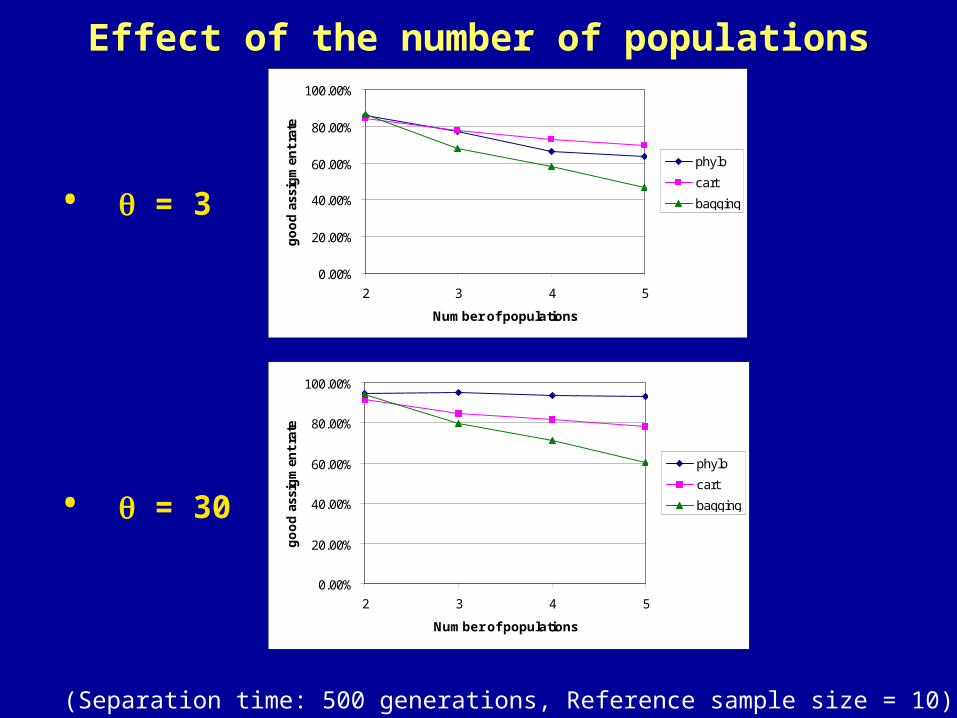
Effect of the number of populations
• = 3
0.00%
20.00%
40.00%
60.00%
80.00%
100.00%
2 3 4 5
Number of populations
go
od
ass
igm
ent
rate
phylo
cart
bagging
• = 30
0.00%
20.00%
40.00%
60.00%
80.00%
100.00%
2 3 4 5
Number of populations
go
od
ass
igm
ent
rate
phylo
cart
bagging
(Separation time: 500 generations, Reference sample size = 10)
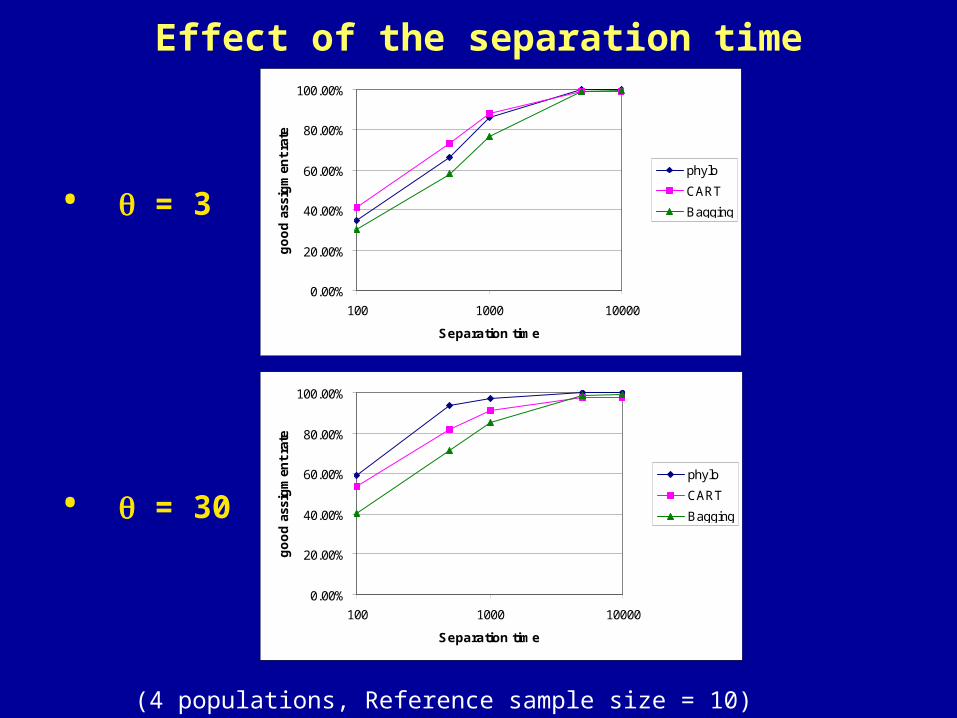
Effect of the separation time
• = 3
• = 30
(4 populations, Reference sample size = 10)
0.00%
20.00%
40.00%
60.00%
80.00%
100.00%
100 1000 10000
Separation time
go
od
ass
igm
ent
rate
phylo
CART
Bagging
0.00%
20.00%
40.00%
60.00%
80.00%
100.00%
100 1000 10000
Separation time
go
od
ass
igm
ent
rate
phylo
CART
Bagging
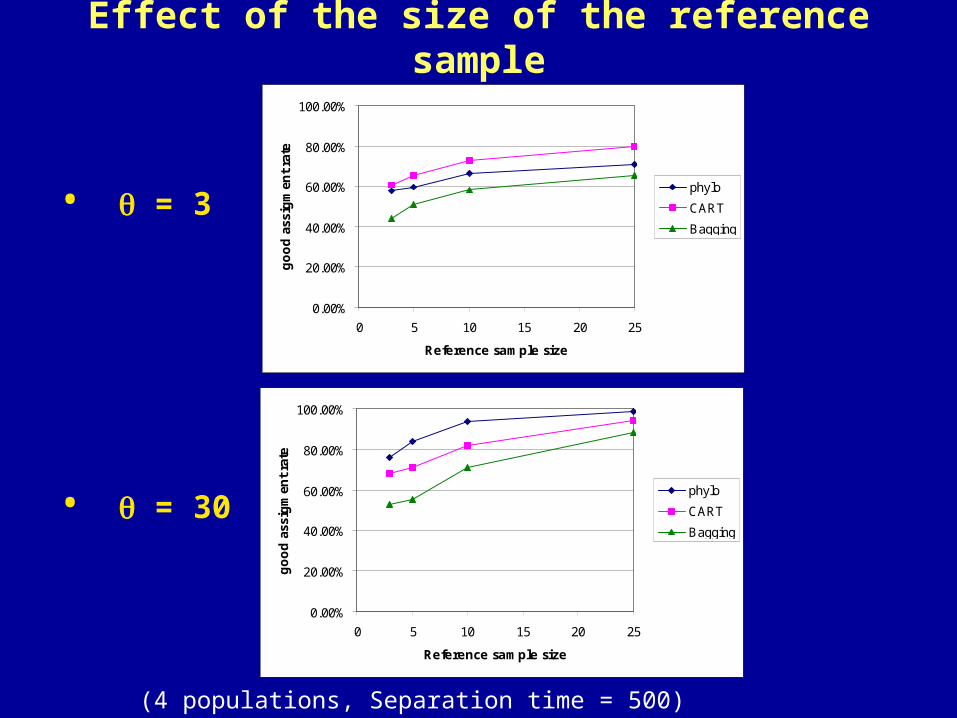
Effect of the size of the reference sample
• = 3
• = 30
(4 populations, Separation time = 500)
0.00%
20.00%
40.00%
60.00%
80.00%
100.00%
0 5 10 15 20 25
Reference sample size
go
od
ass
igm
ent
rate
phylo
CART
Bagging
0.00%
20.00%
40.00%
60.00%
80.00%
100.00%
0 5 10 15 20 25
Reference sample size
go
od
ass
igm
ent
rate
phylo
CART
Bagging

Adding nuclear genes
• We considered the case where polymorphism of nuclear genes are also available.
• We assumed that– these genes were independent.– they all have the same (= 4N) value, equal
to the value for the cytoplasmic genes.– They do not show intragenic recombination or
they show it at a rate equal to the mutation rate (c = , i.e. 4Nc = ).
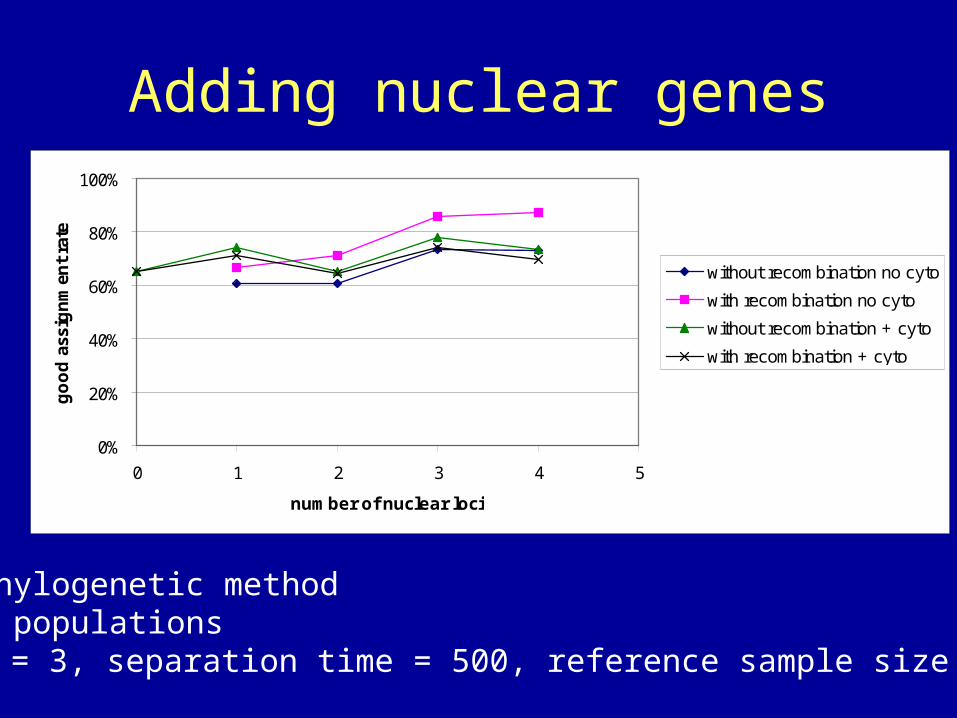
Adding nuclear genes
• Phylogenetic method• 2 populations• = 3, separation time = 500, reference sample size = 10
0%
20%
40%
60%
80%
100%
0 1 2 3 4 5
number of nuclear loci
go
od
ass
ign
men
t ra
te
without recombination no cyto
with recombination no cyto
without recombination + cyto
with recombination + cyto
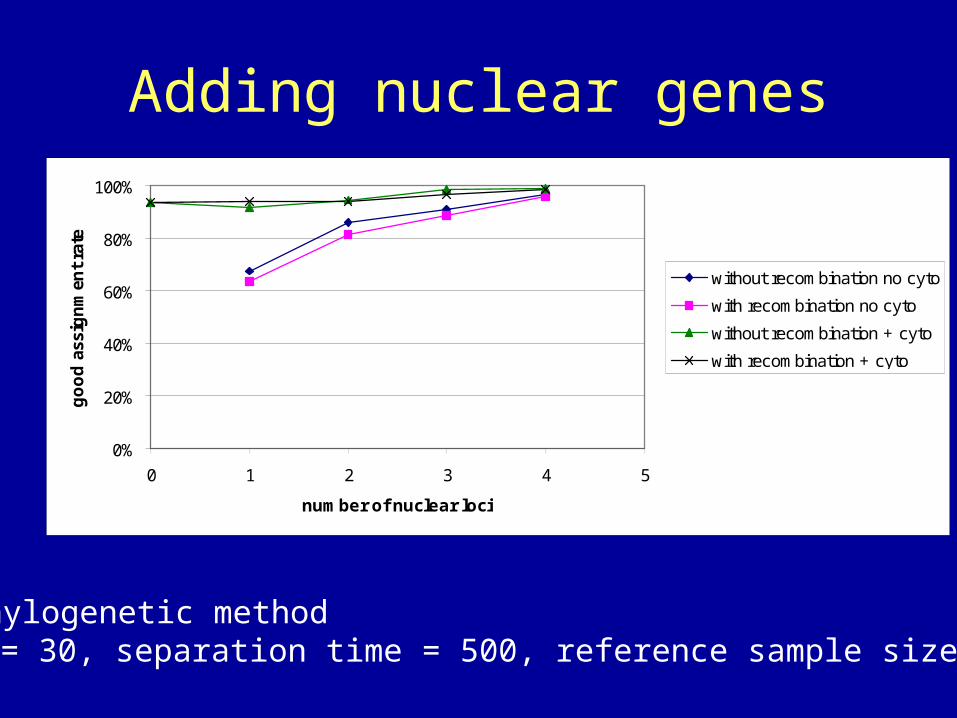
Adding nuclear genes
• Phylogenetic method• = 30, separation time = 500, reference sample size = 10
0%
20%
40%
60%
80%
100%
0 1 2 3 4 5
number of nuclear loci
go
od
ass
ign
men
t ra
te
without recombination no cyto
with recombination no cyto
without recombination + cyto
with recombination + cyto
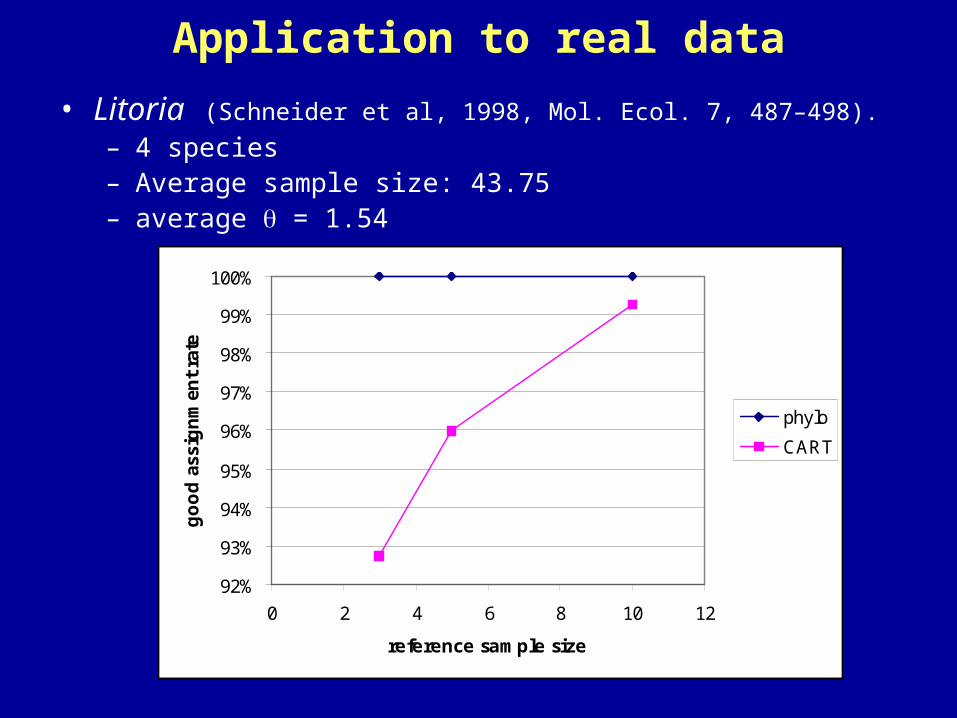
Application to real data
• Litoria (Schneider et al, 1998, Mol. Ecol. 7, 487–498).
– 4 species– Average sample size: 43.75– average = 1.54
92%
93%
94%
95%
96%
97%
98%
99%
100%
0 2 4 6 8 10 12
reference sample size
go
od
ass
ign
men
t ra
te
phylo
CART
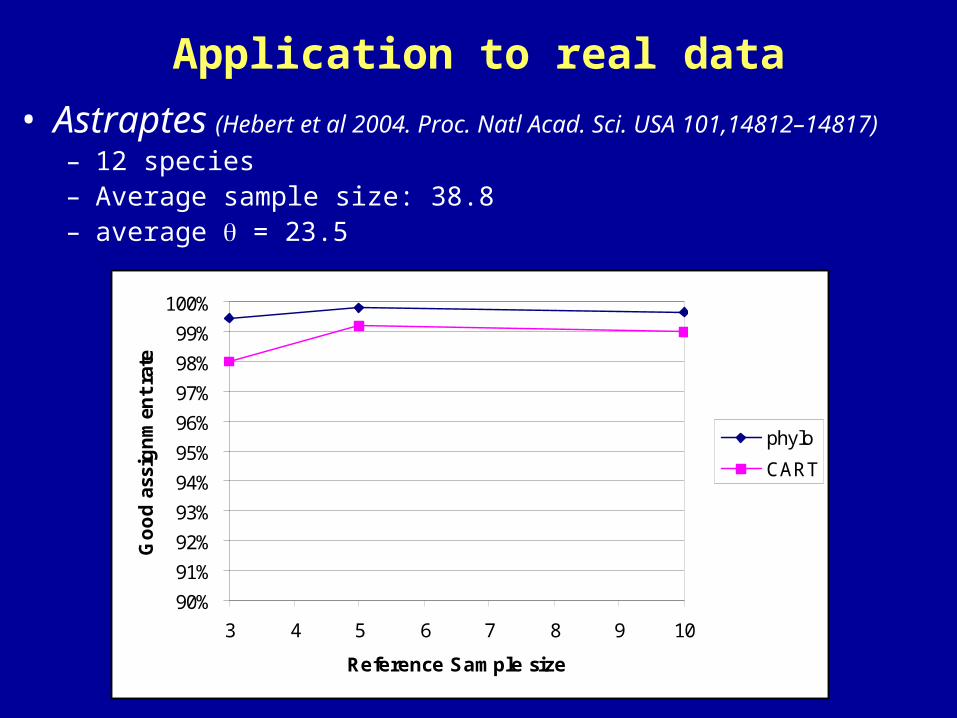
Application to real data
• Astraptes (Hebert et al 2004. Proc. Natl Acad. Sci. USA 101,14812–14817)
– 12 species– Average sample size: 38.8– average = 23.5
90%
91%
92%
93%
94%
95%
96%
97%
98%
99%
100%
3 4 5 6 7 8 9 10
Reference Sample size
Go
od
ass
ign
men
t ra
te
phylo
CART
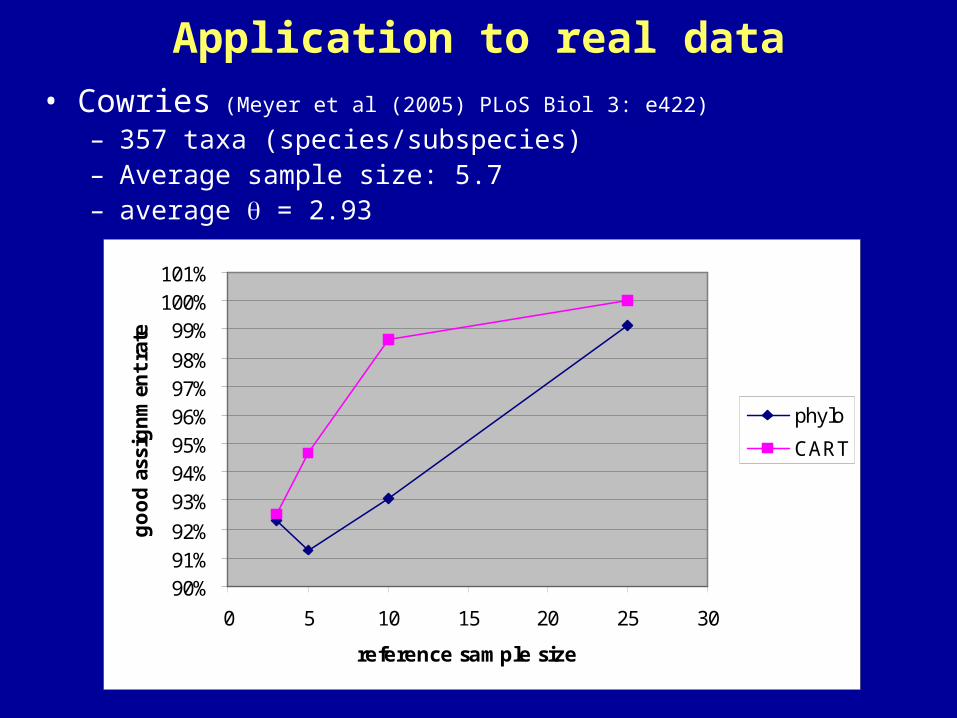
Application to real data• Cowries (Meyer et al (2005) PLoS Biol 3: e422)
– 357 taxa (species/subspecies)– Average sample size: 5.7– average = 2.93
90%91%92%
93%94%95%96%97%98%
99%100%101%
0 5 10 15 20 25 30
reference sample size
go
od
ass
ign
men
t ra
te
phylo
CART

Conclusions• Regarding phylogenetic methods, the maximum
likelihood method performs better than the neighbor joining.
• CART performs better than phylogenetic methods for poorly informative data (low value)
• but not for more polymorphic data (high value)
• Adding nuclear loci can help, but at a quite high cost.
• Recombination improves the phylogenetic method for low values (Ongoing work for CART).

Perspectives
• Developing a statistical method to put a confidence level on a given assignation.
• Evaluating other classification methods (learning methods)





























