Embed Size (px)
Citation preview

1
PowerPoint Slides English Text Spanish Translation
Colorectal Cancer Survivorship: Evaluation of Inherited
Colorectal Cancer Risk
VideoTranscript
Supervivencia al cáncer colorrectal: Evaluación del
riesgo de cáncer colorrectal heredado Transcripción del video
Professional Oncology Education
Colorectal Cancer Survivorship: Evaluation of Inherited
Colorectal Cancer Risk
Time: 39:50
Educación Oncológica Profesional Supervivencia al cáncer colorrectal: Evaluación del
riesgo de cáncer colorrectal heredado Duración: 39:50
Patrick Lynch, J.D., M.D. Professor Gastroenterology, Hepatology, & Nutrition The University of Texas, MD Anderson Cancer Center
Dr. Patrick Lynch, J.D. Profesor Gastroenterología, Hepatología y Nutrición MD Anderson Cancer Center de la Universidad de Texas
Patrick Lynch, J.D., M.D.
Professor
Gastroenterology, Hepatology, & Nutrition
Colorectal Cancer Survivorship:
Evaluation of Inherited Colorectal Cancer Risk
Good day. I’m Pat Lynch from the Department of Gastroenterology, Hepatology, and Nutrition here at the University of Texas MD Anderson Cancer Center. Today’s presentation on Colorectal Cancer Survivorship will focus on evaluation of inherited colorectal cancer risk.
Buenos días. Soy Pat Lynch del Departamento de Gastroenterología, Hepatología y Nutrición en el MD Anderson Cancer Center de la Universidad de Texas. Esta presentación se centrará en la supervivencia y la evaluación del riesgo de cáncer colorrectal heredado.

2
Objectives
At the conclusion of this lesson, the participant will be able to:
• Recognize clinical and family history distinctions between
familial adenomatous polyposis (FAP) and its variants,
hereditary nonpolyposis colorectal cancer (HNPCC), and
“familial colon cancer”
• Be able to understand and apply clinical practice guidelines
that have been developed for each of these conditions
• Be able to appropriately order and interpret genetic
testing results
• Understand the content of genetic counseling and be able
to effectively deliver related material
The objectives: At the end of this lesson, the viewer should be able to recognize clinical and family history distinctions that exist between familial adenomatous polyposis and its variance and hereditary nonpolyposis colorectal cancer or HNPCC to distinguish these from familial colon cancer. We should be able to understand and apply some of the pr --- clinical practice guidelines that have been developed for these conditions; be able to appropriately order and interpret genetic testing; and to understand the content of genetic counseling and be able to effectively deliver related material.
En esta presentación, el participante podrá reconocer las distinciones clínicas y de historial familiar que existen entre la poliposis adenomatosa familiar y sus variantes, y el cáncer colorrectal no polipósico hereditario o HNPCC, para diferenciarlas del cáncer de colon familiar. Debemos entender y aplicar ciertas pautas de práctica clínica que se han desarrollado para estas condiciones; solicitar pruebas genéticas e interpretar apropiadamente sus resultados; y comprender el contenido de la asesoría genética y entregar eficazmente el material relacionado.
Sporadic
(65–85%)
Familial (10–30%)
HNPCC
1-3%
FAP
(including MYH)(<1%)
Rare CRC syndromes:
P-J, JP,
Causes of Colorectal Cancer
The causes of colorectal cancer are many. There certainly are specific conditions such as inflammatory bowel disease that predispose to colorectal cancer. However, most colorectal cancer, well over half, probably at least three-fourths, are sporadic and really of uncertain etiology. A large category of colorectal cancers can be thought of as familial, perhaps 10 to 30 percent. There are likely underlying genetic factors that contribute to this. But, to date, single gene abnormalities have really only been identified in a small number of specific inherited cancer susceptibility conditions shown at the bottom, including some of the rare colorectal cancer syndromes, Peutz-Jeghers syndrome, juvenile polyposis, and a so-called hamartomatous polyposis syndromes. The one familiar probably to most clinicians, familial adenomatous polyposis, including its variant the so-called MYH polyposis that accounts for less than 1 percent of all colorectal cancers. The topic that we’ll be concentrating primarily on today though, hereditary nonpolyposis
Las causas de este cáncer son numerosas. Existen condiciones específicas, como la enfermedad inflamatoria intestinal, que predisponen a este cáncer; sin embargo, entre la mitad y las tres cuartas partes de los casos de cáncer colorrectal son esporádicos y de etiología incierta. Del 10% al 30% de estos cánceres puede considerarse familiares y es probable que contribuyan factores genéticos subyacentes. Hasta la fecha solo se han identificado anomalías genéticas individuales en un pequeño número de condiciones específicas susceptibles al cáncer hereditario, como vemos en la parte inferior de la diapositiva. Esto incluye algunos síndromes infrecuentes como Peutz-Jeghers, la poliposis juvenil y la poliposis hamartomatosa. El más conocido por los médicos clínicos es la poliposis adenomatosa familiar, incluida la variante denominada poliposis MYH, que representa menos del 1% de todos los cánceres colorrectales. Hoy nos concentraremos en el cáncer de colon no polipósico hereditario o HNPCC, que

3
colon cancer or HNPCC, certainly accounts for at least 1 to 3 percent of all colorectal cancers and at one time was thought to probably account for most of the familial category of colorectal cancer. However, investigations done at population levels have failed to show abnormalities in the genes known to be responsible for HNPCC have not identified these in more than about 1 to 3 percent of cases.
representa del 1% al 3% de todos los cánceres de colon. Antes se creía que representaba la mayor parte de los cánceres colorrectales familiares, pero las investigaciones a nivel de población no han demostrado anormalidades en los genes responsables del HNPCC y estas solo se identificaron en el 1% al 3% de los casos.
Hereditary Nonpolyposis Colorectal Cancer
CRC at 80
54
CRC at 22
Endometriumat 41
• 3 or more CRC (or HNPCC-
associated tumors)
• 2 or more generations
• 1 affected age by 50, 1 case
a 10 relative of the other two
• Attenuated FAP excluded
Amsterdam Criteria II
Now, “what are the criteria for HNPCC?” Well, the original Amsterdam Criteria adopted more than 20 years ago called for three or more cases of colorectal cancer or HNPCC associated tumors, we’ll come back to later, occurring over two or more generations in a family with one individual affected by age 50 and with one case being a first-degree relative of the other two and importantly attenuated FAP has to be excluded.
¿Cuáles son los criterios para el HNPCC? Los criterios de Ámsterdam aprobados hace más de 20 años establecían tres o más casos de tumores asociados con cáncer colorrectal ocurridos en dos o más generaciones familiares, con un individuo afectado hacia los 50 años y un caso en un familiar de primer grado de ambos. Es importante excluir la poliposis adenomatosa familiar atenuada, o FAP.
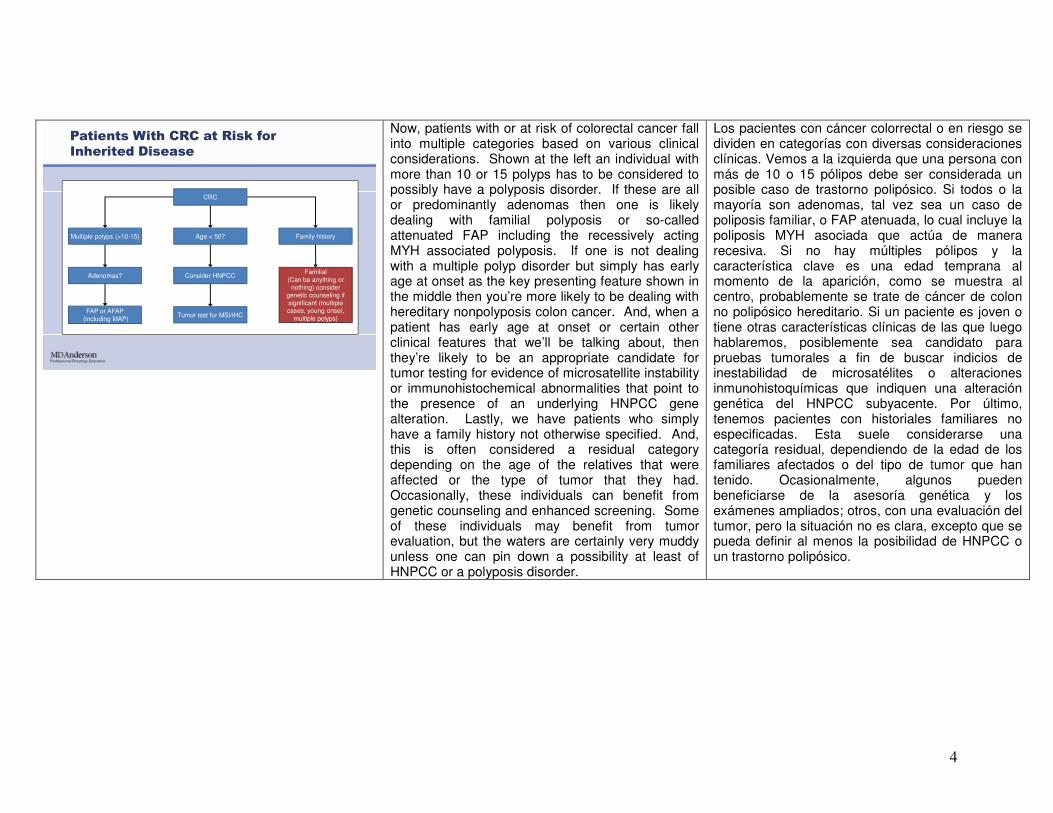
4
CRC
Multiple polyps (>10-15)
Adenomas?
FAP or AFAP(including MAP)
Age < 50? Family history
Consider HNPCC
Tumor test for MSI/IHC
Familial(Can be anything or
nothing) consider genetic counseling if significant (multiple cases, young onset,
multiple polyps)
Patients With CRC at Risk for
Inherited Disease
Now, patients with or at risk of colorectal cancer fall into multiple categories based on various clinical considerations. Shown at the left an individual with more than 10 or 15 polyps has to be considered to possibly have a polyposis disorder. If these are all or predominantly adenomas then one is likely dealing with familial polyposis or so-called attenuated FAP including the recessively acting MYH associated polyposis. If one is not dealing with a multiple polyp disorder but simply has early age at onset as the key presenting feature shown in the middle then you’re more likely to be dealing with hereditary nonpolyposis colon cancer. And, when a patient has early age at onset or certain other clinical features that we’ll be talking about, then they’re likely to be an appropriate candidate for tumor testing for evidence of microsatellite instability or immunohistochemical abnormalities that point to the presence of an underlying HNPCC gene alteration. Lastly, we have patients who simply have a family history not otherwise specified. And, this is often considered a residual category depending on the age of the relatives that were affected or the type of tumor that they had. Occasionally, these individuals can benefit from genetic counseling and enhanced screening. Some of these individuals may benefit from tumor evaluation, but the waters are certainly very muddy unless one can pin down a possibility at least of HNPCC or a polyposis disorder.
Los pacientes con cáncer colorrectal o en riesgo se dividen en categorías con diversas consideraciones clínicas. Vemos a la izquierda que una persona con más de 10 o 15 pólipos debe ser considerada un posible caso de trastorno polipósico. Si todos o la mayoría son adenomas, tal vez sea un caso de poliposis familiar, o FAP atenuada, lo cual incluye la poliposis MYH asociada que actúa de manera recesiva. Si no hay múltiples pólipos y la característica clave es una edad temprana al momento de la aparición, como se muestra al centro, probablemente se trate de cáncer de colon no polipósico hereditario. Si un paciente es joven o tiene otras características clínicas de las que luego hablaremos, posiblemente sea candidato para pruebas tumorales a fin de buscar indicios de inestabilidad de microsatélites o alteraciones inmunohistoquímicas que indiquen una alteración genética del HNPCC subyacente. Por último, tenemos pacientes con historiales familiares no especificadas. Esta suele considerarse una categoría residual, dependiendo de la edad de los familiares afectados o del tipo de tumor que han tenido. Ocasionalmente, algunos pueden beneficiarse de la asesoría genética y los exámenes ampliados; otros, con una evaluación del tumor, pero la situación no es clara, excepto que se pueda definir al menos la posibilidad de HNPCC o un trastorno polipósico.

5
Polyposis
Attenuated Familial Adenomatous Polyposis
(AFAP)
Others (Peutz-Jeghers, Juvenile and
Hyperplastic Polyposis)
Classic Familial Adenomatous Polyposis
(FAP)
Now, polyposis used to be thought of as a very simple straightforward condition. Shown in the far left is a retroflex view in the rectum of a patient with very diffuse involvement with adenomas. This patient clearly has FAP whether they have a family history of the condition or not. And, this patient, of course, went on to have a proctocolectomy because of the extensive polyp burden in the rectum. In the middle panel, we see a patient with a so-called attenuated form of FAP in which indigo carmine dye spray shows many, many very small microadenomas that would not have been evident to the naked eye. It’s important for the traditional clinician who thinks mainly about the category on the left to recognize that patients with attenuated FAP may not present with adenomas until their 50s or 60s. These adenomas may be very few in number and not immediately come to mind as a possible variant of FAP. Nevertheless, many of these patients are found to have mutations in the APC gene and, therefore, clearly are examples of FAP despite the later age and fewer polyps. But it is important always to take a look at the histology of polyps. Because as we see on the right, many patients with multiple polyp disorders have conditions, such as Peutz-Jeghers, juvenile and hyperplastic polyposis, that can look somewhat like familial polyposis except for the fact that the histology of the polyps is altogether different showing hamartoma or perhaps hyperplastic features only.
La poliposis solía considerarse una condición sencilla y muy simple. En el extremo izquierdo tenemos una vista rectal invertida con una afectación muy difusa de adenomas. Claramente existe una FAP, independientemente del historial familiar. Por supuesto, se realizó una proctocolectomía debido a la extensa carga de pólipos en el recto. En el panel central, vemos una forma de FAP denominada “atenuada”, en la cual la tinción con índigo carmín muestra numerosos microadenomas muy pequeños que no serían evidentes a simple vista. Si un médico considera principalmente la categoría de la izquierda, es importante reconocer que los pacientes con FAP atenuada pueden presentar adenomas a partir de los 50 o 60 años. Pueden ser escasos en número y tal vez no se los considere inmediatamente una posible variante de FAP; sin embargo, en muchos pacientes se descubren mutaciones en el gen APC y son claros ejemplos de FAP pese a la edad más avanzada y la menor cantidad de pólipos. Siempre es importante observar la histología de los pólipos. Como vemos a la derecha, muchos pacientes con trastornos de pólipos múltiples tienen condiciones como el síndrome de Peutz-Jeghers, poliposis juvenil e hiperplásica, semejantes a la poliposis familiar, pero la histología de los pólipos es totalmente diferente, ya que muestran hamartoma o solo características hiperplásicas.

6
FAP
This again is a typical case of severe classic FAP. Este es un caso típico de FAP clásica grave.
Attenuated FAP
• Later onset (CRC ~age 50)
• Few colonic adenomas
with CHRPE
• UGI lesions
• Associated with mutations at
5' and 3' ends of APC gene
Attenuated FAP, as I’ve mentioned, shows later age of onset both for polyps and cancer, fairly few adenomas of the colon. Relatively speaking, this can be highly variable. These patients commonly will not have the so-called congenital hypertrophy of the retinal pigment epithelium or otherwise known as pigmented ocular fundic lesions that occur in classic FAP. However, many of the patients with even the attenuated form of FAP will have the characteristic upper GI lesions including fundic gland polyps of the stomach commonly with at least low grade dysplasia but minimal cancer risk and they likely will also have duodenal adenomas, typical plaque-like lesions that may or may not involve the ampulla but certainly require close monitoring. It’s interesting to note that patients with the attenuated form of FAP commonly are found to have mutations at the relatively extreme five prime and three prime ends of the APC gene.
En la FAP atenuada, los pólipos y el cáncer aparecen a una edad más avanzada, con pocos adenomas de colon. En términos relativos, esto puede ser muy variable. Estos pacientes generalmente no tendrán hipertrofia congénita del epitelio pigmentario retinal, las lesiones fúndicas oculares pigmentadas que ocurren en la FAP clásica. Aun con la FAP atenuada, muchos tendrán las características lesiones del tracto gastrointestinal superior, incluidos los pólipos glandulares fúndicos del estómago, usualmente con displasia de bajo grado. El riesgo de cáncer es mínimo y probablemente existan adenomas duodenales, las típicas lesiones similares a placas que pueden o no afectar la ampolla, pero que, por cierto, requieren monitoreo. Es interesante observar que en los pacientes con FAP atenuada comúnmente se detectan mutaciones en los extremos relativos 5 prima y 3 prima del gen APC.

7
• Autosomal dominant (rare)
• Linked to PTEN, SMAD4,
BMPR1A (10q-22-23)
• Juvenile polyps with mixed
adenomatous histology
Juvenile Polyposis
Juvenile polyposis on the other hand presents with technically speaking non-dysplastic, non-adenomatous polyps. These polyps microscopically show cystic dilation of the glands as shown in the photomicrograph at the lower right. This is autosomal dominant but quite a bit more rare than familial polyposis. This has been linked to several different genes; the so-called P10 gene, the SMAD4 gene, and the BMPR1A gene, but all of these are relatively uncommon. One can sometimes have juvenile polyps that are admixed with adenomatous histology and cases of cancer have been described in juvenile polyposis.
Por el contrario, la poliposis juvenil se presenta con pólipos técnicamente no displásicos y no adenomatosos, que microscópicamente muestran una dilatación quística de las glándulas, según la fotomicrografía de la parte inferior derecha. Esta condición autosómica dominante es menos frecuente que la poliposis familiar y se ha vinculado a varios genes diferentes, como el P10, el SMAD4 y el BMPR1A, pero es relativamente infrecuente. Se pueden encontrar pólipos juveniles combinados con histología adenomatosa, y en la poliposis juvenil también se han descrito casos de cáncer.
Peutz-Jegher Syndrome
• Characteristic pigmentation
mutations in STK11GI hamartomas,
not “neoplastic” but some cancer risk
Peutz-Jeghers syndrome is characterized by a very characteristic pigmentation around the lips and buccal mucosa and occasionally fingertips. This shows mutations commonly in the STK11 gene. The mutation detection rate in Peutz-Jeghers syndrome tends to be fairly high. But, genetic testing is not quite as critical because we do have the pigmentation that is a characteristic sign of this condition. As with juvenile polyposis, these polyps are hamartoma and not technically neoplastic, but there is some cancer risk associated with these. And a more common problem, especially in teenagers with Peutz-Jeghers syndrome, is interception or telescoping of the small bowel that can be associated with bowel infarct and a requirement for surgery and occasionally bleeding.
El síndrome de Peutz-Jeghers se caracteriza por una pigmentación típica alrededor de los labios y la mucosa bucal, y a veces en las yemas. Esta condición suele tener mutaciones en el gen STK11. El índice de detección de mutaciones en este síndrome tiende a ser bastante alto, pero las pruebas genéticas no son tan esenciales, ya que la pigmentación es característica de esta condición. Tal como en la poliposis juvenil, estos pólipos son hamartomatosos y no técnicamente neoplásicos, pero se los asocia con ciertos riesgos. Un problema más común, especialmente en los adolescentes con Peutz-Jeghers, es la intususcepción o prolapso del intestino delgado, a veces asociada con infarto intestinal y necesidad de cirugía, con sangrado ocasional.

8
Hyperplastic Polyposis
• More modern term: serrated
polyposis
• Very heterogeneous in number, size
of polyps
• Genetic basis, if any, controversial
• Sessile serrated polyps/adenomas
may be associated with MSI high
CRC
Hyperplastic polyposis is very poorly understurd ---- very poorly understood. The more modern term that’s used for this condition is serrated polyposis and the reason for this is that serrated polyps can have a variety of histologies and most of these are very similar to hyperplastic polyps but with some fairly key differences that are beyond the scope of this discussion. These serrated polyps can be very heterogeneous in number, size, and location. It’s important to emphasize that the genetic basis for hyperplastic polyposis is very controversial and no gene has to date been linked with the development of hyperplastic polyposis in any particular patient or family. It’s also important to note that when sessile serrated polyps or adenomas occur in a particular individual they can be associated with tumors that show microsatellite instability. And, it’s important in this regard that these not be confused with cases of hereditary nonpolyposis colon cancer which also is characterized by microsatellite instability in the tumors.
Hay poca información sobre la poliposis hiperplásica. El término más moderno es “poliposis serrada”, porque los pólipos serrados pueden tener una variedad de histologías y son muy similares a los hiperplásicos, aunque con diferencias notables que están fuera del alcance de esta presentación. Estos pólipos pueden ser muy heterogéneos en número, tamaño y ubicación. Es importante destacar que la base genética de la poliposis hiperplásica es objeto de controversia, y que hasta ahora ningún gen se ha vinculado a su desarrollo en un paciente o una familia. También es importante señalar que los adenomas o pólipos serrados sésiles pueden asociarse a tumores que muestren inestabilidad de microsatélites. En este sentido, estos casos no deben confundirse con el cáncer de colon no polipósico hereditario, que también se caracteriza por esta inestabilidad tumoral.
Clinical Features of HNPCC
• Early but variable age at
CRC diagnosis (~45 years)
• Tumor site in proximal
colon predominates
• Extracolonic cancers:
endometrium, ovary, stomach,
urinary tract, small bowel, bile
ducts, sebaceous skin tumors
• Characteristic pathology: poorly
differentiated, mucinous, tumor-
infiltrating lymphocytes, MSI
Now, the clinical features of HNPCC are very important: Early but variable onset of colorectal cancer with an average of about age 45, a predilection to involving the proximal colon generally to the right of the splenic flexure. Extra colonic tumors are a characteristic of this condition involving first and foremost the colon and rectum as noted but also the endometrium, which can actually be more common in a particular family even than colon cancer. Somewhat less common, but perhaps important because of their more aggressive nature, are tumors involving the ovaries, stomach, bile duct, and pancreas as well as urinary tract, small bowel, and sebaceous skin tumors. These sebaceous skin tumors when they occur characterizing a so-called Muir-Torre variant of HNPCC. These tumors can
Las características clínicas del HNPCC son muy importantes: Aparición de cáncer colorrectal temprana, pero variable, a una edad media de 45 años, así como una tendencia a afectar el colon proximal, generalmente a la derecha de la flexura esplénica. Los tumores extracolónicos caracterizan a esta condición, que ante todo compromete el colon y el recto, pero también el endometrio, lo cual en una familia particular puede ser más común que el cáncer de colon. Menos comunes, pero tal vez más importantes por ser más agresivos, son los tumores de ovarios, estómago, conducto biliar y páncreas, así como de vías urinarias, intestino delgado y los tumores sebáceos. Los tumores sebáceos de piel caracterizan una variante del HNPCC llamada síndrome de Muir-Torre. Pueden
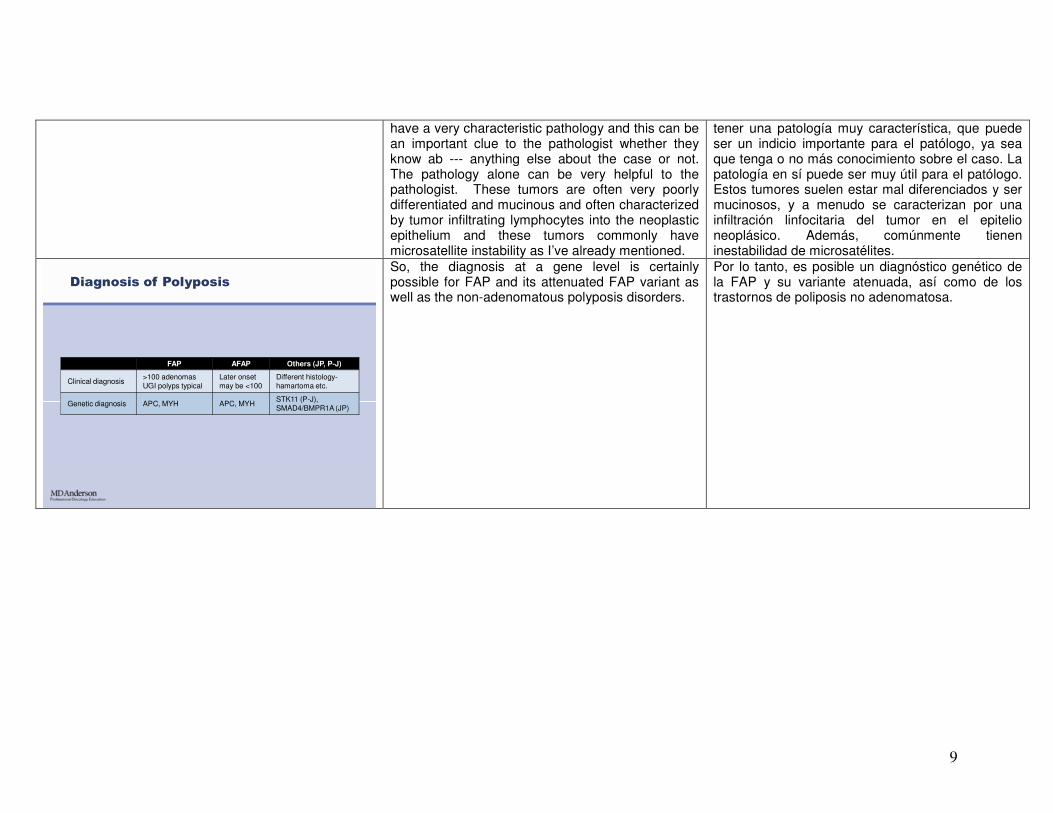
9
have a very characteristic pathology and this can be an important clue to the pathologist whether they know ab --- anything else about the case or not. The pathology alone can be very helpful to the pathologist. These tumors are often very poorly differentiated and mucinous and often characterized by tumor infiltrating lymphocytes into the neoplastic epithelium and these tumors commonly have microsatellite instability as I’ve already mentioned.
tener una patología muy característica, que puede ser un indicio importante para el patólogo, ya sea que tenga o no más conocimiento sobre el caso. La patología en sí puede ser muy útil para el patólogo. Estos tumores suelen estar mal diferenciados y ser mucinosos, y a menudo se caracterizan por una infiltración linfocitaria del tumor en el epitelio neoplásico. Además, comúnmente tienen inestabilidad de microsatélites.
FAP AFAP Others (JP, P-J)
Clinical diagnosis>100 adenomasUGI polyps typical
Later onsetmay be <100
Different histology-hamartoma etc.
Genetic diagnosis APC, MYH APC, MYHSTK11 (P-J), SMAD4/BMPR1A (JP)
Diagnosis of Polyposis
So, the diagnosis at a gene level is certainly possible for FAP and its attenuated FAP variant as well as the non-adenomatous polyposis disorders.
Por lo tanto, es posible un diagnóstico genético de la FAP y su variante atenuada, así como de los trastornos de poliposis no adenomatosa.
10
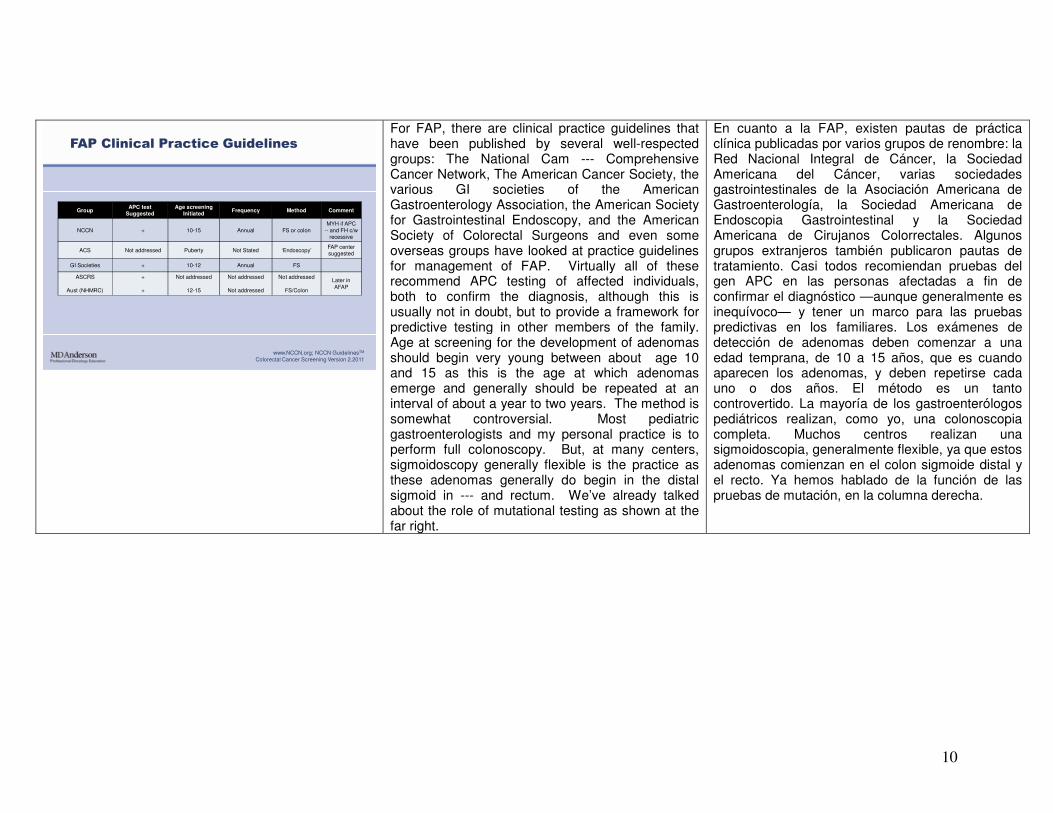
FAP Clinical Practice Guidelines
GroupAPC test
SuggestedAge screening
InitiatedFrequency Method Comment
NCCN + 10-15 Annual FS or colonMYH if APC
-- and FH c/w recessive
ACS Not addressed Puberty Not Stated ‘Endoscopy’FAP center suggested
GI Societies + 10-12 Annual FS
ASCRS
Aust (NHMRC)
+
+
Not addressed
12-15
Not addressed
Not addressed
Not addressed
FS/Colon
Later in AFAP
www.NCCN.org; NCCN GuidelinesTM
Colorectal Cancer Screening Version 2.2011
For FAP, there are clinical practice guidelines that have been published by several well-respected groups: The National Cam --- Comprehensive Cancer Network, The American Cancer Society, the various GI societies of the American Gastroenterology Association, the American Society for Gastrointestinal Endoscopy, and the American Society of Colorectal Surgeons and even some overseas groups have looked at practice guidelines for management of FAP. Virtually all of these recommend APC testing of affected individuals, both to confirm the diagnosis, although this is usually not in doubt, but to provide a framework for predictive testing in other members of the family. Age at screening for the development of adenomas should begin very young between about age 10 and 15 as this is the age at which adenomas emerge and generally should be repeated at an interval of about a year to two years. The method is somewhat controversial. Most pediatric gastroenterologists and my personal practice is to perform full colonoscopy. But, at many centers, sigmoidoscopy generally flexible is the practice as these adenomas generally do begin in the distal sigmoid in --- and rectum. We’ve already talked about the role of mutational testing as shown at the far right.
En cuanto a la FAP, existen pautas de práctica clínica publicadas por varios grupos de renombre: la Red Nacional Integral de Cáncer, la Sociedad Americana del Cáncer, varias sociedades gastrointestinales de la Asociación Americana de Gastroenterología, la Sociedad Americana de Endoscopia Gastrointestinal y la Sociedad Americana de Cirujanos Colorrectales. Algunos grupos extranjeros también publicaron pautas de tratamiento. Casi todos recomiendan pruebas del gen APC en las personas afectadas a fin de confirmar el diagnóstico —aunque generalmente es inequívoco— y tener un marco para las pruebas predictivas en los familiares. Los exámenes de detección de adenomas deben comenzar a una edad temprana, de 10 a 15 años, que es cuando aparecen los adenomas, y deben repetirse cada uno o dos años. El método es un tanto controvertido. La mayoría de los gastroenterólogos pediátricos realizan, como yo, una colonoscopia completa. Muchos centros realizan una sigmoidoscopia, generalmente flexible, ya que estos adenomas comienzan en el colon sigmoide distal y el recto. Ya hemos hablado de la función de las pruebas de mutación, en la columna derecha.

11
Mutationknown
Not tested
APC test those at-risk
APC +
APC --
FS q 12 mostart at 10-15
Average riskscreening
FS start 10-15q y to 24q 2y to 34
q 3y to 44, then q 3-5
FAP Predictive Testing and Screening
www.NCCN.org; NCCN GuidelinesTM
Colorectal Cancer Screening Version 2.2011
So, the --- the approach to combining predictive testing and screening: If we have a patient that has a known mutation in the APC gene and an APC gene mutation will be detected in about 85 to 90 percent of patients with familial polyposis that have had either cancer or multiple polyps that characterize th --- this condition. When such a mutation is identified, we can then test those individuals at risk: sons, daughters, sisters, and brothers, and often more remote family members as well. If these individuals test positive for the same mutation then it’s appropriate to begin sigmoidoscopy or colonoscopy as noted before beginning at about age 10 to 15 and repeated annually. It’s very critical to emphasize, and this is probably the most important reason for even doing mutational testing, is that once we’ve identified a mutation in the index patient and then test an otherwise at risk son or daughter, if that son or daughter is found not to have the mutation that the affected mom or dad has had, then they are not carriers of this condition, they cannot pass on risk to their children, and they do not warrant any enhanced screening. They can be viewed as being at average risk and can be screened just like any other individual in the population. Certainly, if an individual is not tested for some reason, they need to be managed as if they were carriers. So, certainly this is a strong argument in favor of doing genetic testing rather than empiric screening.
Veamos, entonces, el enfoque de combinar pruebas predictivas y exámenes de detección. Consideremos un paciente con una mutación conocida en el gen APC, que se detecta en el 85% al 90% de los pacientes con poliposis familiar que han tenido cáncer, o bien los múltiples pólipos que caracterizan esta condición. Al identificar la mutación, podemos realizar pruebas en las personas en riesgo: hijos, hijas, hermanas y hermanos, y a menudo otros familiares más lejanos. Si estas personas obtienen un resultado positivo para la misma mutación, es conveniente comenzar con la sigmoidoscopia o la colonoscopia a partir de los 10 a 15 años, y repetirla anualmente. Es fundamental destacar —y esta es, probablemente, la razón más importante, aun para las pruebas de mutación— que, una vez que se identifica una mutación en el paciente índice y hacemos pruebas en un hijo o hija en riesgo, si no se le detecta la mutación de la madre o el padre afectados, no es portador de esa condición, no puede transmitir el riesgo a sus hijos y no se justifican los exámenes ampliados. Puede considerarse con riesgo promedio y hacerse exámenes de detección exactamente como cualquier otra persona. Por cierto, si no se le hacen las pruebas por alguna razón, debe ser tratada como si fuera portadora. Este sólido argumento indica pruebas genéticas en lugar de exámenes de detección empíricos.

12
FAP Screening: APC Test Uninformative
MutationUnknown
APC test affected relative
(If APC +follow mutation
known algorithm)
No mutationfound
No affectedavailable to test
FS start 10-15q y to 24, q2 y to 34
q 3y to 44, then q 3-5
Consider testingat-risk
member
If APC +follow mutation
known algorithm
www.NCCN.org; NCCN GuidelinesTM
Colorectal Cancer Screening Version 2.2011
So, once this testing is done if we start with a --- with a individual of uncertain status initially, again we can do APC testing on the affected individual. And most of the time as indicated, it will be informative. But there are occasions even in classic FAP where no mutation is found and in this situation empiric screening of the at-risk sons, daughters, sisters, and brothers has to be considered because we don’t have mutational information that would otherwise more selectively guide the approach to screening.
Así, una vez que se realiza esta prueba en una persona inicialmente de situación incierta, podemos hacer la prueba de APC en el individuo afectado. La mayoría de las veces el resultado será informativo. En ocasiones —incluso con la FAP clásica— no se detecta ninguna mutación. En estos casos, es preciso considerar exámenes de detección empíricos en hijos, hijas, hermanas y hermanos en riesgo, ya que no contamos con información mutacional para orientar más selectivamente el enfoque examinador.
• Key clinical features
• Step-wise approach to genetic diagnosis
• Pitfalls in evaluation
Hereditary Nonpolyposis Colorectal
Cancer (HNPCC)
So, the key features now in --- in thinking about HNPCC are actually very similar to the approach to the polyposis disorders with the key difference that we’re often not quite as sure about the clinical diagnosis of HNPCC in a given case, again, at a clinical level, as we are in FAP. So, we still want to focus on the clinical features. But, generally we adopt a step wise approach to the genetic diagnosis. And I want to emphasize some of the pitfalls in the evaluation of this.
Las claves a tener en cuenta en el HNPCC son muy similares al enfoque de los trastornos polipósicos, con la diferencia fundamental de que a menudo no tenemos la misma seguridad sobre el diagnóstico clínico del HNPCC en un caso determinado, como sí la tenemos con la FAP. Por eso, nos concentramos en las características clínicas, aunque adoptando un enfoque progresivo del diagnóstico genético. Con respecto a esta evaluación, deseo destacar algunos inconvenientes.

13
CRC at 80
54
CRC at 22
Endometrium
at 41
• 3 or more CRC (or HNPCC-
associated tumors)
• 2 or more generations
• 1 affected age by 50, 1 case
a 10 relative of the other two
• FAP excluded
Amsterdam Criteria II
So, these are the Amsterdam Criteria that we’ve already talked about, three or more cases over two or more generations with early onset and exclusion of FAP.
Estos son los criterios de Ámsterdam: tres o más casos en dos o más generaciones, con aparición temprana y exclusión de FAP.
Why Might AC be Obsolete?
• Population studies, with step-wise MSI testing, indicate
that many or most cases of HNPCC do not meet AC
• Yet, many continue to assume, wrongly, that HNPCC is
very rare and that referral of patients for genetic counseling
and testing needs to meet AC
• In many cases, AC can be present but tumors fail to
show MSI: familial colorectal cancer syndrome “X”,
distinct from HNPCC
“Why might these Amsterdam Criteria not be helpful in a particular case?” Well, population studies involving stepwise microsatellite testing of all colorectal cancers regardless of family history have indicated that many or most cases of HNPCC actually do not meet the Amsterdam Criteria. So, these criteria are very, very sensitive, but not terrily --- terribly specific for detecting HNPC. Yet, despite this many continue to assume that HNPCC is a very rare condition and that referral of patients for genetic counseling has to be predicated on the extraordinarily strong Amsterdam Criteria being met. I should also note that in some instances the Amsterdam Criteria can be present but when the tumors fail to show microsatellite instability, about which we’ll say more, we have a condition that we call familial colorectal cancer syndrome “X” in distinction from HNPCC. So, the Amsterdam criteria can be met and you still don’t necessarily have HNPCC.
¿Cuándo no resultarían útiles en un caso particular? Los estudios en poblaciones con pruebas de microsatélites progresivas de todos los cánceres colorrectales, independientemente del historial familiar, indican que la mayoría de los casos de HNPCC no cumplen estos criterios, que son sumamente sensibles, pero no demasiado específicos para detectar el HNPCC. Aun así, muchos consideran que el HNPCC es muy poco frecuente y que el referir a los pacientes a asesoría genética debe basarse en un cumplimiento extraordinariamente acabado de los criterios de Ámsterdam. En algunos casos, estos criterios pueden existir, pero cuando los tumores no muestran inestabilidad de microsatélites, tenemos una condición llamada síndrome “X” de cáncer colorrectal familiar que difiere del HNPCC. Es posible que se cumplan los criterios y que no sea un caso de HNPCC.

14
• Clinical diagnosis
– Amsterdam criteria ideal, less is typical
• Genetic diagnosis
– Microsatellite instability (MSI) in tumor
– Loss of MMR protein expression in tumor by IHC
– Germline mutation testing (following abnormal MSI +/- IHC,
or if strong enough of a prior risk, as by PREMM 1,2,6)
Diagnosis of HNPCC
So at a level of clinical diagnosis Amsterdam criteria are ideal, but they’re less than typical in a particular instance. So, we’re relying more and more on establishing a genetic diagnosis based on microsatellite instability testing in the tumor. This is a PCR based assay. More straightforward probably to clinicians and to pathologists is a surrogate for microsatellite testing that involves evaluation for loss of mismatch repair of protein expression in the tumor by performing immunohistochemistry for evidence of loss of the proteins associated with the mismatch repair genes. When these are informative, either the microsatellite test or the immunohistochemistry, then it’s appropriate to follow with germline mutation testing. And this is why we regard this as a stepwise approach. We start with the clinical suspicion which may be high or low. We try to always evaluate a tumor and when the tumor testing is informative then we take the next step to germline mutation testing. Now, there may be situations where the tumor testing cannot be performed. The key individual is deceased, tissue is not available, and in this situation, if there‘s a strong enough a priori risk of HNPCC, such as demonstrated with some of the various models, the so-called PREMM 1,2,6 model, for example, which is a statistical probability model, it may be appropriate to do germline mutation testing even in the absence of tumor information.
Los criterios de Ámsterdam son ideales a nivel de diagnóstico clínico, pero no resultan tan típicos en una instancia particular. Cada vez nos basamos más en establecer un diagnóstico genético en función de pruebas de inestabilidad de los microsatélites del tumor. Se trata de un ensayo de PCR. Para los clínicos y patólogos, una opción más directa que las pruebas de microsatélites es evaluar la pérdida de reparación de desapareamiento en la expresión de proteínas tumorales por pruebas inmunohistoquímicas. Esto comprueba la pérdida de proteínas asociadas a los genes de reparación de desapareamiento. Cuando la prueba de microsatélites o la inmunohistoquímica son informativas, es conveniente hacer pruebas de mutación de línea germinal. Por eso consideramos que es un enfoque progresivo. Comenzamos con la sospecha clínica, que puede ser alta o baja. Luego intentamos siempre evaluar un tumor y, cuando sus pruebas son informativas, continuamos con la prueba de mutación de línea germinal. Es posible que el tumor no pueda ser sometido a pruebas. La persona clave puede haber fallecido o no se dispone de tejido. En esta situación, si el riesgo de HNPCC es considerable, como en algunos de los diversos modelos —como el PREMM 1,2,6, que es de probabilidad estadística—, en ausencia de información sobre el tumor puede ser conveniente hacer pruebas de mutación de línea germinal.
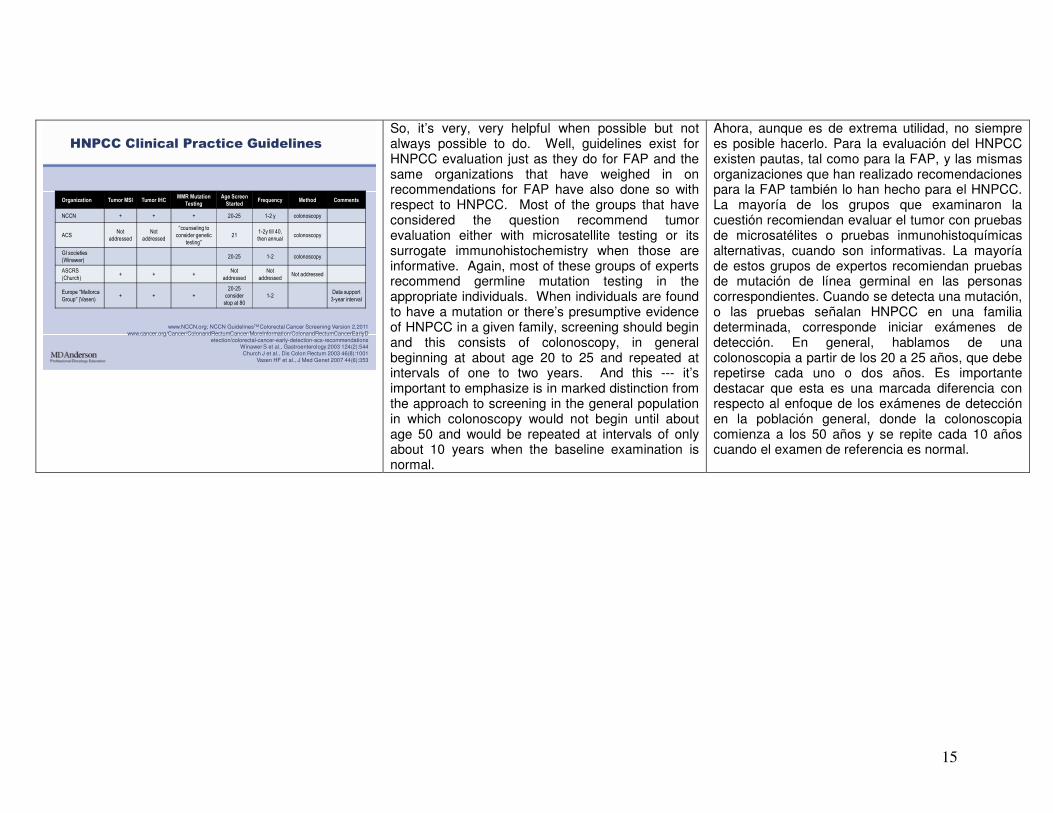
15
Organization Tumor MSI Tumor IHCMMR Mutation
Testing
Age Screen
StartedFrequency Method Comments
NCCN + + + 20-25 1-2 y colonoscopy
ACSNot
addressed
Not
addressed
“counseling to consider genetic
testing”21
1-2y till 40,
then annualcolonoscopy
GI societies
(Winawer)20-25 1-2 colonoscopy
ASCRS
(Church)+ + +
Not
addressed
Not
addressedNot addressed
Europe “Mallorca Group” (Vasen)
+ + +
20-25
consider
stop at 80
1-2Data support
3-year interval
HNPCC Clinical Practice Guidelines
www.NCCN.org; NCCN GuidelinesTMColorectal Cancer Screening Version 2.2011www.cancer.org/Cancer/ColonandRectumCancer/MoreInformation/ColonandRectumCancerEarlyD
etection/colorectal-cancer-early-detection-acs-recommendationsWinawer S et al., Gastroenterology 2003 124(2):544Church J et al., Dis Colon Rectum 2003 46(8):1001
Vasen HF et al., J Med Genet 2007 44(6):353
So, it’s very, very helpful when possible but not always possible to do. Well, guidelines exist for HNPCC evaluation just as they do for FAP and the same organizations that have weighed in on recommendations for FAP have also done so with respect to HNPCC. Most of the groups that have considered the question recommend tumor evaluation either with microsatellite testing or its surrogate immunohistochemistry when those are informative. Again, most of these groups of experts recommend germline mutation testing in the appropriate individuals. When individuals are found to have a mutation or there’s presumptive evidence of HNPCC in a given family, screening should begin and this consists of colonoscopy, in general beginning at about age 20 to 25 and repeated at intervals of one to two years. And this --- it’s important to emphasize is in marked distinction from the approach to screening in the general population in which colonoscopy would not begin until about age 50 and would be repeated at intervals of only about 10 years when the baseline examination is normal.
Ahora, aunque es de extrema utilidad, no siempre es posible hacerlo. Para la evaluación del HNPCC existen pautas, tal como para la FAP, y las mismas organizaciones que han realizado recomendaciones para la FAP también lo han hecho para el HNPCC. La mayoría de los grupos que examinaron la cuestión recomiendan evaluar el tumor con pruebas de microsatélites o pruebas inmunohistoquímicas alternativas, cuando son informativas. La mayoría de estos grupos de expertos recomiendan pruebas de mutación de línea germinal en las personas correspondientes. Cuando se detecta una mutación, o las pruebas señalan HNPCC en una familia determinada, corresponde iniciar exámenes de detección. En general, hablamos de una colonoscopia a partir de los 20 a 25 años, que debe repetirse cada uno o dos años. Es importante destacar que esta es una marcada diferencia con respecto al enfoque de los exámenes de detección en la población general, donde la colonoscopia comienza a los 50 años y se repite cada 10 años cuando el examen de referencia es normal.

16
AGA Panel Guidelines 2003
Winawer S et al., Gastroenterology 2003 124(2):544
Men and Women Symptomatic Diagnostic work-up
Asymptomatic
Age < 50 years Age ≥ 50 years
Positive family historyNegative family history Negative family history
No screening Average risk screening
2 or more first relatives affected or 1 first-degree relative affected at
age < 60 yearsHNPCC or FAP 1 first-degree relative
affected at age ≥ 60 years
Colonoscopy beginning age 40 years, or 10 years earlier than the youngest diagnosis in the family,
whichever comes first
Genetic counseling& special screening
Average risk screening, but beginning at age 40 years
Well, this particular slide I --- I discuss at greater length in my other presentation. I would review --- refer the viewer to that. And this slide is simply included to show that there can be marked heterogeneity --- heterogeneity in a particular family history and sometimes this can clearly indicate the presence of HNPCC. Sometimes it’s simply a positive family history that, even if tumor testing is done, can be uninformative. So, a very heterogeneous group where sometimes empirical approaches, starting colonoscopy a little bit earlier, doing it a little bit more often than we would in the general population, may be warranted even when HNPCC cannot be detected.
Esta diapositiva fue descrita detalladamente en mi otra presentación, a la cual me remito. Se incluye simplemente para demostrar que puede haber una marcada heterogeneidad en un historial familiar particular, que a veces puede indicar claramente la presencia de HNPCC. A veces el historial familiar es positivo, pero las pruebas tumorales no son informativas. Puede ser un grupo muy heterogéneo, donde a veces se justifica adoptar enfoques empíricos, comenzando con la colonoscopia un poco antes que en la población general, aun cuando no pueda detectarse HNPCC.
AGA Panel Guidelines 2003
Men and Women Symptomatic Diagnostic work-up
Asymptomatic
Age < 50 years Age ≥ 50 years
Positive family historyNegative family history Negative family history
No screening Average risk screening
2 or more first relatives affected or 1 first-degree relative affected at
Age < 60 yearsHNPCC or FAP 1 first-degree relative
affected at age ≥ 60 years
Colonoscopy beginning age 40 years, or 10 years earlier than the youngest diagnosis in the family,
whichever comes first
Genetic counseling& special screening
Average risk screening, but beginning at age 40 years
MSI?
Winawer S et al., Gastroenterology 2003 124(2):544
So, one of the new strategies and this is why the MSI with a question mark is shown is to perhaps focus on those situations in which we have two or more relatives with colorectal cancer even if they’re older or perhaps one individual affected at a relatively earlier age and just go ahead and do the microsatellite testing whenever possible. And, this is really an approximation of the so-called Bethesda Guidelines for considering microsatellite testing as a first step screening for the presence of HNPCC.
Una de las nuevas estrategias —y es por esta razón que las pruebas de inestabilidad de microsatélites o MSI se muestran con un signo de interrogación— consiste en centrarse en dos o más familiares con cáncer colorrectal, aunque tengan más edad, o en una persona afectada de menor edad, y hacer pruebas de microsatélites cuando sea posible. Es una aproximación a las denominadas pautas de Bethesda, que sugieren considerar las pruebas de microsatélites como el primer paso en la detección del HNPCC.
17
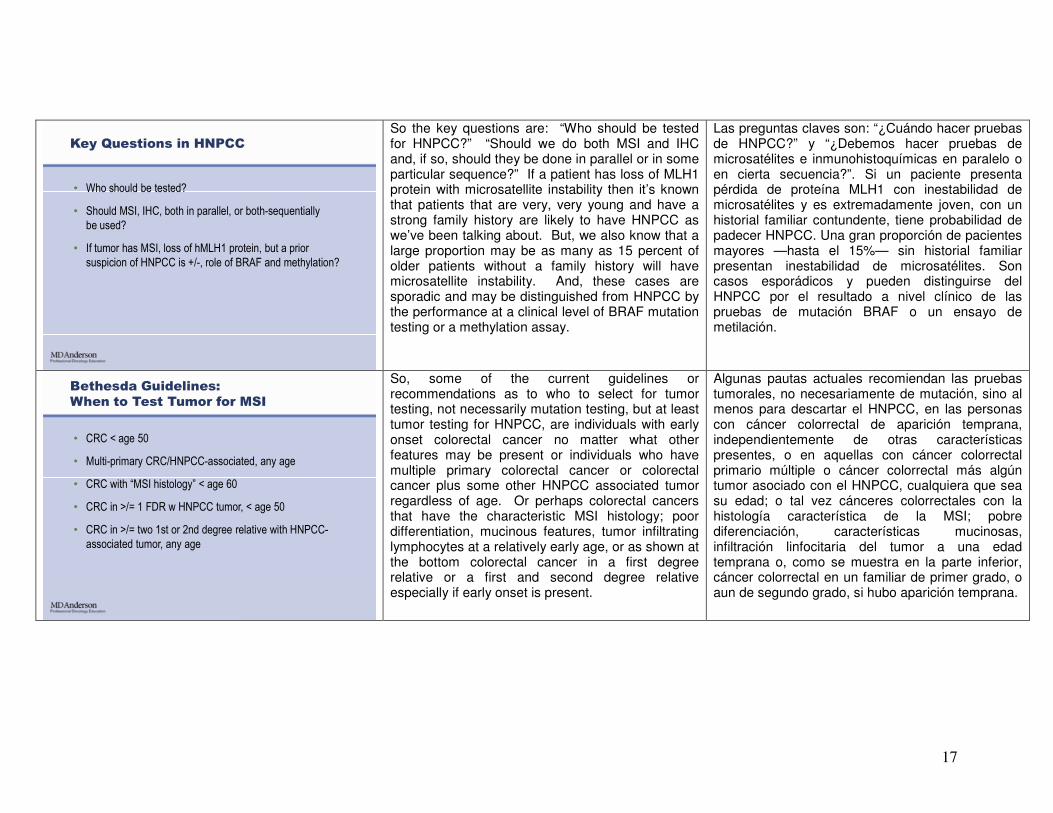
Key Questions in HNPCC
• Who should be tested?
• Should MSI, IHC, both in parallel, or both-sequentially
be used?
• If tumor has MSI, loss of hMLH1 protein, but a prior
suspicion of HNPCC is +/-, role of BRAF and methylation?
So the key questions are: “Who should be tested for HNPCC?” “Should we do both MSI and IHC and, if so, should they be done in parallel or in some particular sequence?” If a patient has loss of MLH1 protein with microsatellite instability then it’s known that patients that are very, very young and have a strong family history are likely to have HNPCC as we’ve been talking about. But, we also know that a large proportion may be as many as 15 percent of older patients without a family history will have microsatellite instability. And, these cases are sporadic and may be distinguished from HNPCC by the performance at a clinical level of BRAF mutation testing or a methylation assay.
Las preguntas claves son: “¿Cuándo hacer pruebas de HNPCC?” y “¿Debemos hacer pruebas de microsatélites e inmunohistoquímicas en paralelo o en cierta secuencia?”. Si un paciente presenta pérdida de proteína MLH1 con inestabilidad de microsatélites y es extremadamente joven, con un historial familiar contundente, tiene probabilidad de padecer HNPCC. Una gran proporción de pacientes mayores —hasta el 15%— sin historial familiar presentan inestabilidad de microsatélites. Son casos esporádicos y pueden distinguirse del HNPCC por el resultado a nivel clínico de las pruebas de mutación BRAF o un ensayo de metilación.
Bethesda Guidelines:
When to Test Tumor for MSI
• CRC < age 50
• Multi-primary CRC/HNPCC-associated, any age
• CRC with “MSI histology” < age 60
• CRC in >/= 1 FDR w HNPCC tumor, < age 50
• CRC in >/= two 1st or 2nd degree relative with HNPCC-
associated tumor, any age
So, some of the current guidelines or recommendations as to who to select for tumor testing, not necessarily mutation testing, but at least tumor testing for HNPCC, are individuals with early onset colorectal cancer no matter what other features may be present or individuals who have multiple primary colorectal cancer or colorectal cancer plus some other HNPCC associated tumor regardless of age. Or perhaps colorectal cancers that have the characteristic MSI histology; poor differentiation, mucinous features, tumor infiltrating lymphocytes at a relatively early age, or as shown at the bottom colorectal cancer in a first degree relative or a first and second degree relative especially if early onset is present.
Algunas pautas actuales recomiendan las pruebas tumorales, no necesariamente de mutación, sino al menos para descartar el HNPCC, en las personas con cáncer colorrectal de aparición temprana, independientemente de otras características presentes, o en aquellas con cáncer colorrectal primario múltiple o cáncer colorrectal más algún tumor asociado con el HNPCC, cualquiera que sea su edad; o tal vez cánceres colorrectales con la histología característica de la MSI; pobre diferenciación, características mucinosas, infiltración linfocitaria del tumor a una edad temprana o, como se muestra en la parte inferior, cáncer colorrectal en un familiar de primer grado, o aun de segundo grado, si hubo aparición temprana.

18
Hereditary Nonpolyposis Colorectal
Cancer: Why Might BG Become Obsolete?
• Population studies, using by step-wise MSI testing, indicate
that many cases of HNPCC do not meet BG
• Surgeons and oncologists are interested in MSI status, even
when sporadic (variation in prognosis, more responsiveness),
leading to proposals for universal MSI testing
Well, many institutions have found even these Bethesda Guidelines to perhaps be insufficiently broad. Population studies using a stepwise microsatellite testing approach in all new diagnoses of colorectal cancer has shown that many cases of HNPCC not only do not meet the Amsterdam Criteria but do not even meet Bethesda Guidelines. So, surgeons and oncologists that are often interested in microsatellite status even when it’s sporadic because there are variations in prognosis and responsiveness or lack of responsiveness to certain chemotherapy agents that can occur in microsatellite unstable tumors. So, for these two reasons, many institutions including our own have very much been considering the possibility of doing universal microsatellite or immunohistochemistry on all new diagnoses of colorectal cancer, certainly on all young patients with colorectal cancer.
Muchas instituciones consideran que las pautas de Bethesda quizás no sean lo suficientemente amplias. Los estudios de población utilizando un enfoque de pruebas de microsatélites progresivas en nuevos diagnósticos de cáncer colorrectal han demostrado que muchos casos de HNPCC no cumplen los criterios de Ámsterdam ni las pautas de Bethesda. Los cirujanos y oncólogos suelen interesarse en la condición de los microsatélites, aunque sea esporádica, porque existen variaciones de pronósticos y respuestas, o ausencia de estas, a determinados antineoplásicos en los tumores con inestabilidad de microsatélites. Por ello, muchas instituciones, incluida la nuestra, consideran la posibilidad de hacer pruebas de microsatélites o inmunohistoquímicas universales en todos los nuevos diagnósticos de cáncer colorrectal, y en todos los pacientes jóvenes con este cáncer.
Genetic Testing for HNPCC
• Mutations occur in mismatch repair (MMR) genes:
hMSH2, hMLH1, hMSH6, hPMS2
• Sensitivity only fair---60-70% mutation detection rate
in optimally selected cases (e.g. AC+ with hMSH2 loss
on IHC)
So, once we have an informative tumor evaluation we know that mutations can occur in either one of four genes, actually a fifth: the MSH2 gene, MLH1 gene, MSH6, and PMS2 and I should add to this list the so-called TACSTD1 or EPCAM gene that is evaluated sequentially in patients that have loss of MSH2 expression by immunohistochemistry but in which an MSH2 mutation is not found. Fairly technical but important consideration. I have to concede that even in cases that clearly have HNPCC, Amsterdam Criteria are met, for example, protein loss for one of the mismatch repair genes occurs. Even under this best case scenario for detecting a mutation, the sensitivity of mutational testing remains only in the range of 60 to 70 percent and in many series lower than that.
Una vez que tenemos una evaluación informativa del tumor, pueden ocurrir mutaciones en uno de cuatro genes, y aun en cinco: los genes MSH2, MLH1, MSH6 y PMS2, y yo agregaría a esta lista el gen TACSTD1 o EPCAM, que se evalúa secuencialmente con pruebas inmunohistoquímicas en los pacientes con pérdida de expresión del gen MSH2, aunque no se detecte su mutación. Es una consideración técnica importante. Aun en los casos evidentes de HNPCC se cumplen los criterios de Ámsterdam, por ejemplo, pérdida de proteínas para uno de los genes de reparación de desapareamiento. En este escenario óptimo para detectar una mutación, la sensibilidad de estas pruebas continúa siendo del 60% al 70%, y en muchas series tiene valores más bajos.

19
HNPCC Testing: For Available Tumor
MSS and IHC staining retained
MSI-H or IHC staining lost
Consider MMRMutation testing
unlikely to benefit from germlineHNPCC testing unless compelling
family history
So the basic approach is if we do microsatellite instability testing or immunohistochemistry and have loss of staining, we would then go on to mismatch repair testing. When we have a microsatellite stable tumor and normal immunohistochemistry, this is a very important negative predictor. These have very strong negative predictive value and a particular patient is unlikely to benefit from germline HNPCC testing unless there are some compelling circumstances otherwise, such as a very strong family history. But, as I’ve already said, many families have been found that manifest Amsterdam Criteria, but have microsatellite stable tumors and are not found to have HNPCC when mutational testing is done. So, very strong negative predictive value when these tests are normal.
El enfoque básico es hacer pruebas de inestabilidad de microsatélites o inmunohistoquímicas, y si hay pérdida de tinción, hacemos pruebas de reparación de desapareamiento. Si el tumor tiene microsatélites estables e inmunohistoquímica normal, tenemos un predictor negativo muy importante. Estos resultados tienen un alto valor predictivo negativo y es improbable que un paciente se beneficie con la prueba de línea germinal para el HNPCC si no hay circunstancias categóricas, como un historial familiar muy contundente. No obstante, se ha comprobado que muchas familias manifiestan los criterios de Ámsterdam, pero tienen tumores con microsatélites estables y no se les detecta HNPCC con las pruebas de mutación. El valor predictivo negativo es muy contundente si estas pruebas son normales.
Microsatellite Instability (MSI)
• 15% of sporadic MSI (hMLH1 methylation, BRAF mutation)
• >95% of HNPCC tumors MSI at multiple loci
• Routine MSI assays available
So a few more words about microsatellite testing: As I‘ve already mentioned about 15 percent of sporadic colorectal cancers will have microsatellite instability. So, as we go to a more population oriented approach, where we’re perhaps testing everyone with colorectal cancer, we will find more and more of these cases of so-called sporadic microsatellite instability. Now, it’s important to note that when a given patient has microsatellite instability, particularly if it involves loss of MLH1 expression and particularly if the patient is older and has little family history, one has to be very much concerned about the possibility that this may be a sporadic. And, there are assays such as methylation assays and BRAF mutation testing that can be done that will show that a particular case is likely a sporadic. But the reason this is so important is that virtually all HNPCC tumors will show microsatellite instability and these MSI assays are routinely available. In the panels shown at the
En cuanto a las pruebas de microsatélites, un 15% de los cánceres colorrectales esporádicos tienen esta inestabilidad. A medida que adoptemos un enfoque más orientado hacia la población, tal vez hagamos pruebas a todas las personas con cáncer colorrectal y encontremos cada vez más casos de inestabilidad esporádica. Cuando un paciente determinado presenta inestabilidad de microsatélites, especialmente si implica pérdida de expresión del gen MLH1, y si el paciente es mayor y tiene poco historial familiar, es preciso considerar la posibilidad de que sea una circunstancia esporádica. Los ensayos de metilación y las pruebas de mutación BRAF permiten demostrar si un caso particular es probablemente esporádico. Esto es importante porque casi todos los tumores del HNPCC presentan inestabilidad de microsatélites y estos ensayos se encuentran rutinariamente disponibles. La gráfica superior muestra el estado normal de un marcador de

20
bottom, the panel at the top shows the normal state for a particular microsatellite marker. And, what happens in the tumor is that the --- the electrophoretic gel actually develops a new peak showing that there is a mutation occurring in the microsatellite marker gene. This is associated with an increase or decrease in the repeat sequences that actually characterize a microsatellite gene. So, evidence of a new peak on an electrophoretic gel is evidence of instability at a particular locus.
microsatélites particular. En el tumor, el gel electroforético desarrolla un nuevo pico, lo que indica una mutación en el gen marcador de microsatélites. Esto se asocia a un aumento o disminución de las secuencias de repetición que caracterizan a un gen de microsatélites. Por lo tanto, un nuevo pico en el gel electroforético demuestra inestabilidad en un locus particular.
Normal hMSH2 somatically mutated(Now, neither allele expressed)
One hMSH2 allele expressed
More easy for the average clinician and pathologist to understand than the microsatellite approach is the use of immunohistochemistry so shown on the left, we have normal mucosa that retains the normal brown immunohistochemical stain against the protein that corresponds to the MSH2 gene. Well, what happens through the process of loss of heterozygosity is that the adjacent cancer shown on the right loses expression of the otherwise normal parental allele for the MSH2 gene in this case. And, so we lose expression altogether. And this is a fairly reliable test and, in addition, and perhaps as importantly, points to the underlying gene of interest. So, when this particular stain is done for MSH2, it shows loss of expression in the tumor. When the same tumor is stained for MLH1, the staining pattern is normal. And, this let’s us know that MSH2 is implicated. That MLH1 is not.
Para el médico y patólogo promedio, la inmunohistoquímica puede resultar más sencilla que los microsatélites. A la izquierda tenemos una mucosa normal que retiene la tinción inmunohistoquímica usual de color marrón sobre la proteína que corresponde al gen MSH2. A través del proceso de pérdida de heterocigosidad, el cáncer adyacente que vemos a la derecha pierde en este caso la expresión del alelo parental normal para el gen MSH2, y así se pierde la expresión por completo. Esta es una prueba bastante confiable que, lo que es igualmente importante, señala además el gen subyacente de interés. Esta tinción particular para el MSH2 muestra la pérdida de expresión en el tumor. Cuando en el mismo tumor se realiza la tinción para el MLH1, el patrón es normal. Esto indica que el gen MSH2 está afectado y que el MLH1 no lo está.

21
MMR Mutation Testing
• Consider germline mutational testing when tumor unavailable
in compelling case (PREMM 1,2,6)
• MSI-H or IHC staining lost in tumor-when HNPCC strongly
suspected, germline mutation testing
• MSI-H or IHC staining lost in tumor --- in older patient with
little or no FH, consider methylation, BRAF
www.NCCN.org; NCCN GuidelinesTM
Colorectal Cancer Screening Version 2.2011
So, as I have already mentioned there can be situations when tumor is unavailable and in those compelling cases, there are computer models that will predict the likelihood of a mismatch repair mutation. And testing may be appropriate when a sufficiently high prior probability is identified through these models. Testing is, as I’ve indicated over and over, is appropriate when we have an informative tumor test based on immunohistochemistry or microsatellite instability but we do have to be very, very careful of the possibility of a sporadic case of microsatellite instability in which case methylation assays and BRAF testing can be helpful.
Puede haber situaciones en que el tumor no esté disponible. En los casos apremiantes, existen modelos por computadora que predicen la probabilidad de una mutación del gen de reparación de desapareamiento. Las pruebas pueden ser apropiadas si se identifica una probabilidad suficientemente alta a través de estos modelos. También son apropiadas cuando tenemos una prueba tumoral informativa por inmunohistoquímica o inestabilidad de microsatélites. Debemos ser extremadamente cautos ante la posibilidad de un caso esporádico de inestabilidad de microsatélites, en el cual son útiles los ensayos de metilación y las pruebas BRAF.
HNPCC Management
Average risk Intensive surveillance(colonoscopy 1-2y intervals)
Segmental resection vs. subtotal colectomy
(controversial!)
Endoscopicpolypectomy, but ??
threshold forprophylactic resection
MSI
IHC
So the approach to HNPCC management --- [excuse me] ---- can be viewed in a somewhat progressive disclosure sort of approach. So, in the upper left, we start with a cancer. This could be a cancer occurring in a patient with a high prior probability, strong family history, early age at onset, and so forth. Or it could be a more population based approach. When we have an informative immunohistochemistry on tumor testing or microsatellite instability, this leads to mutational testing. When that individual is found to have a mismatch repair gene mutation and again this should occur in only ---unfortunately in only about 50 to 60, at best 70 percent, of cases, this helps confirm the diagnosis and provides a firm --- firm foundation for screening at-risk individuals. So, once the mutation is identified in the affected individual, we then recommend similar blood testing for children, siblings, and other at-risk relatives. Once a mutation is known in the index case and we test children, we can only have two pos --- possible
El enfoque de la gestión del HNPCC puede visualizarse como una revelación progresiva de la información. En la parte superior izquierda empezamos con un cáncer. Podría ser en un paciente con alta probabilidad previa, un historial familiar contundente, edad temprana al momento de la aparición, etc. También podría adoptarse un enfoque más general. Luego de una prueba tumoral informativa de inmunohistoquímica o inestabilidad de microsatélites, pasamos a las pruebas de mutación. Si comprobamos una mutación del gen de reparación de desapareamiento —lo que ocurre en apenas un 50%, 60% o 70% en el mejor de los casos—, eso nos ayuda a confirmar el diagnóstico y es una base firme para la detección cuando hay riesgo. Una vez identificada la mutación, recomendamos análisis de sangre similares para los hijos, hermanos y otros familiares en riesgo. Ya determinada la mutación en el caso índice, hacemos pruebas en los hijos y solo hay dos resultados posibles: ausencia de mutación, como

22
outcomes. Either a mutation is absent as shown in the far left, in which case the individual is regarded as average risk and re --- requires no special screening. Or the individual is found to be a mu -- mutation carrier and warrants the intensive surveillance early and frequent colonoscopy. Then when a tumor is identified, generally a recommendation for a more aggressive resection is provided and the role of endoscopic polypectomy is still somewhat debated but, in most hands, probably follows that for individuals found to have sporadic adenomas in the general population.
en el extremo izquierdo, en cuyo caso la persona se considera con riesgo promedio y no requiere exámenes especiales; o se la identifica como portadora de la mutación, lo cual justifica una intensa vigilancia temprana y colonoscopias frecuentes. Cuando se identifica un tumor, suele recomendarse una resección más agresiva y, pese a que la función de la polipectomía endoscópica aún es objeto de debate, la mayoría se inclinaría por esa vía en las personas de la población general a quienes se han detectado adenomas esporádicos.
1: Colon Cancer Diagnosed, Due to Symptoms or
Maybe Age-appropriate or “+FH” Based Screening
• Colon cancer diagnosed,
due to symptoms or maybe
age-appropriate or “+FH”
based screening
So let’s take this one step at a time. We identify a cancer based on symptoms, perhaps age appropriate screening or perhaps family history based screening and so we start off with cancer.
Pero vayamos de a un paso a la vez. Identificamos un cáncer por los síntomas, quizás exámenes de detección apropiados a la edad o basados en el historial familiar. Entonces partimos de un cáncer
23
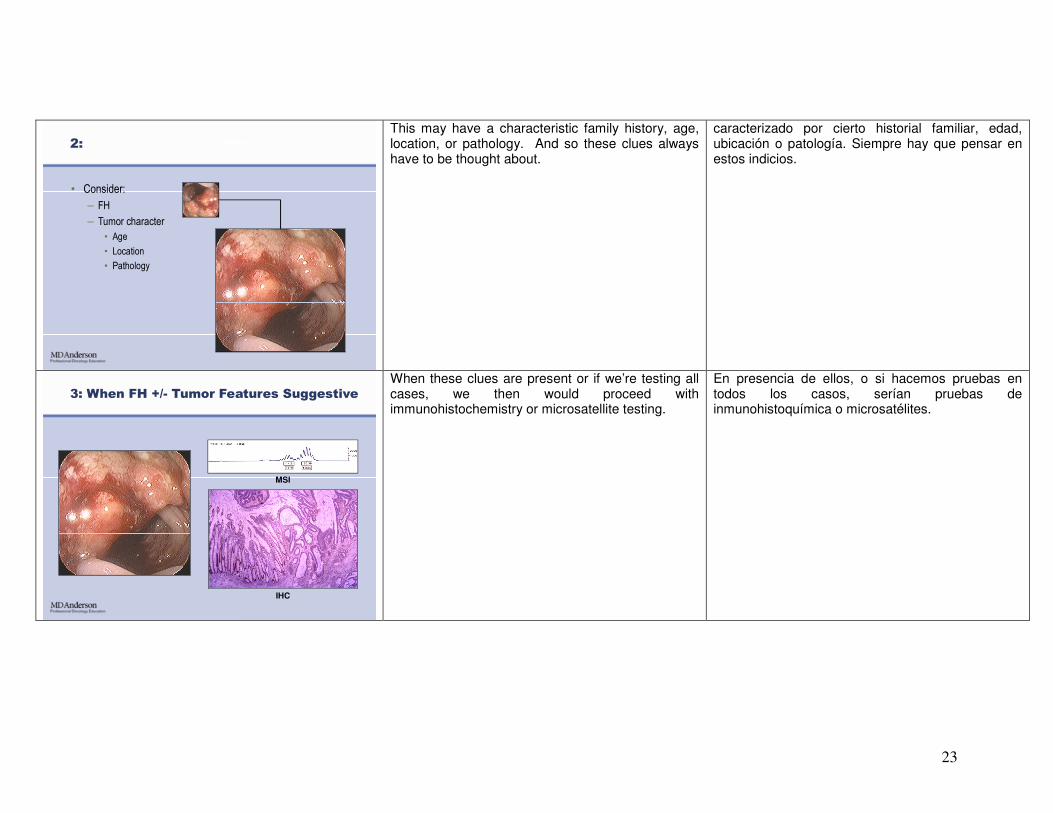
2:
• Consider:
– FH
– Tumor character
• Age
• Location
• Pathology
This may have a characteristic family history, age, location, or pathology. And so these clues always have to be thought about.
caracterizado por cierto historial familiar, edad, ubicación o patología. Siempre hay que pensar en estos indicios.
MSI
IHC
3: When FH +/- Tumor Features Suggestive
When these clues are present or if we’re testing all cases, we then would proceed with immunohistochemistry or microsatellite testing.
En presencia de ellos, o si hacemos pruebas en todos los casos, serían pruebas de inmunohistoquímica o microsatélites.
24
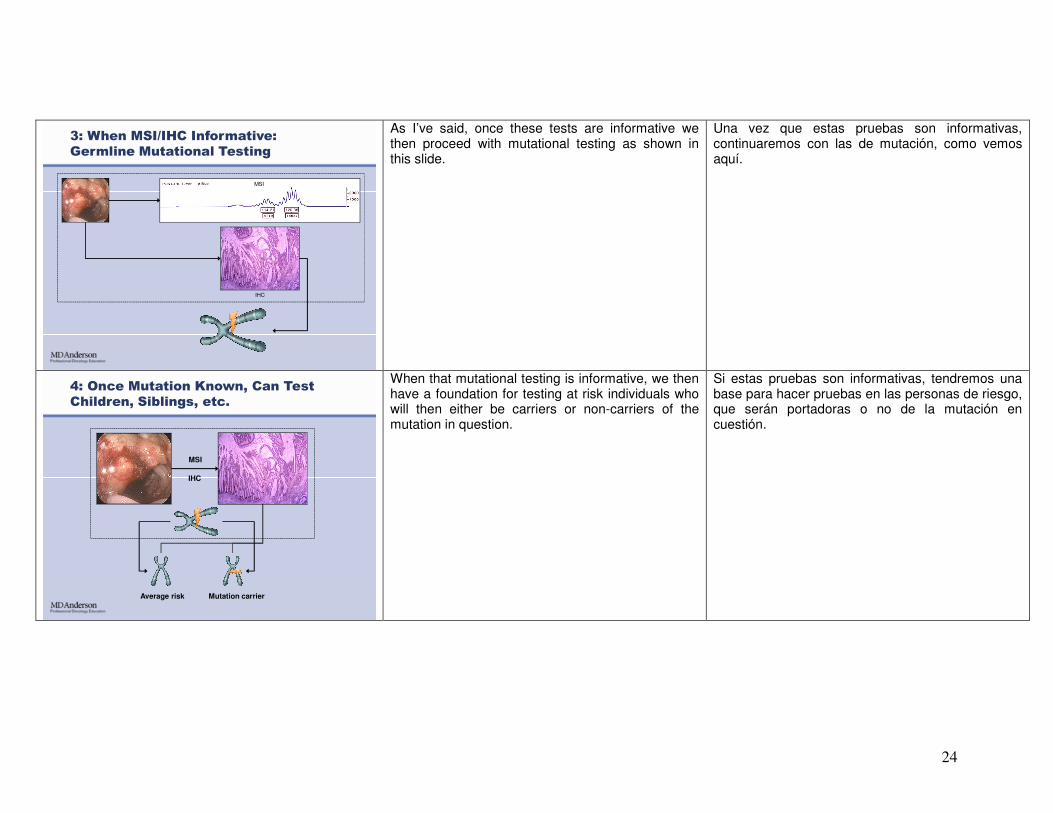
3: When MSI/IHC Informative:
Germline Mutational Testing
MSI
IHC
As I’ve said, once these tests are informative we then proceed with mutational testing as shown in this slide.
Una vez que estas pruebas son informativas, continuaremos con las de mutación, como vemos aquí.
4: Once Mutation Known, Can Test
Children, Siblings, etc.
Average risk Mutation carrier
MSI
IHC
When that mutational testing is informative, we then have a foundation for testing at risk individuals who will then either be carriers or non-carriers of the mutation in question.
Si estas pruebas son informativas, tendremos una base para hacer pruebas en las personas de riesgo, que serán portadoras o no de la mutación en cuestión.
25
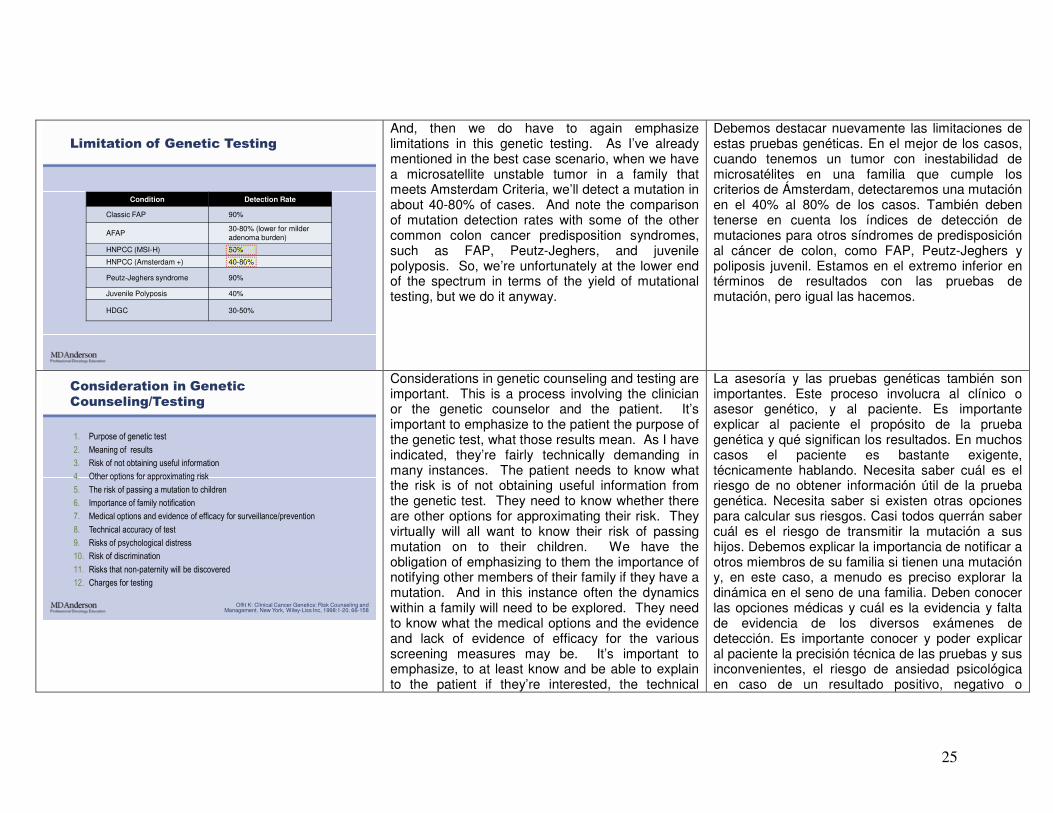
Limitation of Genetic Testing
Condition Detection Rate
Classic FAP 90%
AFAP30-80% (lower for milder adenoma burden)
HNPCC (MSI-H) 50%
HNPCC (Amsterdam +) 40-80%
Peutz-Jeghers syndrome 90%
Juvenile Polyposis 40%
HDGC 30-50%
And, then we do have to again emphasize limitations in this genetic testing. As I’ve already mentioned in the best case scenario, when we have a microsatellite unstable tumor in a family that meets Amsterdam Criteria, we’ll detect a mutation in about 40-80% of cases. And note the comparison of mutation detection rates with some of the other common colon cancer predisposition syndromes, such as FAP, Peutz-Jeghers, and juvenile polyposis. So, we’re unfortunately at the lower end of the spectrum in terms of the yield of mutational testing, but we do it anyway.
Debemos destacar nuevamente las limitaciones de estas pruebas genéticas. En el mejor de los casos, cuando tenemos un tumor con inestabilidad de microsatélites en una familia que cumple los criterios de Ámsterdam, detectaremos una mutación en el 40% al 80% de los casos. También deben tenerse en cuenta los índices de detección de mutaciones para otros síndromes de predisposición al cáncer de colon, como FAP, Peutz-Jeghers y poliposis juvenil. Estamos en el extremo inferior en términos de resultados con las pruebas de mutación, pero igual las hacemos.
Offit K: Clinical Cancer Genetics: Risk Counseling and Management. New York, Wiley-Liss Inc, 1998:1-20, 66-158
Consideration in Genetic
Counseling/Testing
1. Purpose of genetic test
2. Meaning of results
3. Risk of not obtaining useful information
4. Other options for approximating risk
5. The risk of passing a mutation to children
6. Importance of family notification
7. Medical options and evidence of efficacy for surveillance/prevention
8. Technical accuracy of test
9. Risks of psychological distress
10. Risk of discrimination
11. Risks that non-paternity will be discovered
12. Charges for testing
Considerations in genetic counseling and testing are important. This is a process involving the clinician or the genetic counselor and the patient. It’s important to emphasize to the patient the purpose of the genetic test, what those results mean. As I have indicated, they’re fairly technically demanding in many instances. The patient needs to know what the risk is of not obtaining useful information from the genetic test. They need to know whether there are other options for approximating their risk. They virtually will all want to know their risk of passing mutation on to their children. We have the obligation of emphasizing to them the importance of notifying other members of their family if they have a mutation. And in this instance often the dynamics within a family will need to be explored. They need to know what the medical options and the evidence and lack of evidence of efficacy for the various screening measures may be. It’s important to emphasize, to at least know and be able to explain to the patient if they’re interested, the technical
La asesoría y las pruebas genéticas también son importantes. Este proceso involucra al clínico o asesor genético, y al paciente. Es importante explicar al paciente el propósito de la prueba genética y qué significan los resultados. En muchos casos el paciente es bastante exigente, técnicamente hablando. Necesita saber cuál es el riesgo de no obtener información útil de la prueba genética. Necesita saber si existen otras opciones para calcular sus riesgos. Casi todos querrán saber cuál es el riesgo de transmitir la mutación a sus hijos. Debemos explicar la importancia de notificar a otros miembros de su familia si tienen una mutación y, en este caso, a menudo es preciso explorar la dinámica en el seno de una familia. Deben conocer las opciones médicas y cuál es la evidencia y falta de evidencia de los diversos exámenes de detección. Es importante conocer y poder explicar al paciente la precisión técnica de las pruebas y sus inconvenientes, el riesgo de ansiedad psicológica en caso de un resultado positivo, negativo o

26
accuracy of the test and the pitfalls of the testing, risk of psychological distress for positive test, negative test, non-diagnostic test, the risk of discrimination. Many patients are alarmed about the possibility of insurance or employment discrimination. Actual cases of such discrimination are vanishingly rare. And legislation exists both at the state and federal level to protect against this but many patients remain anxious despite all of these assur --- reassurances and the clinician and genetic counselor need to be aware of this. Patients need to know that the possibility of non-paternity can be discovered through such testing. And many patients are curious about the charges for testing. These are not cheap tests but increasingly are covered by standard insurance.
indeterminado, y el riesgo de discriminación. Muchos pacientes se alarman ante la posibilidad de ser discriminados por las compañías de seguros o a nivel laboral. Los casos reales de tal discriminación son muy raros. Además, existe legislación estatal y federal que ofrece protección en ese sentido, pero muchos pacientes siguen ansiosos pese a todas estas garantías, y el médico y el asesor genético deben ser conscientes de ello. Los pacientes deben saber que existe la posibilidad de que esas pruebas revelen la no paternidad. Además, muchos preguntan los costos de las pruebas. El costo no es bajo, pero cada vez hay más cobertura de los seguros estándar.
What Clinicians Need to Know About
Genetics and GI Cancer:
• Guidelines are available (www.guideline.gov)
• Genetic counseling is a lot of work-send patient to
genetic counselor if possible
• HNPCC can be present in patients with no apparent FH
or clinical features
So, what the clinician needs to know about the genetics of GI cancer is that guidelines are available and I have touched on many of these. There are very excellent clearing houses. One I like is www.guideline.gov which is curated by the NIH and provides a quick way to get into the various clinical practice guidelines. Genetic counseling is very, very important, but it is a lot of work. I send my patients to a genetic counselor whenever I possibly can to provide further education and instruction and to arrange genetic testing as warranted. And it’s important to emphasize, as I’ve touched on in several locations, that HNPCC can be present in a patient even with no apparent family history or obvious clinical features of the disease in question.
Así, el médico debe conocer las pautas sobre la genética del cáncer gastrointestinal, a muchas de las cuales ya me he referido. Existen excelentes fuentes de información. Una que recomiendo es www.guideline.gov, administrada por los Institutos Nacionales de la Salud, que ofrece acceso a las diversas pautas de práctica clínica. La asesoría genética es sumamente importante, pero entraña mucho trabajo. Yo suelo referir a mis pacientes a un asesor genético siempre que sea posible, para que los informen y coordinen las pruebas genéticas que se justifique hacer. Es importante recalcar que el HNPCC puede estar presente en un paciente aun sin un historial familiar comprometido o características clínicas evidentes de la enfermedad en cuestión.
27
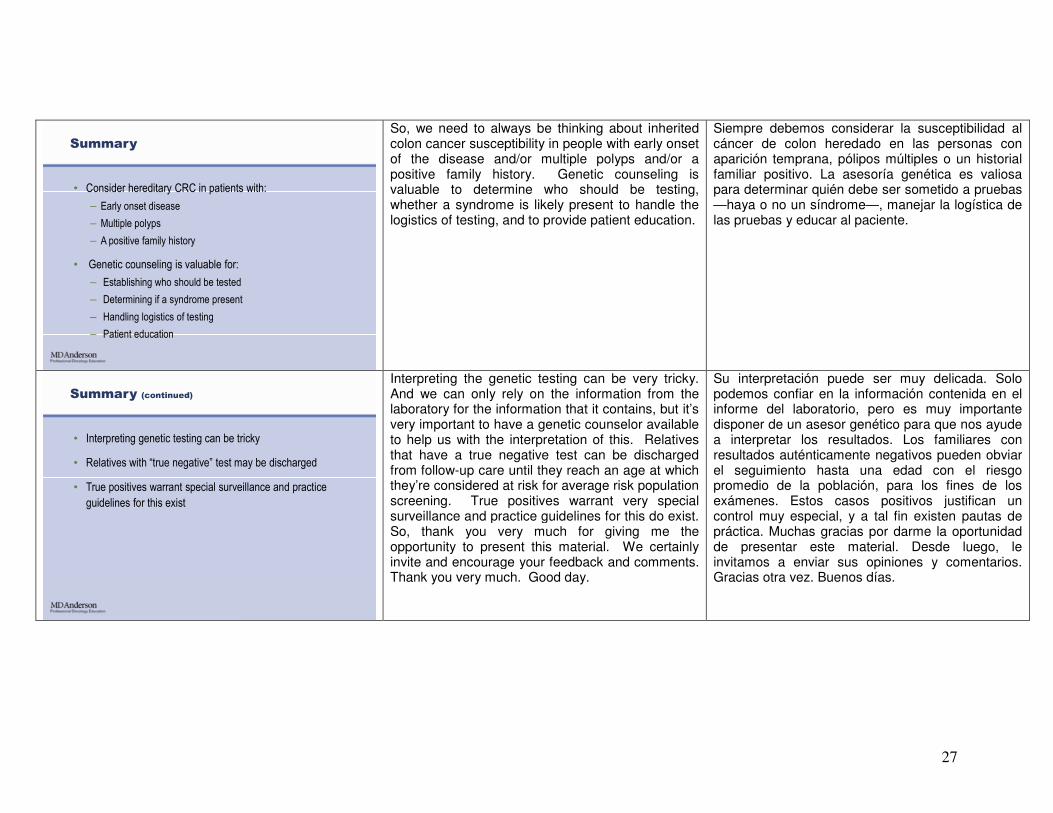
Summary
• Consider hereditary CRC in patients with:
– Early onset disease
– Multiple polyps
– A positive family history
• Genetic counseling is valuable for:
– Establishing who should be tested
– Determining if a syndrome present
– Handling logistics of testing
– Patient education
So, we need to always be thinking about inherited colon cancer susceptibility in people with early onset of the disease and/or multiple polyps and/or a positive family history. Genetic counseling is valuable to determine who should be testing, whether a syndrome is likely present to handle the logistics of testing, and to provide patient education.
Siempre debemos considerar la susceptibilidad al cáncer de colon heredado en las personas con aparición temprana, pólipos múltiples o un historial familiar positivo. La asesoría genética es valiosa para determinar quién debe ser sometido a pruebas —haya o no un síndrome—, manejar la logística de las pruebas y educar al paciente.
Summary (continued)
• Interpreting genetic testing can be tricky
• Relatives with “true negative” test may be discharged
• True positives warrant special surveillance and practice
guidelines for this exist
Interpreting the genetic testing can be very tricky. And we can only rely on the information from the laboratory for the information that it contains, but it’s very important to have a genetic counselor available to help us with the interpretation of this. Relatives that have a true negative test can be discharged from follow-up care until they reach an age at which they’re considered at risk for average risk population screening. True positives warrant very special surveillance and practice guidelines for this do exist. So, thank you very much for giving me the opportunity to present this material. We certainly invite and encourage your feedback and comments. Thank you very much. Good day.
Su interpretación puede ser muy delicada. Solo podemos confiar en la información contenida en el informe del laboratorio, pero es muy importante disponer de un asesor genético para que nos ayude a interpretar los resultados. Los familiares con resultados auténticamente negativos pueden obviar el seguimiento hasta una edad con el riesgo promedio de la población, para los fines de los exámenes. Estos casos positivos justifican un control muy especial, y a tal fin existen pautas de práctica. Muchas gracias por darme la oportunidad de presentar este material. Desde luego, le invitamos a enviar sus opiniones y comentarios. Gracias otra vez. Buenos días.