Embed Size (px)
Citation preview

Thesis on
Cloning of Chicken Anemia Virus VP3 Gene in
Replicase Based Eukaryotic Vector and Study of Its
Apoptic Activity
Submitted for the award of
DOCTOR OF PHILOSOPHY
Degree in
Biotechnology
By
Priyanka Pal
UNDER THE SUPERVISION OF
Dr. Anant Rai Dr. Kusum Agarwal
IBIT, Bareilly Shobhit University, Meerut
Faculty of Biological Engineering
Shobhit University, Meerut-250110
2012

TTTTo my parents, my first teacherso my parents, my first teacherso my parents, my first teacherso my parents, my first teachers

Certificate
This is to certify that the thesis entitled “Cloning of Chicken Anemia Virus VP3
gene in replicase based eukaryotic vector and study of its apoptic activity” submitted
by Ms. Priyanka Pal for the award of Degree of Doctor of Philosophy in the Faculty of
Biological Engineering of Shobhit University, Meerut, is a record of authentic work
carried out by her under my supervision.
To the best of my knowledge, the matter embodied in this thesis is the original
work of the candidate and has not been submitted for the award of any other degree or
diploma to any university or institution.
It is further certified that she has worked with me for the required period in the
Faculty of Biological Engineering (Centre for Biotechnology), Shobhit University,
Modipuram, Meerut.
Date: Prof. Kusum Agarwal
(Internal Supervisor)
Dr. Kusum Agarwal
M.Sc., Ph.D. Professor & Coordinator, Centre for Biotechnology,
Faculty of Biological Engineering, Shobhit University, Meerut
E-mail: [email protected]

Institute of Biotechnology & IT 197, Mudia Ahmadnagar, Bareilly-243122 (UP)
Ph: 09219661948, 0581-2602907, Fax: 0581-2602035
www.ibit.org.in [email protected]
Prof. Anant Rai Ph.D. Director
____________________________________________
Certificate
This is to certify that the thesis entitled “Cloning of Chicken Anemia Virus
VP3 gene in replicase based eukaryotic vector and study of its apoptic activity”
submitted by Ms. Priyanka Pal for the award of Degree of Doctor of Philosophy in
the Faculty of Biological Engineering of Shobhit University, Meerut, is a record of
authentic work carried out by her under my supervision.
To the best of my knowledge, the matter embodied in this thesis is the
original work of the candidate and has not been submitted for the award of any
other degree or diploma.
It is further certified that she has worked with me for a period of 43 months in
the Department of Biotechnology of Institute of Biotechnology and IT, Bareilly.
Date: Prof. Anant Rai (External Supervisor)

Declaration
I, hereby, declare that the work presented in this thesis entitled
“Cloning of Chicken Anemia Virus VP3 Gene in Replicase Based
Eukaryotic Vector and Study of its Apoptic Activity” in fulfillment of
the requirements for the award of Degree of Doctor of Philosophy,
submitted in the Faculty of Biological Engineering at Shobhit
University, Modipuiram, Meerut, is an authentic record of my own
research work carried out under the supervision of Prof. Kusum
Agarwal and Prof. Anant Rai.
I also declare that the work embodied in the present thesis
(i) is my original work and has not been copied from any
Journal/thesis/book, and
(ii) has not been submitted by me for any other Degree or
Diploma of any university/ institution.
(Priyanka Pal)

Acknowledgement
First and foremost I would like to give glory to Almighty for his
grace and help in all my endeavors and for bringing me this far in my
academic career.
I would like to express my deep sense of gratitude to Internal-
Supervisor Dr. Kusum Agarwal, Professor & Coordinator, School of
Biotechnology, Shobhit University, Meerut, for her excellent
cooperation and encouragement. I appreciate all her contributions of
time and ideas to make my Ph.D. experience productive and
stimulating. The joy and enthusiasm she has shown was contagious and
motivational, even during tough times in the pursuit of my Ph.D. work.
I would like to express my sincere gratitude to my External-Supervisor
Dr. Anant Rai, Director, Institute of Biotechnology and IT, Bareilly
(former Principal Scientist & Head, Division of Animal Biotechnology,
Indian Veterinary Research Institute, Izatnagar, Bareilly), for guiding
me with attention and care. He has taken pains in going through the
project and made necessary corrections as and when needed.
I express my thanks to Honorable Chancellor Kunwar Shekhar
Vijendra and Honorable Vice Chancellor Prof. R. P. Agarwal, of
Shobhit University, for providing congenial environment for
conducting research in the university.
I gratefully acknowledge the kind and liberal help from Prof.
D.V. Rai, Dean, Faculty of Biological Engineering, Prof. Ranjit Singh,
Former Director, School of Pharmaceutical Sciences, Dr. Rekha Dixit,
Dr. Jayanand and Dr. Manish Kumar Gupta, Shobhit University,
Meerut, for their valuable suggestions and discussions during my work.

I am extremely thankful to Dr. D.V. Rai, Dean, Faculty of
Biological Engineering, Shobhit University, Meerut, and Mrs. Soni
Gangwar, Assistant Professor (Biotechnology), Institute of
Biotechnology & IT, Bareilly for providing assistance in the laboratory.
I thank my fellow lab mates Usha Tiwari, Rajveer Maurya and
Nitin Sharma for the stimulating discussions, I had with them and for
advising me from time to time for all those days we worked together.
Last but not the least my very special and heartfelt gratitude is
due to my parents, brothers and other family members for their love,
support and encouragement without which this work would have been
a distant reality. I also thank my loving, supportive, encouraging, and
patient husband for his faithful support during the final stage of my
Ph.D. work.
(Priyanka Pal)

Research Papers Published / Accepted / Communicated in Refereed Journals/Proceedings
1. Pal, P., Agarwal, K., Rai, A. and Tiwari, U. (2011): Cloning of Chicken
Anemia Virus VP3 Gene in pSinCMV vector. Biotechnology
International, 4, 16-21. (Online www.bti.org.in).
2. Pal, P., Agarwal, K., Rai, A. and Tiwari, U. (2011): Expression of
Recombinant Plasmid Containing Chicken Anemia Virus VP3 Gene in
HeLa Cells. Research journal of Pharmaceutical Science and
Biotechnology, 1, 41-43. (Online www.rjpsb.info).
3. Pal, P., Agarwal, K., Rai, A. and Tiwari, U. (2010): Isolation of
recombinant plasmid containing Chicken Anemia Virus VP3 gene,
accepted for publication in Transaction of physical & life science.
(Accepted).
4. Pal, P., Agarwal, K., Rai, A. and Tiwari, U. (2012): Apoptic activity of
chicken anemia virus VP3 gene cloned in replicase based eukaryotic
vector. Communicated for publication in The journal of microbiology,
biotechnology and food sciences.

Participation in the Conferences/Workshops/Seminars
1. Attended national Seminar-Cum Workshop on “Biomedical Research
and Clinical Applications of Radioisotopes”, Shobhit University,
Meerut, March 31- 01 April 2012.
2. Attended International Conference on “Indian Civilization Through
the Millennia” held at JambooDweep, Hastinapur, March 2-3, 2012.
3. Attended annual Conference on Vijnana Parishad of India and the
Global Society of Mathematical & Allied Sciences, held at School of
Basic and Applied Sciences, Shobhit University, Meerut, March 24-26,
2011. Presented paper entitled “Expression of Recombinant Plasmid
containing Chicken Anemia Virus VP3 Gene in HeLa Cells.”
4. Attended Conference on “Genes & Genomics: Qualitative &
Quantitative Approach” held at School of Biotechnology, Shobhit
University, Meerut, September 11-12, 2010. Presented paper entitled
“Construction of replicase based Eukaryotic pSinCMV vector
containing Chicken Anemia Virus VP3 Gene” and “Preparation of
Recombinant Plasmid pSinCMV containing Human Erythropoietin
Gene”.
5. Attended Workshop on “Nanomaterials: Recent Techniques and
Applications”, Shobhit University, Meerut, March 27, 2010.

Contents
List of Tables (i)
List of Figures (ii)
Abbreviations (iii)
Chapter 1: Introduction 1-5
Chapter 2: Review of Literature 6-47
2.1 Chicken Anemia Virus 6 2.2 Apoptosis 14 2.3 Apoptin 23 2.4 Apoptin from other viruses 43 2.5 DNA Damage Signaling 46
Chapter 3: Material and Methods 48-65
3.1 Materials 48 3.1.1 Vector 48 3.1.2 Gene 48 3.1.3 Primers 48 3.1.4 Host Bacterial Strains 50 3.1.5 Cell Culture 50 3.1.6 Conjugates 50 3.1.7 Hyper-immune Serum 50 3.1.8 Experimental Animals 51 3.1.9 Glasswares, Plastic wares and Chemicals 51 3.1.10 Solutions and Buffers 51
3.2 Methods 51 3.2.1 Revival of the E. coli culture containing
recombinant plasmid with VP3 gene 51
3.2.2 Isolation of plasmid DNA by TELT method 52 3.2.3 Agarose gel electrophoresis 52 3.2.4 Digestion of the recombinant plasmid with
restriction enzyme to release the gene insert using protocol of Sambrook & Russell (2001)
53
3.2.5 DNA extraction from agarose gel 53 3.2.6 Blunting of EcoRI generated VP3 gene 54

staggered ends 3.2.7 Purification of blunted insert 55 3.2.8 Creation of cloning site in vector 55 3.2.9 Dephosphorylation of 5’ ends 56 3.2.10 Purification of dephosphorylated linearized
vector 57
3.2.11 Blunt end ligation of pSin vector and VP3 gene
57
3.2.12 Transformation of E. coli (DH5α) cells with the ligated product
57
3.2.13 Screening of recombinant clones for VP3 gene in right orientation
58
3.2.14 Transfection of HeLa cells 61 3.2.15 Immunoperoxidase test for DNA expression 61 3.2.16 Sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) 62
3.2.17 DNA fragmentation assay by agarose gel electrophoresis
62
3.2.18 Caspase 3 detection assay 63 3.2.19 Annexin V binding assay 64
Chapter 4: Results 66-80
4.1 Cloning of VP3 gene in pSin vector 66 4.2 Expression of VP3 gene in HeLa cells 71 4.3 Apoptic activity of VP3 in cell line 75
Chapter 5: Discussion 81-92
Chapter 6: Summary and Conclusion 93
Chapter 7: Future Scope 94-95
References 96-115
Appendix 116-120

i
List of Tables
Table No. Title Page No.
1 Primers with their sequences 50
2 Reaction mix for blunting 54
3 Reaction mix for StuI digestion 56
4 Reaction mix for dephosphorylation 56
5 Ligation mixture 57
6 Reaction mix for BglII digestion 59
7 Reaction mix for PCR 60

ii
List of Figures
Fig. No. Title Page No.
1 pSin Vector 49
2 Digestion of recombinant plasmid (pTarget.cav.vp3) with restriction enzyme (EcoRI) to release the vp3 gene insert.
68
3 The digestion of pSin.cav.vp3 with BglII yielded three segments of expected size.
69
4 PCR amplification of cav.vp3 gene. 70
5 HeLa cells transfected with pSin.cav.vp3 rplasmidshowing positive IPT test, indicating expression of gene.
72
6 Healthy control HeLa cells with no color reaction.
73
7 VP3 expressed protein analyzed by SDS-PAGE.
74
8 DNA fragmentation assay. 77
9 Caspase positive HeLa cells showing green fluorescence, indicating positive apoptic reaction.
78
10 Caspase positive HeLa cells showing bright green fluorescence, indicating positive apoptic reaction.
79
11 Annexin-V positive HeLa cells showing bright green fluorescence, indicating positive apoptic reaction.
80

iii
Abbreviations
0C Degree Celsius
% Percent
AA Amino Acid
Ab Antibody
CAV Chicken Anemia Virus
DMEM Dulbecco's Modified Eagle Medium
DNA Deoxyribonucleic acid
dNTP Deoxy ribonucleoside triphosphate
EDTA Ethylene DiamineTetraacetic Acid
Etbr Ethidium Bromide
Fig. Figure
CIAP Calf Intestinal Phosphatase
LB Luria Bertani
MW Molecular Weight
NaCl Sodium Chloride
FCS Fetal calf serum
No. Number
PBS Phosphate Buffered Saline
PCR Polymerase Chain Reaction
pH Log10 hydrogen ion concentration
RE Restriction endonuclease
RNA Ribonucleic acid
Rpm Revolution per minute
TAE Tris-acetate-EDTA
TE Tris-EDTA
UV Ultraviolet
v/v Volume by volume
w/v Weight by volume

iv
Units of Measurement
µg Microgram
µl Mirolittre
0C Degree Celsius
bp Base pair
g Gram
hr Hour
hrs Hours
Kb Kilo base
kDa Kilo Dalton
M Molar
mg Milligram
ml Milliliter
mM Millimolar
N Normal
Ng Nanogram
pmol Picomole
sec Second (s)
U Unit (s)

Introduction
Chapter-1

1
Chapter-1
Introduction
Kerr et al. (1972) described distinct morphologic changes of dying
cells and called this phenomenon as apoptosis. The term was coined on
the basis of the fact that release of apoptic bodies by dying cells resembled
with the picture of falling leaves from decidous trees, called in Greek
“apoptosis.”
The apoptotic downfall of a cell resulted in the formation of small
membrane bound entities known as apoptotic bodies. These bodies pinch
off from the dying cell and were consumed by the phagocytic action of
neighboring cells. This engulfment provided means for the dissemination
of the virus without initiating a concomitant host response, which follows
the discharge of progeny into the extracellular solution (Teodoro and
Branton, 1997).
Apoptosis or programmed cell death is a genetically determined
process to destroy cells for maintaining the cellular homeostasis in the
tissue. There is a family of cysteine proteases known as caspases, or
cysteine-aspartic proteases or cysteine-dependent aspartate-directed
proteases that play a vital role in apoptosis (programmed cell death).
Activation of cysteine called caspases plays the main role in the execution
of apoptosis. These activated caspases selectively cut cellular proteins,
which resulted in apoptotic morphology (internucleosomal fragmentation

2
of DNA into 180-200 base pair pieces, shrinkage of the cell as well as
fragmentation of the cell into apoptic bodies) and death of the cell. Two
pathways of caspase activation were reported. The first was through
triggering of cellular death-receptor superfamily and the second was
mitochondrial pathway induced by the changes of the expression of
proto-and anti-apoptic genes in the cell. It leads to the discharge of
cytochrome c and apoptosis inducing factor from mitochondria
(Lesauskaite and Ivanoviene, 2002).
Schmitz et al. (2000); Lauber et al. (2004) defined apoptosis as a
physiological form of cell death characterized via nuclear chromatin
condensation, cytoplasmic shrinking and memberane blebbing. This form
of programmed cell death was mainly induced by cancer treatment
(Kawanishi and Hiraku, 2004; Wesselborg and Lauber, 2005).
Chicken anemia virus, or CAV, is a virus that affects poultry. CAV
causes anemia, bone marrow atrophy, and severe immunosuppression.
Clinical signs of infection of CAV were mainly found in youthful chicks
due to maternal antibodies present mainly in adult chickens (Sommer
and Cardona, 2003).
The chicken anemia virus protein apoptin induced a p53-
independent, Bcl-2-insensitive type of apoptosis in a variety of human
tumor cells. Apoptin induced apoptosis in human transformed and
malignant cells except in normal cells. It was observed that apoptin failed
to induce programmed cell death in normal lymphoid, dermal, epidermal,
and smooth-muscle cells. Long-term expression of apoptin in normal
human fibroblasts showed that apoptin had no toxic or transforming

3
activity in these cells. In normal cells, apoptin was observed
predominantly in the cytoplasm, whereas in transformed and malignant
cells it was located in the nucleus, which recommended that the
localization of apoptin was related to its activity. These properties make
apoptin a potential agent for the treatment of a large number of tumors
(Oorschot et al., 1997).
It was found that a kinase activity present in cancer cells but
negligible in normal cells regulated the activity of apoptin via
phosphorylation. Apoptin can be activated, in normal cells, by transient
transforming signals conferred by ectopically expressed SV40 LT antigen,
which rapidly induced apoptin's phosphorylation, nuclear accumulation
and the ability to induce apoptosis. In normal cells where such signals
were not received, apoptin became aggregated, epitope-shielded and
were eventually despoiled in the cytoplasm. The mechanism of apoptin-
induced apoptosis could lead to the detection of novel tumor-specific
pathways that might be exploitable as anti-cancer drug targets (Rohn et
al., 2004).
It was observed that apaptin induced tumor specific apoptosis,
which was linked with the nuclear localization of the protein in tumor
cells, while in normal human cells, apoptin was detected mostly in the
cytoplasm and did not induce apoptosis. Using a recombinant adenovirus
expressing apoptin, it was found that apoptin induced G(2)-M cell cycle
arrest and chromatin condensation in cancer cells. Adenovirus mediated
apoptin expression also induced G(2)-M arrest in normal cells. In normal

4
cells apoptin was restricted chiefly in the cytoplasm but was also found in
the nucleus of a subset of cells (He et al., 2005).
Wang et al. (2005) also showed that apoptin could not induce
apoptosis in normal cells, but could induce in transformed cells or tumor
cells. The tumor specificity of apoptin was linked to its subcellular
localization. In tumor cells, apoptin migrated to the nuclei, whereas in
non-transformed cells, it remained chiefly within the cytoplasm.
Phosphorylation was accountable for the nuclear localization of apoptin.
Apoptin was phosphorylated in tumor cells, then translocated into the
nuclei, and induced cell apoptosis. Apoptin-induced apoptosis was
independent of functional p53, and colud not be inhibited by
overexpression of Bcl-2 and Bcl-xL, but caspase-3 activation was essential
for apoptin-induced rapid apoptosis. It was possible that apoptin's ability
to bind DNA was closely linked to its aptitude to induce apoptosis.
Liu et al. (2006) showed involvement of sphingolipids in apoptin
induced cell killing. Apoptin activated acid sphingomylinase (ASMase),
which generated ceramide; in turn ceramide acted as a second messenger
and signaled apoptotic response.
The reaction of the cell to DNA damage was a complex procedure,
which included damage signaling, repair, apoptosis or cell death. It was
connected with serious changes in the cell nucleus, which were caused by
post-translational modifications and active relocalizations of proteins as
well as alterations in the expression of many genes. Those changes were
not limited to the sites of DNA damage, but concerned whole cell nucleus,
including its domains: Promyelocytic leukemia (PML) bodies, Cajal

5
bodies and nucleolus. Promyelocytic leukemia (PML) nuclear bodies were
dynamic macromolecular multiprotein complexes that employ and
release a plethora of proteins. A considerable number of PML nuclear
body components play vital role in apoptosis, senescence regulation and
tumor suppression. Cajal bodies were little nuclear organelles with a
number of nuclear functions (Wysokinski et al., 2010).
Keeping the above facts in view, the present study has been designed
with the following objectives:
1. Cloning of chicken anemia virus VP3 gene in pSin mammalian expression vector.
2. In vitro expression of VP3 gene in cultured cells.
3. Study of apoptic activity of apoptin in cell line.

Review of Literature
2.1 Chicken anemia virus
2.2 Apoptosis
2.3 Apoptin
2.4 Apoptin From Other Viruses
2.5 DNA Damage Signaling
Chapter-2

6
Chapter-2
Review of Literature
2.1 Chicken Anemia Virus
Circoviruses are small, non-enveloped, icosahedral viruses that are
circular, single-stranded DNA genomes. Their genomes are also the
smallest possessed by animal viruses. The circovirus family consists of
three members- chicken anemia virus, porcine circovirus and psittacine
beak and feather disease virus, with pigeon circovirus being classified as
an unsure member. Infections with each of the four circoviruses were
linked with potentially fatal diseases in which destruction to lymphoid
tissue and immunosuppression by a virus were common features.
Knowledge with other animal virus families suggested that additional
animal species will be infected by, and yet undiscovered, circoviruses and
that these might display similar tissue tropism and disease-causing
potential. Novel circoviruses might infect other avian species, including
commercial poultry (Todd, 2000).
Chicken anemia virus (CAV) seemed to have a worldwide
distribution. CAV caused a syndrome in chickens which was identified by
increased mortality, anemia associated with atrophy of the hematopoietic
tissues in the bone marrow, subcutaneous and intramuscular
hemorrhages, and atrophy of the lymphoid system. CAV was found to be

7
spread both vertically and horizontally. Vertical transmission occurred
following primary infection of in-lay breeding stock, and resulted in
clinical disease in their progeny around 2 weeks of age. Horizontal spread
was found to result mostly in subclinical disease. Both clinical and
subclinical diseases caused economic loss as indicated by a vaccine (Todd
et al., 1990; McNulty et al., 1991).
Circular double-stranded replication intermediates were
identified in low-molecular-weight DNA of cells of the avian leukemia
virus-induced lymphoblastoid cell line 1104-X-5 infected with CAV.
Linearized CAV DNA was cloned into the vector pIC20H, to characterize
the genome of CAV. A cytopathogenic effect was caused when
transfection of the circularized cloned insert was done into chicken cell
lines, which was arrested when chicken serum with neutralizing
antibodies directed against CAV was added. Chickens inoculated with
CAV collected from cell lines transfected with cloned CAV DNA
developed clinical signs of CAV. The CAV DNA sequence had three
partially overlapping major reading frames coding for putative peptides
of 51.6, 24.0, and 13.6 kDa. The CAV genome probably contained only
one promoter region and only one poly(A) addition signal. Southern blot
analysis by means of oligomers derived from the CAV DNA sequence
showed that infected cells contained double and single-stranded CAV
DNA, while purified virus contained only the minus strand. It was for
the first time that the genome of one of the three known single-stranded
circular DNA viruses had been completely analyzed (Noteborn et al.,
1991).

8
CAV was initially isolated in Japan and the associated disease
chicken infectious anemia was described in 1979. The virus had a world-
wide distribution and was common in intensive poultry raising areas.
CAV was believed to play an important role in several multiple etiology
disease syndromes; hemorrhagic syndrome; hemorrhagic anemia
syndrome, aplastic anemia, gangrenous dermatitis, anemia dermatitis,
hemorrhagic aplastic anemia syndrome and blue wing disease.
Horizontal spread seemed to be less important than vertical
transmission. The most characteristic lesion was yellow fatty bone
marrow and the most consistent finding was thymic atrophy. Thymic
and bone marrow intranuclear inclusion bodies occurred with infection
but were of limited value diagnostically and were very transient which
were seen rarely (Pope et al., 1991).
The CAV genome contained three partially or completely
overlapping genes. It caused cytopathogenic effects which were fatal
in chicken thymocytes and cultured transformed mononuclear cells via
apoptosis. In transformed chicken cells, the synthesis of the VP3 gene
product apoptin mimiced the CAV-induced apoptosis, which was p53
independent. Apoptin induced apoptosis in human tumor cells but, not
in normal cells. These properties suggested that apoptin might have
potential medical applications (Noteborn et al., 1998).
TT virus (TTV) was the only known human virus with single-
stranded circular DNA, with a relationship to CAV of the
family Circoviridae. A new human virus was designated as TTV-like mini
virus (TLMV) was reported, which resembled TTV and CAV. This non-

9
enveloped virus was smaller but had a similar density to TTV, when a
TLMV/TTV-coinfected plasma was analyzed. Full-length sequencing
revealed that the TLMV genome was a circular DNA consisting 2860 nt,
significantly shorter than TTV but longer than CAV. TLMV was observed
to be similar to both TTV and CAV in genomic organization. The
untranslated region of TLMV resembled CAV in that both had direct
repeats, whereas the sequence homology was more evident between
TLMV and TTV. TLMV was an intermediate between the remotely
related TTV and CAV. Since CAV differed much from other circoviruses,
it might better be classified jointly with TTV and TLMV under a new
family (Takahashi et al., 2000).
Aldair et al. (2000) also analysed a disease in young chicks which
was caused by CAV. They also observed generalised lymphoid atrophy,
increased mortality and severe anemia. The virus targeted erythroid and
lymphoid progenitor cells in bone marrow and thymus respectively. The B
cells in the chicken were not susceptible to CAV infection and were not
directly affected by the virus. Severe anemia, and depletion of
granulocytes and thrombocytes was resulted by destruction of erythroid
progenitors in bone marrow. Destruction of precursor T cells resulted in
depletion of mature cytotoxic and helper T cells with consequent effects
on susceptibility to, and enhancement of, the pathogenicity of secondary
infectious agents, and suboptimal antibody responses. It appeared that
apoptin was a feature of the lymphocyte depletion in the thymic cortex,
which might be mediated by one of the non-structural viral proteins, VP3
(apoptin).

10
A serologic survey in unvaccinated broiler parent and broiler
progeny flocks demonstrated seroconversion against chicken infectious
anemia virus in all parent flocks. The presence of CAV antibodies at
slaughter of broilers was positively correlated with the slaughterhouse
condemnation rates. The results showed that CAV infections were highly
prevalent in both broiler parent and broiler flocks. (De Herdt et al., 2001).
CAV was isolated from a chicken flock in Harbin of China. By
polymerase chain reaction and sequence, the genome of the virus was
cloned and analyzed. The genome was made of 2298 base pairs including
three overlapping open reading frames (vp1, vp2, vp3) and a regulative
region (He et al., 2002).
From different geographical regions of India, four CAV isolates
(CAV-A, -B, -E and -P) were recovered and characterized. The nucleotide
and deduced amino acid sequence of the Indian CAV isolates were
associated and compared with other isolates of CAV from different
countries like Europe, Asia, America and Australia. Phylogenetic
analysis of the Indian CAV isolates was done, based on the nucleotide
and deduced amino acid sequences. It was observed that Indian isolates
were genetically evolved from different parts of the world. The study
showed precious information on molecular epidemiological status of
CAV isolates prevalent in India for the first time (Natesan et al., 2006).
A field strain C14 of CAV was isolated from a 14 day-old broiler
flock with growth runting syndromes. Antibody reactions to inactivated
vaccines to avian influenza viruses (AIV) were suppressed in specific
pathogen free (SPF) chickens inoculated with C14 strain CAV at 1 day-

11
old. When chickens were infected with C14 strain and
reticuloendotheliosis viruses, it demonstrated a synergism in
immunosuppression. The viral genomic DNA was amplified by PCR in 3
overlapped fragments and PCR products were cloned into T-vector
plasmid for sequencing. The sequencing results showed that the total
genome of C14 strain CAV was 2298 bp which contained 3 overlapped
open reading frame and 1 non-coding regulation fragment. Its whole
genome had 97.2% - 99.2% of homogeneity to other several published
CAV reference strains (Li et al., 2007).
A total of 12 CAV isolates from different commercial broiler breeder
farms were isolated and characterized. High level of occurrence of vertical
transmission of viral DNA to the progeny was indicated when detection of
CAV positive embryos by the PCR assay was done in the range of 40 to
100% for different farms. By indirect immunoperoxidase staining, CAV
antigen was detected in the thymus and in the bone marrow but not in
spleen, liver, duodenum, ovary and oviduct. Based on partial sequences of
VP1 gene, the 12 CAV isolates were characterized. Six isolates (MF1A,
M3B5, MF3C, NF4A, P24A and P12B) were found to have maximum
homology with previously characterized Malaysian isolate SMSC-1, four
isolates (NF3A, M1B1, PYT4 and PPW4) with isolate BL-5 and the
remaining two (NF1D and NF2C) had maximum homology both with
isolates 3-1 and BL-5. With amino acid profile of 75-I, 97-L, 139-Q and 144-
Q, seven of the isolates were clustered together in cluster I with other
isolates from different geographical places. The observation demonstrated
that CAV was widespread in the studied commercial broiler breeder farms
(Hailemariam et al., 2008).

12
Cancer remains one of the leading causes of death worldwide. By
combining numerous tumor-specific and enhanced targeting signals into
a single modular multifaceted approach, it might be possible in future to
achieve the desired outcome, without any unwanted bystander effects,
the delivery of cytotoxic drugs/DNA directly to the nucleus specifically
within tumor cells. To achieve this, modules such as the tumor-specific
nuclear targeting signal of the chicken anemia virus apoptin protein
represented exciting possibilities (Wagstaff and Jans, 2009).
CAV has gained much importance as an immunosuppressive and
economically important emerging pathogen of poultry. The virus has
been detected in recent years which was isolated from poultry flocks of
India. The first sero-epidemiological investigation of the presence of
CAV infection was reported in poultry flocks of the country. A total of
404 serum samples were collected from chicken flocks of eleven poultry
farms, which contained a total of 0.34 million birds from four Northern
states and were also suspected of having chicken infectious anaemia
(CIA). By using a commercially available enzyme-linked immunosorbent
assay (ELISA) kit, screening of the sera samples was done which revealed
351 serum samples (86.88%) to be positive for CIAV antibodies. A high
prevalence rate of CIAV recorded in the analysis, along with previous
virus detection reports, indicated the extensive distribution of the virus.
So, CIAV should be considered an economically important pathogen of
poultry, affecting poultry industry of India (Bhatt et al., 2011).
Using PCR, the presence of CAV infection genetic variability of
isolated strains based on restriction of VP1 gene by Mbo1 and apoptotic

13
changes in the CAV positive broiler chickens in upper Egypt, were
investigated. The data indicated that infection with CAV induced
apoptosis in lymphoid cells that might affect on cellular immunity. The
findings highlighted the significance of CAV, therefore, focus should be
made on its epidemiology in the upper Egypt and to further develop and
apply reliable diagnostic tools as well as molecular studies so as to
suggest suitable prevention strategies for the economical important avian
pathogen (Mohamed, 2010).
Previously, the CAV putative intergenotypic recombinants have
been reported. The fact was based on the previous classification of CAV
sequences into three genotypes. Together with a variety of computational
recombination detection algorithms, phylogenetic analysis was used to
investigate CAV approximately full genomes. Significant evidence of
intersubtype recombination was detected in the parent-like and two
putative CAV recombinant sequences. The event was shown to occur
between CAV subgroup A1 and A2 sequences in the phylogenetic trees. It
was revealed that intersubtype recombination in CAV genome sequences
played a role in generating genetic diversity within the natural population
of CAV (Eltahir et al., 2011).
Cancer is a complex progressive multistep disorder that results
from the accumulation of genetic and epigenetic abnormalities, which
show the way to the transformation of normal cells into malignant
derivatives. There were few examples of therapies leading to cure the
cancer. Still cancer remained to be one of the largest causes of death
worldwide. The strategies were analysed which were designed to induce

14
tumor-selective death such as the use of oncolytic virus, tumoricidal
proteins (NS1, Eorf4, apoptin, HAMLET (human α-lactalbumin made
lethal to tumor cells)) and activation of signaling pathways which were
involved in tumor surveillance. The potential of the tumor necrosis factor-
related apoptosis-inducing ligand (TRAIL) pathway, was emphasized. An
essential component of the evolutionary developed defense systems that
eradicated malignant cells. The necessity of targeting tumor-initiating cells
(TICs) was discussed to avoid relapse and increased the chances of
complete remission. The emerging concepts that might provide novel
avenues for cancer therapy were also described (Pavet et al., 2011).
2.2 Apoptosis
The transformed cancer cells were more resistant to apoptosis, so
the ability to survive was enhanced in them. They exhibited resistance to
anticancer drugs, they no longer depend on survival signals, and they
could metastasize. Therefore, cancer progressed as the cancer cells
maintained the proliferative supremacy they acquired from their
oncogenes. In other words, when cancer cells became resistant to
apoptosis, they became resistant to treatment, metastasized, and
proliferated destructively (Kataoka and Tsuruo, 1996).
Kerr, (2000) observed a curious form of liver cell death during his
studies of acute liver injury in rats, and his further study led to an increase
in understanding the role of apoptosis in embryogenesis,
spermatogenesis, cancer growth, and tissue remodelling during healing or
functional regression.

15
Defects in the regulation of apoptosis played crucial role in the
pathogenesis and progression of most cancers and leukemias. Apoptosis
defects figured high resistance to radiotherapy, chemotherapy, hormonal
therapy, and immune-based treatments. Apoptosis was caused by
activation of intracellular proteases, known as caspases that were
responsible for the morphologic and biochemical events that
characterized the apoptotic cell. Numerous proteins which regulated the
cell death proteases have been discovered. Those proteins included
proteins belonging to the Bcl-2, inhibitor of apoptosis, caspase-associated
recruitment domain, death effector domain, and death domain families
(Reed, 2004).
Ghobrial et al. (2005) also defined apoptosis, or programmed cell
death as a mechanism by which cells undergo death to control cell
proliferation or in response to DNA damage. The basis for novel targeted
therapies that could induce death in cancer cells or sensitize them to
establish cytotoxic agents and radiation therapy could be provided by the
understanding of apoptosis. These novel agents include those targeting
the extrinsic pathway such as tumor necrosis factor-related apoptosis-
inducing ligand receptor 1, and those targeting the intrinsic Bcl-2 family
pathway such as antisense bcl-2 oligonucleotides. Numerous pathways
and proteins controlled the apoptosis machinery. Examples included were
p53, the nuclear factor kappa B, the pathway of ubiquitin/proteosome
and the pathway of phosphatidylinositol 3 kinase. These could be
targeted by specific modulators like bortezomib, and mammalian target of
rapamycin inhibitors like CCI-779 and RAD 001. Because these pathways

16
might be preferentially altered in tumor cells, there was potential for
selective effect in tumors sparing the normal tissue.
Apoptosis was known to be coupled to multiple signalling
pathways. Identification of vital points in the regulation of apoptosis was
of major interest both for the understanding of control of cell fate and for
the discovery of new pharmacological targets, mainly in oncology.
Indeed, defects in the implementation of apoptosis were known to
participate in tumour initiation and progression as well as in
chemoresistance. The Bcl-2 family members represented essential
intracellular players in the apoptotic mechanism. Those proteins were
either pro or anti-apoptotic, they interacted with each other to control
apoptosis. Inhibiting the heterodimerisation between pro- and anti-
apoptotic members was sufficient to promote apoptosis in mammalian
cells. Small molecules, antagonists or peptidomimetics inhibiting this
heterodimerisation, represented a therapeutic prototype targeting the
apoptotic cascade. They induced cell death by activating directly the
mitochondrial apoptotic trail (Mazars et al., 2005).
Contrary to earlier assumptions, Bcl-2 and Bcl-xL inhibited apoptin-
induced cell death in several tumor cell lines. In contrast, deficit of Bax
conferred resistance, whereas Bax expression sensitized the cells to
apoptin-induced death. Cell death induction by apoptin was linked with
cytochrome c release from mitochondria as well as with caspase-3 and -7
activation. Benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone, a broad
spectrum caspase inhibitor, was very protective against apoptin-induced
cell death. Apoptosis induced by apoptin required Apaf-1, as

17
immortalized Apaf-1-deficient fibroblasts as well as tumor cells devoid of
Apaf-1 were strongly protected. Thus, data revealed that apoptin-induced
apoptosis was not only Bcl-2- and caspase dependent, but also occupied
an Apaf-1 apoptosome-mediated mitochondrial fatality pathway (Burek
et al., 2006).
The molecules that destroyed cancer cells selectively were: apoptin,
viral protein R (VpR), E4orf4 and Brevinin-2R. Apoptin's cancer-selective
toxicity depended on its nuclear localization both nuclear import and
export (Maddika et al., 2006).
Plasmodium falciparum in a subset of patients could guide to cerebral
malaria (CM), a major provider to malaria-associated mortality. Despite
treatment, CM mortality could be as high as 30%, while 10% of survivors
of the disease might experience short and long-term neurological
complications. The pathogenesis of CM was mediated by alterations in
cytokine and chemokine homeostasis. The hypothesis for this study was
that CM-induced changes in inflammatory, apoptotic and angiogenic
factors. Their recognition enabled the development of new prognostic
markers and adjunctive therapies for preventing CM mortalities (Jain et
al., 2008).
Anticancer drug-induced tumor suppression might involve
mechanisms of protection against neoplastic transformation that were
normally dormant in mammalian cells and consisted in a genetic program
implemented during anti-tumoral defense. This defense program
consequenced in the self elimination of cells harboring potentially
dangerous mutations by triggering cell death through apoptosis and/or

18
autophagy or in the implementation of a program that lead to a
permanent growth arrest known as senescence. These responses were
considered vital tumor suppressive mechanisms and their study appeared
to be necessary to develop therapeutical events based on the enhancement
of the different responses. Interesting evidence showed that different
drugs induced senescence or cell death depending on the genetic features
of the tumor cells as well as on the integrity of the relative pathways
(Chiantore et al., 2009).
While investigating the mechanisms underlying cell death during
wound healing processes, the pro-apoptotic effects of basic fibroblast
growth factor (bFGF) on granulation tissue fibroblasts following
pretreatment with transforming growth factor (TGF)-beta1 in vitro were
undiscovered. Fibroblasts that had been treated and originated from the
uninjured dermis did not show apoptosis, indicating that the
mechanisms underlying apoptosis events in fibroblasts that originate
from normal dermal and wound tissues differ. In this process, it was
established that bFGF inhibited Akt phosphorylation at serine 473 and
induced a rapid failure of phosphorylation of focal adhesion kinase
(FAK) at tyrosine 397 in pretreated GF-1 cells, an incident that coincided
with the dissociation of phosphorylated FAK from the focal adhesions.
Therefore, inhibition of survival signals relayed via the disrupted focal
adhesion structures and inactivated Akt following bFGF treatment might
lead to apoptosis in GF-1 cells pretreated with TGF-beta1. Pretreatment
of GF-1 with TGF-beta1 followed by adding of bFGF resulted in
significantly enhanced inhibition of phosphorylation of Akt and FAK
compared to treatment with TGF-beta1 or bFGF alone. The combinatorial

19
treatment also led to proteolysis of FAK and inhibition of FAK and Akt
protein expression in GF-1 cells. These conclusions demonstrated
important role for the two cytokines in apoptosis of granulation tissue
fibroblasts during wound healing. In vivo studies also established a
marked decline in phosphorylation and protein expression of Akt and
FAK in bFGF-injected skin wounds. These consequences led to the
assumption that temporal activation of TGF-beta1 and bFGF at the injury
site promoted apoptosis in granulation tissue fibroblasts, an event that
was vital for the termination of proliferative granulation tissue formation
(Akasaka et al., 2010).
For chronic hyperglycemia, INS-1 cells were cultured for 5 days
with changes of RPMI 1640 medium containing 33 mM glucose every 12
hours. For irregular hyperglycemia, the medium containing 11 mM
glucose was exchanged with the medium containing 33 mM glucose every
12 hours. Apoptosis was assessed by cleaved caspase 3 and TUNEL assay
Hoechst staining. The expression of Mn-SOD and Bcl-2 was measured by
Western blotting and insulin secretory capacity was assessed. In
comparison to the control group, INS-1 cells uncovered to chronic
hyperglycemia and intermittent hyperglycemia showed an increase in
apoptosis. The apoptosis of INS-1 cells uncovered to irregular
hyperglycemia increased considerably more than the apoptosis of INS-1
cells exposed to chronic hyperglycemia. In comparison to the control
group, the insulin secretory capacity in the two hyperglycemic states was
decreased, and more with intermittent hyperglycemia than with chronic
hyperglycemia. The expression of Mn-SOD and Bcl-2 increased more with
chronic hyperglycemia and less with intermittent hyperglycemia.

20
Intermittent hyperglycemia induced a higher degree of apoptosis and
decreased the insulin secretory capacity more in pancreatic beta cells than
chronic hyperglycemia. This activity might be mediated by the anti-
oxidative enzyme Mn-SOD and the anti-apoptotic signal Bcl-2 (Kim et al.,
2010).
Lin et al. (2010) reported that apoptosis might be closely involved
in diabetes-induced embryonic malformations. The occurrence of
apoptosis at an early stage of development, in oocytes and 2-cell embryos
of streptozotocin-induced diabetic mice and nondiabetic mice was
investigated. Reduced number of growing follicles and delayed oocyte
development were seen when diabetic mouse ovarian sections were
stained with hematoxylin and eosin. Annexin V-positive oocytes were
higher in number in diabetic mice than in nondiabetic mice. Quantitative
RT-PCR and immunofluorescence analysis revealed the expression of Bax
and caspase-3 considerably lesser in nondiabetic oocytes and higher in
diabetic oocytes. Annexin V-positive staining was not seen in 2-cell
embryos of diabetic and nondiabetic mice. Bax expression was elevated in
diabetic 2-cell embryos, but caspase-3 expression did not considerably
differ between diabetic and nondiabetic 2-cell embryos. The results
suggested that maternal diabetes might increase oocyte apoptosis by a
Bax-caspase-3 pathway to play a role in embryonic malformations by late
oocyte development. Development of 2-cell embryos might be favorably
affected through Bax-regulated caspase-3 apoptotic pathway.
Sensitizing radioresistant tumours by combining irradiation with
other therapeutics to induce apoptosis have been widely investigated. It

21
was examined whether chicken anemia virus-derived apoptin protein
would have a useful effect on irradiation of radiosensitive SCC61 and
radioresistant SQD9 human head and neck squamous carcinoma cell
lines. In both cell lines, simultaneous exposure to irradiation and apoptin
resulted in mitochondrial cytochrome c discharge and in cleavage of
caspase-3, whereas irradiation alone of SQD9 cells under identical
conditions did not. Only the synchronized treatment of apoptin and
irradiation resulted in increased cell death in comparison with the
irradiation, especially in the radioresistant SQD9 cells, as measured
through colony survival assay. The data was found to show that apoptin
treatment represented an effective way for enhancing radiotherapy of
tumors responding badly to radiotherapy (Schoop et al., 2010).
Silicosis was a chronic lung disease which was characterized by
granulomatous and fibrotic lesions, which occured due to accumulation of
respirable silica mineral particles. Caspase activation played a central role
in the execution of apoptosis. Silica-induced apoptosis of the alveolar
macrophages could potentially favor a proinflammatory state, which took
place in the lungs of silicotic patients. This result in the activation of
caspase prior to induction of the intrinsic and extrinsic apoptosis
pathways. In recent studies, it was indicated that apoptosis might involve
in pulmonary disorders. In addition, caspase could be a key apoptotic
protein that can be used as an effective biomarker for the study of
occupational diseases. It might provide an important link in
understanding the molecular mechanisms of silica-induced lung
pathogenesis (Tumane et al., 2010).

22
Semiconductor nanoparticles conjugated to photosensitizers have
been shown to increase tumor cell death with ionizing radiation. In an
in vitro system, a molecular probe was used to quantify the component of
photodynamic cell-killing. The intracellular distribution of the
nanoparticle conjugate (NC) was determined by the co-localization of
nanoparticles and the lysotracker. TUNEL assay and western blot analysis
were used to measure the induction of apoptosis of the cleaved caspase-3.
As a result, dose-dependent production was observed with 48 nm
nanoparticle conjugate after irradiating with 6 MV x-rays. A high
geometrical coincidence between the fluorescence emission of the
nanoparticle and lysotracker was observed using confocal microscopy.
Apoptosis, as indicated by the cleavage of caspase-3 and TUNEL stain,
was observed in cells treated by both the nanoparticle conjugate and 6 Gy
of radiation but not in cells treated with radiation alone. The cell death
induced by the nanoparticle conjugate in combination with radiation was
consistent with a supra-additive effect to radiation or nanoparticle
conjugate alone-killing. It was mediated by a nanoparticle conjugate-
induced photodynamic therapy mechanism, which was distinctly
different from that for radiation-killing alone. By providing a second
distinct cell-killing mechanism, this nanoparticle conjugate has great
promise as a targeted physical radiosensitizer aimed at overcoming
radioresistant tumor clonogens or/and reducing normal tissue toxicity by
using a lesser ionizing radiation dose (Wang et al., 2010).
Pancratistatin was a natural compound extracted from
Hymenocallis littoralis, which could selectively induced apoptosis by
various pathways. It showed marked effectiveness on cancer cells.

23
Apoptosis was one of the mechanisms, which detached the cells that were
infected with pathogens or with abnormal cell cycle (Patel and Prajapati,
2011).
2.3 Apoptin
Acetylation of p53 was indispensable for its transcriptional activities
and induction of apoptosis upon DNA damage. Chromatin remodelling
protein SMAR1 inhibited p53 acetylation and p53 dependent apoptosis by
repressing p300 expression in reaction to DNA damage. The repression of
p300 expression by SMAR1 was relieved upon treatment with
proteosomal inhibitors MG132 and Lactacystin. SMAR1 interacted with
p53-p300 transcriptional complex and SMAR1 overexpression
antagonized p300 interaction with p53 and suppressed activation of p53
apoptotic targets and p53 regulated miRNA miR-34a. On the contrary,
knockdown of SMAR1 promoted p300 accumulation and p53 acetylation
while ectopic expression of p300 rescued SMAR1 inhibition on p53.
Collectively, these results indicated that SMAR1 was an important player
in p300-p53 regulated DNA damage signalling pathway and could exert
its effect on apoptosis in a transcription independent manner (Sinha et al.,
2012).
Several viral gene products affect apoptosis by interacting directly
with components of the highly conserved biochemical pathway which
regulated cell death. On the one hand it appeared that viruses blocked
apoptosis to prevent premature death of the host cell and so maximized
virus progeny from a lytic infection or facilitate a persistent infection. On

24
the other hand it appeared that a growing number of viruses actively
promoted apoptosis; viruse may perform both functions, the latter being
the culmination of a lytic infection and serving to spread virus progeny
to neighbouring cells while evading host inflammatory responses.
Apoptosis may then contribute to the cytotoxicity associated with virus
infections (O'Brien, 1998). Apoptin also induced apoptosis in human
osteosarcoma cells, despite of whether they expressed wild-type, mutant
p53, or not even p53. Furthermore, the nuclear position of apoptin
appeared to be important for its most favorable induction of apoptosis.
The reality that apoptin can induce p53-independent apoptosis in human
tumor cells made apoptin a potential candidate for treatment of
frequently occurring types of cancer cells that do not contain functional
p53 (Zhuang et al., 1995).
Pietersen et al. (1999) explained the generation and characterization
of an adenovirus vector, AdMLPvp3, for the expression of apoptin.
Infection with AdMLPvp3 of normal rat hepatocytes in cell culture did
not increase the frequency of apoptosis. On the contrary, in the hepatoma
cell lines Hep3b and HepG2, infection with AdMLPvp3, but not with
control vectors, led to a rapid induction of programmed cell death.
Experiments in rats have shown that AdMLPvp3 could be safely
administered by subcutaneous, intraperitoneal or intravenous injection.
Repetitive intravenous doses of AdMLPvp3 were also well tolerated,
showing that the apoptin-expressing virus can be administered without
severe adverse effects. In an introductory experiment, a single
intratumoral injection of AdMLPvp3 into a xenogeneic tumor (HepG2
cells in Balb/Cnu/nu mice) resulted in a significant reduction of tumor

25
growth. Taken together, the data demonstrated that adenovirus vectors
for the expression of the apoptin gene might constitute a powerful tool for
the treatment of solid tumors.
The presence of caspases was an important component of the
apoptotic machinery present in mammalian cells. By means of a specific
antibody, active caspase-3 was observed in cells expressing apoptin and
undergoing apoptosis. Although apoptin activity was not impacted by
CrmA, p35 inhibited apoptin-induced apoptosis, as observed by nuclear
morphology. Cells expressing apoptin and p35 displayed only a slight
change in nuclear morphology. Though, in most of these cells,
cytochrome c was usually released and the mitochondria were not stained
by CMX-Ros, showing a drop in mitochondrial membrane potential.
These observations implied that although the final apoptotic events were
blocked by p35, apoptin activated parts of the upstream apoptotic
pathway that affected mitochondria. Considering together, these data
illustrated that the viral protein apoptin employed cellular apoptotic
factors for induction of apoptosis. Although initiation of upstream
caspases was not required, initiation of caspase-3 and possibly also other
downstream caspases were vital for rapid apoptin-induced apoptosis
(Danen-van Oorschot et al., 2000). Apoptosis was a very common and
the best understood mechanisms of physiological cell death. It results
from the activation, through any of two primary pathways, of site-specific
proteases called caspases. There were many other routes to cell death,
prominently like autophagy and proteasomal degradation of critical
constituents of cells. Most commonly, autophagic or proteasomal
degradation was used to purge massive cytoplasm of very large cells,

26
particularly post-mitotic cells, and these pathways were prominent even
though caspase genes, messages, and pro-enzymes were found in the
cells. These types of cell death were entirely physiological and not just a
default pathway for a defective cell and they were distinct from necrosis
(Lockshin and Zakeri, 2002).
In order to study the antitumor effect of VP3 protein, particularly
its effect against liver carcinoma in vivo, control vector pcDNA3 and
recombinants pcDNA-vp3 having chicken anemia virus vp3 gene were
mixed with murine liver carcinoma cell lines H22 respectively. The
mixture was inserted subcutaneously into Balb/C mice. After some days,
the mice died and the solid tumor weighed. The antitumor efficiency was
examined. The manners of vp3 protein in vivo inducing tumor cell death
were recognised by using TUNEL assay. All the consequences suggested
that the injection of pcDNA-vp3 and H22 mixture resulted in a major
reduction of tumor growth in mice in comparison with the results of
control groups. TUNEL assay showed that vp3 induced apoptosis in vivo.
All these reflected that cav vp3 might be a promising new gene in
reducing the growth rate of tumor cells in liver carcinoma or in other kind
of solid tumors in vivo (Shen et al., 2003).
Through the use of PCR technique, the vp3 gene of CAV was
cloned into the eukaryotic expression vector pcDNA3 to make a
recombinant pcDNA-vp3. Restriction enzyme digestion and sequencing
analysis showed that cav vp3 gene was correctly inserted into the blank
vector pcDNA3. After LipofectAMINE-mediated transfection in vitro with
pcDNA-vp3 and pcDNA3 respectively, the total mRNA was taken out

27
from liver carcinoma cell lines HepG2 and diploid cell line L-02, and RT-
PCR was conducted afterward. The consequences of RT-PCR showed that
vp3 gene was expressed in these two cell lines. At the same time, using in
situ apoptotic finding assay, TUNEL kits, the apoptotic cells were
observed in pcDNA-vp3 transfected HepG2, but not in mock transfected
cell lines. VP3 could cause cell death by apoptosis in cancer cell lines, but
not in diploid cell lines. All the results showed that cav vp3 gene, a
potential therapeutic agent, has the potential to be used for cancer
treatment (Sun et al., 2003).
Apoptin formed distinct, stable multimeric complex which
was remarkably homogeneous and uniform, when it was produced in
bacteria as a recombinant fusion with maltose-binding protein (MBP-
Apoptin). By the use of cytoplasmic microinjection, recombinant MBP-
Apoptin multimers retained the characteristics of the ectopically expressed
wild-type apoptin; viz., the complexes translocated to the nucleus of
tumor cells and induced apoptosis, though they remained in the
cytoplasm of normal, primary cells and exerted no noticeable toxic effect.
In normal cells the MBP-Apoptin formed gradually large, organelle-sized
globular bodies with time postinjection and ultimately lost the ability to be
detected by immunofluorescence analysis. Costaining with an
acidotrophic marker showed that these globular bodies did not
correspond to lysosomes. Immunoprecipitation studies reflected that
MBP-Apoptin remained entirely antibody-accessible despite of buffer
stringency when microinjected into tumor cells. On the contrary, in
normal cells, MBP-Apoptin was only recoverable under stringent lysis
conditions, while under milder conditions they became entirely shielded

28
with time on two epitopes spanning the entire protein. Moreover
biochemical analysis found that the long-term fate of apoptin protein
aggregated in normal cells was their eventual removal. The outcome
showed that the tumor-specific apoptosis-inducing aggregate was
basically sequestered by factors or conditions present in the cytoplasm of
healthy, nontransformed cells. This characteristic should make known
more about the cellular interactions of this viral protein as well as further
improve its safety as a potential tumor-specific therapeutic agent (Zhang
et al., 2003).
A series of biomedical studies on apoptin have been carried out in
human cell systems, which were revealing about the mechanism of CAV-
induced apoptosis in chicken (transformed) cells. Apoptin contained a
bipartite nuclear localization signal, and one domain that resembled a
nuclear export signal. Elucidation of parts of the apoptin-induced
apoptotic pathway exposed unique characteristics: apoptin-induced
apoptosis was independent of the tumor suppressor p53. The anti-
apoptotic protein Bcl-2 did not inhibit but even accelerated apoptin-
induced apoptosis in tumor cells, while over expression of Bcl-2 in normal
cells has no effect on the apoptin activity. Numerous novel proteins were
shown to interact with apoptin in transformed cells (Noteborn, 2004). It
appeared to have innate tumour-specific p53-independent that Bcl-2-
enhanced pro-apoptotic activity, and therefore might be of great value in
the endeavor to achieve specific and efficient removal of cancer cells,
particularly in cases of drug resistance through Bcl-2 overexpression or
loss of p53 function etc. Apoptin has ability to localise specifically in the
nucleus of transformed but not normal cells. The latter ability,

29
importantly, appeared to be integrally related to its tumour-specific pro-
apoptotic action (Oro and Jans, 2004).
Apoptin was generated and cloned into several mammalian
expression vectors. Microinjection or transfection of apoptin cDNA
resulted in its expression, in the cytoplasm with a filamentous pattern.
apoptin entered the nucleus and efficiently induced apoptosis in several
cancer cell lines. For induction of apoptosis, nuclear localization was
shown to be required. Apoptin expression level was found to be an
important determinant of the efficiency of induction of apoptosis.
Expression of apopti or GFP-apoptin cDNA induced apoptosis in some
normal cells. When fused to the HIV-TAT protein transduction domain
and delivered as a protein, TAT-apoptin was transduced efficiently
(>90%) into normal and tumor cells. However, TAT-apoptin remained in
the cytoplasm and did not kill normal 6689 and 1BR3 fibroblasts. In
contrast TAT-apoptin migrated from the cytoplasm to the nucleus of Saos-
2 and HSC-3 cancer cells resulting in apoptosis after 24 hrs. Apoptin was a
powerful apoptosis-inducing protein with a potential for cancer treatment
(Guelsen et al., 2004).
VP3's anti-cancer activity was strongly linked to its ability to
localize more efficiently in the nucleus of cancer and transformed cells
with a tumor cell-specific nuclear targeting signal located at the C-
terminus of the protein. The VP3 tumor cell-specific nuclear targeting
signal was an exciting prospect to improve non-viral-mediated cancer
cell killing. The understanding of the mechanism responsible for VP3
tumor-specific nuclear localization, including its specific

30
phosphorylation, and the implications for the improvement of anti-
cancer treatment have been discussed. It also proposed alternative
strategies to develop tumor cell-specific nuclear targeting signal for anti-
cancer therapies (Alvisi et al., 2006).
Li et al. (2006) constructed a recombinant fowlpox virus
expressing the apoptin protein (vFV-Apoptin) and compared the tumor-
killing activity of the recombinant virus with that of wild-type fowlpox
virus in the human hepatoma cell line HepG2. It was observed that
although cells were somewhat resistant to the basal cytotoxic effect of
wild-type fowlpox virus, infection with vFV-Apoptin caused a
pronounced, additional cytotoxic effect. Also, cell death and disruption of
tumor integrity were noticeable in the vFV-Apoptin-infected cells. They
also tested whether fowlpox virus-mediated expression of apoptin in
tumor cells could stimulate an antitumor effect by injecting aggressive
subcutaneous tumors derived from H22 mouse hepatoma cells in
C57BL/6 mice with vFV-Apoptin. They observed that fowlpox virus-
mediated intratumoral expression of the apoptin gene can induce
protective and therapeutic antitumor effects and significantly increase
survival. Taken together, these data indicated that infection of tumors
with fowlpox virus expressing apoptin inhibited tumor growth, induced
apoptosis and might be an effective cancer cure.
Neoplastic transformation was the consequences of viruses which
could produce viral oncoproteins that drive multiple genetic alterations.
Viral proteins encoded by onco-related viruses such as Epstein-Barr virus
or polyomavirus SV40 were involved in cellular processes, which resulted

31
in the imbalance between proliferation and cell death. Viruses also
generated viral components that could become a tumor-selective
destroyer by sensing the cellular tumorigenic hallmarks. Apoptin protein
which was derived from the avian virus has been proven to induce
tumor-regression in various pre-clinical animal models without showing
noticeable side effects. Studies were described with representative viral
elements that have contributed to the understanding of significant
tumorigenic processes and have conferred an impact on the growth of
novel anti-cancer therapies (Kooistra et al., 2007).
Lee et al. (2007) investigated the antitumor effect of CAV VP3 gene
in canine mammary tumor (CMT) cells and established primary canine
cell lines that arose from epithelial cells of resected CMTs and
nonneoplastic mammary gland epithelial (MGE) cells. Expression vectors
and lentiviral vectors encoding the VP3 gene from a Taiwan-Ilan isolate of
cav were used to deliver the VP3 gene into CMT cells and nonneoplastic
MGE cells. Ectopic gene expression and the pro-apoptotic effect of the
VP3 gene on CMT and nonneoplastic MGE cells by either viral infection
or transfection were evaluated via western blot analysis,
immunofluorescence microscopy and terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling analysis. Overexpression of
the enhanced green fluorescent protein with VP3 fusion protein was
detected predominantly in the nuclei of CMT cells. In nonneoplastic MGE
cells, the VP3 protein was found to be localized in cytoplasm. Among the
fusion protein-expressing CMT cells, nearly all underwent characteristic
changes of apoptosis, whereas apoptosis was not detected in fusion
protein-expressing, nonneoplastic MGE cells. Induction of apoptosis by

32
VP3 gene overexpression in CMT cells was linked with the caspase-9
mediated apoptosis pathway. The data indicated that the VP3 gene of
CAV did not induce apoptosis in nonneoplastic canine MGE cells but it
induced apoptosis in malignant CMT cells. The VP3 gene might be a
promising agent on the basis of such tumor cell-specific killing, for the
treatment of malignant mammary gland tumors in dog.
Purification of cav vp3 protein which was expressed in a
prokaryotic expression system was established as histidine-tagged fusion
protein. DNA was extracted from the infected liver of chicken and CAV
particle was obtained. By polymerase chain reaction (PCR), the vp3 gene
was amplified from the extracted DNA. This extracted DNA was then
cloned. The recombinant expression construct (pTrc-VP3) was recognized
by PCR and sequencing analysis. The vp3 expressed protein with a
molecular mass of about 21 kDa was confirmed by Western blotting
analysis with cav-specific antibodies. The in vitro expressed VP3 protein
was purified to near homogeneity by elution from the gel, as elucidated
by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis.
The purified VP3 protein was recognized by CAV antibodies in a Western
blotting assay. This finding indicated that recombinant VP3 expressed in
the pTrcHis2 vector system can be used as antigen to detect anti-cav
antibodies (Nogueira-Dantas et al., 2007).
Apoptin interacted directly with the promyelocytic leukemia protein
(PML) intumor cells. Apoptin was accumulated in PML nuclear bodies
(NBs), which were involved in apoptosis induction and viral replication.
Apoptin was sumoylated and that a sumoylation-deficient

33
apoptin mutant was no longer recruited to PML-NBs, but localized in
the nuclear matrix. This mutant failed to attach PML, but could still
induce apoptosis as proficiently as wild-type apoptin.
Moreover, apoptin killed PML cells and promyelocytic leukemia cells
with defective PML expression. The results recommended that apoptin
killed tumor cells independently of PML and sumoylation (Janssen et al.,
2007).
In immunocompetent mice, the effects of apoptin expression in
primary oral tumors were induced by the carcinogen 4-Nitroquinoline-1-
oxide. In vivo, a significant amount of primary oral tumor cells
expressing apoptin cells underwent apoptosis, while synthesis of the
LacZ control product did not. Ectopical expression of apoptin in passage
1 cell cultures derived from these oral tumors also resulted in apoptin-
induced. Both in-vitro and in-vivo treated cells underwent apoptosis via
the activation of caspase-3. Apoptin proved to be a potential therapeutic
agent for the cure of head and neck squamous cell carcinoma from the
fact that apoptin induced apoptosis in primary squamous cell carcinoma
cells (Schoop et al., 2008).
Apoptin phosphorylation, nuclear translocation, and apoptosis
colud transiently be induced in normal cells by cotransfecting SV40 large
T oncogene, indicating that apoptin recognizes early stages of oncogenic
transformation. Apoptin appeared to recognize survival signals in cancer
cells which it was able to redirect into cell death impulses. Apoptin targets
included DEDAF, Nur77, Nmi, Hippi, and the potential drug target
APC1. Apoptin-transgenic mice and animal tumor models have revealed

34
apoptin as a safe and proficient antitumor agent, resulting in signifacant
tumor regression. Future antitumor therapies could use apoptin either as
as an early sensor of druggable tumor-specific processes or as a curative
bullet (Backendorf et al., 2008).
It was studied that multiple genes regulated the initiation and
progression of tumor. Survivin belonged to the inhibitor of apoptosis
protein (IAP) family and was overexpressed in most types of human
tumors. The expression of surviving was silenced by microRNA
(miRNA)-mediated RNA interference (RNAi) and the engineered miRNA
vector was also designed to express apoptin gene. The apoptosis and cell
growth were then examined by MTT assay and flow cytometry. The
miRNA-mediated knockdown of survivin in combination with apoptin
overexpression significantly induced apoptosis and inhibited cell growth.
The combined strategy was more effective on inducing apoptosis and
inhibiting cell growth than either survivin downregulation or apoptin
overexpression only. Taken together, the combined strategy offered
potential advantages in control of tumorigenesis. The combined stategy
offer great advantages in control of tumorogenesis and so deserve further
research as a preferred approach in cancer gene therapy (Liu et al., 2008).
In tumor cells, apoptin caused the nuclear accumulation of
survival kinases including Akt and was phosphorylated by CDK2.
Apoptin redirected survival signals into cell death responses. Apoptin
was also find to bind as a multimeric complex to DNA and interacted
with several nuclear targets, such as the anaphase-promoting complex,
resulting in a G2/M phase arrest. The proapoptotic signal of apoptin was

35
then transduced from the nucleus to cytoplasm by Nur77, which
triggered a p53-independent mitochondrial death pathway. Discoveries
of apoptin's mechanism of action that might provide intriguing insights
for the development of novel tumor-selective anticancer drugs were
described (Los et al., 2009).
The abundantly expressed carcinoembryonic antigen (CEA) on
several cancer types was an attractive target for antibody-directed
therapy. CEA was also present in some normal tissues. It was observed
that a dual functioning protein, designated as CAtin exhibited both
specific binding and killing functions, by fusing a tumor-specific
apoptosis-inducing molecular apoptin to C-terminus of an anti-CEA
single-chain disulfide-stabilized Fv antibody (Yan et al., 2010).
The recombinant lentivirus containing the fused gene of SP-TAT-
Apoptin was packaged to infect HepG2 cell and the efficiency of
apoptosis was measured. The eukaryotic expression vector of SP-TAT-
Apoptin fused gene and other packaging plasmids were transfected into
293FT cells by Lipofectamine(TM);2000 reagent. The supernatant of the
cultured 293FT cells was harvested and real time PCR determined the
virus titration. The expression of the fused gene of SP-TAT-Apoptin in
293FT cells infected by the recombinant lentivirus was examined by
immunofluorescence histochemistry method. Flow cytometer was used to
determine the apoptosis rates of the HepG2 cells infected by the
recombinant lentivirus. The 293FT cells infected by the recombinant
lentivirus could express the fused protein SP-TAT-Apoptin. Annexin-V PI
assay showed that SP-TAT-Apoptin carried by the recombinant lentivirus

36
could cause the HepG2 cell apoptosis, and its apoptosis rate was
considerably more than paired control group and SP-TAT-Apoptin
carried by liposomes only. The recombinant lentivirus of SP-TAT-Apoptin
was successfully packaged and it could induce HepG2 cells to apoptosis
(Han et al., 2010).
A lentiviral vector was developed which encoded a green
fluorescent protein-apoptin fusion gene (LV-GFP-AP). It could efficiently
deliver apoptin into hematopoietic cells. Apoptin selectively killed the
human multiple myeloma cell lines MM1.R and MM1.S, and the leukemia
cell lines K562, HL60, U937, KG1, and NB4. The normal CD34(+) cells
were not killed and maintained their differentiation potential in
multilineage colony formation assays. Dexamethasone-resistant MM1.R
cells were found to be more susceptible to apoptin-induced cell death
than the parental matched MM1.S cells. Death susceptibility was
connected with increased phosphorylation and activation of the apoptin
protein in MM1.R cells. Expression array profiling identified differential
kinase profiles between MM1.R and MM1.S cells. Among these kinases,
protein kinase Cβ (PKCβ) was found by immunoprecipitation and in vitro
kinase studies to be a candidate kinase responsible for apoptin
phosphorylation. Apoptin-mediated cell death proceeded during the
upregulation of PKCβ, cleavage of the PKCδ catalytic domain, activation
of caspase-9/3 and downregulation of the MERTK and AKT kinases. The
results elucidated a novel pathway for apoptin activation involving PKCβ
and PKCδ. It was highlighted that the potential of apoptin and its cellular
regulators was used to purge bone marrow in autologous transplantation
for multiple myeloma (Jiang et al., 2010).

37
In mice, therapeutic effect of adenovirus-mediated apoptin gene
transfer combined with ADM and CDDP on hepatocellular carcinoma
was studied. In c57BL/ 6 mice bearing hepatocellular carcinoma, the
changes of tumor volume, tumor inhibition rate, histomorphology and the
side effects were observed after intratumoral injection of adenovirus
containing apoptin gene, ADM and CDDP. After the treatment of seven
days, the mean volume of the tumor in the mice receiving intratumoral
apoptin-containing adenovirus injection combined with ADM and CDDP
reduced considerably as compared with that in mice treated with
adenovirus vehicle and control group. The tumor inhibition rate in the
combined treatment group was 90.13%, significantly higher than that in
the control group. No adverse effect of the treatment was observed in the
course of the experiment. The adenovirus vectors containing apoptin gene
combined with ADM and CDDP might serve a safe treatment of
hepatocellular carcinoma (Liu et al., 2010).
Li et al. (2010) generated a conditional replication-competent
adenovirus (CRCA), designated as Ad-hTERT-E1a-Apoptin, and
investigated the effectiveness of the CRCA, a gene therapy agent for the
clinical trials. The observation that infection with Ad-hTERT-E1a-Apoptin
significantly supperessed the growth of melanoma cells, protecting
normal human epidermal melanocytes from growth inhibition confirmed
cancer cell selective adenoviral replication, apoptosis induction and
growth inhibition of this therapeutic approach. The ability of the
recombinant adenoviruses was evaluated to prolong the survival of the
tumor bearing mice. When treated with Ad-hTERT-E1a-Apoptin, the
subcutaneous primary tumor volume reduction was observed in

38
intratumoral injection group as well as in systemic delivery mice.
Pulmonary metastatic lesions were effectively suppressed by Ad-hTERT-
E1a-Apoptin, in the lung metastasis model. Mice survival was increased
by the treatment of primary and metastatic models with Ad-hTERT-E1a-
Apoptin. The study showed the engineered CRCA induced apoptosis
selectively in various cancer cells without adverse effects on normal cells.
It was also observed that an adenovirus expressing apoptin was more
effective and advocated the potential applications of Ad-hTERT-E1a-
Apoptin in the treatment of neoplastic diseases in upcoming clinical trials.
It was revealed from structural modeling of pDED-HIPPI that R393
of HIPPI was important for such interaction. R393E mutation in pDED-
HIPPI decreased the interaction with the putative promoter of caspase-1
in cells. Expression of caspase-1 was decreased in cells expressing mutant
pDED-HIPPI in comparison to that observed in cells expressing wild
type pDED-HIPPI. Using HIP-1 knocked down cells as well as over
expressing HIP-1 with mutation at its nuclear localization signal and
other deletion mutations, translocation of HIPPI to the nucleus was
mediated by HIP-1 for the increased expression of caspase-1. HIPPI-HIP-
1 heterodimer was detected in cytoplasm as well as in the nucleus and
was associated with transcription complex in cells. Taking together, the
importance of R393 of HIPPI and the role of HIPPI-HIP-1 heterodimer in
the transcription regulation of caspase-1 were shown (Banerjee et al.,
2010).
The development of molecular technologies has allowed the
elucidation of the multiple mutations and dysregulatory effects of

39
pathways leading to tumorogenesis. Specifically targeted agents
represented a major challenge for current research efforts in oncology to
act against these pathways. As conventional anatomically based
pharmacological endpoints might be inappropriate to monitor the tumor
response to these targeted treatments, the recognition and use of more
appropriate, noninvasive pharmacodynamic biomarkers appeared to be
crucial to optimize the design, dosage and schedule of these novel
therapeutic approaches. An aberrant choline phospholipid metabolism
and enhanced flux of glucose derivatives via glycolysis, which sustained
the redirection of mitochondrial ATP to glucose phosphorylation, were
two main hallmarks of cancer cells. The perspectives of present efforts
addressed to recognize enzymes of the phosphatidylcholine cycle as
possible novel targets for anticancer treatment were summarized (Podo
et al., 2011).
Using yeast two-hybrid and immunoprecipitation approaches, it
was revealed that apoptin interacted with heat shock cognate protein 70
(Hsc70). Apoptin induced the translocation of endogenous Hsc70 in vivo,
from the cytoplasm to the nucleus, and both were co-localized in the
nucleus. In addition, apoptin induced Akt phosphorylation, which was
markedly repressed by Hsc70 knockdown, signifying that Hsc70 might
play a critical role in Apoptin-induced Akt phosphorylation. These
observations helped further to understand the molecular mechanism of
apoptin (Chen et al., 2011).
Recent understanding of the molecular pathogenesis of cancer has
led to the development of targeted novel drugs such as small molecule

40
inhibitors, monoclonal antibodies, mimetics, antisense and small
interference RNA-based strategies, among others. These compounds
acted on specific targets that were believed to contribute to the
development and progression of cancers. Whether delivered individually
or combined with chemo- and/or radiotherapy, such novel drugs have
produced significant responses in certain types of cancer. Among the
most successful novel compounds were those which targeted tyrosine
kinase. However, these compounds could cause severe side-effects as
they inhibited pathways such as epidermal growth factor receptor
(EGFR) or platelet-derived growth factor receptor, which were also
important for normal functions in non-transformed cells. The toxicity by
various proteins was independent of tumor type or specific genetic
changes such as p53, pRB or EGFR aberrations. It showed a current
overview of a selection of proteins with preferential toxicity among
cancer cells and which provide an insight into the
possible mechanism of action, tumor specificity and their potential as
novel tumor-specific cancer therapeutics (Argiris et al., 2011).
Resistance to radiation therapy was one of the hallmarks of
hepatocellular carcinoma. To induce apoptosis, sensitizing radioresistant
cancer by combining radiation with other therapeutics was widely
investigated. The earlier study showed that chicken anemia virus-derived
apoptin protein induced the apoptosis of hepatic carcinoma HepG. It was
observed that apoptin sensitized the cells to radiation-induced apoptosis
using a lentivirus-apoptin expression system in hepatic carcinoma HepG2
cells. The combination of apoptin and radiation up-regulated the p53
expression. Thus, apoptin treatment represented a potential method for

41
enhancing the effectiveness of radiotherapy in badly responding
hepatocellular carcinoma (Han et al., 2011).
Apart from viral approaches, some nonviral approaches have also
been used widely for intracellular gene transfer and gene therapy. A
modified wheat histone H4 protein was able to form stable complexes
with plasmid DNA which increased gene delivery efficiency. H4TL has
been used to deliver apoptin gene into a human ovarian carcinoma cell
line HO8910. Increased expression of apoptin at both mRNA and protein
levels was detected after transfection in HO8910 cells, accompanied by
reduced rate of growth of HO8910 cells in vitro and the loss of
mitochondrial membrane potential in these cells. These data showed that
H4TL-mediated transfer of apoptin initiated mitochondrial death
pathway in ovarian cancer cells and suggested a novel therapeutic
strategy for cancer (Wang and Zhang, 2011).
CAV protein apoptin has the intriguing activity of inducing G(2)/M
arrest and apoptosis specifically in cancer cells by a mechanism that was
independent of p53. At the level of localization, the activity of apoptin
was regulated. The study of apoptin was therefore a unique system for
identifying pathways of apoptosis that were able to kill cancer cells
independent of p53. DNA damage response (DDR) signaling was
required to induce apoptin nuclear localization in primary cells.
Induction of DNA damage in combination with apoptin expression was
able to induce apoptosis in primary cells. Chemical or RNA interference
(RNAi) inhibition of DDR signaling by ATM and DNA-dependent
protein kinase (DNA-PK) was sufficient to cause apoptin to localize in

42
the cytoplasm of transformed cells. The nucleocytoplasmic shuttling
activity of apoptin was required for DDR-induced changes in
localization. The nuclear localization of apoptin in primary cells was able
to inhibit the formation of DNA damage foci containing 53BP1. Apoptin
has been shown to bind and inhibited the anaphase-promoting
complex/cyclosome (APC/C). It was observed that apoptin was able to
inhibit formation of DNA damage foci by targeting the APC/C-
associated factor MDC1 for degradation. These results might point to a
novel mechanism of DDR inhibition during viral infection (Kucharski et
al., 2011).
Panigrahi et al. (2012) calculated a 3D structure of apoptin and
through modeling and docking approaches, they showed its interaction
with Bcr-Abl oncoprotein and its downstream signaling components,
which confirmed some of the newly-found interactions by biochemical
methods. Bcr-Abl oncoprotein was unusually expressed in chronic
myelogenous leukaemia (CML). It had several distinct functional domains
in addition to the Abl kinase domain. The calculated structural
coordinates of apoptin was used to study its interaction with the 3D
structure of CML-associated oncoprotein Bcr-Abl. The SH3 and SH2
domains cooperatively played important roles in autoinhibiting its kinase
activity. Adapter molecules such as Grb2 and CrkL interacted with
proline-rich region and activated multiple Bcr-Abl downstream signaling
pathways that contributed to growth and survival. Therefore, the
oncogenic effect of Bcr-Abl could be inhibited by the interaction of small
molecules with these domains. For therapeutic applications, especially for
CML treatment, Bcr-Abl was an attractive target for rational drug design.

43
Some of the interactions that were first predicted in silico were also
biochemically validated. This structure-property relationship of apoptin
might help in unlocking its cancer-selective toxic properties.
The antibacterial peptide cecropin B mutant (ABPS1) gene has a
broad range of antiproliferative and antibacterial properties. To explore
drug combination in human tumor cells, apoptin and ABPS1 eukaryotic
expression vector pIRES2-EGFP-apoptin and pIRES2-EGFP-ABPS1 were
constructed and their expression effect individually and in combinations
were studied in HepG2 and A375 cells. ABPS1 (23.79% in HepG2 and
8.33% inA375 cells). Moreover, the co-expression of apoptin and ABPS1
showed higher apoptotic rates which were 27.66 and 10.33% in HepG2
and A375 cells respectively. The apoptotic rates obtained in HepG2 cells
treated with ABPS1 and apoptin together were closely similar, but, not in
A375 cells. The results of the study indicated that the combination of
ABPS1 and apoptin had synergistic effect in HepG2 and A375 cell lines
(Birame et al., 2012).
2.4 Apoptin from other viruses
Studies with apoptin and E4orf4 have shown that they induced
G2/M arrest by targeting and inhibiting the anaphase-promoting
complex/cyclosome (APC/C). These two viral proteins induced
apoptosis selectively in transformed cells in a p53-independent manner;
thus pathways affected by these proteins were of significant therapeutic
interest. Further study of the underlying mechanism of G2/M arrest and
subsequent apoptosis induced by viral APC/C inhibitors might shed light
on the mechanisms of current cancer therapies and provide the

44
foundation for developing novel therapeutic targets (Heilman et al.,
2005).
A putative Torque teno viruses (TTVs) open reading frame (ORF) in
TTV genotype 1, named TTV apoptosis inducing protein (TAIP), induced,
like apoptin, p53-independent apoptosis in various human
hepatocarcinoma cell lines to a similar level as apoptin. In comparison to
apoptin, TAIP action was less pronounced in several analyzed human
non-hepatocarcinoma-derived cell lines. Detailed sequence analysis has
revealed that the TAIP ORF was conserved within a limited group of the
heterogeneous TTV population. Its N-terminal half, N-TAIP, was well
conserved in a much broader set of TTV isolates. The similarities between
apoptin and TAIP, and their relevance for the development and treatment
of diseases were discussed (De Smith and Noteborn, 2009).
The correlation between Torque teno virus (TTV) and CAV was
examined. Each had a circular single-stranded (ss) DNA genome with
each one of its known open reading frames (ORF) on its antigenomic
strand. The structure was found to be distinct from those of circoviruses.
TTV and CAV had different genomic sizes which were found to be 3.8 kb
and 2.3 kb, respectively. It was seen that the spectrum of the TTV genome
was enormously diverse and the CAV genome was quite narrow. The
relative allocation of ORFs on each frame in these viruses mimiced each
other. Three or more messenger RNA (mRNAs) were generated by
transcription in both of them. In each virus, the structural protein with the
replicase domain was coded by frame 1, and a nonstructural protein with
a phosphatase domain was coded by frame 2. In transformed cells,

45
apoptosis was induced by a protein on frame 3 in each virus (Hino and
Prasetyo, 2009).
Pseudotype baculovirus was developed as an alternative gene
delivery system. It was used as a vector to express apoptin. The resultant
recombinant baculovirus (BV-Apoptin) efficiently expressed the apoptin
protein and induced apoptosis in HepG2 and H22 cells. Studies in vivo
indicated that intratumoral injection of BV-Apoptin into a xenogeneic
tumor which was derived from H22 murine hepatoma cells in C57BL/6
mice, considerably suppressed tumor growth. It also significantly
prolonged the survival of tumor-bearing mice compared to a control
pseudotype baculovirus that expressed EGFP. The results signified that
apoptin which was expressed from the pseudotype baculovirus vector,
had the potential to become a therapeutic agent for the treatment of solid
tumors (Pan et al., 2010).
Sauvage et al. (2011) identified a new virus in a skin swab sample
from a healthy donor which was named human gyrovirus (HGyV)
because of its similarity to CAV, the only previously known member of
the Gyrovirus genus. This virus encoded a homolog of the CAV apoptin, a
protein that selectively induced apoptosis in cancer cells. The genome of
HGyV presented an overall organization similar to that of CAV. By PCR
screening, HGyV was found in 5 of 115 other nonlesional skin specimens
but in 0 of 92 bronchoalveolar lavages or nasopharyngeal aspirates and in
0 of 92 fecal samples. HGyV might also serve as a tool to decipher
pathways of apoptosis in cancer cells.

46
2.5 DNA Damage Signaling
Polycomb group (PcG) proteins were major determinants of cell
identity, epigenetic gene silencing and stem cell pluripotency during
development. The polycomb repressive complex 1, which contained
BMI1, RING1, and RING2, functions as an E3-ubuiquitin ligase. BMI1 and
RING2 were recruited to sites of DNA double-strand breaks (DSBs) where
they contributed to the ubiquitylation of γ-H2AX. In the absence of BMI1,
several proteins dependent on ubiquitin signaling, including 53BP1,
BRCA1, and RAP80, were impaired in recruitment to DSBs. Loss of BMI1
sensitized cells to ionizing radiation to the same extent as loss of RNF8.
An additive increase in radiation sensitivity was revealed by the
simultaneous depletion of both proteins. The data suggested a possible
role for BMI1 in the pathways of DNA damage response (Ismail et al.,
2010).
Using a synthetic lethal RNA interference screen in human cells,
Cyclin-dependent kinase 9 (CDK9) was identified as a component of the
replication stress response. An increase in spontaneous levels of DNA
damage signalling in replicating cells was caused by the loss of CDK9
activity. This activity was restricted to CDK9-cyclin K complexes and was
independent of CDK9-cyclin T complex. CDK9 accumulated on chromatin
in response to replication stress and limited the amount of single-stranded
DNA in cells under stress. Cyclin K and CDK9 interacted with ataxia
telangiectasia, Rad3-related protein and other checkpoint signalling
proteins. These results revealed an important role for CDK9-cyclin K in
checkpoint pathways that maintained genome integrity in response to
replication stress (Yu et al., 2010).

47
Tumor cells were found as more resistant to apoptosis that were
induced by cytotoxic agents, than normal cells. Resistance to apoptosis
induction could be a direct result of mutations in certain tumor-
suppressor genes (p53) or to certain proto-oncogenes (Bcl-2). Therefore,
new cancer therapies were under development to bypass the resistance to
chemo- and radio-therapy of tumors. Apoptin acted independently of
p53, was stimulated by Bcl-2 and was insensitive to BCR-ABL, which
means that apoptin can induce apoptosis in cases where (chemo)-
therapeutic agents fail. The fact that apoptin induced apoptosis in human
tumorigenic cells but not in normal diploid cells, implied that side-effects
of apoptin treatment were expected to be minor. In-vivo results with anti-
tumor therapy based on expression of apoptin indicated that apoptin had
low acute toxicity and was successful as an anti-tumor agent (Pietersen et
al., 2000).
From the above literature it is evident that apoptosis inducing potential of
apoptin can be advantageously utilized in antitumor therapy.

Materials and Methods
3.1 Materials
3.1.1 Vector
3.1.2 Gene
3.1.3 Primers
3.1.4 Host Bacterial Strains
3.1.5 Cell Culture
3.1.6 Conjugates
3.1.7 Hyper-immune Serum
3.1.8 Experimental Animals
3.1.9 Glass wares, Plastic wares and Chemicals
3.1.10 Solutions and Buffers
Chapter-3

3.2 Methods
3.2.1 Revival of the E. coli culture containing recombinant plasmid with VP3 gene
3.2.2 Isolation of plasmid DNA by TELT method
3.2.3 Agarose gel electrophoresis
3.2.4 Digestion of the recombinant plasmid with restriction enzyme to release the gene insert
3.2.5 DNA extraction from agarose gel
3.2.6 Blunting of EcoRI generated VP3 gene staggered ends
3.2.7 Purification of blunted insert
3.2.8 Creation of cloning site in vector
3.2.9 Dephosphorylation of 5’ ends
3.2.10 Purification of dephosphorylated linearized vector
3.2.11 Blunt end ligation of pSin vector and VP3 gene
3.2.12 Transformation of E. coli (DH5α) cells with the ligated product
3.2.13 Screening of recombinant clones for VP3 gene in right orientation
3.2.14 Transfection of HeLa cells

3.2.15 Immunoperoxidase test for DNA expression
3.2.16 Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
3.2.17 DNA fragmentation assay by agarose gel electrophoresis
3.2.18 Caspase 3 detection assay
3.2.19 Annexin V binding assay

48
Chapter-3
Material & Methods
3.1 Materials
3.1.1 Vector
The pSin vector (Fig. 1) is derived from an alpha virus (Sindbis
virus). The subgenomic promoter of alpha virus is very strong so that it
makes large number of target mRNA from the sequence downstream to
it. In comparative studies of conventional (nonreplicating) plasmid DNA
vectors and alphavirus DNA-based replicon vectors, the latter generally
produce larger quantity of DNA concentrations than do conventional
vectors.
3.1.2 Gene
The VP3 gene of CAV used in this study was cloned in pTarget
vector in Biotechnology laboratory of IBIT, Bareilly.
3.1.3 Primers
Primers used in the study were synthesized from Chromous
Biotech. (India).
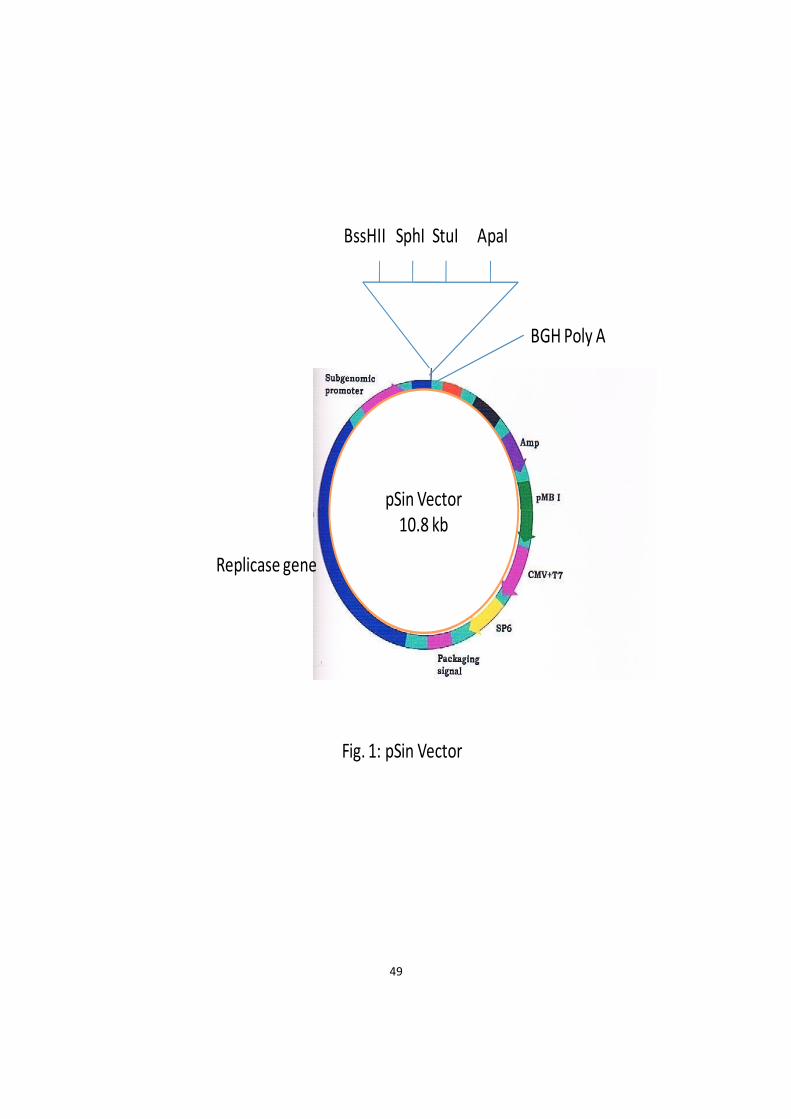
49
pSin Vector
10.8 kb
BssHII SphI StuI ApaI
BGH Poly A
Replicase gene
Fig. 1: pSin Vector

50
Table: 1 Primers with their sequences
S. No. Primer Primer sequence
1. VP3 F 5’ATGAACGCTCTCCAAGAAG3’
2. BGH R 5’TAGAAGGCACAGTCGAGG 3’
3.1.4. Host Bacterial Strains
Escherichia coli (E. coli) DH5α (Proteges, Madison) host strain was
used for transformation with recombinant plasmids.
3.1.5. Cell Culture
HeLa cell lines were obtained from National Centre for Cell
Science (NCCS), Pune. These cell lines were used in the study of
apoptotic activity for recombinant plasmid and was maintained in
DMEM (Gibco, NY) supplemented with 50 µg/ml gentamycin (Amresco,
USA) and 10% fetal calf serum (Hyclone, USA).
3.1.6. Conjugates
Rabbit anti-mouse HRP conjugated antibody was obtained from
Bangalore Genei, Bangalore, (India).
3.1.7. Hyper-immune serum
Anti cav.vp3 hyperimmune sera were raised in mice using
recombinant plasmid by hyper immunization of six mice with
pSin.cav.vp3 plasmid. Fifty µg of plasmid DNA was injected
intramuscularly in lateral region of thigh muscles of each mouse and

51
repeated every week for four weeks consecutively. One week after last
injection, mice were bled through inner canthus of eyes with a capillary
and serum was prepared.
3.1.8. Experimental Animals
Swiss albino mice, 4 weeks old, were procured and kept in animal
room for raising hyper immune sera.
3.1.9. Glasswares, Plasticwares and Chemicals
All the glasswares used were Borosil make. Plasticwares were
from Axygen, Greiner or Eppendorf. All the chemicals used in the study
were procured from Fermentas (UK), Promega, Bangalore Genei
(Bangalore, India), Medox or Qiagen (Germany).
3.1.10. Solutions and buffers
The composition of chemical solutions and buffers used in the study
is given in appendix.
3.2 Methods
3.2.1. Revival of the E. coli culture containing recombinant
plasmid with VP3 gene
The E. coli containing pTarget.cav.vp3 was revived in LB broth
overnight at 370C following the protocol of Sambrook and Russell (2001).
The E. coli containing rplasmid was inoculated by streaking on LB agar
plate containing ampicillin at concentration of 100 µg/ml. The plate was

52
incubated at 370C for 24 hrs. A single colony was picked up from the
plate with the help of a loop and then transferred into 5 ml LB broth
containing 100 µg/ml ampicillin. It was kept in incubator at 370C for 24
hrs.
3.2.2. Isolation of plasmid DNA by TELT method (He et al., 1990).
A 1.5 ml E. coli culture was grown with plasmid of interest in LB
medium containing 100 µg/ml ampicillin for 16 hrs. Cells were pelleted
for 30 sec in microcentrifuge. They were resuspended in 100 µl TELT
solution and an equal volume of 1:1 phenol/chloroform was added. The
sample was vortexed vigorously for 15 sec and spin for 1 minute in
microcentrifuge at 220C. The upper phase of nucleic acid was collected
and mixed with 2 vol of 100% ethanol. After 2 minutes, spin 10 minutes
in microcentrifuge at 40C. The pellet was washed with 1ml of 70%
ethanol, allowed to dry and resuspended in 30 µl TE buffer.
3.2.3. Agarose gel electrophoresis
It was done as per method of Sambrook and Russell, (2001). 1% of
agarose was prepared in 1X TAE buffer and heated till it dissolved. The
solution was cooled to about 450C and then ethidium bromide was
added at a concentration of 0.5 µg/ml. Gel tray was sealed with tape on
its open ends and placed the comb on the tray. Then the above gel was
poured on the sealed tray. It was left to solidify. The comb was removed
and taped. The gel tray was put in electrophoretic tank containing 1X
TAE buffer. The DNA marker and sample DNA were loaded into the

53
slots of submerged gel, using bromophenol dye buffer and then the gel
was run at 50-100 Volts.
3.2.4. Digestion of the recombinant plasmid with restriction
enzyme.
Digestion was done to release the gene insert using protocol of
Sambrook & Russell (2001).
The microfuge tube was labeled to set 50 µl digestion reaction:
DNA (pTarget.cav.vp3)(200 µg/ml) 20 µl
(EcoRI) Enzyme (Promega 12 u/µl) 3 µl
Enzyme buffer (10X) 5 µl
Distilled Water 22 µl
The reaction mixture was mixed by tapping the tube. The tube was
centrifuged at 5000 rpm for 15 sec. Digestion mixture was incubated at
370C for 4 hrs or more in incubator. Agarose gel electrophoresis was run
in 1X TAE buffer using 1.5 % agarose to check restriction digestion.
3.2.5. DNA extraction from agarose gel
The gel extraction of DNA fragment was done using Min Elute gel
extraction kit (Qiagen, Germany) following manufacturers instructions.
Briefly, the agarose gel containing DNA fragment was excised with a
scalpel and weighed in a colorless microfuge tube. Three volumes
of buffer QGH were added to 1 volume of gel and incubated at 50˚C until
gel slice had completely dissolved. One gel volume of isopropanol was

54
added to the melted agarose and mixed by inverting several times. The
sample was applied to the Min Elute column fitted in a collection tube.
The column was centrifuged at 13000 rpm for 1 minute and the flow
through was discarded. 500 µl of QG was applied to the spin column,
centrifuged and the flow through was discarded again. The DNA bound
to the column was washed with 750 µl of buffer PE and the flow through
was discarded. The DNA was eluted from the column in 10 µl nuclease
free water.
3.2.6. Blunting of EcoRI generated VP3 gene staggered ends
For blunting of staggered ends generated by EcoRI enzyme, T4
DNA polymerase (Fermentas) was used. Reaction mixture was prepared
as follows:
Table: 2. Reaction mix for blunting
Component Volume(µl)
VP3 Insert 20
T4 DNA polymerase buffer 10X 5
T4 DNA polymerase (5u/µl) 2
dNTPs mix.(10mM each) 1
Nuclease free water 22
Total 50
The reaction mixture was incubated at 37˚C for 10 minutes.

55
3.2.7. Purification of blunted insert
The blunted VP3 insert was purified by phenol chloroform
precipitation following the protocol of Sambrook and Russell, (2001). In
brief, 50 µl of TE buffer was added to the reaction mixture followed by
addition of equal volume of phenol chloroform isoamyl alcohol (25: 24: 1)
i.e. 100 µl for above reaction mixture. It was centrifuged at 13000 rpm for
10 minutes. The aqueous layer was transformed into a different tube.
Again 50 µl of TE buffer was added to the tube containing phenol
chloroform isoamyl alcohol and centrifuged at 13000 rpm for 10 minutes.
Aqueous layer was transferred to the collection tube. 1/10 volume (5 µl)
of 3M sodium acetate was added to the aqueous collection. Then 2.5
times of 100% ethanol was added to it. The tube was kept at -20˚C
overnight. It was centrifuged at 13000 rpm for 20 minutes. Supernatant
was carefully discarded and pellet was washed in 1ml of 70% ethanol by
centrifuging at 13000 rpm for 10 minutes. The dried pellet was then
dissolved in 12 µl of nuclease free water. The presence of purified
blunted DNA was checked by running 1µl of DNA on 1% agarose gel.
3.2.8. Creation of cloning site in vector
StuI was chosen to create blunt end. A reaction mixture was
prepared with StuI enzyme (Promega 10u/ µl) as per the following
protocol to digest the pSin vector plasmid:

56
Table: 3. Reaction mix for StuI digestion
Component Volume (µl)
StuI enzyme (Promega 10u/ µl) 3
pSin vector DNA (100 µg/ ml) 15
NE Buffer (10X) 5
Nuclease free water 27
Total 50
The reaction mixture was incubated at 37˚C overnight. The lineraized
plasmid was checked on 1% agarose gel electrophoresis. The linearized
plasmid was then extracted from gel.
3.2.9. Dephosphorylation of 5’ ends
The calf intestinal alkaline phosphatase (CIAP) was used to remove
5’ phosphate group from both the ends of linearized plasmid. The
following reaction mixture was prepared in 50 µl volume.
Table: 4. Reaction mix for dephosphorylation
Component Volume(µl)
Linearized vector pSin (100 µg/ ml) 10
CIAP enzyme (Promega 1u/ µl) 1
Buffer (10X) 5
Nuclease free water 34
Total 50
The reaction mixture was incubated at 37˚C for 30 minutes.

57
3.2.10. Purification of dephosphorylated linearized vector
The method used was similar to that used in purification of
blunted insert.
3.2.11. Blunt end ligation of pSin vector and VP3 gene (Sambrook
and Russell, 2001).
A 10 µl reaction mixture was standardized with 30% PEG 8000 for
blunt end ligation. The components were mixed in the following
concentrations:
Table: 5. Ligation mixture
Component Volume (µl)
T4 DNA Ligase ((Promega 1u/ µl) 2
pSin vector (100 µg/ ml) 0.5
Cav.vp3 gene (150 µg/ µl) 4
Ligation buffer (10X) 1
30% PEG 8000 (Amresco) 1.5
Nuclease free water 1
Total 10
The reaction was incubated for overnight at 15˚C.
3.2.12. Transformation of E. coli (DH5α) cells with the ligated
product (Chung et al., 1989).
The single step method of competent cell preparation and
transformation was used.

58
A fresh overnight culture of bacteria was diluted into prewarmed LB
Broth and the cells were incubated at 37˚C in shaking incubator. The cells
were pelleted by centrifugation at 1000xg for 10 minutes at 4˚C,
supernatant removed and resuspended at 1/10th of original in ice cold 1X
TSS. The cell suspension was mixed gently. For transformation, a 0.1 ml
aliquot of cells was pipetted into a cold polypropylene tube containing 1
µl of plasmid DNA and the cell/ DNA suspension was mixed gently. The
cell/ DNA mixture was incubated for 5-7 minutes at 4˚C. A 0.9 ml
aliquot of TSS plus 20 mM glucose was added and the cells were
incubated at 37˚C in shaking incubator at 200 rpm for 1 hr. The above
transformed cells were plated on LB agar plates containing appropriate
antibiotic (ampicillin 50 µg/ ml) and the plates were incubated at 37˚C
for 16 hrs.
3.2.13. Screening of recombinant clones for VP3 gene in right
orientation
Few colonies were picked from the overnight grown
transformants. The individual colonies were inoculated in fresh LB broth
containing ampicillin (50 µg/ ml) and allowed to grow for 18 hrs.
Plasmid was isolated from these colonies by TELT method and checked
on 1.5% agarose gel electrophoresis.

59
3.2.13.1. Restriction digestion of plasmid with BglII enzyme to
check the presence and orientation of CAV-VP3 insert
Isolated plasmids were checked for the presence of insert by
digestion with enzyme BglII. The digestion mixture was prepared by
mixing the components in amounts as mentioned below:
Table: 6. Reaction mix for BglII digestion
Component Volume (µl)
Plasmid DNA (pSin.cav.vp3)( 200 µg/ml) 4
BglII. Enzyme (Fermentas 10 u/µl) 1
Buffer (10X) 1.5
Nuclease free water 8.5
Total 15
The reaction mixture was vortexed and spun; and incubated in a water
bath at 370C overnight. The digested mixture was then electrophoresed
in 1.5% agarose. The released fragments after digestion were compared
against 100 bp marker. One BglII site is present in the vector and one site
in the region of cav.vp3 gene. When the gene is in correct orientation,
three fragments were released viz. 5650 bp, 2180 bp and 3321 bp. The
restriction sites were predicted in pSin.cav.vp3 plasmid using DNASTAR
software.
3.2.13.2. Colony PCR
The presence of gene insert in right orientation in the recombinant
plasmid was confirmed by PCR using VP3 gene specific forward primer

60
and BGH as reverse primer. A part of single colony was picked up and
placed in a microfuge tube containing distilled water. The reaction was
carried as follows:
Table: 7. Reaction mix for PCR
Component Volume (µl)
Autoclaved distilled water 33
r colony (pSin.cav.vp3) 5
Forward primer of VP3 (50 pmole/µl) 2
Reverse primer BGH 2
dNTPs (10 mM) 2
Buffer (10X) 5
Taq DNA polymerase (3 units/ µl) 1
Total 50
The VP3 gene was amplified following initial denaturation at 940C for 5
minutes and 30 cycles of denaturation at 940C for 30 seconds, annealing
at 500C for 50 seconds, and amplification at 720C for 50 seconds and final
extension at 720C for 7 minutes. After amplification, an aliquot of 10
µl was subjected to agarose gel electrophoresis along with 100 bp DNA
molecular weight marker through 1.5% agarose gel at 60 volts for the
analysis of PCR product.
3.2.13.3. Sequencing of cloned chicken anemia virus VP3 gene
The recombinant plasmid selected after above two methods was
sent to Chromous Biotech Ltd. (Bangalore), for sequencing using BGH
reverse primer.

61
3.2.14. Transfection of HeLa cells (Sambrook and Russell 2001).
Cell culture was trypsinised using 0.17% trypsin versenate and
then 4 ml of DMEM containing 10% FCS was added. Then 100 µl of cell
culture suspension was placed in each of 96 well microtitre plate. The
calcium phosphate-DNA coprecipitate was prepared as follows: 50 µl of
2.5M CaCl2 was combined with 10 µg of plasmid DNA and 40 µl distilled
water in a sterile microfuge tube. The calcium phosphate-DNA
suspension was immediately transferred into the wells of 96 well
microtitre plate containing the cell suspension. Transfected cells were
incubated at 370C in a humidified chamber with an atmosphere of 5%
CO2. After 72 hrs of incubation, the cells were assayed for expression of
transfected gene.
3.2.15. Immunoperoxidase test for DNA expression (Gerna et
al., 1976).
The cells were rinsed in 96 well microtitre plate with PBS, pH 7.2.
The cells were fixed with 100 µl of chilled acetone at 40C for 10 minutes
and then air-dried. 10 µl of mouse antiapoptin hyperimmune serum was
added and incubated at 370C for 1 hr. Then they were washed with PBS.
10 µl of Rabbit anti- mouse globulin-HRP conjugate was added and
incubated at 370C for 1 hr. The wells were rinsed three times with PBS.
The preparation was air- dried. Nadi reagent was prepared just before
use as follows: 15 mg alpha naphthol and 22 mg phenylenediamine were
added in 20 ml of phosphate buffer. The solution was then filtered and
few drops of H2O2 were added. 100 µl of Nadi reagent was added in each

62
well. It was allowed to react for 5 minutes. The preparation was rinsed in
PBS. Then it was examined under microscope and photographed.
3.2.16. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE)
HeLa cells were transfected in 96 well plates with pSin.cav.vp3
rplasmid and pSin vector in separate wells. After 48 hrs of transfection,
cells were processed for SDS-PAGE following the method of Rodriguez
and Tait (1983). The lower resolving gel was 12% and the upper spacking
gel was 4%. The gel was run at 40 mA and stained with Coomassie
Brilliant Blue R250.
3.2.17 DNA fragmentation assay
HeLa cells showing 60% monolayer were transfected as described
earlier. After 48 hrs of transfection, the monolayer was trypsinized and
collected in a 1.5 ml tube. The cells were centrifuged at 3000 rpm for 5
min. After centrifugation, the media was removed and the cells were
resuspended in 200 µl PBS and the genomic DNA was isolated using
protocol of Sambrook and Russell (2001).
The cell suspension was transferred to a microfuge tube containing
600 µl of ice-cold cell lysis buffer. 3 µl of proteinase K solution was added
to the lysate to increase the yield of genomic DNA. It was incubated for
30 minutes at 370C. It was allowed to cool to room temperature and then
3 µl RNase was added. It was incubated for 30 minutes at 370C. The
sample was allowed to cool at room temperature. 200 µl of potassium

63
acetate solution was added and the contents of the tube were mixed by
vortexing vigorously for 20 seconds. The precipitated protein was
pelleted by centrifugation at 10000 rpm for 3 minutes. The supernatant
was transferred to a fresh microfuge tube containing 600 µl of
isopropanol. The solution was well mixed and then the tube was
centrifuged at 10000 rpm for 1 minute to recovere the precipitated DNA.
The supernatant was removed and 600 µl of 70% ethanol was added to
the DNA pellet. The tube was centrifuged at 10000 rpm for 1 minute. The
supernatant was removed carefully and the DNA pellet was allowed to
dry in air for 15 minutes. The DNA pellet was redissolved in 10 µl of
TE pH 7.6. The eluted DNA was stored at -20C.
The genomic DNA collected by above procedure was subjected to 2%
agarose gel electrophoresis. The bands were visualized under UV light
and photographed.
3.2.18. Caspase 3 detection assay
Activation of caspases plays an important role in apoptosis.
Caspase 3 was detected using CaspGLOW Fluoroscein Active caspase -3
Staining kit (Biovision, USA). The assay utilizes the caspase 3 inhibitor,
DEVD-FMK, conjugated to FITC (FITC-DEVD- FMK) as a marker. FITC-
DEVD- FMK is cell permeable, nontoxic and irreversibly binds to
activated caspase 3 in apoptotic cells. The FITC label allows for direct
detection of activated caspases in apoptotic cells by fluorescence
microscopy. The following protocol was followed.

64
Apoptosis was induced by transfecting HeLa cells with
pSin.cav.vp3 recombinant plasmid as described earlier. After 48 hrs of
transfection, the cells were trypsinized and collected in a 1.5 ml
microfuge tube. The cells were washed in cold PBS and then centrifuged
at 3000 rpm for 5 minutes. The cells were resuspended in 300 µl of PBS
and then 1 µl of FITC-DEVD- FMK was added into each tube and
incubated for 0.5 -1 hr at 370C incubator with 50% CO2. The cells were
centrifuged at 3000 rpm for 5 minutes and the supernatant was removed.
The cells were resuspended in 0.5 ml of wash buffer and centrifuged
again. This step was repeated once. Finally, the cells were resuspended in
100 µl of wash buffer, observed under a fluorescent microscope and
photographed.
3.2.19. Annexin V binding assay
This test was mainly done to detect plasma membrane alteration,
which occurs during apoptosis. In normal liver cells, phosphatidylserine
(PS) is located on cytoplasmic surface of cell membrane. However, in
apoptotic cells, PS is translocated from the inner to the outer leaflet of the
plasma membrane, thus exposing PS to the external cellular
environment. The human anticoagulant, annexin V, is a 35-36-kDCa
dependent phospholipid binding protein that has a high affinity for
PS. Labeled Annexin V can identify apoptotic cells by binding to PS
exposed on the outer leaflet. This test was performed using
Vybrant Apoptosis Assay Kit # 2 (Invitrogen, USA). The kit contains
recombinant annexin V conjugated to Alexa Flour 488 dye. The kit also

65
contains a ready-to-use solution of the red fluorescent propidium iodide
(PI) nucleic acid binding dye. PI is impermeant to live cells and apoptotic
cells, but stains dead cells with red fluorescence, binding tightly to the
nucleic acids cells in the cell.
The apoptosis was induced by transfecting HeLa cells with
pSin.cav.vp3 recombinant plasmid as described earlier. After 48 hrs of
transfection, cells were collected by trypsinization in a 1.5 ml microfuge
tube and washed in cold PBS. Then 1X annexin binding buffer was
prepared by mixing 200 µl of 5X annexin binding buffer to 800 µl of
distilled water to a total volume of 1 ml. A working solution of
propidium iodide (PI) (100 µg/ ml) was prepared by diluting 5 µl of the
1 mg/ml PI stock solution in 45 µl of 1 x annexin binding buffer. The
washed cells were recentrifuged and the supernatant was discarded. The
cells were resuspended in 1 x annexin binding buffer. 5 µl of annexin V
conjugate and 1 µl of 100 mg /ml PI working solution were added to 100
µl of cell suspension. The cells were incubated at room temperature for
15 minutes and then washed with 1x Annexin Binding buffer. The cells
were deposited onto slides, examined under fluorescent microscope and
photographed.

Results
4.1 Cloning of VP3 gene in pSin vector
4.2 Expression of VP3 gene in HeLa cells
4.3 Apoptic activity of VP3 in cell line
Chapter-4

66
Chapter-4
Results
4.1 Cloning of VP3 gene in pSin vector
E. coli cells containing pTarget.cav.vp3 were grown on LB broth
containing ampicillin (100 µg/ml) and plasmid DNA was isolated and
run on agarose gel electrophoresis which yielded good amount of DNA.
The cav.vp3 gene insert was released from pTarget vector by
digesting with EcoRI enzyme (Fig. 2). Two different bands were seen.
Smaller band was of insert DNA and the larger band was of vactor DNA.
The size of smaller band i.e. vp3 gene was 372 bp and the size of larger
band i.e. pTarget vector was 5670 bp. Smaller band (insert) was cut and
ligated to pSin mammalian expression vector. The recombinant DNA was
designated as pSin.cav.vp3. This was transformed in E. coli DH5 alpha
competent cells using single step method of competent cell preparation
and transformation. In this method, bacteria do not require a heat shock
for optimal uptake of plasmid DNA. The transformed cells were plated on
LB agar plates containing ampicillin 50 µg/ ml and the plates were
incubated at 37˚C for 16 hrs. After 16 hrs incubation at 370C, the plates
were found to contain several colonies from which ten colonies were
selected randomly and grown in LB broth containing ampicillin. The

67
recombinant pSin.cav.vp3 plasmids were isolated from these colonies by
TELT method.
The restriction sites predicted in pSin.cav.vp3 plasmid using
DNASTAR software showed that one BglII site is present in the pSin
vector and one site in the region of cav.vp3 gene. When the gene was in
correct orientation, three fragments were expected viz. 5650 bp, 2180 bp
and 3321 bp. The digestion of pSin.cav.vp3 with BglII yielded three
fragments of expected size (Fig. 3).
Colony PCR using VP3 gene specific forward primer and BGH as
reverse primer yielded 372 bp PCR product showing that the insert was in
right orientation (Fig. 4). The recombinant plasmid selected after above
two methods was sequenced using BGH reverse primer and sequence
confirmed that the gene was in right orientation and the sequence was
also homologous to VP3 gene.

68
M 1
Fig. 2 : Digestion of recombinant plasmid (pTarget.cav.vp3) with restriction enzyme (EcoRI) to release the vp3 gene insert.
Lane M : 100 bp marker
Lane 1 : pTarget vector 5670 bp and cav.vp3 gene 372 bp.
372 bp
5670 bp

69
M 1
Fig. 3: The digestion of pSin.cav.vp3 with BglII yielded three fragments of
expected size.
5650 bp
4.0 kb
3.0 kb
2.0 kb
5.0 kb
3321 bp
2180 bp
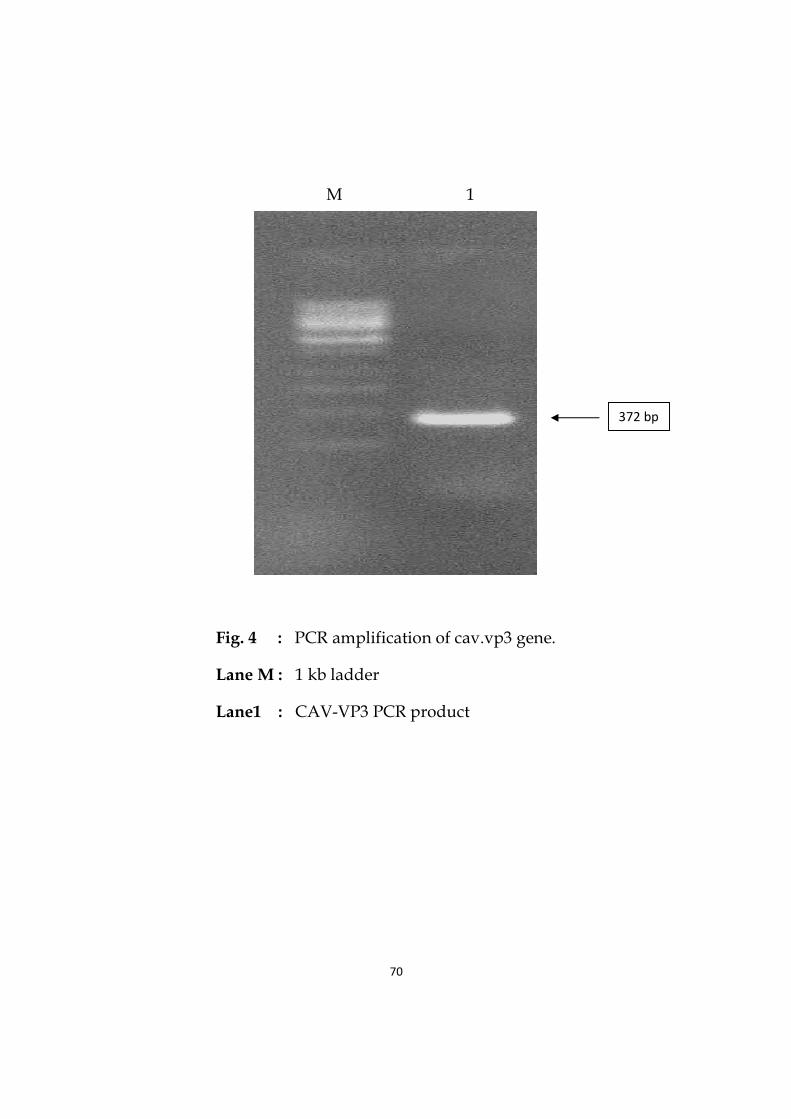
Fig. 4 :
Lane M :
Lane1 :
70
M 1
: PCR amplification of cav.vp3 gene.
: 1 kb ladder
: CAV-VP3 PCR product
372 bp

71
4.2. Expression of VP3 gene in HeLa cells
The cav.vp3 gene cloned in pSin verctor was analysed for its
expression in Hela cells using Immunoperoxidase Test (IPT) and SDS-
PAGE.
The transfection was done into the wells of 96 well microtitre plate.
The transfected cells were incubated at 370C in a humidified chamber
with an atmosphere of 5% CO2. After 72 hrs of incubation, the cells were
assayed for expression of transfected gene. The expression was seen by
Immunoperoxidase Test (IPT), also known as Immunoenzyme technique
(IET). In this test, there occurs a purple/ brown color precipitation
reaction. Immunoperoxidase test of pSin.cav.vp3 plasmid transfected
HeLa cells revealed brown color in transfected cells (Fig. 5), which
confirmed the expression of cav.vp3 gene. However, control showed no
such color (Fig. 6). It was also found that more than 80% of the cells in a
microscopic view showed expression.
The SDS-PAGE (Sodium dodecyl sulphate-polyacrylamide gel
electrophoresis) analysis of expressed protein showed a specific band of
size 13 kDa (Fig. 7). Expression of vp3 in HeLa cells by the gene construct
pSin.cav.vp3 was established and the gene construct was used for
evaluating the apoptic potential of vp3 in HeLa cells.
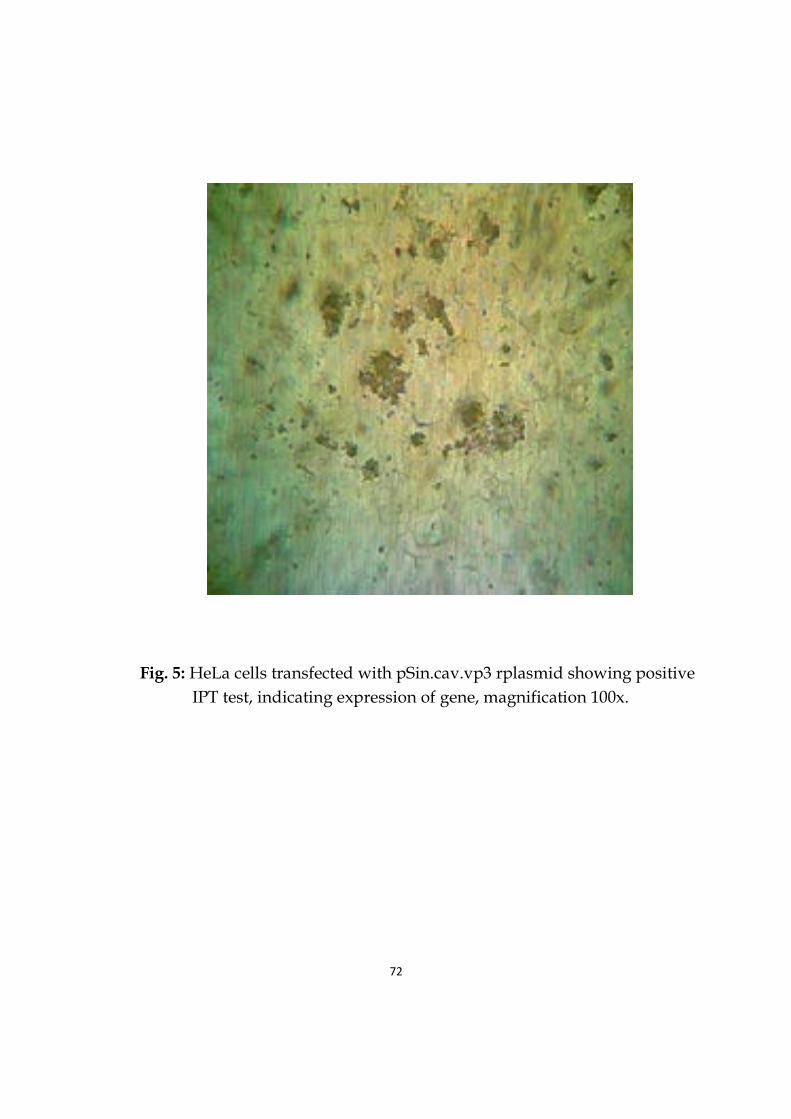
72
Fig. 5: HeLa cells transfected with pSin.cav.vp3 rplasmid showing positive
IPT test, indicating expression of gene, magnification 100x.

73
Fig. 6: Healthy control HeLa cells showing no color reaction, magnification 100x.

74
Fig. 7 : VP3 expressed protein analyzed by SDS-PAGE.
Lane M : Protein molecular weight marker
Lane 1 : 13 kDa expressed protein

75
4.3 Apoptic activity of VP3 in cell line
The apoptosis inducing potential of CAV-VP3 (apoptin) was
evaluated in HeLa cells by the following three assays:
i. DNA laddering assay
ii. Caspase detection assay
iii. Annexin-V-binding assay
DNA laddering assay
In DNA laddering assay, the cleavage of chromosomal DNA occurs
into the oligonucleosomal size fragments, which is an integral part of
apoptosis. When HeLa cells, transfected with rplasmid, were analysed
after 72 hrs, the bands merged and nucleosomal laddering was detected
on agarose gel electrophoresis (Fig. 8) while control cells showed no such
laddering pattern.
Caspase detection assay
The caspases are a family and are one of the main executers of
apoptotic process. These proteins breakdown or cleave key cellular
components that are required for normal; cellular function including
structural proteins in cytoskeleton and nuclear proteins such as DNA
repair enzymes. The caspases can also activate other degrative enzymes
such as DNAses which begin to cleave the DNA in the nucleus. In caspase
detection assay, when HeLa cells transfected with rplasmid, were analysed
after 72 hrs, caspase 3 positive cells showed green fluorescence (Fig. 9 &
Fig. 10). However, control cells showed no such fluorescence.

76
Annexin-V-binding assay
Annexin V was used as a probe in this assay, to detect the cells that
have expressed phosphatidylserine on the cell surface, a feature found
in apoptosis. Phosphatidyl serine at the outer membrane surface of a cell
is a universal process occurring during early apoptosis. Using the
Annexin-V affinity assay, the apoptic cells in suspension was
determined in a fast, simple and sensitive way. Annexin V staining was
specific to apoptic cells and background staining was low in unaffected
cells. In Annexin-V-binding assay, when HeLa cells transfected with
rplasmid were analysed after 72 hrs, apoptic cells showed bright green
fluorescence while control showed yellow fluorescence (Fig. 11).
All these three assays revealed that CAV-VP3 showed good apoptic
activity in cultured cells. It was also observed that majority of the cells
showed apoptic activity.
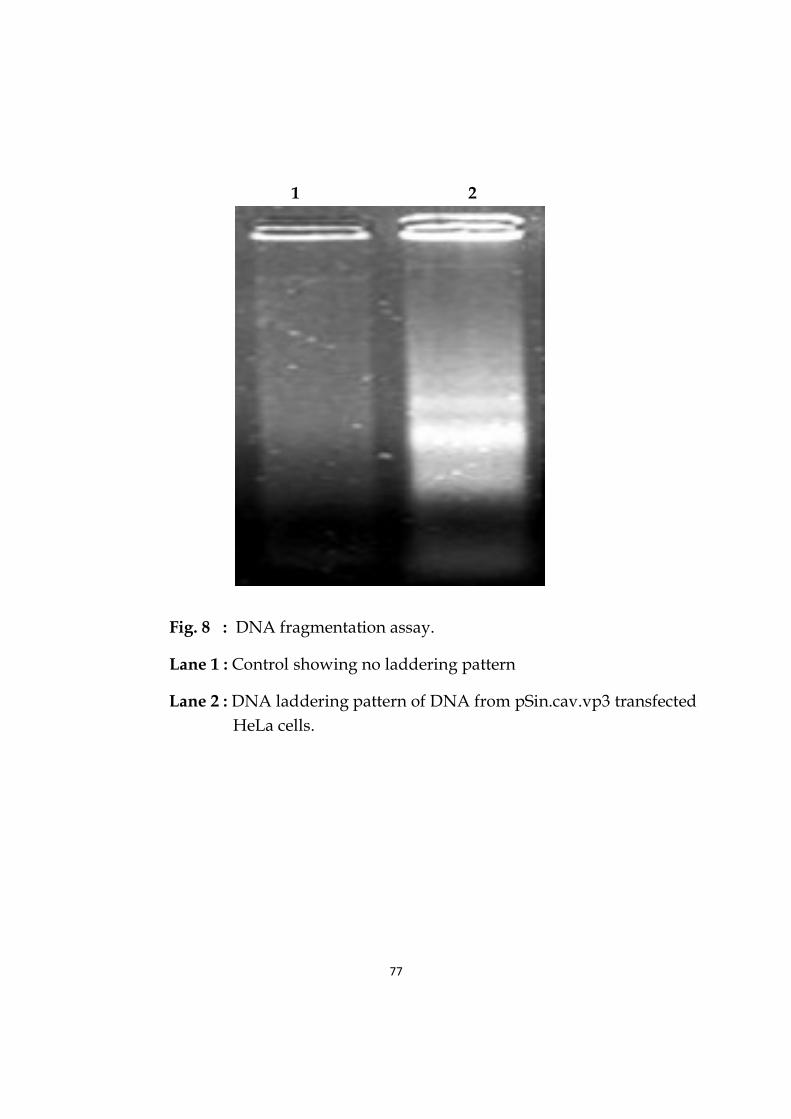
77
1 2
Fig. 8 : DNA fragmentation assay.
Lane 1 : Control showing no laddering pattern
Lane 2 : DNA laddering pattern of DNA from pSin.cav.vp3 transfected
HeLa cells.
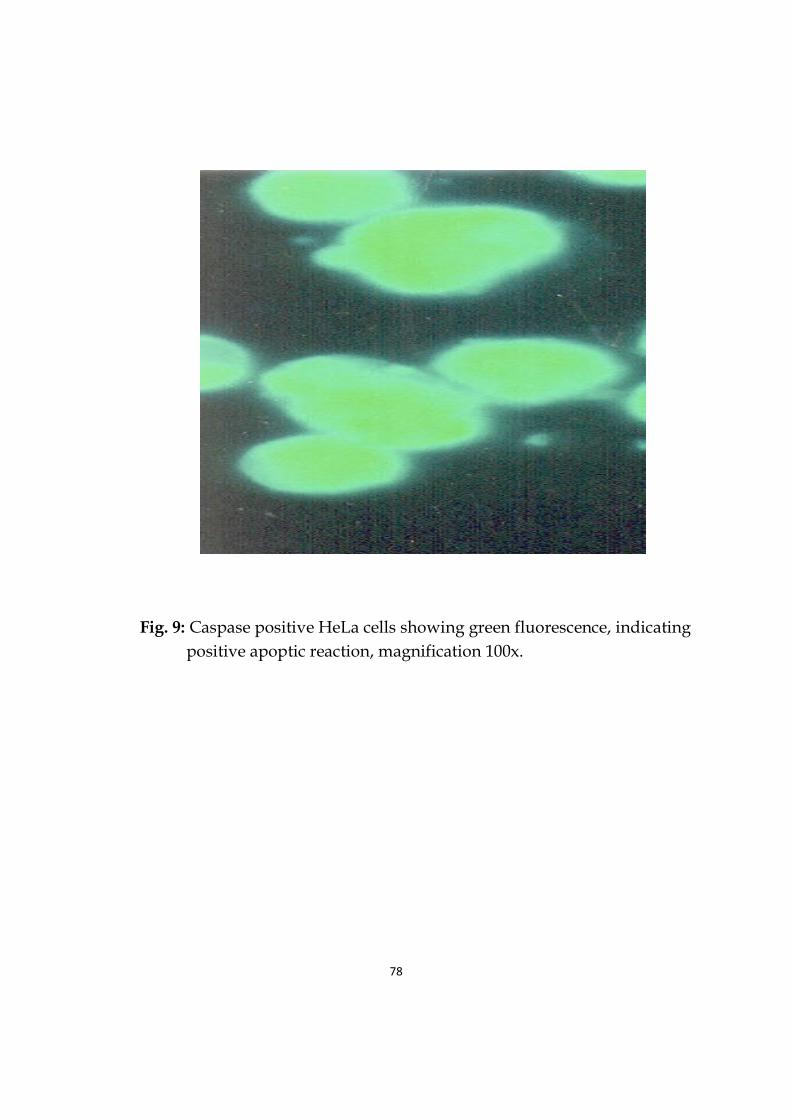
78
Fig. 9: Caspase positive HeLa cells showing green fluorescence, indicating
positive apoptic reaction, magnification 100x.
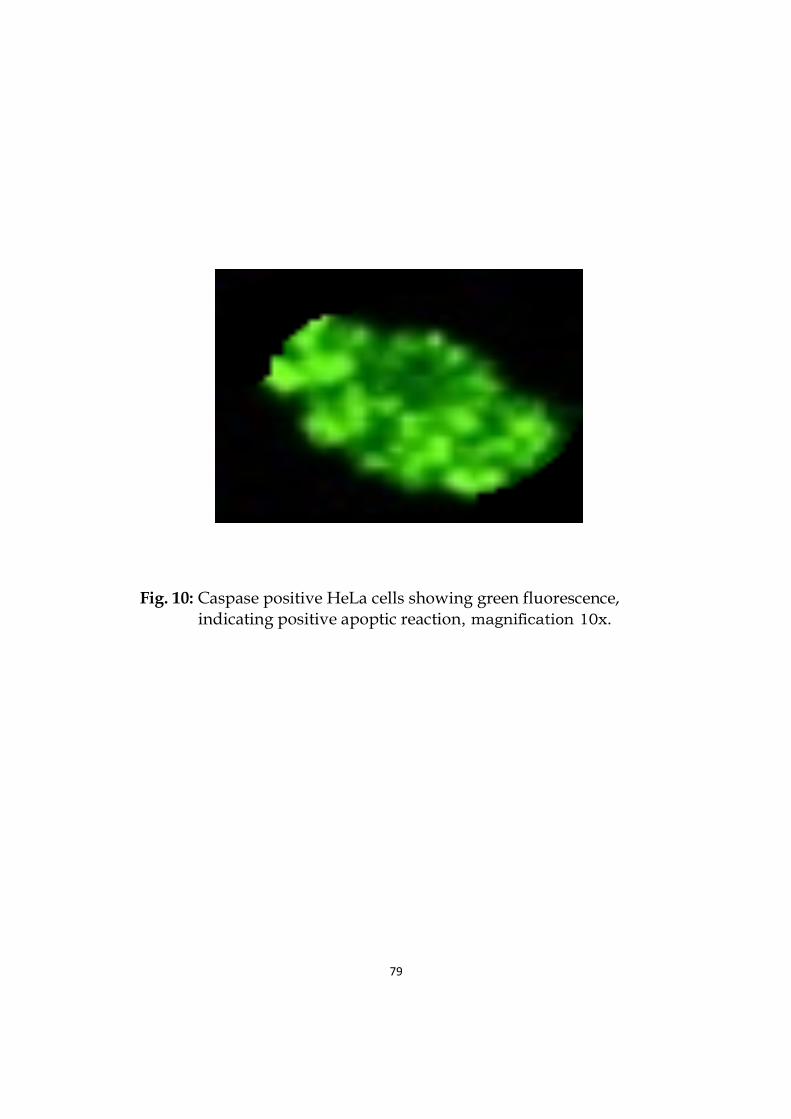
79
Fig. 10: Caspase positive HeLa cells showing green fluorescence, indicating positive apoptic reaction, magnification 10x.

80
Fig. 11: Annexin-V positive HeLa cells showing bright green fluorescence, indicating positive apoptic reaction, magnification 100x

Discussion
Chapter-5

81
Chapter-5
Discussion
The CAV genome has been shown to incorporate three overlapping
open reading frames (ORFs) which code for three viral proteins
designated as VP1, VP2 and VP3, all of which were expressed in cells
infected with CAV (Noteborn and Koch, 1995).
CAV causes severe depletion of some cell types, such as
lymphocytes, by the induction of apoptosis (Noteborn et al., 2004). It
causes cytopathogenic effects in chicken thymocytes and cultured
transformed mononuclear cells through apoptosis. Early after infection of
chicken mononuclear cells, the CAV-encoded protein VP3 exhibited a
finely granular distribution within the nucleus. At a later stage after
infection, VP3 forms aggregated. At this point, the cell became apoptotic
and the cellular DNA was fragmented and condensed. By immunogold
electron microscopy, VP3 was shown to be linked with apoptotic
structures. In vitro, expression of VP3 induced apoptosis in chicken
lymphoblastoid T cells and myeloid cells, which were susceptible to CAV
infection (Noteborn et al., 1994).
The present study, as already explained, was undertaken to clone
the cav.vp3 gene in pSin vector, in vitro expression of VP3 in cultured cells
and the study of apoptotic activity of apoptin (VP3 protein) in cell line.

82
The VP3 gene of CAV was successfully cloned in pSin vector. pSin is
derived from Sindbis virus, which is one of the members of Alphavirus
genus. Several members of the Alphavirus genus, first Sindbis virus
(Bredenbeek et al., 1993; Schlesinger, 1993; Xiong et al. 1989), later
Semliki Forest virus (Berglund et al., 1993; Lilgestorm 1994; Lilgestorm
and Garoff 1991) and other alphavirus members (Davis et al. 1995; Davis
et al., 1989), have received considerable attention for use as virus-based
expression vectors. Typically, these vectors are constructed in a format
known as a replicon, due to the self-amplifying nature of the vector RNA
(Xiong et al., 1989). We have used pSin vector due to the following
reasons. There are many properties which make alphavirus vectors a
desirable alternative to other virus-derived vector systems being
developed. These properties include potential high-level expression of up
to 108 molecules of heterologous protein per cell (Xiong et al., 1989),
infection of nondividing cells and a broad host range (Strauss and Strauss
1994). In addition, replication occurs entirely in the cytoplasm of the
infected cell as RNA molecule, without a DNA intermediate. This is in
contrast to retrovirus and adeno-associated virus vectors, which enter the
nucleus and usually integrate into the host genome for initiation of vector
activity (Jolly 1994; Miller et al., 1993; Samulski et al., 1989). Thus,
retrovirus-associated and adeno-associated virus-derived vectors have
application for long-term expression of foreign proteins, while the
alphavirus vectors are likely better suited for short-term high-level
expression. Furlov et al. (1996) explained the advantages of alphavirus,
which include a broad range of susceptible host cells, high levels of
cytoplasmic RNA and protein expression without splicing the facile

83
construction and manipulation of recombinant RNA molecules using full-
length cDNA clones from which infectious RNA transcripts can be
generated by in vitro transcription. In review of the above, we have seen
the expression of cav.vp3 gene cloned in pSin vector and approximately
more than 80% of cells were found to express the protein. Moreover, it has
the advantage that less amount of DNA is needed to be delivered for
therapeutic purpose.
Earlier cav.vp3 gene was cloned in different vectors by different
authors. Pietersen et al. (1999) used adenovirus vector, AdMLPvp3, for the
expression of apoptin and demonstrated that adenovirus vectors for the
expression of the apoptin gene may constitute a powerful tool for the
treatment of solid tumors. Shen et al. (2003) showed the antitumor effect
of VP3 protein, especially its effect against liver carcinoma in vivo. The
recombinants pcDNA-vp3 containing chicken anemia virus vp3 gene, and
control vector pcDNA3 were mixed with murine liver carcinoma cell lines
H22 respectively. They found that cav vp3 might be a potential new gene
in reducing the growth rate of tumor cells in liver carcinoma or in other
kind of solid tumors in vivo. Nogueira-Dantas et al. (2007) indicated that
recombinant VP3 expressed in the pTrcHis2 vector system can be used as
antigen to detect anti-cav antibodies. Li et al. (2010) generated a
conditional replication-competent adenovirus (CRCA), designated Ad-
hTERT-E1a-Apoptin, and investigated the effectiveness of the CRCA a
gene therapy agent for further clinical trials and observed that infection
with Ad-hTERT-E1a-Apoptin significantly inhibited growth of the
melanoma cells. Jiang et al. (2010) developed lentiviral vector, encoding a
green fluorescent protein-apoptin fusion gene (LV-GFP-AP) that can

84
efficiently deliver apoptin into hematopoietic cells. Saxena et al. (2012)
used pcDNA vector to clone the vp3 gene and observed its oncolytic
property. Birame et al. (2012) constructed eukaryotic expression vector
pIRES2-EGFP-apoptin and pIRES2-EGFP-ABPS1 and observed their
expression effects individually and in combinations with HepG2 and A375
cells. The apoptotic rates obtained in HepG2 cells, treated with apoptin
and ABPS1 together, were closely similar, but, not in A375 cells. Therefore,
the results of their study showed that the combination of apoptin and
ABPS1 has synergistic effect in HepG2 and A375 cell lines.
In the present study, the expression of VP3 gene in HeLa cells was
confirmed by Immunoperoxidase Test (IPT) and SDS-PAGE and thus
indicating that apoptotic action seen in HeLa cells was due to the action of
VP3 protein, which was confirmed by in vitro studies. The
Immunoperoxidase test is easy to detect expressed protein in cell culture.
This test was found to be simple and reliable. It was easy to carry out, it
did not require any special technical equipment and moreover all reagents
were easy to obtain. In SDS-PAGE, the protein of 13kDa was observed.
Earlier expression of vp3 gene was reported by different workers using
different methods like Indirect Immunofluorescence technique (Natesan et
al., 2006). The CAV DNA sequence had three partially overlapping major
reading frames coding for putative peptides of 51.6, 24.0, and 13.6 kDa
(Noteborn et al., 1991). The cell lines MDCC-MSBI and LSCC-HD11 were
transfected with CAV plasmid DNA, the CAV infected cells were anlysed
using immunoblotting and immunoperoxidse assay and the size of VP3
protein was found to be 16 kDa, which is a strong inducer of apoptosis
(Noteborn et al., 1994). Nogueira-Dantas et al. (2007) showed that the in

85
vitro expressed VP3 protein was purified to near homogeneity by elution
from the gel, as judged by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis analysis. The purified VP3 protein with a molecular mass
of approximately 21kDa was confirmed by Western blotting analysis.
Wang et al. (2009) constructed a prokaryotic expression vector
for apoptin and prepared polyclonal antibody of apoptin. The
apoptin protein expression induced by IPTG was analyzed by SDS-PAGE.
Han et al. (2010) showed that the expression of the fused gene of SP-TAT-
Apoptin in 293FT cells infected by the recombinant lentivirus was
examined by immunofluorescence histochemistry method. The
recombinant lentivirus of SP-TAT-apoptin was successfully packaged and
it can induce HepG2 cells to apoptosis. Cao et al. (2010) reported that
Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) and
VP3 genes were cloned into a pBudCE4.1 vector and delivered by
attenuated Salmonella typhimurium into gastric cancer cells, and
their expression and antitumor effects in nude mice were monitored by
Western blot and fluorescence microscopy. Delivery of TRAIL and VP3
genes by attenuated S. typhimurium could significantly inhibit the growth
of gastric cancer cells in vitro and in vivo. Li et al. (2012) investigated the
pro-apoptotic effect of 7, 3'-dimethoxy hesperetin (DMHP) on
synovial apoptosis in vivo. Bcl-2, Bax mRNA and protein expressions in
synovium were determined by western blot and it was found that DMHP
treatment on adjuvant arthritis (AA) rats significantly decreased the
protein ratio of Bcl-2/Bax in synovium. The 16 kDa His-TAT-Apoptin and
the 42 kDa GST-TAT-Apoptin were detected using monoclonal anti-His
antibody and anti-GST antibody respectively (Lee et al., 2012). Saxena et

86
al. (2012) showed expression of VP3 in HeLa cells, transfected with
pcDNA.cav.vp3. Expression was confirmed by flow cytometry, IFAT.
IFAT in cells transfected with pcDNA.cav.vp3 using polyclonal sera
showing specific binding of antibody to the expressed 13.4 kDa vp3
protein when compared to the control untransfected cells. The size of
apoptin protein and apoptin like protein from other viruses, varied due to
size of the recombinant construct used by these workers. In certain cases,
His tag was used in construct, which increased the total size of expressed
protein.
In our study, induction of apoptosis in HeLa cells was analysed by
employing DNA laddering assay, caspase 3 detection assay and Annexin-
V-binding assay. In DNA laddering assay, nucleosomal laddering was
detected on 2% agarose gel electrophoresis. The bands were found to be
merged and a laddering pattern was observed. The nucleosomal laddering
is a hallmark of apoptosis. Li et al. (2006) detected DNA ladder to
investigate the pro-apoptotic effect of 7, 3'-dimethoxy hesperetin (DMHP)
on synovial apoptosis in vivo. Typical DNA ladder formation was found
in DNA extraction of synovium from DMHP treated groups. Cipak et al.
(2007) investigated anticancer activity of newly synthesized 2-
phenoxymethyl-3H-quinazolin-4-one (PMQ). Apoptosis was analysed
using internucleosomal DNA fragmentation assay. DNA ladder formation
assay indicated that PMQ actively induced apoptosis of cells. Ahmed et al.
(2008) investigated whether endosulfan, an organochlorine pesticide was
able to deplete glutathione (GSH) and induce apoptosis in human
peripheral blood mononuclear cells (PBMC) in vitro. Apoptotic cell death
was determined by DNA fragmentation assays. Significant ladder

87
formation was observed at higher concentration, which was indicative of
apoptotic cell death. Mohamed (2010) detected DNA ladder assay with
0.9% agarose. He observed DNA laddering pattern with multiple bands of
180-200 bp and its multiplication that is considered a characteristic feature
of apoptosis. Kucharski et al. (2011) demonstrated that apoptin was able
to inhibit formation of DNA damage foci by targeting the APC/C-
associated factor MDC1 for degradation. They suggested that these results
may point to a novel mechanism of DNA damage response inhibition
during viral infection. Jiang et al. (2012) reported that HepG2 cells were
incubated with tectorigenin at different concentrations, and their viability
was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium
bromide assay. Apoptosis was detected by morphological observation of
nuclear change and agarose gel electrophoresis of DNA ladder.
Tectorigenin at a concentration of 20 mg/L greatly inhibited the viability
of HepG2 cells and induced the condensation of chromatin and
fragmentation of nuclei. Kumar et al. (2012) showed that Ochratoxin A
(OTA) and citrinin (CIT) were nephrotoxic mycotoxins produced mainly
by fungal species Aspergillus ochraceus and Penicillium citrinum,
respectively, which have been found to occur together in various food and
feed commodities. Both OTA and CIT were evaluated for their potential to
induce apoptosis by flow cytometry and gel electrophoresis. The
concentration of MDA was found significantly higher in OTA and
combination-treated groups. OTA and combination-treated groups
revealed more apoptotic cells in flow cytometry when compared with the
CIT-treated group.Characteristic DNA fragmentation, as evidenced
by ladder pattern in electrophoresis appeared in the toxin-treated groups.

88
Steigerova et al. (2012) surveyed the effects of brassinosteroids (BR) on
prostate cancer cells using DNA ladder assays. They observed BRs
inhibited cell growth.
In caspase detection assay, when HeLa cells transfected with
rplasmid were analysed after 72 hrs, caspase 3 positive cells showed green
fluorescence. However, control cells showed no such fluorescence.
Activation of caspase cascade plays a central role in execution of apoptosis
by cleaving a large number of proteins. Danen-van Oorschot et al. (2000)
demonstrated that activation of upstream caspases was not required,
activation of caspase-3 and possibly also other downstream caspases was
essential for rapid apoptin-induced apoptosis. Lesauskaite and
Ivanoviene (2002) showed that activation of caspases plays a major role in
the execution of apoptosis. These activated caspases selectively cleave
cellular proteins, which results in apoptotic morphology. Reed (2004)
showed that numerous proteins that regulate the cell death proteases have
been discovered, including proteins belonging to the Bcl-2, inhibitor of
apoptosis, caspase-associated recruitment domain, death domain, and
death effector domain families. Wang et al. (2005) showed that apoptin-
induced apoptosis does not depend on functional p53, and can't be
inhibited by overexpression of Bcl-2 and Bcl-xL, but caspase-3 activation
was necessary for apoptin-induced rapid apoptosis. Burek et al. (2006)
showed that cell death induction by apoptin was associated with
cytochrome c release from mitochondria as well as with caspase-3 and -7
activation and observed that apoptin-induced apoptosis was caspase
dependent. Schoop et al. (2008) observed that the active form of caspase 3
was present only in apoptin positive cells having an apoptic morphology.

89
Lin. et al. (2010) showed that maternal diabetes might increase oocyte
apoptosis by a Bax-caspase-3 pathway to play a role in embryonic
malformations by delayed oocyte development. Tumane et al. (2010)
observed that silica-induced apoptosis of the alveolar macrophages could
potentially favor a proinflammatory state, occurring in the lungs of
silicotic patients, resulting in the activation of caspase prior to induction of
the intrinsic and extrinsic apoptosis pathways. In addition, caspase could
be a key apoptotic protein that can be used as an effective biomarker for
the study of occupational diseases. It may provide an important link in
understanding the molecular mechanisms of silica-induced lung
pathogenesis. Han et al. (2011) did combination therapy with radiation
and apoptin which dramatically induced mitochondrial cytochrome c
release and the cleavage of caspases -9, -3 and -7 and showed that apoptin
treatment represented a potential method for enhancing the effectiveness
of radiotherapy in poorly responding hepatocellular carcinoma. It is
evident that the caspases are a family that are one of the main executers of
apoptotic process. It was seen that our work is in conformity with other
workers.
In our study, the Annexin-V-binding assay revealed apoptic cells
showeing bright green fluorescence while control cells showed faint green
fluorescence. In double labeling experiments, using Annexin V binding
and counterstaining with the supervital DNA dye Hoechst 33342,
Koopman et al. (1994) in their experiment showed that cells showing
chromatin condensation were annexin v positive. Martin et al. (1995)
showed that annexin-V binds specifically to phosphatidyl serine in a
calcium-dsependent manner and has been used to stain dying cells that

90
expose phosphatidyl serine on their cell surface Van Engeland et al.
(1996) showed that it was feasible to use the bivariate PI/annexin v
analysis for adhering cell in culture. Eijende et al. (1997) used annexin v
assay for the detection of apoptotic cells in situ, biotin- labeled annexin v
was injected into mice. In this way, apoptin could be detected in
developing mouse embryo. So, apoptin can be effectively used for in situ
purpose. Van Engeland et al. (1998) demonstrated that Annexin V
interacted strongly and specifically with phosphatidyl serine (PS) and
could be used to detect apoptosis by targeting for the loss of plasma
membrane asymmetry. Kekre et al. (2005) showed that Annexin-V assay
was carried out at several time-points in order to monitor phosphatidyl
serine flipping to the outer leaflet of the plasma membrane, which is a
characteristic apoptotic event. Annexin-V staining is specific to
apoptotic cells, and background staining is low in unaffected cells Lin. et
al. (2010) showed that apoptosis might be closely involved in diabetes-
induced embryonic malformations. Diabetic mouse ovarian sections
stained with hematoxylin and eosin showed reduced number of growing
follicles and delayed oocyte development. Annexin V-positive oocytes
were higher in number in diabetic mice than in non-diabetic mice. Han et
al. (2010) revealed that Annexin-V PI assay showed that SP-TAT-Apoptin,
carried by the recombinant lentivirus, could cause the HepG2 cell
apoptosis and its apoptosis rate was significantly more than paired control
group and SP-TAT-Apoptin carried by liposomes only. The recombinant
lentivirus of SP-TAT-Apoptin is successfully packaged and it can induce
HepG2 cells to apoptosis. Jiang et al. (2012) showed that HepG2 cells were
incubated with tectorigenin at different concentrations, and their viability

91
was assessed by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium
bromide assay. Apoptosis was detected by Annexin V-EGFP and
propidium iodide staining. Tectorigenin induced apoptosis of HepG2 cells
mainly via mitochondrial-mediated pathway, and produced a slight
cytotoxicity to L02 cells. It was seen that our work is in conformity with
other workers.
Van der Eb et al. (2002) described that multiple AdMLP.apoptin
injections with apoptin gave an evenly distribution throughout the tumor.
Peng et al. (2007) demonstrated that systemic delivery of Asor-apoptinin
mice bearing hepatocarcinomas resulted distribution of apoptin in both
hepatocarcinoma cells and normal liver cells. The hepatocarcinomas showed
clear regression, while normal hepatocytes were not affected. These
remarkable observations can be apotential breakthrough for the current
encountered dilemma of lack of delivery of therapeutic genes in targeted
cells. Further studies with this attractive vehicle linked to apoptin could
allow to choose the most suitable vector for treatment of squamous cell
carcinoma.
Apoptosis is a complex, genetically-determined process involved in
the growth and maintenance of homeostasis in multicellular organisms.
Deregulation of apoptosis has been implicated in a number of diseases,
including cancer and autoimmune disease. Thus, the examination of
apoptotic regulation has evoked considerable interest. Many apoptotic
proteins have been shown to be post-translationally modulated, such as
by protein cleavage, protein-protein interaction, translocation and various
post-translational modifications, which fall specifically within the range of
proteomic analysis. Recently, current proteomic technologies have

92
achieved significant advances and have accelerated research in functional
and chemical proteomics, which have been applied to the field of
apoptosis research and have the potential to be a driving force for the
field (Wang et al., 2011).
The constructed recombinant plasmid has replicase gene which
produces large amount of apoptin protein and thus only small amount of
DNA will be needed to be injected for therapeutic agent. The DNA can be
stored at room temperature. The present study also clearly showed the
apoptotic activity of VP3 protein in HeLa cell line and further studies are
required to exploit its therapeutic potential.

Summary and Conclusion
Chapter-6

93
Chapter-6
Summary and Conclusion
CAV is one of the smallest avian viruses; it is 23–25 nm in size,
icosahedral in shape and non-enveloped, having a 2.3 kb, circular, single-
stranded DNA genome. The genome encodes three viral proteins (vp1,
vp2 and vp3) that are transcribed from a single major transcript of 2.0 kb.
It is believed that the cav genome replicates through the rolling-circle
model. In the present study the vp3 gene was cloned in pSin mammalian
expression vector and the resultant recombinant plasmid pSin.cav.vp3
was then characterized by restriction digestion with BglII and sequencing.
Expression of recombinant clone was confirmed by transfection in HeLa
cell line by Immunoperoxidase test and SDS-PAGE.
The apoptosis inducing potential of cav.vp3 (apoptin) was studied
in cultured HeLa cell line. The present study confirmed that vp3 induced
apoptosis in HeLa cells, which was confirmed by demonstrating the
characteristics changes of apoptosis viz. nuclear condensation, DNA
fragmentation by DNA laddering assay, plasma membrane alteration by
annexin-V binding assy. Caspase 3 was also detected after 48 h of
transfection of Hela cell line by pSin.cav.vp3. Further in vivo trials may be
undertaken to know its therapeutic potential.

Future Scope
Chapter-7

94
Chapter-7
Future Scope
Tumors are commonly prevalent in Indian population. Apoptin
induces apoptosis/ destruction of tumor cells in human transformed and
malignant cells but not in normal cells. The apoptin (VP3 protein) derived
from chicken anemia virus is a potential agent for the treatment of a large
number of tumors. The VP3 protein (apoptin) induces apoptosis in
chicken mononuclear cells. It has attracted great attention, because it
specifically kills tumor cells while leaving normal cells unharmed. Other
tumorogenic agents show resistance when injected in body but apoptin
does not show any such type of resistance. In normal cells, apoptin resides
in the cytoplasm, whereas in cancerous cells it translocates into the
nucleus. Animal tumor models have revealed apoptin as a safe and
efficient antitumor agent, resulting in significant tumor regression. Future
antitumor therapies could use apoptin as a potential therapeutic drug.
The constructed recombinant plasmid has replicase gene which produces
large amount of apoptin protein and thus only small amount of DNA will
be needed to be injected for therapeutic agent. The DNA can be stored at
room temperature.
Future antitumor therapies could use apoptin either as a therapeutic
drug or as an early sensor of druggable tumor-specific processes
(Backendorf et al. 2008). For improved treatments to be developed, the

95
ability to target tumor cells selectively is essential, allowing a greater dose
of therapeutic agent to be delivered without adversely affecting non-
malignant tissue and further studies are required to exploit its therapeutic
potential.

References

96
References
Ahmed, T., Tripathi, A.K., Ahmed, R.S., Das, S., Suke, S.G., Pathak,
R., Chakraboti, A. and Banerjee, B.D. (2008): Endosulfan-induced
apoptosis and glutathione depletion in human peripheral blood
mononuclear cells: Attenuation by N-acetylcysteine. J Biochem Mol
Toxicol, 22: 299-304.
Akasaka, Y., Ono, I., Kamiya, T., Ishikawa, Y., Kinoshita, T., Ishiguro,
S., Yokoo, T., Imaizumi, R., Inomata, N., Fujita, K., Akishima-
Fukasawa, Y., Uzuki, M., Ito, K. and Ishii, T. (2010): The mechanisms
underlying fibroblast apoptosis regulated by growth factors during
wound healing. J Pathol, 221: 285-299.
Aldair B.M. (2000): Immunopathogenesis of chicken anemia virus
infection. Dev Comp Immunol, 24: 247-255.
Alvisi, G., Poon, I.K. and Jans, D.A. (2006): Tumor-specific nuclear
targeting: promises for anti-cancer therapy? Drug Resist Updat, 9: 40-50.
Argiris, K., Panethymitaki, C. and Tavassoli, M. (2011): Naturally
occurring, tumor-specific, therapeutic proteins. Exp Biol Med, 236: 524-
536.
Backendorf, C., Visser, A.E., De Boer, A.G., Zimmerman, R., Visser,
M., Voskamp, P., Zhang, Y.H. and Noteborn, M.H.M. (2008): Apoptin:

97
therapeutic potential of an early sensor of carcinogenic transformation.
Ann Rev Pharmacol Toxicol, 48: 143-169.
Banerjee, M., Datta, M., Majumder, P., Mukhopadhyay,
D. and Bhattacharyya, N.P. (2010): Transcription regulation of caspase-1
by R393 of HIPPI and its molecular partner HIP-1. Nucleic Acids Res, 38:
878–892.
Berglund, P., Sjoberg, M., Atkins, G.J., Sheahan, B.J., Garoff, H. and
Liljestrom, P. (1993): Semliki Forest virus expression system: production
of conditionally infectious recombinant particles. Bio/Technology, 11: 916–
920.
Bhatt P., Shukla, S.K., Mahendran, M., Dhama, K., Chawak, M.M. and
Kataria, J.M. (2011): Prevalence of chicken infectious anaemia virus
(CIAV) in commercial poultry flocks of northern India: a serological
survey. Transbound Emerg Dis, 58: 458-460.
Birame, B.M., Jigui, W., Fuxian, Y., Shuang, W., Li, Y.D., Jiazeng, S.,
Zhili, L., Bao, Y. and Weiquan, L. (2012): Co-expression of apoptin (VP3)
and antibacterial peptide cecropin B mutant (ABPS1) genes induce higher
rate of apoptosis in HepG2 and A375 cell lines. African J Biotech, 11:
10981-10988.
Bredenbeek, P.J., Frolov, I., Rice, C.M. and Schlesinger, S. (1993):
Sindbis virus expression vectors: packaging of RNA replicons by using
defective helper RNAs. J. Virol., 67: 6439–6446.

98
Burek, M., Maddika, S., Burek, C.J., Daniel, P.T., Schulze-Osthoff, K.
and Los, M. (2006): Apoptin-induced cell death is modulated by Bcl-2
family members and is Apaf-1 dependent. Oncogene, 25: 2213-2222.
Cao, H.D., Yang, Y.X., Lü, L., Liu, S.N., Wang, P.L., Tao, X.H., Wang, L.J.
and Xiang, T.X. (2010): Attenuated Salmonella typhimurium carrying
TRAIL and VP3 genes inhibits the growth of gastric cancer cells in vitro
and in vivo. Tumori, 96: 296-303.
Chen, K., Luo, Z., Tang, J. and Zheng, S.J. (2011): A critical role of heat
shock cognate protein 70 in apoptin-induced phosphorylation of Akt.
Biochem Biophys Res Commun, 409: 200-204.
Chiantore, M.V., Vannucchi, S., Mangino, G., Percario, Z.A., Affabris,
E., Fiorucci, G., Romeo, G. (2009): Senescence and cell death pathways
and their role in cancer therapeutic outcome. Curr Med Chem, 16: 287-300.
Chung, C, T., Niemela, S, L. and Miller, R, H. (1989): One step
preparation of competent Escherichia coli: Transformation and storage of
bacterial cells in the same solution. Proc. Natl. Acad. Sci, 86: 2172-2175.
Cipak, L., Repicky, A. and Jantova, S. (2007): Growth inhibition and
apoptosis induced by 2-phenoxymethyl-3H-quinazolin-4-one in HL-60
leukemia cells. Exp Oncol, 29: 13-17.
Danen-van Oorschot, A.A.A.M., Van der Eb, A.J., and Noteborn,
M.H.M. (2000): The chicken anemia virus-derived protein Apoptin
requires activation of caspases for induction of apoptosis in human
tumor cells. J. Virol, 74: 7072-7078.

99
Davis, N.L., Brown, K.W., Caley, I.J., Swanstrom, R.I. and Johnston,
R.E. (1995): Protection against influenza in mice by vaccination with a
Venezuelan equine encephalitis virus vector expressing the HA protein.
J. Cell. Biochem. Suppl., 19: 310.
Davis, N.L., Willis, L.V., Smith, J.F. and Johnston, R.E. (1989): In vitro
synthesis of infectious Venezuelan equine encephalitis virus RNA from a
cDNA clone: analysis of a viable deletion mutant. Virology, 171: 189–204.
De Herdt, P., Van den Bosch, G., Ducatelle, R., Uyttebroek, E. and
Schrier, C. (2001): Epidemiology and significance of chicken infectious
anemia virus infections in broilers and broiler parents under non
vaccinated European circumstances. Avian Dis, 45: 706-708.
De Smit, M.H. and Noteborn, M.H. (2009): Apoptosis-inducing proteins
in chicken anemia virus and TT virus. Curr Top Microbiol Immunol, 331:
131-149.
Eijnde, S.M., Boshart, L., Reutelingsperger, C.P., Zeeuw, C.L. and
Vermeij-Keers, C. (1997): Phosphatidyl serine plasma asymmetry in
vivo: A pancellular phenomenon which alters during apoptosis. Cell
Death Diff, 4: 311-316.
Eltahir, M.Y., Qian, K., Jin, W. and Qin, A. (2011): Analysis of chicken
anemia virus genome: evidence of intersubtype recombination. Virol J, 8:
512.

100
Furlov, I., Hoffman, T.A., Pragai, B.M., Dryga, S.A., Huang, H.V.,
Schlesinger, S. and Rice, C.M. (1996): Aiphavirus-based expression
vectors: Strategies and applications. Proc. Natl. Acad. Sci., 93: 11371-11377.
Gerna, G., McCloud, C. and Chambers, R. W. (1976):
Immunoperoxidase technique for detection of antibodies to human
cytomegalovirus. J Clinical Microbiol, 3: 364-372.
Ghobrial, I.M., Witzig, T.E. and Adjei, A.A. (2005): Targeting apoptosis
pathways in cancer therapy. CA Cancer J Clin, 55: 178-194.
Guelsen, L., Paterson, H., Gäken, J., Meyers, M., Farzaneh, F. and
Tavassoli, M. (2004): TAT-apoptin is efficiently delivered and induces
apoptosis in cancer cells. Oncogene, 23: 1153–1165.
Hailemariam, Z., Omar, R.A., Hair-Bejo, M. and Giap, C.T. (2008):
Detection and characterization of chicken anemia virus from commercial
broiler breeder chickens. Virol J, 5: 128.
Han, S.X., Zhao, J., Ma, J.L., Huang, C., Lv, Y., Ou, W. and Jia, X. (2010):
The effect of the fused gene of SP-TAT-Apoptin transfected by lentivirus
on HepG2 cells. Chinese J Cell Mol Immunol, 26: 301-302.
Han, S.X., Zhu, Q., Ma, J.L., Lv, Y., Zhao, J., Huang, C., Jia, X., Ou, W.
and Guo, H.T. (2011): Apoptin sensitizes radiation-induced cell death via
classic mitochondrial, caspase and p53-dependent signaling in HepG2
cells. Mol Med Report, 4: 59-63.

101
He, C., Ding, N., Li, J. and Li, Y. (2002): Gene cloning and sequencing of
chicken anemia virus (CAV) isolated from Harbin. Wei Sheng Wu Xue
Bao, 42: 436-441.
He, Wilde and Kaderbhai, M.A. (1990): A simple single step procedure
for small scale preparation of Escherichia Coli plasmid. Nucleic Acids Res,
18: 1660.
He, X, Zhang, Q., Liu, Y. and He, P. (2005): Apoptin induces chromatin
condensation in normal cells. Virus Genes, 31: 49-55.
Heilman, D.W., Green, M.R. and Teodoro, J.G. (2005): The anaphase
promoting complex: a critical target for viral proteins and anti-cancer
drugs. Cell Cycle, 4: 560-563.
Hino, S. and Prasetyo, A.A. (2009): Relationship of Torque teno virus to
chicken anemia virus. Curr Top Microbiol Immunol, 331: 117-130.
Ismail, I.H., Andrin, C., McDonald, D. and Hendzel, M.J. (2010): BMI1-
mediated histone ubiquitylation promotes DNA double-strand break
repair. J Cell Biol, 191: 45-60.
Jain, V., Armah, B.H., Tongren, E.J., Ned, M.R., Wilson, O.N.,
Crawford, S., Joel, K.P., Singh, P.M., Nagpal, C.A., Dash, A.P.,
Udhayakumar, V., Singh, N. and Stiles, K.J. (2008): Plasma IP-10,
apoptotic and angiogenic factors associated with fatal cerebral malaria in
India. Malaria Journal, 7: 83.
Janssen, K., Hofmann, T.G., Jans, D.A., Hay, R.T., Schulze-Osthoff K.
and Fischer U. (2007): Apoptin is modified by SUMO conjugation and

102
targeted to promyelocytic leukemia protein nuclear bodies. Oncogene, 26:
1557-1566.
Jiang, C.P., Ding, H., Shi, D.H., Wang, Y.R., Li, E.G. and Wu, J.H.
(2012): Pro-apoptotic effects of tectorigenin on human hepatocellular
carcinoma HepG2 cells. World J Gastroenterol, 18: 1753-1764.
Jiang, J., Cole, D., Westwood, N., Macpherson. L., Farzaneh, F., Mufti,
G., Tavassoli, M. and Gäken J. (2010): Crucial roles for protein kinase C
isoforms in tumor- specific killing by apoptin. Cancer Res, 70: 7242-7252.
Jolly, D. (1994): Viral vector systems for gene therapy. Cancer Gene Ther.,
1: 51–64.
Kataoka, S. and Tsuruo, T. (1996): Physician Education: Apoptosis.
Oncologist, 1: 399-401.
Kawanishi, S. and Hiraku, Y. (2004): Amplification of anti-cancer drug
induced DNA damage and apoptosis by DNA binding compounds. Curr,
Med. Chem. Antican. Agent, 4: 415-419.
Kekre N Griffin, C., McNulty, J. and Pandey, S. (2005): Pancratistatin
causes early activation of caspase-3 and the flipping of phosphatidyl
serine followed by rapid apoptosis specifically in human lymphoma
cells. Cancer Chemother Pharmacol, 56: 29-38.
Kerr, J (2000): Apoptosis. Med. J. Aust, 173: 616–617.

103
Kerr, J.F., Wyllie, A.H. and Currie, A.R. (1972): Apoptosis: a basic
biological phenomenon with wide ranging implications in tissue kinetics.
Br. J. Cancer, 26: 239-257.
Kim, M.K., Jung, H.S., Yoon, C.S., Ko, J.H., Jun, H.J., Kim, T.K., Kwon,
M.J., Lee, S.H., Ko, K.S., Rhee, B.D. and Park, J.H. (2010): The effect of
glucose fluctuation on apoptosis and function of INS-1 pancreatic beta
cells. Korean Diabetes J, 34: 47-54.
Kooistra, K., Zhang, Y.H. and Noteborn, M.H. (2007): Viral elements
sense tumorigenic processes: approaching selective cancer therapy. Mini
Rev Med Chem, 11: 1155-1165.
Koopman, G., Reutelingsperger, C.P., Kuijten, G.A., Keehnen, R, M.,
Pals, S.T. and Van Oers M.H. (1994): Annexin V for flow cytometric
detection of phosphatidyl expression on B cells undergoing apoptosis.
Blood, 84: 1415-1420.
Kucharski, T.J., Gamache, I., Gjoerup, O. and Teodoro, J.G. (2011):
DNA damage response signaling triggers nuclear localization of the
chicken anemia virus protein apoptin. J Virol, 85: 12638-12649.
Kumar, M., Dwivedi, P., Sharma, A.K., Sankar, M., Patil, R.D.
and Singh, N.D. (2012): Apoptosis and lipid peroxidation in ochratoxin
A- and citrinin-induced nephrotoxicity in rabbits. Toxicol Ind Health,
[Epub ahead of print]
Lauber, K., B lumenthal, S.G., Waibel, M. and Wesselborg, S. (2004):
Clearance of apoptotic cells: getting rid of corpses. Mol. Cell, 14: 277-287.

104
Leasauskaite V and Ivanoviene L, (2002): Programmed cell death:
molecular mechanism and detection. Medicina, 38: 869-875.
Lee, J.J., Chen, P.B., Yang, S.H., Cheng, C.H., Chueh, L.L., Pang, V.F.,
Hsiao, M. andLin, C.T. (2007): Effect of the VP3 gene of chicken anemia
virus on canine mammary tumor cells. Am J Vet Res, 68: 411-422.
Lee, M.S., Sun, F.C., Huang, C.H., Lien, Y.Y., Feng, S.H., Lai, G.H., Lee,
M.S., Chao, J., Chen, H.J., Tzen, J.T.C. and Cheng, H.Y. (2012): Efficient
production of an engineered apoptin from chicken anemia virus in a
recombinant E.coli for tumor therapeutic applications. BMC Biotech, 12:
doi: 10. 1186/1472-6750-12-27.
Li, R., Cai, L., Xie, X.F., Peng, L., Wu, T.N. and Li, J. (2012): 7,3'-
dimethoxy hesperetin inhibits inflammation by inducing
synovial apoptosis in rats with adjuvant-induced arthritis.
Immunopharmacol Immunotoxicol. [Epub ahead of print]
Li, X., Jin, N., Mi, Z., Lian, H., Sun, L., Li, X. and Zheng, H. (2006):
Antitumor effects of a recombinant fowlpox virus expressing apoptin in
vivo and in vitro. Int J Cancer, 119: 2948-2957.
Li, X., Liu, Y., Wen, Z., Li, C., Lu, H., Tian, M., Jin, K., Sun, L., Gao, P.,
Yang, E., Xu, X., Kan, S., Wang, Z., Wang, Y. and Jin, N. (2010): Potent
anti-tumor effects of a dual specific oncolytic adenovirus expressing
apoptin in vitro and in vivo. Mol Cancer, 20: 9-10.

105
Li, Y.P. and Cui, Z.Z (2007): Pathogenicity and genomic sequence
comparison of a chicken infectious anemia virus field isolate. Wei Sheng
Wu Xue Bao, 47: 894-898.
Liljestrom, P. (1994): Alphavirus expression systems. Curr. Opin.
Biotechnol., 5: 495–500.
Liljestrom, P. and Garoff, H. (1991): A new generation of animal cell
expression vectors based on the Semliki Forest virus replicon.
Bio/Technology, 9: 1356–1361.
Lin, S., Liu, K., Li, W., Zhou, X. and Huang, T. (2010): Maternal diabetes
increases apoptosis in mice oocytes, not 2-cell embryos. Endocrine, 37:
460-466.
Liu, Q., Fu, H., Xing, R., Tie, Y., Zhu, J., Sun, Z., and Zheng, X. (2008):
Survivin knockdown combined with apoptin overexpression inhibits
cells growth significantly. Cancer Biol. Ther, 7: 1061-1062.
Liu, Q., Luo, Y.S., Geng, L., Wang, W., Lai, Z.and Li, F. (2010):
Therapeutic effect of adenovirus-mediated apoptin gene transfer
combined with ADM and CDDP on hepatocellular carcinoma in mice. J.
of Southern Medical Univ, 30: 538-540.
Liu, X., Zaiden, Y. H., Elojeimy, S., Holman, D. H., El-Zawahry, A. M.,
Guo, G., Bielawska, A., Bielawski, J., Szule, Z., Rubinchik, S., Dong, J.,
Keane, T.E., Tavassoli, M., Hannum, Y.A. and Norris, J.S. (2006):
Involvement of sphingolipids in apoptin induced cell killing. Mol. Ther,
14: 627-636.

106
Lockshin, R.A. and Zakeri, Z. (2002): Caspase-independent cell death.
Curr Opin Cell Biol, 14: 727-733.
Los, M., Panigrahi, S., Rashedi, I., Mandal, S., Stetefeld, J., Essmann, F.,
and Schulze–Osthoff, K. (2009): Apoptin, a tumor–selective killer.
Biochem, Biophys. Acta, 8: 1335-1342.
Maddika, S., Mendoza, F.J., Hauff, K., Zamzow, C.R., Paranjothy, T.
and Los, M. (2006): Cancer-selective therapy of the future: apoptin and
its mechanism of action. Cancer Biol Ther, 5: 10-19.
Martin, J.S., Reutelingsperger, C.P.M., McGahon, A.J., Rader, J.A., Van
Schie, R.C.A., Laface, D.M. and Green, D.R. (1995): Early redistribution
of plasma membrane phosphatidylserine is a general feature of apoptosis
regardless of the initiating stimulus: inhibition by overexpression of Bcl-2
and Abl. J Exp Med, 182: 1545–1556.
Mazars, A., Geneste, O. and Hickman, J. (2005): The Bcl-2 family of
proteins as drug targets. J Soc Biol, 199: 253-265.
McNulty, M.S. (1991): Chicken anaemia agent: a review. Avian Pathol, 20:
187-203.
Miller, A.D., Miller, D.G., Garcia, J.V. and Lynch, C.M. (1993): Use of
retroviral vectors for gene transfer and expression. Methods Enzymol., 217:
581–599.
Mohamed, M.A. (2010): Chicken Infectious Anemia Status in
Commercial Broiler Chickens Flocks in Assiut-upper Egypt: Occurrence,

107
Molecular Analysis Using PCR-RFLP and Apoptosis Effect on Affected
Tissues. International J Poultry Science, 9: 591-598.
Natesan, S., Kataria, J.M., Dhama, K., Bhardwaj, N. and Sylvester, A.
(2006): Anti-neoplastic effect of chicken anemia virus VP3 protein
(apoptin) in Rous sarcoma virus-induced tumours in chicken. J Gen Virol,
10: 2933-2940.
Natesan, S., Kataria, J.M., Dhama, K., Rahul, S. and Baradhwaj, N.
(2006): Biological and molecular characterization of chicken anaemia
virus isolates of Indian origin. Virus Research, 118: 78–86.
Nogueira-Dantas, E.O., Ferreira, A.J., Astolfi-Ferreira, C.S. and
Brentano, L. (2007): Cloning and expression of chicken anemia virus VP3
protein in Escherichia coli. Comp Immunol Microbiol Infect Dis, 30: 133-142.
Noteborn M.H. (2004): Chicken anemia virus induced apoptosis:
underlying molecular mechanisms. Vet. Microbiol, 98: 89-94.
Noteborn, M.H., Todd, D., Verschueren, C.A., de Gauw, H.W., Curran,
W.L., Veldkamp, S., Douglas, A.J., McNulty, M.S., Van der EB, A.J. and
Koch G. (1994): A single chicken anemia virus protein induces apoptosis.
J Virol, 68: 346-351.
Noteborn, M.H., de Boer, G.F., van Roozelaar, D.J., Karreman, C.,
Kranenburg, O., Vos, J.G., Jeurissen, S.H., Hoeben, R.C., Zantema,
A. and Koch, G. (1991): Characterization of cloned chicken anemia virus
DNA that contains all elements for the infectious replication cycle. J.
Virol, 65: 3131 – 3139.

108
Noteborn, M.H.M. and Koch, G (1995): Chicken anaemia virus infection:
Molecular basis of pathogenicity, Avian Pathology, 24: 11-31.
Noteborn, M.H.M., Danen-van Oorschot, A.A.A.M. and Van der Eb,
A.J. (1998): Chicken anemia virus: Induction of apoptosis by a single
protein of a single-stranded DNA virus. Seminars in Virology, 8: 497-504.
O'Brien, V. (1998): Viruses and apoptosis. Gen. Virol, 79: 1833-1845.
Oorschot, A.A.A.M., Fisher, D.F., Gimbergen, J.M., Klien, B., Zuang,
S.M., Falkenburg, J.H.F., Backendorf, C., Quax, P.H.A., Van Der Eb,
A.J., and Noteborn M.H.M. (1997): Apoptin induces apoptosis in human
transformed and malignant cells but not in normal cells. Leiden
University, 94: 5843-5847.
Oro, C. and Jans, D.A. (2004): The tumour specific proapoptotic factor
apoptin Vp3 from chicken anaemia virus. Curr Drug Targets, 5: 179-190.
Pan, Y., Fang, L., Fan, H., Luo, R., Zhao, Q., Chen, H. and Xiao, S. (2010):
Antitumor effects of a recombinant pseudotype baculovirus expressing
apoptin in vitro and in vivo. Int J Cancer, 126: 2741-2751.
Panigrahi, S., Stetefeld, J., Jangamreddy, J.R., Mandal, S., Mandal, S.K.
and Los, M. (2012): Modeling of molecular interaction between apoptin,
BCR-Abl and CrkL--an alternative approach to conventional rational
drug design. PLoS One, 7: 28395.
Patel, R.M. and Prajapati, R.N. (2011): Pancratistatin, an apoptic inducer:
New horizon for targeted therapy in cancer. Biotechnol Mol Biol Rev, 6: 59-
68.

109
Pavet, V., Portal, M.M., Moulin, J.C., Herbrecht, R. and Gronemeyer, H.
(2011): Towards novel paradigms for cancer therapy. Oncogene, 30: 1-20.
Peng, D.J., Sun, J., Wang, Y.Z., Tian, J., Zhang, Y.H., Noteborn, M.H.
and Qu, S. (2007): Inhibition of hepato- carcinoma by systemic delivery
of Apoptin gene via the hepatic asialoglycoprotein receptor. Cancer Gene
Ther, 14: 66-73.
Pietersen, A. and Noteborn, H.M (2000): Apoptin. Adv Exp Med Biol, 465:
153-161.
Pietersen, A.M., van der Eb, M.M., Rademaker, H.J., van den
Wollenberg, D.J., Rabelink, M.J., Kuppen, P.J., van Dierendonck, J.H.,
van Ormondt, H., Masman, D., Van de Velde, C.J., Van der Eb, A.J.,
Hoeben, R.C. and Noteborn, M.H. (1999): Specific tumor-cell killing
with adenovirus vectors containing the apoptin gene. Gene Ther, 6: 882-
892.
Podo, F., Canevari, S., Canese, R., Pisanu, M.E., Ricci, A. and Iorio, E.
(2011): MR evaluation of response to targeted treatment in cancer cells.
NMR Biomed, 24: 648-672.
Pope, C.R. (1991): Chicken anemia agent. Vet. Immunopathol, 30: 51-65.
Reed, J.C. (2004): Apoptosis mechanisms: implications for cancer drng
discovery. Oncology, 18: 11-20.
Rodriguez, R.L. and Tait, R.C. (1983): Recombinant DNA techniques. An
introduction. Addison-Wesley Publ. Co. London.

110
Rohn, J.L. and Noteborn, M.H. (2004): The viral death effector apoptin
reveals tumor- specific processes. Apoptosis, 9: 315-322.
Sambrook, J. and Russell, D.W. (2001): Molecular cloning. A Laboratory
Manual, 3rd ed., Cold Spring Harbor Laboratory Press, edd Spring
Harbor, New York.
Samulski, R.J., Chang, L.S. and Shenk, T. (1989): Helper-free stocks of
recombinant adeno-associated viruses: normal integration does not
require viral gene expression. J. Virol., 63: 3822–3828.
Sauvage, V., Cheval, J., Foulongne, V., Gouilh, M.A., Pariente, K.,
Manuguerra, J.C., Richardson, J., Dereure, O., Lecuit, M., Burguiere, A.,
Caro, V. and Eloit, M. (2011): Identification of the first human gyrovirus,
a virus related to chicken anemia virus. J Virol, 85: 7948-7950.
Saxena, S., Kumar, G.R., Singh, P., Chaturvedi, U., Saxena, L., Kumar,
R., Sahoo, A.P., Doley, J., Rajmani, Kumar, A., Kumar, S. and Tiwari,
A. K. (2012): Prokaryotic expression of chicken infectious anaemia
apoptin protein and characterization of its polyclonal antibodies. Indian J
Exp Biol, 50: 325-331.
Schlesinger, S. (1993): Alphaviruses—vectors for the expression of
heterologous genes. Trends Biotechnol., 11: 18–22.
Schmitz, I., Kirchoff, S. and Krammer, P.H. (2000): Regulation of death
receptor mediator apoptosis pathways. Int. J. Biochem. Cell. Biol, 32: 1123-
1136.

111
Schoop, R.A, Baatenburg de Jong, R.J. and Noteborn, M.H (2008):
Apoptin induces apoptosis in an oral cancer mouse model. Cancer Biol
Ther, 7: 1368-1373.
Schoop, R.A., Verdegaal, E.M., de Jong, R.J. and Noteborn, M.H. (2010):
Apoptin enhances radiation-induced cell death in poorly responding
head and neck squamous cell carcinoma cells. Basic Clin Pharmacol
Toxicol, 106: 130-134.
Shen, Z., Wang, Y., Zong, Y. and Qu, S. (2003): Experimental study on
the antitumor effect of chicken anemia virus vp3 gene against liver
carcinoma in vivo. J Huazhong Univ Sci Technolog Med Sci, 23: 105-107, 115
Sinha, S., Malonia, F.K., Mittal, S.P., Mathai, J., Pal, J.K. and
Chattopadhyay, S. (2012): Chromatin remodelling protein SMAR1
inhibits p53 independent transactivation by regulating acetyl transferase
p300. Int. J Biochem Cell Biol, 44: 46-52.
Sommer, F. and Cardona, C. (2003): Chicken anemia virus in broilers:
dynamics of the infection in two commercial broiler flocks. Avian Dis, 47:
1466–1473.
Steigerova, J., Rarova, L., Oklesťkova, J., Krizova, K., Levkova,
M., Svachova, M., Kolar, Z. and Strnad, M. (2012): Mechanisms of
natural brassinosteroid-induced apoptosis of prostate cancer cells. Food
Chem Toxicol, [Epub ahead of print]
Strauss, J.H. and Strauss, E.G. (1994): The alphaviruses: gene expression,
replication, and evolution. Microbiol. Rev., 58: 491–562.

112
Sun, J., Wang, Y., Zong, Y. and Qu, S. (2003): Cloning of chicken anemia
virus vp3 gene and apoptosis inductive effect of vp3 gene in vitro. J
Huazhong Univ Sci Technolog Med Sci, 23: 329-331, 334.
Takahashi, K., Iwasa, Y., Hijikata, M. and Mishiro, S. (2000):
Identification of a new human DNA virus (TTV-like mini virus, TLMV)
intermediately related to TT virus and chicken anemia virus. Archives
Virol, 145: 979–993.
Teodoro, J.G. and Branton, P.E. (1997): Regulation of apoptosis by viral
gene products. J. Virol, 71: 1739-1746.
Todd, D. (2000): Circoviruses: immunosuppressive threats to avian
species: a review. Avian Pathol, 29: 373-394.
Todd, D., Creelan, J.L., Mackie, D.P., Rixon, F. and McNulty, M.S.
(1990): Purification and biochemical characterization of chicken anemia
agent. J Gen Virol, 71: 819-823.
Tumane, R.G., Pingle, S.K., Jawade, A.A. and Nath, N.N. (2010): An
overview of caspase: Apoptotic protein for silicosis. Indian J Occup
Environ Med, 14: 31-38.
Van der Eb, M.M., Pietersen, A.M., Speetjens, F.M., Kuppen, P.J., Van
De Velde, C.J., Noteborn, M.H. and Hoeben, R.C. (2002): Gene therapy
with apoptin induces regression of xenografted human hepato- mas.
Cancer Gene Ther, 9: 53-61.
VanEngeland, M., Nieland, L.J.W., Ramaekers, F.C.S., Schutte, B.
and Reutelingsperger, C.P.M. (1998): AnnexinV-Affinity Assay: A

113
review on an apoptosis detection system based on phosphatidyl
serine exposure. Cytometry, 31: 1-9.
VanEngeland, M., Ramaekers, F.C.S., Schutte, B., Reutelingsperger,
C.P.M. (1996): A novel assay to measure loss of plasma membrane
asymmetry during apoptosis of adherent cells in culture. Cytometry,
24: 131–139.
Wagstaff, K.M. and Jans, D.A. (2009): Nuclear drug delivery to target
tumour cells. Eur J Pharmacol, 625: 174-180.
Wang, C. and Zhang, Y. (2011): Apoptin gene transfer via modified
wheat histone H4 facilitates apoptosis of human ovarian cancer cells.
Cancer Biotherapy & Radiopharmaceuticals, 26: 121-126.
Wang, J.S., Zhang, M.X., Duan, X.Y., Mo, F., Wang, Q.Y. and Yang, G.X.
(2009): Construction of a prokaryotic expression vector for apoptin and
preparation of polyclonal antibody of apoptin. J Southern Med Univ, 29:
1405-1407.
Wang, L., Yang, W., Read, P., Lamer, J. and Sheng, K. (2010): Tumor cell
apoptosis induced by nanoparticle conjugate in combination with
radiation therapy. Nanotechnology, 21: 475103.
Wang, Q.M., and He, F.C (2005): Molecular mechanism of specific
induction of apoptosis in tumor cells by apoptin. Ai Zheng, 24: 509-512.
Wesselborg, S. and Lauber, K. (2005): Mechanism of anti-cancer drug
action. In Apoptotic Pathways As Target For Novel Therapies in Cancer and

114
Other Diseases (ed. M. Los and S. B. Gibson), New York: Springer
Academic Press, pp 31-56.
Wysokinski, D., Błasiak, J. and Wozniak, K (2010): Involvement of
nuclear domains in the cellular response to DNA damage. Postepy
Biochem, 56: 328-340.
Xiong, C., Levis, R., Shen, P., Schlesinger, S., Rice, C.M. and Huang,
H.V. (1989): Sindbis virus: an efficient, broad host range vector for gene
expression in animal cells. Science, 243: 1188–1191.
Yan, L., Xiangwei, M., Xiao, L., Peng, G., Chang, L., Mingyao,
T., Encheng, Y., Xiaohong, X., Peng, J., Shifu, K., Zhongmei, W.
and Ningyi, J. (2010): Construction, expression and characterization of a
dual cancer-specific fusion protein targeting carcinoembryonic antigen in
intestinal carcinomas. Protein Expr Purif, 69: 120-125.
Yu, D.S., Zhao, R., Hsu, E.L., Cayer, J., Ye, F., Guo, Y., Shyr, Y. and
Cortez, D (2010): Cyclin-dependent kinase 9-cyclin K functions in the
replication stress response. EMBO Rep, 11: 876-882.
Zhang, Y.H., Leliveld, S.R., Kooistra, K., Molenaar, C., Rohn, J.L.,
Tanke, H.J., Abrahams, J.P. and Noteborn, M.H.M. (2003): Recombinant
Apoptin multimers kill tumor cells but are nontoxic and epitope-shielded
in a normal-cell-specific fashion. Exp Cell Res, 289: 36-46.
Zhuang, S.M., Shvarts, A., van Ormondt, H., Jochemsen, A.G., Van der
Eb, A.J. and Noteborn, M.H. (1995): Apoptin, a protein derived from

115
chicken anemia virus, induces p53-independent apoptosis in human
osteosarcoma cells. Cancer Res, 55: 486-489.

Appendix

116
Appendix
A. PLASMID DNA ISOLATION BY TELT METHOD
TELT Solution
Tris Cl, pH 8.0 50 mM
Na2 EDTA 62.5 mM
LiCl 2.5 M
Triton X-100 4% v/v
Phenol/Chloroform 1:1 v/v
B. REAGENTS USED IN AGAROSE GEL LECTROPHORESIS
1. Tris-acetate-EDTA (TAE) buffer 50X
Tris base 24.2 g
Glacial acetic acid 57.1 ml
0.5 M EDTA (pH 8.0) 100 ml
Distilled water was added to make final volume up to 100 ml. A
working solution of 1X was used.
2. Ethidium bromide stock solution (10 mg/ ml)
Ethidium bromide 100 mg
Distilled water 10 ml
The solution was mixed and stored at 40C. A concentration of
0.5 mg/ ml was used in preparing agarose gel.

117
3. Loading Dye (6X)
Bromophenol blue 0.25% (w/v)
Xylene cyanol 0.25% (w/v)
Sucrose 40% (w/v)
C. REAGENTS USED FOR BACTERIOLOGICAL PROCEDURES
1. LB (Luria-Bertani) broth
Bacto tryptone 10 g
Bacto yeast extract 5 g
NaCl 10 g
Distilled water 950 ml
Adjust the pH to 7.0 with 5N NaOH.
Volume was made up to 1 litre and sterilized by autoclaving at 15
psi for 20 minutes and the solution was stored at 40C.
2. LB (Luria-Bertani) Agar Medium
1.5% Agar in LB medium.
Sterilized by autoclaving at 15 psi for 20 minutes and stored at 40C.
3. SOB Medium
Bacto tryptone 20 g
Bacto yeast extract 5 g
NaCl 0.5 g
Nuclease free water 950 ml

118
Dissolve by shaking. Add 10 ml of a 250 mM solution of KCl.
Adjust the pH to 7.0 with 5N NaOH and make volume up to 1000 ml.
Sterilize by autoclaving. Just before use, add 5 ml of a sterile 2M MgCl2.
4. 1X Transformation & Storage Solution (TSS)
LB broth 95 ml
PEG (8000) (w/v) 10 g
MgSO4 (50 mM final conc.) 1.23 g
DMSO 5 ml
DMSO was filtered through 0.22 µ filter and added after
autoclaving of other ingredients at 1210C, 15 psi for 15 minutes.
5. Ampicillin (50 mg/ ml)
Ampicillin 250 mg
Distilled water 5 ml
The mixture was sterilized by filtration through filter and stored at
200C.
D. REAGENTS USED IN CELL CULTURE
1.) PBS
Sodium Chloride (NaCl) 8.00 g
Potassium Chloride (KCl) 0.20 g
Dissodium hydrdrogen phosphate
(Na2HPO42H2O) 1.15 g

119
Distilled water (up to) 1000 ml
The solution was sterilized at 15 psi for 20 minutes and stored at 200C.
2.) DMEM (Gibco)
DMEM powder 13.5 g
with high glucose
With L-glutamine 300 mg
Fungizone 1 ml
Gentamycin 1.2 ml
Sodium bicarbonate 1.5 g
Distilled water (up to) 1000 ml
Distilled water was added to make final volume up to 1000 ml.
Sterilized by filtration.
3. Growth Medium
DMEM 90 ml
FCS 10 ml
4. Trypsin Versene Solution (0.17%)
NaCl 10.0 g
KCl 0.250 g
Na2HPO4 1.9 g
KH2PO4 0.250 g
Trypsin 1.7 g
Versene 1.4 g
0.4% Phenol red 1.0 µl

120
Distilled water was added to make up to 1000 ml
E. REAGENTS USED IN DNA FRAGMENTATION ASSAY
1. Cell lysis buffer
Tris (ph 8.0) 10 mM
EDTA (ph 8.0) 1 mM
SDS 1%
2. Potassium acetate solution
5 M Potassium acetate 60 ml
Glacial acetic acid 11.5 ml
H2O 28.5 ml
This solution is stored at room temperature.











1
Isolation of recombinant plasmid containing Chicken Anemia
Virus VP3 gene.
AUTHORS:
PRIYANKA PAL: Near old chandmari tiwari temple, Street no. 4,
Damodar puram, Subhash nagar, Bareilly. (U.P.)
E-mail: [email protected]. Contact No:
9411217514
PROF. KUSUM AGARWAL: E-mail: [email protected]. Contact No.:
9456089155
PROF. ANANT RAI: E-mail: [email protected]. Contact No.: 9219661948
USHA TIWARI: E-mail: [email protected]. Contact No.:
9917632096
RAJVEER MAURYA: Contact No.: 9457257179
SUBMITTED BY
PRIYANKA PAL
RESEARCH SCHOLAR, BIOTECHNOLOGY
SHOBHIT UNIVERSITY, MEERUT, (U. P.)

2
Isolation of recombinant plasmid containing Chicken Anemia
Virus VP3 gene.
ABSTRACT
Programmed cell death plays critical roles in a wide variety of physiological
processes during fetal development and in adult tissues. Defects in apoptotic cell
death regulation contribute to many diseases, including disorders where cell
accumulation occurs (cancer, restenosis). Knowledge of the molecular mechanisms of
apoptosis is providing insights into the causes of multiple pathologies where aberrant
cell death regulation occurs and is beginning to provide new approaches to the
treatment of human diseases Reed [1]. Apoptin, a small protein from chicken anemia
virus, has attracted great attention, because it specifically kills tumor cells while
leaving normal cells unharmed. In normal cells, apoptin resides in the cytoplasm,
whereas in cancerous cells it translocates into the nucleus Los [2]. For isolation of
plasmid DNA, the procedure described here (originally presented by He [3] for small
isolation of plasmid DNA can also be readily extended for large scale preparation.
Plasmid DNA is obtained from E.coli grown on plates as colonies or in liquid
cultures. The merit of the approach is that it is extremely reliable and rapid- requiring
no more than 20 min of simple and economical operations for a preparation. The final
plasmid DNA preparations are of a purity and quality usable for most biological
application. The yield should be about 30 µg plasmid DNA per preparation. It only
need one (a single) tube per sample!
KEYWORDS
Chicken Anemia Virus, VP3 gene, Apoptin, Apoptosis, Cancer therapy.

3
INTRODUCTION
All human cells have a genetic program that upon activation will cause cell
death, named apoptosis. Cancer cells can grow due to unbalances in proliferation, cell
cycle regulation and their apoptosis machinery: genomic mutations resulting in non-
functional pro-apoptosis proteins or over-expression of anti-apoptosis proteins form
the basis of tumor formation. Surprisingly, lessons learned from viruses show that
cancer cannot be regarded simply as the opposite of apoptosis. For instance,
adenovirus can only transform cells when both its anti- and pro-apoptotic proteins are
produced. Oncolytic viruses are known to replicate selectively in tumor cells resulting
in cell death. Proteins derived from viruses, i.e. chicken anemia virus (CAV)-derived
apoptosis-inducing protein (apoptin), adenovirus early region 4 open reading frame
(E4orf4) and parvovirus-H1 derived non-structural protein 1 (NS1), the human alpha-
lactalbumin made lethal to tumor cells (HAMLET), which is present in human milk or
the human cytokines melanoma differentiation-associated gene-7 (mda-7) and tumor
necrosis factor-related apoptosis-inducing ligand (TRAIL) have all the ability to
induce tumor-selective apoptosis. The tumor-selective apoptosis-inducing proteins
seem to interact with transforming survival processes, which can become redirected
by these proteins into cell death. Transformation-related processes have been
identified, which seem to be crucial for the tumor-selectively killing activity of these
proteins. For instance, the transformation-related protein phosphatase 2A (PP2A)
plays a role in the induction of tumor-selective apoptosis. The proteins mda-7, TRAIL
and HAMLET are already successfully tested in first clinical trials. Proteins harboring
tumor-selective apoptosis characteristics represent, therefore, a therapeutic potential
and a tool for unraveling tumor-related processes. Fundamental molecular and
(pre)clinical therapeutic studies of the various tumor-selective apoptosis-inducing
proteins apoptin, E4orf4, HAMLET, mda-7, NS1, TRAIL and related proteins will be
discussed Noteborn [4].
Caspases belong to a class of cysteine proteases which function as critical
effectors in cellular processes such as apoptosis and inflammation by cleaving
substrates immediately after unique tetrapeptide sites. With hundreds of reported
substrates and many more expected to be discovered, the elucidation of the caspase

4
degradome will be an important milestone in the study of these proteases in human
health and disease. Several computational methods for predicting caspase cleavage
sites have been developed recently for identifying potential substrates. However, as
most of these methods are based primarily on the detection of the tetrapeptide
cleavage sites - a factor necessary but not sufficient for predicting in vivo substrate
cleavage - prediction outcomes will inevitably include many false positives. In this
paper, its shown that structural factors such as the presence of disorder and solvent
exposure in the vicinity of the cleavage site are important and can be used to enhance
results from cleavage site prediction. The multi-factor model augments existing
methods and complements experimental efforts to define the caspase degradome on
the systems-wide basis Wee [5]. Specificity is a prerequisite for systemic gene
therapy of hepatocarcinoma. In vitro, the tumor-specific viral death effector Apoptin
selectively induces apoptosis in malignant hepatic cells. Intratumoral treatment of
xenografted subcutaneous hepatomas with Apoptin results in tumor regression. Here,
a systemic delivery vehicle containing the Apoptin gene linked to asialoglycoprotein
(Asor), which targets asialoglycoprotein receptor (ASGPR) present only on the
surface of hepatocytes. In vitro, the protein-DNA complex Asor-Apoptin induced
apoptosis in HepG2 hepatocarcinoma cells but not in normal L-02 hepatocytes. Non-
hepatocyte-derived tumorigenic human A549 cells lacking the membrane ASGPR
were not affected by Asor-Apoptin. In vivo systemic delivery of Asor-Apoptin via the
tail vein into mice bearing in situ hepatocarcinoma resulted in specific and efficient
distribution of Apoptin in both hepatocarcinoma cells and normal hepatocytes. Five
days after injection of Asor-Apoptin, the in situ hepatocarcinomas showed significant
signs of regression, whereas the surrounding normal hepatocytes did not. Systemically
delivered Asor-LacZ expressing non-apoptotic LacZ gene did not inhibit tumor
growth. Our data reveal that systemic delivery of Asor-Apoptin specifically induces
apoptosis in malignant hepatocytes and thus constitutes a powerful and safe
therapeutics against hepatocarcinomas Peng [6]. The initiation and progression of
tumor is regulated by multiple genes. Survivin belongs to the inhibitor of apoptosis
protein (IAP) family and is overexpressed in most types of human tumors. Apoptin,
originally identified from chicken anemia virus (CAV), can specifically induce
apoptosis of human tumor cells rather than normal cells. In this study, survivin

5
expression was silenced by microRNA (miRNA)-mediated RNA interference (RNAi);
meanwhile, the engineered miRNA vector was also designed to express apoptin gene.
The apoptosis and cell growth were then examined by flow cytometry and MTT
assay. The miRNA-mediated knockdown of survivin in combination with apoptin
overexpression significantly induced apoptosis and inhibited cell growth. Importantly,
the combined strategy was more effective on inducing apoptosis and inhibiting cell
growth than either survivin downregulation or apoptin overexpression alone. Taken
together, the combined strategy offers potential advantages in control of
tumorigenesis, and thus deserves further research as a preferred approach in cancer
gene therapy Liu [7].
MATERIALS AND METHODS
MATERIALS
Host Bacterial strains
Escherichia coli (E.coli) DH5α (Proteges, Madison) host strain was used for isolation
of recombinant plasmid.
Chemicals, glasswares and plastic wares
All chemicals used in this study were either Analar or molecular biology grade from
Sigma (MO), Promega (Madison). Plasticwares and other consumables were from
Axygen, TRP, Nunc, Gneiner, Tarson, Corning or Borosil.
METHODS
Revival of the E.coli culture containing recombinant plasmid with
VP3 gene
Growth of rE.coli cells:
Preparation of LB broth :
2.5gm LB broth in 100ml d/w, sterilized by autoclaving at 15 lbs psi (1210C) for 15
min.
Preparation of LB Agar:
4.0 gm LB Agar powder in 100 ml d/w sterilized by autoclaving as above.
LB Agar Plate: Prepared LB agar plate by dispersing LB agar, cooled to 420C and
added ampicillin to provide concentration of 100µg/ml. Add given rE.coli culture by
streaking method.

6
For preparing E.coli culture in LB broth containing 100µg/ml ampicillin, inoculated
200µl culture in 100 ml broth and kept it in incubator at room temperature (370C) for
24 hours.
Glass test tubes with plastic caps are suitable. Place the tubes at a suitably inclined
angle to achieve good agitation.
Isolation of plasmid DNA containing VP3 gene
Isolation of plasmid DNA miniprep by TELT method-
TELT Solution
50 mM Tris.Cl, pH 8.0
62.5 mM Na2EDTA
2.5 M LiCl
4% (v/v) Triton X-100
1:1 (v/v) phenol/chloroform
Note: All steps are performed at room temperature.
Procedure:
Grown a 1.5 ml E.coli culture with plasmid of interest in LB medium containing
100 mg/ml ampicillin for 16h. Pellet cells 30 sec in micro centrifuge. Resuspend in
100µl TELT solution and add an equal volume of 1:1 phenol/chloroform. Vortex
vigorously 15 sec and spin 1 min in microcentrifuge at 220C. Collect the upper phase
of nucleic acid and mix with 2 vol of 100% ethanol. After 2 minutes, spin 10 minutes
in microcentrifuge at 40C. Wash pellet with 1ml of 70% ethanol, dry under vacuum,
and resuspend in 30µl TE buffer. The pellet may be located as a smeer on one side of
the tube. Dissolve the DNA on a shaker.
Plasmid DNA was isolated and run on agarose gel electrophoresis which yielded
good amount of DNA. Contaminating RNA may interfere with detection of DNA
fragments on the agarose gel; it can be destroyed by adding 1 µl of a 10 mg/ml RNase
soloution (DNase-free) to the digestion mixture.
Agarose gel electrophoresis:
Prepared loading Buffer: TAE (50 X for 100 ml).
Tris Base 24.2 gm
Glatial Acetic Acid 5.71 gm
0.5 M EDTA (pH 8.0) 10 ml

7
Distilled water was added to make the final volume up to 100 ml. A working solution
of 1X was used.
Preparation of loading dye: Bromophenol blue 25% and Sucrose 40% in d/w and mix
it.
Procedure:
Prepared 1% agarose in 1X TAE buffer and heated till it dissolved. Cooled the
solution to about 600C and then added ethidium bromide at a concentration of 0.5
µg/ml. Gel tray was sealed with tape on its open ends and placed the comb on the tray.
Then pour the above gel on the sealed tray. Leave it to solidify. Remove the comb and
tape. The gel tray was put in electrophoretic tank containing 1X TAE buffer. The
DNA marker and sample DNA were loaded into the slots of submerged gel, using
bromophenol dye buffer and run at for about 70-80 volts.
Further inoculated the culture for maintenance.
Preservation of the culture:
It is used for back up and for research work.
It is preserved in glycerol:
Glycerol 10 ml
Autoclave it.
Mixed 200µl glycerol with 800µl of culture and stored at -200C.
RESULTS
The plates containing recombinant E.coli culture were found to contain colonies,
from which ten discrete colonies were selected randomly and grown in LB broth
containing ampicillin and the recombinant pTARGET CAV-VP3 plasmids were
isolated. The DNA Bands were visualized under UV light and photographed.
DISCUSSION
Chicken anemia virus (CAV), etiological agent of a highly immunosuppressive
disease of yong chicken, i.e. chicken infectious anemia (CIV), is economically
important avian pathogen worldwide. It belongs to Gyrovirus genus of the
Circoviridae family having circular single stranded DNA genome of 2.3 kb in size.
The disease is characterized by severe anemia, aplasia of bone marrow and
generalized lymphoid atropy with concomitant immunosuppression. Among the viral
proteins of Chicken anemia virus (CAV), VP3 has the death inducing abilities and so

8
it was renamed as apoptin. Apoptin can induce apoptois in cell lines derived from a
great variety of human tumors.
REFERENCES
Reed, J.C. (2000). Mechanisms of apoptosis, Am. J. Pathol. 157, 1415-1430.
Los, M., Panigrahi, S., Rashedi, I., Mandal, S., Stetefeld, J., Essmann, F., and
Schulze-Osthoff, K. (2009). Apoptin, a tumor-selective killer, Biochem.
Biophys. Acta. 8, 1335-42.
He, M., Wilde, A. and Kaderbhai, M.A. (1990). A simple single step procedure for
small scale preparation of Escherichia Coli plasmid, Nucleic Acids Res., 18,
1660.
Noteborn, H. (2009). Proteins selectively killing tumor cells, Eur. J. Pharmacol. 625,
165-73.
Wee, L.J., Tong, J.C., Tan, T.W. and Ranganathan, S. (2009). A multi-factor model
for caspase degradome prediction, BMC Genomics. 10, Supplements 3.
Peng, D.J., Sun, J., Wan, Y.Z., Tian, J., Zhang, Y.H., Noteborn, M.H. and Qu, S.
(2007). Inhibition of hepatocarcinoma by systemic delivery of Apoptin gene
via the hepatic asialoglycoprotein receptor, Cancer Gene Therapy. 14, 66-73.
Liu, Q., Fu, H., Xing, R., Tie, Y., Zhu, J., Sun, Z., and Zheng, X. (2008). Survivin
knockdown combined with apoptin overexpression inhibits cells growth
significantly, Cancer Biol. Ther. 7, 1053-60.