Embed Size (px)
Citation preview

Clinical and laboratory evidence for a trilineage haematopoietic
defect in patients with refractory Diamond±Blackfan anaemia
N. GIRI,1 E. KAN G,1,2 J. F. TISDA LE,2 D. FOLLMAN,3 M. RIVERA,1 G. N. SCHWA RTZ,4 S. KIM,1 N. S. YOUNG,1
M. E. RICK4
AN D C. E. DUNBA R5 1Hematology Branch, NHLBI, 2Molecular and Clinical Haematology Branch, NIDDK,
3Of®ce of Biostatistics Research, NHLBI, 4Experimental Transplantation and Immunology, and 5Hematology Service,
Clinical Pathology, Clinical Centre, NIH, Bethesda, MD, USA
Received 17 June 1999; accepted for publication 24 September 1999
Summary. Diamond±Blackfan anaemia (DBA) is a constitu-tional pure red cell aplasia presenting in early childhood. Insome patients, neutropenia and/or thrombocytopenia havealso been observed during the course of the disease. We havefollowed 28 patients with steroid-refractory DBA for up to 13years with serial peripheral blood counts and bone marrow(BM) aspirates and biopsies. In 21/28 (75%) patients,moderate to severe generalized BM hypoplasia developed,with overall cellularities ranging from 0% to 30%. Marrowhypoplasia correlated with the development of neutropenia(9/21; 43%) and/or thrombocytopenia (6/21; 29%) in manypatients. No patient had either cytogenetic abnormalities orprogressed to acute leukaemia, although one 13-year-olddeveloped marked marrow ®brosis and trilineage dysplasia.We used the in vitro long-term culture-initiating cell (LTC-IC)assay to quantify multilineage, primitive haematopoietic
progenitors in a representative subset of these patients. LTC-IC assays showed equivalent frequencies of cobblestone area-forming cells (CAFCs) with a mean of 5?42/105 cells 6 1?9SD and 6?13/105 cells 6 2?6 SD in nine patients and sixnormal controls respectively. The average clonogenic celloutput per LTC-IC, however, was signi®cantly lower in DBApatients (mean 2?16 6 1?2 SD vs. 7?36 6 2?7 SD in normalcontrols, P�0?0008). Our results suggest that the under-lying defect in patients with severe refractory DBA may notbe limited to the erythroid lineage, as was evidenced by thedevelopment of pancytopenia, bone marrow hypoplasia andreduced clonogenic cell output in LTC-IC assays.
Keywords: Diamond±Blackfan anaemia, neutropenia,thrombocytopenia, bone marrow hypoplasia, long-termculture-initiating cell assay.
The classic haematological pro®le in patients with Diamond±Blackfan anaemia (DBA) consists of normochromic macro-cytic anaemia presenting at or soon after birth, reticulo-cytopenia and a normocellular marrow with selectivede®ciency of red cell precursors (Diamond & Blackfan,1939; Alter & Young, 1998).The success of allogeneicmarrow transplantation in restoring normal erythroidactivity indicates that the defect is intrinsic to haematopoie-tic progenitor or stem cells (Lenarsky et al, 1988). Theresponse of a majority of patients to prednisone and, lessfrequently, to other immunosuppressive agents is puzzling ina congenital disorder with little other evidence for animmune aetiology. A number of different defects of in vitroerythroid progenitor proliferation, differentiation andcytokine responsiveness have been reported but have not
clari®ed the mechanism of in vivo erythroid failure (Liptonet al, 1986; Halperin et al, 1989; Tsai et al, 1989; Casadevallet al, 1994; McGuckin et al, 1995). DBA is familial in 10±15% of cases, and a molecular defect in a ribosomal proteingene on chromosome 19 has been identi®ed in some familiesand sporadic patients, although the mechanism for this genedefect producing the phenotype is completely unknown(Gustavsson et al, 1997; Draptchinskaia et al, 1999).
Reports of DBA patients developing neutropenia and/orthrombocytopenia and leukaemia suggest that, at least insome cases, the defect may involve multilineage, primitivehaematopoietic precursor cells (Wasser et al, 1978; Bucha-nan et al, 1981; Scho®eld & Evans, 1991; Casadevall et al,1994). As more patients with DBA are effectively chelatedfrom early childhood, the life expectancy of patients witheven steroid-refractory disease has increased, analogous toresults reported for patients with thalassaemia (Brittenhamet al, 1994; Janov et al, 1996). This improvement insurvival could reveal previously unrecognized long-term
British Journal of Haematology, 2000, 108, 167±175
167q 2000 Blackwell Science Ltd
Correspondence: Dr Cynthia Dunbar, Bldg 10/Room 7C103,National Institutes of Health, 9000 Rockville Pike, Bethesda, MD
20892-1652, USA.

consequences of the underlying defect or defects. We havefollowed a cohort of 28 patients with steroid-refractory DBAfor up to 13 years, many into adulthood, with multiplecomprehensive haematological evaluations, including bonemarrow (BM) aspirations and biopsies. We now report thedevelopment of marked BM hypocellularity, accompanied byneutropenia and/or thrombocytopenia in a signi®cantfraction of patients, supporting the hypothesis that thedefect in at least some DBA patients affects more primitivehaematopoietic cells and is not restricted to the erythroidlineage. The results of long-term culture-initiating cell (LTC-IC) assays, performed prospectively on marrow samplesobtained from a representative subset of these patients, alsoindicate a more global haematopoietic defect.
MATERIALS AND METHODS
Patient populationCase records of all 28 patients with an unequivocal diagnosisof DBA followed at the National Institute of Health (NIH)during the 13-year period from 1986 to 1999 werereviewed. All patients met criteria for the diagnosis of DBA,with anaemia, reticulocytopenia and selective severe ery-throid hypoplasia of the marrow (Diamond & Blackfan,1939; Alter & Young, 1998). At the time of initial referral toNIH, all patients were steroid non-responders or unable to bemaintained on non-toxic doses of steroids, and were thus redcell transfusion dependent. Patients were enrolled inInstitutional Review Board-approved protocols for thestudy of iron chelation ef®cacy and/or cytokine or antithy-mocyte globulin treatment protocols. Informed consent forparticipation in research studies at the NIH was obtainedfrom patients or their guardians. All patients underwentevaluations, including complete blood counts, bone marrowaspiration and biopsy, standard cytogenetic studies, diepoxy-butane challenge testing of lymphocytes to exclude thediagnosis of Fanconi's anaemia, ¯ow cytometry of lympho-cyte populations and polymerase chain reaction (PCR) forserum B19 DNA to exclude chronic parvoviraemia as acause of erythroid hypoplasia (Frickhofen & Young, 1989).
Review of bone marrow aspirations and biopsiesAll BM aspirates and biopsies performed at the NIH on the 28DBA patients were reviewed. Marrow aspirates and biopsieswere collected from the posterior superior iliac crest usingstandard techniques. Aspirate clot sections and biopsies were®xed in 10% formalin. Biopsies were further decalci®ed, andboth biopsies and clot sections were then sectioned andstained with haematoxylin and eosin. Aspirate particles weresmeared on a slide and stained with Wright's solution.Patient identity on each biopsy or aspirate clot section slidewas blocked, and the slides were then reviewed indepen-dently by three haematologists. BM cellularity was estimatedby scanning the entire core biopsy (an average of three orfour low-power ®elds) and then visually comparing thecellularity with a standard series of reference photographicexamples of cellularity (Tuzuner & Bennett, 1994). Marrowcellularity was reported as a single percentage value ifsamples were homogenous. In cases of non-homogenous
cellularity, the value reported represented a range, which didnot exceed 20% in any single sample. In two cases in whichthe biopsy was insuf®cient for cellularity determination,aspirate clot cellularity was used and reported similarly.Cellularity estimates from all three observers were used tocalculate a mean cellularity estimate, which was used instatistical analysis. Highly concordant assignments ofcellularity were obtained among the three observers.
Long-term culture-initiating cell (LTC-IC)assayLTC-IC assays were performed according to previouslydescribed methods with slight modi®cations (Gartner &Kaplan, 1980; Sutherland et al, 1990, 1991; Eaves et al,1991; Breems et al, 1994; Pettengell et al, 1994). BMaspirates obtained from six healthy donors and nine DBApatients were subjected to Ficoll density gradient centrifuga-tion using lymphocyte separation medium (ICN Biomedicals,OH, USA). Mononuclear cells (MNCs) recovered from thelow-density fraction were washed twice with phosphate-buffered saline (PBS; Bio¯uids, Rockville, MD, USA), resus-pended in Dulbecco's modi®ed Eagle medium (DMEM; LifeTechnologies, Gaithersburg, MD, USA) supplemented with10 ml/ml penicillin-streptomycin, 5 ml/ml L-glutamine and100 ml/ml fetal calf serum (FCS; Life Technologies) andplated in T75 culture ¯asks at 378C in 5% CO2 in air toremove the adherent cell fraction. The non-adherent cellswere recovered 24 h later and resuspended in MyeloCult(Stem Cell Technologies, Vancouver, BC, Canada) supple-mented with 10ÿ6mol/l hydrocortisone 21-hemisuccinate(Sigma, St Louis, MO, USA). For LTC-IC determination, MNCswere co-cultured with the M2-10B4 stromal cell line, used asa feeder layer in all cultures (Sutherland et al, 1991). Thestroma was irradiated (15 Gy of 250 kV X-rays) before theaddition of MNCs.
The frequency of LTC-IC was estimated by cobblestonearea-forming cell (CAFC) frequency in limiting dilutionassays, which were performed in 96-well ¯at-bottomedmicrowell plates (Costar; Corning, NY, USA) containingpreformed irradiated stroma. The inner 60 wells were seededwith decreasing dilutions (5 ´ 104 to 5 ´ 102 cells/well) oflow-density cells, with ®ve to eight dilutions per sample and30 replicate wells per cell concentration. Cultures wereincubated at 338C in 5% CO2 and fed weekly by semi-depletion. After 5 weeks, the plates were viewed under aphase-contrast microscope. Wells were scored as positive forCAFCs if cobblestone areas consisted of 20 or more smalltightly packed non-refractile cells (Pettengell et al, 1994).
Bulk long-term cultures were used to estimate the colony-forming cell (CFC) capacity of LTC-IC. Two to fourconcentrations of MNCs were plated in duplicate in 35-mmtissue culture plates (Costar; Corning) containing preformedirradiated stroma. Cultures were incubated at 338C in 5%CO2 and fed weekly by semi-depletion. After 5 weeks, thesupernatant containing non-adherent cells from each bulkculture was combined with its corresponding trypsinizedstromal layer, and all cells were plated in duplicate inmethylcellulose, MethoCult (Stem Cell Technologies)supplemented with of 20 ng/ml interleukin-3 (IL-3), 50 ng/ml stem cell factor, 20 ng/ml granulocyte±macrophage
q 2000 Blackwell Science Ltd, British Journal of Haematology 108: 167±175
168 N. Giri et al

169Pancytopenia in DBA
q 2000 Blackwell Science Ltd, British Journal of Haematology 108: 167±175
colony-stimulating factor (GM-CSF), 20 ng/ml interleukin-6(IL-6) and 3 U/ml erythropoietin (all Amgen, ThousandOaks, CA, USA). Cultures were set up in 6-well tissue cultureplates (Costar; Corning) and incubated at 378C in 5% CO2 for14±21 days. Colonies containing more than 50 cells werescored as positive for CFCs. In three normal donors and ®veDBA cultures, secondary CFCs were also assessed for burst-forming unit erythroid (BFU-E) and colony-forming unitgranulocyte macrophage (CFU-GM).
Statistical analysisThe multiplicity of LTC-IC was determined using single-hitkinetics according to Poisson distribution as described byFazekas de St Groth (1982). The statistical program thatperformed the calculations for LTC-IC was established usingMicrosoft Excel 5?0 (Microsoft, Redmond, WA, USA). As thenumber of clonogenic cells (output) present after 5 weeks inLTC has been shown to be a linear function of the number ofcells initially (input) plated, the clonogenic capacity of singleLTC-IC was calculated by dividing the numbers of colonies(secondary CFCs) derived from bulk cultures by thefrequency of LTC-IC generated by limiting dilution assaysat the same concentration (Maciejewski et al, 1996).Unpaired Student's two-tailed t-test (SigmaPlot 97 SPSSInc.) was used to test for differences between normal controlsand DBA patients in LTC-IC frequency and secondary CFCproduction.
Statistical relationships between absolute neutrophilcount, marrow cellularity, platelet counts and age wereestimated using mixed regression models to accommodatemultiple measurements from individual patients (Laird &Ware, 1982). Regression lines were estimated by the methodof Laird & Ware (1982), taking into account variablenumbers of data points for each subject. In each plot, theboundary of the shaded area was two standard deviationsbelow the mean in normal subjects of the same age, asreported in standard references (Hartsock et al, 1965;Dallman, 1977; Friebert et al, 1998). All tests were two-sided.
RESULTS
Patient characteristicsAmong the 28 patients, there were 16 males and 12 females(Table I). All patients presented with anaemia before the ageof 1 year. The median age at diagnosis was 2 months (range0±60 months). All patients were reported to have normalwhite blood cell and platelet counts at the time of diagnosisin infancy. BM aspirates and/or biopsies on all patientsobtained at ®rst presentation showed a selective de®ciency oferythroid precursors with normal myeloid and megakaryo-cytic morphology and maturation. Associated constitutionalanomalies were noted in six patients, including urogenitalanomalies in three (patients 9, 20 and 21), Cathie face in one(patient 5), pancreatic insuf®ciency in one patient who didnot have neutropenia for the ®rst 10 years of life, and thuswas classi®ed as DBA as opposed to Shwachman±Diamondsyndrome (patient 7) and skeletal anomalies in one (patient12). One patient with multiple congenital anomalies was
also mentally retarded (patient 9). Two of the patientsincluded in this analysis were brothers (patients 3 and 4);one (patient 12) had a half-brother and one (patient 20) a®rst cousin with DBA.
Previous treatmentThe age at referral to the NIH ranged from 1 to 23 years(median 6 years). All patients were referred because they hadeither failed prior therapy with prednisone or had experi-enced unacceptable toxicity from corticosteroid dosesnecessary to avoid transfusions. Thus, all were transfusiondependent at the time of referral and initial evaluation. Ofthe 28 patients, eight (29%) had initially achieved acomplete remission (CR) and had needed neither corticoster-oids nor transfusion for 2±6 years until relapse; four (14%)were partial responders with minimal transfusion require-ments on steroids who later relapsed; and 16 (57%) hadnever responded to steroids. Most patients had received othertherapies either previously or during the course of this study,including cyclosporine and prednisone (n�10) (Leonardet al, 1989), antithymocyte globulin and/or gammaglobulins (n�3), GM-CSF (n�6) or IL-3 (n�22) (Dunbaret al, 1991). These treatments were unsuccessful with thefollowing exceptions: one patient (patient 19) responded tocyclosporine and prednisone and has remained in CR forover 9 years; one patient (patient 6) treated with IL-3 for 1month at age 7 years remains in a sustained CR, now formore than 7 years; two others (patients 15 and 1) hadtransient responses to IL-3 for 6 months and 30 monthsrespectively (Dunbar et al, 1991). Neutrophil counts rose inall patients during GM-CSF or IL-3 therapy, and marrowcellularity increased as a result of myeloid hyperplasia.Hence, data on neutrophil and platelet counts and marrowcellularity during or within 3 months after cyclosporine,steroid or cytokine therapy were not included in thisanalysis. One patient (patient 4) had cyclic neutropenia inaddition to red cell aplasia, which responded to ATG and G-CSF therapy. Two patients (patients 14 and 15) underwentbone marrow transplantation, one of whom is alive anddoing well 5 years after matched unrelated donor transplant(patient 14). Six patients have died from various complica-tions, including iron overload (patient 1), chondroblasticosteogenic sarcoma of the jaw (patient 12), hepatic failurefollowing chronic active hepatitis (patient 7), renal failure(patient 9), chronic GVHD (patient 15) and sepsis and corpulmonale (patient 23).
Longitudinal pro®le of peripheral blood countsThe red cell lineage was de®cient in all patients atpresentation, as evidenced by marked reticulocytopeniaand transfusion dependency. Nineteen out of the 22 patientsalive continue to require regular transfusions and chelation.The absolute neutrophil counts (ANCs) at presentation toNIH varied over a wide range from 5 ´ 98 to 6 ´ 5 ´ 109/lwith a median of 2 ´ 88 ´ 109/l (Table I and Fig 1A). Serialdeterminations of ANCs over the 13-year period (range 1±13 years; median 6 years) showed persistent or evolvingneutropenia in 11 (39%) patients (de®ned as below twostandard deviations from the age-speci®c normal values;
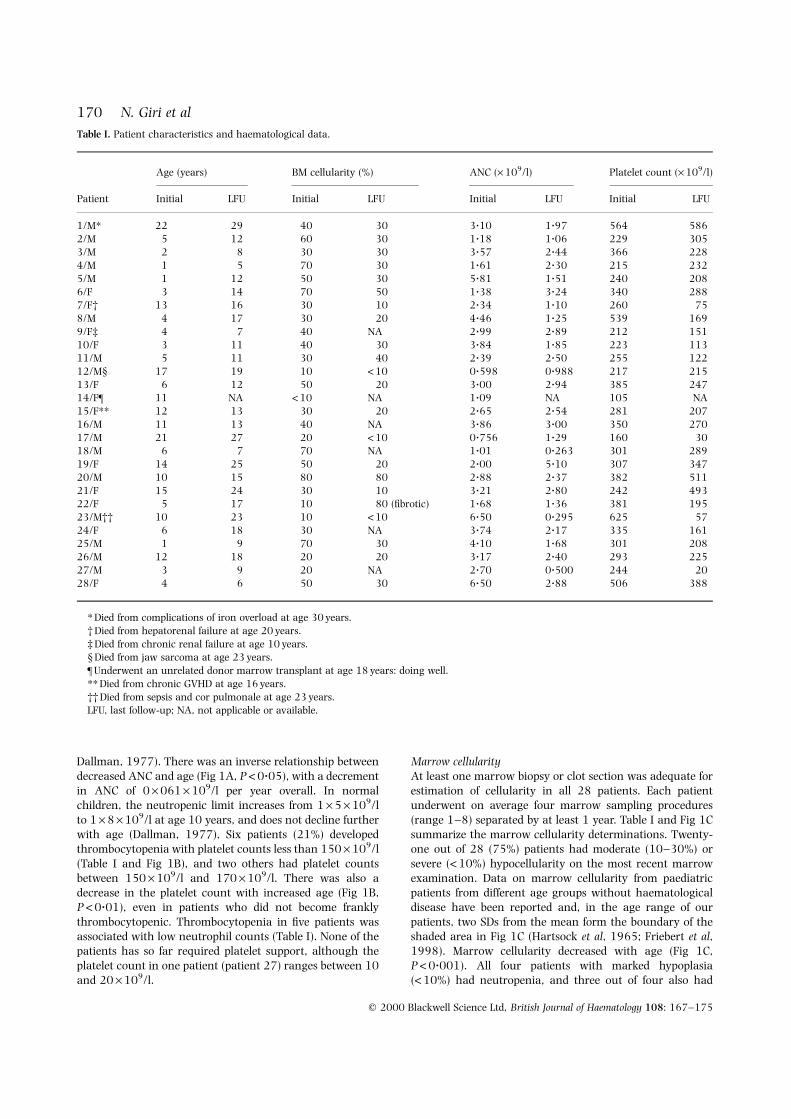
Dallman, 1977). There was an inverse relationship betweendecreased ANC and age (Fig 1A, P<0?05), with a decrementin ANC of 0 ´ 061 ´ 109/l per year overall. In normalchildren, the neutropenic limit increases from 1 ´ 5 ´ 109/lto 1 ´ 8 ´ 109/l at age 10 years, and does not decline furtherwith age (Dallman, 1977), Six patients (21%) developedthrombocytopenia with platelet counts less than 150 ´ 109/l(Table I and Fig 1B), and two others had platelet countsbetween 150 ´ 109/l and 170 ´ 109/l. There was also adecrease in the platelet count with increased age (Fig 1B,P <0?01), even in patients who did not become franklythrombocytopenic. Thrombocytopenia in ®ve patients wasassociated with low neutrophil counts (Table I). None of thepatients has so far required platelet support, although theplatelet count in one patient (patient 27) ranges between 10and 20 ´ 109/l.
Marrow cellularityAt least one marrow biopsy or clot section was adequate forestimation of cellularity in all 28 patients. Each patientunderwent on average four marrow sampling procedures(range 1±8) separated by at least 1 year. Table I and Fig 1Csummarize the marrow cellularity determinations. Twenty-one out of 28 (75%) patients had moderate (10±30%) orsevere (<10%) hypocellularity on the most recent marrowexamination. Data on marrow cellularity from paediatricpatients from different age groups without haematologicaldisease have been reported and, in the age range of ourpatients, two SDs from the mean form the boundary of theshaded area in Fig 1C (Hartsock et al, 1965; Friebert et al,1998). Marrow cellularity decreased with age (Fig 1C,P<0?001). All four patients with marked hypoplasia(<10%) had neutropenia, and three out of four also had
q 2000 Blackwell Science Ltd, British Journal of Haematology 108: 167±175
170 N. Giri et al
Table I. Patient characteristics and haematological data.
Age (years) BM cellularity (%) ANC (´ 109/l) Platelet count (´ 109/l)
Patient Initial LFU Initial LFU Initial LFU Initial LFU
1/M* 22 29 40 30 3?10 1?97 564 586
2/M 5 12 60 30 1?18 1?06 229 3053/M 2 8 30 30 3?57 2?44 366 228
4/M 1 5 70 30 1?61 2?30 215 232
5/M 1 12 50 30 5?81 1?51 240 208
6/F 3 14 70 50 1?38 3?24 340 2887/F² 13 16 30 10 2?34 1?10 260 75
8/M 4 17 30 20 4?46 1?25 539 169
9/F³ 4 7 40 NA 2?99 2?89 212 151
10/F 3 11 40 30 3?84 1?85 223 11311/M 5 11 30 40 2?39 2?50 255 122
12/M§ 17 19 10 <10 0?598 0?988 217 215
13/F 6 12 50 20 3?00 2?94 385 24714/F¶ 11 NA <10 NA 1?09 NA 105 NA
15/F** 12 13 30 20 2?65 2?54 281 207
16/M 11 13 40 NA 3?86 3?00 350 270
17/M 21 27 20 <10 0?756 1?29 160 3018/M 6 7 70 NA 1?01 0?263 301 289
19/F 14 25 50 20 2?00 5?10 307 347
20/M 10 15 80 80 2?88 2?37 382 511
21/F 15 24 30 10 3?21 2?80 242 49322/F 5 17 10 80 (®brotic) 1?68 1?36 381 195
23/M²² 10 23 10 <10 6?50 0?295 625 57
24/F 6 18 30 NA 3?74 2?17 335 16125/M 1 9 70 30 4?10 1?68 301 208
26/M 12 18 20 20 3?17 2?40 293 225
27/M 3 9 20 NA 2?70 0?500 244 20
28/F 4 6 50 30 6?50 2?88 506 388
* Died from complications of iron overload at age 30 years.
² Died from hepatorenal failure at age 20 years.
³ Died from chronic renal failure at age 10 years.§ Died from jaw sarcoma at age 23 years.
¶ Underwent an unrelated donor marrow transplant at age 18 years: doing well.
** Died from chronic GVHD at age 16 years.
²² Died from sepsis and cor pulmonale at age 23 years.LFU, last follow-up; NA, not applicable or available.
171Pancytopenia in DBA
q 2000 Blackwell Science Ltd, British Journal of Haematology 108: 167±175
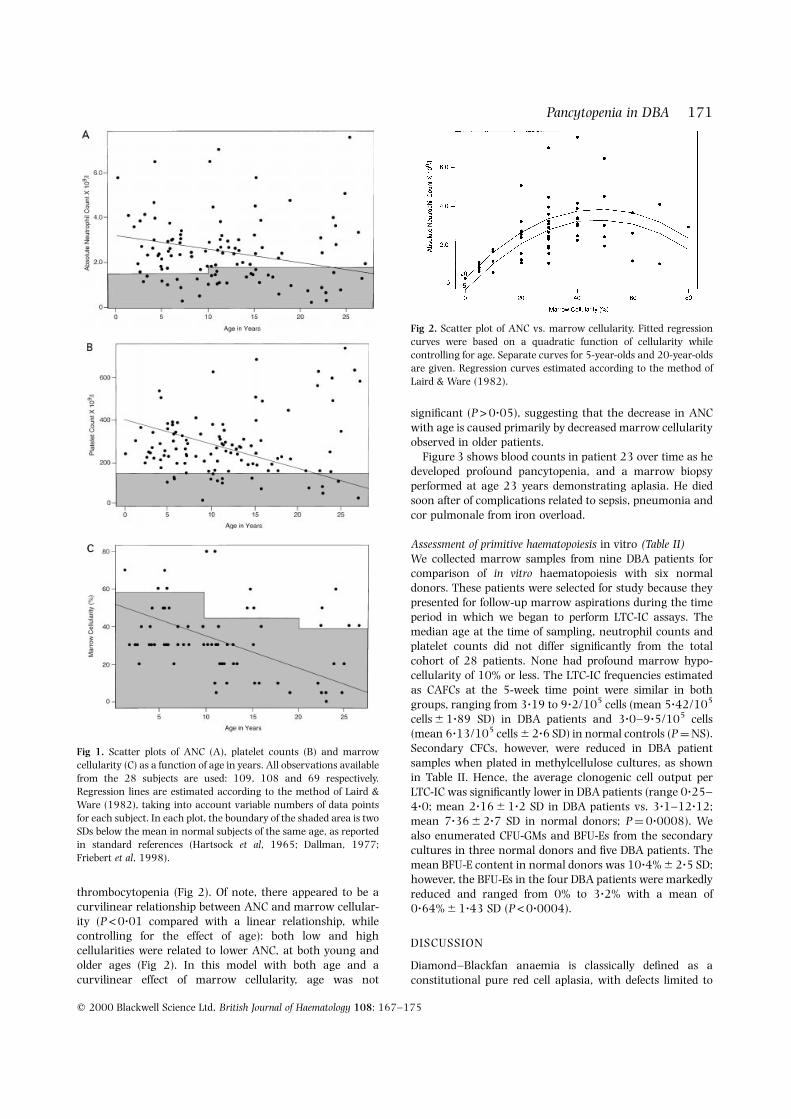
thrombocytopenia (Fig 2). Of note, there appeared to be acurvilinear relationship between ANC and marrow cellular-ity (P <0?01 compared with a linear relationship, whilecontrolling for the effect of age): both low and highcellularities were related to lower ANC, at both young andolder ages (Fig 2). In this model with both age and acurvilinear effect of marrow cellularity, age was not
signi®cant (P>0?05), suggesting that the decrease in ANCwith age is caused primarily by decreased marrow cellularityobserved in older patients.
Figure 3 shows blood counts in patient 23 over time as hedeveloped profound pancytopenia, and a marrow biopsyperformed at age 23 years demonstrating aplasia. He diedsoon after of complications related to sepsis, pneumonia andcor pulmonale from iron overload.
Assessment of primitive haematopoiesis in vitro (Table II)We collected marrow samples from nine DBA patients forcomparison of in vitro haematopoiesis with six normaldonors. These patients were selected for study because theypresented for follow-up marrow aspirations during the timeperiod in which we began to perform LTC-IC assays. Themedian age at the time of sampling, neutrophil counts andplatelet counts did not differ signi®cantly from the totalcohort of 28 patients. None had profound marrow hypo-cellularity of 10% or less. The LTC-IC frequencies estimatedas CAFCs at the 5-week time point were similar in bothgroups, ranging from 3?19 to 9?2/105 cells (mean 5?42/105
cells 6 1?89 SD) in DBA patients and 3?0±9?5/105 cells(mean 6?13/105 cells 6 2?6 SD) in normal controls (P�NS).Secondary CFCs, however, were reduced in DBA patientsamples when plated in methylcellulose cultures, as shownin Table II. Hence, the average clonogenic cell output perLTC-IC was signi®cantly lower in DBA patients (range 0?25±4?0; mean 2?16 6 1?2 SD in DBA patients vs. 3?1±12?12;mean 7?36 6 2?7 SD in normal donors; P�0?0008). Wealso enumerated CFU-GMs and BFU-Es from the secondarycultures in three normal donors and ®ve DBA patients. Themean BFU-E content in normal donors was 10?4% 6 2?5 SD;however, the BFU-Es in the four DBA patients were markedlyreduced and ranged from 0% to 3?2% with a mean of0?64% 6 1?43 SD (P<0?0004).
DISCUSSION
Diamond±Blackfan anaemia is classically de®ned as aconstitutional pure red cell aplasia, with defects limited to
Fig 1. Scatter plots of ANC (A), platelet counts (B) and marrow
cellularity (C) as a function of age in years. All observations available
from the 28 subjects are used: 109, 108 and 69 respectively.Regression lines are estimated according to the method of Laird &
Ware (1982), taking into account variable numbers of data points
for each subject. In each plot, the boundary of the shaded area is two
SDs below the mean in normal subjects of the same age, as reportedin standard references (Hartsock et al, 1965; Dallman, 1977;
Friebert et al, 1998).
Fig 2. Scatter plot of ANC vs. marrow cellularity. Fitted regression
curves were based on a quadratic function of cellularity while
controlling for age. Separate curves for 5-year-olds and 20-year-oldsare given. Regression curves estimated according to the method of
Laird & Ware (1982).

the erythroid lineage (Diamond & Blackfan, 1939; Alter &Young, 1998). Many patients initially respond to steroidsand become transfusion independent. However, about halfeither never respond or cannot be maintained on a tolerabledose of steroids because of side-effects, and thus requirechronic transfusion therapy (Janov et al, 1996). Before theavailability of effective chelation therapy, most DBA patientsdied of complications related to iron overload by the seconddecade of life. Over the past 20 years, desferroxaminechelation has resulted in a majority of patients nowsurviving into adulthood, allowing a more complete assess-ment of the natural history of the disease (Brittenham et al,1994; Janov et al, 1996). In our study, we have followed 28patients with steroid-refractory DBA for prolonged periodsand have evidence for a more global haematopoietic defect,at least in the subset of DBA patients refractory to steroidsand requiring long-term transfusions.
It is of interest that the original report of congenitalhypoplastic anaemia by Diamond & Blackfan (1939) notedthat some patients had mild neutropenia and thrombocyto-penia, although no actual numerical values were given. In amore recent study, three out of 16 patients were noted to bethrombocytopenic with no evidence of qualitative plateletdysfunction (Buchanan et al, 1981). Other case reports andsmall laboratory-based studies have also described thedevelopment of cytopenias or myeloid progenitor growthabnormalities in DBA patients (Diamond & Blackfan, 1939;Buchanan et al, 1981; Scho®eld & Evans, 1991; Casadevallet al, 1994; McGuckin et al, 1996; Santucci et al, 1999). Thelarge long-term study of patients followed at BostonChildren's Hospital by Dr Diamond and his successors did
not include data on neutrophil counts, but total white bloodcell counts were reduced in about a quarter of patients, andplatelets were less than 150 ´ 109/l in 15% (Janov et al,1996). The trend for the development of leucopenia andthrombocytopenia with time did not reach statisticalsigni®cance in that study. The patient cohort included allpatients with a diagnosis of DBA, including those withspontaneous or steroid-induced remissions.
In contrast, our study found that a statistically signi®cantnumber of steroid-refractory DBA patients developed neu-tropenia and/or thrombocytopenia with increasing age. Sixout of the 11 patients followed beyond the age of 15 yearshad marked cytopenias, and one in fact died fromneutropenic sepsis. A unique feature of our study was thesequential examination of bone marrow biopsies. Seventy-®ve per cent of these patients had moderate to severetrilineage hypocellularity. The four patients with profoundhypocellularity (<10%) had neutropenia, and three out offour also had thrombocytopenia. This suggests that thedecrease in platelet and neutrophil counts results from aproduction defect and is not caused by increased clearance inthe spleen resulting from hepatic haemosiderosis andhypersplenism or the development of autoantibodies assuggested previously (Singer et al, 1978; Dianzani et al,1996). Much larger cohorts of patients receiving chronictransfusional therapy for thalassaemias or sickle cellanaemia have not been reported to develop cytopenias,and thus these ®ndings are unlikely to result fromconsequences of iron overload or transfusion-associatedviruses.
None of our patients actually developed leukaemia,
q 2000 Blackwell Science Ltd, British Journal of Haematology 108: 167±175
172 N. Giri et al
Table II. Long-term culture-initiating cell assays.
LTC-IC (105 cells) CFC (105 cells) CFC/LTC-IC BFU-E (%)
Normal donors
1 5?6 17?5 3?11 11?42 3 14?6 4?9 12?3
3 6?6 80 12?12
4 8?5 41 4?85 9?5 71 7?4
6 3?6 29?5 8?2 7?3
Mean 6 SD 6?13 6 2?59 42?26 6 27?54 7?36 6 2?70 10?4 6 2?50
DBA patients
2 5?8 7?8 1?3 0
3 4?6 15?7 3?4 3?24 4?75 1?2 0?25 0
6 5?1 20?4 4?0
10 3?4 10?0 2?9
13 6?1 7?7 1?318 3?19 9?4 2?9 0
19 7?3 12?0 1?6
28 9?2 17?0 1?8 0
Mean 6 SD 5?42 6 1?89 11?24 6 5?7 2?16 6 1?2 0?64 6 1?43
P-value 0?54 0?005 0?0008 0?0004

173Pancytopenia in DBA
q 2000 Blackwell Science Ltd, British Journal of Haematology 108: 167±175
although one patient did develop a ®brotic and dysplasticmarrow and cytogenetic abnormalities (5q±). The follow-uptime and/or the sample size, although relatively large for asingle centre, may have been inadequate for the detection ofan increased leukaemia risk. A previous large long-termfollow-up study did ®nd a statistically signi®cant increase inleukaemia in older DBA patients, and there are case reportsof leukaemias in these patients (Wasser et al, 1978; Mori et al,1992; Janov et al, 1996).
The frequency of global haematopoietic dysfunction inmany of our patients led us to quantify primitive haemato-poietic cell function using the long-term culture-initiatingcell assays. The actual frequency of LTC-IC after more than 5weeks in stromal culture per marrow mononuclear cellplated was not signi®cantly decreased in the nine patientsanalysed. The more striking ®nding was that each primitiveLTC-IC appeared to be hypoproliferative, with fewer CFCs perLTC-IC from patients with DBA. This not only includederythroid colonies, as might be expected, but also myeloidCFU-GMs. It has been shown recently that primitiveprecursors derived from patients with DBA have an impairedcapacity to proliferate and differentiate along not only theerythroid pathway but, in some cases, the granulocyte±
macrophage pathway in long-term bone marrow culturessupported by normal stroma. However, as limiting dilutionanalysis was not carried out, this study could not distinguishbetween decreased LTC-IC frequency and decreased prolif-erative potential per LTC-IC (Santucci et al, 1999).
If DBA does result from a defect that affects the function,differentiation or proliferation of more primitive, multi-potential cells, then why does it primarily manifest as a singlelineage de®ciency in vivo? The requirement for erythroid cellsfar exceeds that of the other lineages, and therefore subtledefects in proliferation may only manifest initially asanaemia. The W mouse strains, with mutations in thereceptor for stem cell factor (c-kit), have marked anaemia butpreserved platelet and neutrophil counts, despite a markedstem cell de®ciency state demonstrated by transplantationassays (Russell, 1979; Abkowitz et al, 1992). These are notactual murine models of DBA, as mutations in c-kit or SCFgenes have not been demonstrated (Mangan et al, 1982;Drachtman et al, 1992; Spritz & Freedman, 1993). Theidenti®cation of a gene defect likely to be responsible for thesyndrome in some DBA patients is exciting and may provideinsight into the cause of the global haematological defect(Gustavsson et al, 1997; Draptchinskaia et al, 1999).
Fig 3. Longitudinal analysis of ANC andplatelets in patient 23 (A) and
photomicrograph of a marrow biopsy obtained
at age 24 years (B); magni®cation 100 ´.

Whatever the aetiology, there are therapeutic implica-tions. If steroid-refractory DBA results from a defect thatcould ultimately lead to pancytopenia and/or furthermutational events resulting in malignancies in a highproportion of patients, it may be reasonable to considerpotentially curative but risky interventions, such as stem celltransplantation earlier and more frequently, especially givenimproving results in transplantation overall using alterna-tive donors. (Lenarsky et al, 1988; Margolis et al, 1996) Therecent formation of a DBA patient registry should help toclarify the frequency of all types of long-term complicationsand give patients, families and physicians better informationto help guide treatment decisions, especially regardingtransplantation.
ACKNOWLEDGMENTS
We thank Anjali Kaushiva and Brian Abbott for technicalassistance in performing the LTC-IC assays.
REFERENCES
Abkowitz, J.L., Broudy, V.C., Bennett, L.G., Zsebo, K.M. & Martin, F.H.(1992) Absence of abnormalities of c-kit or its ligand in two
patients with Diamond±Blackfan anemia. Blood, 79, 25±28.
Alter, B.P. & Young, N.S. (1998) The bone marrow failuresyndromes. In: Hematology of Infancy and Childhood, 5th edn (ed.
by D.G. Nathan & F.A. Oski), p. 237. W. B. Saunders, Philadelphia.
Breems, D.A., Blokland, E.A., Neben, S. & Ploemacher, R.E. (1994)
Frequency analysis of human primitive hematopoietic stem cellsubsets using a cobblestone area forming cell assay. Leukemia, 8,
1095.
Brittenham, G.M., Grif®th, P.M., Nienhuis, A.W., McLaren, C.E.,
Young, N.S., Tucker, E., Allen, C.J., Farrell, D.E. & Harris, J.W.(1994) Ef®cacy of deferoxamine in the prevention of complications
of iron overload in patients with thalassemia major. New England
Journal of Medicine, 331, 567±573.Buchanan, G.R., Alter, B.P., Holtkamp, C.A. & Walsh, E.G. (1981)
Platelet number and function in Diamond±Blackfan anemia.
Pediatrics, 68, 238±241.
Casadevall, N., Croisille, L., Auffray, I., Tchernia, G. & Coulombel, L.(1994) Age-related alterations in erythroid and granulopoietic
progenitors in Diamond±Blackfan anaemia. British Journal of
Haematology, 87, 369±375.
Dallman, P.R. (1977) Blood and blood forming tissues. In: Pediatrics(ed. by A.M. Rudolph), Appleton-Century-Crofts, New York.
Diamond, L.K. & Blackfan, K.D. (1939) Hypoplastic anemia.
American Journal of Diseases of Children, 56, 464±467.
Dianzani, I., Garelli, E. & Ramenghi, U. (1996) Diamond±Blackfananemia: a congenital defect in erythropoiesis. Haematologica, 81,
560±572.
Drachtman, R.A., Geissler, E.N. & Alter, B.P. (1992) The SCF and c-kit genes in Diamond±Blackfan anemia (letter). Blood, 79, 2177±
2178.
Draptchinskaia, N., Gustavsson, P., Anderson, B., Pettersson, M.,
Willig, T.N., Dianzani, I., Ball, S., Tchernia, G., Klar, J., Matsson,H., Tentler, D., Mohandas, N., Carlsson, B. & Dahl, N. (1999) The
gene encoding ribosomal protein S19 is mutated in Diamond±
Blackfan anaemia. Nature Genetics, 21, 169±175.
Dunbar, C.E., Smith, D., Kimball, J., Garrison, L., Nienhuis, A.W. &Young, N.S. (1991) Treatment of Diamond±Blackfan anaemia
with hematopoietic growth factors granulocyte-macrophage
colony stimulating factor and interleukin 3. British Journal of
Haematology, 79, 316±321.
Eaves, C.J., Cashman, J.D. & Eaves, A.C. (1991) Methodology of long-
term culture of human hemopoietic cells. Journal of Tissue CultureMethods, 13, 55±62.
Fazekas de St Groth, S. (1982) The evaluation of limiting dilution
assays. Journal of Immunological Methods, 49, R11±R23.
Frickhofen, N. & Young, N.S. (1989) Persistent parvovirus B19infections in humans. Microbial Pathogenesis, 7, 319±327.
Friebert, S.E., Shepardson, L.B., Shurin, S.B., Rosenthal, G.E. &
Rosenthal, N.S. (1998) Pediatric bone marrow cellularity: are weexpecting too much? Journal of Pediatric Hematological Oncology,
20, 439±443.
Gartner, S. & Kaplan, H.S. (1980) Long-term culture of human bone
marrow cells. Proceedings of the National Academy of Sciences of theUSA, 77, 4756±4759.
Gustavsson, P., Willig, T.N. & Van Haederingen, A. (1997)
Diamond±Blackfan anaemia: genetic homogeneity for a gene on
chromosome 19q13 restricted to 1.8 Mb. Nature Genetics, 16,368±371.
Halperin, D.S., Estrov, Z. & Freedman, M.H. (1989) Diamond±
Blackfan anemia: promotion of marrow erythropoiesis in vitro byrecombinant interleukin-3. Blood, 73, 1168±1174.
Hartsock, R.J., Smith, E.B. & Petty, C.S. (1965) Normal variations
with aging of the amount of hematopoietic tissue in bone marrow
from the anterior iliac crest. American Journal of Clinical Pathology,43, 326±331.
Janov, A., Leong, T., Nathan, D. & Guinan, E. (1996) Diamond±
Blackfan anemia, natural history and sequelae of treatment.
Medicine, 75, 77±87.Laird, J.N. & Ware, J.H. (1982) Random effects models for
longitudinal data. Biometrics, 88, 963±974.
Lenarsky, C., Weinberg, K., Guinan, E., Dukes, P.P., Barak, Y., Ortega,
J., Siegel, S., Williams, K., Lazerson, J., Weinstein, H. & Parkman,R. (1988) Bone marrow transplantation for constitutional pure
red cell aplasia. Blood, 71, 226±229.
Leonard, E.M., Raefsky, E., Grif®th, P., Kimball, J., Nienhuis, A.W. &Young, N.S. (1989) Cyclosporine therapy of aplastic anaemia,
congenital and acquired red cell aplasia. British Journal of
Haematology, 72, 278±284.
Lipton, J.M., Kudisch, M., Gross, R. & Nathan, D.G. (1986) Defectiveerythroid progenitor differentiation system in congenital hypo-
plastic (Diamond±Blackfan) anemia. Blood, 67, 962±968.
McGuckin, C.P., Ball, S.E. & Gordon-Smith, E.C. (1995) Diamond±
Blackfan anaemia: three patterns of in vitro response tohaematopoietic growth factors. British Journal of Haematology,
89, 457.
McGuckin, C.P., Uhr, M.R., Ball, S.E. & Gordon-Smith, E.C. (1996) Invitro progenitor analysis in a Diamond±Blackfan anaemia patient
who responded once but not twice to interleukin-3 therapy using
short-term and long-term cultures and c-kit analysis. British
Journal of Haematology, 93, 319±325.Maciejewski, J.P., Selleri, C., Sato, T., Anderson, S.A. & Young, N.S.
(1996) A severe and consistent de®cit in marrow and circulating
primitive hematopoietic cells (long term culture-initiating cells) in
acquired aplastic anemia. Blood, 88, 1983±1991.Mangan, K.F., Chikkappa, G. & Farley, P.C. (1982) T gamma cells
suppress growth of erythroid colony-forming units in vitro in the
pure red cell aplasia of B-cell chronic lymphocytic leukemia.
Journal of Clinical Investigation, 70, 1148±1156.Margolis, D., Camitta, B., Pietryga, D., Keever-Taylor, C., Baxter-
Lowe, L.A., Pierce, K., Kupst, M.J., French, J.I., Truitt, R., Lawton,
C., Murray, K., Garbrecht, F., Flomenberg, N. & Casper, J. (1996)Unrelated donor bone marrow transplantation to treat severe
q 2000 Blackwell Science Ltd, British Journal of Haematology 108: 167±175
174 N. Giri et al

175Pancytopenia in DBA
q 2000 Blackwell Science Ltd, British Journal of Haematology 108: 167±175
aplastic anaemia in children and young adults. British Journal of
Haematology, 94, 65±72.
Mori, P.G., Haupt, R., Fugazza, G., Sessarego, M., Corcione, A.,
Strigini, P. & Sansone, R. (1992) Pentasomy 21 in leukemiacomplicating Diamond±Blackfan anemia. Cancer Genetics and
Cytogenetics, 63, 70.
Pettengell, R., Luft, T., Henschler, R., Hows, J.M., Dexter, M., Ryder, D.
& Testa, N.G. (1994) Direct comparison by limiting dilutionanalysis of long-term culture initiating cells in human bone
marrow, umbilical cord and blood stem cells. Blood, 84, 3653±
3659.Russell, E.S. (1979) Hereditary anemias of the mouse: a review for
geneticists. Advances in Genetics, 200, 357.
Santucci, M.A., Bagnara, G.P., Strippoli, P., Bonsi, L., Vitale, L.,
Tonelli, R., Locatelli, F., Gabutti, V., Ramenghi, U., D'Avanzo, M.,Paolucci, G., Rosito, P., Pession, A. & Freedman, M.H. (1999)
Clinical investigation: long-term bone marrow cultures in
Diamond±Blackfan anemia reveal a defect of both granuloma-
crophage and erythroid progenitors. Experimental Hematology, 27,9±18.
Scho®eld, K.P. & Evans, D.I.K. (1991) Diamond±Blackfan syndrome
and neutropenia. Journal of Clinical Pathology, 44, 742±744.Singer, J.W., Brown, J.E., James, M.C., Doney, K., Warren, R.P., Storb,
R. & Thomas, E.D. (1978) Effect of peripheral blood lymphocytes
from patients with aplastic anemia on granulocytic colony growth
from HLA-matched and -mismatched marrows: effect of transfu-
sion sensitization. Blood, 52, 37±46.
Spritz, R.A. & Freedman, M.H. (1993) Lack of mutations of the MGFand KIT genes in Diamond±Blackfan anemia. Blood, 81, 3165±
3168.
Sutherland, H.J., Lansdorp, P.M., Henkelman, D.H., Eaves, A.C. &
Eaves, C.J. (1990) Functional characterization of individualhuman hematopoietic stem cells culture at limiting dilution on
supportive marrow stromal layers. Proceedings of the National
Academy of Sciences of the USA, 87, 3584±3588.Sutherland, H.J., Eaves, C.J., Lansdorp, P.M., Thacker, J.D. & Hogge,
D.E. (1991) Differential regulation of primitive human hemato-
poietic cells in long-term cultures maintained on genetically
engineered murine stromal cells. Blood, 78, 666±672.Tsai, P.H., Arkin, S. & Lipton, J.M. (1989) An intrinsic progenitor
defect in Diamond±Blackfan anaemia. British Journal of Haematol-
ogy, 73, 112±120.
Tuzuner, N. & Bennett, J.M. (1994) Letter to the editors: referencestandards for bone marrow cellularity. Leukemia Research, 18,
645±647.
Wasser, J.S., Yolken, R., Miller, D.R. & Diamond, L.K. (1978)Congenital hypoplastic anemia (Diamond±Blackfan syndrome)
terminating in acute myelogenous leukemia. Blood, 51, 991±995.





























