Embed Size (px)
Citation preview

Classification of Mass spectrometry data using
principal components analysis, Bayesian
MCMC modelling and a deterministic peak
finding algorithm.
William Browne (University of Nottingham)
Joint work with Kelly Handley and Ian Dryden
1

Outline
1. Background
2. Data sets and Questions
3. Principal Components
4. MCMC method
5. Peak finding algorithm
6. Cross validation
7. Further Work
2

Mass spectrometry
• Invented by J.J. Thomson in early 20th century.
• Description:
A substance is bombarded with an electron beam having sufficient
energy to fragment the molecule. The positive fragments which are
produced are accelerated in a vacuum through a magnetic field and
are sorted on the basis of mass-to-charge ratio. Since the bulk of
the ions produced in the mass spectrometer carry a unit positive
charge, the value m/z is equivalent to the molecular weight of the
fragment.
3

MALDI and SELDI TOF data
• Two types of mass spectrometry used in proteomics to identify
proteins that are associated with disease states such as cancer.
• MALDI - matrix-assisted laser desorption/ionization
• SELDI - surface-enhanced laser desorption/ionization
• TOF - time of flight
MALDI was developed in 1987 whilst SELDI is an improved
method that was invented in the early 1990s. Both produce mass
spectographs.
4

Example Mass spectrograph
Below is a mass spectrograph for a melanoma patient. Note that
M/Z values below 2000 are removed due to noise.
5000 10000 15000 20000 25000 30000
010
2030
4050
60
mzvalues
data
[1, ]
5

Close up of start of mass spectrograph
Below is a close up of the first 2000 (out of 13951) stored M/Z
values in the mass spectrogram showing how it is a series of
humps/peaks.
2000 2500 3000 3500 4000
02
46
810
12
mzvalues[1:2000]
data
[1, 1
:200
0]
6

Dataset 1: The Breast Cancer Dataset
• consists of 144 SELDI mass spectrographs from 2 independent
experiments.
• each spectrum is either a control or has been treated with the
chemotherapeutic agent Taxol.
• there are 3 drug groups MCF7/ADR, MCF7 and T47D.
• MCF7/ADR is chemoresistant, MCF7 and T47D are
chemosensitive.
• each experiment lasted for 4 days with observations every 24
hours.
• 3 observations (A,B,C) were taken after each 24 hour time period.
7

Dataset 1: The Breast Cancer Dataset
8

Dataset 2: The Melanoma Dataset
• Consists of 205 MALDI mass spectrographs of serum samples.
• The serum samples are taken from melanoma patients.
• Each patient is classified as either an early or late stage
Melanoma.
• There are 101 early stage (AJCC Stage 1) and 104 late stage
(AJCC Stage IV) patients.
9

Dataset 2: The Melanoma Dataset
10

Questions
We are interested in looking for differences between mass
spectrographs for the various groups in our datasets. Our
objectives are primarily dimension reduction and classification
(rather than inference).
We will firstly look at a technique that reduces the spectrographs
of length 13,950 M/Z values to a manageable number of derived
variables that explain most of the variability in the data.
We will then consider other techniques that reduce the
dimensionality to a group of easily interpretable variables. In
particular we are looking for potential biomarkers - individual
peptides that might help in classification
11

Specific Questions
For the breast cancer dataset we are interested in looking for:
• Where are there differences between the control and treated
spectra within the same drug groups?
• Where are there differences between the 3 control spectra?
For the melanoma dataset we only have 2 groups and so interest
lies simply in looking for differences between the early and late
stage spectrographs.
We will start by considering principal components analysis.
12

Principal Components
• Principal components analysis (PCA) tries to describe the
important variability in the data in a reduced number of
dimensions.
• Consider n observation vectors x1,x2, ...,xn each of length p.
The sample mean is
x̄ =1
n
n∑
i=1
xi
• The sample covariance matrix is the n× n matrix
S =1
n
n∑
i=1
(xi − x̄)(xi − x̄)T
13

• The eigenvectors γj of the matrix S are the principal components
(PCs) with corresponding decreasing eigenvalues λj
(j = 1, ..,min(p, n− 1)). The PC score sij for the ith observation
on the jth principal component is
sij = γTj (xi − x̄)
• We will be calculating principal components of a subset of each
dataset and using these to classify the remainder of the dataset
using Fisher’s linear discriminant rule.
14

Problems with a PCA approach
• On the plus side we will see that PCA does well in the
cross-validation exercises we perform later and it does reduce the
dimension of the data used.
• However, classification using PCA uses all of the data to form the
PCs and these PCs are not necessarily easy to interpret.
• What we would like is to be able to identify points in the data
where our groups differ.
Question: Are there ways of classifying using fewer pieces of
information?
15

t tests and False Discovery Rates
• One possibility is simply to compare groups at each of the 13,950
M/Z values via 2 sample t-tests.
• We considered a multivariate version of this approach (Dryden et
al. 2005) using dataset 1 and the 4 day structure to create
4-vectors and using Hotellings T 2 statistic.
• We considered both FDR (Benjamini and Hochberg, 1995) and a
variance offsetting approach to account for the multiple
comparisons problem and found some interesting differences on the
spectrographs between pairs of groups.
• We would however prefer an approach that accounts for more of
the structure in the dataset.
16

A model based approach
• Ideally we would like to fit some functional form to each of our
scans (spectrograms), where the function contains interpretable
parameters.
• The science suggests that a scan will consist of a series of peaks
at differing M/Z values.
• One way to model this is with a mixture of (scaled) normal
curves to represent the peaks. Each normal curve will have a
position (mean), variance and scale factor (height) and the sum of
these normal curves will be used to approximate the observed scan.
17

Too many parameters ?
• We have many scans and so to compare across scans we will need
to have some shared parameters and the non-shared parameters
will then be used for classification.
• Peak means - these will be shared by all scans.
• Peak variances - the scans suggest these increase with increased
M/Z value so we decided to make the peak sds ∝ the peak mean
values.
• Peak heights - these will be allowed to vary across scans and will
hence be used for classification.
18

A model/likelihood
• The model used for each datapoint yij on the spectrum is
yij ∼ N(θij , τ−1) where
θij =K∑
k=1
hkj(ξµ2k)
− 12 exp
(
−1
2(ξµ2
k)−1(mz(i)− µk)
2
)
Here i indexes the M/Z values on each scan, k indexes the peak
number and j indexes the scan number.
We have hkj - the height of the kth peak on the jth scan.
µk - the position of the kth peak and ξ a scale factor that links the
variance of the peak with µ2k. The function mz(i) returns the ith
M/Z value.
19

A Bayesian model
We wish to use MCMC to estimate parameters in this model. We
will assume K the number of peaks is fixed. Then we will need
priors for τ, ξ, hkj and µk.
These will be as follows:
p(τ) ∼ Gamma(0.001, 0.001)
p(ξ) ∼ Uniform(0, 0.01)
p(hkj) ∼ Uniform(0, 100)
p(µ1, . . . , µK) ∝ e−β(∑
i,j |µi−µj |<R)
i.e. a Strauss prior for the means that encourages them to keep
apart and avoids label switching.
20

MCMC algorithm
The MCMC algorithm will update the four groups of parameters,
τ, ξ, hkj and µk in turn. τ has a Gamma conditional posterior
distribution and can be updated using Gibbs sampling but for all
other parameters we use random walk Metropolis sampling.
We use the adapting procedure of Browne and Draper (2000) to
tune the proposal distributions prior to the MCMC burnin. This
method alters the proposal variances by comparing acceptance in
batches of 100 iterations and aims for an acceptance rate of 50%.
We will aim to find the MAP estimate from the MCMC chains that
we produce. The algorithm is still tremendously slow as the y
matrix is of size 13,950 by 144/205(N) for the two datasets and if
we consider K peaks then we have K ∗ (N + 1) + 2 parameters to
estimate.
21

Speeding up the algorithm
1. To speed up the code we store the values of θij , which occur
prominently in the joint posterior at the start. We then modify
them via addition and subtraction rather than recalculating them
from scratch wherever possible during the algorithm.
2. Secondly we consider a ‘truncated’ Normal likelihood where we
ignore the effects of a peak on θij values that are > 4 peak sds from
the peak mean.
22

Application to datasets
We applied the MCMC sampler to small sections of both the breast
cancer and the melanoma datasets. Each section ranged over
roughly the same m/z values. For breast cancer from 6800 Da to
8400 Da and melanoma from 6000 Da to 8400 Da. For each section
we choose K = 8 peaks. The adapting period took 2,100 iterations
for each dataset and we than ran for 1000 burnin iterations and
5000 stored iterations from which we selected the MAP estimates.
23

Tabular summary
Breast cancer
param. µ1 µ2 µ3 µ4 µ5 µ6 µ7 µ8
MAP 6917 7027 7207 7437 7692 7907 8110 8348
with ξ = 0.000048 and τ = 2.1996
Melanoma
param. µ1 µ2 µ3 µ4 µ5 µ6 µ7 µ8
MAP 6056 6461 6650 6762 6864 7793 7963 8165
with ξ = 0.000035 and τ = 6.6729
24

Plots for Breast cancer dataset
7000 7500 8000
−20
−10
010
20
adcon day 1
mzvector
data
MAP
DATA
7000 7500 8000
−20
−10
010
20
adcon day 2
mzvector
data
MAP
DATA
7000 7500 8000
−20
−10
010
20
adcon day 3
mzvector
data
MAP
DATA
7000 7500 8000
−20
−10
010
20
adcon day 4
mzvector
data
MAP
DATA
25

Plots for Melanoma dataset
6000 6500 7000 7500 8000
−50
050
stage 1
m/z value
data
6000 6500 7000 7500 8000
−50
050
stage 4
m/z value
data
26

Problems with MCMC approach
1. The approach is simply not computationally feasible for a
cross-validation exercise.
2. We are currently relying on a fixed K for the number of peaks,
although we could use reversible jump MCMC to cure this.
3. Although the Strauss prior avoids label switching, peaks can be
missed by the MCMC algorithm as it converges to a local
maximum.
4. To this end ‘good’ starting values are essential!
27

Motivation for a simpler approach
The speed of the MCMC method as it stands prohibits its serious
use however if we had a ‘good’ starting position then running
MCMC or something similar for a limited time may be feasible.
We therefore look for an approach to give us such a starting
position. Given the MCMC approach involves a series of peaks at
various locations on the scan with heights varying across scans and
with sd proportional to the mean, we look for a deterministic
approach.
28

Deterministic Peak finding
The MCMC approach considers K peaks for N scans, each peak
defined by three parameters, a position, µk, a variance (ξµ2k) and a
height hkj . We will simplify matters by assuming ξ is a known
constant that we can easily measure by looking at the scans. We
then have a 2 step procedure where we iteratively find the best
position to fit a peak (conditional on existing peaks) and then
calculate the required heights of this peak for each scan so that the
fitted function fits exactly at the peak location.
29

Deterministic Peak finding algorithm
Let yij be the ith location in the jth scan, fk(x) = N(µk, ξµ2k) be a
Gaussian kernel for the kth peak and hkj be the height term for the
kth peak on the jth scan. Then for peak k we form
eij = yij −k−1∑
p=1
hpjfp(mz(i))
where mz(i) returns the M/Z value for the ith location. Here eij is
in effect the residual at location i on scan j when k − 1 peaks have
been fitted. For the first peak we have eij = yij and we now want
to choose a location to place the first peak.
30

Deterministic Peak finding algorithm
For the kth peak we do the following:
Step 1: Find i such that∑
j eij is maximised. The kth peak will
then have mean location mz(i). Note that as we are only
considering positive peaks we prefer to sum the eij rather than
their squares.
Step 2: We now have k peak locations and we wish to calculate the
associated hkj . Here our aim is to fit exactly at the peak locations
for each scan. We will discuss this step further on the next slide.
Step 3: Recalculate the eij = yij −∑k
p=1 hpjfp(mz(i)).
31

Heights calculation
Let us assume we have K peaks and we wish to match at the peak
mean locations. Then let lk be the location of the kth peak with
corresponding M/Z value µk = mz(lk) and we require for scan j
h1jf1(µ1) + h2jf2(µ1) + . . .+ hKjfK(µ1) = yl1j
h1jf1(µ2) + h2jf2(µ2) + . . .+ hKjfK(µ2) = yl2j
. . .
h1jf1(µK) + h2jf2(µK) + . . .+ hKjfK(µK) = ylKj
This is simply K linear equations in K unknowns and in matrix
form the solution is H = F−1Y.
32
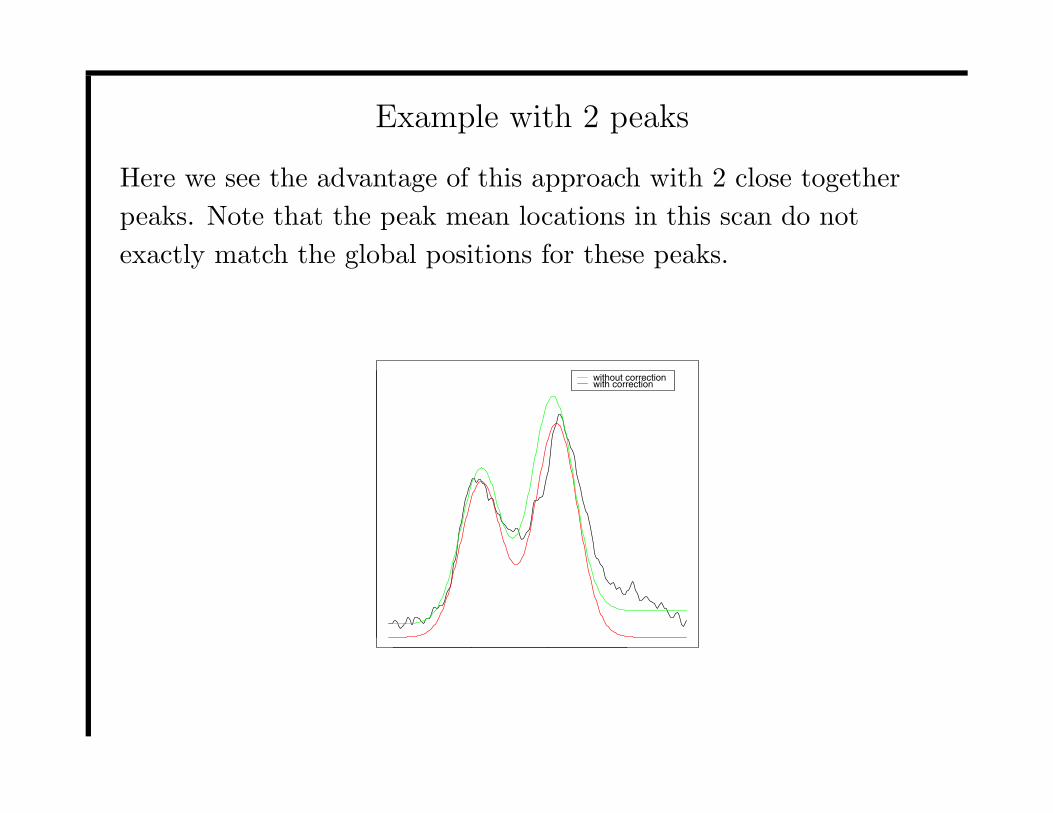
Example with 2 peaks
Here we see the advantage of this approach with 2 close together
peaks. Note that the peak mean locations in this scan do not
exactly match the global positions for these peaks.
6800 6900 7000 7100
05
1015
20
m/z value
data
without correctionwith correction
33

Comments on method
It is worth commenting here that this method was originally
considered only as a way of generating starting values for MCMC.
Although as we will see it is useful in its own right.
As we add the kth peak we have to invert N k × k matrices and so
the method is initially very fast although for a large number of
peaks (k > 100) computation time may be more of an issue.
We tested the method on the same small pieces of each dataset as
MCMC and get the following results:
34

Comparison with MCMC 1
Breast cancer
param. µ1 µ2 µ3 µ4 µ5 µ6 µ7 µ8
Peak 6913 7013 7274 7450 7688 7903 8102 7118
MCMC 6917 7027 7207 7437 7692 7907 8110 8348
Note the broad agreement for 6 of 8 peaks with the peak finding
assessing that 2 peaks around 7200 are more appropriate than an
additional peak at 8348. Note MCMC is constrained by its starting
values and so will not be able to reach the peak finding solution
easily.
35

Plots for Breast cancer dataset
7000 7500 8000
−20
−10
010
20
adcon day 1
m/z values
data
FITTED
DATA
7000 7500 8000
−20
−10
010
20
adcon day 2
m/z values
data
FITTED
DATA
7000 7500 8000
−20
−10
010
20
adcon day 3
m/z values
data
FITTED
DATA
7000 7500 8000
−20
−10
010
20
adcon day 4
m/z values
data
FITTED
DATA
36

Comparison with MCMC 2
Melanoma
param. µ1 µ2 µ3 µ4 µ5 µ6 µ7 µ8
Peak 6543 6451 6646 6744 6856 7777 7937 8154
MCMC 6056 6461 6650 6762 6864 7793 7963 8165
Again we have general agreement between the two approaches with
7 of the peaks in roughly the same position. Looking at the scans
by eye there aren’t really 8 peaks and so the location of the eight
peak is rather arbitrary!
37

Plots for Melanoma dataset
6000 6500 7000 7500 8000
−50
050
stage 1
mz value
6000 6500 7000 7500 8000
−50
050
stage 4
mz value
38

Cross validation experiment
To compare the performance of the 3 approaches discussed we
considered a cross validation experiment. Unfortunately MCMC
was not computationally feasible to be used here.
For each dataset we randomly split the data into two parts, the
training set ( 23 ) and test set ( 13 ).
Note that these splits were performed within each type of scan so
for the breast cancer dataset, the 24 ADC scans were allocated 16
to training and 8 to test etc.
For each dataset 100 such random splits of the dataset were created
and we were interested in average correct classification of the test
data.
39

Assessing the methods - PCA
We firstly take the training set and calculate PCs based on this set.
The resulting principal components are then added in turn and the
classification of the test data based on Fisher’s linear discriminant
rule (applied to the training data) is evaluated after each PC is
added.
This will result in a percentage correct classification for each split,
for each number of PCs and these can be plotted against number of
PCs.
In the graphs that follow we also use the 100 splits to generate a
mean performance and confidence intervals.
40

Assessing the methods - peak finding
Again we firstly take the training set and this time calculate the
first 50 peaks using the peak finding algorithm on the training set.
We then take the test data and evaluate the heights of these 50
peaks for the test data.
It is not clear that the largest peak is the best classifier so we
instead choose the order of introducing the peaks via other rules.
Firstly for both datasets we assess which peaks best predict the
training data using linear discriminant analysis.
Secondly for the melanoma dataset we also consider a simple t test
approach comparing t statistics for the difference between groups in
the training set for each peak.
41

Assessing the methods - peak finding (2)
Having sorted the peaks into an order using one of the above
methods we now add them in one at a time as with the PCs in the
PCA approach.
The classification of the test data is then based on Fisher’s linear
discriminant rule (applied to the training data) which is evaluated
after each peak is added.
This will result in a percentage correct classification for each split
for each number of peaks and these can be plotted against number
of peaks. In the graphs that follow we also use the 100 splits to
generate a mean performance and confidence intervals.
Note the whole procedure is automated in a C program that calls R
several times to perform the bulk of the procedure apart from the
computationally expensive peak finding which is done in C.
42

Results for breast cancer : All 100 runs
0 10 20 30 40 50
020
4060
8010
0
fitted heights − best classification
no of peaks used to classify
perc
enta
ge c
orre
ct c
lass
ifica
tions
Breast Cancer Dataset
0 20 40 60 80
020
4060
8010
0
original PCA − training(2/3)test(1/3)number of datavalues =
13952
no of pc’s used to classify
perc
enta
ge c
orre
ct c
lass
ifica
tions
43
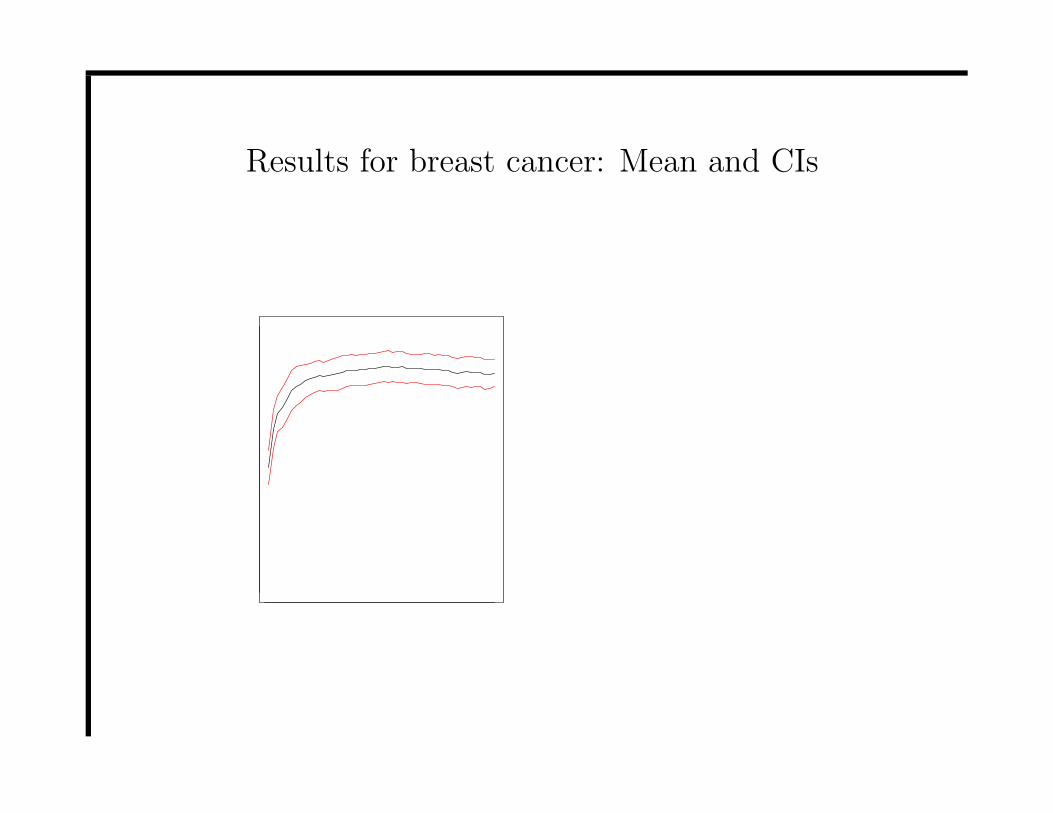
Results for breast cancer: Mean and CIs
0 10 20 30 40 50
020
4060
8010
0
peak fitting − best classification
no of peaks
% c
orre
ct c
lass
ifica
tion
Breast Cancer Dataset
0 20 40 60 80
020
4060
8010
0
pca
no of pc’s
% c
orre
ct c
lass
ifica
tion
44
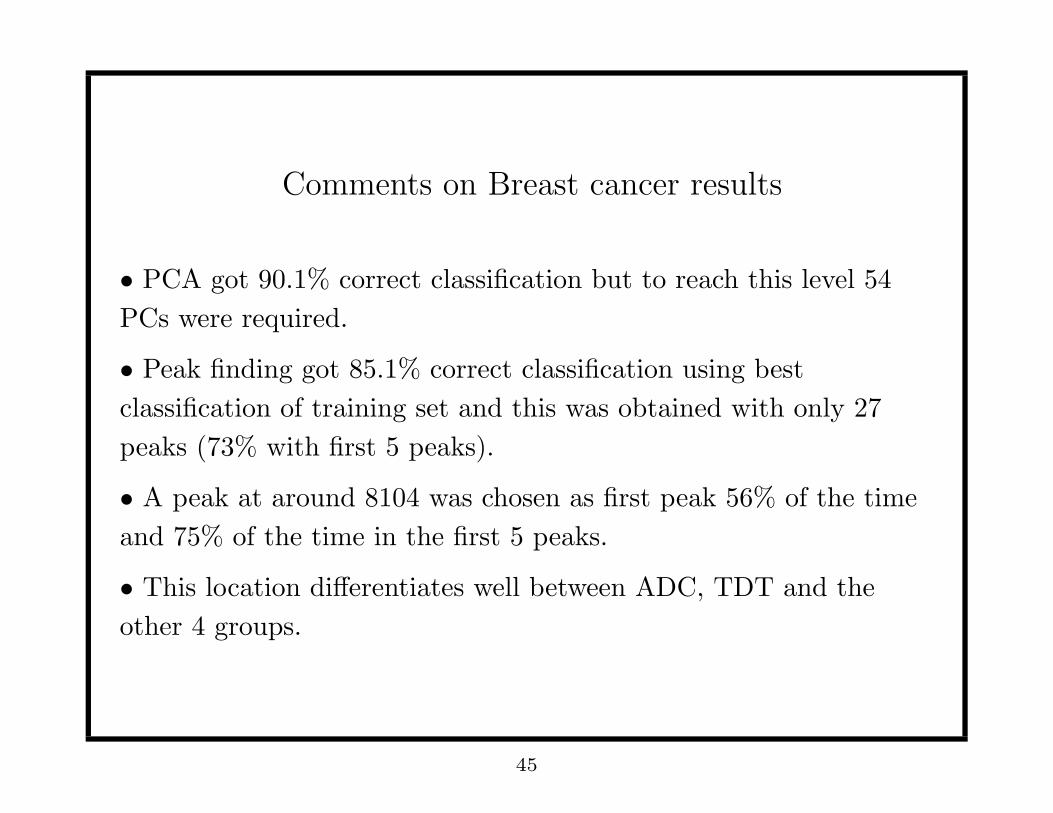
Comments on Breast cancer results
• PCA got 90.1% correct classification but to reach this level 54
PCs were required.
• Peak finding got 85.1% correct classification using best
classification of training set and this was obtained with only 27
peaks (73% with first 5 peaks).
• A peak at around 8104 was chosen as first peak 56% of the time
and 75% of the time in the first 5 peaks.
• This location differentiates well between ADC, TDT and the
other 4 groups.
45
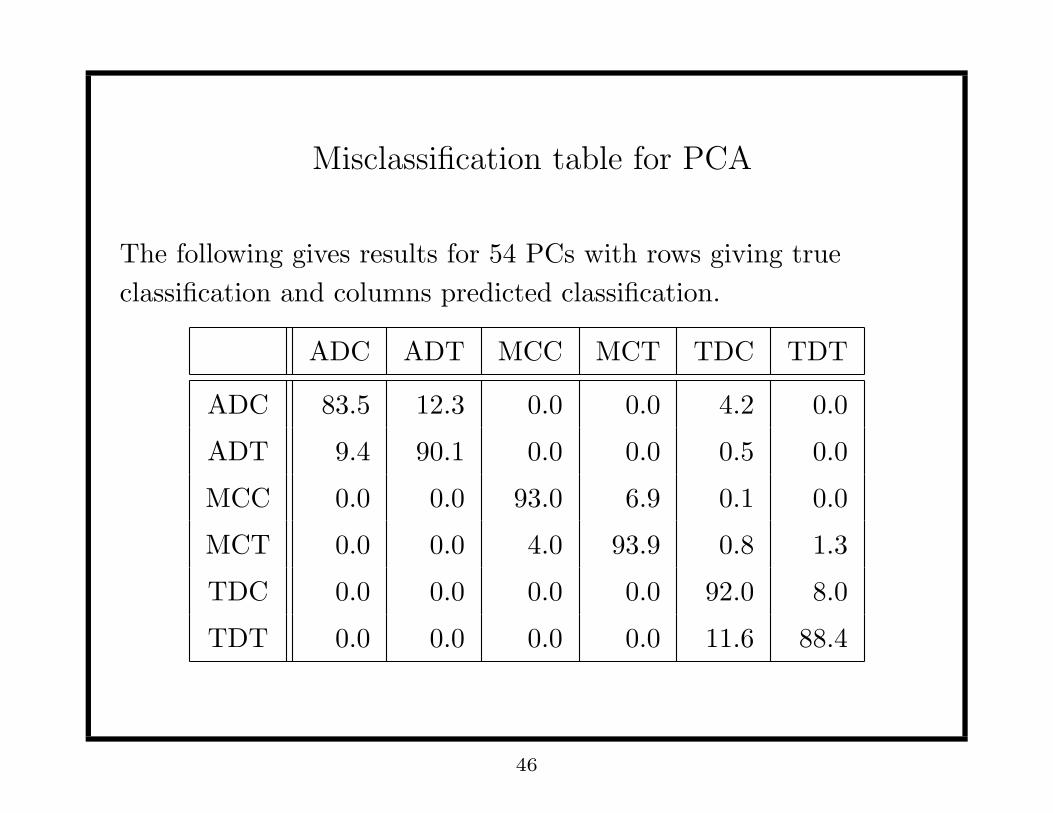
Misclassification table for PCA
The following gives results for 54 PCs with rows giving true
classification and columns predicted classification.
ADC ADT MCC MCT TDC TDT
ADC 83.5 12.3 0.0 0.0 4.2 0.0
ADT 9.4 90.1 0.0 0.0 0.5 0.0
MCC 0.0 0.0 93.0 6.9 0.1 0.0
MCT 0.0 0.0 4.0 93.9 0.8 1.3
TDC 0.0 0.0 0.0 0.0 92.0 8.0
TDT 0.0 0.0 0.0 0.0 11.6 88.4
46

Misclassification table for peak finding
The following gives results for 27 peaks with rows giving true
classification and columns predicted classification.
ADC ADT MCC MCT TDC TDT
ADC 76.8 15.5 0.2 0.0 7.5 0.0
ADT 8.8 87.5 0.0 0.1 3.1 0.5
MCC 0.0 0.9 88.4 10.0 0.1 0.6
MCT 0.0 0.5 10.2 85.8 0.7 2.8
TDC 0.4 0.2 0.0 0.0 89.8 9.6
TDT 0.1 0.0 0.0 0.0 17.3 82.6
47

Chosen peaks
The following table gives the 9 peaks that are chosen most often in
the first 5 peaks in the 100 iterations.
peak M/Z 1st 2nd 3rd 4th 5th Total
1 8104 56 14 2 2 1 75
2 7452 25 34 2 0 1 62
3 5416 0 15 9 10 10 43
4 11133 1 1 14 9 9 34
5 11701 0 0 22 4 5 31
6 4392 16 0 1 5 6 28
7 4345 0 4 4 5 9 22
8 4881 1 9 6 0 1 17
9 10232 0 4 1 6 6 17
48

Pairwise peak plots
Here we see plots for all scans in the 6 groups (ADC - black, ADT -
red, MCC - green, MCT - navy, TDC - cyan, TDT - purple)
0 5 10 15
02
46
810
1214
Peak2
Pea
k1
2 4 6 8 10
02
46
810
1214
Peak3
Pea
k1
2 4 6 8
02
46
810
1214
Peak4
Pea
k1
2 4 6 8 10 12
02
46
810
1214
Peak5
Pea
k1
49

Results for melanoma : All 100 runs
0 10 20 30 40 50
020
4060
8010
0
fitted heights − t−tests
no of peaks used to classify
perc
enta
ge c
orre
ct c
lass
ifica
tions
Melanoma Dataset
0 10 20 30 40 50
020
4060
8010
0
fitted heights − best classification
no of peaks used to classify
perc
enta
ge c
orre
ct c
lass
ifica
tions
0 20 40 60 80 100 120 140
020
4060
8010
0
original PCA − training(2/3)test(1/3)number of datavalues =
13951
no of pc’s used to classify
perc
enta
ge c
orre
ct c
lass
ifica
tions
50

Results for melanoma: Mean and CIs
0 10 20 30 40 50
020
4060
8010
0
peak fitting − t−test
no of peaks
% c
orre
ct c
lass
ifica
tion
Melanoma Dataset
0 10 20 30 40 50
020
4060
8010
0
peak fitting − bestclass
no of peaks
% c
orre
ct c
lass
ifica
tion
0 20 40 60 80 100 120 140
020
4060
8010
0
pca
no of pc’s
% c
orre
ct c
lass
ifica
tion
51

Comments on Melanoma results
• PCA got 88.1% correct classification but to reach this level 132
PCs were required.
• Peak finding got 81.1% correct classification using best
classification of training set and this was obtained with only 26
peaks (78.7% with first peak).
• Peak finding got 84.9% correct classification using t tests and this
was obtained with only 11 peaks (79.1% with first peak).
• A peak at around 3885 was chosen as first peak 92% (96%) of the
time
• This location has mean 4.73 for stage 1 melanoma and 3.03 for
stage 4 melanoma with a threshold of 3.90 correctly predicting
81.4% of scans
52

Conclusions
In this talk we have considered 3 approaches to dimension
reduction and classification:
• PCA is a standard method for dimension reduction which
classifies well but the derived variables used for classification are
difficult to interpret.
• The Model based MCMC approach is interesting but
computationally infeasible for the size of datasets considered.
• The deterministic peak finding method is fast and gives a reduced
set of variables that are still interpretable. It also performs
reasonably well at classifying though not as well as PCA.
53

Further work (1) - Comparison with other approaches
There are many other approaches to model mass spectroscopy data:
• Phil Brown (LASR 2005 presentation) and colleagues have looked
at using a wavelet approach to modelling the data.
• Ball et al. (2002) considered fitting artificial neural network
models to the breast cancer dataset.
• The MCMC approach given here could be extended to a random
effects model which would have benefits if we were interested in
inference.
54

Further work (2)- Extending the deterministic algorithm
Although MCMC seems impractical on speed grounds we can still
consider using the deterministic estimates as starting values for
alternative methods.
• The modes of the conditional log posterior distribution for each
height parameter hkj can be easily evaluated analytically (the
function is a quadratic in hkj) and so we can improve our estimates
by iteratively modifying these height parameters.
• The scale factor ξ that links the position and the variance of the
peaks could also be modified to improve fit. Note here we will need
to use numerical optimisation.
• The locations of the peaks should already be close to optimal and
for use in classification it is sensible to constrain the location to lie
at measured M/Z values however we can consider moving the
location by 1 measured M/Z value in either direction.
55

Further work (3)
The 3 steps suggested could be iterated to give a combined
maximizing algorithm where we iterate until the improvement is
less than some tolerance and then this method could be compared
with the others in the cross-validation.
Other ideas are:
• Consider a nearest neighbour classifier instead of linear
discriminant analysis.
• Use a statistical test to decide on the optimal number of peaks to
fit in the deterministic approach.
• Modify the model to include offset parameters for each peak in
each scan to allow for local variation in the position of the peak in
case such parameters are useful in classification.
56

Further work (4)
• Independents components analysis may be a useful alternative to
PCA and may give more interpretable variables.
• For the breast cancer dataset we might want to incorporate the
day information somehow into the model. Two possibilities are
firstly to try and classify the 24 possible groups. Secondly to use
the peak heights in a (multivariate) regression setting so that we
could classify both type and also time after sample taken.
• Such a regression approach would be more useful in the
melanoma dataset where we might like to predict the stage of
melanoma for a new sample. However for this we would prefer a
dataset containing other stage of the disease.
57





























