Embed Size (px)
Citation preview
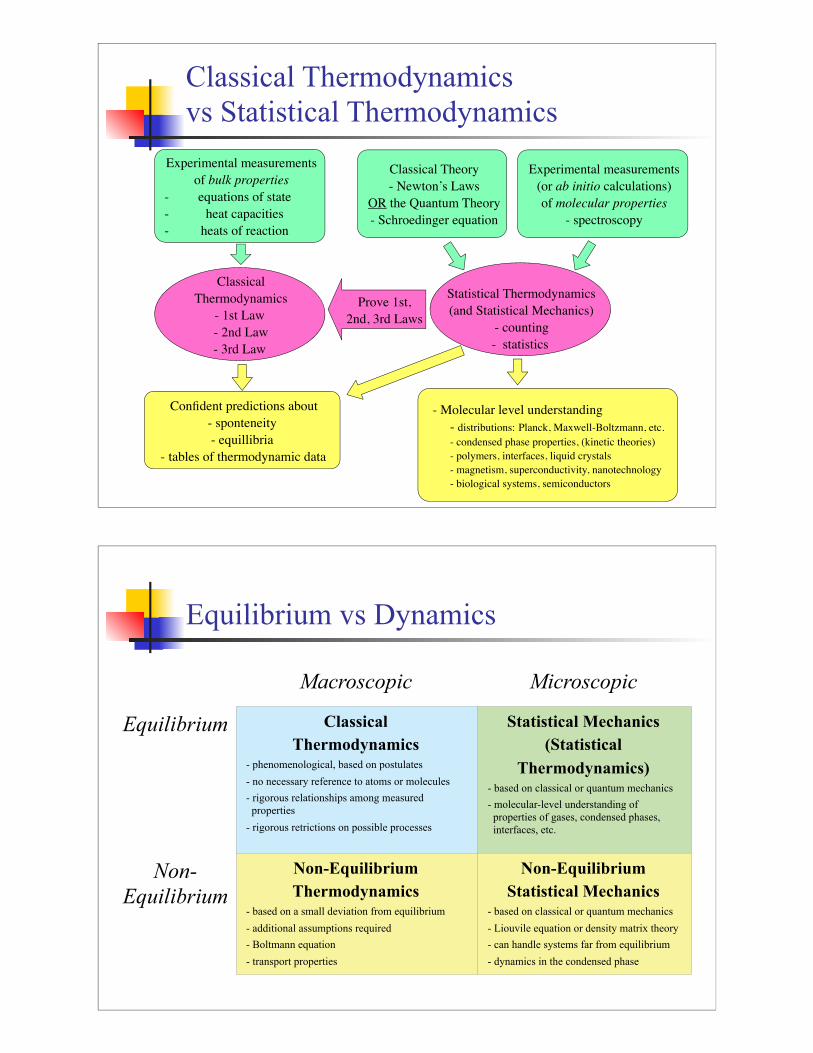
Classical Thermodynamics
vs Statistical Thermodynamics
Experimental measurements
of bulk properties
- equations of state
- heat capacities
- heats of reaction
Classical
Thermodynamics
- 1st Law
- 2nd Law
- 3rd Law
Confident predictions about
- sponteneity
- equillibria
- tables of thermodynamic data
Experimental measurements
(or ab initio calculations)
of molecular properties
- spectroscopy
Classical Theory
- Newton’s Laws
OR the Quantum Theory
- Schroedinger equation
Statistical Thermodynamics
(and Statistical Mechanics)
- counting
- statistics
Prove 1st,
2nd, 3rd Laws
- Molecular level understanding
- distributions: Planck, Maxwell-Boltzmann, etc.
- condensed phase properties, (kinetic theories)
- polymers, interfaces, liquid crystals
- magnetism, superconductivity, nanotechnology
- biological systems, semiconductors
Equilibrium vs Dynamics
Macroscopic Microscopic
Equilibrium
Non-
Equilibrium
Classical
Thermodynamics- phenomenological, based on postulates
- no necessary reference to atoms or molecules
- rigorous relationships among measured
properties
- rigorous retrictions on possible processes
Statistical Mechanics
(Statistical
Thermodynamics)- based on classical or quantum mechanics
- molecular-level understanding of
properties of gases, condensed phases,
interfaces, etc.
Non-Equilibrium
Thermodynamics- based on a small deviation from equilibrium
- additional assumptions required
- Boltmann equation
- transport properties
Non-Equilibrium
Statistical Mechanics- based on classical or quantum mechanics
- Liouvile equation or density matrix theory
- can handle systems far from equilibrium
- dynamics in the condensed phase

3
Statistical Mechanics(based on classical or quantum mechanics)
Classical
Thermodynamics(Three Laws)
Equilibrium
Statistical Mechanics
phenomena far from
equilibrium
Non-equilibrium
Thermodynamics (close to equilibrium)
4
Thermodynamics
! Rules regarding energy conversion.

5
“ Thermodynamics is the only science
about which I am convinced that,
within the framework of applicability
of its basic principles, it will never be
over thrown.”
Albert Einstein
I. Core concepts
• Temperature?
• Heat?
• Work?
• a State Function?
• Equilibrium?
6
Numbered figures and Table in the next 4 slides are from Peter A. Rock, “Chemical Thermodynamics”.
What is

What is temperature
! Ideal gas: PV = nRT
! The volume of an ideal gas extrapolates to zero at -273.12˚C which defines T = 0 kelvin.
! Real gases are ideal in the limit of low pressure.
7
The constant-volume gas thermometer
! Repeat the measurement with lower and lower pressures to obtin the ideal gas limit.
! Thus, ANY gas can be used!
! One fixed point, the triple point of ice, is used to define a temperature scale:
! This is a practical definition of temperature, but is there a more fundamental theoretical definition?
8
!
T
273.16K=
P273.16" 0
limPT
P273.16
#
$ % &
' (

What is heat? “q”
! What units?
! How is it related to temperature?
! What is heat capacity?
! How does q depend on the path “X”
! Related concepts:! internal energy, U
! enthalpy, H
! CV
! CP
9
CX =2T2qX
What is work? “w”
! Sign convention: Work done ON the system is positive
10

What are state functions?
! depend only on the present state of the system
! not on the history of the system
! have defined values for systems at equilibrium
! Examples?
! Examples of non-states functions?
! Exact and inexact differentials?
11
Changes can occur along different paths
! reversible: infinitessimally close to equilibrium
! constant pressure: isobaric
! constant volume: isochoric
! constant temperature: isothermal
! adiabatic
12

Systems can be
! open
! closed
! isolated
13
What is equilibrium?
! mechanical equilibrium ( )
! thermal equilibrium ( )
! chemical equilibrium ( for all chemical substances, i)
14
!
"P = 0 !
!
"T = 0 !
!
"µi= 0
System 1 System 2

15
Extensive and Intensive Quantities
! Extensive quantities increase in proportion to the size (mass) of the sample, (n, V, E, H, G, S, A)
! but intensive quantities remain the same when the size of the system is increased. (T, P)
n, V, E, S,
H, G, A
T, P
n, V, E, S,
H, G, A
T, P
2n, 2V, 2E, 2S,
2H, 2G, 2A
T, P
+ =
The Laws of Thermodynamics
• Application to pure substances
16

First Law
• Internal energy is conserved
• is work done ON the system (e.g., )• is heat absorbed BY the system
Second Law
• There exists a state function, S (which we call entropy). For any change
between states of the system
where the equality holds if the process is reversible and the inequality holds
if the process is irreversible.
Third Law
• Nernst Statement :!
• Planck Statement:!
II. The Three Laws
17
!
dU = "w +"q
!
"w
!
"w = #PdV
!
"q
!
dSsys "dqsys
T
!
T" 0
lim#ST
= 0
!
T" 0
lim ST = 0
! When a crystaline pure solid is heated reversibly from temperature T1 to T2, we can
integrate to get the entropy change
! At constant pressure, !qrev = CPdT, so the above equation becomes
! If we let T1=0, the by the Third Law, S1 = 0, and we get the absolute entropy at
temperature T2:
! If the substance is a liquid at T2, we also need to include the heat of fusion:
18
Absolute Entropies from the Third Law
!
dS ="qrev
T
!
"S = dSS1
S2
# =dqrev
TT1
T2
#
!
S2" S
1=
CP
TdT
T1
T2
#
!
S2
=CP
TdT
0
T2
"
!
S2
=CP
solid
TdT
0
T fusion
" +#H fusion
Tfusion+
CP
liquid
TdT
T fusion
T2
"

19
Absolute Entropies from the Third Law
! If the substance is a gas at T2, we also need to include the heat of vaporization:
!
S2
=CP
solid
TdT
0
T fusion
" +#H fusion
Tfusion+
CP
liquid
TdT
T fusion
Tvap
" +#Hvap
Tvap+
CP
gas
TdT
Tvap
T2
"
Silberberg Chapt 20.
Truth in Advertising
Absolute entropies are not quite as absolute this would
lead you to believe because certain kinds of motion
have not been considered, namely nuclear spins and
the mixing of isotopes in their natural abundances.
However, these things only affect entropy changes at
EXTREMELY low temperatures and for our usual
purposes in chemistry and biochemistry, the tabulated
entropies give accurate results.
Phase Rule
•
!
f = c " p + 2
• consider case of only one kind of work, e.g.,
!
"w = #PdV• For a pure substance there is 1 component (c=1), 1 phase (p=1), so there are two
degrees of freedom (f=2)• degrees of freedom = number of independent variables• change variable at will with the tools of calculus
6
Phase Rule
20

Natural variable for internal energy, U
21
Important Relationships with Natural Variables
• Assume we have only
!
"w = #PdV and no other kind of work.• First Law:!
!
dU = "w +"q
= #PdV +"q
• For a reversible process
!
dq = TdS
• Combine to get
!
dU = "PdV + TdS
• Applies to all process, reversible and irreversible !!
7
(2nd Law)
• dU is an exact differential, so from
!
dU = "PdV + TdS and the general
form!
!
dU ="U
"V
# $
% & S
dV +"U
"S
# $
% & V
dS
we can identify the partial derivatives:
!
"U
"V
# $
% & S
= 'P
"U
"S
# $
% & V
= T
• Mixed second derivative are independent of the order of differentiation
!
"
"S
"U
"V
# $
% & S
#
$ ' %
& (
V
="
"V
"U
"S
# $
% & V
#
$ ' %
& (
S
) *"P
"S
# $
% & V
="T
"V
# $
% & S
(Maxwell’s Relationships)
8
Exact Differentials
22

• Define more variables:
!
H =U + PV ! Enthalpy
!
A =U " TS ! Helmholtz Free Energy
!
G = H " TS ! Gibbs Free Energy
• Get more relationships
!
dH = dU + PdV +VdP
= "PdV + TdS + PdV +VdP
dH = TdS + VdP
• Similarly
!
dA = "PdV " SdT
dG = VdP " SdT
9
Define more thermodynamic functions
23
Criteria for Sponteneity
24
Criteria for Sponteneity
Suppose that we have –PdV work and some other kind of work wother.
!
"w = #PdV + "wother
Then from the 1st Law,
!
dU = "q + "wother # PdV
From the 2nd Law,
!
"q # TdS
!
"dU # TdS $ PdV +%wother
Using A=U-TS,
!
!
dA = dU " TdS " SdT
# TdS " PdV + $wother
= TdS " SdT
!
!
"dA # $SdT $ PdV +%wother
At constant T, V,
!
dA " #wother
! At constant T, V, if there is no other work,
! !
!
dA " 0
10

Similarly using G=A+PV, we find
!
dG " #SdT + VdP +$wother
At constant T, P,
!
!
dG " #wother! ! ! ! ! (1)
At constant T, P, if there is no other work,
!
dG " 0
If the other work is electrical work
!
"EdZwhere E is the electric voltage
and Z is the current,the Eq. (1) becomes at equilibrium
!
!
dG = "EdZ
If one mole of products are formed in an electrochemical reaction when
n moles of current pass through the circuit, this integrates to
!
!
"G = #nFE
where F is the number of coulombs per mole of electrons.
Thus measurement of an electrochemical potential is equivalent to a measuring a
free energy change.
11
Criteria for Sponteneity
25
III. Standard Data
• Standard States
• Chemical Reactions
• Standard Tabulated data
26

Standard States
27
Standard States
• Standard Conditions
- 1 atm pressure
- temperature of interest
• Standard States of Solids and Liquids
- pure substance
- reproducible equilibrium state
- 1 atm pressure & temperature of interest
- REAL state
• Standard States of Gases
- HYPOTHETICAL ideal gas
- 1 atm pressure & temperature of interest
12
Standard Quantities of Formation
28
13
Standard Quantiies of Formation
•
!
"Hf ,T
o for a compound is the enthalpy change for the reaction in which
one mole of the substance is formed from the elements in their standard
states at the temperature of interest
•
!
"Gf ,T
o ,
!
"Sf ,To
, etc. are similarly defined.
•
!
"ST
0 - Entropy changes can be derived from absolute Third Law
entropies
!
ST
0, and are not relative to standard states.
• Superscript “o” means standard conditions
• Examples (at 298 K)
!
H2g( ) + 1
2O2g( ) " H
2O g( )
!
"Hf ,T
o[H2O(g)] = –241.8 kJ/mol
!
H2g( )" H
2g( )
!
"Hf ,T
o[H2(g)] = 0 kJ/mol
!
O2g( )" O
2g( )
!
"Hf ,T
o[O2(g)] = 0 kJ/mol
!
O2g( )" 2O g( )
!
"Hf ,T
o[O (g)] = +247 kJ/mol
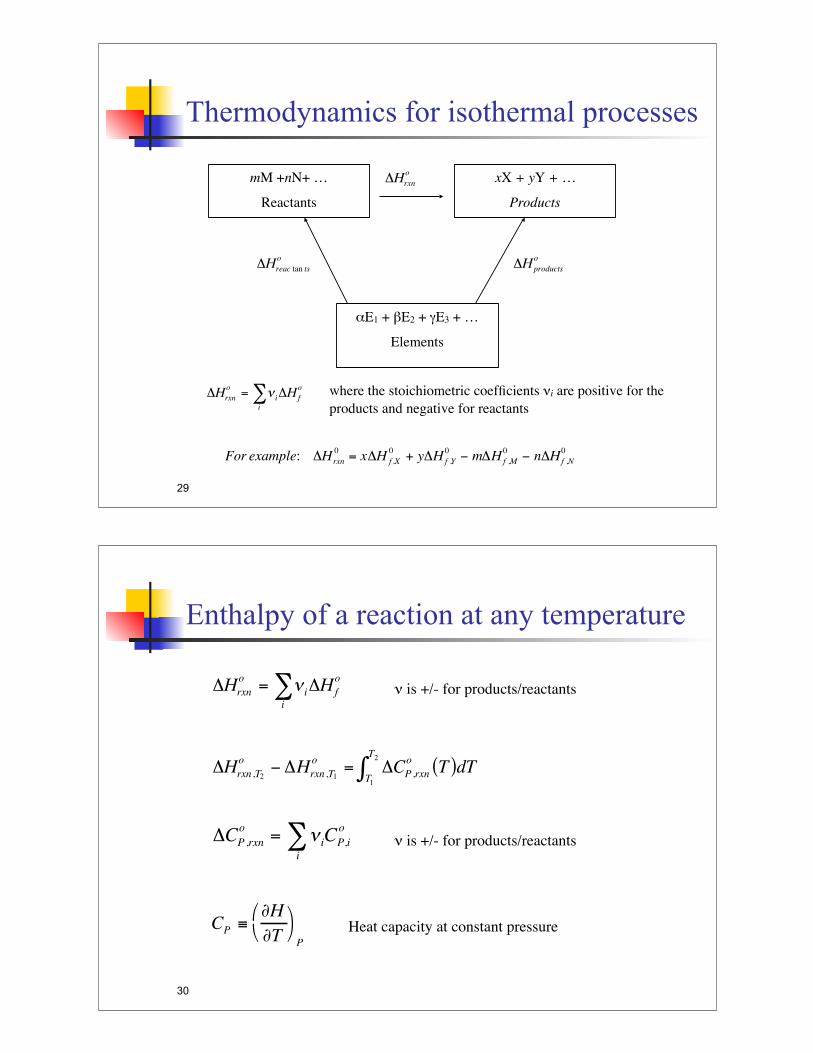
Thermodynamics for isothermal processes
29
Thermodynamics of Any Isothermal Process
aA wherwe
14
mM +nN+ …
Reactants
xX + yY + …
Products
!E1 + "E2 + #E3 + …
Elements
!
"Hrxn
o
!
"Hreac tan ts
o
!
"Hproducts
o
!
"Hrxn
o= # i
i
$ "Hf
o where the stoichiometric coefficients $i are positive for the
products and negative for reactants
!
For example: "Hrxn
0= x"H f ,X
0+ y"Hf ,Y
0# m"Hf ,M
0# n"Hf ,N
0
Enthalpy of a reaction at any temperature
30
Enthalpy of a Reaction
!
"Hrxn
o= # i
i
$ "Hf
o
! ! is +/- for products/reactants
!
"Hrxn ,T2
o# "H
rxn ,T1
o= "C
P ,rxn
o
T1
T2
$ T( )dT
!
"CP ,rxn
o
= #i
i
$ CP,i
o
! ! is +/- for products/reactants
!
CP"
#H
#T
$ %
& ' P
! Heat capacity at constant pressure
15

Entropy of a reaction at any temperature
31
Entropy of a Reaction
!
"Srxn ,T
o= #
iST ,i
o
i
$ ! ! is +/- for products/reactants
!
"Srxn T
o# "S
rxn T
o=
"CP rxn
oT( )
TT
T
$ dT
16
Gibbs Free Energy of a Reaction
!
G = H " TS
!
"Grxn ,T
o= # i"Gf ,i,T
o
i
$ ! ! ! is +/- for products/reactants
For isothermal reaction
!
"Grxn ,T
o= "H
rxn ,T
o# T"S
rxn ,T
o
Noting that all three quantities on the right side of this equation depend
on temperature, we can us the previous results to find
!
"Grxn T
o
# "Grxn T
o
17Gibbs (Free) Energy for a reaction
32
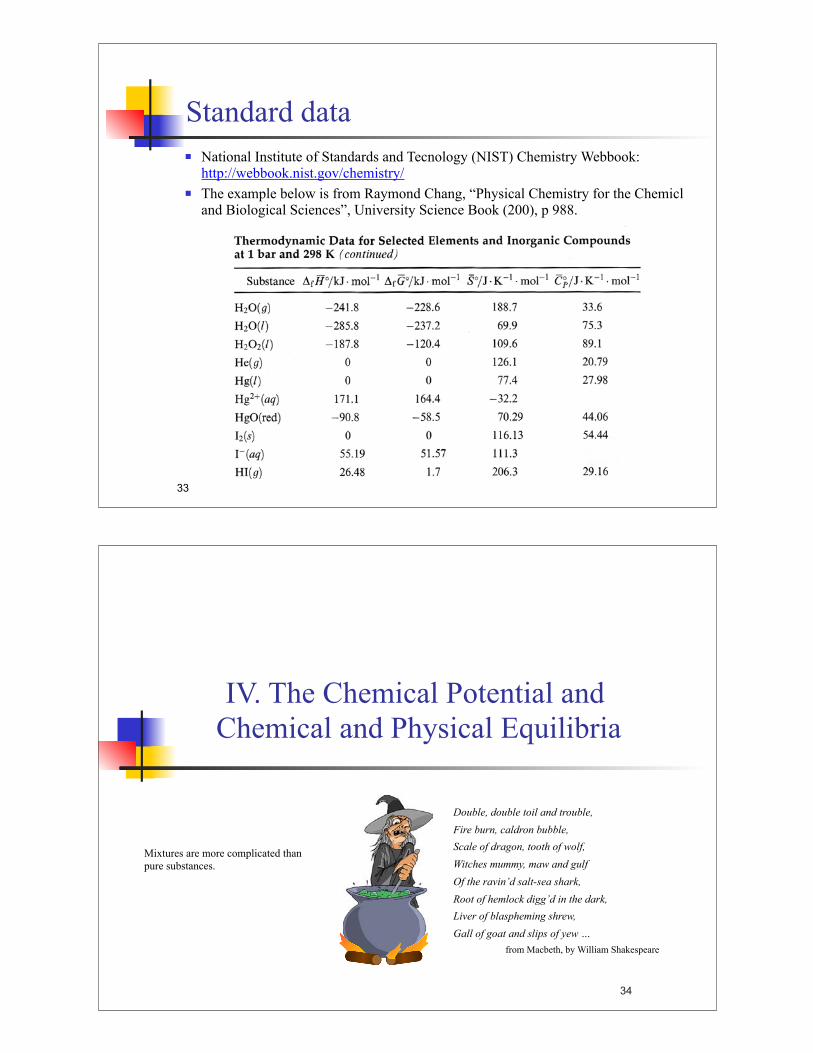
Standard data
! National Institute of Standards and Tecnology (NIST) Chemistry Webbook: http://webbook.nist.gov/chemistry/
! The example below is from Raymond Chang, “Physical Chemistry for the Chemicl and Biological Sciences”, University Science Book (200), p 988.
33
34
Double, double toil and trouble,
Fire burn, caldron bubble,
Scale of dragon, tooth of wolf,
Witches mummy, maw and gulf
Of the ravin’d salt-sea shark,
Root of hemlock digg’d in the dark,
Liver of blaspheming shrew,
Gall of goat and slips of yew …
from Macbeth, by William Shakespeare
IV. The Chemical Potential and
Chemical and Physical Equilibria
Mixtures are more complicated than
pure substances.

35
The Chemical Potential
The chemical potential of component i is
!
µi ="G
"ni
#
$ % &
' (
T ,P,n j
The chemical potential is measure of the tendency species i to emigrate from the
phase in which it is located or to convert to something else by means of a
chemical reaction.
At T, P, the criterion for sponenteity is dG<0 and at equilibrium dG=0. Therefore
at equilibrium between 2 phases, we have dµI=0 and the chemical potential must
be the same on both sides.
18
Activity
36
Activity
A change is the pressure of an ideal gas changes its chemical potential
!
!
dµ = RTd(lnP)
The log part of this equation is somewhat inconvenient so we define a measure of
the chemical potential called the activity ai . For the definition we use the equation
!
!
dµi= RTd(lna
i)
so that the activity of an ideal gas is equal to its pressure in atmospheres,
ai=Pi.
(The pressure of an ideal gas is proportional to its concentration.)
Not all gases are ideal, but for real gases at usual pressures, they are close to ideal
and
! ai! Pi
For other components in other phases, we define the activity so that the
component is its standard state has unit activity and for ideal systems the activity
is equal to its concentration.
19
ininin
ininin
Changes under non-Standard Conditions
Consider the reaction,
! mM + nN ! xX + yY
For each component that is not in its standard state, we must make a correction for
its activity.
!
"Grxn = "Grxn
o+ RT ln
aXxaYy
aMm aN
n
#
$ % &
' (
In terms of the electrochemical potential this becomes
!
!
E = E0
+RT
nFln
aXxaYy
aMm aN
n
"
# $ %
& ' ! ! Nernst Equation
At equilibriium "Grxn=0, which gives
!
!
"Grxn
o
= #RT lnK
where the equilibrium constant K is given as
!
!
K =aXxaYy
aMm aN
n "PXxPY
y
PMmPN
n
Note that K,
!
"Grxn
o
, and
!
Eo
can be determined from each other.
20Changes under non-standard conditions
37
38
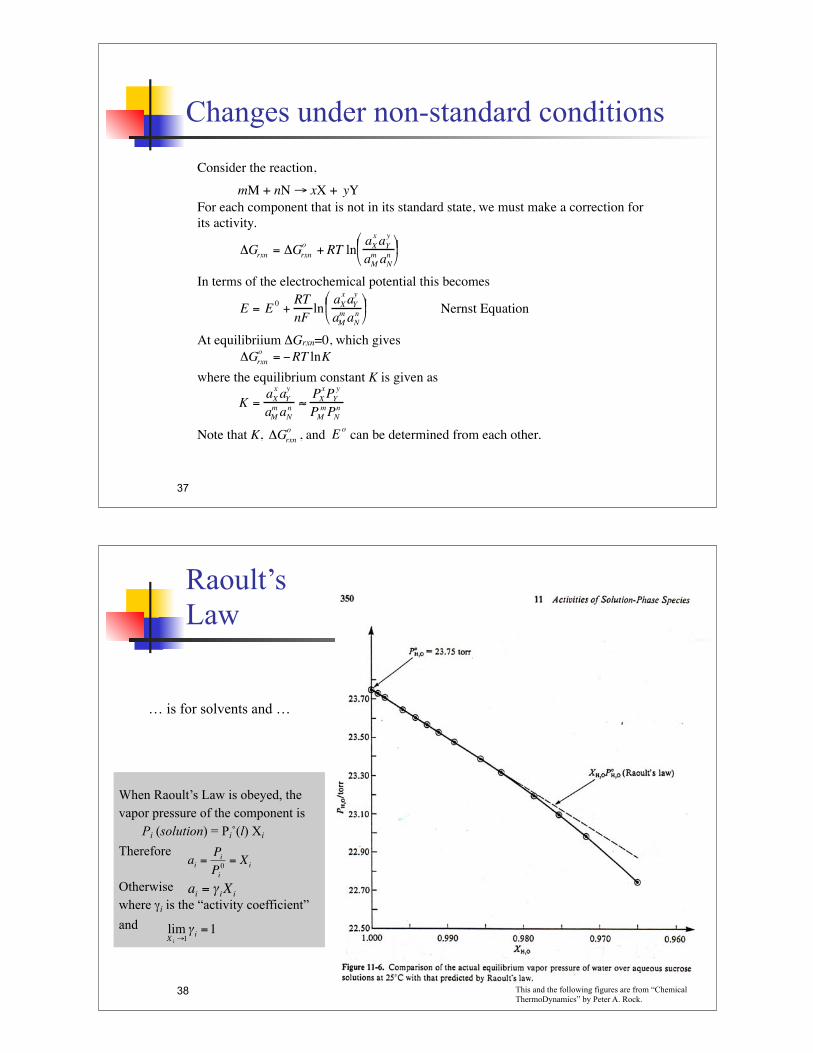
When Raoult’s Law is obeyed, the
vapor pressure of the component is
Pi (solution) = Pi˚(l) Xi
Therefore
Otherwise
where "i is the “activity coefficient”
and
Raoult’s
Law
This and the following figures are from “Chemical
ThermoDynamics” by Peter A. Rock.
… is for solvents and …
!
ai=Pi
Pi
0= X
i
!
ai= "
iXi
!
limXi"1#i=1

39
! Raoult’s Law works best when
the solvent and solute are
similar.
Raoult’s Law
Ideal
40
Raoult’s Law
Non-Ideal
! But Raoult’s Law works
poorly when the solute and
solvent are dissimilar.
! Raoult’s Law still works for
the solvent in the limit of
dilute solution.
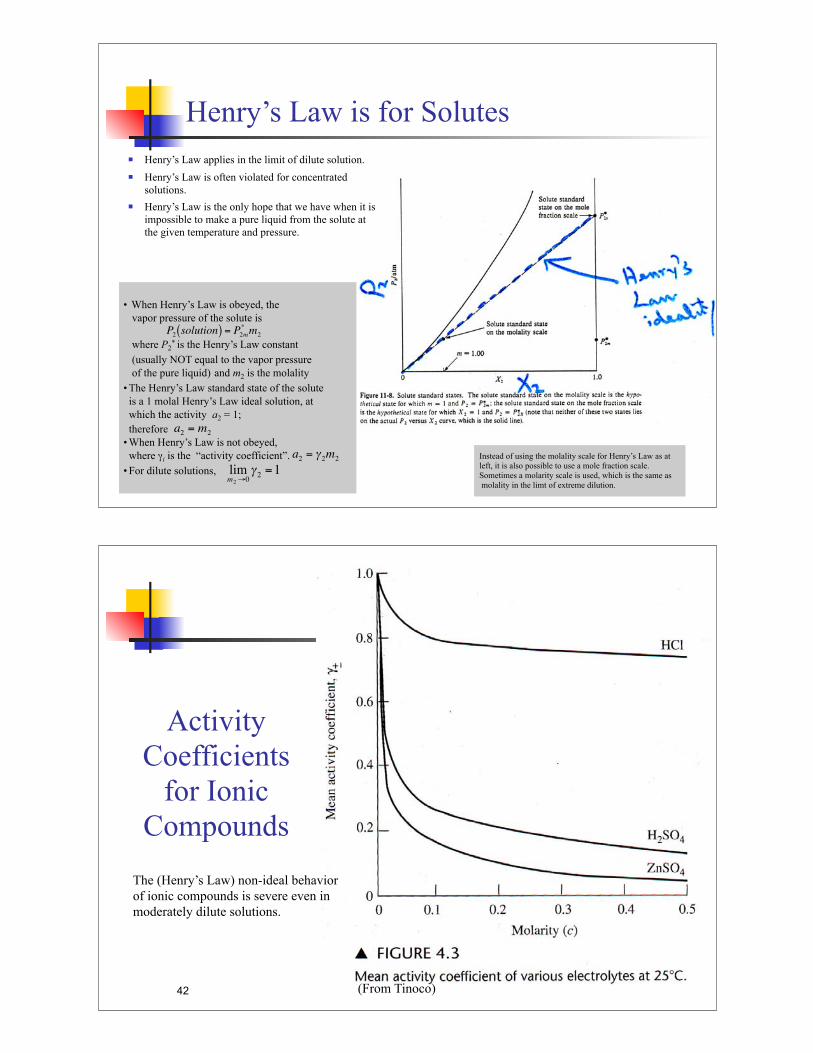
Henry’s Law is for Solutes
! Henry’s Law applies in the limit of dilute solution.
! Henry’s Law is often violated for concentrated
solutions.
! Henry’s Law is the only hope that we have when it is
impossible to make a pure liquid from the solute at
the given temperature and pressure.
• When Henry’s Law is obeyed, the
vapor pressure of the solute is
where P2* is the Henry’s Law constant
(usually NOT equal to the vapor pressure
of the pure liquid) and m2 is the molality
• The Henry’s Law standard state of the solute
is a 1 molal Henry’s Law ideal solution, at
which the activity a2 = 1;
therefore
• When Henry’s Law is not obeyed,
where "i is the “activity coefficient”.
• For dilute solutions,
!
P2solution( ) = P
2m
*m2
!
a2
= "2m2
!
limm2"0#2
=1!
a2
= m2
Instead of using the molality scale for Henry’s Law as at
left, it is also possible to use a mole fraction scale.
Sometimes a molarity scale is used, which is the same as
molality in the limt of extreme dilution.
42
Activity
Coefficients
for Ionic
Compounds
The (Henry’s Law) non-ideal behavior
of ionic compounds is severe even in
moderately dilute solutions.
(From Tinoco)





























