Embed Size (px)
Citation preview
Helicobacter Pylori-Induced Reduction of AcuteLevodopa Absorption in Parkinson’sDisease PatientsMariangela Pierantozzi, PhD,1,6 Antonio Pietroiusti, MD,2
Alberto Galante, MD,2 Giuseppe Sancesario, MD,1
Gianluigi Lunardi, PhD,3 Ernesto Fedele, PhD,4
Patrizia Giacomini, MD,5 and Paolo Stanzione, MD1,6
We occasionally observed a consistent clinical improvementin an advanced Parkinson’s disease (PD) patient (Hoehn andYahr1 [H and Y], stage 4, 10-year disease history) under sta-ble levodopa (L-dopa) therapy, immediately after eradicatingHelicobacter pylori (Hp) infection. We hypothesised that Hp-induced changes of the gastric environment (ie, pH varia-tions2 or increase of oxidant agents3) affecting L-dopa ab-sorption might be reversed by eradicating Hp infection.Therefore, we measured the acute plasma L-dopa concentra-tion–time curve in 7 advanced PD patients (mean age,70.4 6 6.3 years; mean disease duration, 6.8 6 2.4 years; Hand Y $ 2.5) who were Hp-positive (IgG antibody titerhigher than 1,000 Pyloryset; Orion Diagnostica, Espoo, Fin-land), and had no history of previous Hp eradication at-tempts.
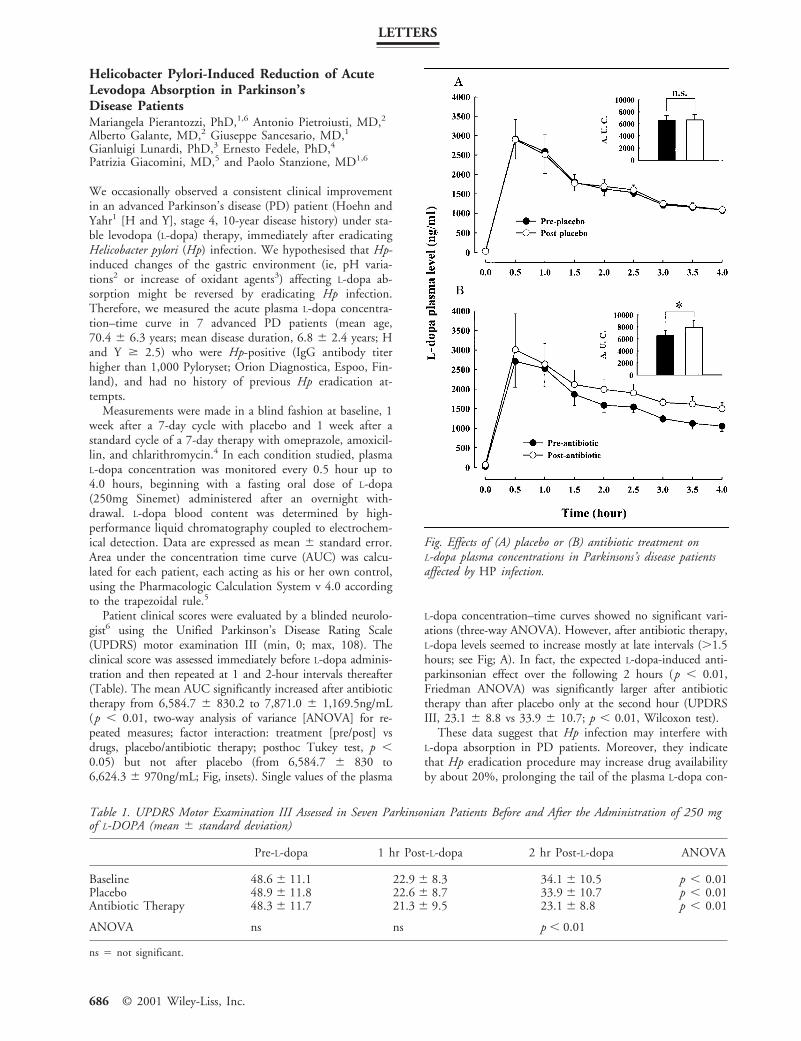
Measurements were made in a blind fashion at baseline, 1week after a 7-day cycle with placebo and 1 week after astandard cycle of a 7-day therapy with omeprazole, amoxicil-lin, and chlarithromycin.4 In each condition studied, plasmaL-dopa concentration was monitored every 0.5 hour up to4.0 hours, beginning with a fasting oral dose of L-dopa(250mg Sinemet) administered after an overnight with-drawal. L-dopa blood content was determined by high-performance liquid chromatography coupled to electrochem-ical detection. Data are expressed as mean 6 standard error.Area under the concentration time curve (AUC) was calcu-lated for each patient, each acting as his or her own control,using the Pharmacologic Calculation System v 4.0 accordingto the trapezoidal rule.5
Patient clinical scores were evaluated by a blinded neurolo-gist6 using the Unified Parkinson’s Disease Rating Scale(UPDRS) motor examination III (min, 0; max, 108). Theclinical score was assessed immediately before L-dopa adminis-tration and then repeated at 1 and 2-hour intervals thereafter(Table). The mean AUC significantly increased after antibiotictherapy from 6,584.7 6 830.2 to 7,871.0 6 1,169.5ng/mL(p , 0.01, two-way analysis of variance [ANOVA] for re-peated measures; factor interaction: treatment [pre/post] vsdrugs, placebo/antibiotic therapy; posthoc Tukey test, p ,0.05) but not after placebo (from 6,584.7 6 830 to6,624.3 6 970ng/mL; Fig, insets). Single values of the plasma
L-dopa concentration–time curves showed no significant vari-ations (three-way ANOVA). However, after antibiotic therapy,L-dopa levels seemed to increase mostly at late intervals (.1.5hours; see Fig; A). In fact, the expected L-dopa-induced anti-parkinsonian effect over the following 2 hours (p , 0.01,Friedman ANOVA) was significantly larger after antibiotictherapy than after placebo only at the second hour (UPDRSIII, 23.1 6 8.8 vs 33.9 6 10.7; p , 0.01, Wilcoxon test).
These data suggest that Hp infection may interfere withL-dopa absorption in PD patients. Moreover, they indicatethat Hp eradication procedure may increase drug availabilityby about 20%, prolonging the tail of the plasma L-dopa con-
Fig. Effects of (A) placebo or (B) antibiotic treatment onL-dopa plasma concentrations in Parkinsons’s disease patientsaffected by HP infection.
Table 1. UPDRS Motor Examination III Assessed in Seven Parkinsonian Patients Before and After the Administration of 250 mgof L-DOPA (mean 6 standard deviation)
Pre-L-dopa 1 hr Post-L-dopa 2 hr Post-L-dopa ANOVA
Baseline 48.6 6 11.1 22.9 6 8.3 34.1 6 10.5 p , 0.01Placebo 48.9 6 11.8 22.6 6 8.7 33.9 6 10.7 p , 0.01Antibiotic Therapy 48.3 6 11.7 21.3 6 9.5 23.1 6 8.8 p , 0.01
ANOVA ns ns p , 0.01
ns 5 not significant.
LETTERS
686 © 2001 Wiley-Liss, Inc.
centration time curve, thus increasing its clinical effect byabout 25%. Considering the large prevalence of Hp infec-tion, it is likely that this factor could play a relevant role inthe reported erratic L-dopa absorption.
This work was supported by Telethon (EC0998) and Ministerodella Sanita (RF99-95).
1Clinica Neurologica and 2Dipartimento di Medicina Interna,Universita di Roma Tor Vergata, Roma; 3Laboratorio diFarmacologia e Tossicologia, Servizio di Farmacologia eNeuroscienze, IST, Genova; 4Dipartimento di MedicinaSperimentale, Sezione di Farmacologia e Tossicologia, Genova;5Instituto di Clinica delle Malattie Nervose e Mentali,Universita di Roma “La Sapienza,” Roma; and 6IRCCSFondazione S. Lucia, Roma, Italy
References1. Hoehn MM, Yahr MD. Parkinsonism: onset, progression and
mortality. Neurology 1967;17:427–442.2. Calam J. Helicobacter pylori modulation of gastric acid. Yale
J Biol Med 1999;72:195–202.3. Drake IM, Mapstone NP, Schoral CJ, et al. Reactive oxygen spe-
cies activity and lipid peroxidation in Helicobacter pylori associ-ated gastritis: relation to gastric mucosal ascorbic acid concentra-tion and effect of H. pylori eradication. Gut 1998;42:768–771.
4. The European Helicobacter pylori Study Group (EHPSG). Cur-rent European concepts in the management of Helicobacter pyloriinfection. The Maastricht Consensus Report. Gut 1997;41:8–13.
5. Tallarida RJ, Murray AB. Manual of pharmacologic calculationswith computer programs. Second edition. New York: SpringerVerlag, 1986.
6. Fahn S, Elton RL, and members of the UPDRS DevelopmentCommittee. Unified Parkinson’s disease rating scale. In: Fahn S,Marsden CD, Goldstein M, Calne DB, eds. Recent develop-ments in Parkinson’s disease, Vol. 2. Florham Park, NJ: Mac-Millan Healthcare Information, 1987:153–163.
Pharmacodynamic Modeling of Oral Levodopa inParkinson’s DiseaseManuela Contin, PharmD, Roberto Riva, MD,Fiorenzo Albani, PharmD, Paolo Martinelli, MD, andAgostino Baruzzi, MD
We read with interest the article by de la Fuente-Fernandezand colleagues1 describing the application of the dopamineD2-type receptor antagonist [11C] raclopride (RAC) in as-sessing progressive changes in time course of oral levodopa(L-dopa)-derived dopamine in patients with Parkinson’s dis-ease. Based on the assumption of a linear relationship be-tween changes in RAC binding potential and synaptic dopa-mine levels, the authors found that patients who developedL-dopa motor response fluctuations over a 3-year follow-upperiod had a more abrupt increase and decrease in estimatedcerebral dopamine concentrations after an oral L-dopa dosethan patients who maintained a “stable” response to chronicdrug therapy. They conclude that the future risk of develop-ing motor fluctuations may be thus predicted by the timecourse of response to oral L-dopa challenges. We have alreadyshown in a series of articles2–4 the potential of a standardizedoral L-dopa test, based on pharmacodynamic modeling of
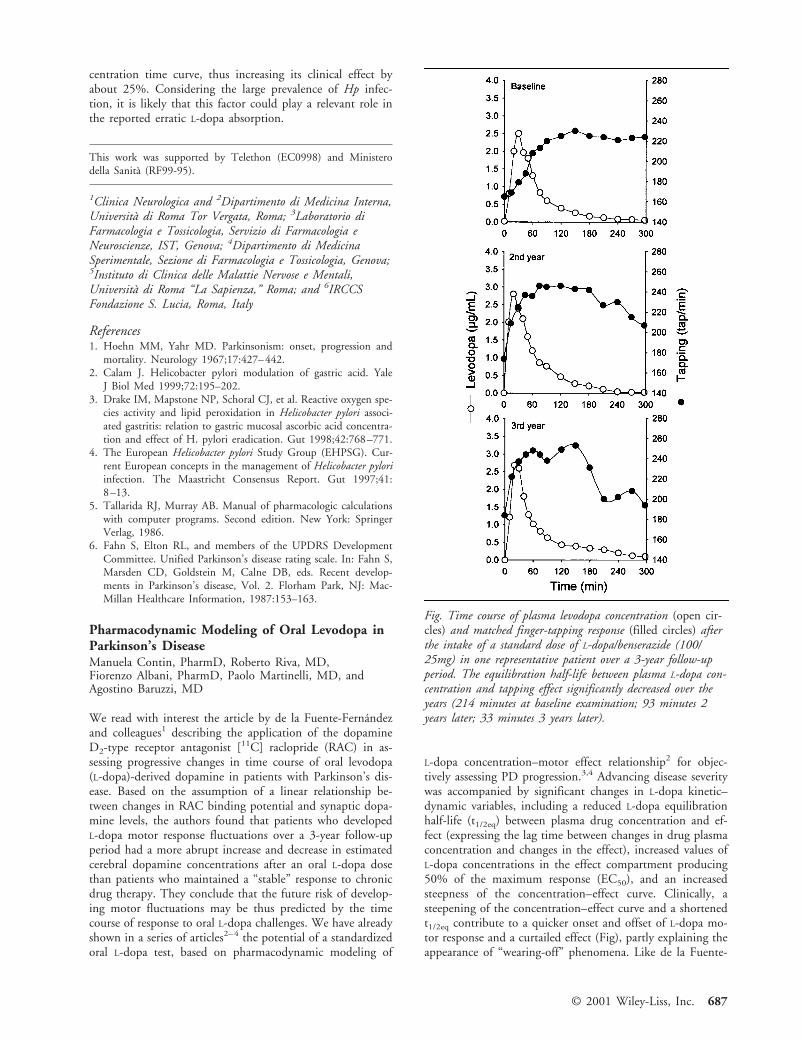
L-dopa concentration–motor effect relationship2 for objec-tively assessing PD progression.3,4 Advancing disease severitywas accompanied by significant changes in L-dopa kinetic–dynamic variables, including a reduced L-dopa equilibrationhalf-life (t1/2eq) between plasma drug concentration and ef-fect (expressing the lag time between changes in drug plasmaconcentration and changes in the effect), increased values ofL-dopa concentrations in the effect compartment producing50% of the maximum response (EC50), and an increasedsteepness of the concentration–effect curve. Clinically, asteepening of the concentration–effect curve and a shortenedt1/2eq contribute to a quicker onset and offset of L-dopa mo-tor response and a curtailed effect (Fig), partly explaining theappearance of “wearing-off” phenomena. Like de la Fuente-
Fig. Time course of plasma levodopa concentration (open cir-cles) and matched finger-tapping response (filled circles) afterthe intake of a standard dose of L-dopa/benserazide (100/25mg) in one representative patient over a 3-year follow-upperiod. The equilibration half-life between plasma L-dopa con-centration and tapping effect significantly decreased over theyears (214 minutes at baseline examination; 93 minutes 2years later; 33 minutes 3 years later).
© 2001 Wiley-Liss, Inc. 687

Fernandez and colleagues,1 we interpreted these findings assuggestive of an altered rate of dopamine turnover, possiblybecause of both the progressive loss of dopamine storage ca-pacity and disruption of modulated release mechanisms bydopaminergic neurons. By applying this oral L-dopa test tothe longitudinal assessment of Parkinson’s disease patients,3,4
we were able to detect subtle changes in L-dopa pharmaco-dynamics even preceding the appearance of clinically evidentdrug motor fluctuations (Fig). While we cast some doubt onthe clinical availability of RAC positron emission tomogra-phy as an objective measure of Parkinson’s diasease evolu-tion,5 we think that L-dopa pharmacodynamic modeling canoffer a simple and clinically practical tool for indirect assess-ment of the functional integrity of the nigrostriatal dopami-nergic system in Parkinson’s disease patients.3,4
Laboratory of Clinical Neuropharmacology, Institute ofNeurology, University of Bologna, Bologna, Italy
References1. de la Fuente-Fernandez R, Lu JQ, Sossi V, et al. Biochemical
variations in the synaptic level of dopamine precede motor fluc-tuations in Parkinson’s disease: PET evidence of increased do-pamine turnover. Ann Neurol 2001;49:298–303.
2. Contin M, Riva R, Martinelli P, et al. Pharmacodynamic mod-eling of oral levodopa: clinical application in Parkinson’s dis-ease. Neurology 1993;43:367–371.
3. Contin M, Riva R, Martinelli P, et al. Longitudinal monitoringof the levodopa concentration-effect relationship in Parkinson’sdisease. Neurology 1994;44:1287–1292.
4. Contin M, Riva R, Martinelli P, et al. A levodopa kinetic-dynamic study of the rate of progression in Parkinson’s disease.Neurology 1998;51:1075–1080.
5. Frey AK. The neurochemistry of therapeutics: levodopa phar-macodynamics in Parkinson’s disease. Ann Neurol 2001;49:285–287.
ReplyRaul de la Fuente-Fernandez, MD,and A. Jon Stoessl, MD, FRCPC
We thank Contin and colleagues for their interest in ourwork.1 The story of levodopa (L-dopa)-related motor compli-cations in Parkinson’s disease is as old as that of L-dopa itself.Literally hundreds of studies have been published on thistopic. Theories on the pathogenesis of “wearing off” fluctu-ations ranging from changes in storage capacity to down-stream postsynaptic changes have been elegantly presentedover the years by several groups of researchers, includingContin and associates.2–4 We cited some excellent reviews inan attempt to adequately cover key aspects of this field.
Contin and colleagues used an objective test of motor per-formance (finger tapping) to gain some insight into thepathogenesis of motor fluctuations. They found that al-though plasma L-dopa kinetics remain unchanged, the dura-tion of the response to L-dopa shortens as disease progresses.From this observation, they infer a change in cerebral levo-dopa kinetics.3 The authors proposed that “increased rate ofsynthesis and release of dopamine by the surviving nigrostri-atal projections as the disease progresses, up to a potentialconversion of levodopa to dopamine outside aminergic neu-rons” may account for their observations.3 They also re-
ported that “wearing off” fluctuations could be the result of“both an impaired efficiency of presynaptic mechanisms . . .and postsynaptic-receptor hyposensitivity.”2 It is naturallydifficult to infer how the brain handles L-dopa based only ona simple test of finger tapping; a great number of assump-tions are required, and the conclusions are necessarily veryimprecise.
We agree with Contin and colleagues that positron emis-sion tomography (PET) is not a practical technique for as-sessing the future risk of motor fluctuations. However, thisapproach allowed us to specifically examine how the esti-mated synaptic levels of dopamine change during the first 4hours following L-dopa administration. All patients were sta-ble responders at the time of the study (most of the patientshad objective finger-tapping measurements). We found thatonly those patients with no significant L-dopa-induced in-crease in the synaptic levels of dopamine 4 hours after L-dopaadministration developed early “wearing off” fluctuationsduring the follow-up.1 Even using a sophisticated techniquelike PET, and looking at only one of the components of thecomplex pharmacodynamics of L-dopa, we were forced tomake a number of assumptions that, while reasonable, couldjeopardize the conclusions.1,5 Nevertheless, our observationrepresents the first direct evidence for differences in dopa-mine turnover unrelated to disease severity between fluctua-tors and stable responders. We are pleased that our findings,based on more indirect measures, are compatible with thoseof Contin and colleagues.
Neurodegenerative Disorders Centre, University of BritishColumbia, Vancouver, BC, Canada
References1. de la Fuente-Fernandez R, Lu JQ, Sossi V, et al. Biochemical
variations in the synaptic level of dopamine precede motor fluc-tuations in Parkinson’s disease: PET evidence of increased dopa-mine turnover. Ann Neurol 2001;49:298–303.
2. Contin M, Riva R, Martinelli P, et al. Pharmacodynamic mod-eling of oral levodopa: Clinical application in Parkinson’s disease.Neurology 1993;43:367–371.
3. Contin M, Riva R, Martinelli P, et al. Longitudinal monitoringof the levodopa concentration-effect relationship in Parkinson’sdisease. Neurology 1994;44:1287–1292.
4. Contin M, Riva R, Martinelli P, et al. A levodopa kinetic-dynamic study of the rate of progression in Parkinson’s disease.Neurology 1998;51:1075–1080.
5. Frey AK. The neurochemistry of therapeutics: levodopa pharma-codynamics in Parkinson’s disease. Ann Neurol 2001;49:285–287.
Are the “Newly Discovered” Paraneoplastic Anti-Collapsin Response-Mediator Protein 5 AntibodiesSimply Anti-CV2 Antibodies?Jerome Honnorat, MD, PhD,1
Jean-Christophe Antoine, MD,2
and Marie-Francoise Belin, PhD3
Yu and colleagues1 recently reported that anti-Ulip6/collap-sin response-mediator protein 5 (CRMP5) antibodies are a“new” marker of paraneoplastic neurological syndromes andclaimed that these were different from anti-CV2 antibodiesdescribed long ago by our group. This claim relied on three
688 © 2001 Wiley-Liss, Inc.

points: (1) the cell types recognized are different; (2) ourlabeling of oligodendrocytes was an artifact; and (3) the onlyUlip/CRMP recognized by these “new” antibodies wasUlip6/CRMP5, whereas anti-CV2 antibodies recognize otherUlip/CRMPs.
Historically, the first case that led to the discovery of anti-CV2 antibodies was published in 1993,2 and the immuno-histochemical and Western blotting characteristics were de-finitively established in 1996.3 If most patients with anti-CV2 antibodies (70%) develop small cell lung cancer,4,5 therare association with thymoma was already underlined,4 aswas the frequent occurrence of optic manifestations.4
As their first point, Yu and colleagues claim that their an-tibody “clearly binds to neurons” with “minor binding topresumptive glia” and state (incorrectly) that “anti-CV2 isthe only paraneoplastic autoantibody reported to bind exclu-sively to oligodendrocytes.” In fact, although anti-CV2 anti-bodies bind to an oligodendrocyte subpopulation in theadult brain,3,5 we clearly showed that they also react withneurons and neuronal fibers, particularly during embryogen-esis and in regions that retain brain plasticity in the adult,such as the dentate gyrus and the rostral migratory stream ofneuron precursors of the olfactory bulb.2,3,5 They also staincells in the retina2 and the peripheral nervous system6 withlight diffuse labeling of the neuropile.2
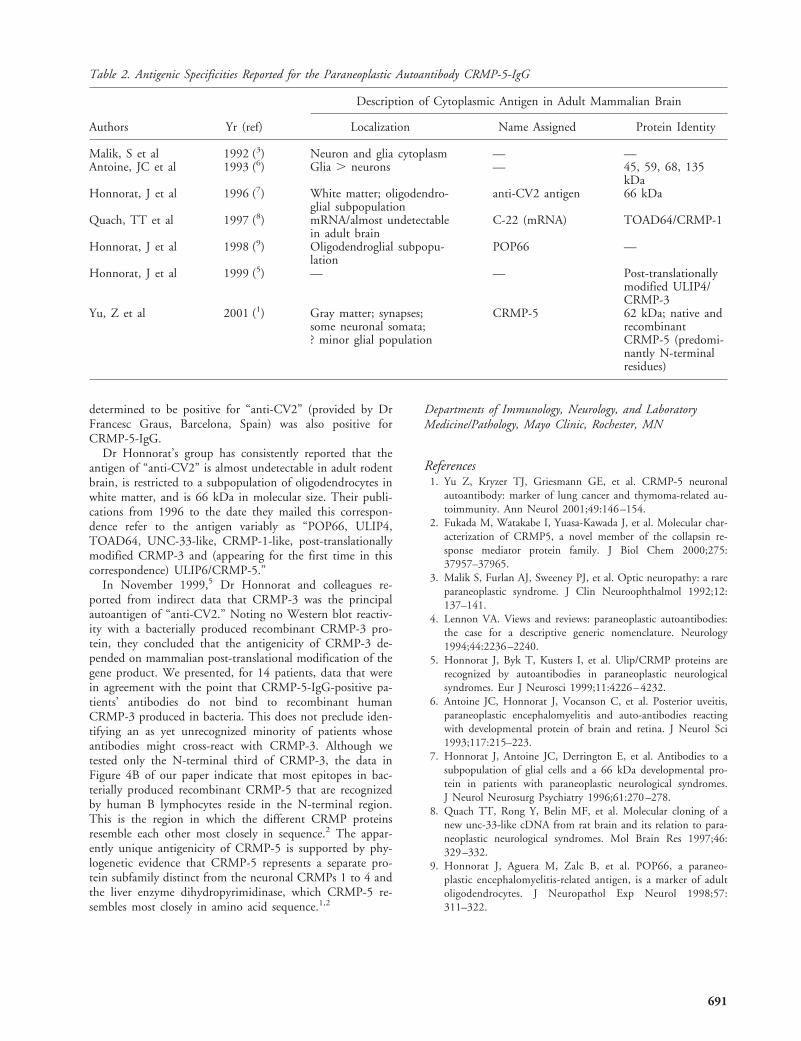
In their second point, Yu and colleagues state that ouroligodendrocytic labeling was an artifact resulting from fixa-tion by intracardiac perfusion of 4% paraformaldehyde. Thiswidely used procedure is considered superior to immersionfixation for neuroanatomy, resulting in good tissue preserva-tion and stabilization of cellular organization, preventingdegradation and loss of soluble components. As the anti-CV2 antigen is quite soluble,3 in situ fixation is optimal forits detection and the postmortem fixation by immersion informalin used by Yu and associates might have underesti-mated labeling of certain cells. It is strange that their anti-Ulip6/CRMP5 antibodies do not recognize oligodendrocytesbecause, using a rabbit polyclonal anti-Ulip6/CRMP5 anti-serum (Fig, C) and in situ hybridization with Ulip6/CRMP5RNA probes (see Fig, B), we have demonstrated that in theadult brain a subpopulation of oligodendrocytes similar tothat recognized by anti-CV2 antibodies, express high levelsof Ulip6/CRMP5 (Ricard et al, unpublished research). Fur-thermore, Western blots of purified adult rat brain oligoden-drocytes confirm that these cells express Ulip6/CRMP5 (seeFig, D). Interestingly, Ulip2/CRMP2 and, to a lesser extent,Ulip4/CRMP3 are also expressed by oligodendrocytes,7 sug-gesting a particular role of Ulip/CRMPs in oligodendrocytes.
Their third point is that their antibodies only react withUlip6/CRMP5, but they only tested full-length Ulip6/CRMP5 and Ulip2/CRMP2 and another construct repre-senting one-third of Ulip4/CRMP3. They wrongly claimedthat our laboratory “assigned two different molecular identi-ties to the anti-CV2 antigen” and that we “first concludedthat the antigen was Ulip3/CRMP1” then “later revised itsidentity to Ulip4/CRMP3.” In fact, characterization of theCV2 antigen(s), using anti-CV2 antibodies, initially allowedus to identify several peptide sequences from members of theUlip/CRMPs8 and to clone and sequence Ulip3/CRMP1.9
At this time, when only four Ulip/CRMPs were known, weshowed that most, but not all, anti-CV2 antibodies recog-
nized Ulip4/CRMP3 or Ulip3/CRMP1.8 Subsequently, us-ing anti-CV2 antibodies negative with both of these proteins,we cloned Ulip6/CRMP5 (GenBank AF 264015) at thesame time as other groups.10,11 Of 23 sera from patientswith anti-CV2 antibodies, 33% recognized Ulip3/CRMP1,40% Ulip4/CRMP3, and almost all ULIP6/CRMP5. Wealso found two sera that recognize Ulip2/CRMP2 (see Fig,A). Thus, it is clear that anti-CV2 antibodies react with atleast four of the five known Ulip/CRMPs, and that virtuallyall anti-CV2 sera recognize Ulip6/CRMP5.
Fig. (A) Western-blot analysis of anti-CV2 sera reactivity onrecombinant Ulip/CRMP proteins produced in HeLa cells.(rU1/C4 5 Ulip1/CRMP4, rU2/C2 5 Ulip2/CRMP2, rU3/C1 5 Ulip3/CRMP1, rU4/C3 5 Ulip4/CRMP3, rU6/C5 5Ulip6/CRMP5). Each recombinant Ulip/CRMP protein beingrecognized by the specific corresponding rabbit polyclonal anti-serum (lane 1), anti-CV2 sera from 7 patients (lanes 2–8)recognized different Ulip/CRMP proteins. Patients 2 and 8had also anti-Hu antibodies. (B) and (C) Sagittal section ofadult rat spinal cord hybridized with Ulip6/CRMP5 cRNAprobes (B) and frontal section of adult rat brainstem immuno-labeled with specific anti-Ulip6/CRMP5 rabbit polyclonal an-tibodies (C). Both Ulip6/CRMP5 mRNA and protein wereclearly detected in oligodendrocytes of the white matter. (D)Detection of Ulip6/CRMP5 protein on Western blot from pu-rified oligodendrocyte extracts using specific anti-Ulip6/CRMP5 rabbit polyclonal antibodies (lane 2). Preimmuneserum (lane1) showed no reactivity.
689

A paraneoplastic antibody can only be described as “new”when the cellular and molecular characteristics of the antigenare clearly different from those of previously described anti-bodies. As suggested by Posner and Dalmau in their edito-rial12 we believe that this “new” paraneoplastic antibody re-flects only one aspect of the Ulip/CRMPs recognition byanti-CV2-antibodies.
1Neurologie B, Hopital Neurologique, Lyon; 2Neurologie B,Hopital Bellevue, St-Etienne; and 3INSERM U 433, FaculteLaennec, Lyon, France
References1. Yu Z, Kryzer TJ, Griesmann GE, et al. CRMP-5 neuronal
autoantibody: marker of lung cancer and thymoma-related au-toimmunity. Ann Neurol 2001;49:146–154.
2. Antoine JC, Honnorat J, Vocanson C, et al. Posterior uveitis,paraneoplastic encephalomyelitis and auto-antibodies reactingwith a developmental protein of brain and retina. J Neurol Sci1993;117:215–223.
3. Honnorat J, Antoine JC, Derrington E, et al. Antibodies to asubpopulation of glial cells and a 66 kDa developmental pro-tein in patients with paraneoplastic neurological syndromes.J Neurol Neurosurg Psychiatry 1996;61:270–278.
4. Rogemond V, Honnorat J. Anti-CV2 autoantibodies and para-neoplastic neurological syndromes. Clin Rev Allergy Immunol2000;19:51–59.
5. Honnorat J, Aguera M, Zalc B, et al. POP66, a paraneoplasticencephalomyelitis-related antigen, is a marker of adult oligoden-drocytes. J Neuropathol Exp Neurol 1998;57:311–322.
6. Antoine JC, Honnorat J, Camdessanche JP, et al. Paraneoplas-tic anti-CV2 antibodies react with peripheral nerve and are as-sociated with a mixed axonal and demyelinating peripheral neu-ropathy. Ann Neurol 2001;49:214–221.
7. Ricard D, Stankoff B, Bagnard D, et al. Differential expressionof collapsin response mediator proteins (CRMP/ULIP) in sub-sets of oligodendrocytes in the postnatal rodent brain. Mol CellNeurosci 2000;16:324–337.
8. Honnorat J, Byk T, Kuster I, et al. Ulip/CRMP proteins arerecognized by autoantibodies in paraneoplastic neurologicalsyndromes. Eur J Neurosci 1999;11:4226–4232.
9. Quach TT, Rong Y, Belin MF, et al. Molecular cloning of anew unc-33-like cDNA from rat brain and its relation to para-neoplastic neurological syndromes. Brain Res Mol Brain Res1997;46:329–332.
10. Inatome R, Tsujimura T, Hitomi T, et al. Identification ofCRAM, a novel unc-33 gene family protein that associates withCRMP3 and protein-tyrosine kinase(s) in the developing ratbrain. J Biol Chem 2000;275:27291–27302.
11. Fukada M, Watakabe I, Yuasa-Kawada J, et al. Molecular char-acterization of CRMP5, a novel member of the collapsin re-sponse mediator protein family. J Biol Chem 2000;275:37957–37965.
12. Posner JB, Dalmau JO. Yet another paraneoplastic antibody.Ann Neurol 2001;49:141–142.
ReplyVanda A. Lennon, MD, PhD, Zhiya Yu, MD, PhD,Thomas J. Kryzer, AS, and Guy E. Griesmann, MS
To objectively clarify the historical points raised by DrsHonnorat, Antoine, and Belin, we made two tables. Table 1documents the chronology for discovery of the humanCRMP-5 protein and Table 2 the evolving definition of theautoantibody that we recognize as CRMP-5-specific.CRMP-5 is related more distantly to the family of collapsinresponse-mediator proteins (involved in neural development)than CRMPs 1–4.1,2 As cited in our study,1 a paraneoplasticautoantibody that was in all likelihood CRMP-5-specific wasfirst described by Malik and colleagues in 19923 in a patientwith optic neuropathy and cerebellar ataxia related to small-cell lung carcinoma. IgG in that patient’s serum bound toboth neuronal and glial cytoplasm in human cerebral cortex,cerebellum, and optic nerve but not to the patient’s neo-plasm.
We reported CRMP-5-IgG as a “newly defined” neuronalautoantibody related to small-cell lung carcinoma and thy-moma. We did not claim discovery but rather definitionof a new neuronal autoantibody specificity. We cited fourreports from Dr Honnorat and colleagues (see references12,13,26,27),1 in addition to citing the report of Malik andcolleagues (reference 25),1 as likely but imprecisely-definedprecedents for the CRMP-5 autoantibody. The importanceof our study1 is that it defines, for the first time, the molec-ular identity of a new onconeuronal antigen (CRMP-5) andunambiguously assigns it a predominantly neuronal localiza-tion in the adult central and peripheral nervous systems, inaddition to being a universal cytoplasmic antigen of small-cell lung carcinomas. It also defines the spectrum of CRMP-5-IgG-related neurological disorders in 116 patients, with aremarkable 12% frequency of subacute chorea/athetosis, anda systemic context that includes 14% frequency of vasculiticand thromboembolic phenomena. In keeping with a genericnomenclature,4 we named this paraneoplastic autoantibody“CRMP-5-IgG.” CRMP-5 is the name independently as-signed by three of the five laboratories that submitted perti-nent human DNA sequence data to GenBank following ouroriginal submission of the CRMP-5 sequence (see Table 1).
We summarize the major differences between our dataand characteristics reported by Dr Honnorat’s group for theautoantibody that they call “anti-CV2” (see Table 2). Werecently had the opportunity to verify that a patient’s serum
Table 1. Chronological Order of GenBank Submissions for Human CRMP-5 Protein
Submission Date Accession Number Authors Name
09-Jun-1999 AF 157634 Yu Z et al CRMP-502-Dec-1999 AJ 251275 Horiuchi M et al Hypothetical protein05-May-2000 AF 264015 Aguera M et ala ULIP-602-Nov-2000 NM_020134 Inatome R et al CRMP-505-Feb-2001 BC 002874 Strausberg R et al CRMP-516-Apr-2001 XM_010830 NCBI Annotation Project CRMP-5
aDr. Honnorat’s group.
690 © 2001 Wiley-Liss, Inc.
determined to be positive for “anti-CV2” (provided by DrFrancesc Graus, Barcelona, Spain) was also positive forCRMP-5-IgG.
Dr Honnorat’s group has consistently reported that theantigen of “anti-CV2” is almost undetectable in adult rodentbrain, is restricted to a subpopulation of oligodendrocytes inwhite matter, and is 66 kDa in molecular size. Their publi-cations from 1996 to the date they mailed this correspon-dence refer to the antigen variably as “POP66, ULIP4,TOAD64, UNC-33-like, CRMP-1-like, post-translationallymodified CRMP-3 and (appearing for the first time in thiscorrespondence) ULIP6/CRMP-5.”
In November 1999,5 Dr Honnorat and colleagues re-ported from indirect data that CRMP-3 was the principalautoantigen of “anti-CV2.” Noting no Western blot reactiv-ity with a bacterially produced recombinant CRMP-3 pro-tein, they concluded that the antigenicity of CRMP-3 de-pended on mammalian post-translational modification of thegene product. We presented, for 14 patients, data that werein agreement with the point that CRMP-5-IgG-positive pa-tients’ antibodies do not bind to recombinant humanCRMP-3 produced in bacteria. This does not preclude iden-tifying an as yet unrecognized minority of patients whoseantibodies might cross-react with CRMP-3. Although wetested only the N-terminal third of CRMP-3, the data inFigure 4B of our paper indicate that most epitopes in bac-terially produced recombinant CRMP-5 that are recognizedby human B lymphocytes reside in the N-terminal region.This is the region in which the different CRMP proteinsresemble each other most closely in sequence.2 The appar-ently unique antigenicity of CRMP-5 is supported by phy-logenetic evidence that CRMP-5 represents a separate pro-tein subfamily distinct from the neuronal CRMPs 1 to 4 andthe liver enzyme dihydropyrimidinase, which CRMP-5 re-sembles most closely in amino acid sequence.1,2
Departments of Immunology, Neurology, and LaboratoryMedicine/Pathology, Mayo Clinic, Rochester, MN
References1. Yu Z, Kryzer TJ, Griesmann GE, et al. CRMP-5 neuronal
autoantibody: marker of lung cancer and thymoma-related au-toimmunity. Ann Neurol 2001;49:146–154.
2. Fukada M, Watakabe I, Yuasa-Kawada J, et al. Molecular char-acterization of CRMP5, a novel member of the collapsin re-sponse mediator protein family. J Biol Chem 2000;275:37957–37965.
3. Malik S, Furlan AJ, Sweeney PJ, et al. Optic neuropathy: a rareparaneoplastic syndrome. J Clin Neuroophthalmol 1992;12:137–141.
4. Lennon VA. Views and reviews: paraneoplastic autoantibodies:the case for a descriptive generic nomenclature. Neurology1994;44:2236–2240.
5. Honnorat J, Byk T, Kusters I, et al. Ulip/CRMP proteins arerecognized by autoantibodies in paraneoplastic neurologicalsyndromes. Eur J Neurosci 1999;11:4226–4232.
6. Antoine JC, Honnorat J, Vocanson C, et al. Posterior uveitis,paraneoplastic encephalomyelitis and auto-antibodies reactingwith developmental protein of brain and retina. J Neurol Sci1993;117:215–223.
7. Honnorat J, Antoine JC, Derrington E, et al. Antibodies to asubpopulation of glial cells and a 66 kDa developmental pro-tein in patients with paraneoplastic neurological syndromes.J Neurol Neurosurg Psychiatry 1996;61:270–278.
8. Quach TT, Rong Y, Belin MF, et al. Molecular cloning of anew unc-33-like cDNA from rat brain and its relation to para-neoplastic neurological syndromes. Mol Brain Res 1997;46:329–332.
9. Honnorat J, Aguera M, Zalc B, et al. POP66, a paraneo-plastic encephalomyelitis-related antigen, is a marker of adultoligodendrocytes. J Neuropathol Exp Neurol 1998;57:311–322.
Table 2. Antigenic Specificities Reported for the Paraneoplastic Autoantibody CRMP-5-IgG
Authors Yr (ref)
Description of Cytoplasmic Antigen in Adult Mammalian Brain
Localization Name Assigned Protein Identity
Malik, S et al 1992 (3) Neuron and glia cytoplasm — —Antoine, JC et al 1993 (6) Glia . neurons — 45, 59, 68, 135
kDaHonnorat, J et al 1996 (7) White matter; oligodendro-
glial subpopulationanti-CV2 antigen 66 kDa
Quach, TT et al 1997 (8) mRNA/almost undetectablein adult brain
C-22 (mRNA) TOAD64/CRMP-1
Honnorat, J et al 1998 (9) Oligodendroglial subpopu-lation
POP66 —
Honnorat, J et al 1999 (5) — — Post-translationallymodified ULIP4/CRMP-3
Yu, Z et al 2001 (1) Gray matter; synapses;some neuronal somata;? minor glial population
CRMP-5 62 kDa; native andrecombinantCRMP-5 (predomi-nantly N-terminalresidues)
691

Classic Rett Syndrome in a Boy as a Result ofSomatic Mosaicism for a MECP2 MutationJudith Armstrong, MsC,1 Pilar Poo, MD, PhD,2
Merce Pineda, MD, PhD,2 Elena Aibar, MsC,1
Esther Gean, MsC, MD,1 Vicenc Catala, MsC, PhD,3
and Eugenia Monros, MsC, PhD1
Rett syndrome (RTT) is a neurodevelopmental disorder af-fecting females in a sporadic manner. In a high proportion ofpatients the disease is caused by de novo dominant muta-tions at MECP2 gene (Xq28).1,2 The existence of RTT maleshas been discussed extensively, and less restrictive diagnosticcriteria have been proposed to include this variant, whichshould be considered when evaluating boys with idiopathicdevelopmental regression, autistic features, and loss of handfunction. Nevertheless, no MECP2 analysis has been re-ported from Rett-like males to date. Analysis of 2 familialcases showed that boys carrying the same MECP2 mutationthat caused RTT in their sisters suffered from severe–fatalneonatal encephalopathy.2,3 Recent data, however, demon-strate that the clinical spectrum of MECP2 mutations iswider than previously expected. With a frequency compara-ble to that of fragile X syndrome, recessive nonspecificX-linked mental retardation can be caused by missense mu-tations at MECP2, different than those causing RTT.4 Mu-tations have also been described in patients with congenitalnonprogressive encephalopathy and in some cases of An-gelman syndrome (AS), the only reported AS boy being asomatic mosaic for a MECP2 truncating mutation.5
We document the first MECP2 analysis of a boy withclassic RTT and a normal 46,XY karyotype. The patient is14 years of age and fulfils eight of nine necessary criteria,seven of eight supportive criteria, and no exclusion criteria,according to the Rett Syndrome Diagnostic Criteria WorkGroup. Genetic informed consent was obtained and thestudy was approved by the Ethical and Investigation Com-missions of our hospital.
MECP2 sequencing of two independent patients’ DNAsamples from peripheral lymphocytes revealed the presenceof a heterozygous change 398G3A, causing an R133H sub-stitution. The mutation had been previously described in 2RTT female patients.6 As the boy had a normal karyotype,heterozygosity could be explained by (1) a low frequencymosaicism for a Klineffelter syndrome, discarded by FISH onprophasic nuclei; (2) a MECP2 locus duplication, rejected byhigh-resolution karyotype and observation of hemizygosityfor X-linked markers and two intragenic MECP2 polymor-phisms; or (3) somatic mosaicism for the mutation. To testthis last hypothesis, DNA was prepared from the patient’soral mucosa and sequenced. The normal sequence, with onlya small amount of the mutated allele, was observed. Theseresults demonstrated that the RTT boy is a somatic mosaicfor the R133H mutation and seemed to indicate that themutation is present in a high proportion of lymphocytes butat a lower frequency in oral mucous. To specifically test andsemiquantify the heterozygous status of the mutation, anamplification–refractory mutation system (ARMS) experi-ment was designed. Both the normal and mutated alleleswere amplified in the patient’s lymphocytes and oral mucosa
(Fig). Relative differences in band intensities corroboratedthe different degree of mosaicism between the two tissues.
The infrequent clinical picture of this patient is thus ex-plained by the existence of somatic mosaicism for an RTT-causing MECP2 mutation. Concerning X-linked diseases,this phenomenon in males mimics the result of lyonizationnaturally occurring in females and may explain the classicRett syndrome phenotype of the boy.
This work was supported by the Asociacion Valenciana y Catalanade Sındrome de Rett, and by Fondo de Investigacion Sanitaria(FIS99/0235 to J.A.).
Sequencing runs were done at Serveis Cientificotecnics, Universitatde Barcelona, and at Sistemas Genomicos, Valencia.
1Genetics Section and 2Neurology Service, HospitalUniversitari Sant Joan de Deu, Barcelona; and 3PrenatalGenetics, Barcelona, Spain
References1. Amir RE, Van den Veyver IB, Wan M. Tran CQ, et al. Rett
syndrome is caused by mutations in X-linked MECP2, encodingmethyl-CpG-binding protein 2. Nature Genet 1999;23:185–188.
2. Wan M, Sung Jae Lee S, Zhang X, et al. Rett Syndrome andbeyond: recurrent spontaneous and familial MECP2 mutations atCpG hotspots. Am J Hum Genet 1999;65:1520–1529.
3. Villard L, Kpebe A, Cardoso C, et al. Two affected boys in aRett syndrome family. Neurology 2000;55:1188–1193.
4. Couvert P, Bienvenu T, Aquaviva C, et al MECP2 is highly mu-tated in X-linked mental retardation. Hum Mol Genet 2001;10:941–946.
5. Watson P, Black G, Ramsden S, et al. Angelman syndrome phe-notype associated with mutations in MECP2, a gene encoding amethyl CpG binding protein. J Med Genet 2001;38:224–228.
6. Hoffbuhr K, Devaney J, LaFleur B, et al. Genotype and pheno-type evaluation of MeCP2 mutations in Rett syndrome. Neurol-ogy 2001;56:1486–1495.
Fig. ARMS analysis of the 398G3A mutation. The 350 bpband corresponds to fragment 2b from exon 2 of MECP2(according to Amir et al),1 used as an internal control of am-plification in a multiplex PCR reaction. The 181 bp fragmentwas designed to specifically amplify the normal or the mutatedbase at position 398. Pairs of adjacent lines correspond tonormal (N) and mutated (M) specific amplifications of thesame DNA sample: (1) patient’s DNA from peripheral bloodlymphocytes, first blood extraction; (2) patient’s DNA obtainedfrom peripheral blood lymphocytes, second blood extraction; (3)patient’s DNA from oral mucous; (4) DNA from a controlmale individual.
692 © 2001 Wiley-Liss, Inc.





























