Embed Size (px)
Citation preview

Chymase Inhibition Reduces Infarction and MatrixMetalloproteinase-9 Activation and Attenuates Inflammationand Fibrosis after Acute Myocardial Ischemia/Reperfusion□S
Shizu Oyamada, Cesario Bianchi, Shinji Takai, Louis M. Chu, and Frank W. SellkeCardiovascular Research Center, Division of Cardiothoracic Surgery, Rhode Island Hospital and Alpert Medical School ofBrown University, Providence, Rhode Island (S.O., C.B., L.M.C., F.W.S.); and Department of Pharmacology, Osaka MedicalCollege, Osaka, Japan (S.T.)
Received January 20, 2011; accepted July 26, 2011
ABSTRACTChymase is activated after acute myocardial ischemia/reperfu-sion (AMI-R) and is associated with an early activation of matrixmetalloproteinase-9 (MMP-9), which increases infarct size afterexperimental AMI, and late fibrosis. We assessed the effect ofchymase inhibition on myocardial protection and early signs offibrosis after AMI-R. Fourteen pigs underwent AMI-R and re-ceived intravenously either vehicle (V; n � 7) or chymase inhib-itor (CM; n � 7). Separately, rat myocardial fibroblast wasincubated with vehicle (n � 4), low-dose chymase (n � 4),high-dose chymase (n � 4), or high-dose chymase plus chy-mase inhibitor (n � 4). Infarct size (V, 41 � 5; CM, 24 � 5; P �0.01) and serum troponin T (P � 0.03) at the end of reperfusionwere significantly reduced in CM. Chymase activity in both thearea at risk (AAR) (P � 0.01) and nonischemic area (P � 0.02)was significantly lower in CM. Myocardial levels of pro, cleaved,
and cleaved/pro-MMP-9 in the AAR were significantly lower inCM than V (P � 0.01, � 0.01, and � 0.02, respectively),whereas phospho-endothelial nitric-oxide synthase (eNOS)(P � 0.01) and total eNOS (P � 0.03) were significantly higherin CM. Apoptotic cells (P � 0.05), neutrophils (P � 0.05), andMMP-9-colocalizing mast cells (P � 0.05) in the AAR weresignificantly reduced in CM. Interleukin-18 (P � 0.05) and in-tercellular adhesion molecule-1 (P � 0.05) mRNA levels weresignificantly lower in CM. In cultured cardiac fibrosis, Ki-67-positive cells were significantly higher in the high-dose chy-mase groups (P � 0.03). This study demonstrates that chymaseinhibition plays crucial roles in myocardial protection related toMMP-9, inflammatory markers, and the eNOS pathway. It mayalso attenuate fibrosis induced by activated chymase afterAMI-R.
IntroductionChymase is a chymotrypsin-like serine protease abundant in
the secretory granules of mast cells. Chymase has been shownto be a key enzyme in the local renin-angiotensin system (RAS)that generates angiotensin II (Ang II) independently from an-giotensin-converting enzyme (ACE). Chymase is stored in mastcells in an inactive form and is released as an active enzymewhen mast cells are stimulated by injury or inflammation. Thedensity of cardiac mast cells is remarkably increased in patientswith heart failure, and cardiac chymase may play an important
role in the development of several cardiovascular diseases (Pa-tella et al., 1998; Kumar et al., 2009; Pejler et al., 2010). Re-cently, we found that chymase activation was increased in isch-emic myocardium after acute myocardial ischemia/reperfusion(AMI-R) compared with nonischemic and sham myocardial tis-sue (Oyamada et al., 2010). Chymase is also known to activatematrix metalloproteinase (MMP)-9 by cleaving a specific site ofthe catalytic domain of MMP-9 (Fang et al., 1996, 1997; Tchou-gounova et al., 2005). MMP-9, known as a 92-kDa gelatinase, iscorrelated with an increase in infarct size and left ventricle (LV)fibrosis after experimental AMI (Heymans et al., 1999; Rohde etal., 1999; Ducharme et al., 2000; Kelly et al., 2007).
Thus, we hypothesized that chymase inhibition might havean effect on myocardial protection and fibrosis after AMI-R. Inthis study, we assessed the early effects of chymase inhibition
Article, publication date, and citation information can be found athttp://jpet.aspetjournals.org.
doi:10.1124/jpet.111.179697.□S The online version of this article (available at http://jpet.aspetjournals.org)
contains supplemental material.
ABBREVIATIONS: RAS, renin-angiotensin system; Ang II, angiotensin II; ACE, angiotensin-converting enzyme; CM, chymase inhibitor; AMI-R,acute myocardial ischemia/reperfusion; MMP, matrix metalloproteinase; LV, left ventricle; LAD, left anterior descending artery; AAR, area at risk;TTC, triphenyl tetrazolium chloride; CCL, chemokine (CC-motif) ligand; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling; V,vehicle; eNOS, endothelial nitric-oxide synthase; IL, interleukin; ICAM, intercellular adhesion molecule; RT-PCR, reverse transcription-polymerasechain reaction; TNF, tumor necrosis factor; CRP, C-reactive protein; NLV, normal left ventricle (nonischemic area); TY51469, 2-[4-(5-fluoro-3-methylbenzo[b]thiophen-2-yl)sulfonamide-3-methanesulfonylphenyl]-thiazole-4-carboxylic acid; PBS, phosphate-buffered saline.
0022-3565/11/3391-143–151$25.00THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 339, No. 1Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics 179697/3720856JPET 339:143–151, 2011 Printed in U.S.A.
143
http://jpet.aspetjournals.org/content/suppl/2011/07/27/jpet.111.179697.DC1Supplemental material to this article can be found at:
at ASPE
T Journals on July 21, 2018
jpet.aspetjournals.orgD
ownloaded from

with 2-[4-(5-fluoro-3-methylbenzo[b]thiophen-2-yl)sulfon-amide-3-methanesulfonylphenyl]-thiazole-4-carboxylic acid(TY51469; molecular weight 526.60), a specific chymase inhib-itor (CM), on a clinically relevant swine model of AMI-R. A60-min ischemia followed by a 120-min reperfusion was chosento assess an early effect of chymase inhibition (it is the shortertime point where necrosis can be determined with accuracy andreproducibility) and to allow comparison with previous experi-ments performed by our group.
Materials and MethodsReagents. The specific chymase inhibitor TY51469 was synthe-
sized at Toa Eiyo Ltd. (Tokyo, Japan). TY51469 has been shown to bea highly specific and stable inhibitor of chymase (Koide et al., 2003).
Animals and Surgery. Animals were housed individually andprovided with standard chow and water ad libitum. All experimentswere approved by the Beth Israel Deaconess Medical Center (Boston,MA) animal care and use committee and the Harvard Medical Area(Boston, MA) standing committee on animals. The experiments con-formed to the National Institutes of Health guidelines regulating thecare and use of laboratory animals (Institute for Laboratory AnimalResources, 2010).
Fourteen intact male Yucatan mini-swine (20 weeks old, 27 � 2kg; Sinclair Research Center, Columbia, MO) were used. All animalswere subjected to regional LV ischemia by left anterior descendingartery (LAD) occlusion distal to the second diagonal branch for 60min, followed by release of the artery and reperfusion for 120 min asdescribed previously (Osipov et al., 2009; Oyamada et al., 2010).Animals received intravenously either vehicle (V; n � 7) or CM (n �7) as a bolus of 2.0 mg/kg 50 min into the occlusion period (ischemia),followed by a continuous infusion of 2.0 mg/kg/h during the entireperiod of reperfusion (Harvard Apparatus Inc., Holliston, MA).
Measurement of Global and Regional Function. Mean arte-rial blood pressure, heart rate, developed LV pressure, and globalsystolic LV function as determined from dP/dt were measured atbaseline and subsequently at 30-min intervals to the end of reper-fusion as described previously (Osipov et al., 2009; Oyamada et al.,2010).
Quantification of Myocardial Infarct Size. The area at risk(AAR), the nonischemic area (NLV), and the infarcted/noninfarctedAARs were determined as described previously (Osipov et al., 2009;Oyamada et al., 2010) with minor modifications. In brief, at the endof 120 min of reperfusion, AAR was delineated by religating the LADand injecting � 30 ml of a 1:150 dilution in PBS of monastryl bluepigment (phthalocyanine blue; Engelhard Corp., Louisville, KY) intothe root of a cross-clamped aorta (between the cross-clamp and theaortic valve). The AAR lacked the blue dye. The heart was sliced,from the LV apex, into four �1-cm-thick slices (perpendicular to theLAD) and slices 1, 3, and 4 starting from the apex were immersedinto 1% TTC in PBS at 37°C for 10 min to determine myocardialinfarcted and noninfarcted AAR. Slice 2 was used to collect tissue foradditional studies (below). The delimited areas were measured by com-puterized planimetry (Scion Image; Scion Corporation, Frederick, MD).
Chymase Activity Assay. Myocardial tissue (AAR, n � 5; NLV,n � 5) was minced and homogenized in 20 mM Tris-HCl buffer, pH8.0. The homogenate was centrifuged (10,000 rpm, 30 min) and thesupernatant was discarded. The pellet was homogenized in 10 mMTris-HCl buffer, pH 8.0, containing 2 M KCl and 0.1% Triton X-100.The homogenate was stored overnight at 4°C and centrifuged (14,000rpm, 30 min). The supernatant was used as the tissue extract thatcontains chymase. Protein concentration was measured spectrophoto-metrically at 595 nm with a BCA Protein Assay Kit (Thermo FisherScientific, Waltham, MA) and standardized to 3.1 �g/�l with 10 mMTris-HCl buffer, pH 8.0, containing 2 M KCl and 0.1% Triton X-100.
Chymase activity was measured using the specific synthetic sub-strate Suc-Ala-Ala-Pro-Phe-pNA (Bachem California, Torrance, CA)
in a total volume of 100 �l of 100 mM Tris-HCl buffer, pH 8.0, 200mM NaCl, 2.5 mM substrate, and 31 �g of lysate. The initial rates ofnitroaniline release were measured spectrophotometrically at 405nm every 3 min. Chymase activity was calculated by subtractingbackground from the peak value. Human purified chymase (Merckand Co., Inc., Whitehouse Station, NJ) was used as positive control.
Western Blotting. Lysates from AAR and NLV tissues (V, n � 7;CM, n � 7) and postreperfusion serum (V, n � 7; CM, n � 7) wereprepared as described previously (Osipov et al., 2009; Oyamada etal., 2010). Twenty to 60 �g of total protein were fractionated by 4 to20, 8 to 16, or 12% SDS polyacrylamide gel electrophoresis (Invitro-gen, Carlsbad, CA) and transferred to polyvinylidene difluoridemembranes (Millipore Corporation, Billerica, MA). Each membranewas incubated overnight at 4°C with the following antibodies: ang-iotensinogen (1:1000; R&D Systems, Minneapolis, MN), chymase(1:200; Abcam plc, Cambridge, UK), MMP-9 (1:500), total eNOS(1:50), phosphor-eNOS (Ser1177; 1:50) (Cell Signaling Technology,Danvers, MA), renin (1:1000), ACE (1:50), angiotensin II type 1receptor (1:500), angiotensin II type 2 receptor (1:500), vitronectin(1:100), fibronectin (1:100), thrombin (1:100), elastase (1:100), plas-minogen (1:100) (Santa Cruz Biotechnology Inc., Santa Cruz, CA),mast cell tryptase (1:100; Leica Microsystems Inc., Bannockbum, IL),and troponin T (1:500; United States Biological, Swampscott, MA).The membranes were subsequently incubated for 45 min with theappropriate peroxidase-conjugated secondary antibody (1:1000;Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Im-mune complexes were visualized with enhanced chemiluminescence(GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK), recordedusing a charge-coupled device system (G-Box; Syngene, Frederick,MD) and quantified by microdensitometry (ImageJ 1.4; NationalInstitutes of Health, Bethesda, MD). Ponceau S staining and/or�-tubulin (BD Biosciences, San Jose, CA) were used to confirm eventransfer and equivalent protein loading.
Enzyme-Linked Immunosorbent Assay (Ang II, TNF-�, IL-6,CCL-2). Commercial porcine enzyme-linked immunosorbent assaykits for Ang II (Peninsula Laboratories, Belmont, CA), TNF-�(Thermo Fisher Scientific), IL-6 (R&D Systems), and CCL-2 (BethylLaboratories, Montgomery, TX) were used to obtain serum levels(n � 5 per group), as well as tissue levels of Ang II (AAR; n � 5).Myocardial lysates from AAR (3 �g/�l) and serum samples at bothbaseline and after reperfusion (10 �g/�l) were prepared as describedpreviously (Oyamada et al., 2010).
TUNEL Staining. TUNEL-positive cells in the AAR (n � 5 pergroup) were identified in the AAR as described previously (Oyamadaet al., 2010).
Immunohistochemistry. Frozen sections (10 �m) from bothAAR and NLV (V, n � 5; CM, n � 5) were stained for mast cellchymase, MMP-9, and myeloperoxidase. Primary antibodies againstchymase (1:100; Abcam Inc., Cambridge, MA), myeloperoxidase (1:100; Athens Research and Technology, Athens, GA), Ki-67 (SantaCruz Technology Inc.), and MMP-9 (1:100; Cell Signaling Technol-ogy) were incubated at 4°C overnight, then incubated with the ap-propriate DyLight-labeled secondary antibodies (1:100; Jackson Im-munoResearch Laboratories) for 30 min at room temperature.Sections incubated only with secondary antibody were used as neg-ative controls. Stained sections were imaged by confocal microscopy.Confocal images were acquired with a Nikon C1si confocal micro-scope (Nikon Inc., Mellville, NY) using diode lasers 402, 488, and561. Serial optical sections were performed with EZ-C1 computersoftware (Nikon Inc.). Each wavelength was acquired separately byinvoking frame lambda. Deconvolution and projections were per-formed in Elements version 3.1 computer software (Nikon Inc.).
Gel Zymography. Tissue extract (100 �g; V, n � 5; CM, n � 5)were resolved on a 10% polyacrylamide gel containing 0.1% gelatin(Bio-Rad Laboratories, Hercules, CA). Gels were renatured in 50 mMTris-HCl, pH 7.5, containing 100 mM NaCl and 2.5% Triton X-100for 90 min to remove SDS and then incubated with 50 mM Tris-HCl,pH 7.5, containing 10 mM CaCl2 for 20 h at 37°C. Gels were stained
144 Oyamada et al.
at ASPE
T Journals on July 21, 2018
jpet.aspetjournals.orgD
ownloaded from
with Coomassie Brilliant Blue and gelatinolytic activity was quan-tified as described above.
Serum CRP Measurement. Serum CRP levels at the end ofreperfusion were measured at the Chemistry Laboratory, RhodeIsland Hospital.
Reverse Transcription-Polymerase Chain Reaction. A re-verse transcription (RT) reaction followed by polymerase chain reac-tion (PCR) was performed using the One-Step RT-PCR kit (QIAGEN,Valencia, CA) with 2 �g of RNA isolated from both AAR and non-ischemic myocardium in V (n � 4) and CM (n � 4) and specific geneprimers for porcine chymase, MMP-9, fibronectin, vitronectin,TNF-�, IL-1�, IL-6, IL-18, ICAM-1, CCL-2, caspase-1, caspase-3,Smad-3, connective tissue growth factor, and cAMP-dependent tran-scription factor 3 (Supplemental Table 1). PCR-specific primers weredesigned with the PrimerQuest (Integrated DNA Technologies Inc.,Coralville, IA). Primer for 18S RNA was used to correct for mRNAloading. PCR conditions were 50°C (30 min), 94°C (10 min) (cDNAsynthesis), 94°C denaturation, 55°C annealing, and 72°C extending(1 min each) for a total of 32 cycles. PCR products were subjected toelectrophoresis on 1% agarose gel, visualized with ethidium bromide,and quantified as described above. Amplicon size was confirmed bycomparing with a DNA ladder (Fermentas, Glen Burnie, MD). Onlyamplicons corresponding to the expected size were analyzed.
In Vitro Fibroblast Proliferation. Neonatal ventricular fibro-blasts were isolated from 2-day-old Sprague-Dawley rats by enzy-matic digestion, separated from cardiomyocytes on a discontinuous
Percoll gradient, and plated in Dulbecco’s modified Eagle’s mediumcontaining 10% fetal bovine serum. Cells were divided into fourgroups and seeded until 80% confluence was reached. Cells werecultured with serum-free CS-C medium for 48 h and then 300 �l ofthe supernatant was injected onto glass slides with either vehicle(PBS), low-dose human purified chymase (10 ng/ml), high-dose chy-mase (30 ng/ml), or high-dose chymase plus chymase inhibitor (10�M). Slides were incubated at 37°C for 24 h, then fixed in 4%formalin in PBS, and stained for Ki-67 as described above. Cellproliferation was evaluated by percentage of Ki-67-positive nuclei tototal nuclei.
Statistical Methods. All results were expressed as mean �S.E.M.), and a P value of less than 0.05 was considered statisticallysignificant (Systat Software, Inc., San Jose, CA). Comparisons be-tween two groups were performed using unpaired Student’s t testand �2 test for categorical variables. Functional and microvasculardata were analyzed using two-way repeated-measures analysis ofvariance. Western blots were expressed as a ratio of protein toloading band density and analyzed after digitization and quantification.
ResultsArterial Blood Gas, Hematocrit, and Core Tempera-
ture. No significant differences were observed betweengroups in arterial pH, partial pressures of CO2 and O2, he-
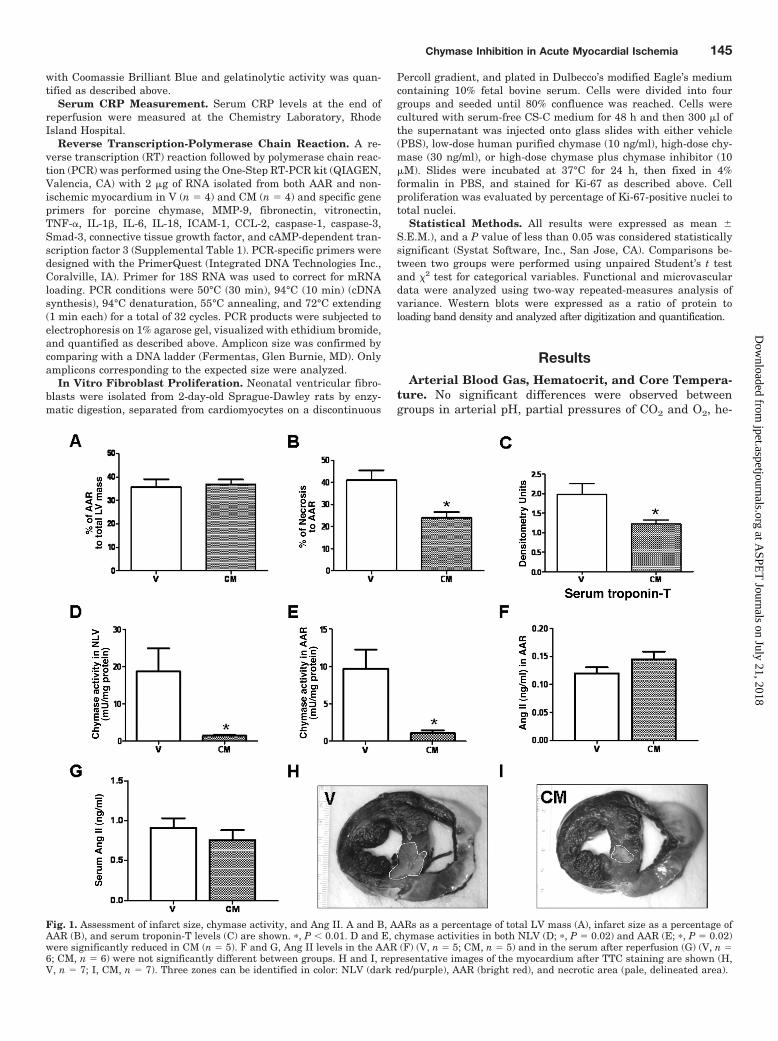
Fig. 1. Assessment of infarct size, chymase activity, and Ang II. A and B, AARs as a percentage of total LV mass (A), infarct size as a percentage ofAAR (B), and serum troponin-T levels (C) are shown. �, P � 0.01. D and E, chymase activities in both NLV (D; �, P � 0.02) and AAR (E; �, P � 0.02)were significantly reduced in CM (n � 5). F and G, Ang II levels in the AAR (F) (V, n � 5; CM, n � 5) and in the serum after reperfusion (G) (V, n �6; CM, n � 6) were not significantly different between groups. H and I, representative images of the myocardium after TTC staining are shown (H,V, n � 7; I, CM, n � 7). Three zones can be identified in color: NLV (dark red/purple), AAR (bright red), and necrotic area (pale, delineated area).
Chymase Inhibition in Acute Myocardial Ischemia 145
at ASPE
T Journals on July 21, 2018
jpet.aspetjournals.orgD
ownloaded from
matocrit, or core temperature at any time point in the pro-tocol.
Global and Regional Left Ventricular Function.Mean arterial blood pressure, heart rate, developed LV pres-sure, and global systolic LV function as determined from LVdP/dt and from the baseline to the end of reperfusion werenot significantly different between groups (Fig. 1, A–D).There were no significant differences in percentage of seg-mental shortening on either the horizontal or vertical axes(Supplemental Fig. S1).
Incidence of Ventricular Fibrillation/VentricularTachycardia. There was no difference in incidence of ven-tricular fibrillation/ventricular tachycardia during ischemia(V, 4/7 animals; CM, 6/7 animals; �2, P � 0.22), or duringreperfusion (V, 0/7 animals; CM, 1/7 animals; �2, P � 0.65).All dysrhythmias were successfully terminated with intrave-nous lidocaine and electrical cardioversion.
Myocardial Infarct Size and Serum Troponin TLevel. The size of the ischemic AAR, expressed as a percent-age of total LV mass, was not significantly different amonggroups (V, 36 � 3%; CM, 37 � 2%, P � 0.77), whereas the sizeof the infarcted area expressed as a percentage of the AARwas significantly smaller in CM than V (V, 41 � 5%; CM,24 � 5%; P � 0.01) (Fig. 1, A and B). Serum troponin T afterreperfusion was significantly lower in CM (P � 0.03) (Fig. 1C).
Chymase Activity. Chymase activity (mU/mg protein) inboth AAR (V, 9.7 � 2.6; CM, 1.1 � 0.3; P � 0.01) and NLV (V,18.7 � 6.1; CM, 1.3 � 0.2; P � 0.02) were significantly lowerin CM than V (Fig. 1, D and E).
Myocardial and Serum Ang II. Ang II levels in both theAAR (P � 0.65; Fig. 1F) and the serum after reperfusion (P �0.44; Fig. 1G) were not modified by chymase inhibition. Fig-
ure 1, H and I illustrates heart TTC staining in V- andCM-treated pigs. Notice the smaller area of necrosis com-pared with the AAR. The areas delineated represent thenecrotic left ventricle.
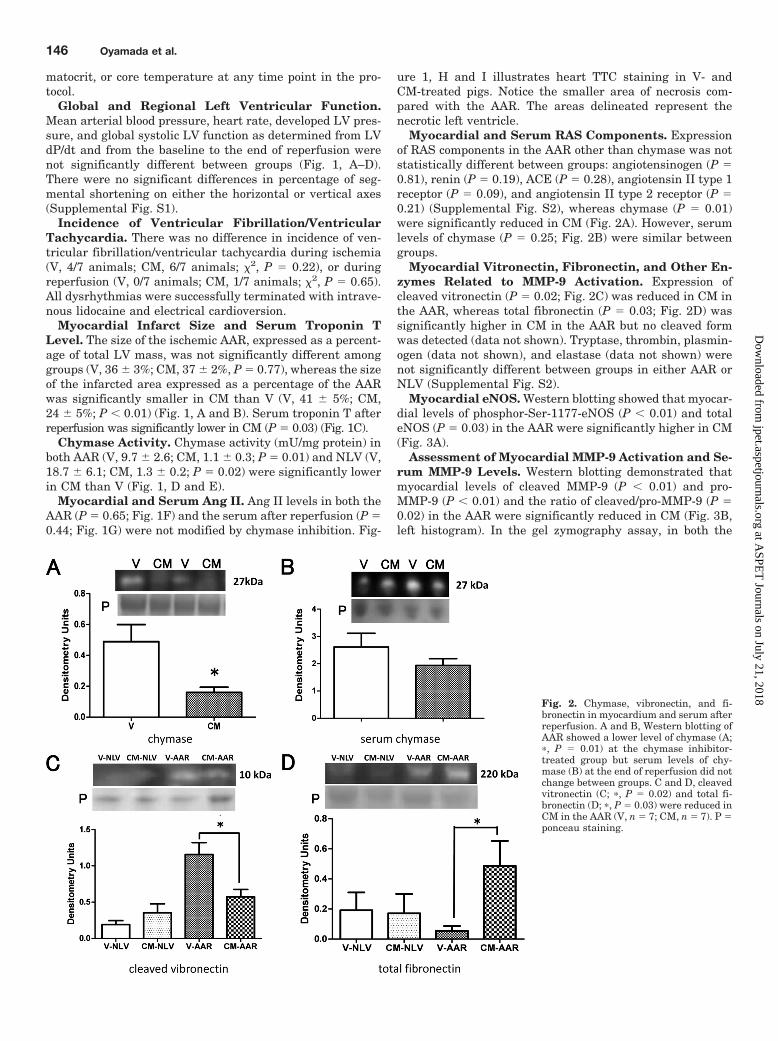
Myocardial and Serum RAS Components. Expressionof RAS components in the AAR other than chymase was notstatistically different between groups: angiotensinogen (P �0.81), renin (P � 0.19), ACE (P � 0.28), angiotensin II type 1receptor (P � 0.09), and angiotensin II type 2 receptor (P �0.21) (Supplemental Fig. S2), whereas chymase (P � 0.01)were significantly reduced in CM (Fig. 2A). However, serumlevels of chymase (P � 0.25; Fig. 2B) were similar betweengroups.
Myocardial Vitronectin, Fibronectin, and Other En-zymes Related to MMP-9 Activation. Expression ofcleaved vitronectin (P � 0.02; Fig. 2C) was reduced in CM inthe AAR, whereas total fibronectin (P � 0.03; Fig. 2D) wassignificantly higher in CM in the AAR but no cleaved formwas detected (data not shown). Tryptase, thrombin, plasmin-ogen (data not shown), and elastase (data not shown) werenot significantly different between groups in either AAR orNLV (Supplemental Fig. S2).
Myocardial eNOS. Western blotting showed that myocar-dial levels of phosphor-Ser-1177-eNOS (P � 0.01) and totaleNOS (P � 0.03) in the AAR were significantly higher in CM(Fig. 3A).
Assessment of Myocardial MMP-9 Activation and Se-rum MMP-9 Levels. Western blotting demonstrated thatmyocardial levels of cleaved MMP-9 (P � 0.01) and pro-MMP-9 (P � 0.01) and the ratio of cleaved/pro-MMP-9 (P �0.02) in the AAR were significantly reduced in CM (Fig. 3B,left histogram). In the gel zymography assay, in both the
Fig. 2. Chymase, vibronectin, and fi-bronectin in myocardium and serum afterreperfusion. A and B, Western blotting ofAAR showed a lower level of chymase (A;�, P � 0.01) at the chymase inhibitor-treated group but serum levels of chy-mase (B) at the end of reperfusion did notchange between groups. C and D, cleavedvitronectin (C; �, P � 0.02) and total fi-bronectin (D; �, P � 0.03) were reduced inCM in the AAR (V, n � 7; CM, n � 7). P �ponceau staining.
146 Oyamada et al.
at ASPE
T Journals on July 21, 2018
jpet.aspetjournals.orgD
ownloaded from
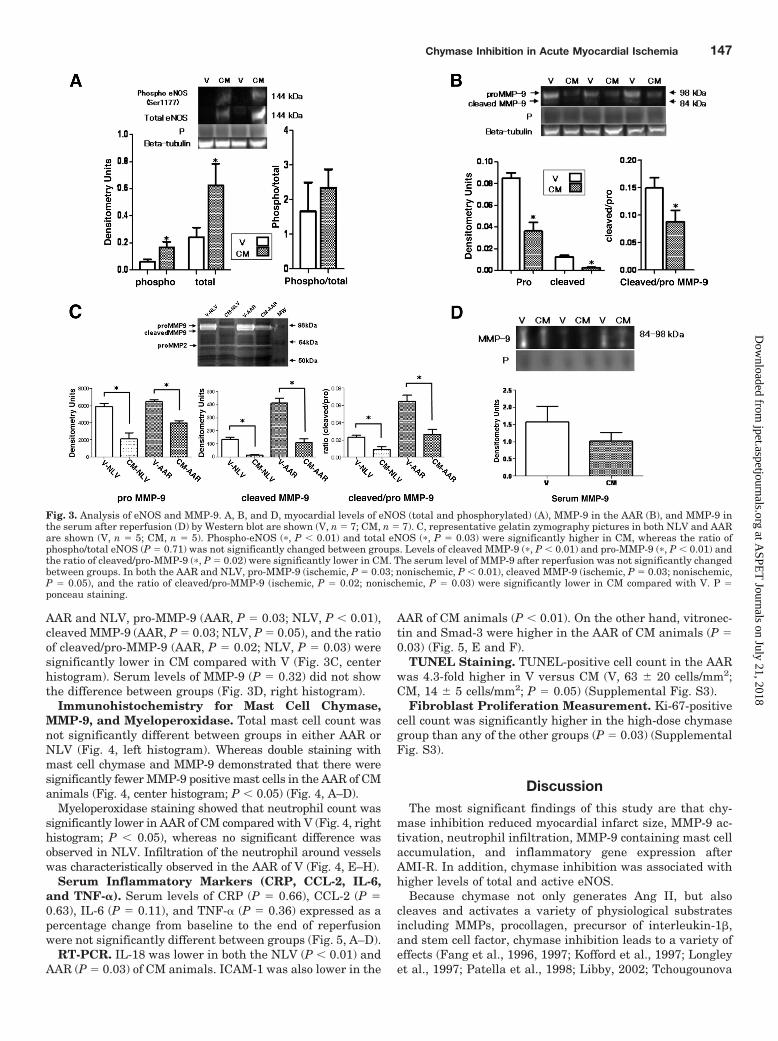
AAR and NLV, pro-MMP-9 (AAR, P � 0.03; NLV, P � 0.01),cleaved MMP-9 (AAR, P � 0.03; NLV, P � 0.05), and the ratioof cleaved/pro-MMP-9 (AAR, P � 0.02; NLV, P � 0.03) weresignificantly lower in CM compared with V (Fig. 3C, centerhistogram). Serum levels of MMP-9 (P � 0.32) did not showthe difference between groups (Fig. 3D, right histogram).
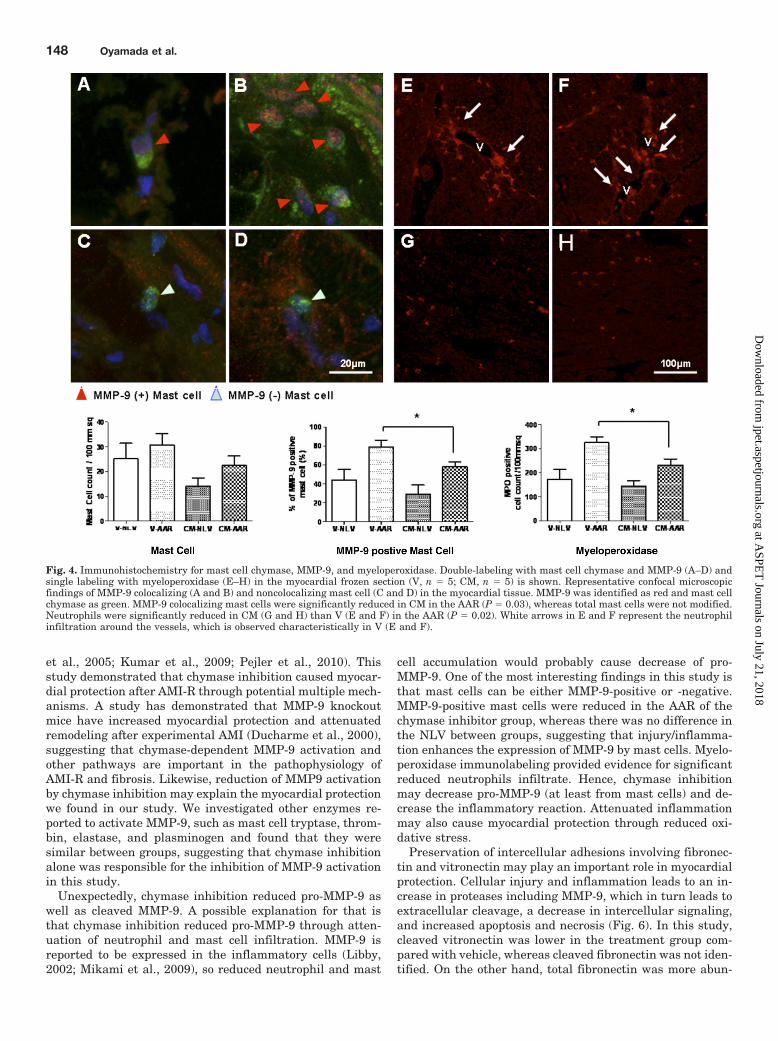
Immunohistochemistry for Mast Cell Chymase,MMP-9, and Myeloperoxidase. Total mast cell count wasnot significantly different between groups in either AAR orNLV (Fig. 4, left histogram). Whereas double staining withmast cell chymase and MMP-9 demonstrated that there weresignificantly fewer MMP-9 positive mast cells in the AAR of CManimals (Fig. 4, center histogram; P � 0.05) (Fig. 4, A–D).
Myeloperoxidase staining showed that neutrophil count wassignificantly lower in AAR of CM compared with V (Fig. 4, righthistogram; P � 0.05), whereas no significant difference wasobserved in NLV. Infiltration of the neutrophil around vesselswas characteristically observed in the AAR of V (Fig. 4, E–H).
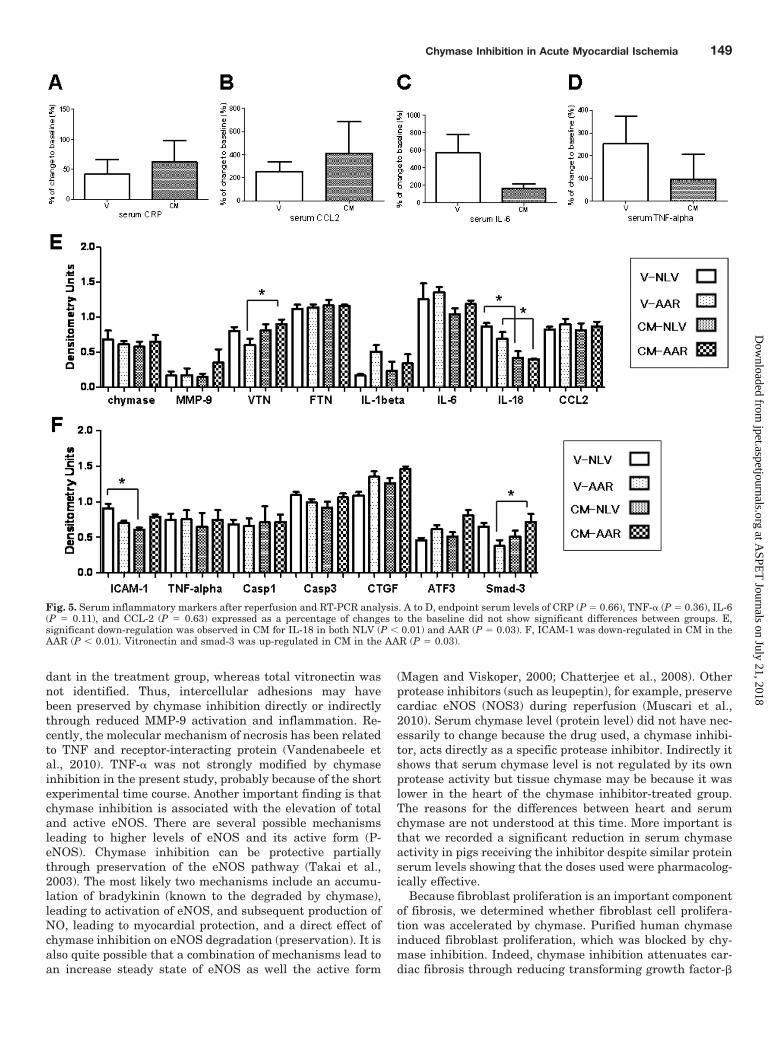
Serum Inflammatory Markers (CRP, CCL-2, IL-6,and TNF-�). Serum levels of CRP (P � 0.66), CCL-2 (P �0.63), IL-6 (P � 0.11), and TNF-� (P � 0.36) expressed as apercentage change from baseline to the end of reperfusionwere not significantly different between groups (Fig. 5, A–D).
RT-PCR. IL-18 was lower in both the NLV (P � 0.01) andAAR (P � 0.03) of CM animals. ICAM-1 was also lower in the
AAR of CM animals (P � 0.01). On the other hand, vitronec-tin and Smad-3 were higher in the AAR of CM animals (P �0.03) (Fig. 5, E and F).
TUNEL Staining. TUNEL-positive cell count in the AARwas 4.3-fold higher in V versus CM (V, 63 � 20 cells/mm2;CM, 14 � 5 cells/mm2; P � 0.05) (Supplemental Fig. S3).
Fibroblast Proliferation Measurement. Ki-67-positivecell count was significantly higher in the high-dose chymasegroup than any of the other groups (P � 0.03) (SupplementalFig. S3).
DiscussionThe most significant findings of this study are that chy-
mase inhibition reduced myocardial infarct size, MMP-9 ac-tivation, neutrophil infiltration, MMP-9 containing mast cellaccumulation, and inflammatory gene expression afterAMI-R. In addition, chymase inhibition was associated withhigher levels of total and active eNOS.
Because chymase not only generates Ang II, but alsocleaves and activates a variety of physiological substratesincluding MMPs, procollagen, precursor of interleukin-1�,and stem cell factor, chymase inhibition leads to a variety ofeffects (Fang et al., 1996, 1997; Kofford et al., 1997; Longleyet al., 1997; Patella et al., 1998; Libby, 2002; Tchougounova
Fig. 3. Analysis of eNOS and MMP-9. A, B, and D, myocardial levels of eNOS (total and phosphorylated) (A), MMP-9 in the AAR (B), and MMP-9 inthe serum after reperfusion (D) by Western blot are shown (V, n � 7; CM, n � 7). C, representative gelatin zymography pictures in both NLV and AARare shown (V, n � 5; CM, n � 5). Phospho-eNOS (�, P � 0.01) and total eNOS (�, P � 0.03) were significantly higher in CM, whereas the ratio ofphospho/total eNOS (P � 0.71) was not significantly changed between groups. Levels of cleaved MMP-9 (�, P � 0.01) and pro-MMP-9 (�, P � 0.01) andthe ratio of cleaved/pro-MMP-9 (�, P � 0.02) were significantly lower in CM. The serum level of MMP-9 after reperfusion was not significantly changedbetween groups. In both the AAR and NLV, pro-MMP-9 (ischemic, P � 0.03; nonischemic, P � 0.01), cleaved MMP-9 (ischemic, P � 0.03; nonischemic,P � 0.05), and the ratio of cleaved/pro-MMP-9 (ischemic, P � 0.02; nonischemic, P � 0.03) were significantly lower in CM compared with V. P �ponceau staining.
Chymase Inhibition in Acute Myocardial Ischemia 147
at ASPE
T Journals on July 21, 2018
jpet.aspetjournals.orgD
ownloaded from
et al., 2005; Kumar et al., 2009; Pejler et al., 2010). Thisstudy demonstrated that chymase inhibition caused myocar-dial protection after AMI-R through potential multiple mech-anisms. A study has demonstrated that MMP-9 knockoutmice have increased myocardial protection and attenuatedremodeling after experimental AMI (Ducharme et al., 2000),suggesting that chymase-dependent MMP-9 activation andother pathways are important in the pathophysiology ofAMI-R and fibrosis. Likewise, reduction of MMP9 activationby chymase inhibition may explain the myocardial protectionwe found in our study. We investigated other enzymes re-ported to activate MMP-9, such as mast cell tryptase, throm-bin, elastase, and plasminogen and found that they weresimilar between groups, suggesting that chymase inhibitionalone was responsible for the inhibition of MMP-9 activationin this study.
Unexpectedly, chymase inhibition reduced pro-MMP-9 aswell as cleaved MMP-9. A possible explanation for that isthat chymase inhibition reduced pro-MMP-9 through atten-uation of neutrophil and mast cell infiltration. MMP-9 isreported to be expressed in the inflammatory cells (Libby,2002; Mikami et al., 2009), so reduced neutrophil and mast
cell accumulation would probably cause decrease of pro-MMP-9. One of the most interesting findings in this study isthat mast cells can be either MMP-9-positive or -negative.MMP-9-positive mast cells were reduced in the AAR of thechymase inhibitor group, whereas there was no difference inthe NLV between groups, suggesting that injury/inflamma-tion enhances the expression of MMP-9 by mast cells. Myelo-peroxidase immunolabeling provided evidence for significantreduced neutrophils infiltrate. Hence, chymase inhibitionmay decrease pro-MMP-9 (at least from mast cells) and de-crease the inflammatory reaction. Attenuated inflammationmay also cause myocardial protection through reduced oxi-dative stress.
Preservation of intercellular adhesions involving fibronec-tin and vitronectin may play an important role in myocardialprotection. Cellular injury and inflammation leads to an in-crease in proteases including MMP-9, which in turn leads toextracellular cleavage, a decrease in intercellular signaling,and increased apoptosis and necrosis (Fig. 6). In this study,cleaved vitronectin was lower in the treatment group com-pared with vehicle, whereas cleaved fibronectin was not iden-tified. On the other hand, total fibronectin was more abun-
Fig. 4. Immunohistochemistry for mast cell chymase, MMP-9, and myeloperoxidase. Double-labeling with mast cell chymase and MMP-9 (A–D) andsingle labeling with myeloperoxidase (E–H) in the myocardial frozen section (V, n � 5; CM, n � 5) is shown. Representative confocal microscopicfindings of MMP-9 colocalizing (A and B) and noncolocalizing mast cell (C and D) in the myocardial tissue. MMP-9 was identified as red and mast cellchymase as green. MMP-9 colocalizing mast cells were significantly reduced in CM in the AAR (P � 0.03), whereas total mast cells were not modified.Neutrophils were significantly reduced in CM (G and H) than V (E and F) in the AAR (P � 0.02). White arrows in E and F represent the neutrophilinfiltration around the vessels, which is observed characteristically in V (E and F).
148 Oyamada et al.
at ASPE
T Journals on July 21, 2018
jpet.aspetjournals.orgD
ownloaded from
dant in the treatment group, whereas total vitronectin wasnot identified. Thus, intercellular adhesions may havebeen preserved by chymase inhibition directly or indirectlythrough reduced MMP-9 activation and inflammation. Re-cently, the molecular mechanism of necrosis has been relatedto TNF and receptor-interacting protein (Vandenabeele etal., 2010). TNF-� was not strongly modified by chymaseinhibition in the present study, probably because of the shortexperimental time course. Another important finding is thatchymase inhibition is associated with the elevation of totaland active eNOS. There are several possible mechanismsleading to higher levels of eNOS and its active form (P-eNOS). Chymase inhibition can be protective partiallythrough preservation of the eNOS pathway (Takai et al.,2003). The most likely two mechanisms include an accumu-lation of bradykinin (known to the degraded by chymase),leading to activation of eNOS, and subsequent production ofNO, leading to myocardial protection, and a direct effect ofchymase inhibition on eNOS degradation (preservation). It isalso quite possible that a combination of mechanisms lead toan increase steady state of eNOS as well the active form
(Magen and Viskoper, 2000; Chatterjee et al., 2008). Otherprotease inhibitors (such as leupeptin), for example, preservecardiac eNOS (NOS3) during reperfusion (Muscari et al.,2010). Serum chymase level (protein level) did not have nec-essarily to change because the drug used, a chymase inhibi-tor, acts directly as a specific protease inhibitor. Indirectly itshows that serum chymase level is not regulated by its ownprotease activity but tissue chymase may be because it waslower in the heart of the chymase inhibitor-treated group.The reasons for the differences between heart and serumchymase are not understood at this time. More important isthat we recorded a significant reduction in serum chymaseactivity in pigs receiving the inhibitor despite similar proteinserum levels showing that the doses used were pharmacolog-ically effective.
Because fibroblast proliferation is an important componentof fibrosis, we determined whether fibroblast cell prolifera-tion was accelerated by chymase. Purified human chymaseinduced fibroblast proliferation, which was blocked by chy-mase inhibition. Indeed, chymase inhibition attenuates car-diac fibrosis through reducing transforming growth factor-�
Fig. 5. Serum inflammatory markers after reperfusion and RT-PCR analysis. A to D, endpoint serum levels of CRP (P � 0.66), TNF-� (P � 0.36), IL-6(P � 0.11), and CCL-2 (P � 0.63) expressed as a percentage of changes to the baseline did not show significant differences between groups. E,significant down-regulation was observed in CM for IL-18 in both NLV (P � 0.01) and AAR (P � 0.03). F, ICAM-1 was down-regulated in CM in theAAR (P � 0.01). Vitronectin and smad-3 was up-regulated in CM in the AAR (P � 0.03).
Chymase Inhibition in Acute Myocardial Ischemia 149
at ASPE
T Journals on July 21, 2018
jpet.aspetjournals.orgD
ownloaded from

activation in an in vivo experiment (Murphy and Steenber-gen, 2008).
Gene expression of IL-18 and ICAM-1, well known proin-flammatory and profibrotic markers (Benson et al., 2007; Fixet al., 2011), were down-regulated in the chymase inhibitorgroup, indicating that chymase inhibition may also affect theinflammation-related gene expression immediately afterAMI-R. IL-18 is induced by ischemia and/or infarction, andhigher levels of IL-18 are associated with increased heartfailure (Woldbaek et al., 2003; Mallat et al., 2004; Wang etal., 2009). On the other hand, several serum inflammatorymarkers did not show remarkable changes that may be ex-plained, in part, by the tissue and blood harvesting at only asingle time point of 2 h after reperfusion.
Chymase inhibition did not result in any significant differ-ence in global and regional LV function or coronary micro-vascular reactivity. It is noteworthy that chymase inhibitoradministered intravenously does not seem to change circu-lating RAS components, including Ang II, which regulatesblood pressure, even though it did result in significantchanges to the myocardial infarct size, which probably affectthe LV function. There are two possible explanations for thisapparent discrepancy. First, our time course may be too shortto see LV functional improvement caused by decreased in-farct size. It is known that reperfusion causes stunned myo-cardium immediately after reperfusion and a longer period isneeded to regain myocardial function. Second, the infarct sizemight be small enough to not cause significant differences inLV function even though there was significant difference inthe infarct size between groups. Because the AAR was simi-lar, it may be that the percentage of ischemic, non-necroticmyocardium salvaged in the chymase inhibitor group wasstunned and hence not functional, explaining the lack of LVfunctional improvement 2 h after reperfusion.
There are several limitations to this study that must betaken into account. The ischemic myocardial tissue we as-sessed in this study included both necrotic and non-necroticmyocardium. In addition, our time course for tissue harvest(3 h after the onset of ischemia) does not account for long-term effects of the drug on myocardial function and infarctextension and, conversely, may have been long enough to
miss rapid changes in the activation/phosphorylation statusof other signaling pathways.
In conclusion, this study demonstrates that chymase inhi-bition plays a crucial role in myocardial protection involvingMMP-9, inflammatory markers, and the eNOS pathway andmay attenuate fibrosis induced by activated chymase afterAMI-R.
Acknowledgments
We thank the staff of the Animal Research Facility at the BethIsrael Deaconess Medical Center for assistance; Dr. Satoh Shoji ofToa Eiyo, Ltd. for chymase inhibitor; and Dr. Peng Zhang (Cardio-vascular Research Center, Rhode Island Hospital/Alpert MedicalSchool of Brown University) for isolated neonatal rat fibroblast.
Authorship Contributions
Participated in research design: Oyamada, Bianchi, Takai, andSellke.
Conducted experiments: Oyamada, Bianchi, and Chu.Contributed new reagents or analytic tools: Oyamada, Bianchi, and
Takai.Wrote or contributed to the writing of the manuscript: Oyamada,
Bianchi, Takai, Chu, and Sellke.
ReferencesBenson VL, McMahon AC, and Lowe HC (2007) sICAM-1 measurements are inde-
pendent of processing method and sampling site in patients with coronary arterydisease. J Thromb Thrombolysis 24:307–308.
Chatterjee A, Black SM, and Catravas JD (2008) Endothelial nitric oxide (NO) andits pathophysiologic regulation. Vascul Pharmacol 49:134–140.
Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, KellyRA, Werb Z, Libby P, et al. (2000) Targeted deletion of matrix metalloproteinase-9attenuates left ventricular enlargement and collagen accumulation after experi-mental myocardial infarction. J Clin Invest 106:55–62.
Fang KC, Raymond WW, Blount JL, and Caughey GH (1997) Dog mast cell �-chy-mase activates progelatinase B by cleaving the Phe88-Gln89 and Phe91-Glu92bonds of the catalytic domain. J Biol Chem 272:25628–25635.
Fang KC, Raymond WW, Lazarus SC, and Caughey GH (1996) Dog mastocytomacells secrete a 92-kD gelatinase activated extracellularly by mast cell chymase.J Clin Invest 97:1589–1596.
Fix C, Bingham K, and Carver W (2011) Effects of interleukin-18 on cardiac fibro-blast function and gene expression. Cytokine 53:19–28.
Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, DyspersinGD, Cleutjens JP, Shipley M, Angellilo A, et al. (1999) Inhibition of plasminogenactivators or matrix metalloproteinases prevents cardiac rupture but impairstherapeutic angiogenesis and causes cardiac failure. Nat Med 5:1135–1142.
Institute for Laboratory Animal Resources (2010) Guide for the Care and Use ofLaboratory Animals 8th ed. Institute for Laboratory Animal Resources, Divisionon Earth and Life Sciences, National Research Council, Washington DC.
Kelly D, Cockerill G, Ng LL, Thompson M, Khan S, Samani NJ, and Squire IB (2007)
Fig. 6. Diagram of the effect of chymaseinhibition on myocardial protection andfibrosis. Chymase inhibition may be pro-tective mainly through attenuating in-flammatory markers including MMP-9activation, resulting in less oxidativestress, more preservation of intercellularadhesins, thus contributing to preservedintercellular signal, less apoptosis andnecrosis, and partially through increas-ing eNOS pathway. Chymase inhibitionmay also contribute to less myocardialfibrosis by attenuating the effect of chy-mase on fibroblastic cell proliferation.
150 Oyamada et al.
at ASPE
T Journals on July 21, 2018
jpet.aspetjournals.orgD
ownloaded from

Plasma matrix metalloproteinase-9 and left ventricular remodelling after acutemyocardial infarction in man: a prospective cohort study. Eur Heart J 28:711–718.
Kofford MW, Schwartz LB, Schechter NM, Yager DR, Diegelmann RF, and GrahamMF (1997) Cleavage of type I procollagen by human mast cell chymase initiatescollagen fibril formation and generates a unique carboxyl-terminal propeptide.J Biol Chem 272:7127–7131.
Koide Y, Tatsui A, Hasegawa T, Murakami A, Satoh S, Yamada H, Kazayama S, andTakahashi A (2003) Identification of a stable chymase inhibitor using a pharma-cophore-based database search. Bioorg Med Chem Lett 13:25–29.
Kumar R, Singh VP, and Baker KM (2009) The intracellular renin-angiotensinsystem in the heart. Curr Hypertens Rep 11:104–110.
Libby P (2002) Inflammation in atherosclerosis. Nature 420:868–874.Longley BJ, Tyrrell L, Ma Y, Williams DA, Halaban R, Langley K, Lu HS, and
Schechter NM (1997) Chymase cleavage of stem cell factor yields a bioactive,soluble product. Proc Natl Acad Sci U S A 94:9017–9021.
Magen E and Viskoper RJ (2000) Interactions of angiotensin-converting enzyme,kinins and nitric oxide in circulation and the beneficial effects of ACE inhibitors incardiovascular diseases. Isr Med Assoc J 2:929–934.
Mallat Z, Heymes C, Corbaz A, Logeart D, Alouani S, Cohen-Solal A, Seidler T,Hasenfuss G, Chvatchko Y, Shah AM, et al. (2004) Evidence for altered interleukin18 (IL)-18 pathway in human heart failure. Faseb J 18:1752–1754.
Mikami Y, Dobschutz EV, Sommer O, Wellner U, Unno M, Hopt U, and Keck T(2009) Matrix metalloproteinase-9 derived from polymorphonuclear neutrophilsincreases gut barrier dysfunction and bacterial translocation in rat severe acutepancreatitis. Surgery 145:147–156.
Murphy E and Steenbergen C (2008) Mechanisms underlying acute protection fromcardiac ischemia-reperfusion injury. Physiol Rev 88:581–609.
Muscari C, Capanni C, Giordano E, Stefanelli C, Bonavita F, Stanic I, Bonafe F,Caldarera CM, and Guarnieri C (2010) Leupeptin preserves cardiac nitric oxidesynthase 3 during reperfusion following long-term cardioplegia. J Surg Res 164:e27–e35.
Osipov RM, Robich MP, Feng J, Clements RT, Liu Y, Glazer HP, Wagstaff J, BianchiC, and Sellke FW (2009) Effect of thrombin fragment (TP508) on myocardialischemia-reperfusion injury in hypercholesterolemic pigs. J Appl Physiol 106:1993–2001.
Oyamada S, Bianchi C, Takai S, Robich MP, Clements RT, Chu L, and Sellke FW(2010) Impact of acute myocardial ischemia reperfusion on the tissue and blood-borne renin-angiotensin system. Basic Res Cardiol 105:513–522.
Patella V, Marino I, Arbustini E, Lamparter-Schummert B, Verga L, Adt M, andMarone G (1998) Stem cell factor in mast cells and increased mast cell density inidiopathic and ischemic cardiomyopathy. Circulation 97:971–978.
Pejler G, Ronnberg E, Waern I, and Wernersson S (2010) Mast cell proteases:multifaceted regulators of inflammatory disease. Blood 115:4981–4990.
Rohde LE, Ducharme A, Arroyo LH, Aikawa M, Sukhova GH, Lopez-Anaya A,McClure KF, Mitchell PG, Libby P, and Lee RT (1999) Matrix metalloproteinaseinhibition attenuates early left ventricular enlargement after experimental myo-cardial infarction in mice. Circulation 99:3063–3070.
Takai S, Jin D, Sakaguchi M, Katayama S, Muramatsu M, Sakaguchi M, Mat-sumura E, Kim S, and Miyazaki M (2003) A novel chymase inhibitor, 4-[1-([bis-(4-methyl-phenyl)-methyl]-carbamoyl)3-(2-ethoxy-benzyl)-4-oxo-a zetidine-2-yloxy]-benzoic acid (BCEAB), suppressed cardiac fibrosis in cardiomyopathichamsters. J Pharmacol Exp Ther 305:17–23.
Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, and Pejler G(2005) A key role for mast cell chymase in the activation of pro-matrix metallo-protease-9 and pro-matrix metalloprotease-2. J Biol Chem 280:9291–9296.
Vandenabeele P, Declercq W, Van Herreweghe F, and Vanden Berghe T (2010) Therole of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal 3:re4.
Wang M, Tan J, Wang Y, Meldrum KK, Dinarello CA, and Meldrum DR (2009) IL-18binding protein-expressing mesenchymal stem cells improve myocardial protec-tion after ischemia or infarction. Proc Natl Acad Sci U S A 106:17499–17504.
Woldbaek PR, Tønnessen T, Henriksen UL, Florholmen G, Lunde PK, Lyberg T, andChristensen G (2003) Increased cardiac IL-18 mRNA, pro-IL-18 and plasma IL-18after myocardial infarction in the mouse; a potential role in cardiac dysfunction.Cardiovasc Res 59:122–131.
Address correspondence to: Dr. Frank W. Sellke, Department of Surgery/Division of Cardiothoracic Surgery, Rhode Island Hospital and Alpert MedicalSchool of Brown University, 2 Dudley Street, Suite 360, Providence, RI 02903.E-mail: [email protected]
Chymase Inhibition in Acute Myocardial Ischemia 151
at ASPE
T Journals on July 21, 2018
jpet.aspetjournals.orgD
ownloaded from
![Within the Brain: The Renin Angiotensin System€¦ · Renin angiotensin system (RAS) research has a long and rich history dating back to the discovery of renin in 1898 [1]. In the](https://img.pdfslide.us/doc/110x75/6060b8ac7a8e2361e061e02e/within-the-brain-the-renin-angiotensin-system-renin-angiotensin-system-ras-research.jpg)















![INDEX [jpet.aspetjournals.org]](https://img.pdfslide.us/doc/110x75/629818f027424e7e5e6aa348/index-jpet-.jpg)












