Embed Size (px)
Citation preview

Chronic Anthracycline Cardiotoxicity: Molecular and FunctionalAnalysis with Focus on Nuclear Factor Erythroid 2-RelatedFactor 2 and Mitochondrial Biogenesis Pathways□S
Eduard Jirkovsky, Olga Popelova, Pavla Krivakova-Stankova, Anna Vavrova, Milos Hroch,Pavlína Haskova, Eva Brcakova-Dolezelova, Stanislav Micuda, Michaela Adamcova,Tomas Simunek, Zuzana Cervinkova, Vladimír Gersl, and Martin SterbaDepartments of Pharmacology (E.J., O.P., M.H., E.B.-D., S.M., V.G., M.S.), Physiology (P.K.-S., M.A., Z.C.), and Medical Biochemistry(M.H.), Faculty of Medicine in Hradec Kralove, and Departments of Biochemical Sciences (A.V., P.H., T.S.) and Biological and MedicalSciences (E.B.-D.), Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Hradec Kralove, Czech Republic
Received July 11, 2012; accepted August 21, 2012
ABSTRACTAnthracycline anticancer drugs (e.g., doxorubicin or daunoru-bicin) can induce chronic cardiotoxicity and heart failure (HF),both of which are believed to be based on oxidative injury andmitochondrial damage. In this study, molecular and functionalchanges induced by chronic anthracycline treatment with pro-gression into HF in post-treatment follow-up were analyzedwith special emphasis on nuclear factor erythroid 2-relatedfactor 2 (Nrf2) and peroxisome proliferator-activated receptor-�coactivator-1� (PGC1�) pathways. Chronic cardiotoxicity wasinduced in rabbits with daunorubicin (3 mg/kg, weekly for 10weeks), and the animals were followed for another 10 weeks.Echocardiography revealed a significant drop in left ventricular(LV) systolic function during the treatment with marked pro-gression to LV dilation and congestive HF in the follow-up.Although daunorubicin-induced LV lipoperoxidation was found,it was only loosely associated with cardiac performance. Fur-
thermore, although LV oxidized glutathione content was in-creased, the oxidized-to-reduced glutathione ratio itself re-mained unchanged. Neither Nrf2, the master regulator ofantioxidant response, nor the majority of its target genesshowed up-regulation in the study. However, down-regulationof manganese superoxide dismutase and NAD(P)H dehydroge-nase [quinone] 1 were observed together with heme oxygenase1 up-regulation. Although marked perturbations in mitochon-drial functions were found, no induction of PGC1�-controlledmitochondrial biogenesis pathway was revealed. Instead, es-pecially in the post-treatment period, an impaired regulation ofthis pathway was observed along with down-regulation of theexpression of mitochondrial genes. These results imply thatglobal oxidative stress need not be a factor responsible for thedevelopment of anthracycline-induced HF, whereas suppres-sion of mitochondrial biogenesis might be involved.
IntroductionIntroduction of anthracycline (ANT) antibiotics to clinical
practice in the 1960s represented a significant milestone in
cancer treatment. High antitumor efficacy and a broad clin-ical spectrum made ANTs one of the most useful chemother-apeutics ever developed (Minotti et al., 2004). Unfortunately,the benefits from ANTs have been found to be notably coun-terbalanced by the risk of cardiac toxicity. Major clinicalconcerns are related to the chronic type of ANT cardiotoxicityassociated with dilated cardiomyopathy and heart failurewith either early- or late-onset forms (Lipshultz et al., 2008;Ewer and Suter, 2010). Unlike other complications of cancerchemotherapy, chronic ANT cardiotoxicity usually stays si-
This study was supported by the Czech Science Foundation [Grant 305/09/0416];Charles University [Grant SVV 264901/2012 and the program PRVOUK P37/5].
Article, publication date, and citation information can be found athttp://jpet.aspetjournals.org.
http://dx.doi.org/10.1124/jpet.112.198358.□S The online version of this article (available at http://jpet.aspetjournals.org)
contains supplemental material.
ABBREVIATIONS: ANT, anthracycline; ANOVA, analysis of variance; ARE, antioxidant response element; COX, cytochrome c oxidase; CTR,control; CuZnSOD, copper zinc superoxide dismutase; DAU, daunorubicin; DTT, dithiothreitol; FS, fractional shortening; FU, follow-up; GCLC,glutamate-cysteine ligase catalytic subunit; GPx, glutathione peroxidase; GR, glutathione reductase; GSH, reduced glutathione; GSSG, oxidizedglutathione; GST, glutathione transferase; HO1, heme oxygenase 1; HPLC, high-performance liquid chromatography; LMV, last measured value;LV, left ventricular; LVEDD, LV end-diastolic diameter; MDA, malondialdehyde; MnSOD, manganese superoxide dismutase; mtDNA, mitochondrialDNA; nDNA, nuclear DNA; NQO1, NAD(P)H dehydrogenase [quinone] 1; NRF1, nuclear respiratory factor 1; Nrf2, nuclear factor erythroid 2-relatedfactor 2; PCR, polymerase chain reaction; qPCR, quantitative PCR; PGC1�, peroxisome proliferator-activated receptor � coactivator 1�; ROS,reactive oxygen species; smtCK, sarcomeric mitochondrial creatine kinase; TFAM, mitochondrial transcription factor A.
1521-0103/12/3432-468–478$25.00THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 343, No. 2Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics 198358/3802912JPET 343:468–478, 2012
468
http://jpet.aspetjournals.org/content/suppl/2012/08/22/jpet.112.198358.DC1Supplemental material to this article can be found at:
at ASPE
T Journals on Septem
ber 2, 2018jpet.aspetjournals.org
Dow
nloaded from

lent during cancer treatment, whereas it can strike backweeks, months, or even years later to significantly affect themorbidity and prognosis of cancer survivors (Ewer and Suter,2010). Chronic cardiotoxicity is known to be a class effecttypical of all ANT derivatives introduced to clinical practiceso far (Minotti et al., 2004). Although many modifiable andnonmodifiable risk factors have been described, the mostimportant one is the lifetime cumulative dose of the drug(Lipshultz et al., 2008; Ewer and Suter, 2010).
Despite many experimental and clinical studies performedthroughout the last 40 years, the unequivocal explanation ofmolecular basis for ANT cardiotoxicity development remainselusive and is still a matter of debate and controversy (Gi-anni et al., 2008; Simunek et al., 2009; Menna et al., 2012).The prevailing mechanistic concept highlights ANT-inducedand iron-catalyzed formation of reactive oxygen species(ROS), resulting in direct oxidative damage to the myocar-dium (Keizer et al., 1990; Simunek et al., 2009). In addition,anthracyclines have been shown to interfere with the normalregulation of cellular iron homeostasis in cardiomyocytes(Kwok and Richardson, 2003, 2004). Other hypotheses pointto ANT-induced impairment in mitochondrial bioenergetics(Wallace, 2003; Tokarska-Schlattner et al., 2006), damage tomitochondrial DNA (Berthiaume and Wallace, 2007; Leb-recht and Walker, 2007) with subsequent perturbations inexpression of mitochondria-encoded genes (Lebrecht et al.,2003), disruption in mitochondrial and cellular Ca2� homeo-stasis (Wallace, 2007; Lebrecht et al., 2010), and alterationsin expression and stability of cardiac myofilaments (Lim etal., 2004).
Indeed, in the clinical setting it is very difficult to directlyassess ANT-induced molecular changes in the myocardiumand their further development in the post-treatment period.Furthermore, it is also challenging to appropriately mimicthe nature of chronic ANT cardiotoxicity in vitro or by usingsingle high doses in vivo models (Gianni et al., 2008). Thiscan partially explain our poor insight into molecular eventsdetermining the development of chronic ANT cardiotoxicityand its transition into heart failure in the post-treatmentfollow-up (FU). In this regard, it might be particularly im-portant to understand what happens with ANT-induced myo-cardial oxidative stress, mitochondrial damage, perturba-tions in bioenergetics, and cell death. Furthermore, becauseoxidative stress is the most frequently cited mechanism ofchronic ANT cardiotoxicity development, it is rather surpris-ing that there is no information on the response of a keyantioxidant and cytoprotective pathway regulated by nuclearfactor erythroid 2-related factor 2 (Nrf2). Cellular oxidativestress is known to stabilize Nrf2 and induce its translocationin to the nucleus where it markedly up-regulates expressionof a battery of antioxidant response element (ARE)-associ-ated target genes (Baird and Dinkova-Kostova, 2011). Inaddition to its own expression, Nrf2 up-regulates the expres-sion of superoxide dismutase [both copper zinc superoxidedismutase (CuZnSOD) and manganese superoxide dismutase(MnSOD)], inducible heme oxygenase 1 (HO1), NAD(P)Hdehydrogenase [quinone] 1 (NQO1), numerous enzymes in-volved in the glutathione system, and many others (Bairdand Dinkova-Kostova, 2011).
Furthermore, mitochondria have been repeatedly suggestedas the main target for chronic ANT cardiotoxicity (Wallace,2003; Tokarska-Schlattner et al., 2006). However, involvement
of the mitochondrial biogenesis pathway remains uncertain inthe chronic ANT cardiotoxicity setting. The master regulationof mitochondrial biogenesis is performed by peroxisome prolif-erator-activated receptor-� coactivator-1� (PGC1�), which isinduced by numerous conditions including energy starvationand mitochondrial oxidative stress (Ventura-Clapier et al.,2008; Rimbaud et al., 2009). Once activated, PGC1� acts as akey coactivator up-regulating the expression of numerous im-portant mitochondrial proteins. With respect to the respiratorychain subunits, there is a requirement for coordination of ex-pression of components encoded by both nuclear and mitochon-drial genome, and this is mediated by downstream pathwaymembers, nuclear respiratory factors (NRFs) and mitochon-drial transcription factor A (TFAM), respectively (Rimbaud etal., 2009). Despite the potential importance of this pathway, itsrole in the development of chronic anthracycline cardiotoxicityand transition into heart failure remains to be determined.
Therefore, in this study we analyzed left ventricular (LV)molecular and functional changes induced by repeated ad-ministration of ANT and their further progress in the post-treatment follow-up with a focus on the involvement of oxi-dative stress and mitochondrial damage. Moreover, weinvestigated the response of two logically related endogenousprotective machineries, the Nrf-2 regulated antioxidant re-sponse pathway and the mitochondrial biogenesis pathway,to assess their role in chronic ANT cardiotoxicity.
Materials and MethodsAll chemicals were purchased from Sigma-Aldrich (St. Louis, MO)
unless otherwise stated.Animals and Experimental Design. The study was performed
by using the previously well established animal model of chronicANT cardiotoxicity (Simunek et al., 2004; Popelová et al., 2009;Sterba et al., 2011). Adult male Chinchilla rabbits (�4 months old;�3.5 kg; n � 46; Velaz, Kolec u Kladna, Czech Republic) were housedunder a 12-h light cycle with constant temperature and humidityand free access to tap water and a standard laboratory pellet diet. Allprocedures were performed according to the Guide for the Care andUse of Laboratory Animals (Institute of Laboratory Animal Re-sources, 1996) as approved and supervised by the Animal Experi-mentation Committee of the Faculty of Medicine, Charles Universityin Prague, Hradec Kralove. Cardiotoxicity was induced in rabbitswith daunorubicin (DAU; 3 mg/kg i.v., once weekly for 10 weeks; n �27; Daunoblastina, Pfizer, Rome, Italy), whereas control (CTR) ani-mals received saline (1 ml/kg i.v.; n � 19). A week after the last drugadministration, the animals were randomized into two groups. Thefirst group was sacrificed, whereas the second was followed for thenext 10 weeks and designated as the FU group (DAU FU, n � 11;CTR FU, n � 10). Mortality was determined in the treatment period.During the FU animals were sacrificed whenever weekly echocardi-ography examination showed LV fractional shortening (FS) to belower than 20% (indicating decompensated heart failure) to avoidloss of myocardial samples because of sudden deaths.
All experimental procedures were performed under light anesthe-sia consisting of ketamine (30 mg/kg i.m.; Narketan, Vetoquinol AG,Ittigen, Switzerland) and midazolam (2.5 mg/kg i.m.; MidazolamTorrex, Torrex Chiesi Pharma, Vienna, Austria), and freshly pre-pared pentobarbital solution [4% (w/w), i.v.] was used for animaloverdose.
During autopsy hearts were rapidly excised and briefly retrogradelyperfused with ice-cold saline. A piece of the LV free wall was removedfor analysis of myocardial bioenergetics, and the rest was shock-frozen,homogenized under liquid nitrogen, and stored at �80°C.
Chronic Anthracycline Cardiotoxicity, Nrf2, and PGC1� 469
at ASPE
T Journals on Septem
ber 2, 2018jpet.aspetjournals.org
Dow
nloaded from

Examination of Cardiac Functions. Echocardiographic mea-surements were performed by using Vivid 4 equipped with a 10-MHzprobe (GE Medical Systems Ultrasound; GE Healthcare, Chalfont St.Giles, Buckinghamshire, UK) during the treatment period andweekly in the FU period. Guided M-mode measurements were per-formed at the tips of the mitral valve to obtain the LV end-systolicand end-diastolic diameters (LVESD and LVEDD, respectively, inthe equation). LV FS as an index of the systolic function was deter-mined as follows:
LVFS(%) �LVEDD � LVESD
LVEDD � 100.
Plasma Troponin T. Cardiac troponin T was determined inplasma samples by using Elecsys Troponin T high sensitivity assay(Roche Diagnostics, Basel, Switzerland) with a limit of detection of0.003 �g/l. Blood samples were collected before the first, fifth, sev-enth, eighth, and 10th drug administration, a week after the lastadministration of DAU and weekly in the FU period. Area under thecurve of plasma troponin T was determined by using Prism 5.0(GraphPad Software, Inc., San Diego, CA).
Oxidative Damage to the Myocardium. Markers of oxidativedamage were analyzed in LV samples homogenized in ice-cold buffer[25 mM Tris and 0.1% (v/v) Triton X-100, pH 7.6] on ice, and super-natants were collected and stored in �80°C. For measurement of LVglutathione content, the low-spin supernatant was treated with 10%(w/w) metaphosphoric acid on ice and further centrifuged at 15,000gto yield final supernatants.
LV malondialdehyde (MDA) content was measured as a marker oflipoperoxidation in supernatants prepared as described above usinga HPLC method according to Pilz et al. (2000) with minor modifica-tions (Popelova et al., 2009). This TBARS-independent technique isbased on the derivation of MDA with 2,4-dinitrophenylhydrazineyielding a fluorescent compound, which is then selectively detectedby using a HPLC system.
For the measurement of individual oxidized and reduced glutathioneforms (GSSG and GSH, respectively) in the LV myocardium a selectiveHPLC method was used (Kand’ar et al., 2007). The method is based onthe derivation of GSH with o-phthalaldehyde yielding a HPLC-detectable fluorescent derivative, whereas for selective GSSG measure-ment the samples were incubated with N-ethylmaleimide to removeGSH from the sample before the o-phthalaldehyde derivation.
Activity of Glutathione System Enzymes. Activity of glutathi-one system enzymes were analyzed in the LV samples homogenizedin ice-cold buffer [5 mM HEPES, 1 mM EGTA, 1 mM DTT, and 0.1%(w/v) Triton X-100, pH 8.7]. Enzyme-coupled spectrophotometric as-says for detection of activity of glutathione peroxidase (GPx) andglutathione reductase (GR) were performed as described previously(Vavrova et al., 2011). Glutathione transferase (GST) activity wasdetermined by using the SensoLyte GST Activity Assay FluorimetricKit (Anaspec, Inc., San Jose, CA) according to the manufacturer’sinstructions.
Mitochondrial Function Measurement. Fresh LV myocardiumwas homogenized in ice-cold buffer (25 mM sucrose, 75 mM sorbitol, 100mM KCl, 10 mM H3PO4, 5 mM MgCl2, 10 mM Tris, 20 mM EDTA, and1 mg/ml bovine serum albumin) by using a glass/Teflon Potter-Elve-hjem grinder (P-LAB, Prague, Czech Republic), and the homogenatewas passed through a nylon filter. Mitochondrial respiration was mea-sured by using a high-resolution Oxygraph 2K (Oroboros, Innsbruck,Austria) in a MiR05 respiration buffer (Oroboros) at 30°C. Oxygenconsumption was induced by glutamate (10 mM), malate (2.5 mM), andADP (1 mM). The status of the mitochondrial outer membrane wasevaluated by the addition of cytochrome c (10 �M), and an increase ofoxygen consumption by approximately 69% suggested partial damageto the membrane, which occurred during the preparation of the tissuehomogenate. No significant differences were found between the treat-ment groups. Rotenone (1 �M) was used for the inhibition of the com-plex I, whereas succinate (10 mM) was used for the activation of com-
plex II. The activity of cytochrome c oxidase (COX) was evaluatedseparately by using cytochrome c (10 �M) as a substrate, N,N,N�,N�-tetramethyl-p-phenylenediamine (0.5 mM) as an artificial electron do-nor, and ascorbate (2 mM) as a regenerating system for reducedN,N,N�,N�-tetramethyl-p-phenylenediamine. Oroboros software (Dat-Lab 4.1) was used for evaluating oxygen consumption. The data werecorrected for by the rate of chemicals autoxidation and are expressed asrelative fold over the control group.
Additional rotenone-sensitive complex I activity assay was per-formed according to Long et al. (2009) with minor modification in themitochondria-enriched fraction as described previously (Sterba etal., 2011). Citrate synthase activity in the LV samples was analyzedin the LV homogenates prepared as described under Activity ofGlutathione System Enzymes by using an enzyme-coupled spectro-photometric method as described previously (De Sousa et al., 1999).
Active Form of Nrf2 in the Nuclear Fraction. The amount oftranscriptionally active form of Nrf2 in the LV nuclear extract wasanalyzed by using a commercial kit (TransAM Nrf2; Active Motif,Inc., Carlsbad, CA) according to the manufacturer’s recommenda-tions. The assay contains 96-well plates coated with immobilizedoligonucleotides containing the ARE consensus binding site, whichspecifically binds with the active form of Nrf2 contained in thenuclear extract. Ten micrograms of nuclear extract were loaded intoeach well, and samples were incubated for 1 h. Nrf2 bound to theimbedded ARE oligonucleotides on the 96-well plates was detectedcolorimetrically at 450 nm by incubation with a primary antibodyagainst Nrf2 and secondary antibody conjugated to horseradish per-oxidase. Nuclear extracts from COS-7 cells transfected with Nrf2were used as a positive control for Nrf2 binding activity. The foldincrease in Nrf2 binding activity was determined by comparison ofthese results with the levels determined in the samples from controlanimals. Nuclear extract were isolated from LV myocardium byusing Dignam’s protocol (Dignam et al., 1983). In brief, approxi-mately 50 mg of the tissue was homogenized in ice-cold buffer A [10mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.005%nonidet P-40 (v/v), and protease inhibitor cocktail, pH 7.9]. After cen-trifugation (3000g, 10 min), the pellet was collected and dissolved in amixture of ice-cold buffer B [5 mM HEPES, 1.5 mM MgCl2, 0.2 mMEDTA, 0.5 mM DTT, and 26% (v/v) glycerol, pH 7.9] and 4.6 M NaCl.
Quantitative Real-Time PCR. Total RNA from LV myocardiumwas isolated by using TRIzol reagent. For analysis of the expressionof ND1, ND4, NDUFS2, COX1, and COX4I1, the isolated total RNAwas further treated with rDNase and cleaned with a NucleospinRNA XS kit (Macherey-Nagel, Duren, Germany) according to themanufacturer’s protocol. Isolated RNA was converted into cDNA viaa High Capacity cDNA Reverse Transcription Kit (Applied Biosys-tems, Foster City, CA). Commercially available qPCR assays basedon the combination of two primers and sequence-specific hydrolysisprobe (Supplemental Table 1) were obtained from Generi Biotech(Hradec Kralove, Czech Republic). The amplification was performedin triplicate with TaqMan Fast Universal PCR Master Mix (AppliedBiosystems) by using a 7500HT Fast Real-Time PCR System (Ap-plied Biosystems). The assay was performed in fast mode of 50 cycleswith the following time-temperature profile: 95°C for 3 min, 95°C for5 s, and 60°C for 25 s. All results were normalized by geometric meanof hypoxanthine phosphoribosyltransferase 1 expression.
For analysis of the change of mitochondrial DNA (mtDNA)/nu-clear DNA (nDNA), total DNA was extracted from the LV myocar-dium by using a DNeasy Blood and Tissue Kit (QIAGEN, Valencia,CA). Commercial qPCR assays (Generi Biotech) for three mtDNA-encoded genes (ND1, ND4, and COX1) were used to analyze ex-tracted DNA to assess the relative abundance of mtDNA. Averagedresults were normalized over the results of qPCR analysis of nDNA-encoded leptin. The assay was performed in fast mode of 50 cycleswith following time-temperature profile: 95°C for 3 min, 95°C for10 s, and 60°C for 30 s.
Determination of Protein Concentration. Protein concentrationwas determined by using a BCA Protein Assay Kit (Sigma-Aldrich).
470 Jirkovsky et al.
at ASPE
T Journals on Septem
ber 2, 2018jpet.aspetjournals.org
Dow
nloaded from

Statistical Analyses. The results are shown as individual valuesalong with their means/medians according to data character unlessotherwise stated. Statistical significance was determined by usingone-way ANOVA/ANOVA on Ranks or paired t test/Wilcoxon SignedRank Test according to the data character by using Sigmastat 3.5(SPSS, Chicago, IL). Correlation analyses were performed by usingPearson’s or Spearman’s methods.
ResultsDAU-Induced Cardiotoxicity and Its Progression
into Dilated Cardiomyopathy and Heart Failure in theFU. The repeated administration of DAU induced a moder-ate mortality in the treatment period (5 of 27 animals), and 9of 11 DAU-treated rabbits were gradually sacrificed accord-
ing to the protocol in the post-treatment FU because of theprogression of cardiac dysfunction (Fig. 1A). No prematuredeaths occurred in the control groups. There was no signifi-cant body weight gain in the DAU group, neither at the endof the treatment nor in the FU, which contrasted with sig-nificant body weight gain in both intervals of the controlgroup (Fig. 1B). A heart weight-to-body weight ratio wassignificantly higher in the DAU-treated animals comparedwith the control group, both at the end of the treatment(2.72 0.21 versus 2.06 0.06 g/kg, respectively) and in theFU (2.69 0.08 versus 1.80 0.07 g/kg; p � 0.001).
Echocardiographic examination revealed a significant de-crease of the LV FS at the end of the DAU treatment, whichwas followed by marked and significant progression in the
Fig. 1. General toxicity, cardiac func-tion, and troponin T as a marker ofcardiac damage. A, animal survival:mortality was determined in thetreatment period, in the FU the ani-mals were sacrificed when left ven-tricular fractional shortening fell be-low 20% (data shown as survival withFS 20%). B, body weight change. C,left ventricular systolic function as-sessed by echocardiography. D, leftventricular end-diastolic dimensionassessed by echocardiography. E,plasma concentrations of cardiac tro-ponin T. All data are shown as eithermeans S.E.M. or medians with boxand whiskers representing 5 to 95percentiles. # and $ indicate statisti-cal significance in comparison withthe initial and end-of-treatment val-ues, respectively (paired t test; p �0.01). c indicates statistical signifi-cance in comparison with the controlgroup (one-way ANOVA/ANOVA onRanks; p � 0.01). Statistical signifi-cance of the troponin T data is shownas: ��, p � 0.01; ���, p � 0.001 (one-way ANOVA on Ranks). FU, 10-weekpost-treatment follow-up; LMV, lastmeasured value.
Chronic Anthracycline Cardiotoxicity, Nrf2, and PGC1� 471
at ASPE
T Journals on Septem
ber 2, 2018jpet.aspetjournals.org
Dow
nloaded from

FU (to �48% of the initial value; p � 0.001; Fig. 1C). Thiscorresponded with a higher incidence of pleural effusions(1.0 ml) in the FU than at the end of the DAU treatment (55versus 13%, respectively). The LVEDD was not changed atthe end of the DAU treatment, whereas it rose significantlyin the FU compared with the control group in initial as wellas end-of-treatment values (Fig. 1D). These data describemoderate LV dysfunction with no change in the LV dimen-sion by the end of the treatment, whereas there was amarked progression into dilated cardiomyopathy and conges-tive heart failure in the FU period.
Plasma concentrations of cardiac troponin T were signifi-cantly elevated in the DAU group by the fifth week, and theycontinued to rise further until the end of the treatment in the11th week (Fig. 1E). It is noteworthy that the concentrationsremained similarly elevated in the FU for at least another 3weeks with slow decreases thereafter. In addition, the areaunder the curve of troponin T plasma concentrations wasfound to be markedly higher in the DAU group than in thecontrol group during treatment (0.64 0.14 versus 0.09 0.02 �g/l; p � 0.001, respectively) and increased further inthe FU (1.12 0.02 versus 0.16 0.02 �g/l; p � 0.001,
respectively). Because cardiac troponin T has a short plasmahalf-life, these data indicate that cardiomyocyte degenera-tion and cell death take place during the time course of ANTtreatment, but also in the post-treatment period.
DAU-Induced Myocardial Oxidative Stress and Re-sponse of Nrf2 Pathway. Analyses of oxidative stress mark-ers in the LV myocardium revealed significantly increasedMDA content at the end of the DAU treatment (Fig. 2A) andfurther significant elevation in the post-treatment period.Although a significant association (p � 0.05) between indi-vidual values of MDA and LV FS was identified (Fig. 2B), therelationship was relatively weak (R � �0.417). Furthermore,there was a significant increase in the content of oxidizedglutathione caused by the treatment persisting in the FUperiod (Fig. 3A). Nevertheless, the GSSG/GSH ratio was notaltered (Fig. 3C), because the reduced glutathione contentalso increased in the DAU-treated animals (Fig. 3B). Corre-lation analysis found no association between the GSSG/GSHratio and cardiac function (Fig. 3D). Activities of Nrf2-regulated enzymes involved in glutathione system mainte-nance, such as glutathione transferase, glutathione reductase,and glutathione peroxidase, showed no changes caused by theDAU treatment (Fig. 3, E–G) with the exception of a slightincrease of the latter enzyme in the FU period.
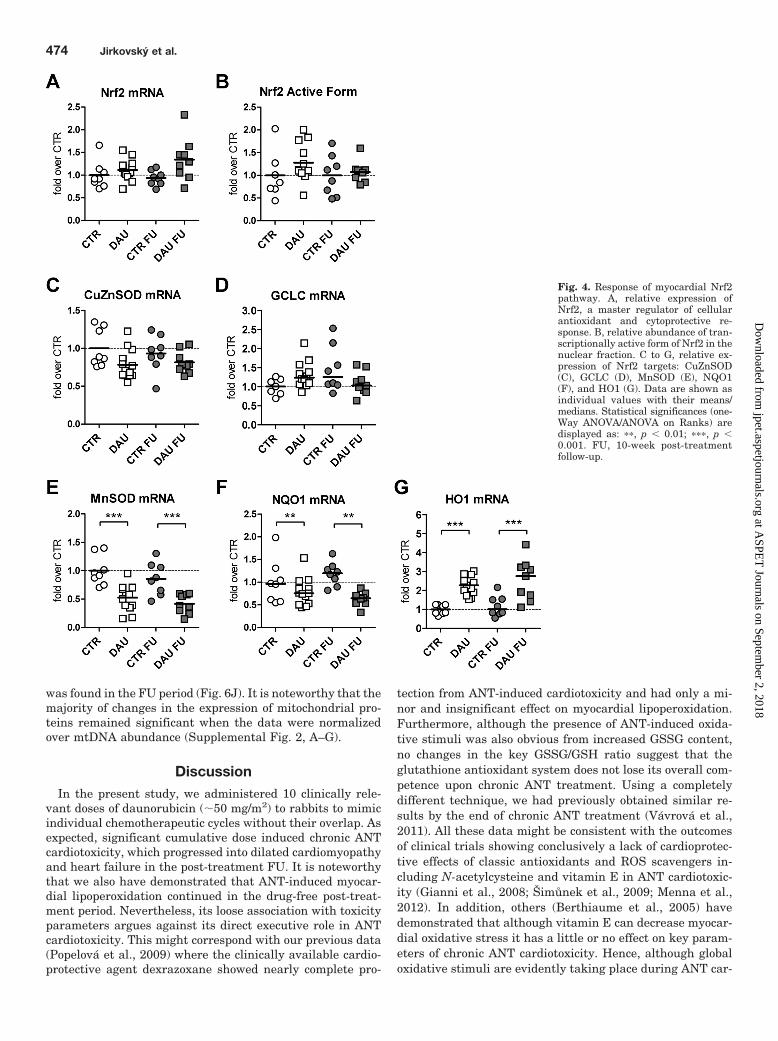
We were surprised that no significant changes caused bythe DAU treatment were found in the expression of the masterregulator of the oxidative stress response, Nrf2 (Fig. 4A).The relative gene expression of Nrf2 was without significantchanges both during the treatment and in subsequent FU. Inaddition, determination of the transcriptionally active formof Nrf2 in the myocardial nuclear fraction revealed no signif-icant changes caused by the treatment in either interval (Fig.4B). Gene expression of key Nrf2 target genes showed nocoordinated induction caused by DAU treatment. AlthoughCuZnSOD and glutamate-cysteine ligase catalytic subunit(GCLC) gene expression showed no significant change causedby DAU treatment (Fig. 4, C and D), MnSOD and NQO1 weremarkedly and significantly down-regulated at the end of theDAU treatment, and this change did not progress in the FUperiod (Fig. 4, E and F). In contrast, HO1 (Fig. 4G) wasmarkedly up-regulated (up to 2.3- and 2.6-fold, respectively)in the same periods. Correlation analyses of Nrf2 pathwaycomponents with the LV systolic function (LV FS) showedonly insignificant or weak association in individual animals,with the exception of MnSOD, HO1, and NQO1 (Supplemen-tal Fig. 1, A-C). The relationship was particularly strong forthe former two genes (r � 0.75 and R � �0.77, respectively),albeit they showed opposite regulation caused by the treat-ment.
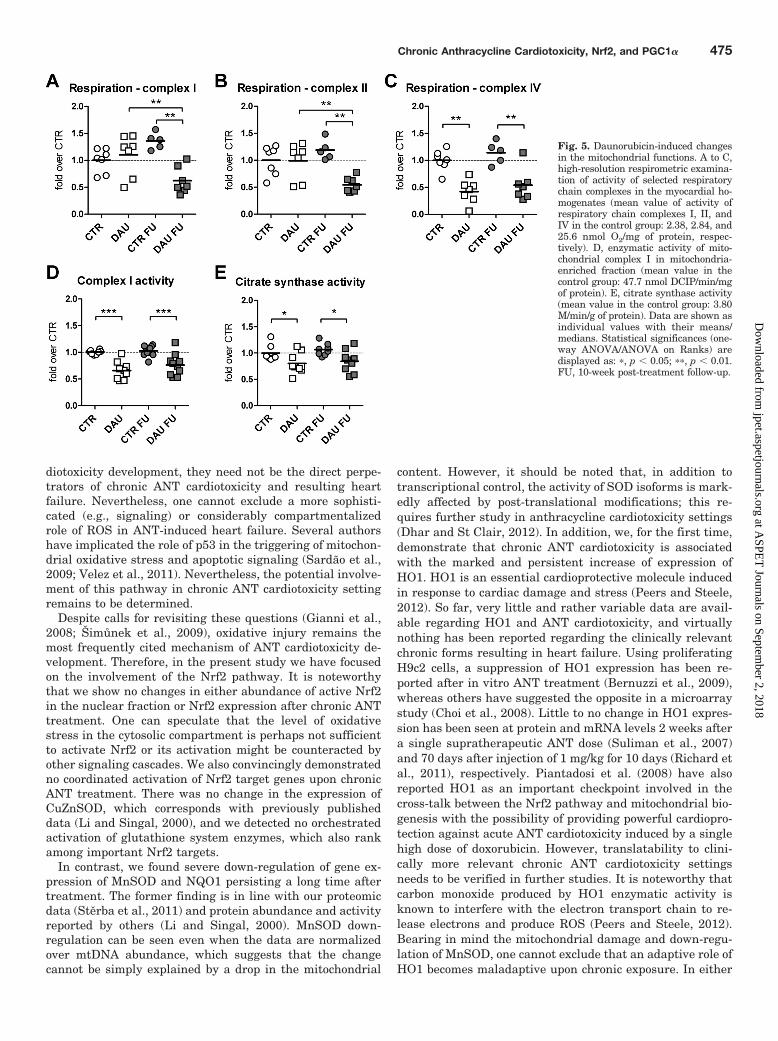
DAU-Induced Mitochondrial Damage and Responseof Mitochondrial Biogenesis Pathway. High-resolutionrespirometric measurements revealed significantly de-creased activity of complex I and II in the post-treatment FU,whereas the values at the end of the treatment showed nochange in either case (Fig. 5, A and B). Furthermore, complexIV function was significantly decreased by DAU treatment inboth periods (Fig. 5C). Rotenone-sensitive enzymatic func-tion of complex I determined in mitochondria-enriched frac-tions of LV myocardium was significantly decreased both bythe end of the DAU treatment and in the FU (by 34 and 26%,respectively, Fig. 5D). Moreover, citrate synthase activity(general marker of mitochondria content) was decreased sig-
Fig. 2. Daunorubicin-induced myocardial lipoperoxidation. A, HPLC de-termination of total MDA (a marker of lipoperoxidation) in the left ven-tricular myocardium (TBARS-independent method). Data are shown asindividual values along with their medians. Statistical analysis wasperformed by using ANOVA on Ranks. Significance is shown as: ���, p �0.001. FU, 10-week post-treatment follow-up. B, correlation of left ven-tricular performance with lipoperoxidation (malondialdehyde content) inindividual animals as determined by Spearman correlation and regres-sion analysis. Correlation coefficient and statistical significance areshown.
472 Jirkovsky et al.
at ASPE
T Journals on Septem
ber 2, 2018jpet.aspetjournals.org
Dow
nloaded from

nificantly at the end of the treatment by 19.7 and 20.9% inthe FU period (Fig. 5E) compared with the correspondingcontrols.
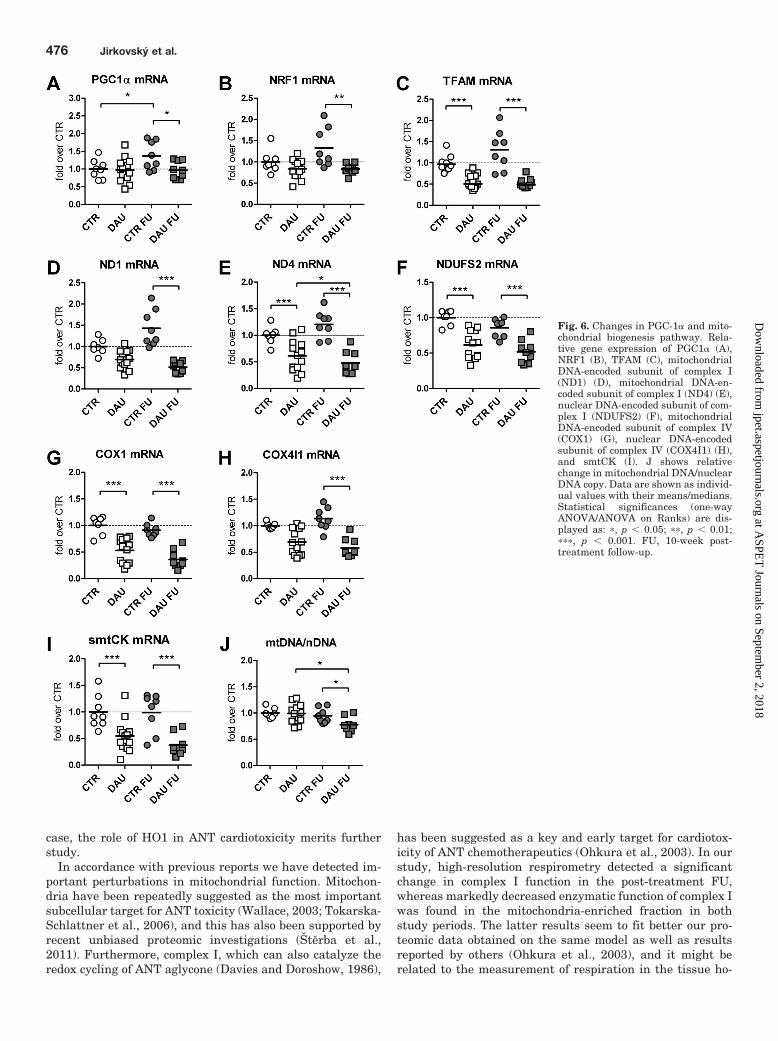
Subsequent analyses of mitochondrial biogenesis pathwayshowed either no changes or significant down-regulation ofkey pathway components caused by the chronic DAU treat-ment (Fig. 6). Gene expression of PGC1�, a master regulatorof mitochondrial biogenesis, did not show any change causedby DAU administration by the end of the treatment (Fig. 6A).The levels of PGC1� transcripts in the control group showeda slight, but significant, relative increase in the FU period.However, PGC1� mRNA levels did not follow the same trendin the DAU FU group; hence, a significant difference wasfound between the groups. A similar pattern was found in thegene expression of nuclear respiratory factor 1 (NRF1), al-though with somewhat lower values in the DAU groups andinsignificant rise in the controls in the FU (Fig. 6B). On theother hand, TFAM (Fig. 6C), exhibited a profound drop ingene expression (by more than 50%) caused by DAU treat-ment (p � 0.001) with no further progression in the FUperiod. Expression of the essential mtDNA-encoded subunit
of complex I (ND1) showed an insignificant tendency towarddown-regulation by the end of the treatment, whereas ex-pression of another mtDNA-encoded subunit (ND4) was sig-nificantly suppressed by nearly 40%. The expression of bothcomplex I subunits was evidently severely depressed in theFU with significant difference between both DAU-treatedgroups in the case of ND4 (Fig. 6, D and E). A nuclear-encoded complex I subunit (NDUFS2) exhibited a markeddown-regulation at the end of the treatment to a similarextent to ND4 (Fig. 6F). In addition, the apparent tendencyto decrease was found in the gene expression of the complexIV subunits encoded by both mtDNA and nDNA (Fig. 6, Gand H, respectively), although it did not reach statisticalsignificance in the latter case at the end of the treatment.Furthermore, analysis of gene expression of another nuclear-encoded mitochondrial protein outside the oxidative phos-phorylation machinery, sarcomeric mitochondrial creatinekinase (smtCK), also revealed profound down-regulation(Fig. 6I) in both study periods (by nearly 50 and 75%, respec-tively). No change in mtDNA/nDNA copy ratio was found atthe end of treatment, whereas a significant drop in this ratio
Fig. 3. Myocardial glutathione systemand daunorubicin treatment. A and B,HPLC determination of the left ven-tricular content of oxidized (GSSG)(A) and reduced (GSH) (B) glutathi-one. C, left ventricular GSSG/GSH ra-tio. D, correlation of left ventricularperformance with GSSG/GSH ratio inindividual animals as determined bySpearman correlation and regressionanalysis. Correlation coefficient andstatistical significance are shown. Eto G, relative enzymatic activity ofGPx (mean value in the control group:24.9 U/g of protein) (E), GR (meanvalue in the control group: 13.6 U/g ofprotein) (F), and GST (mean value inthe control group: 4.63 U/mg of pro-tein) (G) in the left ventricular myo-cardium. Data are shown as individ-ual values with their means/medians.Statistical significances (one-wayANOVA/ANOVA on Ranks) are dis-played as: �, p � 0.05; ��, p � 0.01;���, p � 0.001. FU, 10-week post-treatment follow-up.
Chronic Anthracycline Cardiotoxicity, Nrf2, and PGC1� 473
at ASPE
T Journals on Septem
ber 2, 2018jpet.aspetjournals.org
Dow
nloaded from
was found in the FU period (Fig. 6J). It is noteworthy that themajority of changes in the expression of mitochondrial pro-teins remained significant when the data were normalizedover mtDNA abundance (Supplemental Fig. 2, A–G).
DiscussionIn the present study, we administered 10 clinically rele-
vant doses of daunorubicin (�50 mg/m2) to rabbits to mimicindividual chemotherapeutic cycles without their overlap. Asexpected, significant cumulative dose induced chronic ANTcardiotoxicity, which progressed into dilated cardiomyopathyand heart failure in the post-treatment FU. It is noteworthythat we also have demonstrated that ANT-induced myocar-dial lipoperoxidation continued in the drug-free post-treat-ment period. Nevertheless, its loose association with toxicityparameters argues against its direct executive role in ANTcardiotoxicity. This might correspond with our previous data(Popelova et al., 2009) where the clinically available cardio-protective agent dexrazoxane showed nearly complete pro-
tection from ANT-induced cardiotoxicity and had only a mi-nor and insignificant effect on myocardial lipoperoxidation.Furthermore, although the presence of ANT-induced oxida-tive stimuli was also obvious from increased GSSG content,no changes in the key GSSG/GSH ratio suggest that theglutathione antioxidant system does not lose its overall com-petence upon chronic ANT treatment. Using a completelydifferent technique, we had previously obtained similar re-sults by the end of chronic ANT treatment (Vavrova et al.,2011). All these data might be consistent with the outcomesof clinical trials showing conclusively a lack of cardioprotec-tive effects of classic antioxidants and ROS scavengers in-cluding N-acetylcysteine and vitamin E in ANT cardiotoxic-ity (Gianni et al., 2008; Šimunek et al., 2009; Menna et al.,2012). In addition, others (Berthiaume et al., 2005) havedemonstrated that although vitamin E can decrease myocar-dial oxidative stress it has a little or no effect on key param-eters of chronic ANT cardiotoxicity. Hence, although globaloxidative stimuli are evidently taking place during ANT car-
Fig. 4. Response of myocardial Nrf2pathway. A, relative expression ofNrf2, a master regulator of cellularantioxidant and cytoprotective re-sponse. B, relative abundance of tran-scriptionally active form of Nrf2 in thenuclear fraction. C to G, relative ex-pression of Nrf2 targets: CuZnSOD(C), GCLC (D), MnSOD (E), NQO1(F), and HO1 (G). Data are shown asindividual values with their means/medians. Statistical significances (one-Way ANOVA/ANOVA on Ranks) aredisplayed as: ��, p � 0.01; ���, p �0.001. FU, 10-week post-treatmentfollow-up.
474 Jirkovsky et al.
at ASPE
T Journals on Septem
ber 2, 2018jpet.aspetjournals.org
Dow
nloaded from
diotoxicity development, they need not be the direct perpe-trators of chronic ANT cardiotoxicity and resulting heartfailure. Nevertheless, one cannot exclude a more sophisti-cated (e.g., signaling) or considerably compartmentalizedrole of ROS in ANT-induced heart failure. Several authorshave implicated the role of p53 in the triggering of mitochon-drial oxidative stress and apoptotic signaling (Sardao et al.,2009; Velez et al., 2011). Nevertheless, the potential involve-ment of this pathway in chronic ANT cardiotoxicity settingremains to be determined.
Despite calls for revisiting these questions (Gianni et al.,2008; Šimunek et al., 2009), oxidative injury remains themost frequently cited mechanism of ANT cardiotoxicity de-velopment. Therefore, in the present study we have focusedon the involvement of the Nrf2 pathway. It is noteworthythat we show no changes in either abundance of active Nrf2in the nuclear fraction or Nrf2 expression after chronic ANTtreatment. One can speculate that the level of oxidativestress in the cytosolic compartment is perhaps not sufficientto activate Nrf2 or its activation might be counteracted byother signaling cascades. We also convincingly demonstratedno coordinated activation of Nrf2 target genes upon chronicANT treatment. There was no change in the expression ofCuZnSOD, which corresponds with previously publisheddata (Li and Singal, 2000), and we detected no orchestratedactivation of glutathione system enzymes, which also rankamong important Nrf2 targets.
In contrast, we found severe down-regulation of gene ex-pression of MnSOD and NQO1 persisting a long time aftertreatment. The former finding is in line with our proteomicdata (Sterba et al., 2011) and protein abundance and activityreported by others (Li and Singal, 2000). MnSOD down-regulation can be seen even when the data are normalizedover mtDNA abundance, which suggests that the changecannot be simply explained by a drop in the mitochondrial
content. However, it should be noted that, in addition totranscriptional control, the activity of SOD isoforms is mark-edly affected by post-translational modifications; this re-quires further study in anthracycline cardiotoxicity settings(Dhar and St Clair, 2012). In addition, we, for the first time,demonstrate that chronic ANT cardiotoxicity is associatedwith the marked and persistent increase of expression ofHO1. HO1 is an essential cardioprotective molecule inducedin response to cardiac damage and stress (Peers and Steele,2012). So far, very little and rather variable data are avail-able regarding HO1 and ANT cardiotoxicity, and virtuallynothing has been reported regarding the clinically relevantchronic forms resulting in heart failure. Using proliferatingH9c2 cells, a suppression of HO1 expression has been re-ported after in vitro ANT treatment (Bernuzzi et al., 2009),whereas others have suggested the opposite in a microarraystudy (Choi et al., 2008). Little to no change in HO1 expres-sion has been seen at protein and mRNA levels 2 weeks aftera single supratherapeutic ANT dose (Suliman et al., 2007)and 70 days after injection of 1 mg/kg for 10 days (Richard etal., 2011), respectively. Piantadosi et al. (2008) have alsoreported HO1 as an important checkpoint involved in thecross-talk between the Nrf2 pathway and mitochondrial bio-genesis with the possibility of providing powerful cardiopro-tection against acute ANT cardiotoxicity induced by a singlehigh dose of doxorubicin. However, translatability to clini-cally more relevant chronic ANT cardiotoxicity settingsneeds to be verified in further studies. It is noteworthy thatcarbon monoxide produced by HO1 enzymatic activity isknown to interfere with the electron transport chain to re-lease electrons and produce ROS (Peers and Steele, 2012).Bearing in mind the mitochondrial damage and down-regu-lation of MnSOD, one cannot exclude that an adaptive role ofHO1 becomes maladaptive upon chronic exposure. In either
Fig. 5. Daunorubicin-induced changesin the mitochondrial functions. A to C,high-resolution respirometric examina-tion of activity of selected respiratorychain complexes in the myocardial ho-mogenates (mean value of activity ofrespiratory chain complexes I, II, andIV in the control group: 2.38, 2.84, and25.6 nmol O2/mg of protein, respec-tively). D, enzymatic activity of mito-chondrial complex I in mitochondria-enriched fraction (mean value in thecontrol group: 47.7 nmol DCIP/min/mgof protein). E, citrate synthase activity(mean value in the control group: 3.80M/min/g of protein). Data are shown asindividual values with their means/medians. Statistical significances (one-way ANOVA/ANOVA on Ranks) aredisplayed as: �, p � 0.05; ��, p � 0.01.FU, 10-week post-treatment follow-up.
Chronic Anthracycline Cardiotoxicity, Nrf2, and PGC1� 475
at ASPE
T Journals on Septem
ber 2, 2018jpet.aspetjournals.org
Dow
nloaded from
case, the role of HO1 in ANT cardiotoxicity merits furtherstudy.
In accordance with previous reports we have detected im-portant perturbations in mitochondrial function. Mitochon-dria have been repeatedly suggested as the most importantsubcellular target for ANT toxicity (Wallace, 2003; Tokarska-Schlattner et al., 2006), and this has also been supported byrecent unbiased proteomic investigations (Sterba et al.,2011). Furthermore, complex I, which can also catalyze theredox cycling of ANT aglycone (Davies and Doroshow, 1986),
has been suggested as a key and early target for cardiotox-icity of ANT chemotherapeutics (Ohkura et al., 2003). In ourstudy, high-resolution respirometry detected a significantchange in complex I function in the post-treatment FU,whereas markedly decreased enzymatic function of complex Iwas found in the mitochondria-enriched fraction in bothstudy periods. The latter results seem to fit better our pro-teomic data obtained on the same model as well as resultsreported by others (Ohkura et al., 2003), and it might berelated to the measurement of respiration in the tissue ho-
Fig. 6. Changes in PGC-1� and mito-chondrial biogenesis pathway. Rela-tive gene expression of PGC1� (A),NRF1 (B), TFAM (C), mitochondrialDNA-encoded subunit of complex I(ND1) (D), mitochondrial DNA-en-coded subunit of complex I (ND4) (E),nuclear DNA-encoded subunit of com-plex I (NDUFS2) (F), mitochondrialDNA-encoded subunit of complex IV(COX1) (G), nuclear DNA-encodedsubunit of complex IV (COX4I1) (H),and smtCK (I). J shows relativechange in mitochondrial DNA/nuclearDNA copy. Data are shown as individ-ual values with their means/medians.Statistical significances (one-wayANOVA/ANOVA on Ranks) are dis-played as: �, p � 0.05; ��, p � 0.01;���, p � 0.001. FU, 10-week post-treatment follow-up.
476 Jirkovsky et al.
at ASPE
T Journals on Septem
ber 2, 2018jpet.aspetjournals.org
Dow
nloaded from

mogenate. It is noteworthy that complex IV function wasfound compromised by the end of the treatment, and it per-sisted further in the post-treatment period, which may cor-respond with decreased enzymatic function of this complex inthe late-onset form of chronic ANT cardiotoxicity (Lebrecht etal., 2003). Our data also suggested a decline in the LV mito-chondria content with no recovery in the drug-free period.Hence, our findings confirm that chronic ANT cardiotoxicityis associated with marked mitochondrial alterations, whichpersist or deteriorate further in the post-treatment period.
Therefore, we addressed the function of mitochondrial bio-genesis pathway, which might provide an endogenous pro-tective response against such impairment (Ventura-Clapieret al., 2008). Earlier reports provided only limited insightsinto the role of mitochondrial biogenesis in ANT cardiotoxic-ity. Although two studies (Suliman et al., 2007; Miyagawa etal., 2010) reported down-regulation of this pathway causedby the ANT treatments, opposing findings have been recentlypublished (Marechal et al., 2011). It is noteworthy that all ofthese investigations have been performed on acute ANT car-diotoxicity models using single rather supratherapeutic ANTdoses. Thus, the connection to chronic ANT cardiotoxicityand heart failure remains uncertain.
In the present study, we demonstrate for the first time thatPGC1� as well as its downstream transcription factor NRF1show no change by the end of chronic ANT treatment, al-though evident mitochondrial alterations were in place. Thefact that TFAM was markedly depressed in the same periodmay imply more complex regulation of some parts of mito-chondrial biogenesis. Gene expression of PGC1� showed aslight, but significant, tendency toward increase in the con-trol group comparing FU and end-of-treatment values and asimilar, but insignificant, trend was observed in severaldownstream pathway members. Although the reasons forthese observations are unknown, considering a significantlength of the FU period, different factors such as animalaging or challenge by the scheduled experimental procedurescould be involved. In either case DAU-treated animals wereunable to follow this trend, which might imply different reg-ulation than in the controls. On the other hand, TFAMshowed marked down-regulation in both study periods,which was evidently caused by the treatment. Most impor-tantly, a severe decrease in gene expression was found in allinvestigated subunits of complex I and IV without markedpreference of either mtDNA or nDNA-encoded ones. Becausethis trend has been observed even after the normalization ofthe data on mtDNA abundance, this change may be inter-preted as down-regulation caused by the treatment.
We have not found any change in mtDNA/nDNA ratio atthe end of the chronic treatment, whereas the significantdrop that was identified in the FU period suggests continuingmitochondrial damage in the post-treatment period. Previ-ously reported data seem to depend on this model, becauseLebrecht et al. (2003) have also shown a decrease in mtDNA/nDNA ratio with a late-onset chronic ANT cardiotoxicitymodel, whereas others have reported either decreases (Miya-gawa et al., 2010) or even increases (Marechal et al., 2011)after acute ANT dosing.
It should be noted that decreased mitochondrial contentcaused by the treatment evidenced by significant decrease ofthe citrate synthase activity (by �20%) may have an impacton some mitochondria-related parameters. However, the
mean relative changes of all significantly affected mitochon-dria-related parameters seem to be too high (average change47.4%) to be solely determined by this event. Furthermore,normalization of the data over mtDNA argues against theloss of mitochondria in surviving cardiomyocytes as the maindriving force for these observations. This is in line withmarkedly decreased complex I activity, which was found evenin the mitochondria-enriched tissue fractions. Future studiesshould work out the present results to encounter the proteinlevel and follow the changes identified in this study through-out the whole time course of cardiotoxicity development. Fur-thermore, to draw firm mechanistic conclusions it will benecessary to use a genetic manipulation approach. In addi-tion, although the induction of mitochondrial biogenesisthrough the administration of CO, CO-releasing molecules,or metformin treatment seemed to be helpful in acute andsubacute anthracycline cardiotoxicity settings (Piantadosiand Suliman, 2006; Asensio-Lopez et al., 2011; Soni et al.,2011; Ashour et al., 2012), it will be important to revealwhether this intervention may have a therapeutic valueagainst chronic anthracycline cardiotoxicity.
In conclusion, in the present study we describe molecularand functional changes associated with the development ofchronic ANT cardiotoxicity and its progression to heart fail-ure in the post-treatment FU. Our data strongly suggest thatANT-induced global oxidative stress is not a key factor di-rectly responsible for heart failure development. Further-more, we demonstrate that the Nrf2 pathway coordinatingantioxidant response is not induced by chronic ANT treat-ment. It is noteworthy that we have observed Nrf2-indepen-dent regulation of expression of MnSOD, NQO1, and HO1,which merits further study. Despite evident damage to mi-tochondria that persists or further develops in the FU,chronic ANT cardiotoxicity was not associated with the in-duction of the mitochondrial biogenesis pathway controlledby PGC1�. Instead, this pathway seemed impaired, whichwas associated with the severe down-regulation of gene ex-pression of numerous mitochondrial proteins encoded by bothnuclear and mitochondrial genomes. This is particularly ev-ident in the FU, and it may be connected with ongoing myo-cardial damage and transition to heart failure.
Acknowledgments
We thank Dr. Rene Endlicher for help with tissue preparation forrespiration experiments; Jitka Pohorska for excellent assistance;and Dr. Catherine McGrath for reading the manuscript, helpfulsuggestions, and correction of English.
Authorship Contributions
Participated in research design: Jirkovsky, Popelova, Vavrova,Micuda, Simunek, Cervinkova, Gersl, and Sterba.
Conducted experiments: Jirkovsky, Popelova, Krivakova-Stankova,Vavrova, Hroch, Haskova, Brcakova-Dolezelova, Adamcova, andSterba.
Performed data analysis: Jirkovsky, Popelova, Krivakova-Stankova, Vavrova, Hroch, Haskova, Brcakova-Dolezelova, Micuda,Adamcova, Simunek, Cervinkova, Gersl, and Sterba.
Wrote or contributed to the writing of the manuscript: Jirkovsky,Popelova, Krivakova-Stankova, Vavrova, Micuda, Adamcova,Simunek, Cervinkova, Gersl, and Sterba.
ReferencesAsensio-Lopez MC, Lax A, Pascual-Figal DA, Valdes M, and Sanchez-Mas J (2011)
Metformin protects against doxorubicin-induced cardiotoxicity: involvement of theadiponectin cardiac system. Free Radic Biol Med 51:1861–1871.
Chronic Anthracycline Cardiotoxicity, Nrf2, and PGC1� 477
at ASPE
T Journals on Septem
ber 2, 2018jpet.aspetjournals.org
Dow
nloaded from

Ashour AE, Sayed-Ahmed MM, Abd-Allah AR, Korashy HM, Maayah ZH, AlkhalidiH, Mubarak M, and Alhaider A (2012) Metformin rescues the myocardium fromdoxorubicin-induced energy starvation and mitochondrial damage in rats. OxidMed Cell Longev 2012:434195.
Baird L and Dinkova-Kostova AT (2011) The cytoprotective role of the Keap1-Nrf2pathway. Arch Toxicol 85:241–272.
Bernuzzi F, Recalcati S, Alberghini A, and Cairo G (2009) Reactive oxygen species-independent apoptosis in doxorubicin-treated H9c2 cardiomyocytes: role for hemeoxygenase-1 down-modulation. Chem Biol Interact 177:12–20.
Berthiaume JM, Oliveira PJ, Fariss MW, and Wallace KB (2005) Dietary vitamin Edecreases doxorubicin-induced oxidative stress without preventing mitochondrialdysfunction. Cardiovasc Toxicol 5:257–267.
Berthiaume JM and Wallace KB (2007) Adriamycin-induced oxidative mitochondrialcardiotoxicity. Cell Biol Toxicol 23:15–25.
Choi EH, Lee N, Kim HJ, Kim MK, Chi SG, Kwon DY, and Chun HS (2008)Schisandra fructus extract ameliorates doxorubicin-induce cytotoxicity in cardio-myocytes: altered gene expression for detoxification enzymes. Genes Nutr 2:337–345.
Davies KJ and Doroshow JH (1986) Redox cycling of anthracyclines by cardiacmitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J BiolChem 261:3060–3067.
De Sousa E, Veksler V, Minajeva A, Kaasik A, Mateo P, Mayoux E, Hoerter J, BigardX, Serrurier B, and Ventura-Clapier R (1999) Subcellular creatine kinase altera-tions. Implications in heart failure. Circ Res 85:68–76.
Dhar SK and St Clair DK (2012) Manganese superoxide dismutase regulation andcancer. Free Radic Biol Med 52:2209–2222.
Dignam JD, Martin PL, Shastry BS, and Roeder RG (1983) Eukaryotic gene tran-scription with purified components. Methods Enzymol 101:582–598.
Ewer MS and Suter TM (2010) Diagnostic aspects of cardiovascular toxicity ofantitumor drugs, in Cardiotoxicity of Non-Cardiovascular Drugs (Minotti G ed) pp201–221, John Wiley & Son Ltd, Padstow, UK.
Gianni L, Herman EH, Lipshultz SE, Minotti G, Sarvazyan N, and Sawyer DB(2008) Anthracycline cardiotoxicity: from bench to bedside. J Clin Oncol 26:3777–3784.
Institute of Laboratory Animal Resources (1996) Guide for the Care and Use ofLaboratory Animals 7th ed. Institute of Laboratory Animal Resources, Commis-sion on Life Sciences, National Research Council, Washington, DC.
Kand’ar R, Zakova P, Lotkova H, Kucera O, and Cervinkova Z (2007) Determinationof reduced and oxidized glutathione in biological samples using liquid chromatog-raphy with fluorimetric detection. J Pharm Biomed Anal 43:1382–1387.
Keizer HG, Pinedo HM, Schuurhuis GJ, and Joenje H (1990) Doxorubicin (adriamy-cin): a critical review of free radical-dependent mechanisms of cytotoxicity. Phar-macol Ther 47:219–231.
Kwok JC and Richardson DR (2003) Anthracyclines induce accumulation of iron inferritin in myocardial and neoplastic cells: inhibition of the ferritin iron mobiliza-tion pathway. Mol Pharmacol 63:849–861.
Kwok JC and Richardson DR (2004) Examination of the mechanism(s) involved indoxorubicin-mediated iron accumulation in ferritin: studies using metabolic inhib-itors, protein synthesis inhibitors, and lysosomotropic agents. Mol Pharmacol65:181–195.
Lebrecht D, Kirschner J, Geist A, Haberstroh J, and Walker UA (2010) Respiratorychain deficiency precedes the disrupted calcium homeostasis in chronic doxorubi-cin cardiomyopathy. Cardiovasc Pathol 19:e167–174.
Lebrecht D, Setzer B, Ketelsen UP, Haberstroh J, and Walker UA (2003) Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain de-fects in chronic doxorubicin cardiomyopathy. Circulation 108:2423–2429.
Lebrecht D and Walker UA (2007) Role of mtDNA lesions in anthracycline cardio-toxicity. Cardiovasc Toxicol 7:108–113.
Li T and Singal PK (2000) Adriamycin-induced early changes in myocardial antiox-idant enzymes and their modulation by probucol. Circulation 102:2105–2110.
Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, Suter TM,Liao R, and Sawyer DB (2004) Anthracyclines induce calpain-dependent titinproteolysis and necrosis in cardiomyocytes. J Biol Chem 279:8290–8299.
Lipshultz SE, Alvarez JA, and Scully RE (2008) Anthracycline associated cardiotox-icity in survivors of childhood cancer. Heart 94:525–533.
Long J, Ma J, Luo C, Mo X, Sun L, Zang W, and Liu J (2009) Comparison of twomethods for assaying complex I activity in mitochondria isolated from rat liver,brain and heart. Life Sci 85:276–280.
Marechal X, Montaigne D, Marciniak C, Marchetti P, Hassoun SM, Beauvillain JC,Lancel S, and Neviere R (2011) Doxorubicin-induced cardiac dysfunction is atten-uated by ciclosporin treatment in mice through improvements in mitochondrialbioenergetics. Clin Sci 121:405–413.
Menna P, Paz OG, Chello M, Covino E, Salvatorelli E, and Minotti G (2012) Anthra-cycline cardiotoxicity. Expert Opin Drug Saf 11 (Suppl 1):S21–S36.
Minotti G, Menna P, Salvatorelli E, Cairo G, and Gianni L (2004) Anthracyclines:
molecular advances and pharmacologic developments in antitumor activity andcardiotoxicity. Pharmacol Rev 56:185–229.
Miyagawa K, Emoto N, Widyantoro B, Nakayama K, Yagi K, Rikitake Y, Suzuki T,and Hirata K (2010) Attenuation of doxorubicin-induced cardiomyopathy by en-dothelin-converting enzyme-1 ablation through prevention of mitochondrial bio-genesis impairment. Hypertension 55:738–746.
Ohkura K, Lee JD, Shimizu H, Nakano A, Uzui H, Horikoshi M, Fujibayashi Y,Yonekura Y, and Ueda T (2003) Mitochondrials complex I activity is reduced inlatent adriamycin-induced cardiomyopathy of rat. Mol Cell Biochem 248:203–208.
Peers C and Steele DS (2012) Carbon monoxide: a vital signalling molecule andpotent toxin in the myocardium. J Mol Cell Cardiol 52:359–365.
Piantadosi CA, Carraway MS, Babiker A, and Suliman HB (2008) Heme oxygenase-1regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional con-trol of nuclear respiratory factor-1. Circ Res 103:1232–1240.
Piantadosi CA and Suliman HB (2006) Mitochondrial transcription factor A induc-tion by redox activation of nuclear respiratory factor 1. J Biol Chem 281:324–333.
Pilz J, Meineke I, and Gleiter CH (2000) Measurement of free and bound malondi-aldehyde in plasma by high-performance liquid chromatography as the 2,4-dinitrophenylhydrazine derivative. J Chromatogr B Biomed Sci Appl 742:315–325.
Popelová O, Sterba M, Hasková P, Simunek T, Hroch M, Guncová I, Nachtigal P,Adamcová M, Gersl V, and Mazurová Y (2009) Dexrazoxane-afforded protectionagainst chronic anthracycline cardiotoxicity in vivo: effective rescue of cardiomy-ocytes from apoptotic cell death. Br J Cancer 101:792–802.
Richard C, Ghibu S, Delemasure-Chalumeau S, Guilland JC, Des Rosiers C, ZellerM, Cottin Y, Rochette L, and Vergely C (2011) Oxidative stress and myocardialgene alterations associated with doxorubicin-induced cardiotoxicity in rats persistfor 2 months after treatment cessation. J Pharmacol Exp Ther 339:807–814.
Rimbaud S, Garnier A, and Ventura-Clapier R (2009) Mitochondrial biogenesis incardiac pathophysiology. Pharmacol Rep 61:131–138.
Sardao VA, Oliveira PJ, Holy J, Oliveira CR, and Wallace KB (2009) Doxorubicin-induced mitochondrial dysfunction is secondary to nuclear p53 activation in H9c2cardiomyoblasts. Cancer Chemother Pharmacol 64:811–827.
Simunek T, Klimtová I, Kaplanová J, Mazurová Y, Adamcová M, Sterba M, HrdinaR, and Gersl V (2004) Rabbit model for in vivo study of anthracycline-inducedheart failure and for the evaluation of protective agents. Eur J Heart Fail 6:377–387.
Simunek T, Sterba M, Popelová O, Adamcová M, Hrdina R, and Gersl V (2009)Anthracycline-induced cardiotoxicity: overview of studies examining the roles ofoxidative stress and free cellular iron. Pharmacol Rep 61:154–171.
Soni H, Pandya G, Patel P, Acharya A, Jain M, and Mehta AA (2011) Beneficialeffects of carbon monoxide-releasing molecule-2 (CORM-2) on acute doxorubicincardiotoxicity in mice: role of oxidative stress and apoptosis. Toxicol Appl Phar-macol 253:70–80.
Sterba M, Popelová O, Lenco J, Fucíková A, Brcáková E, Mazurová Y, Jirkovský E,Simunek T, Adamcová M, Micuda S, et al. (2011) Proteomic insights into chronicanthracycline cardiotoxicity. J Mol Cell Cardiol 50:849–862.
Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, and PiantadosiCA (2007) The CO/HO system reverses inhibition of mitochondrial biogenesis andprevents murine doxorubicin cardiomyopathy. J Clin Invest 117:3730–3741.
Tokarska-Schlattner M, Zaugg M, Zuppinger C, Wallimann T, and Schlattner U(2006) New insights into doxorubicin-induced cardiotoxicity: the critical role ofcellular energetics. J Mol Cell Cardiol 41:389–405.
Vávrová A, Popelová O, Sterba M, Jirkovský E, Hasková P, Mertlíková-Kaiserová H,Gersl V, and Simunek T (2011) In vivo and in vitro assessment of the role ofglutathione antioxidant system in anthracycline-induced cardiotoxicity. Arch Toxi-col 85:525–535.
Velez JM, Miriyala S, Nithipongvanitch R, Noel T, Plabplueng CD, Oberley T,Jungsuwadee P, Van Remmen H, Vore M, and St Clair DK (2011) p53 regulatesoxidative stress-mediated retrograde signaling: a novel mechanism for chemother-apy-induced cardiac injury. PLoS One 6:e18005.
Ventura-Clapier R, Garnier A, and Veksler V (2008) Transcriptional control ofmitochondrial biogenesis: the central role of PGC-1�. Cardiovasc Res 79:208–217.
Wallace KB (2003) Doxorubicin-induced cardiac mitochondrionopathy. PharmacolToxicol 93:105–115.
Wallace KB (2007) Adriamycin-induced interference with cardiac mitochondrialcalcium homeostasis. Cardiovasc Toxicol 7:101–107.
Address correspondence to: Dr. Martin Sterba, Department of Phar-macology, Faculty of Medicine in Hradec Kralove, Charles University inPrague, Simkova 870, Hradec Kralove, 500 38, Czech Republic. E-mail:[email protected]
478 Jirkovsky et al.
at ASPE
T Journals on Septem
ber 2, 2018jpet.aspetjournals.org
Dow
nloaded from





























