Embed Size (px)
Citation preview

Chm 118Fall 2016
Problem Solving in Chemistry
Mr. Linck
Version: 1.6.August 15, 2016

Written for and dedicated to MCAugust, 2016
i

Contents
1 Introduction 1
2 A Simple Structural Model 2
2.1 Basis Principles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
2.1.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
2.2 Quantum Issues . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
2.2.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
2.3 Basis of Simple Structural Model . . . . . . . . . . . . . . . . . . . . . . . . 6
2.3.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
2.4 The Simplest Model to Count Electrons for VSEPR Structures . . . . . . . 7
2.4.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
2.5 A Slight Expansion of the Simple Model . . . . . . . . . . . . . . . . . . . . 10
2.5.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2.6 Patching the Lewis Structure Model when Needed . . . . . . . . . . . . . . 11
2.6.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
3 Using Lewis Structures to Understand Structure and Reactivity 14
3.1 Bond Lengths . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
3.1.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
3.2 Bond Energies and Enthalpy . . . . . . . . . . . . . . . . . . . . . . . . . . 15
3.2.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
ii

0.0 iii
3.3 Polarization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
3.3.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
4 Acidity 20
4.1 Definitions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
4.1.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
4.2 Estimating Acidity, Part I: Nature of Element . . . . . . . . . . . . . . . . . 21
4.2.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
4.3 Estimating Acidity, Part II: Charge . . . . . . . . . . . . . . . . . . . . . . . 23
4.3.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
4.4 Estimating Acidity, Part III: Resonance . . . . . . . . . . . . . . . . . . . . 24
4.4.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
4.5 Estimating Acidity, Part IV: Inductive Effects . . . . . . . . . . . . . . . . . 25
4.5.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
4.6 Hydration Energy and the Acidity of Metal Ions . . . . . . . . . . . . . . . 26
4.6.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
4.7 Distribution Diagrams . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
4.7.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
5 Symmetry 30
5.1 Definitions and Proper Symmetry Operations . . . . . . . . . . . . . . . . . 30
5.1.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
5.2 Improper Symmetry Operations: Planes of Symmetry . . . . . . . . . . . . 32
5.2.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
5.3 Improper Symmetry Operations: Center of Inversion . . . . . . . . . . . . . 33
5.3.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
5.4 Improper Symmetry Operations: Rotation-Reflection Axis . . . . . . . . . . 33
5.4.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
Chm 118 Problem Solving in Chemistry

0.0 iv
5.5 The Group of Symmetry Operations . . . . . . . . . . . . . . . . . . . . . . 34
5.5.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
5.6 The Behavior of Single Objects Under Symmetry Operations . . . . . . . . 35
5.6.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
5.7 The Behavior of Several Objects Under Symmetry Operations . . . . . . . . 39
5.7.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
5.8 Classifying the Symmetry for Multiple Objects; the Combinations . . . . . 40
5.8.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
6 Quantum Mechanics 45
6.1 Probability and Quantum Measurement . . . . . . . . . . . . . . . . . . . . 45
6.1.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
6.2 Waves and Interference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
6.2.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
6.3 Simple Version of the Rules of Quantum Mechanics . . . . . . . . . . . . . . 49
6.3.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
6.4 A Simple but Useful Quantum Problem: The Parve on a Pole . . . . . . . . 50
6.4.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
6.5 Quantum Mechanics of the Hydrogen Atom I. Energy Levels . . . . . . . . 52
6.5.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
6.6 Quantum Mechanics of the Hydrogen Atom II. Radial Wave Function . . . 53
6.6.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
6.7 Quantum Mechanics of the Hydrogen Atom III. Angular Wave Function . . 59
6.7.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
6.8 Multi-electronic Atoms, Configurations . . . . . . . . . . . . . . . . . . . . . 63
6.8.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64
6.9 Multi-electronic Atoms, Ionization Energies . . . . . . . . . . . . . . . . . . 65
Chm 118 Problem Solving in Chemistry

0.0 v
6.9.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
6.10 Multi-electronic Atoms, Valence Orbital Ionization Energies . . . . . . . . . 68
6.10.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
6.11 Multi-electronic Atoms, Transition Metal Ions . . . . . . . . . . . . . . . . . 69
6.11.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
7 Transition Metal Compounds and Color 70
7.1 Energy of Orbitals in Electrostatic Fields . . . . . . . . . . . . . . . . . . . 70
7.1.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
7.2 Metal Ions in an Octahedral and Tetrahedral Environments . . . . . . . . . 72
7.2.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73
7.3 Metal Ions in Other Environments: Lowering of Symmetry . . . . . . . . . 74
7.3.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
7.4 The Configuration of Metal Ion Compounds . . . . . . . . . . . . . . . . . . 75
7.4.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
7.5 Configuration of Metal Ion Compounds, Spin, Field, and Ligand Strength . 77
7.5.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
7.6 Color of Metal Ion Compounds. I. Octahedral Compounds. . . . . . . . . . 79
7.6.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
7.7 Color of Metal Ion Compounds. II. Low Symmetry Compounds. . . . . . . 81
7.7.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
7.8 Color of Metal Ion Compounds. III. Intensities of Color. . . . . . . . . . . . 84
7.8.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
7.9 Magnetism in Metal Ion Compounds. . . . . . . . . . . . . . . . . . . . . . . 87
7.9.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
7.10 Crystal Field Stabilization Energies . . . . . . . . . . . . . . . . . . . . . . . 88
7.10.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
Chm 118 Problem Solving in Chemistry

0.0 vi
8 Absorbance and Kinetics 92
8.1 Using Light to See . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
8.1.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
8.2 Basic Kinetic Expressions and Definitions . . . . . . . . . . . . . . . . . . . 94
8.2.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
8.3 First Order Reactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 96
8.3.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 96
8.4 Dealing with More Complex Rate Laws . . . . . . . . . . . . . . . . . . . . 97
8.4.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
9 Bonding 100
9.1 Storage of Energy in Molecules . . . . . . . . . . . . . . . . . . . . . . . . . 100
9.1.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
9.2 Using Atomic Orbitals to Make Molecular Orbitals . . . . . . . . . . . . . . 103
9.2.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 108
9.3 A Nomenclature for Molecular Orbitals . . . . . . . . . . . . . . . . . . . . 110
9.3.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
9.4 Molecular Orbital Energy Diagrams . . . . . . . . . . . . . . . . . . . . . . 113
9.4.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113
9.5 Heteronuclear Diatomics and the First Row Diatomics with s/p Mixing . . 114
9.5.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 116
9.6 Hybridization as a Simplifying Tool. Part I: sp Hybridization . . . . . . . . 118
9.6.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 118
9.7 Hybridization as a Tool. Part II: sp2 and sp3 Hybridization . . . . . . . . . 120
9.7.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
9.8 Using Symmetry to Determine the Orbitals that can Bind. Part I. SimpleSystems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
Chm 118 Problem Solving in Chemistry

0.0 vii
9.8.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122
9.9 Using Symmetry to Determine the Orbitals that can Bind. Part II. Degen-erate Systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
9.9.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 124
9.10 Exploring a Model for Solids . . . . . . . . . . . . . . . . . . . . . . . . . . 125
9.10.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
9.11 Molecular Orbital Theory of Hypervalent Compounds . . . . . . . . . . . . 126
9.11.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 126
9.12 Bonding in Metal Compounds. Part I. σ Donors . . . . . . . . . . . . . . . 128
9.12.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 129
9.13 Bonding in Metal Compounds. Part II. σ and π Donors . . . . . . . . . . . 130
9.13.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 131
9.14 Bonding in Metal Compounds. Part III. σ Donors/π Acceptors . . . . . . . 132
9.14.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132
9.15 The Eighteen Electron Rule . . . . . . . . . . . . . . . . . . . . . . . . . . . 134
9.15.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 134
10 Entropy 136
10.1 Definitions, and the Number of Microstates in a Configuration . . . . . . . 136
10.1.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 138
10.2 A Way to Count Microstates in a Configuration and Predominant Configu-rations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 140
10.2.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 141
10.3 A Detour to Define Some Thermodynamic Quantities: The First Law . . . 142
10.3.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 143
10.4 Adding Heat to the Predominant Configuration. Part I. Qualitative . . . . 143
10.4.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 144
10.5 What Does a Predominant Configuration Look Like? The Boltzmann Equation145
Chm 118 Problem Solving in Chemistry

0.0 viii
10.5.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 146
10.6 Adding Heat to the Predominant Configuration. Part II. Quantitative. . . . 147
10.6.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 149
10.7 Disguising our Finding in a Word: Entropy . . . . . . . . . . . . . . . . . . 149
10.7.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
10.8 Entropy Defined in Terms of W; The Third Law . . . . . . . . . . . . . . . 152
10.8.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153
10.9 Disorder is a Poor Word to Describe Entropy . . . . . . . . . . . . . . . . . 153
10.9.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154
10.10Entropy Change in Reactions . . . . . . . . . . . . . . . . . . . . . . . . . . 154
10.10.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155
11 The Free Energy: Entropy in Another Guise 157
11.1 Using the First Law of Thermodynamics at Constant P and T: Enthalpy . . 157
11.1.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158
11.2 Using Enthalpy of the System to Understand the Surrounding’s EntropyChange . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 159
11.2.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 159
11.3 Getting Rid of the Universe Altogether: Free Energy . . . . . . . . . . . . . 161
11.3.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
11.4 The Pressure (or Concentration) Dependence of G . . . . . . . . . . . . . . 162
11.4.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
11.5 Free Energy and Equilibrium . . . . . . . . . . . . . . . . . . . . . . . . . . 166
11.5.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167
12 Equilibrium and Other Uses of the Free Energy 169
12.1 Equilibrium in Acid Solutions . . . . . . . . . . . . . . . . . . . . . . . . . . 169
12.1.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
Chm 118 Problem Solving in Chemistry

0.0 ix
12.2 Mixtures of Acids and Salts: Buffer Solutions . . . . . . . . . . . . . . . . . 172
12.2.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 173
12.3 Solubility Equilibrium, including Common Ion Effect . . . . . . . . . . . . . 173
12.3.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 174
12.4 Electrochemical Cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 174
12.4.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 176
12.5 Free Energy, Other Work, and the Nernst Equation . . . . . . . . . . . . . . 177
12.5.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 178
12.6 Using Electrochemical Cells to Solve Problems . . . . . . . . . . . . . . . . 178
12.6.1 Exercises . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 179
Chm 118 Problem Solving in Chemistry

Chapter 1
Introduction
There are three large subject areas in chemistry that this course deals with. These arethe structure, color, and reactivity of molecules. Structure means we want to understandthe arrangement in space of the nuclei and learn what we can about where the electronsare to be found between those nuclei. Also, how those structures influence the chemistryof the materials. Color is of interest to us because it teaches us what the structures are,and how easily electrons are moved around. The latter leads directly to asking questionsabout reactivity: Which molecules will react with each other, what energy changes takeplace during that reaction, and how fast do those reactions take place?
These are the topics in this course. We shall work our way to knowledge in each of theseareas in many small steps, coming back to a more sophisticated vision with each round ofsteps. Repitition is the heart of learning.
Note to the reader: A skill that is often overlooked in scientific education is the ability tosense when data appear weird; when they are wrong by some human error, or when they aretelling the observer to modify her understanding because it does not work. To teach thisskill there are numbers in this document that have been made weird intentionally. Look atwhat you see critically and ask ”Can that be?”
1

Chapter 2
A Simple Structural Model
2.1 Basis Principles
The simplest question to ask about the structure of a molecule is ”Where are the nucleilocated in space?” If all we consider is the nuclei, then the answer is clear: as far apart asthey can get, since all nuclei are positively charged, and hence repel each other accordingto Coulomb’s law, which is at the heart of all chemical understanding:
U =1
4πε0
Z1Z2
r1,2(2.1)
where U is the potential energy, Z1 and Z2 are the charges on the two species of concern,r1,2 is the distance separating them, and the first term is just there to convert from units ofcharge, Coulombs, and distance, to energy units. Note when both the charges are the same,either positive or negative, then the potential energy is more positive, less stable, when thedistance is small.
So why do molecules have nuclei relatively close together? The answer is, of course, thatelectrons are also present, and these negatively charged species attract nuclei, holding themcloser together in a favorable arrangement. Our question is what is that arrangement? Sincethe cause is the electrons, it is not surprising that we should look to where the electronsare in order to understand the location of the nuclei. We will see our model requires theexamination of electron pairs.
2.1.1 Exercises
2.1.1.1
Will two +1 charges have a greater potential energy at a distance of 1.0A or 2.0A.
2

2.2 3
Figure 2.1: Random ping-pong balls descending to two slits.
2.1.1.2
For this problem, assume 14πε0
is equal to 1. Plot the value of U as a function of ri,j for apair of positive charges of unit value.
2.1.1.3
For this problem, assume 14πε0
is equal to 1. Plot the value of U as a function of ri,j for anegative charge and a positive charge, each of unit value.
2.2 Quantum Issues
To learn where electrons are, and what they do, requires a knowledge of quantum mechanics,which is the collection of the rules that dictate the behavior of absolutely small things1. Wewill talk extensively, but mostly non-mathematically, about quantum issues in this course;there are two aspects that we need here to proceed. The first is to acknowledge that thebehavior of electrons is ”weird,” often thought to be ”other worldly.” One aspect of this is
1Dirac defines absolutely small things as those that cannot be observed without a change in their prop-erties, usually position or momentum.
Chm 118 Problem Solving in Chemistry

2.2 4
Figure 2.2: Probability distribution for ping-pong balls.
that quantum mechanics tells us often that the best we can do is to find the probability ofthe result of a measurement, not a determined value.
Consider dropping a bunch of randomly positioned ping-pong balls toward a floor with twoslits (holes) in it, as illustrated in Figure 2.1. Those that fall through the holes will hitdetectors below and will be counted as doing so. The distribution of ping-pong ball hitsat the detectors will be as shown in Figure 2.2, where the vertical axis is the number ofhits and the horizontal axis is the position of the hit. This is classical behavior and offersnothing your experience does not anticipate.
If we carry out the same experiment with electrons or any other small object going throughtwo slits, the distribution of hits is different. That which is observed is shown in Figure 2.3.Notice that there are regions of high probablilty of an electron hitting and regions of lowprobability. The probablity of hits now looks nothing like the physical arrangement of theslits. Rather, it appears like a diffraction pattern that would be exhibited by something likewater waves. A consequence of this is that quantum mechanics will use a wave function todescribe the behavior of electrons. This wave function is a function of the position in spaceand, in general, of time. It is often written as ψ(x,y,z,t). A typical example would be
ψ(x, y, z, t) = 2√
2Sin[πx]Sin[πy]Sin[πz] (2.2)
which is a time-independent wave function. The probability issue we talked about abovecomes about by squaring of this descriptive wave function,
P = [ψ(x, y, z, t)]2 (2.3)
where P is the probability of finding the electron at the point x, y, z at time t. We willdescribe electrons in atoms and molecules by describing the corresponding wave functions.Note there is nothing here about those descriptions, just an establishment of where we willultimately go. We will begin this with very few details in the next section.
The second feature that we need to deal with in a simple structural model is the fact thatelectrons exhibit a feature usually called spin. This is a pure quantum mechanical feature
Chm 118 Problem Solving in Chemistry

2.3 5
Figure 2.3: Probability distribution for electrons through two slits
and has no classical analogue. The spin of electrons can take only one of two values, spin“up”, spin of 1/2, or spin “down”, spin of -1/2. Electrons of each of these two kinds ofspin can be separated by appropriate magnetic devices. What is important to us here is thePauli Principle, which can be expressed in a number of equivalent ways, one of which is:“No two electrons of the same spin can come close to each other.” Electrons are held apartfrom each other because of their charge via the operation of Coulomb’s Law, equation 2.1.Electrons of the same spin are held apart from each other by the additional operation ofthe Pauli Principle.
2.2.1 Exercises
2.2.1.1
A wave function is just a recipe for finding a value at some point in space. Find the valueof ψ of equation 2.2 at the point x = 0.1, y = 0.2, z = 0.6.
2.2.1.2
Find the probability that a particle with the ψ given in equation 2.2 can be found in a smallvolume centered on the point x = 0.2, y = 0.1, z = 0.6.
2.2.1.3
For an electron described by the wave function ψ = 2√
2 Sin[2πx] Sin[πy] Sin[πz], find theprobability that the electron will be found in a small volume centered on the point x = 0.5,y= 0.2, z = 0.2. Note: Regions of space where the probability of finding the electron iszero2 are called “nodes.”
2A favorite student question is ”If there is a region where there is no probability of finding the electron,how does it get from one side to the other?” Don’t ask. Such a question is applying classical behavior tothings that do not behave that way.
Chm 118 Problem Solving in Chemistry

2.4 6
2.3 Basis of Simple Structural Model
Imagine a nucleus with four electrons around it, two of which are of up spin and two ofwhich are of down spin. Each set of electrons will, according to the Pauli Principle, stayas far apart from each other as possible. If we now bring another atom up to the first,we need electrons between the nuclei to keep them from repelling each other according toequation 2.1. So an up spin and down spin of each set will come reasonably close (to holdthe nuclei together), but the second of up spin will stay as far as it can from the first electronof up spin, and similarly for those of down spin. This simple model is called the VSEPRmodel (for valence shell electron pair repulsion).
2.3.1 Exercises
2.3.1.1
What will be the location of two electrons of up spin near a nucleus if you require them tobe as far apart from each other as possible?
2.3.1.2
In BeCl2 there are four electrons around the Be, two of up spin and two of down spin.Where will you find the two chlorine atoms?
2.3.1.3
What will be the arrangement for six electrons, three of each spin, in order to stay as farapart from each other as possible?
2.3.1.4
In BF3 there are six electrons around the B, three of up spin and three of down spin. Wherewill you find the three fluorine atoms?
2.3.1.5
The arrangements talked about in the earlier problems are for two dimensional structures.In three dimensions, it is a little harder to use intuition. For eight electrons, four of each spin,the optimium arrangement of each set of four is the tetrahedron. Look up a tetrahedronon the web and be able to draw this arrangement.
2.3.1.6
In CH4 there are eight electrons around the C, four of up spin and four of down spin. Wherewill you find the four hydrogen atoms?
2.3.1.7
Make of drawing of CH4 which shows the real geometry in space.
Chm 118 Problem Solving in Chemistry

2.4 7
Figure 2.4: Schematic representation of nitrogen atom bonding
2.4 The Simplest Model to Count Electrons for VSEPR Struc-tures
The arguments just given require that we know how many electrons are around a givenatom to determine how the atoms attached to it will be arranged. The simplest modelfor where the electrons are in a molecule is called a Lewis structure. This is well knownto students in this course, but here is a concise summary. Lewis structures of compoundsmade from elements in groups IV (14) through VII (17) in the periodic table have theirelectrons arranged in pairs (for reasons discussed above) and there are (usually–we paya lot of attention to the exceptions below) eight electrons around any given atom. (Theubiquitous hydrogen atom has only a pair of electrons.) There is a simple consequence ofthis. Consider a nitrogen atom with five electrons in the valence shell, the layer importantin bonding. One set of up spin and down spin electrons form a lone pair on the nitrogen.Since the nitrogen atom needs three further electrons to reach the set of eight, nitrogen mustinteract with three one electron donors to give, for example, NH3; or with a one electrondonor and an atom that donates two electrons to give, for example, HNO; or with a singleatom that donates three electrons to give, for example, N2–see the schematic representationin Figure 2.4. In all cases three bonds are formed; in the first case three single bonds, inthe second a single and a double bond, and in the third, a triple bond. So a nitrogen atomalways has three bonds and a lone pair. Similar arguments pertain to the other atomsleading to the conclusion that the number of bonds around the elements in groups IV (14)through VII (17) are four, three, two, and one, respectively.
These rules are violated if an atom carries charge, either as a result of being in a moleculewith net charge, or having formal charge. The former is illustrated by NH+
4 where thenitrogen atom has four bonds and OH– where the oxygen atom has only one bond. Formalcharge is the charge on an atom calculated by adding to the valence shell nuclear charge (apositive number) the charge caused by the number of lone pair electrons and one-half thenumber of bonding electrons.
There is still another issue to consider to make this simple model applicable. In compoundswhere there is an ambiguity, which atom is the “central” atom in the structure? Thatis, should one write for CO2 the structure OOC or the structure OCO? Formal chargeanswers this question. Usually one of the choice of structures will have a formal charge thatis unfavorable. A comment on what constitutes unfavorable is needed here (and will bedeveloped later): Elements to the right and top of the periodic table should be negativelycharged and those to the left and bottom should be positively charged. If formal charge in amolecule is the other direction, the structure drawn is not favorable. For those of you likinga rule, there is one that usually, but not always, works: the element of lower ionization
Chm 118 Problem Solving in Chemistry

2.4 8
energy–the energy to remove an electron from a gaseous atom–or electronegativity is in thecentral position.
What to do in the VSEPR model when there are both bonding pairs of electrons and lonepairs of electrons present in the Lewis structure? To a first approximation, one treats thetwo kinds of electrons equivalently. So if the structure of CH4 is a tetrahedron, that of NH3
will also have electrons pairs arranged at the corners of a tetrahedron, but the hydrogenatoms only occupy three of the vertices of the tetrahedron, so the shape of NH3 is trigonalpyramidal. Likewise in H2O, the arrangement of electron pairs is tetrahedral, but thehydrogen atoms only occupy two of the vertices, so the shape of the molecule is “bent”.There is an extension of the VSEPR model to predict bond angles in compounds containinglone pairs that works reasonably, but not always; it is discussed in some of the exercises.
Finally, there is the issue of how VSEPR handles double and triple bonds. As we shallsee when we discuss bonding in a complete fashion, double and triple bonds should beviewed as a normal bond accompanied by one or two less efficient bonds. These latter are“not active” in a VSEPR sense. For instance, in the compound CH2O, there is a doublebond between the carbon atom and the oxygen atom. Therefore the number of “pairs” ofelectrons around the carbon atom from a VSEPR point of view is three; the geometry isplanar and triangular.
2.4.1 Exercises
2.4.1.1
Give the Lewis structure of H2O, SCl2, PH3, ClF, and CH3OH; convince yourself that allformal charges are zero in these compounds. Use line structures and not “dot” structures.Include lone pairs.
2.4.1.2
Show that the rule about the number of bonds holds for the compounds in the last exercise.
2.4.1.3
Draw the Lewis structure of urea, (NH2)2CO.
2.4.1.4
Draw the VSEPR structure of the compound in the last exercise. HINT: Consider eachnon-hydrogen atom separately.
2.4.1.5
Draw the Lewis structure of AlCl3, SiH4, CF4, H2S, H3O+, and NH–2.
2.4.1.6
Draw the VSEPR structure of the compounds in the last exercise.
2.4.1.7
Write Lewis structures for GeH4, AsCl3, and SeF2. Use VSEPR to determine structure.
Chm 118 Problem Solving in Chemistry

2.5 9
2.4.1.8
Write Lewis structures for NCS– where the atoms are attached as indicated and the lefthand nitrogen atom is double bonded to the carbon. Note formal charges.
2.4.1.9
Write Lewis structures for NCS– where the atoms are attached as indicated and the lefthand nitrogen atom is triple bonded to the carbon. Note formal charges.
2.4.1.10
Use the results of the last two exercises and that of exercise 5 to formulate a rule for thenumber of bonds to an element that is formally charged positively. For one the is formallycharged negatively.
2.4.1.11
Is OF2 likely to have an oxygen atom or a fluorine atom in the center? Why?
2.4.1.12
The Lewis structure of CO2 is not C=O=O, where some electrons are missing: you fill themin. What’s wrong with this Lewis structure? What is the correct Lewis structure? Why isthe one we call “correct” correct?
2.4.1.13
Is the structure of NOF as written, or is it ONF, or perhaps OFN? Prove it.
2.4.1.14
Give the VSEPR structure of your answer to the last exercise.
2.4.1.15
Consider the lone pair electrons near a nitrogen atom. Do you anticipate they will be closerto or further from the nucleus than a bonding pair of electrons between a nitrogen atomand a fluorine atom? Which then will be more dominant in pushing other electron pairsaway in a VSEPR sense?
2.4.1.16
Consider the bonding pair electrons between a nitrogen atom and a chlorine atom. Doyou anticipate they will be closer to or further from the nucleus than a bonding pair ofelectrons between a nitrogen atom and a fluorine atom? Which then will be more dominantin pushing other electron pairs away in a VSEPR sense?
2.4.1.17
Use your results from the last two exercises to predict which bond angle will be larger,the F-N-F in NF3 or the Cl-N-Cl in NCl3. The observed values are 102.2o and 107.1o,respectively.
2.4.1.18
Predict which bond angle will be larger, the F-C-F in CHF3 or the Cl-C-Cl in CHCl3. Theobserved values are 108.8o and 110.4o, respectively.
Chm 118 Problem Solving in Chemistry

2.5 10
2.4.1.19
Predict which bond angle will be larger, the Cl-C-Cl in CHCl3 or the Cl-C-Cl in CH2Cl2.The observed values are 110.4o and 111.8o, respectively.
2.5 A Slight Expansion of the Simple Model
The observant student will already have noted that we introduced BeCl2 and BF3 as ex-amples with two and three pairs of electrons, yet this is completely impossible if one insistsupon the rule of eight, the octet, of the normal Lewis structure. In this section we addressthe deviations from the octet structure that occur on both the low side (less than eight)and the high side (more than eight).
Atoms to the left of the carbon column in the periodic table often have less than eightelectrons around the central atom in compounds. Generally the number of electrons istwice the group number, so boron has six, magnesium four, etc. Also note that Lewisstructures are not particularly valid for compounds in which there is substantial chargeseparation between the atoms in the bond: Sodium chloride is not well represented by asimple Lewis structure. The reason for the deviation from the rule of eight is that thenuclear charge is small for boron (compared to C, N, O, F) and hence fewer electrons canbe attracted to that nucleus. The octet rule is “out the window” for compounds of B, Be,etc.
A more serious issue concerns compounds that violate the octet rule on the high side,have more than eight electrons in the valence shell–see the exercises. Table 2.1 gives someexample compounds. The VSEPR model for these compounds involves determining themost stable arrangement for five or six pairs of electrons around a central atom. The issueof six is easy: it is the octahedron. For five pairs of electrons, the arrangement is thetrigonal bipyramid. If this is not familiar to you, look it up on the web. The tbp is moredifficult in some cases because there are two geometrically different kinds of positions, twoenvironments. Generally the more electronegative element goes on the axial position. Ourbonding model for these compounds, developed later, will account for this rule.
2.5.1 Exercises
2.5.1.1
Give the Lewis structure and the VSEPR of BF3.
2.5.1.2
Give the Lewis structure and the VSEPR of SO2–4 (with some cation), SF5, and SiF2–
6 (withsome cation).
2.5.1.3
Give the Lewis and VSEPR structures for Cl3PO, SF4, and SF4O. HINT: In the case ofSF4 you should think about the rule for the position of the most electronegative element.
Chm 118 Problem Solving in Chemistry

2.6 11
Cmpd Color Properties Cmpd Color Properties
PF5 colorless gas, mp −84.5oC PCl5 colorless l, bp 150oCPBr5 red-yellow dec, 84oC AsF5 colorless gas, bp −52.8oCAsCl5 colorless dec. −50oC SbF5 colorless l, bp (with d) 140oCSF6 colorless gas, mp −50.8oC SF4 colorless gas, bp −40oC
SF5Cl colorless gas, mp −64oC SF5Br colorless gas, mp −79oCSCl4 colorless s, dec. −30oC SO3, colorless mp 16.8oCSO2 colorless gas bp −10oC SO3 colorless s, bp 44.4oCSeF6 colorless gas, mp −46.6oC SeF4 colorless gas, bp 102oCSeCl4 colorless solid, sublimes with dec 196oC SeBr4 orange solid and liquid only, mp 123oCTeF4 colorless solid, mp 130oC TeBr4 yellow dec with boiling at 414oCTeI4 black solid, dec with boiling at 283oC
Table 2.1: Properties of Compounds that Violate the Octet.
2.5.1.4
Give the Lewis and VSEPR structures for SiF2–6 , XeF2, and XeOF4.
2.5.1.5
In Table 2.1 are several compounds that violate the octet. See if you can find some consistent“rule” that gives you some ability to predict what species will violate the octet rule andwhat species will not.
2.5.1.6
Some more compounds that exist (i.e., can be put into a bottle) but violate the Lewis “octetrule” on the high side are SO2–
4 (with some anion), IF5, ClF3, SiF2–6 (with some cation).
Compounds that violate the Lewis “octet rule” and (therefore?) do not exist are: SI4,SiH2–
6 , S2–5 (tetrahedral form), PH5, F–
3, NF5 (all negative ions with some cation). See if youformulation from the last exercise works. If your rule needs modification, do so.
2.6 Patching the Lewis Structure Model when Needed
A situation exists where the Lewis model breaks down completely. A good example is ozone,O3. The structure of ozone is a bent molecule; it is not cyclic. If you try to do a Lewisstructure of ozone (as you should) you will find that in order to preserve the sacred octet,you must have one of the external oxygen atoms double bonded to the central oxygen atom,and the other single bonded. Should you choose the left one as the double bonded one? orthe right one? As we shall see more extensively in the next section of this course, Lewisstructures do a reasonable job of understanding relative bond lengths. The facts are thatthe single bond in H2O2 (draw Lewis structure, always) is 1.47A whereas the double bondin O2 is 1.21A; but the two bond lengths in O3 are the same, 1.278A. Note this last valueis between the value for a single bond and that for a double bond. There is no method bywhich a single Lewis structure can deal with these facts.
Chm 118 Problem Solving in Chemistry

2.6 12
Figure 2.5: Resonance in Lewis structures
The solution to this problem is to introduce a concept called resonance. The contention isthat the ozone structure with the left hand oxygen double bonded and that with the righthand oxygen double bonded should be “averaged” to get the true structure of ozone. This“averaging” takes place in your head.3 In the case of ozone, we end up saying that thebonding of an external oxygen with the central atom is by “one + two bonds divided bytwo or a bond and a half,” as illustrated with the average signs in Figure 2.5. Likewise,look at the two pictures and average the charge on the external oxygen atom. What do youget?
You must use this simplest application of resonance whenever there are two different butequivalent ways to write a structure. Try it on NO–
2. Or on cyclic C6H6, a hexagon ofcarbon atoms, each of which has one hydrogen atom attached to it. And resonance has animportant aspect other than accounting for the equivalency of the bond lengths: resonanceis stabilizing; compounds with resonance are more stable than they would be if there wereno resonance. We explore the reasons for this later in the course, but it is important tokeep this in mind as we progress. There are some situations in which the resonance occursbetween structures that are not equivalent. And example is in the ion NCS– as also shown inFigure 2.5. These require us to make judgments about which structure is more important,a process that is not always easy.
2.6.1 Exercises
2.6.1.1
Give the Lewis and VSEPR structures for NO–3, CH3C(O)O– (where the (O) means off the
chain of elements, in this case the chain is C-C-O ), and CO2–3 . HINT: If you end up with a
negative formal charge on a carbon atom in the last structure, you probably are not correct.
2.6.1.2
Draw the Lewis and VSEPR structures of NO2Cl.
3In some simple cases like the ozone case, there are “pictures” that do the averaging for you, but theseare open to mis-interpretation and confusion. Best to do it in your head.
Chm 118 Problem Solving in Chemistry

2.6 13
Figure 2.6: Structures for exercise.
2.6.1.3
Are structures 5 and 6 in Figure 2.6 resonance structures of each other? If so, which doyou think is “more important” and why?
2.6.1.4
Write Lewis structures for HOClO3, HOClO2, HOClO, and ClOH, where the chlorine atomis the central atom in the first three. HINT: The hydrogen atom is bonded to an oxygenatom in all structures.
2.6.1.5
Write Lewis structures for ClO–4, ClO–
3, ClO–2, and ClO–.
Chm 118 Problem Solving in Chemistry

Chapter 3
Using Lewis Structures toUnderstand Structure andReactivity
3.1 Bond Lengths
The distance between two atoms in a molecule is referred to as the “bond length.” Thesevary according to the Lewis structure and the position in the periodic table; since atomslower in the table have more shells of electrons; they are bigger. The Lewis structureinfluences the bond length depending on the nature of the bond between the elements,single, double or triple. Of course, as discussed above, resonance can modify these values.For instance, Table 3.1 gives the bond length of the compounds with nitrogen-nitrogenbonds.
3.1.1 Exercises
3.1.1.1
The structures of species ClOn2 for n = -1, 0, and 1 are given in Table 3.2. Do these values
make sense? Why or why not? HINT: Use chemical intuition; we have no model for one ofthese.
Molecule Bond Length, A
N2H4 1.45N2H2 1.21
N2 1.098
Table 3.1: Bond Length Data for Nitrogen Compounds.
14

3.2 15
Molecule Bond Length, A Bond Angle
ClO+2 1.408 119o
ClO2 1.475 165o
ClO–2 1.57 110o
Table 3.2: Structural Data for Various Chlorine Dioxides.
Bond Bond Energy Bond Bond Energy Bond Bond Energy Bond Bond Energy
H-H 436 C-H 416 N-H 391 O-H 463F-H 565 S-H 362 Cl-H 429 Br-H 365I-H 497 C-C 331 C-N 305 C-O 358C-F 489 F-F 158 Si-C 306 C-I 214N-N 163 O-O 146 Cl-Cl 242 Si-Si 226
Table 3.3: Bond Energies in kJ/mole for Various Single Bonds.
3.1.1.2
Predict the approximate nitrogen to nitrogen bond lengths in tetrazene, NH2NNNH2.
3.1.1.3
The P-P bond length in PH2PH2 is about 2.2A and that in elemental white phosphorous,which exists as tetrameric units of formula P4, is the same. Deduce a structure for P4 andcomment on the observation concerning the bond lengths. HINT: Be inventive and makeuse of the facts: the bond lengths in the two materials are the same, therefore the bondsare of the same kind.
3.1.1.4
The bond length found in P2 is 1.893A. Explain the difference between that and the bondlength in P4, which is about 2.2A.
3.1.1.5
The S-N single bond is about 1.74A and the S-N double bond is about 1.54A. There are fourequivalent S-N bonds in S2N2 with a bond length of about 1.6A. Formulate a reasonableLewis structure for S2N2. HINT: One of the sulfur atoms in any Lewis structure you candraw has five electron pairs around it.
3.2 Bond Energies and Enthalpy
The bond energy is the energy required to break homolytically (evenly) a chemical bondand separate the two fragments from each other. This might be represented in the generalcase as:
X2 = 2X (3.1)
Chm 118 Problem Solving in Chemistry

3.2 16
Double BondsC-C 615 C-N 616 C-O 729 O-O 498
Triple BondsC-C 811 C-N 892 C-O 1077 N-N 945
Table 3.4: Bond Energies in kJ/mole for Various Double and Triple Bonds.
This, of necessity, produced atoms or fragments with an odd number of electrons. Suchcompounds, at least when they contain atoms in the right side of the periodic table, arecalled radicals. Generally radicals are unstable species, although there are some exceptions.Two other terms useful in this discussion are diamagnetic, which for our purposes meanscompounds “with only pairs of electrons,” and paramagnetic, which means “with moreelectrons of one spin than the other.” Radicals are paramagnetic; most Lewis structures,by the very nature of having eight electrons in four pairs, are diamagnetic.
The bond energy required to break the H-H bond in dihydrogen is 436 kJ/mole. Data forsome other compounds with single bonds in the Lewis structure are given in Table 3.3.Data for compounds with double and triple bonds are in Table 3.4.
For careful work, we need to be more precise about our language concerning rupture of achemical bond. (The following is our first of many examinations of the beautiful construc-tion of the human mind called thermodynamics.) If we separate two hydrogen atoms indihydrogen from each other, we are pulling apart a stable compound; this requires thatwe put energy into the compound. Heat is one way of changing the energy of a system.The heat added to a system at constant pressure is so useful to chemists that it has a spe-cific name: Enthalpy is the heat added at constant pressure. Enthalpy has units of energyper mole, kJ/mole usually. A useful measure of enthalpy is called the enthalpy of forma-tion. This is the enthalpy change when one mole of a compound is made (formed) fromits constituent elements in their standard states. This latter is a form that has generallybe agreed upon; for instance, H2(g) at one atmosphere pressure is the standard state ofdihydrogen, and for C(s) the standard state is graphite. Any element in its standard statehas an enthalpy of formation of zero, by definition.
There are many cases where an element exists in more than one form. For instance, solidcarbon can exist as diamond. We then ask how much heat is required at constant pressure(usually of one atmosphere) to convert graphite to diamond. This quantity would be theheat of formation of diamond:
C(s, graphite) = C(s, diamond) ∆Hof = 1.9kJ/mole (3.2)
A table of enthalpies of formation is very useful in determining the enthalpy change ofchemical reactions. Because the enthalpy of a compound is independent of how we got tothat compound, one can imagine two different paths for mixing solid MgO and gaseous CO2
to form solid MgCO3. This first is direct reaction:
MgO(s) + CO2(g, 1atm) = MgCO3(s) (3.3)
Chm 118 Problem Solving in Chemistry

3.2 17
and the second is a convoluted path:
MgO(s) = Mg(s) +1
2O2(g, 1atm) (3.4)
CO2(g, 1atm) = C(s, graphite) + O2(g, 1atm) (3.5)
Mg(s) + C(s, graphite) +3
2O2(g, 1atm) = MgCO3(s) (3.6)
Imagine we desire the enthalpy change for the reaction in equation 3.3. Hess’s law statesthat the total enthalpy change for a series of reactions is just the sum of the enthalpychanges for the individual reactions–enthalpy does not depend on path. Therefore, sincethe sum of reactions in equations 3.4 to 3.6 is the reaction in equation 3.3, we have:
∆Ho3.3 = ∆Ho
3.4 + ∆Ho3.5 + ∆Ho
3.6 (3.7)
But we can write this in terms of enthalpies of formation as follows:
∆Ho3.3 = −∆Ho
f (MgO)−∆Hof (CO2) + ∆Ho
f (MgCO3) (3.8)
This is the familiar statement that the enthalpy change of a reaction is the enthalpy offormation of products minus the enthalpy of formation of reactants.
3.2.1 Exercises
3.2.1.1
Why is it harder to break the N-N bond in dinitrogen than it is to break the H-H bond indihydrogen?
3.2.1.2
What patterns do you see in the data in Table 3.3? Do you believe enough in your patternsto doubt any data?
3.2.1.3
Use the bond energies in the Tables 3.3 and 3.4 to compute the energy of the process
H2CCH2 + H2 → H3CCH3
Is the process energetically downhill? Why? HINT: Lewis structures.
3.2.1.4
The energy (actually enthalpy—heat at constant pressure) required to take P4(g) to 2 molesof P2(g) is 289 kJ/mole of P4. That required to take a mole of P2(g) to 2 moles of P(g) is485.3 kJ/mole of P2. What is the energy required to take one mole of P4(g) to four molesof P(g)? What property of energy (enthalpy) did you use?
Chm 118 Problem Solving in Chemistry

3.3 18
3.2.1.5
Compounds of the formula CaP and SrP, which are diamagnetic, have been isolated. De-scribe the bonding within these compounds. HINTS: A diamagnetic compound is one inwhich there are no unpaired electrons. Also, there is most likely a dominant ionic bondbetween the element on the left of the periodic table (charged positively) and that on theright (charged negatively). Finally, you might want to work with emphpolyatomic anions.
3.2.1.6
Which of the following molecules are unlikely to be “found in a bottle?” SFn where n runsfrom 2 to 6. Give your reasoning.
3.2.1.7
Which of the species in the last exercise are unlikely to be diamagnetic?
3.2.1.8
If you had a compound of empirical formula SF that was diamagnetic, how would youformulate the molecular structure? That is, give a valid Lewis structure.
3.2.1.9
Use enthalpy of formation data (https://en.wikipedia.org/wiki/Standard_enthalpy_of_formation) to find the enthalpy change for the process
BaSO4(s) = BaO(s) + SO3(g, 1atm) (3.9)
which is the thermal decomposition of barium sulfate.
3.2.1.10
Find the enthalpy change for the reaction
N2H4(g, 1atm) + H2(g, 1atm) = 2NH3(g, 1atm) (3.10)
HINT: The enthalpy of formation of hydrazine gas is 50.6 kJ/mole.
3.2.1.11
If you did the last exercise correctly, you should be able to articulate the last sentence of3.2 more completely and carefully. Do so.
3.3 Polarization
Looking at Lewis structures is one way to get an handle on the reactivity of molecules. Forinstance, the Lewis structure of ozone, O3, has a positive charge on the central oxygen.Having a positive charge on an atom with high ionization energy (or, if you prefer, highelectronegativity) is not a energy stabilizing situation. High ionization potential atoms wantelectrons, not a deficiency of them. In a similar fashion, reactions take place more readily,release more energy, when they occur between an atom of high ionization energy and oneof low. This is because in the bond between those atoms, say a C-F bond, the electronsare not distributed equally, but are pulled toward the fluorine atom (see the last exercises
Chm 118 Problem Solving in Chemistry

3.3 19
Reaction ∆H per mole of F atoms, kJ/mole of F
F2(g) + Cl2(g) = 2FCl(g) -25.32F2(g) + C(s, graphite) = CF4(g) -244
F2(g) + Be(s) = BeF2(s) -51312F2(g) + Li(s) = LiF(s) -612
Table 3.5: Enthalpy changes for some reactions of F2.
in section 2.4.1); this polarization is the unequal sharing of electrons in a bond. It makesthe fluorine atom negatively charged, negatively polarized, and the carbon atom positivelypolarized. This arrangement is stabilizing because of the difference in nuclear charges. Notethe Lewis structure does not convey this information. Periodic position is used to determinepolarization. Upper right elements are stronger electron acceptors than lower left ones are.
3.3.1 Exercises
3.3.1.1
What is the direction of the polarization in the following bonds: C-O, B-O, F-I, C-F, O-S?
3.3.1.2
The reaction of half a mole of O2 with Be to form BeO has an enthalpy change of -610kJ/mole, whereas that of one-third a mole of O3 with Be to form BeO has ∆H of -658kJ/mole. Comment.
3.3.1.3
Table 3.5 contains enthalpy data for reactions of gaseous F2. Comment on the data.
Chm 118 Problem Solving in Chemistry

Chapter 4
Acidity
4.1 Definitions
A substance is a strong acid if the reaction of the substance to produce a hydrogen ionand some anion (there are more general definitions of acidity, which we neglect here) occursreadily:
HA(aq) = H+(aq) + A−(aq) (4.1)
where the reaction is, in this case and most we consider in this document, in aqueoussolution, in water. Expressed in terms of the concentrations of materials at equilibrium, astrong acid is one in which the concentration of protons, [H+], and those of anions, [A–],are large compared to the concentration of the original acid, [HA]. This can be madequantitative by consideration of the so-called equilibrium constant, which is defined as:
Ka =[H+][A−]
[HA](4.2)
where the subscript “a” stands for “acidity” constant. A substance is a strong acid if Ka islarge and weak if Ka is small (both, of course, vague, relative words). Usually the featurethat contributes most to the value of Ka is the enthalpy change of the reaction.1
Values of Ka vary from about 1×108 to 1×10−50. This large range can be more easily talkedabout by using the concept of a pKa. The pKa is the negative logarithm (to the base 10)of the Ka. Therefore, a Ka of 1.0×10−5 has a pKa = 5 and a Ka of 5.6×10−7 has a pKa =6.25.
Many reactions are processes in which an acid reacts with a substance that accepts thehydrogen ion, a base, to produce an new acid base pair:
HA(aq) + B− = HB(aq) + A−(aq) (4.3)
In such reaction HA is clearly the acid and B– the base. The other useful language is thatHB is called the conjugate acid of the base B and A– is called the conjugate base of the acidHA.
1The other factor, entropy, will be introduced later in this document.
20

4.2 21
4.1.1 Exercises
4.1.1.1
What is an acid? What is a base? HINT: There are several different definitions of an acid(base). Let’s use the easiest one for now.
4.1.1.2
Write Lewis structures for SO2–2 and SO2–
3 .
4.1.1.3
Would you expect that the materials in the last exercise to be called “acids” or “bases”?
4.1.1.4
How can H2O be an acid and a base?
4.1.1.5
One tenth of a mole of an acid HX is dissolved in a liter of water and at equilibrium the[H+] = 1.33×10−3M. What is the Ka of the acid?
4.1.1.6
What is the conjugate base of the acid NH+4 ?
4.1.1.7
What is the pKa of an acid with a Ka of 4.4×10−3?
4.1.1.8
What is the Ka of an acid with a pKa of 5.9?
4.1.1.9
The general form of any equilibrium constant is the concentrations (or pressures) of theproducts, raised to their stoichiometric power, divided by the concentrations (or pressures)of reactants, similarly powered. Write the equilibrium constant for the reaction of Cu2+
with NH3 to form Cu(NH3)2+, all species in water.
4.1.1.10
Write the equilibrium constant for the reaction of Cu2+ with NH3 to form Cu(NH3)2+2 , all
species in water.
4.1.1.11
How is the equilibrium constant for the reaction of Cu(NH3)2+ to form Cu(NH3)2+2 and
Cu2+ (all in water) related to the equilibrium constants of the last two exercises?
4.2 Estimating Acidity, Part I: Nature of Element
To see what factors influence the reaction that defines acidity, let’s break it down into severalsteps for the acid HA, taking advantage of Hess’s law so that the sum of the equations given
Chm 118 Problem Solving in Chemistry

4.2 22
Group IV Group V Group VI Group VII
CH4 NH3 H2O HF55 35 15.7 3.2
SiH4 PH3 H2S HCl35 27 7.1 -7
GeH4 AsH3 H2Se HBr25 23 3.8 -8
H2Te HI2.6 -9
Table 4.1: pKa Values for Binary Hydrogen-Element Compounds.
below equals the equation for acidity of HA:
HA(aq) = HA(g) (4.4)
HA(g) = H(g) + A(g) (4.5)
H(g) = H+(g) + e−(g) (4.6)
A(g) + e−(g) = A−(g) (4.7)
H+(g) = H+(aq) (4.8)
A−(g) = A−(aq) (4.9)
We are interested in the change in the enthalpy of these processes as “A” changes, soequations 4.6 and 4.8 do not matter. Further, equation 4.4 is likely to be small, so theimportant terms are those in equations 4.5, 4.7, and 4.9. If we examine a series of compoundsof approximately the same size and shape, say HnA, then as A is changed from left to rightin the periodic table (from CH4 to NH3, ...) bond energies (and hydration (equation 4.9)should be approximately constant and those species that can tolerate negative charge moreeasily, those with the more negative values for the enthalpy change of equation 4.7, shouldbe the stronger acid. Atoms that can tolerate negative charge are those to the right in theperiodic table, with large valence shell nuclear charges, or, high electronegativity. We wouldtherefore expect that HF would be a stronger acid than H2O, which would be stronger thanNH3, etc.
If we ask what would happen to the enthalpies of equations 4.5, 4.7, and 4.9 if we movevertically in the periodic table, the situation is more complicated. Bond energies (equa-tion 4.5) decrease, become less positive, as we move down the periodic table as does theaffinity for an electron (equation 4.7), factors that increase the acidity. Hydration energies(equation 4.9) become less negative, which decreases the acidity. One way to remember thefacts is to say there are more terms increasing the acidity than decreasing it. Hence, HCl isa stronger acid that HF and H2S is a stronger acid than H2O. Acid strength when a protonis lost from an element in the periodic table increases as you move to the right and downin the periodic table. Same data are given in Table 4.1.
Chm 118 Problem Solving in Chemistry

4.3 23
4.2.1 Exercises
4.2.1.1
Is HCl or H2O the strongest acid?
4.2.1.2
The compound GeH4 reacts with NH3 in liquid ammonia to form NH+4 and GeH–
3; methanedoes not undergo a similar reaction. Which is the stronger acid, germane or methane?
4.2.1.3
If a substance HA had a particularly strong bond between the H and the A, how would theacidity of HA be affected?
4.2.1.4
If a substance HA had an A– species that was strongly solvated, how would the acidity ofHA be affected?
4.2.1.5
Which would you expect to be the stronger acid, CH3CH2OH or CH3CH2NH2? HINT:First determine which proton is likely to be removed in each species.
4.2.1.6
Which would you expect to be the stronger acid, CH3CH2OH or CH3CH2SH?
4.3 Estimating Acidity, Part II: Charge
In the reaction for an acid giving up a proton, equation 4.1, the positively charged protonis being pulled away from a negatively charged A–. Imagine the original acid was H2O.Then the proton is being removed from OH– in the process defining the acidity of water.Now consider what would happen if we thought about taking a proton away from the OH–:What charge would be left on the species left behind if a proton was removed from OH–?The answer to that question tells you the effect of charge on acidity.
4.3.1 Exercises
4.3.1.1
Which material would you expect to be the strongest acid, H3O+ (going to what?), H2O(going to what?), or OH– (and you know the extra question)?
4.3.1.2
Which material would you expect to be the strongest base, H2O (going to what?), OH–, orO2–?
Chm 118 Problem Solving in Chemistry

4.4 24
Acid pKa Acid pKa
H3PO4 2.23 H3AsO4 2.22HClO4 -7.0 HClO3 -1.30HClO2 2.95 HOCl 7.53H3BO3 9.14 HNO2 3.37HIO3 0.77 H2SO4 -2.0
Table 4.2: Some Acid Dissociation Constants for Oxyacids.
4.3.1.3
Is NH+4 or NH3 the strongest acid?
4.3.1.4
What factors should you consider to determine if H2O or NH+4 is the stronger acid?
4.3.1.5
Which would you expect to be the stronger acid, HOCH2CH2O– or OH–? HINT: DrawLewis structures.
4.3.1.6
Which would you expect to be the stronger acid, HOCH2CH2O– or HOCH2CH2CH2O–?
4.4 Estimating Acidity, Part III: Resonance
We learned earlier (see 2.6) that resonance has a stabilizing effect on the energy of a com-pound. Since the arguments we have been discussing about acidity are based on enthalpyconsiderations, something that stabilizes compounds will place an important role. Considerthe equation defining the acidity, equation 4.1. If the material HA has resonance stabilityand substance A– does not, then reactants are stabilized and HA is a weaker acid thanyou would otherwise expect. If the material A– has resonance stability and substance HAdoes not, then products are stabilized and HA is a stronger acid than you would otherwiseexpect. If both substances, HA and A– are stabilized, then you must make a judgmentabout which is stabilized the most.
4.4.1 Exercises
4.4.1.1
Which would you expect is the strongest acid, CH3CH2OH or CH3C(O)OH? Why?
4.4.1.2
Which material is the stronger acid, H2SO2, H2SO3, or H2SO4? Why? HINT: The S atomis central in all Lewis structures.
Chm 118 Problem Solving in Chemistry

4.5 25
4.4.1.3
Which material is the stronger acid, HSO–2 or HSO–
3? Why?
4.4.1.4
What is a pKa?
4.4.1.5
Table 4.2 shows the pKa for a number of “oxy acids”. Can you find a pattern that allowsa rough prediction of the acidity?
4.4.1.6
Predict the pKa of H4SiO4.
4.4.1.7
Given the information in Table 4.2, if you saw data that said that arsenous acid, H3AsO3
has a pKa of 9.23 whereas phosphorous acid, H3PO3 has a pKa of 2.00, what might youconclude?
4.4.1.8
The pK of the two acids in the last exercise really are what is listed there. They are true,not intentionally wrong. Which of these materials is “normal”?
4.4.1.9
Phosphorous acid (see exercise 4.4.1.7) forms only monoanions and dianions when reactedwith bases whereas arsenous acid form both of these and trianions. Can you account forthis and the acid strength of these species (for data see exercise 4.4.1.7)? HINTS: You needa different topology for one of them. To review, which one is odd?
4.5 Estimating Acidity, Part IV: Inductive Effects
The least important factor influencing acidity is the inductive effect. Consider the moleculesCH3CH2OH and CH2FCH2OH. The hydrogen ion that is easy to remove is on the oxygenatom in both cases. The molecules differ in that one hydrogen in the latter has been replacedby a fluorine atom. That fluorine atom attracts electrons toward itself in the bond to thecarbon atom, polarization, and that makes the carbon more positive. It therefore pullselectrons from the carbon to which it is attached, making it a little more positive. Thatsecond carbon pulls electrons better from the oxygen than normal, making the oxygen alittle more positive than normal. Hence the hydrogen ion will come off more readily.
4.5.1 Exercises
4.5.1.1
Which is the stronger acid, CF3CH2OH or CH3CH2OH? Why?
Chm 118 Problem Solving in Chemistry

4.6 26
Ion ∆H Ion ∆H
Li+ -123 Be2+ -594Na+ -97 Mg2+ -459K+ -77 Ca2+ -381Rb+ -71 Mn2+ -441Cs+ -63 Zn2+ -489Fe2+ -459 Co2+ -491Ni2+ -503 Cu2+ -502
Table 4.3: Enthalpies of Hydration (kcal/mole) for Various Cations.
4.5.1.2
Which is the stronger acid, CF3CH2C(O)OH or CCl3CH2C(O)OH?
4.5.1.3
Which is the stronger acid, CF3CH2CH2OH or CF3CH2OH?
4.6 Hydration Energy and the Acidity of Metal Ions
We saw above–section 4.2–that hydration energy plays a role in the determining of acidity.Generally the interaction of a solvent, often water, with species, especially charged ones,has a dramatic effect on chemical processes. In this section we examine the behavior ofmetal ions when they are hydrated:
Mn+(g) = Mn+(aq) (4.10)
Some data for the enthalpy change for this process are given in Table 4.3.
4.6.1 Exercises
4.6.1.1
What trends do you see in the data in Table 4.3?
4.6.1.2
Metal ions in aqueous solution typically exist as aquated species, M(H2O)n+6 . From where
would a proton be taken if the aquated metal ion were to act as an acid?
4.6.1.3
If the metal ion, M(H2O)n+6 , donates a proton to some base, what are the ligands that
remain on the resulting metal ion “complex”?
4.6.1.4
Write a chemical reaction that illustrates how a metal ion in aqueous solution, M(H2O)n+6 ,
acts as an acid.
Chm 118 Problem Solving in Chemistry

4.7 27
4.6.1.5
Write the expression for the equilibrium constant for the process in the last exercise.
4.6.1.6
Which would you expect to be the stronger acid, Cu(H2O)+6 or Ag(H2O)+
6 ? Why?
4.6.1.7
Which would you expect to be the stronger acid, K(H2O)+6 or Fe(H2O)3+
6 ? Why?
4.6.1.8
How do you account for the fact that the dominant form of V(II) in dilute acidic aqueoussolution is V(H2O)2+
6 whereas that of V(IV) is VO(H2O)2+5 ?
4.6.1.9
The changes in free energy, the energy that can be converted into work other than that ofthe pressure-volume kind, for the process
H+(g) + X−(g) = H+(aq) + X−(aq)
where X is a halogen, are -1537, -1409, -1382, and -1347 kJ/mole for X = F, Cl, Br, and I,respectively. Plot these data versus the inverse of the ionic radius of the halide ion. Canyou guess why the plot is approximately a straight line? HINT: You can treat these dataas if they were an enthalpy for now.
4.6.1.10
Think about the extrapolation to a zero value of 1/r for the plot in the last exercise. Whatphysical property is that?. Give a meaning to that intercept. HINT: Think about what youare plotting.
4.7 Distribution Diagrams
A useful tool for visualizing what is occuring in acidity reactions is a distribution diagram.To deal with this, we need to remind ourselves about the equilibrium constant for an acid:
Ka =[H+][A−]
[HA](4.2 revisited)
If we take the negative of the log (to base 10) of each side, we get
pKa = pH − log(
[A−]
[HA]
)(4.11)
where pH is the common measure of [H+], -log([H+]). The ratio of [A−][HA] clearly changes
as the pH changes. A distribution diagram shows this variation in an explicit manner byplotting the fraction of the total amount of A (sum of [A–] + [HA]) as a function of pH. Atypical plot is shown for a metal ion with a pKa of 5.0 in Figure 4.1.
Chm 118 Problem Solving in Chemistry
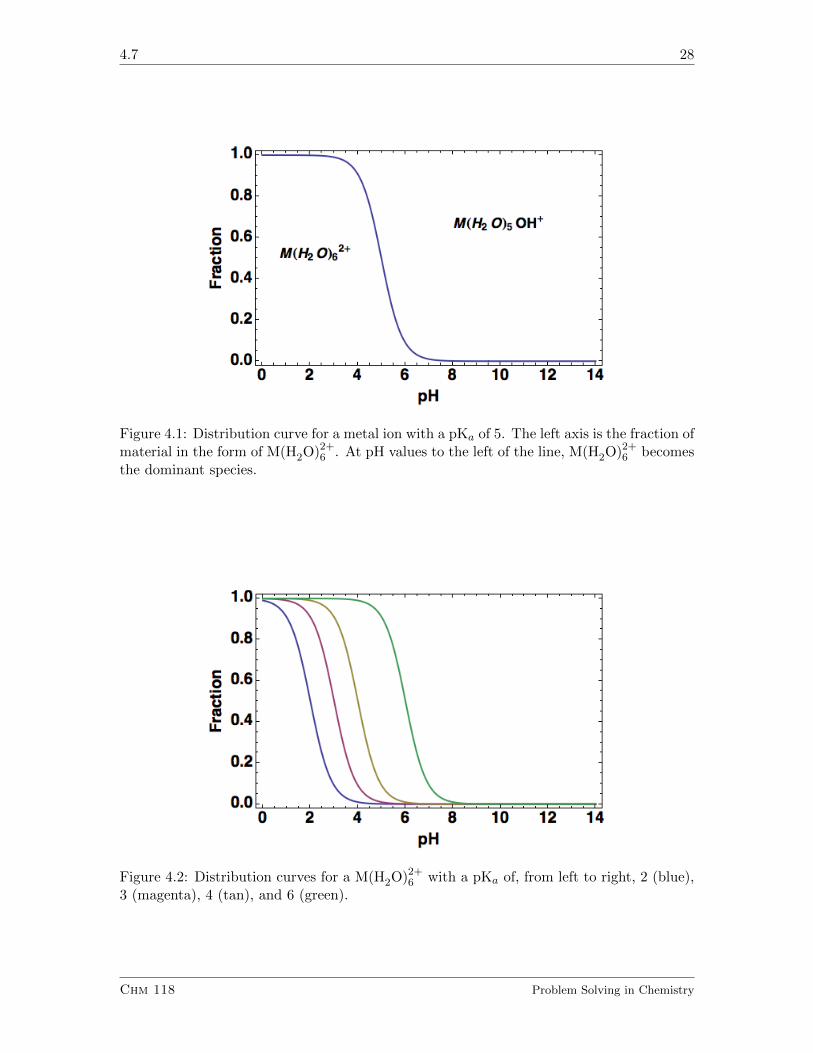
4.7 28
Figure 4.1: Distribution curve for a metal ion with a pKa of 5. The left axis is the fraction ofmaterial in the form of M(H2O)2+
6 . At pH values to the left of the line, M(H2O)2+6 becomes
the dominant species.
Figure 4.2: Distribution curves for a M(H2O)2+6 with a pKa of, from left to right, 2 (blue),
3 (magenta), 4 (tan), and 6 (green).
Chm 118 Problem Solving in Chemistry

4.7 29
4.7.1 Exercises
4.7.1.1
Looking at Figure 4.1, what fraction of the metal ion is in the form M(H2O)2+6 at a pH of
4?
4.7.1.2
Looking at Figure 4.2, see if you can determine the pH at which there are equal amounts ofM(H2O)2+
6 and M(H2O)5(OH)+ as a function of the pK. HINT: You could use the definingequation as well.
4.7.1.3
Articulate why as the concentration of hydrogen ion increases, the concentration of M(H2O)5(OH)+
decreases. HINT: You need the equation, not a distribution diagram for this.
Chm 118 Problem Solving in Chemistry

Chapter 5
Symmetry
5.1 Definitions and Proper Symmetry Operations
Symmetry is an important tool for understanding chemistry; the application of symmetrysimplifies complicated problems. At the heart of using symmetry is the symmetry operation.A practical definition of a symmetry operation is as follows. You look at an object and thenclose your eyes. Then I do something to that object while your eyes are closed. You openyour eyes. If you cannot tell anything was done, what I did was a symmetry operation.There are two kinds of symmetry operations, proper and improper. A proper symmetryoperation is one that you can do with your fingers on a model, a rotation, for instance. Animproper symmetry operation is one you can imagine, but not physically carry out, such asa internal reflection plane.
Imagine a water molecule, a bent species with one of the hydrogens to the lower left and theother to the lower right. There is a rotation by 180o about the vertical axis going throughthe oxygen atom; this rotation puts the hydrogen atom that was lower left where the lowerright one was and reciprocally. You cannot tell anything was done. This rotation is calleda C2 rotation; the “C” indicating the rotation and the “2” telling you it is one of (360/2)o.
There is another proper operation that seems a little silly, but for the theory of symmetry(called group theory) it is very important. It is the do nothing, or identity rotation. Thesymbol for this rotation is “E.”
5.1.1 Exercises
5.1.1.1
A proper symmetry operation of rotation by 180o about the z axis changes the point { x yz } to the point { -x -y z }. What does a rotation about the z axis of 90o do to the point {x y z }? See Figure 5.1 for an illustration.
30

5.2 31
Figure 5.1: Rotation of 90o about the z axis (vertical axis) in S2+4 . The sulfur atoms are
labeled with numbers so that the motion can be seen.
Figure 5.2: Improper symmetry operation, σ, on S2+4 . The sulfur atoms are labeled with
numbers so that the motion can be seen.
5.1.1.2
What would you name the rotation by 90o from the last exercise?
5.1.1.3
I contend if there is a rotation of 90o there must also be a rotation of −90o. Comment.
5.1.1.4
Find a proper symmetry operation in NO–2 other than E.
5.1.1.5
Find a proper symmetry operation in NH3 other than E. Is there a second proper operation?
5.1.1.6
Demonstrate that ethylene, C2H4, has a C2 symmetry operation; does it have more thanone? HINT: You must know where the nuclei are in space to answer this type of question.
5.1.1.7
What are the proper symmetry operations found in the square planar compound PtCl2–4 ?
Chm 118 Problem Solving in Chemistry

5.3 32
Figure 5.3: Another improper symmetry operation, σ, on S2+4 . The sulfur atoms are labeled
with numbers so that the motion can be seen.
5.2 Improper Symmetry Operations: Planes of Symmetry
There are three kinds of improper symmetry operations, which are operations that cannotbe done with your fingers on a model, but must be imagined. The first is a plane ofsymmetry. You must specify the plane (or use the your flattened hand to show where itis). If the plane is the yz plane, then a reflection plane, usually labeled σ changes the point{x y z} to {-x y z}. Consider NO–
2. This has a plane of symmetry in the molecular plane.Also, it has a plane of symmetry perpendicular to the molecular plane and containing theC2 axis.
5.2.1 Exercises
5.2.1.1
What is the plane of symmetry as illustrated in Figure 5.2? The vertical axis could be calledthe z axis. It takes one other axis to define the plane. HINT: You could use the labels onthe sulfurs to help define your plane.
5.2.1.2
What is the plane of symmetry as illustrated in Figure 5.3? The vertical axis could be calledthe z axis. It takes one other axis to define the plane. HINT: You could use the labels onthe sulfurs to help define your plane.
5.2.1.3
Is there a plane of symmetry in NH3? Specify where it is relative to a drawing (or, if youprefer, in words).
5.2.1.4
Is there a plane of symmetry in C2H4? Label it so a reader will understand, either in apicture or by words.
Chm 118 Problem Solving in Chemistry

5.4 33
Figure 5.4: Improper symmetry operation, i, on the trans form of H2O2. The atoms arelabeled with numbers so that the motion can be seen.
5.3 Improper Symmetry Operations: Center of Inversion
The second kind of improper symmetry operation is called a center of inversion, usuallygiven the symbol i. This operation takes a point with coordinates of {x,y,z} and moves itto the point {-x,-y,-z}. Or, in more prosaic terms, takes a point that is up, right, and outand converts it to a point that is down, left, and back. In Figure 5.4 is an illustration of ani on the trans form of hydrogen peroxide, H2O2.
5.3.1 Exercises
5.3.1.1
Is there a center of inversion in S2+4 , see Figure 5.2?
5.3.1.2
Is there a center of inversion in PtF2–4 , a square planar compound?
5.3.1.3
Is there an i in NH3?
5.3.1.4
Is there an i in staggered ethane, C2H6? HINT: Staggered ethane appears, looking downthe C-C bond, to have the hydrogen atoms on front carbon at, say, 0, 120o, and 240o; andhydrogen atoms on the back carbon at 60o, 180o, and 300o.
5.3.1.5
Is there an i in eclipsed ethane, C2H6? HINT: Eclipsed ethane appears, looking down theC-C bond, to have the hydrogen atoms on front carbon at the same angles as those on theback carbon.
5.4 Improper Symmetry Operations: Rotation-Reflection Axis
The last kind of improper symmetry operation is called a rotation-reflection axis, usuallygiven the symbol Sn. In this process you first rotate by an angle of 360/n, then reflect in
Chm 118 Problem Solving in Chemistry

5.5 34
Figure 5.5: Improper symmetry operation, S4, on the B2Cl4. The chlorine atoms are labeledwith numbers so that the motion can be seen.
a plane perpendicular to that axis of rotation. This procedure is illustrated in Figure 5.5.Consider a S4 about the z axis: The rotation leaves z alone and converts x to y and y to -x.Then the reflection leaves the xy coordinates of the points alone, but inverts the z. ThusS4 converts the point {x,y,z} to {y,-x,-z}.
5.4.1 Exercises
5.4.1.1
Use the logic concerning the point {x,y,z} as expressed above to show that S2 is the sameas i.
5.4.1.2
Is there a S4 axis in PtF2–4 , a square planar compound?
5.4.1.3
Is there a S3 axis in NH3?
5.4.1.4
Is there a S6 in staggered ethane, C2H6?
5.4.1.5
Is there a S6 in eclipsed ethane, C2H6?
5.5 The Group of Symmetry Operations
Molecules have a certain collection, set, of symmetry operations. That set is mathematicallycalled a group. For instance, a water molecule has E, C2 down an axis bisecting the H-O-Hbond and containing the oxygen atom, and two planes of symmetry, one in the plane of themolecule and the other a plane perpendicular to the molecular plane and containing the C2
axis. This set of four symmetry operations is labeled the C2v group, and is a set found formany molecules–we will encounter it often. Note in this set there are two proper operationsand two improper ones. The general rule is that if there are any improper operations,there will be as many improper operations as there are proper operations. Identification of
Chm 118 Problem Solving in Chemistry

5.6 35
the set of symmetry operations in a molecule is critical to applying symmetry argumentsto the molecule’s properties. For further aid in symmetry operations, see the web sitehttp/symmetry.otterbein.edu/galley/index.html.
5.5.1 Exercises
5.5.1.1
Does B2H6, called diborane, whose structure is indicated in Figure 5.6, have a plane ofsymmetry? Does it have a C2 axis of rotation? Is anything wrong with the Lewis structureof this molecule?
5.5.1.2
Find all the symmetry operations in diborane. HINT: There are eight.
5.5.1.3
Find the symmetry operations in H3PO. HINT: You may need a Lewis structure and aVSEPR analysis on this to get the structure; if so, do so, as you always should.
5.5.1.4
Comment on the sentence taken from The Little Book of Symmetry, published in 1879 byBoniface Beebe: “The following molecules all contain a C3 axis of rotation: CH4, SF6, NH3,CH2Cl2.”
5.5.1.5
What are the symmetry operations present in non-cyclic O3?
5.5.1.6
What are the symmetry operations present in cyclic O3? This group of symmetry operationsis called D3h.
5.5.1.7
Find the proper symmetry operations in benzene, a planar C6H6 molecule. HINT: Don’tforget resonance in this case, which is very important for assignment of symmetry. Why?
5.5.1.8
Find the improper symmetry operations in benzene, a planar C6H6 molecule.
5.6 The Behavior of Single Objects Under Symmetry Oper-ations
To use symmetry to learn about molecules, we have to understand how “objects” in themolecule behave when the symmetry operations are applied to the molecule. Reflect uponthis for a moment: A symmetry operation leaves the molecule unchanged; an object in themolecule, whether it is a vector, a function, a motion, are not necessarily left unchanged.
Chm 118 Problem Solving in Chemistry
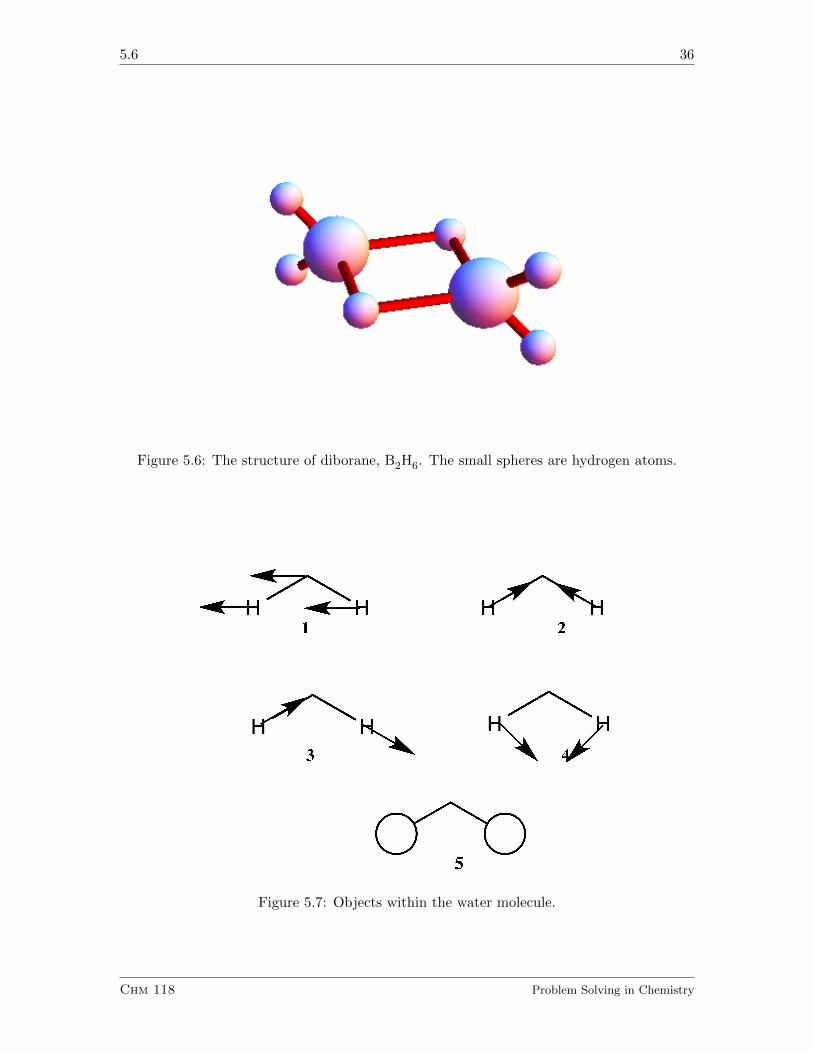
5.6 36
Figure 5.6: The structure of diborane, B2H6. The small spheres are hydrogen atoms.
Figure 5.7: Objects within the water molecule.
Chm 118 Problem Solving in Chemistry

5.6 37
C2v E C2z σplane σperpObject 1 1 -1 1 -1
Table 5.1: Table of characteristic numbers for motion of water molecule to the left.
To get correct answers about the behavior of the objects requires, foremost, that you keeptrack of how many objects you have.
As a first example, consider the water molecule and its C2v point group. In Figure 5.7,structure 1, are three vectors that I want to consider as one object. These vectors describethe motion of the water molecule as a whole to the left; so the “object” is motion of themolecule to the left. What we want to know is what happens to that object under thesymmetry operations of the water molecule? Under the identity operation, the object isleft alone; it “goes into itself.” We want to associate a number that corresponds to whathappens to the object. Under the identity operation, that number would be a one. Wewrite that as a 1 under the identity, see Table 5.1. Consider now the C2 rotation. Thisturns all three vectors of the object into minus themselves, or the object goes into minusitself. The appropriate number is “-1,” see Table 5.1. You can finish the arguments for theother two symmetry operations and get the values in the table.
5.6.1 Exercises
5.6.1.1
Find the set of characteristic numbers for the single object show in Figure 5.7 given as 2.Fill out a table as done above.
5.6.1.2
Find the set of characteristic numbers for the single object show in Figure 5.7 given as 3.Fill out a table as done above.
5.6.1.3
Find the set of characteristic numbers for the single object show in Figure 5.7 given as 4.Fill out a table as done above.
5.6.1.4
A molecule’s shape determines the symmetry operations it has. Objects in the molecule,the vectors on the hydrogen atoms, the spheres, of the last few exercises, determine thecharacteristic numbers we have been talking about. A table of the characteristic numbersfor C2v symmetry, that is, the symmetry of a water molecule, is in Table 5.2. The namesof the sets of characteristic numbers, A1, . . . B2, are the normal ones used; however, theimportant point is that they are just names and for our purposes in this course it does notmatter if you use lower case or capital letters. Use a1 as a name, nothing more (althoughthat particular name is always given to the set of numbers that are all “ones”). Experssyour answers in the last several problems like a professional: “b2 symmetry”, for instance.
Chm 118 Problem Solving in Chemistry

5.7 38
C2v E C2z σplane σperpA1 1 1 1 1A2 1 1 -1 -1B1 1 -1 1 -1B2 1 -1 -1 1
Table 5.2: Table of characteristic numbers for C2v symmetry.
C3v E 2C3 3σvA1 1 1 1A2 1 1 -1E 2 -1 0
Table 5.3: Table of characteristic numbers for C3v symmetry.
5.6.1.5
Consider the NH3 molecule. Find the symmetry operations. Do you agree with those givenin the C3v table, Table 5.3? HINT: In that table the nomenclature 2C3 implies that thereare two different rotations of 120o, both of which have the same “number” characterizingthem within any given row.
5.6.1.6
For the symmetric stretch in the NH3 molecule–where all three hydrogen atoms move awayfrom the nitrogen atom along the bond axis–treated as one object, what is the symmetry?
5.6.1.7
Sketch the structure of CH2Cl2 and find the name of the set of symmetry operations thatthis molecule has.
5.6.1.8
Find the C-H stretch in CH2Cl2, last problem, that has b1 symmetry. HINTS: Clearly theanswer must be one of the sets of characteristic numbers that you have in the table. Letthe plane defined by the carbon and the two hydrogens be the xz plane.
5.6.1.9
Find the bend of the two C-H bonds in the plane defined by HCH in CH2Cl2 that has a1
symmetry. HINTS: Clearly the answer must be one of the sets of characteristic numbersthat you have in front of you. A bend changes an angle but not a bond length. Hence thevectors must be perpendicular to bonds and, in this case, centered on the hydrogen atoms.
5.6.1.10
Find the bend (or some call it a wag) of the two C-H bonds in the plane defined by HCHin CH2Cl2 that has b1 symmetry. HINTS: Clearly the answer must be one of the sets ofcharacteristic numbers that you have in front of you. A bend changes an angle but not abond length. Hence the vectors must be perpendicular to bonds and, in this case, centeredon the hydrogen atoms. Also, as a “bend” it must change an angle.
Chm 118 Problem Solving in Chemistry

5.7 39
C2v E C2z σplane σperpObject 5 2 0
Table 5.4: Beginning of table of characteristic numbers for two spheres on the hydrogenatoms of a water molecule.
D4h E 2C4z C2z 2C‘2 2C“
2 i 2S4z σh 2σv 2σdA1g 1 1 1 1 1 1 1 1 1 1A2g 1 1 1 -1 -1 1 1 1 -1 -1B1g 1 -1 1 1 -1 1 -1 1 1 -1B2g 1 -1 1 -1 1 1 -1 1 -1 1Eg 2 0 -2 0 0 2 0 -2 0 0A1u 1 1 1 1 1 -1 -1 -1 -1 -1A2u 1 1 1 -1 -1 -1 -1 -1 1 1B1u 1 -1 1 1 -1 -1 1 -1 -1 1B2u 1 -1 1 -1 1 -1 1 -1 1 -1Eu 2 0 -2 0 0 -2 0 2 0 0
Table 5.5: Characteristic numbers for D4h symmetry.
5.7 The Behavior of Several Objects Under Symmetry Op-erations
If we have more than one object in a molecule, things are a little different than they were inthe last section. To use symmetry in these cases we have to ask how many of the objects gointo themselves, or minus themselves, and add up those numbers; and then ask how manyobjects get turned into another, in which case the object does not go into plus or minusitself, so contributes a zero to the set. Consider the two spheres, one on each hydrogenatom, show in Figure 5.7 given as 5. Clearly each of the two spheres go into themselvesunder the identity operation, so the number under that operation is a “2.” On the otherhand, under a C2 rotation, the left hand sphere goes to the right, a zero, and the right handsphere goes to the left, another zero; total, zero. These data are given in Table 5.4.
5.7.1 Exercises
5.7.1.1
Find the set of characteristic numbers for the pair of objects, two spheres, one on eachhydrogen atom, show in Figure 5.7 given as 5. Fill out the rest of Table 5.4. Note that thisset of numbers is not one of the characteristic sets for C2v symmetry.
5.7.1.2
Find the set of characteristic numbers for the pair of object show in Figure 5.7 given as 2.This is the same picture as exercise 5.6.1.1 except we are assuming we have two objects,
Chm 118 Problem Solving in Chemistry

5.8 40
Figure 5.8: Objects for vibration of atoms in a square planar molecule. See exercise 5.7.1.4for a description.
not one. Fill out a table as done above. Note that this set of numbers is not one of thecharacteristic sets for C2v symmetry
5.7.1.3
I contend that the number under the “E” symmetry operation is the number of “pictures”that you are dealing with. See if you can offer a rationalization for this contention.
5.7.1.4
Figure 5.8 illustrates two motions of atoms in a square planar molecule such as PtF2–4 .
Structure 1 contains two arrows that are one object; both the indicated atoms must moveas indicated at the same time. Structure 2 also contains two arrows that is a single object.You are to consider 1 and 2 together as two objects. The symmetry of this molecule is calledD4h and has the symmetry operations given in Table 5.5. Find the set of characteristicnumbers generated by the two objects.
5.8 Classifying the Symmetry for Multiple Objects; the Com-binations
We learned in section 5.6 that we could label the “symmetry” of an object in a moleculeif all symmetry operations turned that object into either itself (+1) or minus itself (-1) byfinding the set of numbers that those operations produced. In the last section, we foundthat multiple objects, which always produce a integer greater than one under the identitysymmetry operation, sometimes produce a set of numbers that are not to be found in thelist of all possible characteristic numbers–exercise 5.7.1.1; and sometimes produce a set thatis found in the list of all possible characteristic numbers–exercise 5.7.1.4. In the latter case,there is no issue; since the set of characteristic numbers from exercise 5.7.1.4 is that of the“Eu” row of the table, the vibration of the molecule shown in Figure 5.8 is of Eu symmetry.The contention is that in the former case, exercise 5.7.1.1, that the answer you obtained is
Chm 118 Problem Solving in Chemistry

5.8 41
Figure 5.9: Spherical objects for determination of combinations in a square planar molecule.See section 5.8 for a description.
the addition of two (or more, if the number under E is greater than 2) rows of the table.We say that the two spheres on the hydrogen atoms can be combined (in a way we shallspecify) to form an A1 combination and a B1 combination.
In group theory there is a more direct way to find the characteristicnumbers within a set of arbitrary numbers rather than by just thetrial and error procedure of adding various rows. Let χC,R be thenumber generated by the multiple objects, such as those in exer-cise 5.7.1.1 by the symmetry operation labeled C; and let χC,i bethe characteristic number for the same symmetry operation for theith row in the table of characteristic numbers. Then the numberof times that the ith entry is in the set generated by the multipleobjects, ni, is given by:
ni =1
h
∑i
nCχC,RχC,i (5.1)
where h is the total number of symmetry operations in the groupand nC is the number in front of the equivalent symmetry operationsin the characteristic table (i.e., the “2” in front of C3 in the tablefor C3v group).
The idea behind the combinations are most easily seen from what we have already done.Consider the two spheres on the hydrogen atoms of the water molecule. They generateda set of numbers that you determined in exercise 5.7.1.1. The number under the identitysymmetry operation was a 2. Whenever this is true, the combinations of the two objectsis simply the sum and the difference. In this case the sum would give a sphere on eachhydrogen atom with the same sign, say the implied positive sign that you have been dealingwith naturally. On the other hand, the difference is two spheres of equal size, but one ofwhich is positive and the other negative in sign. (We will see how important this is whenwe deal with wave functions, where the function has a sign at a given point in space.) Thecombination with both spheres positive is A1, that with one positive and the other negativeis B1.
Chm 118 Problem Solving in Chemistry

5.8 42
D3h E 2C3z 3C2 σh 2S3 3σvA′1 1 1 1 1 1 1
A′2 1 1 -1 1 1 -1
E′
2 -1 0 2 -1 0 (x, y)
A′′1 1 1 1 -1 -1 -1
A′′2 1 1 -1 -1 -1 1 z
E′′
2 -1 0 -2 1 0
Table 5.6: Characteristic numbers for D3h symmetry.
There is a formula in group theory to find the combinations. Itrequires that you take one of the multiple objects, any one, anddetermine what happens to that object under all the symmetryoperations. For instance, imagine a square with four spheres on it,such as show in Figure 5.9. What happens to sphere 1 under theidentity? It goes to sphere 1, itself; write that down. What happensto sphere 1 under a clockwise C4? It goes to sphere 2; write thatdown. Under C−4? To sphere 4. Etc. You now have this list ofspheres that we will label as Oi(s1) (which stands for symmetryoperation on sphere 1). You now use that list in the following sum,where Y is the desired combination of symmetry Γ:
YΓ =∑i
χOi,ΓOi(s1) (5.2)
and χOi,Γ is the characteristic number for the symmetry of interestfor the symmetry operation of interest. This looks complicated, butall the quantities you need are entries in the table you prepared asdirected above or in the characteristic table for the symmetry ofinterest.
5.8.1 Exercises
5.8.1.1
Show that the statement that the numbers generated in exercise 5.7.1.1 are produced byadding the A1 and B1 characteristic numbers of the C2v point group.
5.8.1.2
Place a sphere where each of the fluorine atoms is in the square planar compound PtF2–4 .
Find the set of numbers generated by these four objects.
5.8.1.3
Find the symmetries of the combinations of the four spheres from the last exercise. Useeither the trial and error method or the formula in equation 5.1. HINT: You do not needto know what the combinations look like to do this problem; but see below.
Chm 118 Problem Solving in Chemistry

5.8 43
Figure 5.10: A motion of SO3. All arrows representing motion of oxygen atoms are equalin length
5.8.1.4
Use equation 5.2 to find the combinations of two spheres (exercise 5.7.1.1) and see if itagrees with the answer we stated in section 5.8.
5.8.1.5
Use equation 5.2 to find the combinations of four spheres (exercise 5.8.1.3).
5.8.1.6
Consider the SO3 molecule. Find the symmetry operations. Do you agree with those givenin Table 5.6 for D3h symmetry? HINTS: To find symmetry operations you must know thestructure of the molecule in space. In the D3h table the nomenclature 2C3z implies that thereare two different rotations of 120o, both of which have the same “number” characterizingthem within any given row.
5.8.1.7
In Figure 5.10 is a motion of the SO3 molecule. This is one object. What is the symmetryof this (single) motion? How would you describe what is happening to the SO3 molecule?
5.8.1.8
Label the three (identical) oxygens of SO3 a, b, and c. Now for each symmetry operationfor SO3 determine how many of these oxygen atoms retain their label after the symmetryoperation has been performed. HINT: For instance, for a C2 (180o) rotation about the S-O(a) bond, O atoms b and c are not where they were, but O(a) is. So the number associatedwith that rotation is “1”.
Chm 118 Problem Solving in Chemistry

5.8 44
5.8.1.9
I contend that your answer for the last problem is made up of the sum of two of the rows ofthe D3h table. That is, under each symmetry operation, if you add the numbers associatedwith that operation for row X and row Y of the table, you will get your answer from the lastexercise. See if you can determine which those rows are, either by trial and error or by theuse of equation 5.1. Speaking professionally, you would say “The characteristic numbersgenerated by the three oxygen atoms are a
′2 and e
′′where you should substitute the correct
answers.
Chm 118 Problem Solving in Chemistry

Chapter 6
Quantum Mechanics
6.1 Probability and Quantum Measurement
Quantum mechanics is the method to determine the properties of small objects. This scienceis weird because the actions of small objects do not fit our notions of proper behavior, whichwere hewn from what we can readily observe. Two of these actions are the probabilisticnature of quantum measurements and the effect of the experiment on the result.
To illustrate these we use a Stern-Gerlach apparatus. For our purposes this is a box–seeFigure 6.1–with an inlet hole on the left and two exit ports on the right of the box, one ontop and one on the bottom. (We will want to turn the box on its side for some experiments,in which case the exit holes are in front or back.) Inside the box is a magnet (and someplumbing) that generates an inhomogeneous magnetic field. We are not going to talk aboutthis field except to say that a classical (big) magnet (with a property called a magneticmoment) passing along the magnetic field of the apparatus would be deflected either up ordown by some small or large amount: a bunch of classical (big) magnets would exit in a fanshape, including some that pass straight through. When silver atoms are passed into theStern-Gerlach apparatus via the left hand hole, they exit either from the top hole or thebottom hole. In particular, none of these silver atoms try to pass straight through and runinto the wall of the box. So here is a difference between a big and small particle right away;big ones can take any value of the magnetic moment whereas small ones seem to chooseonly one of two values. This is, of course, the quantization for which quantum mechanicsis famous.
Figure 6.1: The Stern-Gerlach apparatus
45

6.1 46
Figure 6.2: Three Stern-Gerlach magnets located in series; see section 6.1.
Now set up the following experiment. Run some silver atoms through a Stern-Gerlach (SG)device and send those that exit the top hole of the device (lets call those “spin up”) andput them into the entrance hole of a second SG device. All of them, 100%, exit the tophole of the second SG device. That makes sense. If we use the first device to choose spinup silver atoms, and then measure their spin with a second up/down SG device, we find itis spin up. The reproducibility of science; duh!
Consider a similar experiment. Take the output from the top hole of a first SG device andinput that beam into a second SG device turned on its side (so that the holes are front andback). We observe that 50% of the silver atoms come out the front hole and 50% out theback, in a random fashion. For instance, if we let one silver atom at a time go throughthe device, we would observe hits on the front and back detectors that might be like this:f,f,b,f,f,b,b,f,b,b,b,b,f,f. Over a long enough time, equal hits; random probability for eithera front hit or back hit for any given silver atom. All of this could be explained classically.We simply would say that the first silver atom (in the list just above) has spin up (hencethrough the upper hole of the first SG device) and spin front (hence through the front holeof the second SG device).
If we do the experiment shown in Figure 6.2 life gets a little more messy. Here we select upspin silver atoms from an initial SG magnet, pass these into a second SG magnet alignedfront/back, and select back spin silver atoms and pass those through a third SG magnetoriented up/down. If our conclusion at the end of the last paragraph was correct, we wouldexpect the silver atoms exiting the second SG magnet to have spin up/back and to come outof the upper hole of the third SG magnet. What happens is that there is a 50% probabilityof getting a silver atom out of the upper hole, but also a 50% chance of getting one out ofthe bottom hole of the third magnet. In the language of quantum philosophy, making thefront/back measurement in the second magnet removes all traces of the results of the upspin resulting from the first magnet.
6.1.1 Exercises
6.1.1.1
A silver atom is directed to a Stern-Gerlach apparatus arranged in the up/down direction.What is the probability that the Ag atom will come out of the upper tube?
6.1.1.2
A silver atom that came out of the upper tube of the apparatus in the last exercise isdirected to another Stern-Gerlach apparatus arranged in the up/down direction. What isthe probability that the Ag atom will come out of the upper tube?
Chm 118 Problem Solving in Chemistry
6.2 47
Figure 6.3: Two cosine waves
6.1.1.3
A silver atom that came out of the upper tube of a SG apparatus is directed to a SGapparatus arranged in the front/back direction. What is the probability that the Ag atomwill appear from the “front” tube?
6.1.1.4
Quantum mechanics is weird, but not illogical. Try this: A silver atom from the upper tubea SG apparatus in the up/down orientation is directed into a SG apparatus that is tilted0.001 degrees toward the front/back position; that is, it is almost an up/down apparatus.What is the probability a Ag atom will come out of the upper tube of the second apparatus?HINT: I am not looking for a numerical answer, but a statement showing understanding.
6.1.1.5
Quantum mechanics is weird, but not illogical. Try this: A silver atom from the uppertube of an up/down SG apparatus is directed into a SG apparatus that is tilted 45 degrees(toward “front/back” with the “upper” hole becoming the “front” hole) relative to the firstSG magnet; that is, it is between a situation where the probability of the silver atom comingout the upper tube is 1.0 and the situation where the apparatus is in a front/back positionand the probability is 0.5 from either hole. What would you guess that the probability a Agatom will come out of the “upper” tube (or “front” tube, depending on your perspective)of the second apparatus? HINT: See the last hint.
6.2 Waves and Interference
The mathematics of quantum mechanics use a wave equation to describe a particle. A waveis something that extends throughout a region of space and has values at various points inspace. For instance, the wave function Sin[π x], has a value of zero at x = 0; a value of 1 atx = 0.5, and a value of zero at x = 1. In fact, we could simply plot Sin[π x] as a functionof x and see what value it has at any point in space. Note a function is just a recipe for
Chm 118 Problem Solving in Chemistry

6.3 48
Figure 6.4: The wave obtained by adding Cos[x] + Cos[2x]
finding a value at a point in space. The wave is not anything “real” for this description of aquantum particle, but the square of that wave gives the probability of finding the particleat the respective point in space.
Where the distinction between wave and probability becomes critical is when there are twosources of the “particle,” say from one or the other of the two slits in a two slit experiment.The quantum mechanical rules for dealing with this situation (which are supported ascorrect because they always give the correct answer) is to add the waves and then squareto get the probability. This procedure gives the possibility of interference, where the twowaves add either to make a larger wave or to partially or completely cancel. Figure 6.3 isa plot of the wave function, the wave amplitude, versus x for two cosine waves; the solidline is that for Cos[x] and the dashed is for Cos[2x]. Look at x = 0.5; at this point thereis constructive interference as both waves are positive. On the other hand, at x = 3 thereis dominantly destructive interference. We can show this directly by actually plotting thesum of the two waves as is done in Figure 6.4.
6.2.1 Exercises
6.2.1.1
What is the principle way in which waves that you have seen in life (those are for mostpeople waves in water) differ from a particle?
6.2.1.2
Do you understand the most fundamental problem with quantum mechanics when one saysthat particles are described by waves?
6.2.1.3
Consider the wave functions Sin[π x] and Sin[π x + π]. What is the nature of the interferenceof these waves, constructive or destructive?
Chm 118 Problem Solving in Chemistry

6.4 49
6.3 Simple Version of the Rules of Quantum Mechanics
The rules for the application of quantum mechanics require that the wave function discussedabove be manipulated by an operator. Operators, O, in the mathematical sense used hereare objects that change functions. For instance, a trivial operator could be the number “3,”which triples a function,
O(2x+ 1) = 3(2x+ 1) (6.1)
while a more subtle operator could be the differential operator,
O(2x+ 1) =∂
∂x(2x+ 1) = 2 (6.2)
Often in quantum mechanics we are looking for an operator that changes a function into amultiple of itself, for instance,
Oψ = cψ (6.3)
where ψ is the wave function and c is a number. When we have an operator and a functionthat fulfill this relationship, it is called an eigenfunction/eigenvalue problem. In particular,a satisfactory wave function for a one dimensional problem where the energy is constantsatisfies the eigenfunction/eigenvalue relationship
− ~2
2m
∂2
∂x2ψ + V (x)ψ = Eψ (6.4)
where ~ is Planck’s constant divided by 2π, m is the mass of the object, the quantumobject, a parve,1 V(x) is the potential energy the parve experiences, which may change asthe parameter x is changed, and E is the stationary state energy. This equation is calledSchrodinger’s equation in one dimension.
6.3.1 Exercises
6.3.1.1
Show that the operator ∂∂x on the function e−kx, where k is a constant, is an eigenfunc-
tion/eigenvalue problem. What is the eigenvalue? HINT: If you don’t know calculus, skipthis exercise.
6.3.1.2
Show that the operator ∂∂x on the function Sin(k x), where k is a constant, is not an
eigenfunction/eigenvalue problem. HINT: If you don’t know calculus, skip this exercise.
6.3.1.3
Show that the operator ∂2
∂x2 on the function Sin(k x), where k is a constant, is an eigenfunc-tion/eigenvalue problem. HINT: If you don’t know calculus, skip this exercise.
1A parve is a name unique to this document
Chm 118 Problem Solving in Chemistry

6.4 50
6.4 A Simple but Useful Quantum Problem: The Parve ona Pole
In this section we solve a quantum problem. It is a particularly easy problem to solve, yeta number of the features of quantum behavior are illustrated by it. Further, the results ofthis problem will be useful to us throughout the rest of this course, reaching all the wayinto thermodynamics. The problem at hand is a particle, a parve, moving in one dimensionalong a pole of length L with no potential energy present. Schrodinger’s equation for thissituation is given by equation 6.4 with V(x) = 0. In addition, I want to demand that theparve stay on the pole, so I am going to insist that the potential energy outside the endsof the pole be infinite, so there is no possibility that the parve be there. We need to findfunctions that satisfy this equation. I suggest that you learned in exercise 6.3.1.3 that Sin(kx) would satisfy Schrodinger’s equation in this case, as will the functions Cos(k x) and e−kx,where k is a constant. Which of these three solutions is proper for the quantum problem?
That the wave function must satisfy Schrodinger’s equation is one consideration. Thesecond, and an important one, is that the boundary conditions must be met. Here ishow that plays out in the parve on a pole, POP, problem. A second demand of quantummechanics is that the wave functions must vary smoothly in space, from one point to another.We know from our demand that there is no probability for the parve to be outside the polethat Px<0 = 0. The probability is given by the square of the wave function for the parve,ψ, so Px<0 = ψ2 = 0; which requires the ψx<0 be zero. Since the wave function must varysmoothly with a change in x, this demands that ψx=0 = 0. That is the boundary condition.As you will show in the exercises, of the three functions that satisfy Schrodinger’s equation,only one satisfies this boundary condition.
There is a second boundary to our pole, the end where x = L. Outside this end of the pole,the probability must also be zero, as must, therefore, the wave function. This demands asecond requirement on our function, that Sin(k x) = 0 at x = L; or (k L) = nπ, wheren is an integer. Look at that! The boundary condition introduces a quantum number,n. We conclude that a legal (in that it obeys quantum rules) wave function for a POP isψ = Sin(n π x/L). A quantum problem solved!
6.4.1 Exercises
6.4.1.1
Show that the wave function Cos(k x) and e−kx do not satisfy the boundary condition thatψ = 0 at x = 0.
6.4.1.2
Take equation 6.4, set V = 0, use the ψ from the end of the last section, and show that theenergy for a POP is given by n2~2π2
2mL2 . HINT: If you don’t know calculus, skip this exercise.
Chm 118 Problem Solving in Chemistry

6.5 51
6.4.1.3
The lowest energy wave function for a parve on a pole of length L is a sine wave runningbetween x = 0 and x = L and spanning half a wavelength in this interval. Use the deBroglie relationship, p = h
λ , where p is the momentum, p = m v, and solve the equationsfor the energy of this parve. HINTS: All educated people (that means you!) should knowdeBroglie’s equation. Also, all the energy of this parve is kinetic energy (E = 1/2 m v2).
6.4.1.4
A parve is trapped on a pole with zero potential energy. What happens to the lowestallowed energy of the parve if the length of the pole is doubled?
6.4.1.5
Sketch the n = 5 wave function for a parve on a pole.
6.4.1.6
Given the de Broglie postulate in problem 6.4.1.3, what can you say about the kinetic energyof a parve on a pole in the n = 5 level compared to the n = 1 level? Explain without acalculation by thinking about the de Broglie postulate.
6.4.1.7
Compute the energy an electron would have if it was trapped on a pole of length 4.2 A(about the length of a four carbon fragment) in the n = 1 level using the POP model.HINTS: You need to look up some constants and expand units like the Joule in terms ofkg, m, and sec.
6.4.1.8
What energy would it require to excite the electron from the n = 1 to the n = 2 level inthe system of the last problem?
6.4.1.9
What wavelength of light must be absorbed to provide the energy to excite the electronfrom the n = 1 level to the n = 2 level in the system of the last two problems? HINT:You should know (or learn and then know) the relevant equation, one of Einstein’s severalequations.
6.4.1.10
Given that you know what the wave function for the ground state of a POP looks like,make a guess about what the wave function for the ground state of a parve on the surfaceof a square table looks like. If you are a reasonable artist, you might be able to draw theshape of the wave function in the third dimension given the surface is defined by x and y;you certainly should be able to move your hands over a table to outline roughly the shapeof the wave function for a parve on a surface.
6.4.1.11
Make a guess for the wave function for a parve in a room, a three dimensional problem.You will not be able to draw it (since that requires four dimensions), but should be able todescribe it. How many quantum numbers will be necessary for this wave function?
Chm 118 Problem Solving in Chemistry

6.6 52
6.5 Quantum Mechanics of the Hydrogen Atom I. EnergyLevels
The wave functions and the energy levels for a hydrogen atom can be solved exactly usingquantum mechanics. This solution is mathematically messy, so we won’t do it here, butthe results are worth discussing for their use in understanding all the other atoms of theperiodic table. Two features of importance to us come out of any quantum problem, theenergy of the allowed quantum levels and the nature of the wave functions. In this section,we discuss allowed energy levels.
The allowed energy levels of the hydrogen atom are given by a particularly easy equation,
En = −[m
2
] [ e2
4πε0~
]21
n2(6.5)
where e is the charge on the electron and m is the mass of the electron. Notice that all theconstants in the two square brackets are known, so this equation can be written numericallyas:
En = −13.6011
n2(6.6)
in units of electron volts, eV. The energy of the allowed levels in the hydrogen atom dependinversely on the square of the quantum number n, which is an integer varying from oneupward. Thus the most stable level is -13.601 eV (n = 1), the next most stable -3.4 eV (n= 2), etc. These energies differences agree perfectly with the positions of the wavelengthsof absorbance and emission of energy of the hydrogen atom (which move an electron to orfrom higher energy levels to lower ones), verifying the results of the quantum calculation.
6.5.1 Exercises
6.5.1.1
How many quantum numbers are there in the hydrogen atom? Why? HINT: Think aboutwhat introduces quantum numbers.
6.5.1.2
Light from the sun is partially absorbed at 656 nm. Show that this is consistent withhydrogen atoms on the surface of the sun being excited from the n = 2 level to the n = 3level.
6.5.1.3
There are a whole “series” of wavelengths that fail to reach earth from the sun because ofexcitations from the n = 2 level of hydrogen atoms to more excited levels. Calculate a fewof these. NOTE: This series is called the Balmer series.
6.5.1.4
The ionization energy is the energy necessary to remove an electron from an atom in thegas phase. What is the normal ionization energy of a hydrogen atom?
Chm 118 Problem Solving in Chemistry

6.6 53
6.6 Quantum Mechanics of the Hydrogen Atom II. RadialWave Function
The discussion of wave functions for a hydrogen atom is difficult because of our inabilityto handle multidimensional space. We can deal with one or two dimensional spaces easily,and with three dimensional space if we are careful. However, to visualize a hydrogen wavefunction we would need to plot the value of the function, ψ, as a function of x, y, and zpositions in space: that takes four dimensions, with which few of us can easily accommodate.The method of choice to avoid this problem is to plot the wave functions under some sortof restrictive condition and then to let your imagination run free to accommodate the truefunction. In this section we give one view of the hydrogen atom wave functions. It is fromthese pictures that we approximate more complicated atoms. We note here that the threequantum numbers for hydrogen atoms are usually given by a combination of numbers andletters. The principle quantum number, n, is an integer starting at 1. The second quantumnumber, usually called `, has values running from 0 to n-1, but is usually symbolized, forhistorical reasons, by a letter code: The code is, ` = 0, s; ` = 1, p; ` = 2, d; ` = 3, f.The third quantum number, m, has values from -` to `, but is often discussed in a mannerbeyond what we cover here, in terms of Cartesian coordinate symbols. For instance, thethree m values for ` = 1, which rigorously are -1, 0, and 1, are often labeled x, y, and z; andthe five m values for ` = 2, which are -2, -1, 0, 1, and 2, are called xy, xz, yz, x2-y2 and z2.
We will not deal with any of this with mathematical rigor, but foryour enlightenment we discuss one wave function in detail. A typicalwave function is that with quantum numbers (n, l, m) of (3, 1, 0),or what is usually called the 3pz wave function. It has the followingform:
ψ3pz = C(z/r)(1/a)3/2(1− r
6a)r
ae−r3a (6.7)
where C and a are constants, r is the distance from the nucleusto the electron, and z is the z component of that vector length.Notice that the z direction is favored by this function (when z =0, the function disappears) and that the function gets smaller asthe distance from the nucleus increases because of the exponentialdecrease in the last term.
Radial wave functions give how the value of the wave function changes as you move in astraight line away from the nucleus in some arbitrary direction (although as you will see inthe exercises, certain choices of direction lead to odd results). The results given here aresimply what comes out of solving Schrodinger’s equation; They are by no means intuitive.Radial wave functions depend on the quantum numbers n and ` but not on the quantumnumber m. There are distinct patterns that evolve as n and ` are changed. The wavefunctions are shown in Figures 6.5 to 6.8, and are best viewed in color.
Another important feature of radial functions involves the square of the function, which, aswe have discussed, gives the probability. Want we will need, especially for our discussion ofmulti-electronic atoms is the radial distribution curves which give the probability of findingthe electron at a given distance from the nucleus. Since the number of possible volumes
Chm 118 Problem Solving in Chemistry

6.6 54
Figure 6.5: The radial wave function for the n = 1 electron of hydrogen atom. Disregardthe vertical scale.
Figure 6.6: The radial wave functions for the n = 2 electron of hydrogen atom. Disregardthe vertical scale
Figure 6.7: The radial wave functions for the n = 3 electron of hydrogen atom. Disregardthe vertical scale
Chm 118 Problem Solving in Chemistry
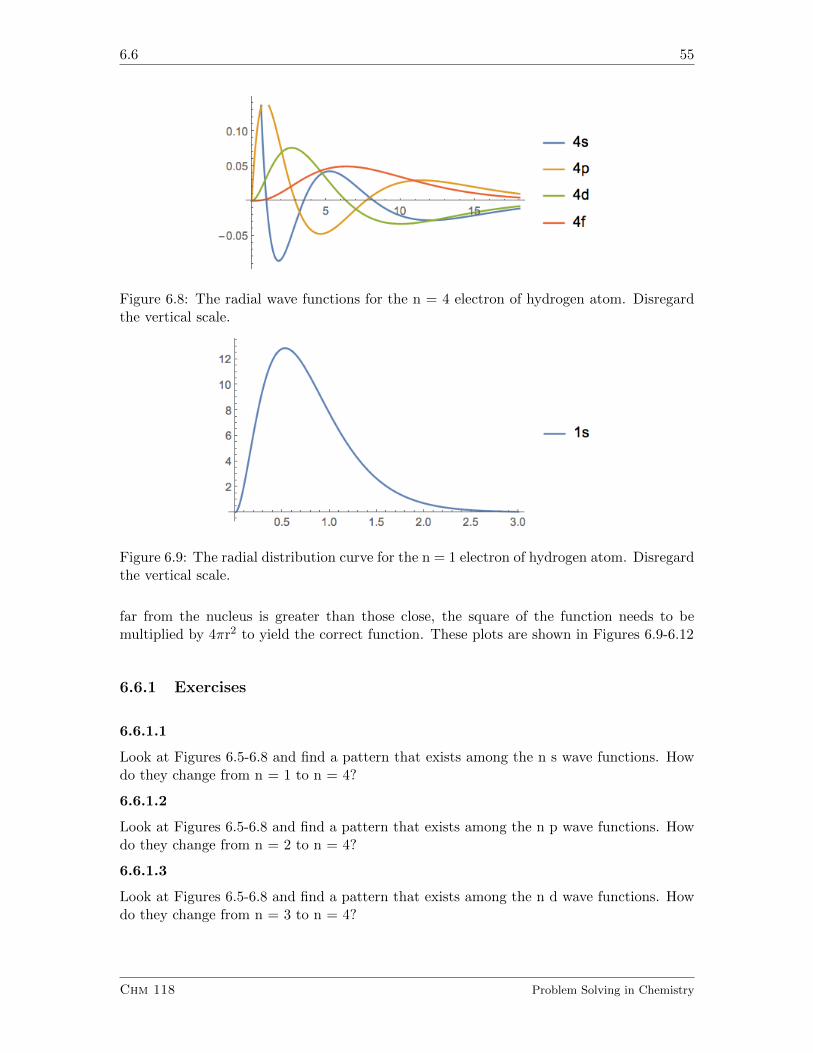
6.6 55
Figure 6.8: The radial wave functions for the n = 4 electron of hydrogen atom. Disregardthe vertical scale.
Figure 6.9: The radial distribution curve for the n = 1 electron of hydrogen atom. Disregardthe vertical scale.
far from the nucleus is greater than those close, the square of the function needs to bemultiplied by 4πr2 to yield the correct function. These plots are shown in Figures 6.9-6.12
6.6.1 Exercises
6.6.1.1
Look at Figures 6.5-6.8 and find a pattern that exists among the n s wave functions. Howdo they change from n = 1 to n = 4?
6.6.1.2
Look at Figures 6.5-6.8 and find a pattern that exists among the n p wave functions. Howdo they change from n = 2 to n = 4?
6.6.1.3
Look at Figures 6.5-6.8 and find a pattern that exists among the n d wave functions. Howdo they change from n = 3 to n = 4?
Chm 118 Problem Solving in Chemistry
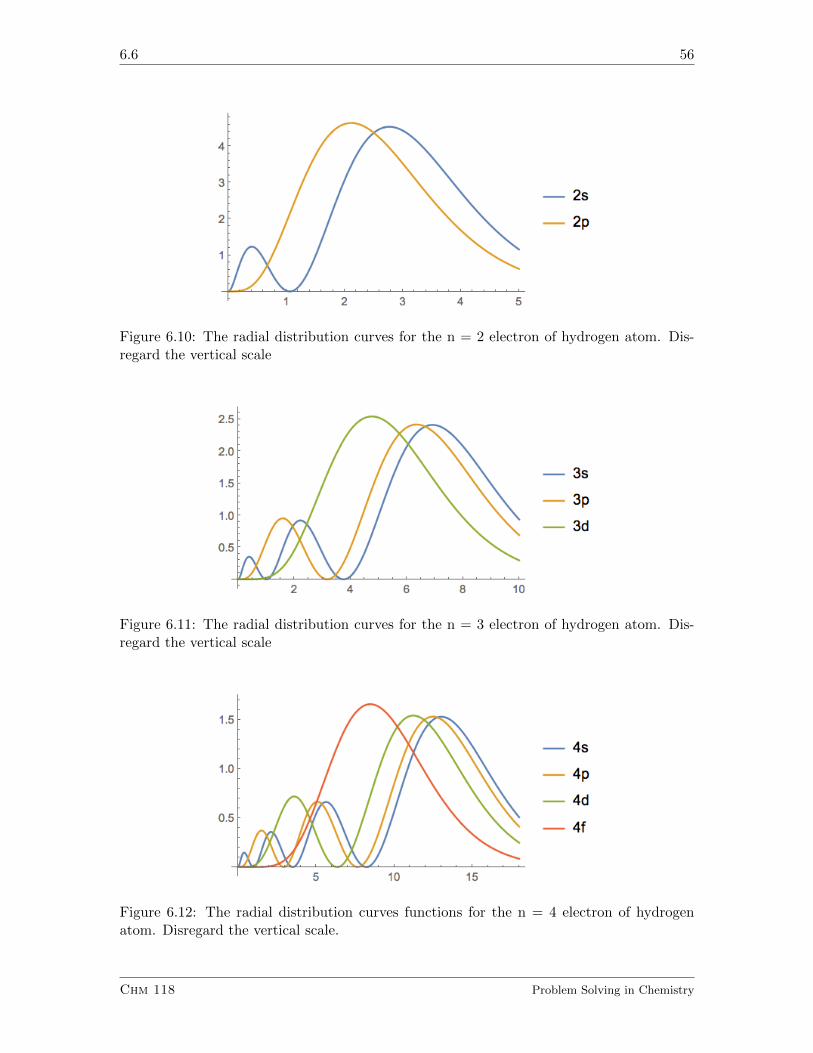
6.6 56
Figure 6.10: The radial distribution curves for the n = 2 electron of hydrogen atom. Dis-regard the vertical scale
Figure 6.11: The radial distribution curves for the n = 3 electron of hydrogen atom. Dis-regard the vertical scale
Figure 6.12: The radial distribution curves functions for the n = 4 electron of hydrogenatom. Disregard the vertical scale.
Chm 118 Problem Solving in Chemistry
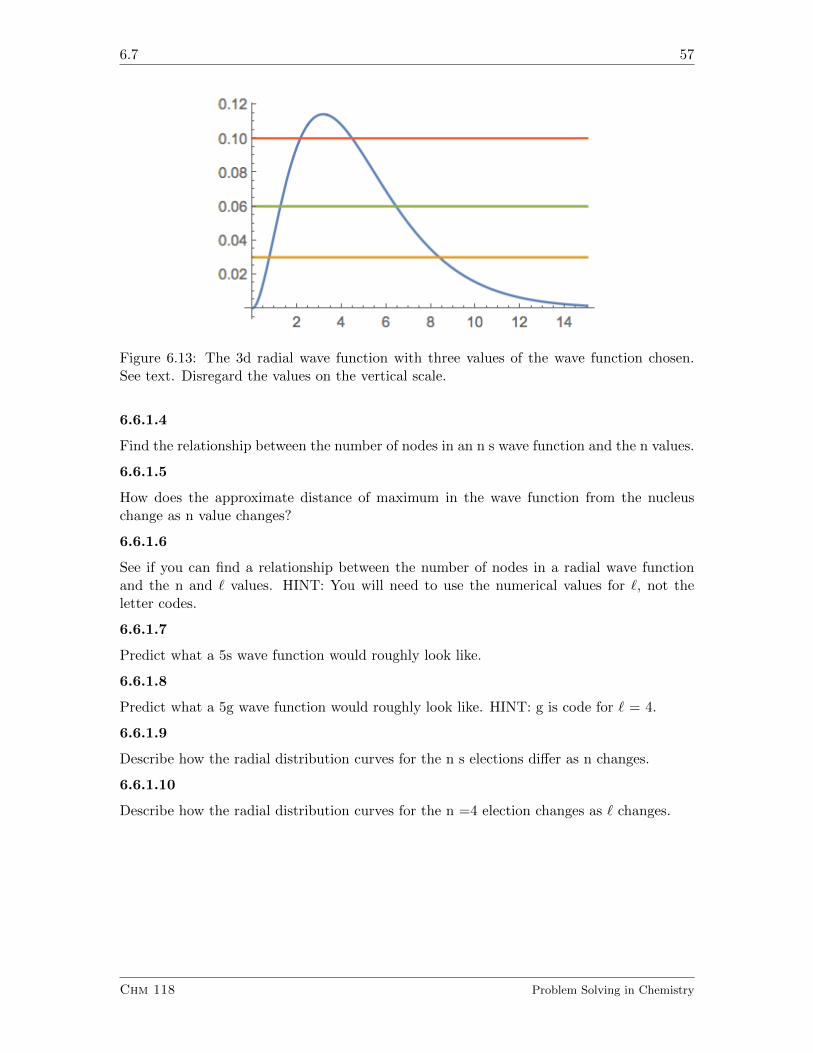
6.7 57
Figure 6.13: The 3d radial wave function with three values of the wave function chosen.See text. Disregard the values on the vertical scale.
6.6.1.4
Find the relationship between the number of nodes in an n s wave function and the n values.
6.6.1.5
How does the approximate distance of maximum in the wave function from the nucleuschange as n value changes?
6.6.1.6
See if you can find a relationship between the number of nodes in a radial wave functionand the n and ` values. HINT: You will need to use the numerical values for `, not theletter codes.
6.6.1.7
Predict what a 5s wave function would roughly look like.
6.6.1.8
Predict what a 5g wave function would roughly look like. HINT: g is code for ` = 4.
6.6.1.9
Describe how the radial distribution curves for the n s elections differ as n changes.
6.6.1.10
Describe how the radial distribution curves for the n =4 election changes as ` changes.
Chm 118 Problem Solving in Chemistry

6.7 58
Figure 6.14: The surface with all values equal to a small number, both positive (yellow)and negative (blue), for a 3dx2−y2 angular wave function.
Figure 6.15: The surface with all values equal to a moderate number, both positive (yellow)and negative (blue), for a 3dx2−y2 angular wave function.
Chm 118 Problem Solving in Chemistry

6.7 59
Figure 6.16: The surface with all values equal to a large number, both positive (yellow) andnegative (blue), for a 3dx2−y2 angular wave function.
6.7 Quantum Mechanics of the Hydrogen Atom III. AngularWave Function
The radial wave function shows how the wave function changes magnitude and sign as onemoves out from the nucleus. The angular wave function gives a sense of how the wavefunction changes magnitude as one moves around the nucleus in an angular sense. Thereare several ways of illustrating this. For our purposes, we are going to look at all places inspace where the wave function has the same value and connect those points with a surface;we will do this for both positive and negative values of the same magnitude. Again, theseresults come from solution to Schrodinger’s equation; you simply must learn them as theyare the key to bonding in chemical compounds. The angular functions depend on thequantum numbers ` and m, and not on the quantum number n.
The concept is illustrated by looking at the function named dx2−y2 . In Figure 6.13 we givethe radial function for a 3d electron. On that diagram are three lines indicating variousvalues of the function. Note that each line crosses the function twice. Consider now the linewith the lowest value on the absissa; we take that value, call it η, as the one of interest. Ifwe now change the angle and find the coordinates where the value of the function is η, andthen connect all the points of that value, we get a picture like shown in Figure 6.14. Notethat the surface is “large” because the values of r where the function has value of η are farapart. Now consider the middle line in Figure 6.13 and the surface corresponding to itsvalue, shown in Figure 6.15. The surface now is somewhat smaller. Finally, the upper linein Figure 6.13 gives the surface shown in Figure 6.16. It is possible to plot these isosurfacesbecause we ask where in three dimensional space the value of the function has a certainfixed value. This is the nature of the angular functions that we will use in this course.
So what do the angular functions look like? The s function is spherical, as shown in
Chm 118 Problem Solving in Chemistry

6.7 60
Figure 6.17: The surface with all values equal to a number for an s wave function.
Figure 6.17. Sometime people say this function is like an onion in that there are the layerswe discussed above–Figures 6.14-6.16–one after another as you move toward the nucleus.There are three p functions, usually called px, py, and pz. Figure 6.18 gives the shape ofa px function. It doesn’t take much imagination to determine what py and pz look like; infact, the subscript on the value of the letter corresponding to ` exactly gives the directionin which the function is big.
This last relationship holds for most of the d orbitals as well. There are five of these, dxy,dxz, dyz, dx2−y2 , and dz2 , For instance, the dxz orbital is big where the function xz is big,along the diagonals of the in the xz plane. Figure 6.19 illustrates this function. The otherfunctions that are simple product functions of two variables are similar, and dx2−y2 has beenillustrated above in Figure 6.16. The function that appears unique (it is not really, but toshow why not requires more math than we want to do here, although later in the coursewe shall attack it) is dz2 , which is illustrated in Figure 6.20. All of the angular functionsthen are defined by the “letters” that are used to describe the m quantum number. Eventhe correct signs flow from these values.
6.7.1 Exercises
6.7.1.1
Make a sketch of the 2py angular wave function. Make the sketch when you are standingon the positive z axis.
6.7.1.2
Make a sketch of the 2py angular wave function. Make the sketch when you are standingon the positive y axis. HINT: The ability to do such drawings will be useful when we takeup bonding.
Chm 118 Problem Solving in Chemistry

6.7 61
Figure 6.18: The surface with all values equal to a number, both positive (yellow) andnegative (blue), for a px angular wave function.
Figure 6.19: The surface with all values equal to a number, both positive (yellow) andnegative (blue), for a dxz angular wave function.
Chm 118 Problem Solving in Chemistry

6.8 62
Figure 6.20: The surface with all values equal to a number, both positive (yellow) andnegative (blue), for a dz2 angular wave function.
6.7.1.3
Make a sketch of the 3dxy angular wave function. Make the sketch when you are standingon the positive z axis.
6.7.1.4
Make a sketch of the 3dxy angular wave function. Make the sketch when you are standingon the positive y axis.
6.7.1.5
How many angular nodes does a 3dxy wave function have? A 2pz? Describe the surfacesthat defines these nodes. Do the same for a 3dz2 wave function. What is the relationshipbetween the number of angular nodes and the ` quantum number?
6.7.1.6
Is there any relationship between the total number of nodes in a wave function and somequantum number or combination thereof?
6.7.1.7
What is the difference between a 2px and a 2py wave function?
6.7.1.8
Describe the “shape” of a fxyz orbital. HINT: My intent is that you use the “name” to figureout the shape; looking up the shape on the web is ok, but then you need to memorize it.
Chm 118 Problem Solving in Chemistry

6.8 63
6.7.1.9
Describe the difference between the dxy orbital and the dx2−y2 orbital.
6.8 Multi-electronic Atoms, Configurations
We use the quantum mechanical results for the hydrogen atom to generate a model tounderstand multi-electronic atoms. There are two factors that go into this model: thewave functions that the last couple of sections have described, and the Pauli principle. Thefeature of the wave functions that we accented previously is that the quantum number nis a rough measure of how far an electron is from the nucleus; this will remain important.However, we will use a statement of the Pauli principle that differs from our previous one(section 2.2): “No two electrons can have all four quantum numbers the same.”
We have seen the three quantum numbers that characterize a hydrogen atom electron, n, `,and m. The fourth quantum number that we need for multi-electronic atoms is the spin ofthe electron, usually called mS . Our application then goes as follows. The element with twoelectrons, He, can put both electrons close to the nucleus, n = 1, into the 1s wave function(called by chemists an “orbital”) if one of those electrons has spin up and one has spindown. We cannot put a third electron this close to the nucleus because we have “run out ofquantum numbers,” so that next electron must go into the n = 2 level and we get a lithiumatom with the configuration of 1s22s1. (We postpone putting forward the argument whythe configuration favors 2s1 rather than 2p1 for a while.) As we continue to add anothernuclear charge and another electron we move horizontally across the periodic table and getthe configurations of the elements from Be 1s22s2 to B 1s22s22p1 to Ne 1s22s22p6 where inthe last case we have populated all three of the 2p orbitals, 2px, 2py, and 2pz, each with twoelectrons, one of spin up and one of spin down. The Pauli principle now forbids any moreelectrons with n = 2, so we are forced to go to the n = 3 level and get Na, 1s22s22p63s1,and add electrons and nuclear charges as before until we arrive at Ar with the configuration1s22s22p63s23p6. Going further requires that we understand the filling of 2s before 2p, sowe turn to that next.
The concept we need to apply to understand why the 2s level is filled before 2p in theelectron configuration is called penetration. It is based on Gauss’ law: To a test chargeoutside the surface of a sphere, the charge on that surface acts as if it was located at thecenter of the sphere. Now consider Figure 6.21 for a lithium atom and imagine a 2s electronis three units away from a nucleus that has two 1s electrons. That pair of electrons arebetween the 2s electron and the nucleus, so they act as if they were at the nucleus; thus thenet charge on the nucleus is +3 -2 = 1. The same is true of a 2p electron at this distance.Now consider a 2s electron at a distance of 0.5 units from the nucleus. Some of the chargeprobability for the pair of 1s electrons is “outside” that distance, and so has no effect onthat 2s electron. It therefore “sees” a much greater positive charge. Looking at the 2pcurve, we observe that it does not get very close to the nucleus, and is effectively screenedby the pair of 1s electrons. State this whatever way you wish: 2s penetrates better andhence experiences a greater positive charge and a more stable orbital than 2p; or, the 2pelectron is better screened from the nuclear charge by the pair of 1s electrons and hence
Chm 118 Problem Solving in Chemistry

6.8 64
Figure 6.21: The radial probability distributions for 1s, 2s, and 2p wave functions showinghow a 2s electron has a larger probability of getting into the volume occupied by a 1selectron than does a 2p electron. The shaded area under the 1s curve is present just tohelp illustrate the penetration. Note that the 1s function has been plotted for an effectivenuclear charge of 3.
experiences a smaller positive charge and is held less tightly. The conclusion is that a 2selectron is more stable than a 2p electron in a multi-electronic atom. This is the reasonthat the configuration of a lithium atom is 1s22s1 rather than 1s22p1; or, as is often said,2s fills before 2p.
A similar argument holds for the 3s electron in sodium filling before the 3p. Here the pene-tration is beneath both the pair of electrons in the n = 1 shell and the eight electrons in then = 2 shell; and from Figure 6.11 the 3s electron penetrates better than 3p with penetratesbetter than 3d. So these arguments account for the configurations of the neutral atoms upto argon. The next element in the periodic table is potassium, which has the configuration[Ar]4s1 rather than [Ar]3d1. Here a reasonable explanation is that 4s penetrates so muchbetter than 3d does that it overcomes the inherent stability of 3d over 4s because of close-ness to the nucleus (measured just by n value). We could not predict this. However, itis reasonably easy to understand. The net result is that we have in the periodic table anelectron configuration map, see Figure 6.22.
6.8.1 Exercises
6.8.1.1
When we say that a 2s electron penetrates better than a 2p, what do we mean?
6.8.1.2
Which penetrates the most, 2s or 2p in B?
6.8.1.3
Which penetrates the most, 2p or 3p in H? HINT: Careful!
Chm 118 Problem Solving in Chemistry

6.9 65
Figure 6.22: A rough sketch of the periodic table showing where electrons of various ` valuesare filling in the atoms.
6.8.1.4
What is the electronic configuration of B? of N? of S?
6.8.1.5
Why is the electron configuration of Li 1s22s1 rather than 1s22p1?
6.8.1.6
According to the Pauli principle, which would be most stable? C (1s22s22p1x(↑)2p1
y(↓))or C(1s22s22p1
x(↑)2p1y(↑))? How would the energies of these two compare to the energy
of C(1s22s22p2x)? HINT: Strictly speaking, these do not describe legitimate multielectron
wave functions, but they will suffice to illustrate the point at this stage in your education.
6.8.1.7
What is the energy ordering for the 3s, 3p, and 3d orbitals in K? What causes that order?
6.9 Multi-electronic Atoms, Ionization Energies
There is no single property more important in understanding chemistry and being ableto solve chemical problems than ionization energy, IE. Electron motion is at the heart ofchemistry and it is the ease of the movement of electrons, which is partially determinedby ionization energy, that dictates electron motion. Ionization energies of the atoms arelargely determined by the four concepts that we explore in this section.
Since Coulomb’s law (equation 2.1) is inverse in distance, the stability of an electron withsmall n is much greater than that with large n. In the hydrogen atom this is very obvious,as show by equation 6.6: the IE of hydrogen atoms from various excited states drops veryfast as n increases. For other essentially one electron species, such as lithium ion or sodiumion, the change in IE with a change in n is damped somewhat by penetration, see below.Thus the IE of sodium is less than that of lithium, but not by much. The second factor
Chm 118 Problem Solving in Chemistry

6.9 66
Figure 6.23: Ionization energies for the first and second row elements Li through Ne andNa through Ar.
comes into play when we compare two atoms with the same n value. that factor is thevalence shell nuclear charge , VSNC. This is the charge that a non-penetrating electron inthe valence shell would experience if no other electrons are present in the valence shell. Wecan get a sense of this factor by looking at a beryllium atom compared to a lithium atom.In lithium the n = 2 electron has the nuclear charge of +3 partially shielded by the innershell (n = 1) of two electrons, so the VSNC is +1. In a beryllium atom, the nuclear chargeof +4 is also shielded by the inner shell of electrons; classically, the two electrons in the n =2 shell would be opposite each other, which means that neither shields the other very muchfrom the nuclear charge. Hence the charge “seen” by one of the n = 2 electrons is about +2,double that of the value “seen” in a lithium atom. The experimental IE of Li and Be are5.39 eV and 9.32 eV, nearly the doubling that this crude model predicts. Similar reasoningleads us to conclude that as we move across a row in the periodic table the VSNC increases,leading to the general trend of higher IE to the right in the periodic table. Figure 6.23 plotsthe ionization energies for the first and second row elements from Li to Ne and Na to Ar toillustrate the general increasing IE as one move to the right in the periodic table.
Although the general trend in Figure 6.23 is a increasing IE as we move from left to rightin the periodic table, there is clearly some other factor(s) playing a role in determiningthe IE because of the “glitches” in the data. The first one occurs as a drop of the IEfrom beryllium to boron even though the VSNC increases; a similar feature occurs betweenmagnesium and aluminum in the second row. Inspection of the electronic configurationsof these elements suggest the cause: beryllium is 1s22s2 and boron is 1s22s22p1. In thefirst case the ionization process removes a 2s electron, which penetrates well. In the secondcase, the process removes a 2p electron that does not penetrate as well. We conclude thatalthough the VSNC increases from Be to B, the change in penetration is sufficient to causethe IE to drop. A similar effect occurs at Mg/Al involving 3s and 3p electrons.
The second “glitch” in Figure 6.23 occurs between nitrogen and oxygen in the first row, andphosphorous and sulfur in the second. In all cases it is a p electron that is being removedfor both elements, so penetration differences cannot be the cause. What is changing thenormal increase with VSNC in these cases? Again, examination of the electronic configu-rations suggests the cause, although this time we look closely at the configuration. In thenitrogen atom, the configuration is 1s22s22p3, which, because of the electron-electron re-
Chm 118 Problem Solving in Chemistry

6.10 67
pulsion should probably be written as 1s22s22p1x2p1
y2p1z. This configuration keeps electrons
far apart because of their spatial position along the x, y, and z axis, but also because of thePauli principle: all three 2p electrons have the same spin. At oxygen, the detailed configu-ration is 1s22s22p2
x2p1y2p1
z (or with the paired electrons in either of the other two equivalentorbitals) and that last electron is easier to remove because it is paired with another in theorbital. Hence the IE of oxygen is less than expected by the change in the VSNC.
6.9.1 Exercises
6.9.1.1
What are the four factors than influence ionization energy?
6.9.1.2
Explain why the ionization energy (generally) decreases down a column in the periodictable. HINT: There are two opposing factors.
6.9.1.3
Explain why the ionization energy (generally) increases across a row in the periodic table.
6.9.1.4
How can you rationalize the relative IE’s of Mg and Al?
6.9.1.5
The first IE of sodium is less than that of magnesium. The second IE, which is the energynecessary to remove an electron from a gaseous ion of charge +1, of sodium is greater thanthat of magnesium. Comment.
6.9.1.6
Which has the lower second IE, Ca or Mg?
6.9.1.7
The IE of Rb[Kr 5s] is 4.176 eV whereas that of the excited state of H, H[5s] is 0.544 eV.Account for the difference. HINT: The second case is the ionization of an excited state ofthe hydrogen atom.
6.9.1.8
The IE of the excited state of Rb[Kr 5f] is 0.547 eV whereas that of the excited state of H,H[5f] is 0.544 eV. Account for the lack of a substantial difference.
6.9.1.9
Successive ionization energies for Si are 8.15, 16.34, 33.46, 45.13, and 166.73, all in electronvolts. Take each value and divide it by the charge on the species after the electron is removed(for example, divide 8.15 by 1 since the charge on the ion after the first ionization energyis +1). Explain the resulting data, both similarities and differences. HINT: Don’t expectexact agreement but be startled at how nicely the numbers arrange during this procedure;even though I have seen it before, I always am.
Chm 118 Problem Solving in Chemistry

6.11 68
Element s orbital p orbital Element s orbital p orbital
H 13.6 — — — —He 24.5 — — — —Li 5.45 — Na 5.21 —Be 9.30 — Mg 7.68 —B 14.0 8.30 Al 11.3 5.95C 19.5 10.7 Si 15.0 7.81N 25.5 13.1 P 18.7 10.2O 32.3 15.9 S 20.7 11.7F 46.4 18.7 Cl 25.3 13.8Ne 48.5 21.5 Ar 29.2 15.9
Table 6.1: Valence orbital ionization energy for low atomic number elements in eV.
6.10 Multi-electronic Atoms, Valence Orbital Ionization En-ergies
In discussion of bonding there will be the need to know the average energy of the bondingof an electron to an element. This is because in bonding, many of the special features thatoccur in an atomic species are muted. For instance, in a bonding situation, there are eightelectrons around an oxygen atom, including six in the 2p shell. The peculiar feature of theelectron-electron repulsion that we discussed in the last section is thus absent. A methodto asses how the other factors only influence the IE is needed. This method exists and iscalled the Valence Orbital Ionization Energy, VOIE. In the VOIE method the energy of a2p electron in an atom is computed as the average value over all possible arrangements ofthe electrons. Data are given in Table 6.1.
6.10.1 Exercises
6.10.1.1
From the data in Table 6.1 is the 2s VOIE or the 2p VOIE most sensitive to VSNC?
6.10.1.2
Give a reasonable explanation for your answer to the last exercise.
6.10.1.3
The VOIE of the 4p orbital of Ga is 5.93 eV and that of Kr is 14.22. Make a guess for thevalue of the 4p orbital of As.
Chm 118 Problem Solving in Chemistry

6.11 69
6.11 Multi-electronic Atoms, Transition Metal Ions
One final topic concerning multi-electronic atoms, or rather ions. The configuration of Kand Ca are [Ar]4s1 and [Ar]4s2, respectively. The configuration of most transition metal ionsof the first row, is [Ar]3dn4s2 where n runs from 1 at Sc to 10 at Zn. A simple interpretationof this result would suggest the filling of the 4s level before the 3d. It is easy to offer arationalization for this behavior. The conflict to be dealt with it that the 4s penetrates verywell whereas 3d is closer to the nucleus. If we assume that penetration “wins” this battle,we can “account” for the data. This is not a predictable result, but consistent with what isobserved.
The real situation is much more complicated. One issue is that thewave equation for the system is a multi-electronic wave equation anda discussion of individual electrons is just not appropriate. Also, ananalysis2 suggests that there is an interplay between the differencein the apparent energy of the 3d and 4s orbitals (for the reasonstalked about above) and electron repulsion differences between a 4sand 3d electron with the other electrons in the molecule.
What is clear in all analyses of this situation is the response to an increase in the effectivenuclear charge that occurs when one goes from a neutral atom to a charged ion. The has theeffect of stabilizing small n values more than large ones, so the 3d orbital is stabilized morethan the 4s in the ions. (One way to look at this is that the compression of the inner shellsby the increased charge shrinks the inner orbitals a lot and hence lowers the importanceof penetration.) Thus if we have an atom with the configuration [Ar]3dn4s2, its dipositiveion will have the configuration [Ar]3dn. The conclusion is this: The chemistry of transitionmetal ions (and of transition metal compounds) is dominantly 3d chemistry, with the ionsformed by “taking out the 4s electrons first.”
6.11.1 Exercises
6.11.1.1
What is the electron configuration of V?
6.11.1.2
What is the electron configuration of V2+?
6.11.1.3
What is the electron configuration of Cr3+? of Mn4+?
2Vanquickenborne, L. G.; Pierloot, K.; Devoghel, D.; Inorg. Chem., 1989, 28, 1805-1813.
Chm 118 Problem Solving in Chemistry

Chapter 7
Transition Metal Compounds andColor
7.1 Energy of Orbitals in Electrostatic Fields
We have in the Lewis structure model a simple picture of bonding in compounds containingelements in the upper right hand side of the periodic table. That model does not workwell for compounds involving transition metal atoms, those containing elements in the “d-block.” In this chapter we deal with a simple scheme for bonding in transition metal ions(tmi). This model is usually called crystal field theory.
The essence of the model involves thinking about two electrostatic interactions, governedby Coulomb’s law (section 2.1). The first is that the central ion in our picture, the ion weare mostly interested in and almost always positively charged, attracts negatively chargedions, say F–, or the negative end of a dipole (in, say NH3), toward itself. These ions ordipoles attracted are called ligands. The second interaction is the response of the electronsin various orbitals of the central ion to the field generated by those ligands. Generallyspeaking, the various orbitals of the central ion that were degenerate, had the same energyin the absence of the ligands, split into various new energy levels because of the ligands.Preferential placement of electrons into the more stable of these split orbitals then causesthe compound to be more stable than before splitting.
As mentioned, this model is applied almost exclusively to tmi, but for purposes of intro-duction, we apply it to an ion with p orbitals. Imagine an ion at the origin of a coordinatesystem with two negatively charged ligands surrounding it: one is located at (x, y, z) =(0.0, 0.0, 1.0)A and the other at (x, y, z) = (0.0, 0.0, -1.0)A, see Figure 7.1. Let the ionat the center have an electron in a 5pz orbital and let the ligands be represented by thenegative point charges. Since the 5pz orbital points at the negative charges, an electron init will be repelled rather severely, will have a high energy. In contrast, an electron in a 5pxorbital or a 5py orbital, which are oriented away from the point charges, will not be repelledas severaly. Hence the degeneracy of the three 5p orbitals is lifted; two of them are morestable than the third (5pz).
70

7.2 71
Figure 7.1: Two orbitals in a linear crystal field arrangement.
7.1.1 Exercises
7.1.1.1
How many p electrons would iodine have in the molecule IF–2 assuming that the bonding is
ionic, that is, that the two fluorine nuclei are both ions, F–?
7.1.1.2
Show how the p orbitals would split in energy in a linear compound, say IF–2, under the
crystal field model where you treat the fluoride ions as point charges. HINT: Pay attentionto degeneracies.
7.1.1.3
What would be the occupancy of the various split orbitals for the iodine in the last twoexercises?
7.1.1.4
Show how the p orbitals would split in energy in a square planar complex IF–4 under the
crystal field model.
7.1.1.5
The molecule in the last problem has D4h symmetry. Find the name of the characteristicnumbers for the pz and the px, py wave functions on the iodine in this symmetry.
7.1.1.6
Speak like a professional and describe the splitting of exercise 7.1.1.4 in terms of the lastexercise. For example, “The iodine p orbitals are split into a high energy b1g and a morestable eg by the ligands.”
7.1.1.7
What would be the splitting of the p orbitals in an octahedral compound such as SF6,under, as usual, the crystal field approximation?
7.1.1.8
Which orbital, d2z or dx2−y2 would be of highest energy is two negatively charged ligands
were placed the ±z axis?
7.1.1.9
We have two negative charges along the positive and negative z axis. Two of the d orbitalsare in Figure 7.2. Which are they? What are their relative energies under a crystal fieldapproximation.
Chm 118 Problem Solving in Chemistry

7.2 72
Figure 7.2: Two of the d orbitals for consideration in exercise 7.1.1.9.
7.2 Metal Ions in an Octahedral and Tetrahedral Environ-ments
Transition metal ions exist in a number of different environments, the most common beingoctahedral, in which the metal ion is surrounded by six ligands along the positive and nega-tive Cartesian coordinates, ±x, ±y, ±z. Convince yourself that this octahedral environmentof point charges will cause dxy, dxz, and dyz to be repelled equally. We say that these threeorbitals remain degenerate. They are named the t2g set because of their characteristic num-bers. Although not so obvious (some exercises below explore the arguments), the other twod orbitals, d2
z and dx2−y2 are also degenerate. Since they point right at the ligands, theyare of higher energy than the t2g set. These two orbitals are labeled eg. To summarize, anoctahedral field of ligands split the five d orbitals into two sets, a more stable set of three,t2g and a less stable set of two, eg.
A second common geometry for transition metal ion compounds is tetrahedral. The relativeenergies are most easily seen by placing a tetrahedron within a cube. In Figure 7.3 we givea cube with axes going through the center of the faces of the cube. Put the four ligandson corners of the cube such that they are diagonally opposite each other on the faces ofthe cube. Convince yourself that dxy, dxz, and dyz have the same energy. In tetrahedralsymmetry, these three orbitals are labeled t2. The other two d orbitals, d2
z and dx2−y2 arealso degenerate. They have e symmetry. As suggested in an exercise, you should be able todeduce that the t2 set is of high energy and the e set of low energy, more stable.
Chm 118 Problem Solving in Chemistry

7.2 73
Figure 7.3: A cube with axis drawn.
7.2.1 Exercises
7.2.1.1
Sketch the five angular d orbitals as they would look if you stood on the positive x axis.HINT: You have to be able to draw these orbitals, accurately and quickly, to work withcrystal field problems.
7.2.1.2
Sketch the five angular d orbitals as they would look if you stood on the positive z axis.
7.2.1.3
Three of the d orbitals are dxy, dxz, and dyz. Note that these orbitals occur with one Carte-sian coordinate times another. There are three more possible combinations of coordinatestaken pairwise, xx, yy, and zz. These are usually considered in three combinations, x2-y2,z2-x2, and z2-y2. Sketch each of these functions. HINT: Use the name to guide you toascertain (1) where the function is big, where it is zero; and (2) where it is positive andwhere it is negative.
7.2.1.4
Which orbital is repelled the most by negative charges on the three (x, y, and z) plus andminus Cartesian axes (octahedral symmetry), dx2−y2, dz2−x2, dz2−y2? HINT: Use yourpictures from the last exercise.
7.2.1.5
We now do something sneaky. Since quantum mechanics demands that there are only fivepossible d orbitals and there are six possible combinations of a pair of Cartesian coordinates,
Chm 118 Problem Solving in Chemistry

7.3 74
we have to combine them in some fashion. We choose to add the last two (of the list in thelast exercise) and then divide by two since we don’t want to change the size. Add z2-x2 toz2-y2 and then divide by two. What do you get?
7.2.1.6
The answer to the last exercise, z2-12x2-1
2y2 is the real form of the function we usuallyabbreviate as dz2. Since it is composed of those two orbitals that are equivalent to dx2−y2
but which would have double the interaction with ligands, but then is divided by two, wehave the same interaction. Does this argument convince you that dz2 is equivalent in energyto dx2−y2? If not, it is the best I can do; you will have to take a quantum course.
7.2.1.7
Can you make an argument for why the t2 set of orbitals is of higher energy in a tetrahedronthan is the e set? HINT: This is a problem in solid geometry, but can be approximated bylooking at just the wave function lobe closest to the point charges.
7.2.1.8
Which geometry, octahedral or tetrahedral, do you anticipate will have the largest gapbetween the two sets of split orbitals? REMARK: Real solid geometry suggests the gap inthe tetrahedral environment is 4/9 that in the octahedral.
7.3 Metal Ions in Other Environments: Lowering of Symme-try
Both the octahedral and tetrahedral environments are of “high” symmetry, meaning that thesets of symmetry operations are large (48 and 24, respectively). As symmetry is “lowered,”as the number of symmetry operations decreases, degeneracies generally are dispatched.Hence a particularly easy way to determine the energy ordering of the d orbitals in acompound of low symmetry is to start with one of higher symmetry and carry out a motionof ligands that changes that high symmetry to the lower one. As an example, consider atetrahedral compound that is distorted as indicated in Figure 7.4. This has the effect ofchanging the tetrahedral compound to a distorted tetrahedron, and ultimately, to a squareplanar compound. If we sketch how the various d orbital energy change as the distortionoccurs, we generate a correlation diagram; you are asked to do this in the exercises.
7.3.1 Exercises
7.3.1.1
What happens to the energy of the dxz orbital as the distortion in Figure 7.4 occurs?
7.3.1.2
What can you say about the energy of the dyz orbital (in Figure 7.4) compared to that ofdxz?
Chm 118 Problem Solving in Chemistry

7.4 75
Figure 7.4: Distortion for problems ?? and ??.
7.3.1.3
What happens to the energy of the dxy orbital as the distortion in Figure 7.4 occurs?
7.3.1.4
Sketch how the d orbitals on a metal ion at the center of the cube shown in Figure 7.4would change in energy as the distortion shown occurred.
7.3.1.5
A tetragonal distortion of an octahedral compound is one in which the ligands on the z axismove away from the metal ion and those in the xy plane move toward the metal ion. Makea correlation diagram for this distortion.
7.4 The Configuration of Metal Ion Compounds
To deal with transition metal compounds efficiently it is useful to devise a way to talkabout the electronic configuration. These are the same words that we use to describe theconfiguration of atoms, 1s22s22p2 for a carbon atom, for instance. The difference being thatwe want to describe the orbitals in which the electrons reside, and in compounds these areno longer s, p, d, etc. Rather the names of the characteristic sets of numbers that are usedto describe the orbitals are used. Thus the configuration of diagram “A” in figure 7.5 is a2
1
a12. The configuration for diagram “B” in Figure 7.5 is a2
2g b21u.
When dealing with octahedral environments, a similar notation is used. This would be, forinstance, for a d3 metal, t3
2g. For a a d7 metal complex in a tetrahedral environment, the
Chm 118 Problem Solving in Chemistry

7.4 76
Figure 7.5: Using characteristic number names for configurations; see problem ??.
configuration would be e4t32.
7.4.1 Exercises
7.4.1.1
What is the “d count” for each of these transition metal species in bonding situations:Cr(III), Mn(II), V(II), Cu(II), Ag(I), Ag(III), Ni(III), Mo(0), W(VI), Ru(II), Os(III),Mn(V)? HINT: Remember that the configuration of transition metal ions in compoundsis sans 4s electrons.
7.4.1.2
How many d electrons do these transition metal species have in bonding situations: V(III),Ti(IV), Mo(V), Ni(0), Co(II), Co(III), Rh(III), Ir(IV), Fe(II), Mn(II)? HINT: “In bondingsituations” means sans 4s electrons.
7.4.1.3
Give the electronic configuration (in the tn2g/emg notation) for an octahedral Cr(III) com-pound; for an octahedral Ni(II) compound.
7.4.1.4
Give the electronic configuration (in the tn2g/emg notation) for an octahedral Cu(II) com-pound.
7.4.1.5
There is an ambiguity in electronic configuration for some octahedral metal compounds.The issue is this for a d4 system. If the energy separation between t2g and the eg levelsis small, then rather than paying the price of putting two electrons in the same orbital, itis energetically more favorable to place the fourth electron in the eg level (and gain somestability from the Pauli principle). On the other hand, if the energy separation (often called“the gap”) between the t2g and the eg levels is large, then more stability is gained by puttinga fourth electron in the t2g, even at the cost of putting two electrons in the same orbital.So there are two configurations possible for a d4 system depending on the gap. Give theelectronic configuration (in the tn2g/emg notation) for an octahedral Mn(II) compound if the
Chm 118 Problem Solving in Chemistry

7.5 77
splitting is small; for an octahedral Mn(II) compound if the splitting is large.
7.4.1.6
Give the electronic configuration (in the tn2g/emg notation) for an octahedral Fe(II) compoundif the splitting is small; for an octahedral Fe(II) compound if the splitting is large.
7.4.1.7
Give the electronic configuration (in the em/tn2 notation) for small gap tetrahedral com-pounds of Cu(II), Fe(II), and Co(II).
7.5 Configuration of Metal Ion Compounds, Spin, Field, andLigand Strength
In exercise 7.4.1.5, and those that followed it, we discussed how the size of the gap betweenthe more stable and less stable levels in octahedral and tetrahedral compounds influencedthe configuration. There are two notations for this situation that are used interchangeably.If the gap is small, then the electrons all tend to have their spins in the same direction (sothat Pauli avoidance can occur). If we assign a spin along the z axis of 1
2 to an electronwith spin up, then we can get a total spin along the z axis, called MS , by adding up thesevalues. For a d4 metal ion with a t3
2g/e1g configuration (with a small gap), the value of MS
would then be 2. On the other hand, if the gap was large for this d4 metal ion, then theconfiguration would be t4
2g/e0g and the total value of MS would be only 1. (We shall see
shortly there is an easy way to measure “spin.”) Compounds of the former type are called“high spin” complexes and those of the latter are “low spin” complexes. The other notationfor these two kinds of compounds reflect the gap size. We call the compound with the 3
2g/e1g
configuration a “low field” compound because the gap (caused by the field of the ligands) issmall. Contrariwise, the t4
2g/e0g configuration would be called a “high field” compound. It
is unfortunate that these two notations are opposite, but note that they are: “High field”compounds are of “low spin.”
As implied above, what causes the difference between a high field compound and a lowfield compound is the field generated by the ligands. Remember that there are two fields incrystal field theory, that which attracts the ligands to the metal (not of concern to us now)and the field produced by the ligands that causes the metal d orbitals to have differenceenergies. The stronger the field created by the ligands, the greater the gap (in an octahedralcompound) between the t2g and eg levels. What ligands create a strong field? We wouldanticipate, according to Coulomb’s law, section 2.1, that ligands close to the metal ion (smallr) and those with large charge (big Z1) would create the largest fields; we might expect, forinstance, that O2– >S2– >F– >Br– >NH3. The first because of the high charges, and thelast because it has only a dipole and not a full charge. This expectation turns out to becompletely wrong. Here is a serious flaw in the crystal field theory.
Chm 118 Problem Solving in Chemistry

7.5 78
Compound S
Cr(CH3)(H2O)2+5
32
FeCl3(H2O)352
Co(NH3)4Cl+2 0
MnF3–6 2
Table 7.1: Spin data for some compounds
The order that is observed for the strength of the ligands is called the spectrochemicalseries. We will figure out later in the course why this order is correct, but for now let usjust take it as a experimental observation for the strength of ligands. It is presented here:
I− < Br− < NCS–, S bonded < Cl− < NO–3, O bonded < F− < OH− <
C2O2–4 , O bonded < H2O < SCN−, N bonded < NH3, about the same as py
< en < NO–2, N bonded << CN−, C bonded ∼= CO, C bonded.
According to this series, the gap will be smaller in CrCl3–6 than in Cr(H2O)3+
6 , even thoughthe former has a real charge and the latter only a dipole. Likewise, we would expectthat generally speaking, compounds such as MBr3–
6 would be low spin compounds, whereasM(CN)3–
6 would be high spin.
7.5.1 Exercises
7.5.1.1
In Table 7.1 are the values of the spin quantum number for some compounds. Verify thesevalues.
7.5.1.2
Is the compound Mn(CN)4–6 with configuration t5
2g/e0g high spin or low spin? high field or
low field?
7.5.1.3
What would be the MS value for a low spin octahedral Mn(II) compound? for a high spinoctahedral Mn(II) compound?
7.5.1.4
Almost all tetrahedral compounds are high spin. Comment.
7.5.1.5
For what n values in the dn configurations is there a possibility of high spin and low spincompounds in octahedral geometry?
Chm 118 Problem Solving in Chemistry

7.6 79
7.5.1.6
Which compound will have the greater splitting, MoI3–6 or Mo(H2O)3+
6 ?
7.5.1.7
Which compound will have the larger splitting between the t2g orbitals and the eg orbitalsof an octahedral complex, Cr(NH3)3+
6 or Cr(NCS)3–6 when the NCS− is nitrogen bonded.
7.5.1.8
A solid compound of formula MCl2 is dissolved in water, presumably to form the ionM(H2O)2+
6 . This compound has a spin of 52 . When treated with KCN, presumably to
form M(CN)4–6 , the spin changes to 1
2 . What is M?
7.5.1.9
There are two fields in crystal field theory. The first is created by the charge on the metal.What would you expect would happen to the metal ligand distance in a complex with ametal with charge of +2 as opposed to one in which the metal is charged +3?
7.5.1.10
To continue from the last exercise, in which of the two compounds above would the ligandexert the greater field? Which, therefore, would have the greater gap?
7.5.1.11
Think about the two electric fields that operate in the crystal field model and make anargument for whether the energy of splitting will be larger in FeF3–
6 or FeF4–6 ? Articulate
an eloquent answer.
7.6 Color of Metal Ion Compounds. I. Octahedral Com-pounds.
How do we know that any of the words written in this chapter are true? Certainly notjust because they are written. One of the most dramatic aspects of crystal field theoryis its ability to understand a striking feature of transition metal compounds. Whereasmaterials such as NaCl, CaO, CCl4, O2 are colorless, most transition metal ions have color.For instance Cr(H2O)3+
6 is steel blue, Co(H2O)2+6 is pink, and Ni(H2O)2+
6 is green. Whatcauses these colors? Quantum mechanics says that if light has energy (given by the Einsteinequation, E = hν = hc
λ ), that exactly matches the energy gap between two quantum levels,then there is a possibility for that light to be absorbed by the sample and excite the systemfrom the lower state to the upper state. If one color of visible light is removed from whitelight, then it appears colored to our eyes. It turns out the the gap between the t2g and eglevels of octahedral compounds has energy corresponding to visible light. So if we excitea t2g electron to the eg level, we can expect the compound to be colored. This transitionof an electron from a t2g level to an eg one is called a d to d transition, since both of thelevels originated from d orbitals. Because the size of the gap for a given metal ion dependson the nature of the ligand (through the spectrochemical series), we can often understandwhat is happening in the immediate environment of a metal ion by observing its color.
Chm 118 Problem Solving in Chemistry

7.6 80
Compound λmax, nm Compound λmax, nm
CrF3–6 671 MoCl3–
6 520
CrCl3–6 729 MoBr3–
6 546
Cr(H2O)3+6 575 MoI3–
6 725
Cr(NH3)3+6 464 Mo(H2O)3+
6 383
Table 7.2: Spectra data for the lowest energy t2g to eg transition in some d3 compounds.Data from A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, 1984.
7.6.1 Exercises
7.6.1.1
In Table 7.2 are the values of the highest wavelength of absorption for some octahedralCr(III) and Mo(III) compounds. Are these values consistent with the spectrochemicalseries?
7.6.1.2
What would you expect to happen to the wavelength of absorption if Mn(Cl)4–6 were con-
verted to MnF4–6 ?
7.6.1.3
Which compound would absorb at smaller wavelength (that is, in the “blue”), Co(NO2)3–6
or Co(NCS)3–6 , where both ligands are nitrogen bonded to the Co(III).
7.6.1.4
Which compound would you expect has the lowest energy of absorbance, Ni(H2O)2+6 or
Ni(H2O)2+4 , where the latter is presumably tetrahedral?
7.6.1.5
An octahedral compound of Cr(III) with six ligands, L1, has an absorption peak near 550nm. The ligand is changed to L2 (but the symmetry is still octahedral) and the peak is nowat 640 nm. What can you say about the ligand field strength of L1 and L2?
7.6.1.6
The lowest energy peak in CrF3–6 occurs at 14900 cm−1 and that in Cr(en)3+
3 at 22300 cm−1.Which ligand has the larger crystal field splitting parameter, F− or en? HINT: It probablywould be nice to know what en is.
7.6.1.7
The lowest energy peaks in solid VCl2, VBr2, and VI2 (where the coordination is approx-imately octahedral by sharing of anions between two or more metal ions) are 9300, 8600,and 7870 cm−1, respectively. Calculate the wavelength of the light associated with theseabsorbances and explain the results.
Chm 118 Problem Solving in Chemistry

7.7 81
Figure 7.6: Orbital energy shifts on changing ML2+6 to trans-ML4W2+
2 and transitions.
7.6.1.8
VBr2 is a orange brown solid. (Brown is typically the color associated with something thatabsorbs over most of the visible region.) When it is dissolved in water and then treatedwith KCN, the solution turns pale yellow. What color of light is removed from white lightto make a yellow color? Is the light that is removed of high or low energy? Explain thecolor changes described above.
7.6.1.9
The lowest energy peak in Ni(py)2+6 (where py is pyridine) occurs at 11700 cm−1. Describe
the change in the position of the electron that occurs as a result of this absorption of light.HINTS: From what orbital to what orbital is the electron moving? How do the positions ofthese orbitals differ from one another? This is an important concept to use to distinguishfrom other sources of color in compounds.
7.7 Color of Metal Ion Compounds. II. Low Symmetry Com-pounds.
The discussion in the last section involves octahedral compounds. There are many morecompounds that, although six coordinate, are not strictly octahedral, such as Cr(H2O)5Cl2+.There are two extreme approximations to handling such compounds (and then the difficultin-between, which we will not deal with). One is the separate orbital approach and theother is the average field approach.
Let’s talk about the separate orbital approach first. Imagine we have an octahedral com-pound of the type ML2+
6 where L is some ligand. Now we take the two L ligands on the zaxis and replace them with a weaker ligand, W. Our compound is now trans-ML4W2+
2 . Weuse the simplest approximation to determine what happens to the d orbital energy levels.Since the ligands in the xy plane are not changes, dx2−y2 is not changed in energy; andbecause the ligand W is weak and is on the z axis, d2
z is lowered in energy. We also assumethat the orbitals that avoid the ligands, dxy, dxz, and dyz, are barely changed in energy.This is shown in Figure 7.6. We see that the single transition found in the octahedral parentcompound becomes two transitions in the substituted compound, one at the same energyas that found in the octahedral compound, and the other at lower energy. This situationwill pertain if the ligands L and W are widely separated in the spectrochemical series.
Before dealing with the average field model, it is useful to examine a property of the ab-
Chm 118 Problem Solving in Chemistry

7.7 82
Figure 7.7: All spectral lines in a tetragonally distorted compound from t2g derived levelsto eg derived levels.
sorbance of light in transition metal compounds. Recall our model that the strength of aligand depends on the distance it is from the metal ion. Also, note that all atoms are alwaysmoving back and forth in vibrations that change the bond length. In a metal complex, onesuch motion is for all of the ligands to move in and out in phase with each other. Whenthe ligands are all close to the metal ion, the gap is larger; when they are all far from themetal ion, the gap is smaller. Since a photon of light could hit the metal complex at anytime, it might catch the ligands out (small gap) or it might catch the ligands in (big gap)or anywhere in-between. As a result, a whole range of colors is absorbed rather than just asingle wavelength. It is usual for the “peaks” of transition metal compounds to be ratherbroad, covering a range of wavelengths.
We deal with the average field model by looking carefully at the same tetragonal splittingthat we examined above for the separate orbital model. The difference will be that thistime we look at all possible transitions from levels that were originally t2g in the octahedralparent compound to the levels that were originally eg. This is shown in Figure 7.7. Thereare four possible transitions as shown there, all with very similar energies. The averagefield contends that these will show up as one transition with average energy as shown onthe right side of Figure 7.7. To illustrate exactly how this works, consider Figure 7.8 wherebroadened spectral lines–see last paragraph–are positioned at each of the four energies givenin the left side of Figure 7.7. Of course, these would not be seen as individual peaks, asshown in Figure 7.8, but would add together to give a net absorbance peak. This is shownin blue curve in Figure 7.9–the lower curve at 17000 cm−1 if you are looking in black andwhite. We can compare this to the orange curve in that Figure which is the curve for theaverage peak from the right hand side of Figure 7.7. This argument suggests that whenwe have ligands that do not differ in their strength by too much, we expect a peak atthe average position of the strength of the various ligands. This is called the average fieldapproximation.
Chm 118 Problem Solving in Chemistry
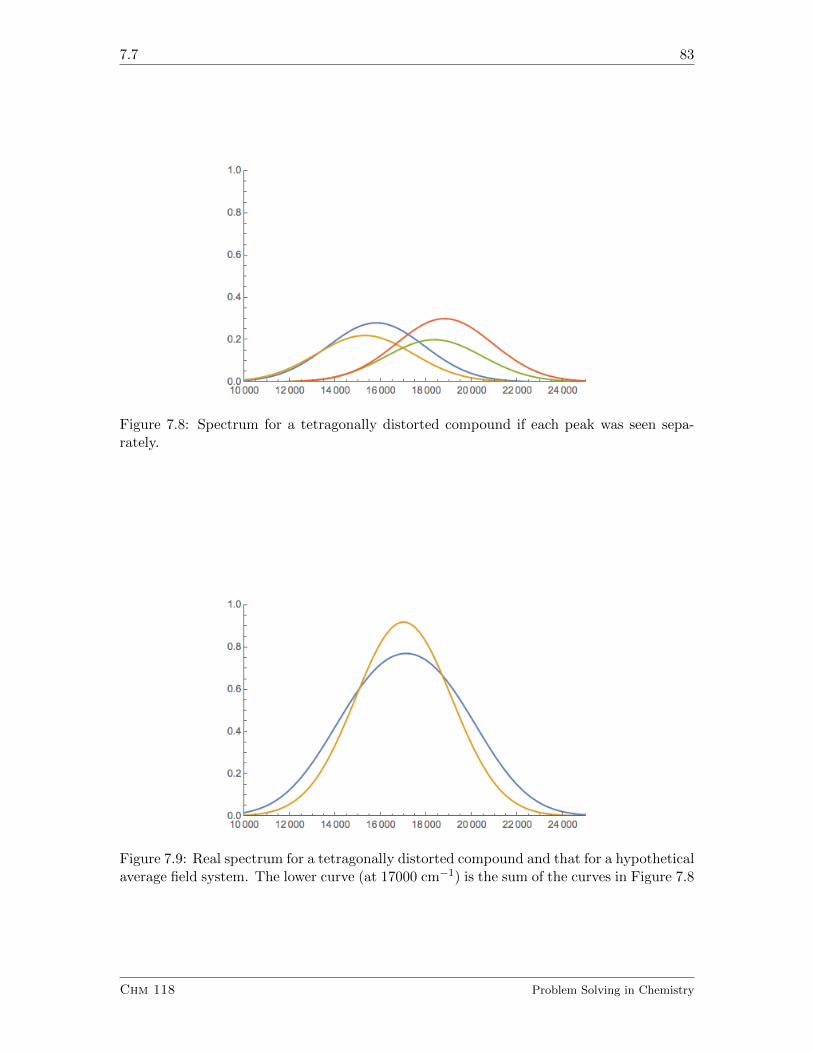
7.7 83
Figure 7.8: Spectrum for a tetragonally distorted compound if each peak was seen sepa-rately.
Figure 7.9: Real spectrum for a tetragonally distorted compound and that for a hypotheticalaverage field system. The lower curve (at 17000 cm−1) is the sum of the curves in Figure 7.8
Chm 118 Problem Solving in Chemistry

7.8 84
Compound, X λmax, nm Compound, X λmax, nm
SCN− 620 I− 650Br− 322 Cl− 609F− 595 NCS− 570
NNN− 585 NO 559NC− 560 NH3 545
Table 7.3: Spectral data for Cr(H2O)5X2+ compounds.
7.7.1 Exercises
7.7.1.1
When Cr(H2O)2+6 reacts with Co(NH3)5(NCS)2+ one of the products is Cr(H2O)5(NCS)2+.
Can you tell whether or not the thiocyanate ion is bonded to the chromium ion throughthe nitrogen atom or the sulfur atom by using spectroscopy? What data would you need?HINT: Apply an average field argument.
7.7.1.2
Linkage isomerization occurs when a ligand can bind to a metal ion in more than one way.Which of the following ligands might exhibit linkage isomerization? NO–
2, NCS–, S2O2–3 .
Use Lewis structures to make your point.
7.7.1.3
The ion Cr(H2O)5SCN2+ has an absorption peak in the visible region of the spectrum at620 nm. The linkage isomer of this material, Cr(H2O)5NCS2+ has a peak at 570 nm. Givean explanation. HINT: Use the average field concept.
7.7.1.4
When two pyridine ligands along the z axis in Ni(py)2+6 – see exercise 7.6.1.9 – are replaced
by Br− ligands, two peaks appear, one at 8430 cm−1 and the other at 11550 cm−1. Accountfor this behavior. HINT: This is NOT an average field problem because we see two peaks,rather it is a separate orbital problem.
7.7.1.5
Table 7.3 gives the lowest energy (spin-allowed) spectral peak for some compounds of theform Cr(H2O)5X2+. From these data construct a spectrochemical series. HINT: Use theaverage field concept.
7.8 Color of Metal Ion Compounds. III. Intensities of Color.
Our discussion on the color of metal ion compounds has thus far been concerned with thewavelength of the absorbance. There is a second parameter that is also important. Howintense is the color? To measure the intensity of the color of a sample in solution we putthe liquid in a container of fixed length, generally one cm, and irradiate it with light of
Chm 118 Problem Solving in Chemistry

7.8 85
wavelength λ and intensity I0. On the backside of the sample, we place a detector thatmeasures the intensity of the beam after it has passed through the sample and find anintensity of I. The transmittance is defined as the ratio of these numbers,
T = I/I0 (7.1)
and varies between 0 and 1. The absorbance is defined as A = -log[T] and varies between0 (for T = 1) and a large number when T approaches 0. An intensely absorbing compoundthus has a small T and a large A. Clearly these parameters depend on the concentrationof the material, c, generally in M, and the length of the cell, `. This dependence is givenby Beer’s law:
A = ελc` (7.2)
The parameter of interest to us is ελ, the molar absorptivity, which is a measure of theintensity of the color, or, in words that will serve us better, the allowedness of the transition:how easy it is for the molecule to grab the photon of light and use its energy to promotethe electron to a higher energy state.
The question before us is what causes a compound to have a large ελ. Be clear that ελ cannever be large unless the λ at which you measure it corresponds to light of the right energyto match the energy gap in the molecule. Beyond that, however, there are other factors thatinfluence the size of ελ. We consider two of these with associated flavors. The first is thatελ is small unless the value of the spin of the compound does not change upon excitation.This is called the “spin selection rule,” and we talk about “spin-allowed transitions,” where∆S = 0; and “spin-forbidden transitions,” where ∆S 6= 0. For metal ion complexes thismeans a transition from the configuration t3
2g/e1g to t2
2g/e2g could be spin-allowed, but that
a transition from t32g/e2
g to t22g/e3
g must be spin-forbidden.
The second factor that we consider to determine allowedness, the size of ελ, is called parity.For systems that contain the symmetry operation i, the sets of characteristic numberseither have a positive number under i or a negative number. Those with a positive numberare called (and labeled) “g” functions; and those with a negative number are called “u”functions. For reasons of simple calculus (but not proven here), the only transitions thatare possible are between a g function and a u function; both g to g and u to u transitions areforbidden. Said in words, transitions are allowed when they proceed from an even functionto an odd one, or in reverse. Otherwise, they are forbidden. Here is a simple example. Theatomic functions have the following parity, which you should prove to yourself: s (even),p (odd), d(even), f(odd). Thus a s to p transition is allowed, but a p to f transition isforbidden. Likewise, and importantly, a d to d transition, such as the transitions we havebeen talking about, are forbidden.
The last sentence in the last paragraph seems rather ridiculous. Why have we been talkingabout something that is forbidden. The answer is that nothing is totally forbidden. Whenwe say something is strongly forbidden, we simply mean that ελ is small. And when wesay something is allowed, we mean ελ is large. To give us a handle on how to use our tworules for allowedness, look at Table 7.4. Across the top are three categories of the whetheror not the parity rule holds–more explanation in a moment for the middle one. Down theside are the two possibilities for ∆S. At each intersection is a rough (very) guide to theapproximate size of ελ. The first column indicates the ελ values when the transition is not
Chm 118 Problem Solving in Chemistry

7.9 86
g to u? No g to u? Remembers it to be No g to u? Yes
∆S = 0 1 to 100 100 to 1000 greater than 1000
∆S not 0 less than 1 less than 10 less than 100
Table 7.4: Approximate values of the molar absorptivity under different conditions of ∆Sand odd/even nature of the functions; the units are M−1cm−1. Note that “Remembers ...”refers to an orbital in an environment that does not have the center of inversion symmetryoperation, but the starting function and final function are both even (or odd). Use of thistable without thinking will result in incorrect results; intensity is hard to predict.
officially g to u, that is, is forbidden when the molecule contains a center of inversion inits symmetry. The middle column indicates the ελ value when the molecule does not havea center of inversion, but when the transition is, in essence, g to g or u to u. An exampleis the d to d transition in a tetrahedral compound. This symmetry does not have a centerof inversion, but the orbitals “remember” that they came from an orbital, a d orbital, thatin spherical symmetry did have a center of inversion. The last column is for compoundswhere the transition if from a g orbital to a u one, either in actuality, or by “remembering.”Apply the appropriate ∆S to each by choosing the right row and you get an approximationto the value of ελ.
7.8.1 Exercises
7.8.1.1
A compound has a molar absorptivity of 15100 M−1cm−1. Using Table 7.4, what can yousay about the nature of the transition?
7.8.1.2
The complex ion Co(H2O)2+6 has absorbance peaks, nm, (molar absorptivity, M−1 cm−1)
at 12340 (2), 625 (shoulder), and 515 (4.6). Given the intensities of these peaks, what kindof transition are they associated with? HINT: Remember the warning in Table 7.4; thereare a couple of possibilities and you need to use other thoughts and other data to sort outwhich you want to use.
7.8.1.3
Compounds of Mn(II) that are high spin are generally almost colorless (which probablymeans that ελ is less than 1). Why?
7.8.1.4
A solution of Fe(H2O)3+6 is nearly colorless. When some NCS– is added, the solution turns
blood red with a peak near 440 nm with an ελ of about 4600 M−1 cm−1. Comment.
Chm 118 Problem Solving in Chemistry

7.9 87
Figure 7.10: A Gouy balance. A diamagnetic substance is repelled from the magnetic fieldand hence weighs less. A paramagnetic substance is pulled into the field and thereforeweighs more.
7.8.1.5
The ion VO3–4 is colorless to our eye, but has a peak at 276 nm with an intensity of about
8000 M−1 cm−1. Give the “d count” of this species and comment on the spectrum.
7.9 Magnetism in Metal Ion Compounds.
We have talked about the spin of transition metal compounds. To review, compounds withnet spin of 0 are said to be diamagnetic. and compounds with a net spin other than zeroare called paramagnetic. When a solid sample of a diamagnetic compound is placed in amagnetic field, as illustrated in Figure 7.10 and weighted, it is found to weigh less than itweighs in the absence of the field. On the other hand, a paramagnetic substance weighsmore in the presence of the magnetic field. From the difference in apparent weight, themagnetic moment of the material, µ, can be found. Theory indicates an approximation canbe calculated from the spin via the “spin only magnetic moment” :
µS.O. = 2√S(S + 1) (7.3)
where S is the net sum of the spins of the electrons and the units of the moment are BohrMagnetons, B.M. (This equation is only an approximation because orbital motion of theelectrons also contributes to the magnetic moment; it is harder to quantify.) For instance,a compound with a configuration of t3
2g/e0g would have a magnetic moment of about 3.87
B.M.
7.9.1 Exercises
7.9.1.1
What will the magnetic moment be for Cr(NH3)3+6 ?
Chm 118 Problem Solving in Chemistry

7.10 88
7.9.1.2
A octahedral complex of Mn(II) is made and has a measured magnetic moment of 6.0. Isit high spin or low spin?
7.9.1.3
A compound of Co(II) is synthesized and found to have a magnetic moment of 1.7. In asimple model where it is assumed to be either octahedrally or tetrahedrally coordinated,which is it?
7.9.1.4
VBr2 has a magnetic moment of about 3.8 B.M. When it is dissolved in water and thentreated with KCN, the color of the solution changes dramatically, but the magnetic momentis largely unchanged. Explain.
7.9.1.5
When CrBr2 is treated in a manner similar to that of VBr2 in the last exercise, the magneticmoment decreases. Explain.
7.9.1.6
The complex CoF3–6 is paramagnetic. What is its electronic configuration?
7.9.1.7
The complex Co(NH3)3+6 is diamagnetic. What is its electronic configuration?
7.9.1.8
Do the results of the last two exercises make sense based on the spectrochemical series?
7.9.1.9
Although Ni(NH3)2+6 is paramagnetic, Pt(NH3)2+
4 is diamagnetic. Explain. HINTS: First,both have the same ”d count.” What is the value of that? The latter compound is fourcoordinate; that means it is probably either tetrahedral or square planar; we looked atsquare planar energy levels in exercise 7.3.1.5.
7.10 Crystal Field Stabilization Energies
There are energetic consequences that are driven by the splitting of the d orbitals in acrystal field; that is the topic of this section. First let’s take the six negative changes in anoctahedron and smash them with a hammer into little fractional charges that are spreadevenly on the surface of a sphere surrounding the ion. Now imagine an electron in oneof the d orbitals of an ion at the center of the sphere. The energy of the electron in thed orbital is increased because of the negative charges on the sphere. Since the symmetryremains spherical, all five of the d orbitals are repelled the same, as shown in change from“Free Ion” to “Spread Out Charge” in Figure 7.11. As we are free to choose any arbitraryenergy as our zero of energy, we take the spread out charge value to be zero. Now we let thelittle fragments of charge on the sphere come back together to form charges at the cornersof the octahedron. This will increase the energy of the orbitals that point at the corners
Chm 118 Problem Solving in Chemistry

7.10 89
Figure 7.11: Energetic consequence of the splitting of the d orbitals by an octahedral crystalfield.
of the octahedron, the eg levels, as the charge increases there. And decrease the energy ofthe orbitals that avoid the corners, the t2g. This situation is labeled “octahedral charge”in Figure 7.11.
If all the orbitals were filled in the “spread out charge” state, the net energy would be, bydefinition, zero. If all the orbitals remain filled in the “octahedral charge“ state, the energymust still be zero since there has been no change in the net charge on the sphere. Thismeans that the stability of an electron in the t2g set, x, times the six electrons in that setmust equal the instability of an electron in the eg set, y, times the four electrons in it. Thisequation, and the one that sets the total gap between the t2g and the eg levels, energy ofeg minus energy of t2g, as ∆0 are:
−6x = 4y (7.4)
x+ y = ∆0 (7.5)
These equations solve to yield the destabilization of the eg level as 3/5∆0 and the stabi-lization of the t2g level as -2/5∆0, both relative to the zero of energy, which is the sphericalenvironment of the ligand charge. This means that a d1 metal ion compound in the t1
2g/e0g
configuration would be more stable by -2/5∆0 than it should be (if the charges produced bythe ligands were spherical). This is called crystal field stabilization energy, often abbreviatedCFSE. CFSE has a significant influence on the properties of many metal complexes.
7.10.1 Exercises
7.10.1.1
For Co(II) compounds, chloride ion has a value of ∆0 of 6600 cm−1. Find the CFSE inkcal/mole for a six coordinate CoCl2. HINT: Chloride ions are shared by various Co(II)ions in this material.
7.10.1.2
The lowest energy absorbance peak for Rh(NH3)3+6 occurs at 304.8 nm. This compound
has a magnetic moment of zero. Find the CFSE for Rh(NH3)3+6 .
Chm 118 Problem Solving in Chemistry

7.10 90
7.10.1.3
What is the change in CFSE (in units of ∆0) upon moving a Co(II) ion from an octahedralenvironment to a tetrahedral one? HINT: Recall the tetrahedral splitting is just the inverseof the octahedral one, so stability of the e set is -3/5∆t and instability of the t2 set is 2/5∆t.Also, recall that ∆t = 4/9 ∆0.
7.10.1.4
The heat of hydration, the heat evolved in taking a gas phase metal ion (spherical) andputting it into a octahedral environment of water molecules, is more negative for Mn(II)than for Ca(II) (presumably because of the smaller radius of Mn(II)) and smaller yet forZn(II). In fact the points for those three ions lie on roughly a straight line when plottedagainst the atomic number. The value for V(II) is considerably more negative than the linewould suggest. Give an explanation.
7.10.1.5
Look up closest packing of anions or examine Figure 7.12. The spheres on top (bold or redin color) are in depressions of the first layer. There are two kinds of holes in this packing,tetrahedral and octahedral. Which is “A” and which is “B?”
7.10.1.6
A spinel is a solid metal oxide with closest packed oxides of formula M(II)M(III)2O4 wherethe three M need not be the same. In a spinel, one-third of the metal ions are in tetrahedralsites, and the others are in octahedral sites. A normal spinel has the M(II) in the tetrahedralsites. Why would this be “normal”? HINT: Think about the stability of a dipositive andtripositive cation in the presence of differing numbers of oxygen anions via Coulomb’s Law.
7.10.1.7
An “inverted” spinel has M(III) in the tetrahedral sites and the remainder of the M(III)and all the M(II) in the octahedral sites. Fe3O4 is an inverted spinel and Mn3O4 is normal.Why? HINTS: Something is over-riding the result of the last exercise, which is charge.Could that something be CFSE?
7.10.1.8
Account for the fact that the spinel CoFe2O4 is inverted.
Chm 118 Problem Solving in Chemistry

7.10 91
Figure 7.12: An example of closest packing. Red spheres are on top of black one.
Chm 118 Problem Solving in Chemistry

Chapter 8
Absorbance and Kinetics
8.1 Using Light to See
How do we know what we write is true? By probing chemical systems with light we canlearn a lot about what is going on at the molecular level. We take advantage of Beer’s law,
A = ελc` (7.2 revisited)
to learn about the concentration of a material in solution. Note in this equation andthroughout this course, the symbol A is a symbol for an absorbance, while “A” itself mightstand for the name of a molecule. For instance, in Figure 8.1 is given the absorbance curvesof a solution of Cr(H2O)5SCN2+ as a function of time. Since the absorbance is changingwith time, some “chemistry” must be going on with the compound. Further, we can gainsome knowledge of what is happening from our understanding of the absorbance spectra oftransition metal ions. Since the peak at about 620 nm is disappearing and one is building at570, the average strength of the ligand is going up. This would be consistent with the NCS–
being replaced by a water molecule, since water is higher on the spectrochemical series thansulfur bonded NCS– is. More careful observation suggests even more information. Sincethe peak for Cr(H2O)3+
6 , the product we just postulated, occurs at 575 nm, and the newpeak is lower in wavelength than this, some ligand with greater field strength than watermust be replacing the sulfur bonded NCS–. Other experiments prove this to be true. Thedominant process occurring here is a linkage isomerization, as Cr(H2O)5SCN2+ is convertedinto Cr(H2O)5NCS2+. This example shows how powerful the color of a solution can be indetecting chemical change.
Another feature in Figure 8.1 is important. The absorbance at a wavelength of about 593.5does not change with time. Such points are called isosbestic points and are importantfor understanding the chemistry behind the change. In the exercises you will derive theconditions for isosbestic behavior. To do this derivation requires the equation for twoabsorbing species in solution; since they act independent of each other, this is just a Beer’slaw sum:
A = ελ,A[A]`+ ελ,B[B]` (8.1)
For a system that is reacting, say conversion of molecule A to molecule B, there are a
92
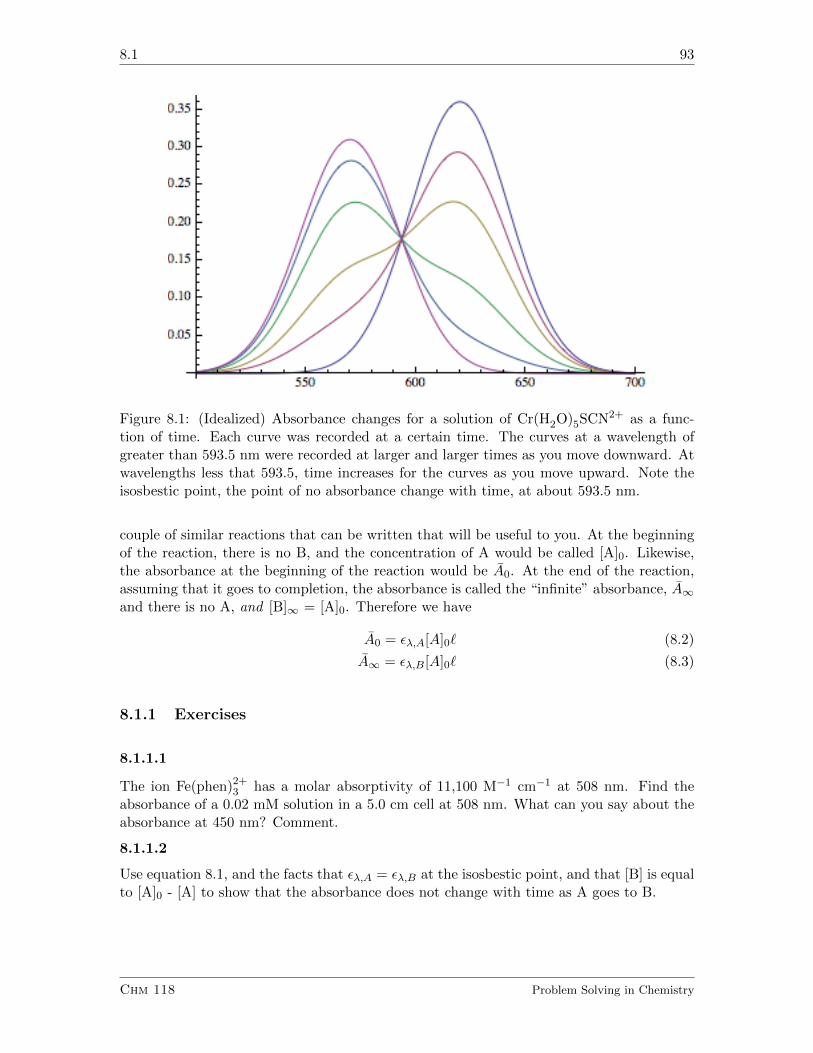
8.1 93
Figure 8.1: (Idealized) Absorbance changes for a solution of Cr(H2O)5SCN2+ as a func-tion of time. Each curve was recorded at a certain time. The curves at a wavelength ofgreater than 593.5 nm were recorded at larger and larger times as you move downward. Atwavelengths less that 593.5, time increases for the curves as you move upward. Note theisosbestic point, the point of no absorbance change with time, at about 593.5 nm.
couple of similar reactions that can be written that will be useful to you. At the beginningof the reaction, there is no B, and the concentration of A would be called [A]0. Likewise,the absorbance at the beginning of the reaction would be A0. At the end of the reaction,assuming that it goes to completion, the absorbance is called the “infinite” absorbance, A∞and there is no A, and [B]∞ = [A]0. Therefore we have
A0 = ελ,A[A]0` (8.2)
A∞ = ελ,B[A]0` (8.3)
8.1.1 Exercises
8.1.1.1
The ion Fe(phen)2+3 has a molar absorptivity of 11,100 M−1 cm−1 at 508 nm. Find the
absorbance of a 0.02 mM solution in a 5.0 cm cell at 508 nm. What can you say about theabsorbance at 450 nm? Comment.
8.1.1.2
Use equation 8.1, and the facts that ελ,A = ελ,B at the isosbestic point, and that [B] is equalto [A]0 - [A] to show that the absorbance does not change with time as A goes to B.
Chm 118 Problem Solving in Chemistry

8.2 94
8.1.1.3
Write the equation 8.1 for the reaction in Figure 8.1 for a wavelength of 610nm, with Abeing Cr(H2O)5SCN2+ and B being Cr(H2O)5NCS2+, where both ελ,A and ελ,B have value;do this for some arbitrary time and label the time dependent parameters with a “sub t.”Now put into your equation the stoichiometric relationship for [B], the expression [A]0 - [A];and then subtract from both sides equation 8.3. Show that the result is:
[A]t =At − A∞
ελ,A`− ελ,B`(8.4)
8.1.1.4
Write equation 8.1 for zero time, appropriately labeled, where [B] = 0. Write this equationfor infinite time, with appropriate labels, where [A] = 0 and [B] = [A]0. Subtract the infinitetime value of absorbance from the zero time value and show, using equations 8.2-8.3, thatthe difference is ελ,A` - ελ,B`.
8.1.1.5
Use the results of the last two exercises to prove that the following ratio holds:
[A]t[A]0
=At − A∞A0 − A∞
(8.5)
This is a very important and useful equation for using light to examine the time course ofreactions.
8.2 Basic Kinetic Expressions and Definitions
We looked at how color indicated that something was happening in the last section. Here webecome more precise in our language of describing change in a chemical system by discussingthe field of kinetic studies. First we get some sense of the issue of how fast reaction occursby considering the rate of reaction. This is a change in concentration of our reactant dividedby a change in time, or, in the calculus, d[A]
dt .
To get a sense of what happens in order for reaction to occur, imagine the space in abeaker is divided up into a bunch of small compartments and imagine time divided into abunch of small increments. There are energy fluctuations in our space, so there is a finiteprobability that a given compartment will have a slight excess of energy in any given timeincrement. We are interested in the molecule A reacting. Let’s presume if the compartmenthas that excess of energy and contains a molecule of A reaction occurs. Imagine the rate(early in time when there are lots of reactants) is 0.00001 mole/sec. What do you thinkthe magnitude of the rate will be at a larger time when an energy fluctuation in a givencompartment might occur, but the concentration of A is lower? Clearly the rate will besmaller. We learn that rate is concentration dependent, and it seems plausible that the ratewill depend on the concentration of A. We have what is called a rate law,
−d[A]
dt= k1[A] (8.6)
Chm 118 Problem Solving in Chemistry

8.3 95
where the constant (for a given reaction and temperature), k1, is called the rate constant .In essence, the parameter k1 means how much energy is required for the reaction: large k1,little energy; small k1, lots of energy.
Let’s change our scenario a little. Imagine the same beaker with its compartments andenergy fluctuations. Now let the reaction be between a molecule of A and a molecule ofB to form some product. In this case assume that for reaction to take place, you musthave not only the energy, but also a molecule of A and a molecule of B. Assume when thecontainer has a reasonable concentration of A and B, the rate is 0.00001 moles/sec. Now, ifthe container has a reasonable concentration of A, but is very dilute in B, what is the rate?You would expect it to be lower since the three conditions needed, energy, an A, and a B,is not met very often if [B] is small. Also, if very dilute in A but with a reasonable B, whatcould we say about the rate? The rate law in this case must depend on both concentrations:
−d[A]
dt=−d[B]
dt= k2[A][B] (8.7)
where k2 is the second order rate constant, and again is an (inverse) measure of the energyrequired.
A final case. Molecule A reacts with itself to form products. What has to be in thecompartment for reaction to take place is two molecules of A and energy, so the rate law is:
−d[A]
dt= k2[A]2 (8.8)
How are these three types of rate laws distinguished? Equation 8.6 is called a first orderor overall first order rate law because the concentration of A is raised to the first power.Equation 8.7 is a second order or mixed second order rate law because there are two con-centrations, each raised to the first power. The language “first order in A and first orderin B” would apply to equation 8.7. Equation 8.8 is a pure second order rate law. Thelanguage “second order in A” would apply to equation 8.8.
8.2.1 Exercises
8.2.1.1
What is the interpretation of a rate law of the form −d[X]dt = k [X]2? What is the order of
this rate law?
8.2.1.2
What is the interpretation of a rate law of the form −d[X]dt = k [X]? What is the order of
this rate law?
8.2.1.3
There are almost no known reactions that are zero order. That is, have a rate law of theform −d[X]
dt = k? Use our interpretation of how a reaction proceeds to understand why thisis so.
Chm 118 Problem Solving in Chemistry

8.3 96
8.3 First Order Reactions
Let’s focus first on how to identify a first order reaction such as −d[A]dt = k[A]. The rate is
concentration (or time) dependent, so it is the slope of a plot of [A] versus time. Slopes arehard to evaluate accurately, so another method is needed. We take advantage of the factthat we can integrate a first order rate law and get an equation that can readily be plotted.Here’s the math:
d[A]
dt= −k[A] (8.9)
d[A]
[A]= −kdt (8.10)∫ [A]
[A]0
d[A]
[A]=
∫ t
0−kdt (8.11)
ln[A]
[A]0= −kt (8.12)
We conclude that if we plot the logarithm of the ratio of [A] to [A]0 versus time, we will geta straight line whose slope will be the negative of the first order rate constant, k. Note thatequation 8.5 allows a plot of an absorbance function rather than the concentration ratioitself.
8.3.1 Exercises
8.3.1.1
Here is some kinetic data for a reaction in which the reagent being measured, A, starts outat a concentration of 0.01 M. Show that the data are consistent with a first order rate lawand evaluate k.
t, min [A], M t, min [A], M
1 0.00885 2 0.008063 0.00750 4 0.006785 0.00600 6 0.005397 0.00473 8 0.004419 0.00402 10 0.00397
8.3.1.2
Take the integrated form of the first order rate law, equation 8.12, and evaluate the timeat which the reaction is half finished, i.e., [A] = [A]0/2.
8.3.1.3
Consider a first order reaction that starts with a concentration of A of magnitude [A]0/2.Take the integrated form of the first order rate law, equation 8.12, and evaluate the timeat which the reaction is half finished, i.e., [A] = [A]0/4.
Chm 118 Problem Solving in Chemistry

8.4 97
8.3.1.4
Compare your results from the last two exercises. We say the “half-life” of a first orderreaction is independent of the concentration.
8.3.1.5
The great natural philosopher of rural Arkansas, Boniface Beebe, carried out a rate mea-surement. He weighed a portion of solid compound A, took some water from the distilledwater bottle, mixed the solid in the solvent and placed the sample into the temperatureequilibrated compartment of a spectrophotometer. The initial concentration of A was 0.01M and the experiment was carried out at 50oC. Determine if the data are consistent witha first order rate law. If they are not, suggest a cause for the deviation. HINT: Thinkcarefully about what Bonnie did in the experiment; and what he might have done.
t, min [A], M t, min [A], M
2 0.00985 4 0.009266 0.00798 8 0.0067810 0.00600 12 0.0053914 0.00473 16 0.0044118 0.00402 20 0.00397
8.4 Dealing with More Complex Rate Laws
Integration of more complex rate laws than first order is possible, but except for the secondorder rate law, equation 8.8, which gives a linear plot of 1/[A] versus time (with a slope ofk), such integrations are difficult and sometimes impossible. Another method is needed todeal with complex rate laws. That method is presented in this section where we use themixed second order rate law as an example.
Consider the rate law given in equation 8.7 for a reaction in which one mole of A reactswith one mole of B. We run the reaction under a special set of conditions that allow thesimplification of the rate equations. Imagine that we start with a concentration of A of0.0001 M and of B of 0.1 M. At the end of the reaction, when all of the limiting reagent,A, is used up, the [B] = 0.0999 M, which is very close to the starting concentration of 0.1M. Using this initial conditions we have managed to keep the concentration of B essentiallyconstant. The math becomes:
−d[A]
dt= k2[A][B] (8.13)
−d[A]
dt≈ (k2[B]0)[A] (8.14)
−d[A]
dt= kpfo[A] (8.15)
which suggests that we can make a normal first order plot and get as a slope the quantitykpro, which equals the true rate constant times the concentration of B. This is called thepseudo-first-order approach because we have converted our more complex rate equation into
Chm 118 Problem Solving in Chemistry

8.4 98
a first order one by having all concentrations except one in large excess. To get the truerate constant requires a second experiment at a different concentration (but still in excess)of B.
8.4.1 Exercises
8.4.1.1
What will happen to the value of kpfo if the experiment described above is run with aninitial concentration of B of 0.2 M?
8.4.1.2
If the value of kpfo from the experiment described in the last exercise is 1.0×10−4 sec−1,what is the true rate constant?
8.4.1.3
Let the rate law for a given reaction of A with B be first order in [A] and second order in[B]; you run a pseudo first order reaction with [A]0 = 0.0001 and [B]0 = 0.1 and get a kpfoof 0.01. What will the kpfo be if the initial conditions are [A]0 = 0.0001 and [B]0 = 0.2?
8.4.1.4
Here are some data for the reaction of A with B with a one to one stoichiometry. The initialconditions are: [A]0 = 0.003 M, [B]0 = 0.06 M. Show the reaction is pseudo-first-order byplotting ln[A]t versus time. The [A]t values as a function of time are given in the table.HINT: What is the concentration of B at the end of the reaction?
t, min [A], M t, min [A], M
2 0.00251 4 0.002056 0.00177 8 0.0014010 0.00119 12 0.0018514 0.000971 16 0.0008418 0.00054 20 0.00058
8.4.1.5
What is kpfo in the last exercise?
8.4.1.6
For the same reaction as in exercise 8.4.1.4, but with [A]0 = 0.003, [B]0 = 0.096. Show thereaction is still pseudo-first-order. The [A] values as a function of time are given for integralsteps of time from 2 to 20 in steps of 2 min: 0.00232, 0.00171, 0.00134, 0.000996, 0.000789,0.000520, 0.000388, 0.000294, 0.000155, 0.000147.
8.4.1.7
For the same reaction as in exercise 8.4.1.4, but with [A]0 = 0.003, [B]0 = 0.12. Show thereaction is still pseudo-first-order. The [A] values as a function of time are given for integralsteps of time from 2 to 20 in steps of 2 min: 0.00207, 0.00146, 0.00106, 0.000660, 0.000618,0.000292, 0.000238, 0.000146, 0.0000645, 0.000190.
Chm 118 Problem Solving in Chemistry

8.4 99
8.4.1.8
Establish that the order with respect to [B] is one. Find the true rate constant for thereaction in the last several exercises.
8.4.1.9
A reaction under pseudo-first-order conditions of [H+] has kpfo as a function of [H+] asfollows in pairs of [H+], kpfo: 0.1M, 1.3 x 10−3 sec−1; 0.2M, 5.2 x 10−3 sec−1; 0.5 M, 3.2 x10−2 sec−1. What is the order of the reaction with respect to [H+]?
8.4.1.10
What is the true rate constant for the reaction in the last exercise?
8.4.1.11
Use our method of “what has to collide” to understand what must be involved in a rate lawof the following form:
Rate = (k1 + k2[A])[B] (8.16)
Chm 118 Problem Solving in Chemistry

Chapter 9
Bonding
9.1 Storage of Energy in Molecules
Energy can be stored in a molecule. It is convenient for us to somewhat arbitrarily dividethat energy into a number of components. In this section we discuss these, from those ofthe lowest energy to those of the maximum. However, since these energies are of molecules,all of them are quantized.
The smallest energy gaps are those due to the translation of the molecule through space,motion in the x, y, or z direction. A very reasonable model for this motion is that of aparve on a pole, section 6.4, expanded to three dimensional space. Recall that such energydepends inversely on the square of the length of the pole which gives a dependence of theenergy levels on the volume of the container in three dimensions. This means the energylevels for a translating parve are much closer together in a large container than in a smallone. Also, the energy levels are inverse in the mass of the parve.
The next two kinds of motion that store energy do so exclusively in molecules, but not inatoms. The first of these leads to a larger energy level separations than translation: it isrotation of the molecule as a whole. The quantized energy levels for a three dimensionalrotating diatomic molecule is given by the equation:
EJ =~2J(J + 1)
2I(9.1)
where J is the quantum number and can take values of 0, 1, 2, . . . , ~ is Planck’s constantdivided by 2π, and I is the moment of inertia, the rotational equivalent of mass in linearprocesses, and is given by the expression
I =∑i
mir2i (9.2)
where the sum is over all atoms, mi is the mass of the atom, and ri is the distance the atomis from the center of mass of the molecule. The inverse term in I means that small moleculeshave large energy gaps. The next important motion, leading to larger gaps, is vibrational
100

9.1 101
motion: stretching and bending of the bonds in a molecule. Quantum vibrational energylevels are given by the expression
En = ~
√k
µ
(n+
1
2
)(9.3)
where k is the force constant, related to the stiffness of the bond and roughly related to thebond strength, and µ is the reduced mass (in a diatomic molecule, equal to m1m2
m1+m2), and n
is the vibrational quantum number, n = 0, 1, 2, . . . . Once again, the energy levels dependinversely on the mass, and directly on the strength of the bond (though both through thesquare root dependency). This means that low mass, reasonably bonded atoms, such as H,dominate the high energy portion of the vibrational energy levels.
The fourth, and largest, energy that an atom of molecule can have is electronic energy. Wemight estimate a typical value of this kind of energy (as you are asked to do in the exercises)but moving an electron from the n = 1 level of a hydrogen atom, close to the nucleus, tothe n = 2 level, further from the nucleus. What it is important to grasp is that the energyneeded to move an electron around is considerably larger than the other kinds of energiesthat we considered. A pictorial summary of the magnitudes of these energies, which youwill explore in the exercises, is given in Figure 9.1.
9.1.1 Exercises
9.1.1.1
Calculate the energy gap between the first and second energy levels for a parve, which welet here be a Ne atom, on a pole of length 10 cm. HINT: Don’t forget to get the mass peratom, not per mole.
9.1.1.2
Calculate the spacing between the first two translational energy levels for a CO2 molecule.Use a POP approximation with a length of the pole of 10 cm. HINT: Watch your massunits; not per mole!
9.1.1.3
Find the energy between the J = 0 and J = 1 rotational energy levels for CO which has anI of 1.4 x 10−46 kg m2.
9.1.1.4
Generally speaking, how would an increase in mass (and hence, usually in ri), affect thespacing of the rotational energy levels?
9.1.1.5
For CO, the force constant is 1.860 x 103 N/m. Find the energy gap between the n = 0 andn = 1 vibrational energy levels.
9.1.1.6
What will happen to the spacing of vibrational energy levels as the atoms increase in mass?
Chm 118 Problem Solving in Chemistry

9.1 102
Figure 9.1: Schematic energy level diagram. The long, thick solid lines are electronic energylevels; two are shown. Each of these electronic energy levels has associated with it severalvibrational levels, shown as (next to longest) lines, three in each of the electronic levels–thelowest in on top of the electronic energy line. The rotational levels are shown (only for thetwo lowest vibrational levels of the lowest electronic level) as the shortest lines. The grayrectangles are the closely spaced translational levels, shown only for the lowest rotationallevels.
Chm 118 Problem Solving in Chemistry

9.2 103
9.1.1.7
What will happen to the spacing of vibrational energy levels as the bond becomes weaker?
9.1.1.8
Compute the energy required to excite a hydrogen atom from the n = 1 to the n = 2 level.Recall that the energy of the hydrogen atom is given by En = 13.601/n2 eV. Express youranswer in Joules.
9.1.1.9
For the four motions: translation, rotation, vibration, and electronic, order them in termsof the size of the energy level gaps.
9.1.1.10
The compound Ru(NH3)4bip2+ has an electronic energy absorption peak at 525 nm with anε of 3950 M−1 cm−1. What is the energy gap between the two levels? What is the origin ofthis transition? HINTS: The metal is Ru(II), a rather lowly charged metal; bip, bipyridyl,has low lying empty orbitals composed mostly of p orbitals on the carbons and nitrogens.
9.1.1.11
Imagine a polyatomic gaseous molecule in a box at very low temperature. Now warm thesides of the box a little and get the molecules in the box to start vibrating a little. Whenone of our molecules of gas comes along and hits the wall, it may be given a push by thevibrating wall. What will happen to the energy of the gaseous molecule after that push?What kind of excitation will take place at low temperature?
9.1.1.12
Continuation of the last exercose. Warm the walls of the box more. Now we can givea greater translational velocity to our gaseous molecule when it hits the wall. If it thenhits another gaseous molecule off center, it could get that second molecule to rotate moreviolently. What happens to the energy of the gaseous molecules?
9.1.1.13
Continue the scenario of the last two exercises, adding more and more heat to the walls ofthe vessel. What happens to the gaseous molecules? Do they translate? How? Do theyrotate? How? Do they vibrate? How?
9.2 Using Atomic Orbitals to Make Molecular Orbitals
We have a model of atomic orbitals derived from the solution to Schrodinger’s equation.These orbitals have a shape dictated by the spherical field of the hydrogen nucleus. In amolecule, the field is no longer spherical, but rather distorted in the shape of the molecule.However, one might imagine that this undulating molecular field would not affect a wavefunction very much near any given nucleus, where the dominant force is still the sphericalattraction to the nucleus. A mathematical approach that achieves this is called a linearcombination of atomic orbitals, abbreviated LCAO. In such an approach, we add theatomic wave functions on various atoms to make a molecular orbital. Since the atomic
Chm 118 Problem Solving in Chemistry

9.2 104
functions decrease rapidly with distance from the nucleus, the dominate effect of the additionoccurs between nuclei, which is exactly where one would anticipate bonding electrons wouldcongregate. However, the electronic charge that concentrates between the nuclei must comefrom somewhere. As we shall see, it comes and goes from behind the nuclei.
To see the essence of the LCAO method of bonding, we consider only a one dimensionalradial hydrogen atom 1s orbital on each of two nuclear centers, one at -1 units and theother at +1 units. These two wave functions are show in Figure 9.2. Note that there is aregion in space, shaded pink in the Figure, where the two functions overlap in space. Thisis called the overlap region and will be important to us because it is here that constructive(or destructive) interference can occur. If we add these two functions as drawn they willinteract constructively since both are positive near zero. We must insure that the areaunder the curve for the square of this new wave function generated by adding the twoatomic functions is unity (called normalization and it makes the total probability of findingthe electron somewhere equal to one). Adding and normalizing gives us the blue (solidline) curve in Figure 9.3. The language we use to describe this blue curve is that it is theprobability resulting when the two waves interact. What should a non-interacting situationlook like? In this case we would have a 50% chance of finding the electron in the wavefunction on the right nucleus and a 50% chance on the left. If we plot this non-interactingcurve we get the brown (dotted) line in Figure 9.3. Notice that the interaction leads to anincreased electron probability in the internuclear region, just as we hypothesized. Becauseit is hard to see what happens behind the nuclei, that is, in the region of space from 1 to3 (or from -1 to -3), I have plotted in Figure 9.4 an enlarged plot between 1.5 and 2.5.Note that the brown dashed curve is above the blue curve in this region, meaning there isless electron probability for the interacting system behind the nuclei than there is in thenon-interacting system. Electron density moves from behind the nuclei to between them inthe addition of the functions, in this constructive-interaction-wave function.
We made what is called a bonding molecular orbital by the addition of the two hydrogenatom 1s wave functions. There is another way to combine the two functions, that is tosubtract them. This is shown in Figure 9.5 and has destructive interaction in the internu-clear region. Not only is this a second way of “adding,” but it is also a necessary process.The verbal way to describe the process is to say we must have conservation of orbitals.We started with two hydrogen 1s wave functions, two orbitals, and when we get done wemust also have two orbitals. The addition of the two functions is one of the new ones;the subtraction is the other. The logic behind this verbal description is that an orbital, awave function, is the only way of describing an electron. If we initially had two electrons todescribe, and did so with the two hydrogen 1s wave functions, φ1 and φ2, we must also havethe ability to describe the two electrons when we finish. The addition and subtraction arethose two ways:
ψb = B(φ1 + φ2) (9.4)
ψa = A(φ1 − φ2) (9.5)
where A and B are normalizing constants. In the case presented here the addition leads toa buildup of electron density between the nuclei and hence is label with a “sub-b” to reflectthat this is a bonding molecular orbital. In contrast, as we now develop, the subtraction isactually an antibonding orbital, hence the subscript “a.”
Chm 118 Problem Solving in Chemistry
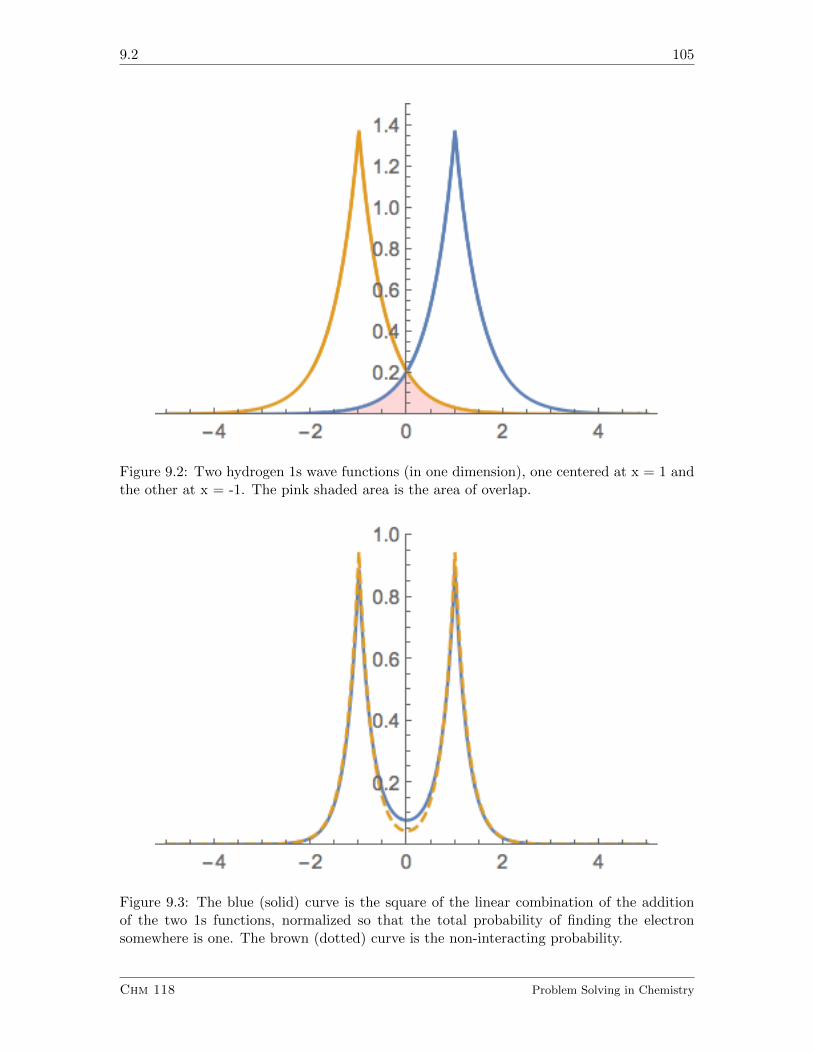
9.2 105
Figure 9.2: Two hydrogen 1s wave functions (in one dimension), one centered at x = 1 andthe other at x = -1. The pink shaded area is the area of overlap.
Figure 9.3: The blue (solid) curve is the square of the linear combination of the additionof the two 1s functions, normalized so that the total probability of finding the electronsomewhere is one. The brown (dotted) curve is the non-interacting probability.
Chm 118 Problem Solving in Chemistry
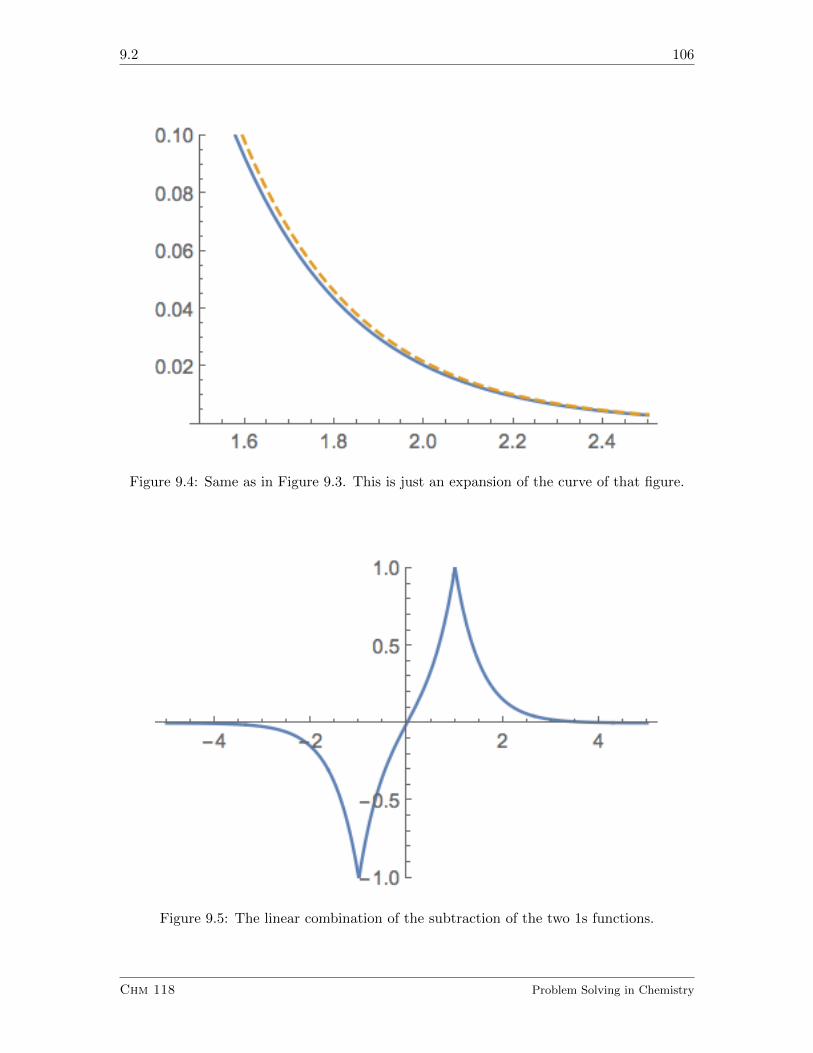
9.2 106
Figure 9.4: Same as in Figure 9.3. This is just an expansion of the curve of that figure.
Figure 9.5: The linear combination of the subtraction of the two 1s functions.
Chm 118 Problem Solving in Chemistry
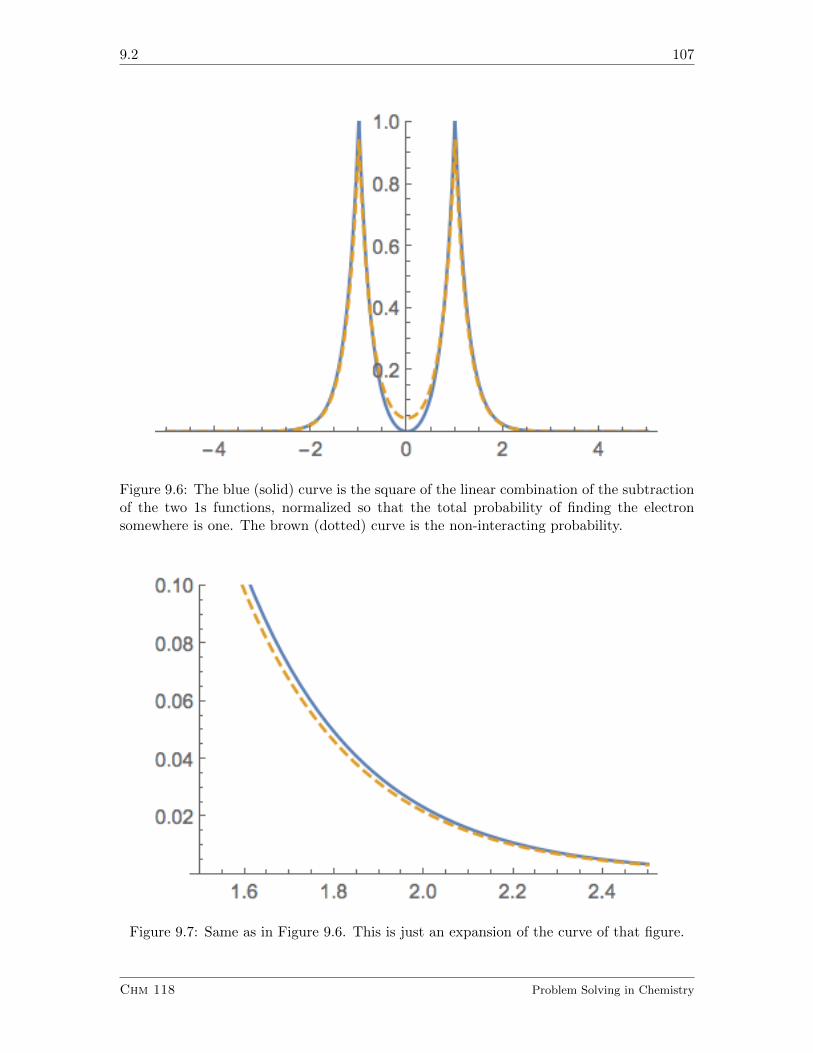
9.2 107
Figure 9.6: The blue (solid) curve is the square of the linear combination of the subtractionof the two 1s functions, normalized so that the total probability of finding the electronsomewhere is one. The brown (dotted) curve is the non-interacting probability.
Figure 9.7: Same as in Figure 9.6. This is just an expansion of the curve of that figure.
Chm 118 Problem Solving in Chemistry

9.2 108
Figure 9.8: Overlap of two s orbitals using angular functions. The line is the internuclearaxis.
The destructive interaction resulting from the subtraction of the two functions actuallylowers the electron density between the two nuclei, relative to the non-interacting proba-bility, as shown in Figure 9.6. Where does that electron density go? Figure 9.7 shows anenlargement of the region behind the nuclei for the antibonding combination: the antibondhas greater probability there than does the non-interacting probability. In other words,in the antibonding combination, electron density flows from the internuclear region to theregion behind the nuclei. This flow would tend to pull the nuclei away from each other:truly antibonding.
The following exercises deal with the same issues just described, but use angular functionsinstead of radial ones.
9.2.1 Exercises
9.2.1.1
Let atom A be at the origin of the coordinate system. Let atom B be at z = 1; y = 0; x =0. Let the x axis be pointing “in and out”, the y axis “up and down”, and the z axis “leftand right.” Use an angular wave function picture to show the overlap of two s orbitals, oneon each atom. Compare your picture to Figure 9.8.
9.2.1.2
Same coordinate system as in exercise 9.2.1.1. Show, using angular functions, the overlapof a pz orbital on A with an s orbital on B. HINT: Don’t forget that you have to “shade”one side of a p orbital.
Chm 118 Problem Solving in Chemistry

9.3 109
Figure 9.9: Overlap of two p orbitals. The line is the internuclear axis.
9.2.1.3
Same coordinate system as in exercise 9.2.1.1. Show the overlap of a pz orbital on A with allthree of the p orbitals on B. Does one of your pictures look like that in Figure 9.9? Whichp orbital on B? Added or subtracted to that on A?
9.2.1.4
Same coordinate system as in exercise 9.2.1.1. Show the overlap of a py orbital on A witha py orbital on B. Compare your answer with Figure 9.10.
9.2.1.5
Same coordinate system as in problem 9.2.1.1. Show the overlap of a s orbital on A withall five of the d orbitals on B.
9.2.1.6
Same coordinate system as in problem 9.2.1.1. Show the overlap of a pz orbital on A withall five of the d orbitals on B.
9.2.1.7
We have seen the overlap of functions on two separate atoms. When that overlap is construc-tive, what happens to the amplitude of the wave (function) in the region of overlap? Whathappens to the square of the wave function? What happens to the electronic probability?
9.2.1.8
If the electronic probability (electron density) is greater between the two nuclei because ofthe occupation of the molecular orbital, will the two nuclei be attracted or repelled?
9.2.1.9
We have seen the overlap of functions on two separate atoms. When that overlap is destruc-tive, what happens to the amplitude of the wave (function) in the region of overlap? Whathappens to the square of the wave function? What happens to the electronic probability?
9.2.1.10
If the electronic probability (electron density) is smaller (than the non-interacting function)between the two nuclei because of the occupation of the molecular orbital, will the twonuclei be attracted or repelled?
Chm 118 Problem Solving in Chemistry

9.3 110
Figure 9.10: Overlap of two p orbitals perpendicular to the molecular axis. The line is theinternuclear axis.
9.3 A Nomenclature for Molecular Orbitals
For the simplified molecular orbitals in this course, formed as they are from linear combi-nation of atomic orbitals, we need a nomenclature system. As we shall see, there are twoaspects to our naming of the functions, the first is the dominant way the overlap occurs,which is the subject of this section; the second we shall return to involves the symmetry ofthe functions.
The overlap of two 1s orbitals in a hydrogen molecule is schematically pictured in Figure 9.8.In that figure is a faint black circle drawn in a plane perpendicular to the internuclear axis.What happens to the sign of the wave function as you travel around that circle? HINT:Another view of this same circle is given in Figure 9.11. When the sign of a wave functiondoes not change as you “circle around,” the bonding is said to be a sigma bond (or a sigmaanti-bond), designated σ (or σ∗). On the other hand, when the sign of a wave functionchanges twice upon a complete “circle around,” that is, from “plus” to “minus” and backto “plus,” the bonding is said to be a pi bond (or a pi anti-bond), designated π (or π∗)–seethe exercises. (We see it seldom except in transition metal ions, but when the sign changesfour times, the bond is a delta bond, designated δ (or δ∗).)
Chm 118 Problem Solving in Chemistry
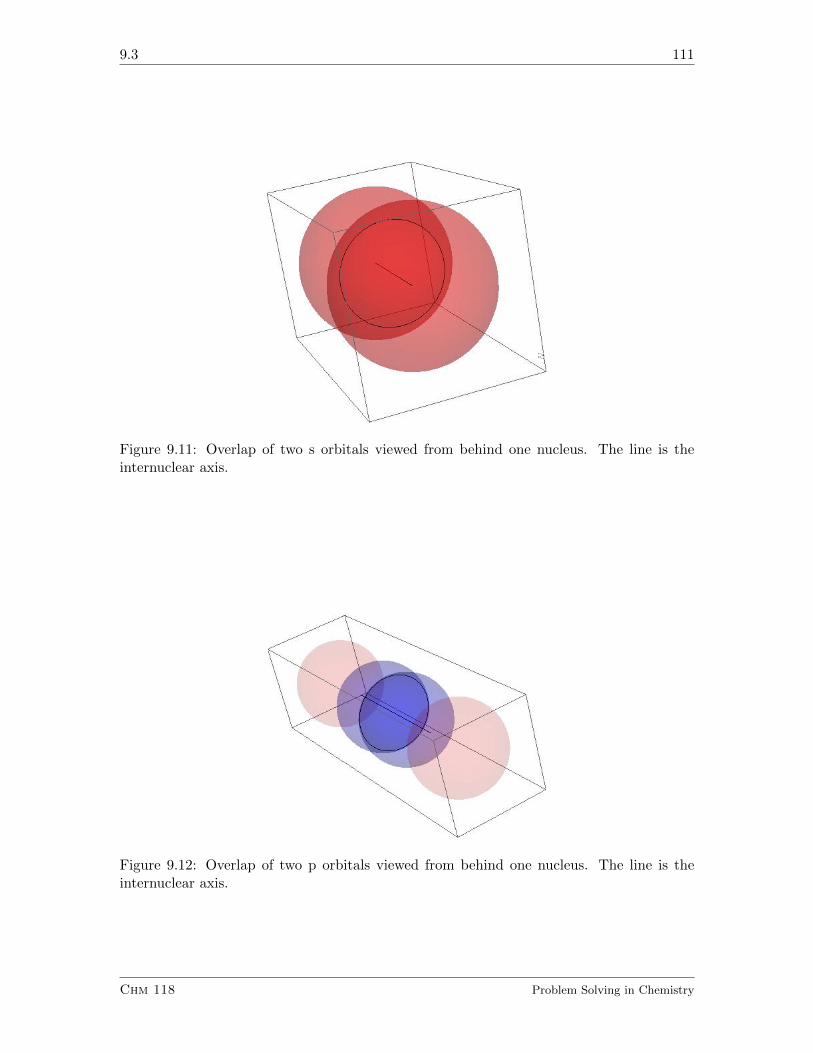
9.3 111
Figure 9.11: Overlap of two s orbitals viewed from behind one nucleus. The line is theinternuclear axis.
Figure 9.12: Overlap of two p orbitals viewed from behind one nucleus. The line is theinternuclear axis.
Chm 118 Problem Solving in Chemistry

9.4 112
Figure 9.13: Overlap of Two p Orbitals Viewed from Behind One Nucleus. The line is theinternuclear axis.
9.3.1 Exercises
9.3.1.1
A picture of two p orbitals overlapping is given in Figure 9.9; another view is given inFigure 9.12. Is this a σ overlap?
9.3.1.2
Another picture of two p orbitals overlapping is given in Figure 9.10; another view of thissame overlap is given in Figure 9.13. Does the wave function change sign as you “circlearound?” Would you call this overlap a σ overlap? Or would you call it a π overlap?
9.3.1.3
Bring two hydrogen atoms with their 1s wave functions together and form the constructiveinteraction. Also form the destructive interaction. What would you name those two differentmolecular wave functions? Which one is more stable?
Chm 118 Problem Solving in Chemistry

9.4 113
Figure 9.14: MO diagram for H2.
9.4 Molecular Orbital Energy Diagrams
We need a way to symbolize the relative energies of the bonding and antibonding orbitalswe have been discussing. Such a representation is made using what is called a MO diagram.One for the H2 case is shown in Figure 9.14. The vertical axis is energy, with the morestable orbitals being at the bottom. The energy of the orbitals are represented by lines, andthe lines on the left and right sides of the diagram represent the energies of the atomic levelswe started with. Just below those lines are given the atomic functions (with a small dotindicating the position of the other nucleus for perspective). In the center of the diagramare the two molecular orbitals, again indicated by lines; the stable bonding one, and theunstable antibonding one. Beneath each of these is a “picture” of the orbital.
9.4.1 Exercises
9.4.1.1
The diagram in Figure 9.14 refers to H2 only because it has a pair of electrons in the σbond. The same diagram would work for for H+
2 , He+2 , and He2. Write the configurations
(in the sense of σaσ∗b (a and b are numbers) for each of these species.
9.4.1.2
The bond energies of H+2 , H2, He+
2 , and He2 are 61, 103, 55, and 0 kcal/mole, respectively.Explain using MO theory.
9.4.1.3
If the H-H distance in H2 is 0.74 A, and that in H+2 is 1.06 A, what would you expect for
He+2 ?
Chm 118 Problem Solving in Chemistry

9.5 114
9.4.1.4
Imagine an atom with only a pz orbital. Bring two of these together with the internuclearaxis the z axis and make the corresponding MO diagram. HINT: Read carefully: this is anartificial problem, but an easy one.
9.4.1.5
Imagine an atom with only a pz orbital. Bring two of these together with the internuclearaxis the x axis and make the corresponding MO diagram. HINT: Read carefully: this is anartificial problem, but an easy one.
9.4.1.6
Is there a difference between the strength of the bonding in the last two exercises? If so,make sure your diagrams reflect that before you do the next exercise.
9.4.1.7
Imagine an atom with only p orbitals, three of them. Bring two of these together with theinternuclear axis the z axis and make the corresponding MO diagram.
9.4.1.8
Assume your molecule from the last exercise has 6 electrons. Give the configuration.
9.4.1.9
Build an MO energy diagram for a homonuclear diatomic molecule having 2s and 2p atomicorbitals. Do not let any interaction occur between an s orbital on one center and a p orbitalon the other; This is called “ignore s/p mixing.”
9.4.1.10
Write the configurations and state the number of bonds in the homonuclear diatomics fromLi2 to Ne2 using your MO diagram from the last exercise.
9.5 Heteronuclear Diatomics and the First Row Diatomicswith s/p Mixing
In the last section you found the MO diagrams for homonuclear diatomics without s/pmixing, that is, with no orbital interaction between an s orbital and a p orbital. Yet weknow from exercise 9.2.1.2 that an s orbital overlaps quite well with a p orbital. So thereis no justification for ignoring the interaction except that the s orbital is more stable thanthe p. This is not a trivial issue and we pause to investigate it first.
Let’s consider the interaction of an 1s orbital on a hydrogen atom with the more stable 1sorbital on a He atom. These atomic orbitals are shown in Figure 9.15 with the hydrogenatom on the left and the He on the right. The two s orbitals still overlap and form thebonding (constructive) and antibonding (destructive) MO. But think about the situation.The helium 1s level is more stable than that of the hydrogen atom and if we added equalamounts of the two functions together, as is implied in equation 9.5, then the electrons wouldhave equal probability of being on the hydrogen (less stable) atom and the helium (more
Chm 118 Problem Solving in Chemistry

9.5 115
Figure 9.15: MO diagram for HHe+.
stable) atom. Why should electrons leave the comfortable environment of a dipositve chargeto wander over to a unipositive one? So the bonding MO should be more concentrated onthe helium atom. We write this as:
ψb = cHφH + cHeφHe (9.6)
where cHe > cH . Using our concept of orbital conservation, if we used a lot of φHe in thebonding orbital, we only have a little left to use in the antibonding one. Conversely, weused only a little of φH in the bonding orbital, we have a lot left to use in the antibondingone. Therefore the antibonding orbital is:
ψa = c∗HφH + c∗HeφHe (9.7)
where c∗H > c∗He. The drawings at the sides of the molecular orbitals in the center of ourMO diagram reflect this argument.
In a similar manner, we need to allow s orbitals (stable) and p orbitals (less stable) onhomonuclear diatomics to “mix.” The easiest way to do this is to take the answer withoutmixing and make a correction for what mixing does; such a treatment, a two step treatment,is called perturbation theory. The left hand side of Figure 9.16 gives us the results for ahomonuclear diatomic without s/p mixing. The first feature to note is an important resultwe can use because of our development earlier in the course: only orbitals of the samesymmetry can interact. Application of symmetry to homonuclear diatomics is a little messyin that the set of symmetry operations is infinite. You can see this by noting that a rotationof 17.7 degrees about the internuclear axis is a symmetry operation, as is one of 17.8 degrees.
Chm 118 Problem Solving in Chemistry

9.5 116
Figure 9.16: MO diagram for homonuclear diatomic without and with s/p mixing.
To use symmetry we will just select certain symmetry operations and demand that if twoorbitals are going to interact, they (as “objects” in the previous discussion) must do thesame thing under those selected symmetry operations. The exercises lead you through theprocess.
9.5.1 Exercises
9.5.1.1
Let the z axis of our diatomic molecule be the internuclear axis. We consider three symmetryoperations, C2z, σxz, and σxy, the plane bisecting the molecule and perpendicular to theinternuclear axis. Determine what happens to all eight orbitals on the left side of Figure 9.16under these three symmetry operatons. Making a little table might be useful.
The choice of symmetry operations in this exercise are such thatthey do not establish a critical feature of the π and the π∗ levels. Incontrast to all the σ levels, the π levels are turned into each otherunder certain symmetry operations, such as C4z. From our previousdiscussion of objects in symmetric environments, this means theymust be “considered together.” A real physical consequence is thatwhen certain objects are “considered together” it leads to degener-ate systems. That is what is happening here. The pair of π levelsbelong to a set of characteristic numbers in this infinite group thatrequires them to be degenerate in energy.
9.5.1.2
In the last exercise you presumably found that the level σ1 and the level σ2 had the samesymmetry. Hence they are capable of interacting. The red (dotted) lines in Figure 9.16 showthe interaction. Such interactions always stabilize the more stable level and destablize the
Chm 118 Problem Solving in Chemistry

9.5 117
least stable. It is as if the orbitals “repel” each other. Another way to look at this: Thelower orbital takes whatever “good” things the upper one has and ships back to the upperany “bad” things it has. At this stage it is worth noting that how far the orbitals move,whether the upper level is pushed above the π levels, as indicated in the Figure, is notknowable at the level we are doing things.
9.5.1.3
It is interesting to see why the orbitals “repel” as indicated in the last problem. To do so,we could draw the new orbital by adding and (of course) subtracting the starting functions.Try it. Add in a purely graphical way σ1 to σ2 and see what you get.
9.5.1.4
Now subtract in a purely graphical way σ1 from σ2 and see what you get.
9.5.1.5
From the pictures in the last two exercises, can you guess which one new picture, theaddition or the subtraction, is the more stable orbital?
9.5.1.6
Do any other pair of orbitals on the left-hand side of Figure 9.16 have the same symmetryand hence are capable of interacting? If so, draw in the interaction.
9.5.1.7
Add and subtract σ∗1 and σ∗2 and see what the functions look like. Can you determine whichis lower in energy?
9.5.1.8
The diagram you have now build is not so easily analyzed as the one without s/p mixingbecause of the “good/bad character” trade off in the mixing. But there are interestingconsequences. What conclusion did you draw about the stability of Be2 in exercise 9.4.1.10?What conclusion do you now draw? The fact is that Be2 is very slightly stable rather thanbeing like He2.
9.5.1.9
Although this exercise concerns interesting aspects of s/p mixing, it involves very unstablecompounds. Nevertheless, the data support the model we have developed. If we accept thatσ2 is pushed above π, predict the magnetic characteristic–diamagnetic or paramagnetic–ofB2 for both the simple model of bonding and the model with s/p mixing. Do the same forC2. Experimentally, both answers agree with the model with s/p mixing.
9.5.1.10
Draw a Lewis structure for O2. One of the great triumphs of MO theory is that it naturallypredicted that O2 was parmagnetic. Use the MO diagram to show that this is true.
9.5.1.11
How many net bonds are there in N2? in N2+2 ? HINTS: Do this without s/p mixing. Give
your answer professionally: “There are two σ bonds and four π bonds.”
Chm 118 Problem Solving in Chemistry

9.6 118
9.5.1.12
How many bonds (and of what type) are there in N2–2 ?
9.5.1.13
The bond lengths in O+2 , O2, O–1
2 , and O2–2 are 1.122, 1.21, 1.26, and 1.49 A. Comment.
9.5.1.14
The bond lengths in C2, N2, O2, and F2 are 1.243, 1.298, 1.208, and 1.412 A. Comment.
9.5.1.15
Build the m.o. diagram for V2+2 considering only d orbitals on the atom. HINT: Remember
that a V(I) compound exhibits chemistry of d electrons only: Count all valence electrons,but then put all of the appropriate number into the d orbitals of the vanadium.
9.6 Hybridization as a Simplifying Tool. Part I: sp Hy-bridization
It is relatively easy to deal with diatomic molecules, but they are of limited interest. Morecompelling are polyatomic species. As the number of atoms increases, the molecular orbitalprocedure becomes harder (unless there is lots of symmetry, as we shall see), and less infor-mative for a chemist. To deal with this situation, chemists have adopted an approximationthat works reasonably well in a number of cases. This approximation makes the answers toMO problems closer to the intuitive Lewis structures. This method is called hybridization.It fits in at this position in our development because for diatomics, hybridization allows usto mix s and p orbitals on an atom before we build the molecular orbitals.
The basis of hybridization is simple. We add and (as always) subtract functions on the sameatom. This process generates orbitals that are not solutions to Schrodinger’s equation, butas we shall see, are useful in getting the bonding of molecules. The result of hybridization isalways orbitals that point in some specific direction; contrast this with an s orbital, whichpoints everywhere, a p orbital that points left and right, or a d orbital that points in fourdirections. Orbitals that point in a specific direction simplifies problems because only atomsin that direction bond well to the hybrid.
9.6.1 Exercises
9.6.1.1
Draw the z axis from an atom to the right and left on the paper. Draw an atomic s orbitalon an atom at the origin and let it be positive everywhere. Do you agree that this orbitalcould have a value of 0.03 at the point x = 0, y = 0, z = 1? What would the value be atthe point (0,0,-1)? Draw a pz orbital at the same atomic center. Do you agree that thissecond orbital could have a value of 0.02 at the point (0,0,1)? What would the value beat the point (0,0,-1)? Now if we add these two orbitals, what would the net value be at
Chm 118 Problem Solving in Chemistry

9.6 119
the points (0,0,1) and (0,0,-1)? Choose the correct words in the following sentence: “Theorbital formed from adding the s and the p atomic orbitals is (bigger or smaller) when z ispositive and is (bigger or smaller) when z is negative.” The orbital that you have formed iscalled an sp hybrid.
9.6.1.2
Do the last exercise again, except this time subtract the p from the s. HINT: Recall, “ifyou add two orbitals, you must . . . ”.
9.6.1.3
Construct an sp hybrid so that it maximizes in the positive x direction. To do this, youmust understand which orbital in the last two exercises gave the directional properties.Show that the second sp hybrid must maximize in the negative x direction. HINT: Use theconservation of orbitals rule: if you add two functions to make one new one, you must alsosubtract them to make another new one.
9.6.1.4
When you build an sp hybrid you use one s orbital and one p orbital. What orbitals are lefton the atom that “have not been used?” You will find as you progress that these orbitalsare always used for producing π bonds.
9.6.1.5
Given your answer to the last exercise, complete the following statement: “An atom inwhich we assume sp hybridization will have π bonds.
9.6.1.6
Build the m.o. diagram for N2. Use sp mixing via hybridization before bonding. Once youset these up, divide them into those that point at the other nitrogen atom and those that donot. Let each of these pairs interact. The purpose of the sp hybrid on one nitrogen atom isto get a big orbital pointing at the other nitrogen orbital, and the other way around. Thesetwo interact strongly. On the other hand, the other sp hybrid on each nitrogen points awayfrom the other orbital so it does not bond very well; give them small interactions.
9.6.1.7
Build the m.o. diagram for CO. Use sp mixing via hybridization on the carbon atom beforebonding, but leave the oxygen atom “unhybridized” and use only p orbitals on the oxygenatom.
9.6.1.8
How does the m.o. diagram of CO differ from the isoelectronic N2?
9.6.1.9
In going from CO to CO+ the bond length changes only slightly, from 1.128 to 1.115 A.How could you account for this observation? HINT: Think about what must be true of theelectron that you remove for the bond length to change very little.
9.6.1.10
It is much easier to ionize NO than to ionize CO. Why? HINT: Build an MO diagram.
Chm 118 Problem Solving in Chemistry

9.7 120
9.6.1.11
The advantage of hybrid orbitals is that bonds are localized and therefore, most of the time,only one overlap has to be considered for each orbital. Take advantage of this truth to buildan mo diagram for C2H2. HINT: You will need a structure with geometry.
9.7 Hybridization as a Tool. Part II: sp2 and sp3 Hybridiza-tion
Hydribization is used extensively in organic chemistry, the chemistry of carbon compounds.There are two other hybridization schemes that are commonly used, although neither is aseasy to comprehend as the sp hybrid case discussed above. The first is sp2 hybridization,which mixes an s orbital with two p orbitals. These three hybrids point at the corners of atriangle. When you want to bond a carbon atom to three other atoms, sp2 hybrids shouldbe used. The other hybridization mixes all three p orbitals with the s orbital; this producessp3 hybrids that point at the vertexes of a tetrahedron. They are used to bond a carbonatom to four other atoms.
9.7.1 Exercises
9.7.1.1
When you build an sp2 hybrid you use one s orbital and two p orbitals. What orbitals areleft on the atom that “have not been used?” For what can you use this left over p orbital?
9.7.1.2
Complete the following statement: “An atom in which we assume sp3 hybridization willhave π bonds.
9.7.1.3
The advantage of hybrid orbitals is that bonds are localized and therefore, most of the time,only one overlap has to be considered for each orbital. Take advantage of this truth to buildan mo diagram for BH3.
9.7.1.4
The advantage of hybrid orbitals is that bonds are localized and therefore, most of the time,only one overlap has to be considered for each orbital. Take advantage of this truth to buildan mo diagram for C2H4.
9.7.1.5
Build an mo diagram for C2H6 using hybrid orbitals.
9.7.1.6
Some molecules are best treated with a combination of methods, using hybrid orbitals andsymmetry. The second we turn to next, but first let’s do a separation of a problem into twoparts to prepare for later. Consider the molecule C3H+
5 in the form CH2CHCH+2 . Draw
Chm 118 Problem Solving in Chemistry

9.8 121
Figure 9.17: MO diagram for water molecule.
the Lewis structure and use VSEPR to determine structure. What hybridization do youuse for the three carbon atoms? Consider only the sp2 orbitals on each carbon and thehydrogen atoms; that is, neglect the three pz orbitals on the three carbon atoms. Make anmo diagram for the σ part of the bonding scheme.
9.8 Using Symmetry to Determine the Orbitals that canBind. Part I. Simple Systems
We have seen previously only orbitals of the same symmetry can bind to each other. Inpolyatomic molecules, use of symmetry greatly simplifies the approach to MO diagram. Thetrick to using symmetry is to produce symmetry adapted linear combinations of atomicorbitals, SALCAO. Consider a water molecule as our first example. This molecule has C2v
symmetry with a table of characteristic numbers given here:
where the molecular plane is the xz plane, as indicated in molecular sketch in Figure 9.17.Let’s classify the orbitals on the oxygen atom according to their symmetry. You should dothis. What I got is indicated in the last column of Table 9.1; for instance, the pz orbitalhas a1 symmetry and the px orbital has b2 symmetry. Notice in Figure 9.17 that these are
Chm 118 Problem Solving in Chemistry

9.8 122
C2v E C2 σyz σxzA1 1 1 1 1 Os, Opz , Ha + Hb
A2 1 1 -1 -1B1 1 -1 1 -1 Opy
B2 1 -1 -1 1 Opx , Ha + Hb
2 0 0 2 (Ha, Hb)
Table 9.1: Characteristic number table for water molecular orbitals.
labeled as such in the right hand column. These symmetry assignments are straightforwardbecause the oxygen atom orbitals are turned only into plus or minus themselves.
The hydrogen atom orbitals are not so simple. The orbital on Ha is turned into that onHb and hence the two must be considered together, as two objects. This results in thenumbers given in the last row of Table 9.1; prove this to yourself. To find the combinationof hydrogen orbitals that have characteristic numbers from the named rows of the table, weadd and subtract the two hydrogen 1s functions, generating what are called ha1 = ha + hband hb2 = ha - hb. Show that these combinations do have the indicated symmetry.
Bonding can occur between orbitals of the same symmetry. To start off, we note thatthere is only one b1 orbital, the py on the oxygen atom; it is therefore non-bonding and“goes straight across” in the diagram–see Figure 9.17. There are only two orbitals ofb2 symmetry, one of the oxygen p orbitals and the antisymmetric linear combination ofhydrogen orbitals we called hb2. These therefore “repel” each other and we get one bondingb2 and one antibonding b2. The only remaining symmetry is a1; there are three orbitals ofthis symmetry, and that situation is hard to deal with in a qualitative manner, as we aredoing. The easiest way out of this problem is to remember that orbitals that differ in energya lot don’t interact very much. So to approximate what is happening, we neglect the verystable oxygen atom 2s orbital, and then let the oxygen pz interact with ha1. Figure 9.17shows all of this. Work your way through the arguments and be sure you understand whatis happening.
9.8.1 Exercises
9.8.1.1
Find the names of the characteristic numbers for the s and p orbitals on the sulfur in SF2.
9.8.1.2
Find the names of the characteristic numbers for the F pin orbitals in SF2. What do thetwo combinations look like? HINT: A pin orbital has the shape of a p orbital and pointsfrom the F to the S on both F’s; hence its name “in.”
9.8.1.3
Build an m.o. diagram for SF2 molecule using only pin orbitals on the F and only p orbitalson the S.
Chm 118 Problem Solving in Chemistry

9.9 123
9.8.1.4
What is the symmetry of NO–2? Let the plane of a NO–
2 ion be the xz plane. Then thereare three py orbitals on the three atoms. Sketch these.
9.8.1.5
Find the three symmetry adapted orbitals that result from the three orbitals you describedin the last exercise.
9.8.1.6
Combine the SALCOs of the last problem to make an MO diagram for nitrite ion.
9.8.1.7
There are four electrons in the π system of NO–2. (Do you see where those are from a Lewis
structure?) Is there a π bond in NO–2? On what atoms is it located?
9.9 Using Symmetry to Determine the Orbitals that canBind. Part II. Degenerate Systems
As we saw previously, when two or more functions belong to a characteristic set of numberswith a two or greater under the identity operator, manipulation of them is a little moredifficult. We work through an example here. Imagine an s orbital on one of the hydrogenatoms in NH3. What happens to that s orbital when you carry out a C3 symmetry opera-tion? Clearly it gets carried into another hydrogen atom. That means we have to considerthe two together. However, a C−3 symmetry operation takes the initial hydrogen atominto the third one. So all three must be considered together. Using the C3v table given inTable 9.2, we can find the numbers associated with these three functions as indicated inthe last row of the table. Examining this set of numbers, it is clearly composed of a1 and echaracteristic sets. Verify this for yourself. Since we have three functions, the easy rule ofadding and subtracting doesn’t work and we need to be more innovative (or use equation 5.2of section 5.8). It is easy, always, to get the a1 function as it has to have the same signeverywhere, and all functions of equal magnitude. In this case that function would be:
ψa1 = h1sa + h1sb + h1sc (9.8)
Since the pz function on the nitrogen atom of the NH3 has a1 symmetry (we will ignore thenitrogen atom 2s function as we did previously to make matters simpler), these two orbitalsbind to each other to make a bonding and antibonding pair.
What does the e set of the hydrogens interact with? The answer, although it is somewhatmessier to show, is the N px and py orbitals, which have characteristic numbers labeled “e”.Here’s a start to showing that: If we let one of the H atoms in NH3 have y coordinates of 0(i.e., it is in the xz plane), then the σv operation in the xz plane takes px and py into px and-py, respectively, hence giving a total characteristic number of 0. Also, it is clear that theidentity operator takes px and py each into themselves, generating a characteristic numberof 2. That gives us two of the characteristic numbers of the “e” set. Close enough unlessyou want to be hardcore:
Chm 118 Problem Solving in Chemistry

9.10 124
C3v E 2C3 3σvA1 1 1 1A2 1 1 -1E 2 -1 0
3 0 1
Table 9.2: The C3v table for NH3.
The operation C3 also works, but since it is a 120o rotation, theorbital px goes, as does the vector x, into -1/2 of itself (and -
√3/2
of py, which is why the two functions are mixed up together) as youcan show with simple plain geometry. Likewise for the vector y andhence py. The total “functions” that go into themselves is then -1/2-1/2 = -1, as required by the characteristic number.
9.9.1 Exercises
9.9.1.1
Build an MO energy level diagram for NH3 using the symmetry adapted functions fromabove. HINT: Use only the p orbitals on the nitrogen atom.
9.9.1.2
Consider a square planar compound of a transition metal. Let the ligands have only pinorbitals and the metal have only d orbitals. Find the symmetry of the ligand orbitals.HINT: The D4h characteristic numbers table is given in Table 9.3.
9.9.1.3
What symmetry do the metal d orbitals have in the system defined in the last exercise?HINTS: Some metal orbital may get turned into another under a given symmetry operation.If so it is possible that the two together may generate a set of numbers in the characteristicnumber table. We would then say, speaking professionally, that “function x and functiony have Eu symmetry. Also, pay attention to the number under the center of inversionoperator; it is often easy to distinguish between Eu and Eg on the basis of this operator.
9.9.1.4
Make an MO diagram of the square planar molecule defined in the last two exercises.
9.9.1.5
If the metal is Ni2+ and the ligands each have two electrons in their pin orbitals, put theappropriate number of electrons into your MO diagram of the last exercise.
Chm 118 Problem Solving in Chemistry

9.10 125
D4h E 2C4 C2 2C′2 2C”
2 i 2S4 σh 2σv 2σdA1g 1 1 1 1 1 1 1 1 1 1A2g 1 1 1 -1 -1 1 1 1 -1 -1B1g 1 -1 1 1 -1 1 -1 1 1 -1B2g 1 -1 1 -1 1 1 -1 1 -1 1Eg 2 0 -2 0 0 2 0 -2 0 0A1u 1 1 1 1 1 -1 -1 -1 -1 -1A2u 1 1 1 -1 -1 -1 -1 -1 1 1B1u 1 -1 1 1 -1 -1 1 -1 -1 1B2u 1 -1 1 -1 1 -1 1 -1 1 -1Eu 2 0 -2 0 0 -2 0 2 0 0
Table 9.3: The D4h table of characteristic numbers.
9.10 Exploring a Model for Solids
In this section we take what we know about MO diagrams and examine a long chain ofatoms, bonded together. This is a one-dimensional model of a solid. It is based on conceptsalready known to you.
9.10.1 Exercises
9.10.1.1
This problem is fictitious to make it easier to do. Imagine a set of atoms of type A in theplane of the paper with the x axis from left to right. These A atoms have only one p orbitalemphperpendicular to the x direction, in our case, “up and down.” We do all our liningup of atoms along the x direction. Build a MO diagram for a dimer of A, each with oneorbital. Sketch the two molecular orbitals.
9.10.1.2
Now bring two of the dimers from the last exercise together along the x axis, each withits two molecular orbitals. First we let orbitals close to each other (in energy) interact;construct the m.o. scheme for the interaction of the two lowest molecular orbitals. Do soalso for the two high molecular orbitals. This molecule with the four molecular orbitals isour tetramer. Sketch each of the four orbitals.
9.10.1.3
The tetramer of the last exercise has a plane of symmetry down the middle of the moleculeand perpendicular to the x axis. The molecular orbitals that you got there are eithersymmetric (go into themselves) or asymmetric (go into minus themselves) upon reflectionin that plane. If two molecular orbitals have the same symmetry, they can interact (if theydid not previously). Do the appropriate interaction between the low levels and the highlevels from the last problem.
Chm 118 Problem Solving in Chemistry

9.11 126
Orbital E σxy σxz σyzI pxI pyI z
2 F pin, added2 F pin, subtracted
Table 9.4: Selected symmetry operations for MO diagram for IF–2.
9.10.1.4
Bring two of the simple tetramers (exercise 9.10.1.2) together to make an octamer. Whatdo your molecular orbitals look like? Do you see a pattern in how they wave? Do yousee why the more stable ones are stable? why the ones in the middle are there? why theunstable ones are unstable?
9.10.1.5
What happens if you keep bringing two of what you made in this set of exercises together?What do you get for a molecular orbital diagram? You will have to describe this ratherthan draw it out. This set of molecular orbitals is called a band of bonds (and antibonds)and is used in a theory of metal bonding called band theory.
9.11 Molecular Orbital Theory of Hypervalent Compounds
When we discussed the exceptions to the Lewis octet theory earlier (see section 2.5) weconcluded that expansion beyond the octet required electronegative elements on the outsideof an element in the second row or higher of the periodic table. Herein we establish why thiscondition is needed. We use as our example molecule IF–
2, a linear molecule with the iodineatom at the center. The exercises will lead you through a MO approach to understandingthe bonding in this species.
9.11.1 Exercises
9.11.1.1
Since the symmetry of IF–2 has an infinite number of symmetry operations (and that is a
lot to deal with), we use only selected operations. Consider only the p orbitals on the I andlet the internuclear axis be the z axis. Fill in the top part of the chart in Table 9.4.
9.11.1.2
On the fluorine atoms, let’s consider only the pin orbitals. Since there are two orbitals, wemust add them and subtract them. Describe the symmetry of the addition of the two pinorbitals on the two fluorine atoms. Put you entry in the chart in Table 9.4.
Chm 118 Problem Solving in Chemistry

9.11 127
9.11.1.3
Describe the symmetry of the subtraction of the two pin orbitals on the two fluorine atoms.Finish filling in the chart in Table 9.4.
9.11.1.4
Start an MO diagram by putting in the atomic orbitals on the sides. HINT: Which is morestable, 2p on fluorine or 5p on iodine?
9.11.1.5
Can the function made by adding the two fluorine pin orbitals interact with any orbital?Draw the appropriate line to its energy in the center of your picture.
9.11.1.6
Can px on I interact with any orbital? Draw the appropriate line to its energy in the centerof your picture.
9.11.1.7
Make an argument for why the py on I will have the same energy as the px orbital does.
9.11.1.8
In there any orbital on I that can interact with any orbital on the fluorine atoms? Indicateyour answer on your MO diagram.
9.11.1.9
Electrons are hard to count when atoms used in an MO diagram which does not use all theorbitals of a given type. In the exercises in this section we ignored the electrons in two ofthe three p orbitals of the fluorine atoms. In which of those three orbitals should we placethe “hole” present in a fluorine atom? To avoid this difficulty, it is useful to think aboutthe fluorine atoms as having a complete shell (six electrons in the p orbitals and hence a-1 charge) and assign the appropriate charge to the other atom, the iodine atom. Then weknow since all orbitals on the fluorine atom are filled, that pin is filled. What charge shouldwe put on the iodine atom in IF–
2 if we say the fluorine atoms have a charge of -1 each? Howmany electrons does the iodine atom them contribute to the MO diagram? Fill in your MOdiagram with electrons.
9.11.1.10
How many “bonds” are holding the IF–2 molecule together?
9.11.1.11
How many electrons would you have to put into an m.o. diagram for IF–4 if you considered
only pin orbitals on the fluorine atoms and only p orbitals on the iodine atom?
9.11.1.12
How many electrons would you have to put into an m.o. diagram for IF–4 if you considered
only pin orbitals on the fluorine atoms and both s and p orbitals on the iodine atom?
Chm 118 Problem Solving in Chemistry

9.12 128
9.11.1.13
How many electrons would you have to put into an m.o. diagram for IO–4 if you considered
only pin orbitals on the oxygen atoms and only p orbitals on the iodine atom?
9.11.1.14
Consider the bond between the two hydrogen atoms in H2. There are a pair of electronsin that bond. If you were to try to assign those electrons as “belonging” to one atom oranother, how would you do it? HINT: Very easy problem.
9.11.1.15
Assume the pair of electrons in any bonding orbital (for the MO diagram in problem 9.11.1.10)can be assigned 50% to each of the two atomic orbitals (or SALCAOs) and that electrons inlone pairs belong to the appropriate atom. Under this assumption, what will be the chargeson the atoms in IF–
2? Do this slowly and carefully. It is just a mechanical count of electrons.
9.11.1.16
Do the fluorine atoms in IF–2 end up with charge? Does the iodine end up with charge?
Does this result agree with the conclusion we made long ago that the external atoms shouldbe electronegative, should want electrons relative to the central atoms?
9.11.1.17
Build an molecular orbital diagram for linear H–3; place the appropriate number of electrons
in the molecular orbitals.
9.11.1.18
Is the central atom or an external atom of H–3 more negative? The MO diagram of hyper-
valent compounds always places negative charge on the outside atoms; that’s the source ofour rule.
9.12 Bonding in Metal Compounds. Part I. σ Donors
In this and the next three sections we put together a molecular orbital model of bondingin transition metal compounds. This model is a more realistic model than the crystal fieldtheory that we dealt with earlier, section 7.1 and following sections. It will, of course,support the splitting of the d orbitals in an octahedral field into a lower energy set of threea higher energy set of two. In addition, it will be able to account for the spectrochemicalseries, something that crystal field theory could not do. It will lead us to consider animportant model for stability of a certain class of compounds, the “eighteen electron rule.”
We use a fairly simple model to understand the pertinent features. Consider the moleculeM(NH3)2+
6 where M is some transition metal ion. To deal with the bonding we considerthe lone pair electrons on the ammonia ligands, and treat it as a pin orbital (we would getthe same symmetry results if it was an s orbital or an sp3 hybrid). On the metal ion, weconsider only the metal d orbitals. The symmetry of the octahedral molecule is high (whichis beneficial), but that means that the manipulations are not as simple as those we havedone. I ask you believe what I am going to tell you. The symmetry of the six ligand orbitals
Chm 118 Problem Solving in Chemistry

9.12 129
Oh E 8C3 6C2 6C4 3C′2 i 8S3 6S4 6σd 3σh
A1g 1 1 1 1 1 1 1 1 1 1A2g 1 1 -1 -1 1 1 1 -1 -1 1Eg 2 -1 0 0 2 2 -1 0 0 2T1g 3 0 -1 1 -1 3 0 -1 1 -1T2g 3 0 1 -1 -1 3 0 1 -1 -1A1u 1 1 1 1 1 -1 -1 -1 -1 -1A2u 1 1 -1 -1 1 -1 -1 1 1 -1Eu 2 -1 0 0 2 -2 1 0 0 -2T1u 3 0 -1 1 -1 -3 0 1 -1 1T2u 3 0 1 -1 -1 -3 0 -1 1 1
Table 9.5: The Oh table of characteristic numbers.
in octahedral symmetry–see Table 9.5–is a1g + eg + t1u. The appropriate SALCAOs areschematically indicated in Figure 9.18. The symmetry of the metal d orbitals is, as indicatedlong ago, eg and t2g. Clearly the only interaction is between the two eg orbitals.
Since the metal ion’s orbitals are less stable than the ligand orbitals, the metal eg levelsget pushed up in energy and the ligand eg levels get stabilized. That is to say, for the twomolecular orbitals of eg symmetry, the upper one, closest in energy to the original metal dorbitals, is primarily metal in character–see section 9.5 and Figure 9.15. The lower eg orbitalis primarily ligand in character. All other orbitals “go straight across.” The ammonia ligandis called a σ donor: Electrons move from ligand to metal because of charge, so the ligandis a donor. The only orbital available to the ammonia to donate the electrons is cylindricalabout the metal-ammonia axis, hence the σ designation. There are relatively few pure σdonor ligands as most ligands have more than one lone pair, which, as we will see in thenext section, changes the bonding pattern.
9.12.1 Exercises
9.12.1.1
Using the information given above, make an MO diagram for the M(NH3)2+6 compound.
9.12.1.2
The ligand orbitals are of σ type, symmetrical about the ligand-metal bond. So the molec-ular orbital that is mostly metal eg is in character. Choices are σ, σ∗, π, π∗, non-bonding.
9.12.1.3
The metal t2g level is in character. Choices are σ, σ∗, π, π∗, non-bonding.
9.12.1.4
All of the ammonia orbitals are filled. Assume that the metal ion brought in three electrons.Put the electrons into your MO diagram.
Chm 118 Problem Solving in Chemistry

9.13 130
Figure 9.18: Schematic of ligand orbitals for an octahedral environment. Symbols give thesign of the wave function at the indicated position. The “double plus” is bigger than a“plus.”
9.12.1.5
Compare the configuration of the d3 compound of the last exercise with the answer youwould get with crystal field theory.
9.12.1.6
In crystal field theory we introduced a parameter called ∆ that was the difference in energybetween the eg levels and the t2g levels. Since that same difference exists in the molecularorbital approach (although now the eg level is just “mostly” metal in character), it wouldseem reasonable to use the same nomenclature for this gap. For the compound M(NH3)2+
6 ,what determines the size of ∆? Remember your answer because we shall use it later.
9.13 Bonding in Metal Compounds. Part II. σ and π Donors
We now deal with the bonding in a compound of the type MF4–6 . This analysis follows
closely the one in the last section; in fact, half of the work is identical to that in the lastsection. Consider fluoride ion as a typical σ donor, through the pin orbital, and a π donorthrough the filled p⊥ orbitals, those perpendicular to the metal-ligand bond. There are twoof these orbitals per fluorine ion, for a total of 12 orbitals. As you will show below, the p⊥orbitals can interact with dxy, dxz, and dyz, the t2g set of orbitals. The symmetry agrees,in that the SALCAOs generated by the 12 p⊥ orbitals are t1g + t1u + t2g + t2u. Clearlythe symmetry allowed interaction between the t2g on the metal and that on the fluoride
Chm 118 Problem Solving in Chemistry

9.14 131
ligands forms a bonding/antibonding pair. The σ aspect of bonding is identical to that inthe last section.
9.13.1 Exercises
9.13.1.1
Sketch a p⊥ on one fluoride ion interacting with dxz on the metal. This is typical of thetype of interaction found between the t2g of the metal and those of the ligands.
9.13.1.2
What kind of bond (or antibond) did you draw in the last exercise, σ or π?
9.13.1.3
Using the information given above, make an MO diagram for the MF4–6 compound. HINT:
Be careful about how far things gets pushed up or down. Remember that the overlap isbetter for a σ interaction than for a π one.
9.13.1.4
The ligand orbitals that are of σ type, are symmetrical about the ligand-metal bond. Sothe molecular orbital that is mostly metal eg is in character. Choices are σ, σ∗, π,π∗, non-bonding.
9.13.1.5
The ligand orbitals that are of π type, p⊥, make π bonds and antibonds. Therefore, themostly metal t2g level is in character. Choices are σ, σ∗, π, π∗, non-bonding.
9.13.1.6
All of the fluoride orbitals are filled. Assume that the metal ion brought in three electrons.Put the electrons into your MO diagram.
9.13.1.7
Compare the configuration of the d3 compound of the last exercise with the answer youwould get with crystal field theory.
9.13.1.8
In crystal field theory we introduced a parameter called ∆ that was the difference in energybetween the eg levels and the t2g levels. Since that same difference exists in the molecularorbital approach (although now both the eg level and the t2g level are “mostly” metal incharacter), it would seem reasonable to use the same nomenclature for this gap. For thecompound MF4–
6 , what determines the size of ∆? This is more complicated than the answerto the similar question in the last section. Remember your answers because we shall usethem later.
Chm 118 Problem Solving in Chemistry

9.14 132
9.14 Bonding in Metal Compounds. Part III. σ Donors/πAcceptors
In order to examine the third kind of bonding between metal and ligand we need to under-stand ligands with π bonds. We use as our example CN–. This is a heteronuclear diatomicand will have an energy level diagram roughly like that seen previously, Figure 9.16; refer tothat diagram for the following arguments. In the case of CN– we have ten valence electronsso the configuration of the molecule is σ2
1σ∗21 π
4σ22. Since the nitrogen atom has a greater
VOIE, the π level is distorted toward the nitrogen atom whereas π∗ is distorted toward thecarbon atom. The former, being filled, is dominately between the nuclei and, since it isdistorted toward the nitrogen atom, does not overlap well with metal orbitals; we neglectit in our treatment. The latter, the π∗, is on the outside of nuclei and localized on thecarbon atom, which is the atom which the cyanide ion uses to bind to metal. We must payattention this high energy empty orbital in our treatment. It is of note that this orbitalis the lowest occupied molecular orbital , called the LUMO. Since these π∗ levels have thesame symmetry as any other π orbital on a ligand, we know that symmetry from the lastsection: t1g + t1u + t2g + t2u. The only orbital of importance for bonding with the metal dorbitals then is t2g. Since this orbital is empty on the ligand it serves the role of acceptingelectron density from the metal. We call ligands like cyanide ion π acceptors.
Within the σ electrons, the most important ones are those in the highest occupied molecularorbital, the HOMO, which is σ2. Careful analysis shows this orbital is dominately an orbitalon carbon atom pointing away from the nitrogen atom; ideal for σ donation to the metal.As before the symmetry of these σ donor orbitals is a1g + eg + t1u and the only componentthat is important for bonding to the d orbitals of the metal is the eg one.
The situation simplifies down to this if we ignore all the orbitals on the ligands that are ofthe wrong symmetry to bind to the metal: We have a stable eg level on the ligands filledwith electrons, the usual eg and t2g on the metal (with as many electrons as the metal has),and a high energy t2g on the ligands that is empty. In the exercises you will build an MOdiagram from these orbitals.
9.14.1 Exercises
9.14.1.1
Sketch a π∗ on one cyanide ion ligand interacting with dxz on the metal. This is typical ofthe type of interaction found between the t2g of the metal and those of the ligand.
9.14.1.2
What kind of bond (or antibond) did you draw in the last exercise, σ or π?
9.14.1.3
Using the information given above, make an MO diagram for the M(CN)4–6 compound.
HINT: Be careful about how far things gets pushed up or down. Remember that theoverlap is better for a σ interaction than for a π one.
Chm 118 Problem Solving in Chemistry

9.14 133
9.14.1.4
The ligand orbitals that are of σ type, are symmetrical about the ligand-metal bond. Sothe molecular orbital that is mostly metal eg is in character. Choices are σ, σ∗, π,π∗, non-bonding.
9.14.1.5
The ligand orbitals that are of π type, π∗, make π bonds and antibonds. In this case themostly metal t2g level is in character. Choices are σ, σ∗, π, π∗, non-bonding.
9.14.1.6
All of the cyanide ion orbitals of type σ2 are filled and all of those of type π∗ are empty.Assume that the metal ion brought in three electrons. Put the electrons into your MOdiagram.
9.14.1.7
Compare the configuration of the d3 compound of the last exercise with the answer youwould get with crystal field theory.
9.14.1.8
In crystal field theory we introduced a parameter called ∆ that was the difference in energybetween the eg levels and the t2g levels. Since that same difference exists in the molecularorbital approach (although now both the eg level and the t2g level are “mostly” metal incharacter), it would seem reasonable to use the same nomenclature for this gap. For thecompound M(CN)4–
6 , what determines the size of ∆? This is more complicated than theanswer to the similar question in the last two sections.
9.14.1.9
In this and the last two sections we now have three kinds of ligand systems, all of which areσ donors. They differ in being π neutral (NH3), a π donor (F–), and a π acceptor (CN–).One can roughly look at the three classes of ligands as having the same effect on the egorbitals of the metal (though see exercise 9.14.1.13). They differ in what they do to the t2g
levels. Articulate that difference.
9.14.1.10
Look at the spectrochemical series and see if you can see how the conclusions from the lastexercise determine the size of ∆ as the ligand is changed.
9.14.1.11
What properties does a ligand have that makes it occur low in the spectrochemical series?
9.14.1.12
What properties does a ligand have that makes it occur high in the spectrochemical series?
9.14.1.13
Which would you expect would be the better σ donor, F– or CN–? Why?
9.14.1.14
Does your result from the last exercise enhance the effect of π difference, or subdue it?
Chm 118 Problem Solving in Chemistry

9.15 134
9.14.1.15
For a metal complex with π accepting ligands, such as CN– or CO, how many electrons areneeded to fill all the stable orbitals? For purposes of counting here, we only consider thoseorbitals on the ligands that are σ donor, π donor, or π acceptor with respect to the metalatom or ion; also, a stable orbital might be said to be any orbital found in a MO diagramto be below the original energy of the metal orbitals.
9.15 The Eighteen Electron Rule
In the last exercise of the last section you established that 12 electrons are needed to fill allof the σ levels of the ligands, including those that are non-bonding as well as those that arebonding. On the metal, you found that if the t2g were stabilized by a π accepting ligand,those bonding orbitals on the metal held 6 electrons. We conclude that for the most stablecompound (with π accepting ligands, 18 electrons are needed. This analysis is valid enoughthat there is a rule called the eighteen electron rule for metal containing compounds. Theexercises illustrate the extent and range of its validity.
9.15.1 Exercises
9.15.1.1
To use the eighteen electron rule you have to decide what charge to give to the ligandsaround the metal. You can make this assignment arbitrarily, but you must be consistent.Usually, it is best to make a reasonable assignment: Cl– is negative; CO and NH3 areneutral. All of these ligands contribute two electrons to the complex. How many electronswould H– contribute?
9.15.1.2
Does the compound Mn(CO)5H obey the eighteen electron rule if you consider the H atomas an H– ion?
9.15.1.3
Does the compound Mn(CO)5H obey the eighteen electron rule if you consider the H atomas an atom? How can the answer to both this question and the one of the last exercise be“yes?”
9.15.1.4
Which of the following are valid 18 electron rule compounds? Cr(CO)6, Fe(CO)6, Mn(CO)5Cl,Fe2(CO)9, (HINT: think metal/metal bond), Ni(CO)5, HCo(CO)4? If it is not valid, whatclosely related compound would be valid?
9.15.1.5
Compounds having NO as a ligand are tricky, although those having NO+ are easy. Com-ment.
Chm 118 Problem Solving in Chemistry

9.15 135
9.15.1.6
Which of the following are valid 18 electron rule compounds? Mn(CO)5(NO), Fe(CO)3Cl3,Ni(CO)4, Co2(CO)8? If it is not valid, what closely related compound would be valid?
9.15.1.7
Should the compound CrF3–6 obey the eighteen electron rule? It so, we are in trouble,
because this is a perfectly stable compounds. Comment.
Chm 118 Problem Solving in Chemistry

Chapter 10
Entropy
10.1 Definitions, and the Number of Microstates in a Con-figuration
Entropy is a very confusing concept. Although there are strong voices at work attemptingto wean people away from the concept that it is “disorder,” that concept is still prevalent.Even if it were valid to accept such a definition, how do you measure “disorder?” In thisChapter we will look at a way of assessing entropy that has a strong conceptual foundationeven if it is impractical in practice; then we will tie the strong foundation to the experimentalmeasure, gaining the best of both worlds.
We start by considering a set of oscillators with their associated quantum levels; theseoscillators are localized in space so we can tell which one is the first, A, which the second,B, etc. Although it is not essential to the development, it makes it easier for us if we dealwith oscillators rather than parves moving through space because the quantum levels inoscillators are evenly spaced, as shown in Figure 10.1. One of our oscillators is in one oranother of those quantum levels, but only one level; this is not to be confused by the figureswe will use soon which portray a collection of oscillators. The first question we ask is howmany different ways can we give a total of 2 units of energy to four oscillators. One waywould be to give oscillator A 2 units of energy and all the rest zero units. This is shownin Figure 10.2. We shall call this picture of one way to give four particles (the number ofparticles is N) two units of energy a microstate of the system.
A second way for an N = 4/ET = 2 system to exist is shown in Figure 10.3 where it isparticle B that has the two units of energy. If you step back far enough the labels on theoscillators (balls) are no longer visible and we have a picture that corresponds to both of themicrostates at once. Such a picture that shows the arrangement of the oscillators withouttheir labels is called a configuration, Figure 10.4. Be sure you see the difference betweena microstate and a configuration. One important question we want to ask is: How manydifferent microstates lead to this same configuration? In this case it is relatively easy todetermine the answer by just counting. This configuration has one oscillator, the one withtwo units of energy, in a unique position. That one oscillator is either A or B or C or D. So
136
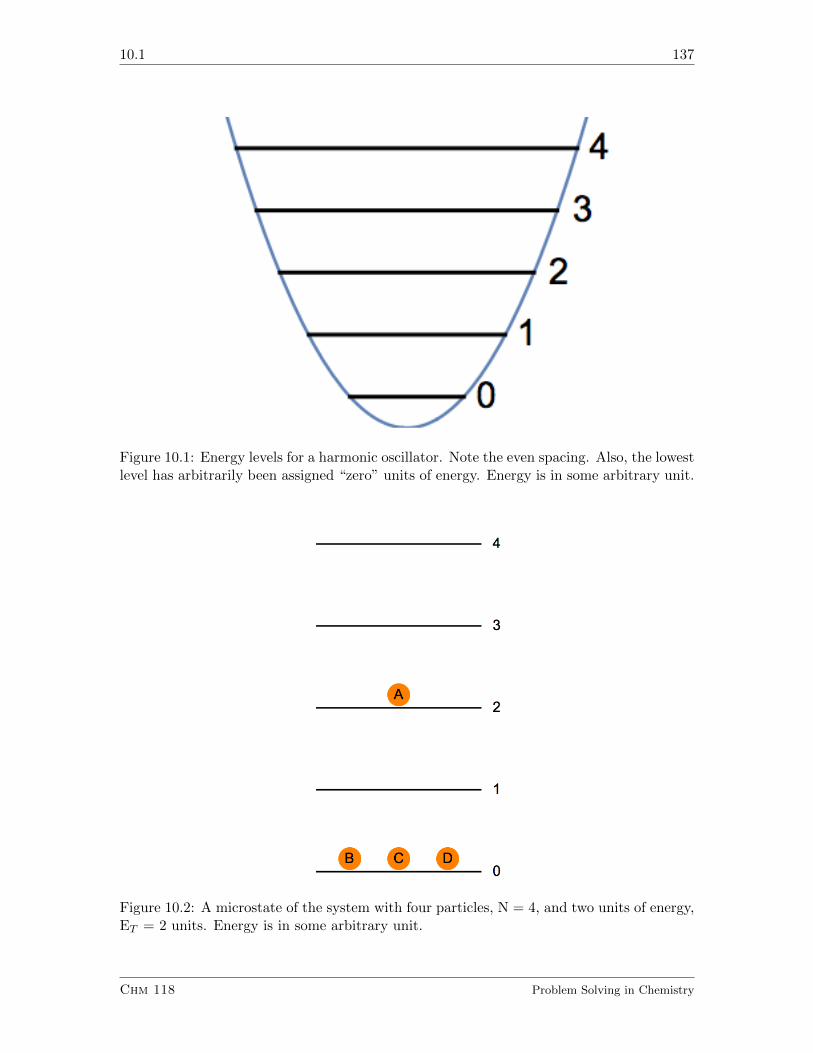
10.1 137
Figure 10.1: Energy levels for a harmonic oscillator. Note the even spacing. Also, the lowestlevel has arbitrarily been assigned “zero” units of energy. Energy is in some arbitrary unit.
Figure 10.2: A microstate of the system with four particles, N = 4, and two units of energy,ET = 2 units. Energy is in some arbitrary unit.
Chm 118 Problem Solving in Chemistry

10.1 138
Figure 10.3: Another microstate of the system with four particles, N = 4, and two units ofenergy, ET = 2 units. Energy is in some arbitrary unit.
there are four microstates in this configuration.
The second important question is: Are there any other configurations with the same N andthe same ET ? A little thought will give you the answer. We could give oscillators A andB each one unit of energy and let C and D be without any energy. Another way of sayingthat, consistent with our pictures, is that we put oscillator A and B in level e1 and C andD in level e0. Is this configuration different? How many microstates are in this secondconfiguration. We can count these, as you are asked to do in the exercises.
Finally, an important fact. Each microstate is as probable as any other microstate. Here weenter the statistical issue of probability that will play such an important part of what follows.If we throw a unit of energy at our four oscillators, any one of them is as likely as the otherto pick it up. So each microstate is equally probable. It follows if two configurations differin the number of microstates that the configuration with the higher number of microstatesis more probable. Configurations differ in probability. To summarize: Each microstate isequally probable; configurations differ in their probability.
10.1.1 Exercises
10.1.1.1
Craps is a game in which rolling a “7” as the sum of the two dice has consequences. Whatis the probability that one will roll a “7” with two dice? What is the probability that one
Chm 118 Problem Solving in Chemistry

10.2 139
Figure 10.4: The configuration for four particles, N = 4, and two units of energy, ET = 2units. Energy is in some arbitrary unit.
will roll a “2” with two dice?
10.1.1.2
What is most probable in craps, rolling a “7” or rolling a “2” (with two dice)?
10.1.1.3
What is the difference between a microstate and a configuration?
10.1.1.4
How many microstates are in the configuration (with N = 4/ET = 2) in which two oscillatorseach have one unit of energy and two oscillators have zero units of energy. Brute forcecounting. We will get a formula soon.
10.1.1.5
Are there any other configurations for the N = 4/ET = 2 system?
10.1.1.6
How many configurations are there for N = 4/ET = 3 system? HINT: Don’t do the numberof microstates at this point.
10.1.1.7
How many configurations are there for N = 10/ET = 3 system? HINT: Don’t do the numberof microstates at this point.
Chm 118 Problem Solving in Chemistry

10.2 140
10.2 A Way to Count Microstates in a Configuration andPredominant Configurations
In the last section we determined the number of microstates in some configuration by bruteforce counting. This method becomes increasingly harder as the number of oscillatorsincreases (and we will want to increase the number up to Avogadro’s number, No!). Wehence switch to a mathematical formula that evaluates the number of microstates in aconfiguration. This formula uses factorials. Recall that N! is one times two times threetimes ... up to N. Two features: Factorials get big very quickly. My calculator bombsout at 70!. Imagine No! Secondly, 0! is defined as one.1 Given these two statements, theformula that correctly tells us the number of microstates in a configuration for our row ofoscillators is:
W =N !
n0!× n1!× n2!× . . .(10.1)
where W is the number of microstates in the configuration, N is the total number of oscil-lators and n0 is the number in the e0 level, n1 the number in the e1 level, etc. So to do thecalculation in the last section:
W =4!
3!× 0!× 0!× 1!=
24
6= 4 (10.2)
With the equation in hand, we can calculate the number of microstates in configurationseasily (as long as the numbers don’t get too large: the bane of factorials). Let’s agree ona shorthand to designate the configuration. We list a set of numbers with slashes betweenthem. The numbers are the number of oscillators in the various energy levels, startingat the lowest energy level, the one with zero units of energy. In this nomenclature, theconfiguration given in Figure 10.4 is 3/0/1/0/0.
For any listing of the N/ET , one can write the configurations that are possible. We cannever do this with large numbers of oscillators because it becomes impossible to imagine allof the possible configurations. We are going to use a small number of oscillators to establishfeatures that are strictly true only for a large number of oscillators. If you make up your ownexamples, beware that sometimes the general truths, valid for a large number of oscillators,do not hold for a small number. To get started, let’s examine the configurations possiblefor the system N = 10/ET = 3. The three configurations and the number of microstatesfor each is given in the first row of Table 10.1. You should verify these results are correct.In the following rows of this table are the number of microstates in the three configurationsfor an increasing number of particles, up to 70, holding the total energy constant at 3units. This example shows that a configuration becomes predominant as the number ofparticles increases. In this case, it is configuration C, which with ten particles is only slightlyfavored over configuration B, but becomes increasingly favored as N goes up. This is showngraphically in Figure 10.5. Notice that a N = 10, configurations B and C are almost equallyprobable, but by N = 70, configuration C occurs over 91 percent of the time. Our rule: Asthe number of oscillators goes up, a configuration (or a set of configurations closely related inthe case of a large number of oscillators), becomes overwhelmingly predominant. This means
1There are web pages that address why this has been defined this way and why that definition is useful.
Chm 118 Problem Solving in Chemistry

10.2 141
N A B C
(N-1)/0/0/1 (N-2)/1/1/0 (N-3)/3/0/010 10 90 12020 20 380 114030 30 870 406040 40 1560 988050 50 2450 1960060 40 3540 3422070 70 4830 54740
Table 10.1: The number of microstates in the various configurations for the N = N/ET =3 system.
Figure 10.5: The fraction of the microstates in the various configurations for N particleswith three units of energy, ET = 3 units; see text.
that for practical purposes we are always working with the predominant configuration; theothers are so overwhelmingly disfavored that we never find the collection in them.
10.2.1 Exercises
10.2.1.1
Find the number of microstates, W, in the configuration 5/1/1/1/0.
10.2.1.2
Find the number of microstates, W, in the configuration 6/3/1/0/0.
10.2.1.3
Consider 10 particles with a total energy of 4 units. Find all the configurations and the Wof each. Identify the most probable configuration and note W for it.
Chm 118 Problem Solving in Chemistry

10.3 142
10.2.1.4
Consider 20 particles with a total energy of 4 units. Find all the configurations and the Wof each. Identify the most probable configuration and note W for it.
10.2.1.5
Consider 70 particles with a total energy of 4 units. Find all the configurations and the Wof each. Identify the most probable configuration and note W for it.
10.2.1.6
From the last three exercises, what is the predominant configuration? What do you concludeabout how W for the most probable configuration changes as N goes up?
10.3 A Detour to Define Some Thermodynamic Quantities:The First Law
We are about to ask the question: What happens to the value of W of the predominantconfiguration when we add energy to the system? Adding energy to the system occurseither in the form of heat or work. In that sentence are three words that we must be carefulto define in a precise manner. First, the system. The system is that part of the universethat we are studying. It could be a beaker of liquid or a rock or a room with its contents.The rest of the universe, that part we are not studying, is called the surroundings. Youdefine the system; nature does not. Heat is something we all think we know about, but itis a hard concept to define. I like the definition given by Fenn2: “Heat is the interaction(what happens) between a hot object and a cold object that are in contact with eachother.” To make sense of this, we need to understand what is meant by an interaction.This is when there is relation between a observable change in the system and one in thesurroundings. The most obvious such change relative to heat is, of course, temperature.When a hot object (high temperature) is placed next to a cold object (low temperature),the temperature of both change in the expected direction. The interaction that causes thosechanges in temperature is heat. Fenn points out that one way to test for a heat interchangeis to insulate the system and ask if the rate of the change in the property is lowered. Wewill use the symbol “q” for the total heat absorbed by the system in a given process, andδq for the amount of heat absorbed in a given small step of that process.
Work is defined as a force operating over a distance. Also, any interaction that is not heatis work. If we restrict ourselves to systems of gases, as we shall mostly do, then work is theexternal pressure (force per unit area) times the change in volume (where the distance ofour definition comes in because of the volume divided by the area of the pressure term) ofthe gas. For gases then, the work done on the gas for a small change in volume is
δw = −PextdV (10.3)
where the minus sign arises because a positive change in volume means the gas did work,not the other way around. Please note that the external pressure may be a function of the
2Fenn, J. B., “Engines, Energy, and Entropy. A Thermodynamic Primer,” W. H. Freeman and Co., 1982,p. 5.
Chm 118 Problem Solving in Chemistry

10.4 143
volume of the gas; we will find it necessary for this to be true later. Integration of thisexpression gives us the total work, “w.”
This restriction to gases also allows us to examine how the interaction called work affectsthe kind of systems that we are investigating in this chapter. Since for gases work involvesa change in volume, and a change in volume influences the quantum levels for translationdirectly: A smaller volume (shorter pole for a POP) causes the separation of the quantumlevels to become larger, but, importantly for us, to change the spacing. On the other hand,one can add energy by heat interaction and, if there is no volume change, there is no changein energy level spacings. It is this last kind of process that we want to talk about in ourdiscussion of configurations.
The discussion above suggests a relationship that is known as the first law of thermodynam-ics. If the only two methods of interaction to give energy to a system are heat and work,then we should be able to express the energy change in terms of those variables. That isthe first law in differential form:
dE = δq + δw (10.4)
where δq is the heat absorbed by the system, and δw is the work done on the system. Interms of finite differences
∆E = q + w (10.5)
10.3.1 Exercises
10.3.1.1
If the external pressure is constant at 1.0 atm, calculate the work done on the system whena gas is compressed from 2.0 L to 1.0 L. Your units are peculiar here, L atm. It turns out1 L atm is about 101 J.
10.3.1.2
In the last exercise, comment on the sign of your answer.
10.3.1.3
Imagine a system where after a process in which an external pressure of 1.0 atm compressesan ideal gas from 2.0 L to 1.0 L there is no change in energy. (The energy of an ideal gas isa function of temperature only; no change in temperature, no change in energy.) Find theheat absorbed by the system during the process.
10.4 Adding Heat to the Predominant Configuration. PartI. Qualitative
We now return to our discussion of configurations and ask the question: What happens tothe number of microstates in the predominant configuration if heat is added to the system.From the last section, we know that addition of energy as heat changes the population ofthe levels, but does not change the energy level spacings. So we can use our equally spaced
Chm 118 Problem Solving in Chemistry

10.4 144
Energy Predominant Configuration W WiWi−1
1 19/1/0/0/0 20 —2 18/2/0/0/0 190 9.53 17/3/0/0/0 1140 6.04 16/4/0/0/0 4845 4.255 16/3/1/0/0 19380 4.00
Table 10.2: The change in the number of microstates in the predominant configuration asheat is added to a system. Energy units arbitrary.
oscillator levels (convenient) and work out the answer by brute force. We start with a N= 20 system with ET = 1. The only configuration for this system is 19/1/0/0 and thishas a W = 20. Then we add one unit of energy in the form of heat (no change in energyspacings) and get two possible configurations, 18/2/0/0/0 and 19/0/1/0/0. The first is thepredominant configuration. (Remember, we are doing this with a small number of particlesto illustrate the principle. With a large number, there would be a configuration that isoverwhelming predominant.) This has a W = 190. We continue this process as show inTable 10.2. The W values of the predominant configuration for each case up to ET = 5 aregiven in the table along with, in the last column, the ratio of the W for a given amount ofenergy to that with one unit less energy. Notice that this ratio starts large and becomessmaller as the amount of energy increases. Since we are adding heat to our system, theenergy content is indicative of the “temperature” of the system. This allows the followingimportant conclusion: The number of microstates in the predominant configuration goes upwhen heat is added, but it goes up by a larger factor at low temperature than it does at hightemperature.
10.4.1 Exercises
10.4.1.1
Imagine twelve particles in the set of energy levels. Find the configuration that is mostprobable for a system with a total of two units of energy.
10.4.1.2
Same number of particles as in exercise 10.4.1.1. Find the configuration that is most prob-able for a system with a total of three units of energy.
10.4.1.3
Same number of particles as in exercise 10.4.1.1. Find the configuration that is most prob-able for a system with a total of four units of energy.
10.4.1.4
Show that W increases in going from the most probable configuration of exercise 10.4.1.1 tothat of 10.4.1.2.
Chm 118 Problem Solving in Chemistry

10.5 145
10.4.1.5
Show that the ratio of W10.4.1.3/W10.4.1.2 is less than W10.4.1.2/W10.4.1.1, where the subscriptsrefer to exercise numbers, and the W’s are for the most probable configuration, hereafterWmpc, and then latter still, just W for reasons that will become clear.
10.4.1.6
How do you state the conclusion of the exercises in this section in general terms? HINT:There are two issues concerning the Wmpc, how it changes with an increase in E by a constantamount, and how it changes at a low E (temperature) versus a high E (temperature).
10.5 What Does a Predominant Configuration Look Like?The Boltzmann Equation
We have been dealing with predominant configurations by trial and error. It would be niceif we could find a property of predominant configurations so that we could assess how thepopulations of the individual levels are related when a configuration is predominant. Tobe concrete, let’s examine the N = 26/ET = 7 situation. I claim that the predominantconfiguration of this system is 20/5/1/0/0. See if you can verify that this configuration hasa larger W than 19/7/0/0/0 or 21/3/2/0/0. In what follows, I want to continue to use thesymbol “n” for the number of particles in a given level; therefore, I am going to use theletter “v” to denote the level name. Now examine the ratio nv=1/nv=0 which is the ratio ofthe number of particles in two levels separated by one unit of energy. Compare that to theratio nv=2/nv=0 where the levels are separated by two units of energy. Those ratios differ,suggesting that the ratio of the number of particles in two levels depends on the differencein the energy between those two levels,
nv=1
nv=0= f(e1 − e0) (10.6)
This result leads to an interesting conclusion: We can write the following:
nv=2
nv=0=nv=2
nv=1× nv=1
nv=0(10.7)
which upon insertion of equation 10.6 into equation 10.7 gives
f(e2 − e0) = f(e2 − e1)× f(e1 − e0) (10.8)
Now look at this required functional form. It is of the type
ea+b = ea × eb (10.9)
which suggests thatnv=1
nv=0= e−β(e1−e0) (10.10)
with β some constant. Since this holds for any pair of values (with the appropriate energydifference), the parameters “1” and “0” can be replaced by “i” and “‘j.”
Chm 118 Problem Solving in Chemistry

10.5 146
A book by Nash3 derives equation 10.10 in an elegant fashion fromconsideration of the moment of oscillators from one level to another.Nash also does an neat calculation showing that the predominantconfiguration for Avogadro’s number of oscillators is more predom-inant than one that differs from it by about 1 part in 1010 (in eachenergy level) by a factor of about 10430!! That is predominant.
Using several methods that we won’t discuss here, we can show that β = 1kbT
where kbis Boltzmann’s constant, 1.38×10−23 J/K. So our final equation for the appearance of thepredominant configuration is what is know as (one of) Boltzmann’s equations,
nv=j
nv=0= e
−(ej−e0)
kbT (10.11)
This equation gives the relative populations in two levels in the predominant configuration,so it defines what the predominant configuration looks like. Note that for our equallyspaced energy level system of oscillators, that the population goes down by the same factorfor each increase in v value. Therefore a configuration such as 50/10/2/0/0 would be apredominant configuration as the decrease is by a factor of 5 for each increase in level,whereas 50/4/5/0/0 would not be.
10.5.1 Exercises
10.5.1.1
You have a set of equally spaced energy levels, the lowest of which has zero units of energyand each energy level gap is one unit. If this system is in the predominant configurationand there are 1920 particles in the e=0 level and 480 in the e=1 level, how many are in thee=5 level? HINT: Don’t try to calculate this by evaluating factorials!
10.5.1.2
Consider a predominant configuration. If you have 1000 particles in the n = 0 level, thespacing between the levels is 4.14×10−21 J/K, and the temperature is 300 K, what is thenumber of particles in n = 1? in n = 2?
10.5.1.3
For ease of this problem, imagine a system with translational energy spacing of two units,comprised of 20/2/0 particles for a total energy of 4 units. Hold the energy constant, butincrease the size of the container such that the spacing between the levels becomes one unit.What configuration is roughly consistent with 4 units of energy now?
10.5.1.4
What is W for each of the configurations of exercise 10.5.1.3?
10.5.1.5
Does W increase in going from the small container of exercise 10.5.1.3 to the large one?
3Nash, L. K., “A Statistical Approach to Classical Chemical Thermodynamics,” Addison-Wesley Pub-lishing Co., 1971.
Chm 118 Problem Solving in Chemistry

10.6 147
10.5.1.6
If we make the argument that systems will evolve to the predominant configuration (becauseit has the greatest number of equally probable microstates), will a gas expand to occupy alarger volume if given the opportunity? In view of the last problem, do you see why?
10.6 Adding Heat to the Predominant Configuration. PartII. Quantitative.
We follow Nash4 to get a quantitative relationship between adding heat energy to a pre-dominant configuration and the change in the number of microstates of that configuration.The actual derivation is of secondary importance. What is critical to our ultimate goal ofbeing able to think about entropy are the two assumptions that go into the derivation. Thefirst of these involves the constancy of β, of temperature, while the energy is added; thesecond demands a constancy of the energy level spacing, which, in turn, means that theinteraction that increases the energy is heat, not work.
To proceed with the math, we first take the logarithm of both sides of equation 10.1 anddifferentiate it for changes in the number of oscillators in various levels, noting that d ln(N!)= 0. We then use Stirling’s approximation5 to get rid of the factorials in the differentialterm:
ln(W ) = ln(N !)−∑i
ln(ni!)
dln(W ) = −∑i
dln(ni!)
= −∑i
(ln(ni)dni + nidni/ni − dni)
= −∑i
ln(ni)dni
(10.12)
At this point we introduce Boltzmann’s equation in the form of equation 10.10 to produce
dln(W ) = −∑i
(ln(n0)dni) +∑i
(βeidni)
= −ln(n0)∑i
dni + β∑i
(eidni)(10.13)
where the transformation from the first equation of 10.13 to the second is only valid if βis a constant ; that is, temperature is a constant. This is a critical point for our future useof this equation. The first term in the last equation of 10.13 is zero since the sum of thechanges of all oscillators in all levels must be zero since the number of oscillators is constant.Also because we have an expression for the total energy,
ET =∑i
(eini) (10.14)
4Ibid.5Stirling’s approximation is that ln (a!) = a ln (a) - a
Chm 118 Problem Solving in Chemistry

10.6 148
and differentiation of both sides of equation 10.14 this gives the sum in the second term ofthe second equation in 10.13 if the energy levels are constant!. Our final equation is:
dln(W ) = βdET (10.15)
or, if we specifically recognize that the energy change is heat, we can substitute the symbolfor a small amount of heat into the expression to give us
dln(W ) = βδq (10.16)
The change of the logarithm of the number of microstates in the predominant configurationis equal to the heat absorbed at constant temperature times β (or the heat absorbed atconstant temperature divided by kbT). The last two conditions because of the conditionsused in the derivation: constant temperature so that we can factor the β out and heat asthe interaction so that the spacing of the energy levels does not change.
Our knowledge to here: There are a bunch of configuration possible for a given N and agiven ET . We know one of these (or a collection of very closely related ones) is the predom-inant configuration, which is the one that systems will adopt because it is overwhelminglypredominant. We also know what the predominant configuration “looks like.” Further, weknow how the value of W in the predominant configuration changes with an input of heatenergy.
The highly restrictive nature of our model thus far causes somedoubt about whether it will be useful in “real life.” Again we canuse the arguments advanced by Nash6 to lift the specific restrictions.We note that if we imagine a system, call it C (for complex), wherethe spacings of the levels do change during the process, and put it inthermal contact with a system of the type described above, called S(for simple), then at thermal equilibrium we know that there will beno change in Wtotal upon moving a few particles to different levels.Therefore
dln(Wtotal) = dln(WC) + dln(WS) = 0
dln(WC) = −dln(WS)
= −βδqS(10.17)
However, qS = - qC , and hence the same equation holds for thechange in the number of microstates in the predominant configura-tion of the complex system as is true for the simple system.
Now imagine that we consider a system (that part of the universe that we are interested in)and its surroundings, which we imagine are very large, in essence the rest of the universe.Further these surroundings are so large that no matter what happens to the temperatureof the system, that of the surroundings is constant. We know that the condition for aspontaneous process is that the ln Wtotal will increase. Therefore
dln(Wtotal universe) = dln(Wsys) + dln(Wsurr) ≥ 0 (10.18)
Here is our basic equation for a spontaneous process.
6Ibid.
Chm 118 Problem Solving in Chemistry

10.7 149
10.6.1 Exercises
10.6.1.1
One conclusion of this section is equation 10.15. What assumptions are made to arrive atthis critical equation?
10.6.1.2
Carbon tetrachloride, CCl4, is a liquid at room temperature. Gaseous CCl4 surely has alarger W that does the liquid because of the translational energy levels in the gas. BonifaceBeebe, the great natural philosopher from rural Arkansas, thought about these facts andsaid ”Because W is greater for gaseous CCl4 than for the liquid, CCl4 is always a gas.”Comment.
10.6.1.3
Show that equation 10.18 requires that a cup of hot chai will cool if left in a room at ambienttemperature.
10.7 Disguising our Finding in a Word: Entropy
Equation 10.18 is the condition for a spontaneous process. In concept it is perfectly clearwhat is happening in this equation–the probability of population of energy levels in thepredominant configuration rather than others–but in practice equation 10.18 is not veryuseful if we want numbers to express the process since for real systems W is too large tocalculate easily. What we do is to define a new concept that is related to W in order tomake calculations of numerical values easier. That new concept is entropy, which we define:
S = kbln(W ) (10.19)
This definition lowers the tremendous value of W by taking the logarithm of it, and by multi-plying by the very small value of Boltzmann’s constant. Note also that from equation 10.16that the change in entropy is given by
dS = kbdln(W )
= kbβδq
=δq
T
(10.20)
where all the conditions on equation 10.16 hold for the last equation of the set 10.20.
Using this definition of entropy allows us to recast equation 10.18 in terms of entropychanges:
dSuniverse = dSsys + dSsurr ≥ 0 (10.21)
for a spontaneous process.
Out choice of a system and big surroundings, which maintain a constant temperature (be-cause of their size) allows an easy calculation of the ∆Ssurr. It is merely the heat flow
Chm 118 Problem Solving in Chemistry

10.7 150
Figure 10.6: The expansion of a gas from V = 1 L, P = 3 atm. to V = 3 L against anexternal pressure of 1 atm.
divided by the temperature of the surroundings. For instance, if during a process a systemabsorbs 25 J of heat from the surroundings held at 298K, then the entropy change of thesurroundings is -25J/298K or -0.084 J/K; note the negative sign occurs because the heatabsorbed by the surroundings is the negative of the heat absorbed by the system.
Determining the entropy change for the system is more of a challenge. The cause of thischallenge is the requirement from the last sections that the temperature remain constantwhen we use equation 10.16. Figure 10.6 shows an ideal gas at a pressure of 3 atm ina volume of 1 L expanding against an external pressure of 1 atm to a final volume of 3L, all in a temperature bath at 298K. This happens very fast and the time course of thevolume of the gas in shown schematicallly by the blue curve in in Figure 10.7. Since thegas does work nearly instantaneously on the surroundings, the energy of the gas must drop.The surroundings slowly supply heat to the gas to raise the temperature back to the initialvalue of 298; the time dependence of the temperature is shown (schematically, no scale oftemperature is given!) in the purple curve in Figure 10.7. The real heat absorbed by thegas in the piston is not at a constant temperature and hence cannot be used to calculatethe entropy change of the gas.
One important feature. Entropy is proportional to W and W depends on the state ofthe system, not where it was some instant ago. Hence entropy itself is a state function,dependent only on the state of the system and not its history. So if we could figure out away to expand the gas from the initial conditions of 1 L, 3 atm to the final condition of 3L,1 atm, such that the temperature did not change, then we could use that heat to calculatethe entropy change and that entropy change would be the same no matter how we actuallycarry out the expansion. There is a way to do this in principle and Sir Issac Newton showedus how to do the calculation. The way is very slowly. If we expand the gas a very smallamount, say by dV against an external pressure, Pext which is essentially the same as theinternal pressure, P, then the temperature change will be very small, and we can calculatethe work of that small expansion, and since we have an ideal gas at constant temperature(∆E = 0), we can calculate the heat absorbed by the system in that very small expansion
Chm 118 Problem Solving in Chemistry
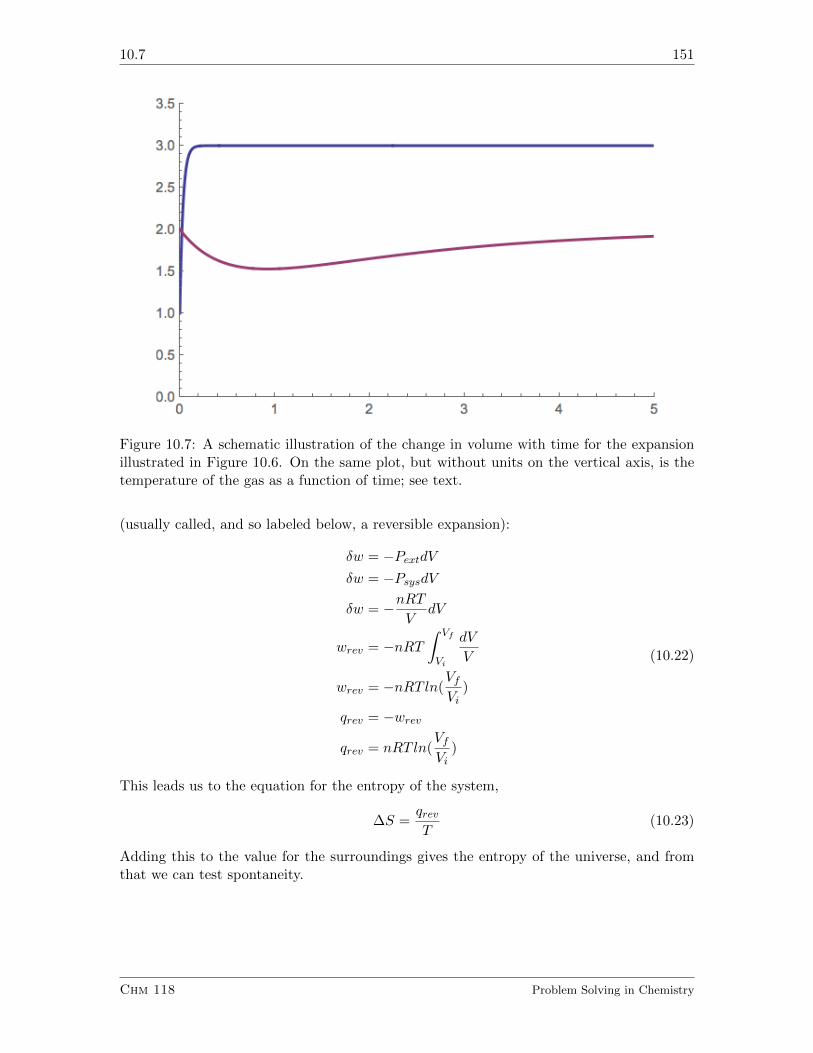
10.7 151
Figure 10.7: A schematic illustration of the change in volume with time for the expansionillustrated in Figure 10.6. On the same plot, but without units on the vertical axis, is thetemperature of the gas as a function of time; see text.
(usually called, and so labeled below, a reversible expansion):
δw = −PextdVδw = −PsysdV
δw = −nRTV
dV
wrev = −nRT∫ Vf
Vi
dV
V
wrev = −nRTln(VfVi
)
qrev = −wrev
qrev = nRTln(VfVi
)
(10.22)
This leads us to the equation for the entropy of the system,
∆S =qrevT
(10.23)
Adding this to the value for the surroundings gives the entropy of the universe, and fromthat we can test spontaneity.
Chm 118 Problem Solving in Chemistry

10.8 152
10.7.1 Exercises
10.7.1.1
Boniface Beebe, the wisest of the Arkansarian philosophers, noted that the entropy ofCCl4(g) was 309.6 J/(K mole) and that of CCl4(l) was 214.4 J/(K mole). He said: ”ClearlyCCl4 will exist as a gas since the entropy is greater for a gas.” Comment.
10.7.1.2
Calculate the work done on an ideal gas (in a piston and cylinder) when 1.0 moles at apressure of 20.0 atm. and T = 200K is expanded against an external pressure of 1.0 atmisothermally. Find the heat.
10.7.1.3
Do the expansion in the last exercise, but in this case let the external pressure be 10.0 atmuntil the piston stops moving, then reduce the external pressure to 1.0 atm. Find the workand heat for the overall process.
10.7.1.4
Do the expansion in the last exercise, but in this case let the external pressure be 15.0 atmuntil the piston stops moving, then reduce the external pressure to 10 atm until the pistonstops moving, then reduce the pressure to 1.0 atm. Find the work and heat for the overallprocess.
10.7.1.5
In view of the last several exercises, are work and heat dependent upon the path?
10.7.1.6
Expand the gas in exercise 10.7.1.2 from the same initial state to the same final state, butdo it reversibly. What is the work? What is the reversible heat? What can you say aboutthe reversible work compared to that found in the last several exercises?
10.7.1.7
Compute the entropy change of the system for the gas in exercise 10.7.1.2.
10.7.1.8
Compute the entropy change of the surroundings and of the universe for the gas in ex-ercise 10.7.1.2. Does experience tell you that the process is spontaneous? Does entropyagree?
10.8 Entropy Defined in Terms of W; The Third Law
The definition of entropy:S = kbln(W ) (10.24)
where kb is Boltzmann’s constant and the W is for the predominant configuration createsa situation that is unusual. Entropy has an absolute zero. When W = 1, S = 0 J/K. We
Chm 118 Problem Solving in Chemistry

10.9 153
get a W = 1, of course, when all the particles are in their lowest possible energy level.That occurs at low temperature, specifically 0K. The third law of thermodynamics statesthis: the entropy of any pure substance is zero at absolute zero. This leads to the conceptof absolute entropies. Since entropy increases as you add heat to a system (and addingheat to a system generally raises its temperature), entropy of a substance can be given anabsolute value dependent upon the temperature. Tables of absolute entropies exist, butread section 10.10 before using.
10.8.1 Exercises
10.8.1.1
Determine which system has the highest entropy, Ag(s) at 298K or Ag(s) at 340K? Howdid you reach your conclusion?
10.8.1.2
The entropy of CCl4 is about 210 J/(mole K) at 65oC, 214 J/(mole K) at 75oC, but is 309J/(mole K) at 80oC. Explain these data.
10.8.1.3
Which would have the higher entropy at 298oK, NH3(g) or Ne(g)? Why? HINT: As always,think population of energy levels.
10.8.1.4
The entropies of NaF(s), MgO(s), and AlN(s) are, respectively, 51.5, 26.8, and 20.2 J/(moleK) at 298oK. Give a rationalization.
10.8.1.5
Determine which system has the highest entropy Na(s) at 371K or Na(`) at 371K? How didyou reach your conclusion?
10.9 Disorder is a Poor Word to Describe Entropy
For many years entropy has been described by the word “disorder,” and pictures in textbooks have shown a student’s very messy dorm room (or one could use a picture of my desk)as an example of high entropy. This word is fraught with difficulties if used to describeentropy and should be avoided. Look at Figure 10.8 and determine which side is most“disordered.” I say that the right hand picture has roughly even spacing of the particles,and looks “ordered” to me, whereas the left hand side has all the particles bunched tothe left and is thereby “disordered”. But if you use a simple calculation of W (divide thecontainer into two parts and determine the probability all the particles will be in the lefthand part compared to the probability they will be equally distributed) you will find thatthe right hand picture has the highest entropy. Entropy is better described as the dispersionof energy among various energy levels.
Chm 118 Problem Solving in Chemistry

10.10 154
Figure 10.8: The Folly of Using “Disorder” after Lambert, F. L.; J. Chem. Ed, 2002, 79,187-192.
10.9.1 Exercises
10.9.1.1
If you have a bottle of Li(g) and a bottle of the same size of Cs(g), both at the sametemperature, determine which is moving more rapidly. HINT: All the energy is kinetic andtotal energy is proportional to temperature.
10.9.1.2
Given your answer to the last exercise, which would you imagine is more chaotic, lessordered, Li(g) or Cs(g). What does “disorder” predict for the highest entropy?
10.9.1.3
Now consider Li(g) and Cs(g) from the point of view of microstates, not “disorder.” If youuse a POP model, which has energy levels more closely spaced (in the same size container),Li(g) or Cs(g)? Which do you predict has the highest W and the highest entropy?
10.9.1.4
Look up some data to determine which system has the highest entropy Cs(g) or Li(g) at298K, both at the same pressure?
10.10 Entropy Change in Reactions
We need to talk about properties of systems that depend on the amount of material present,an extensive property. Volume is extensive as is energy. Properties that do not depend uponthe amount of material present are call intensive, such as temperature or pressure. Whatkind of property is entropy? As chemists we most often want to express the value of athermodynamic quantity for a reaction of some sort. Since the quantity of material thatwe deal with is variable, the most useful kind of quantity is an intensive one. What wouldhappen if we expressed entropy per mole of material transferred from the left of our equationto the right? What kind of property—intensive or extensive—would that quantity be? Wehave a symbol for the change in entropy of a substance, dS or ∆S. Now we need one for thequantity we just invented, one to be used with reactions. There are several suggestions forsuch symbols in use, including ∆rS, which is used in advanced books, but is often regardedas rather clumsy, ∆S, which was suggested but never adopted, and even ∆S, which is oftenused in introductory texts (and courses) and which leaves it up to the user to understand
Chm 118 Problem Solving in Chemistry

10.10 155
the context–probably seldom the case. What would I mean if I told you the hunk of gold ingoing from state a to state b has ∆S of -20 J/K? Am I giving you an intensive or extensiveproperty? Work you way through the following exercises to get a feel for this dual use ofthe same symbol.
A second issue we need to address is that changes in chemical reactions depend upon theconditions of the reagents and products, their pressures, temperatures, etc. Generally, it hasbeen agreed that in the standard state all materials are pure gases are at one atmospherepressure and all concentration are equal to one molar, and, usually, the temperature is298.15K If this is so, the thermodynamic parameter is given a “super o”.
10.10.1 Exercises
10.10.1.1
What would I mean if I told you that ∆rS for
H2O(s, T=273K) = H2O(`, T=273K)
was 22 J/(mole K)?
10.10.1.2
Given the information in the last exercise, how would you interpret the following sentence:“The entropy change, ∆S, for melting water at 273K is 11 J/K.”
10.10.1.3
What would you conclude with the information that “∆S for the melting of water at 273Kis 22 J/(mole K)”?
10.10.1.4
Would the entropy change of the following reaction at 298.15K be characterized by a ∆So?
C(s) + 2Cl2(g, P = 0.1 atm) = CCl4(`)
10.10.1.5
Use a table of absolute entropies to predict the value of ∆So for
2K(s) + F2(g) = 2KF(s)
Give a rationalization for your answer. HINT: If the text you are using does not have agood table of thermodynamic values, try the web page at
http://chemistrytable.webs.com/enthalpyentropyandgibbs.htm
or other sites found by searching for “enthalpy of formation” or “absolute entropy”.
Chm 118 Problem Solving in Chemistry

10.10 156
10.10.1.6
Use a table of absolute entropies to predict the value of ∆So for
NH3(g) + HBr(g) = NH4Br(s)
Give a rationalization for the sign of your answer.
10.10.1.7
Use a table of absolute entropies to predict the value of ∆So for
CH3CHCH2(g) = cyclic-C3H6(g)
Give a rationalization for the sign of your answer.
10.10.1.8
Use a table of absolute entropies to predict the value of ∆So for
3 H2(g) + Fe2O3(s) = 2Fe(s) + 3H2O(g)
Give a rationalization for the sign of your answer.
10.10.1.9
Comment on which direction the reaction
2NO(g) + Cl2(g) = 2NOCl(g)
will proceed if all reagents are under standard conditions and you are at some “high enough”temperature. HINT: You need to apply your thoughts about ∆Suniv since you are beingasked about spontaneity.
10.10.1.10
Comment on which direction the reaction
SrSO4(s) = SrO(s) + SO3(g)
will proceed at some “high enough” temperature.
10.10.1.11
Comment on which direction the reaction
SO3(g) = SO2(g) + 12O2(g)
will proceed at some “high enough” temperature.
Chm 118 Problem Solving in Chemistry

Chapter 11
The Free Energy: Entropy inAnother Guise
11.1 Using the First Law of Thermodynamics at Constant Pand T: Enthalpy
We saw in section ?? that the first law of thermodynamics says that the change in energyof a system is equal to the heat absorbed by the system plus the work done on the system.To remind you, energy is a state function–it does not depend on the path taken to get fromthe initial state to the final state–whereas heat and work are not state functions. For ourpurposes here, we consider systems in which the only work is “pressure-volume” work, thatof pushing back the atmosphere, δw = - PextdV. Since most experiments are done on theopen bench top in which the external pressure is constant, we are going to assume thatcondition. Also, we assume that the system is in a freely floating piston so that the externalpressure is the same as the internal. This essentially defines the path. Here is the algebra:
∆E = δq + δw
= δqP −∫PextdV
= δqP − Pext∫dV
= δqP − P∆V
∆E + P∆V = δqP
(11.1)
where we labeled the δq value for the constant pressure in the second equation, took ad-vantage of the constancy of the external pressure in the third and set the internal pressureas equal to the external in the fourth. The final equation has δqP equal to combination ofa bunch of state functions, and hence must also be a state function. We label this statefunction the change in enthalpy, ∆H, which was introduced in section 3.2: δqP is ∆H.More generally the enthalpy, H, is defined as E + PV, which, under conditions of constantpressure leads to our definition of the change in enthalpy. The words exothermic (giving
157

11.1 158
off heat) and endothermic (absorbing heat) are commonly used to describe the sign of theenthalpy change.
11.1.1 Exercises
11.1.1.1
What is the change in enthalpy equal to?
11.1.1.2
The enthalpy change for a reaction is -5.4 kJ/mole. Is the process exothermic? Is theprocess spontaneous? HINT: Second is a trick question.
11.1.1.3
Use tables of enthalpies of formation to determine the enthalpy change for the process
2SO2(g) + O2(g) = 2SO3(g)
under standard conditions. What do those numbers tell you about the spontaneity of thisreaction?
11.1.1.4
Give a reason why the enthalpy change is what it is in the reaction in the last exercise.HINT: Think about what is happening.
11.1.1.5
What is the sign of the enthalpy of vaporization of a liquid, any liquid? Give a molecularexplanation for your answer.
11.1.1.6
The enthalpy of vaporization of water is 40.7 kJ/mole and that of fusion of water is 6.01kJ/mole. Assuming these values are independent of temperature, what is the enthalpy ofsublimation of ice to water vapor at -10oC?
11.1.1.7
State Hess’s Law. Be articulate. (We covered this in section 3.2.)
11.1.1.8
Describe the process of combustion in terms of what chemicals come together and whatproducts are formed. What would the enthalpy of combustion be?
11.1.1.9
The standard enthalpy of combustion of propane is -2220; of C(gr), -394; and of dihydrogen,-286; all in kJ/mole of the substance named. Find the enthalpy change for the conversionof graphite into propane by reaction with dihydrogen. HINT: Use the last two exercises.
11.1.1.10
Find the enthalpy change for the reaction (not balanced)
Chm 118 Problem Solving in Chemistry

11.2 159
B2O3(s) + CaF2(s) = BF3(g) + CaO(s)
HINT: You will need to look up some numbers.
11.1.1.11
Describe how a calorimeter works. How would you calibrate a calorimeter?
11.2 Using Enthalpy of the System to Understand the Sur-rounding’s Entropy Change
There are two factors that allow us to use a system parameter, the enthalpy change, toassess a surrounding parameter, the entropy change of the surroundings. The first is thatat constant pressure the enthalpy change is the heat absorbed by the system. Since what isabsorbed by the system came from the surroundings, then the negative of the heat absorbedby the system is the heat absorbed by the surroundings. Secondly, remember that we definedthe surroundings so that it is big. Therefore the heat that is absorbed by the surroundings isabsorbed at constant temperature and we can use equation ?? to compute the surroundingsentropy.
This allows us to use equation 10.21 to get the entropy change of the universe for someprocess:
∆Suniverse = ∆Ssys + ∆Ssurr
= ∆Ssys +∆Hsurr
T
= ∆Ssys +−∆Hsys
T
= ∆S − ∆H
T
(11.2)
where in going from the third to the fourth equation we have understood the lack of asubscript to mean a system parameter.
11.2.1 Exercises
11.2.1.1
Calculate the entropy change of the surroundings if we start with one mole of an ideal gasin a cylinder with a piston at a pressure of 10 atm and a temperature of 298K held in placewith a pin. We remove the pin and let it expand against an external pressure of one atmin a large temperature bath at 298K.
11.2.1.2
Calculate the entropy change of the system for the apparatus described in the last exercise.
11.2.1.3
Calculate the entropy change of the universe for the last two exercises.
Chm 118 Problem Solving in Chemistry

11.3 160
11.2.1.4
Calculate the entropy change of the universe when a mole of liquid water is converted to amole of gaseous water at a pressure of one atm against an external pressure of 1 atm anda temperature of 298K. HINTS: Remember that the temperature does not change duringsuch a conversion. You will have to look up a number.
11.2.1.5
Repeat the last exercise—find the entropy change of the universe–at a temperature of 373Kassuming that the enthalpy and entropy of the conversion are not a function of temperature.
11.2.1.6
Seldom are we interested in gases expanding. In most instances of interest to a chemist, weare focussed on a reaction. The concept of spontaneity in a reaction depends, as we shallsee, on the states of the various components. For a while we will deal with the question ofspontaneity when all chemicals are in their standard states, one atm pressure and pure. Todo this, we can proceed in one of two equivalent thinking processes: (1) We can imaginea very large vessel of reactants and products, with all species at one atm pressure andthen allow a mole of reactants to move to products without that conversion causing anysignificant change in pressure of any reagent or product; or (2) we can move a small amountof reactants to products so that pressures do not change, compute the entropy changefor that small amount, and convert it to a “per mole” number. The description of thesesystems is “all species in their standard state”. Compute the enthalpy change when CaCO3
decomposes into CaO and CO2(g) with all species in their standard state. If this occurs at25oC, what is the change in the entropy of the surroundings?
11.2.1.7
Find the entropy change of the system for the reaction in the last exercise.
11.2.1.8
Is the reaction of the last two exercises spontaneous at room temperature? What wouldhappen at a higher temperature? Show your answer to this second question mathematicallyassuming that the system properties, ∆S and ∆H, are independent of temperature.
11.2.1.9
The reaction to produce formaldehyde is
H2(g) + CO(g) = H2CO(g)
At T = 25oC, ∆Ho = 1.96 kJ/mole and ∆So = -109.6 J/mole. Find the change in entropyof the surroundings when one mole of formaldehyde is produced at 25oC under standardconditions.
11.2.1.10
If the temperature for the formation of formaldehyde (see last problem) is raised to 50oC,what will happen to the value of the change in entropy of the surroundings provided ∆Ho
and ∆So are approximately independent of temperature over this range.
Chm 118 Problem Solving in Chemistry

11.3 161
11.2.1.11
Is the production of formaldehyde (see exercise 11.2.1.9) spontaneous at 25oC?
11.3 Getting Rid of the Universe Altogether: Free Energy
We now are going to manipulate the last equation in the set 11.2 so that there is no mentionof the universe. We first multiply by a constant T and then by negative one, which changethe sign on the inequality for spontaneous processes:
∆Suniverse = ∆S − ∆H
T≥ 0
T∆Suniverse = T∆S −∆H ≥ 0
−T∆Suniverse = ∆H − T∆S ≤ 0
∆G = ∆H − T∆S ≤ 0
(11.3)
where in going from the third equation to the fourth we have defined ∆G as -T∆Suniv; ∆Gis a state function since it is equal to a combination of state function values. We call ∆Gthe free energy change and it is less than zero for a spontaneous process.
Another approach (somewhat more sophisticated) is as follows. Let’sinvent a new function called G that is composed of previously de-fined functions:
G = E + PV − TS (11.4)
We take the total differential of this equation and then assert theconditions that T and P are constant to get an expression for dG,
dGP,T = dE + PdV − TdST (11.5)
Insert the definition of dH at constant P,
dH = dE + PdV (11.6)
and we getdGP,T = dHP − TdST (11.7)
or, for a finite change in the standard state
∆Go = ∆Ho − T∆So (11.8)
There are tables of free energies of formation, the free energy change when a compound ismade from its elements in their standard state.
11.3.1 Exercises
11.3.1.1
Take equation 11.8 and change the enthalpy change from a system value to one for thesurroundings, then divide all terms by T. Show that this equates -∆Go/T with ∆Souniv.
Chm 118 Problem Solving in Chemistry

11.4 162
11.3.1.2
What is the criterion for spontaneity in terms of WPC?
11.3.1.3
What is the criterion for spontaneity in terms of ∆Suniv?
11.3.1.4
What is the criterion for spontaneity in terms of ∆Go?
11.3.1.5
For the reaction
2 CuCl(s) + Cl2(g) = 2 CuCl2(s)
compute the values of ∆Ho and ∆So by looking up the appropriate values from tables ofthermodynamic parameters. Use these and equation 11.8 to compute ∆Go. Compute ∆Go
from data in thermodynamic tables and show agreement. HINT: The value of ∆Ho and∆Go for Cl2 are zero, but the value for the So is not.
11.4 The Pressure (or Concentration) Dependence of G
Clearly entropy is a function of pressure. You can see this in our original model, the numberof microstates in the predominant configuration: To increase the pressure requires that weadd particles to a fixed volume which will increase W. To see how to deal with the pressuredependency, we work with the free energy function. There are some tricky concepts in thisdevelopment; if you miss those, be sure to appreciate the end result, equation 11.17.
We start with equation 11.4 and take the total differential, then usethe first law for a reversible process,
dG = dE + PdV + V dP − TdS − SdT
dG = δqrev − PdV + PdV + V dP − T δqrevT− SdT
dG = V dP − SdT
(11.9)
where the substitution of dS of the system for its equivalent, δqrevT
was used in going from the first equation to the second.
We now take the third equation in the set 11.9 and integrate itbetween the limits of pressure = Po and some arbitrary pressure, Pat constant T (so the second term drops out), for an ideal gas, forwhich we can substitute nRT/P for V. This gives us
G = Go +RTln
[P
P o
](11.10)
Chm 118 Problem Solving in Chemistry

11.4 163
where Go is the G when P = Po, the pressure in some “standard”state (usually 1 atm). This free energy is a state property of the sys-tem and hence must be dependent on those parameters that changethe state of the system. If we consider a mixture of chemicals, sayA, B, and C, then G must be a function of T and P, as always, butalso the number of moles of each of the various components. Wecould write this as
G = G(T, P, nA, nB, nC) (11.11)
If we take the total differential of this equation at constant T andP we get
dG =∑i
(∂G
∂n i
)nj ,T,P
dni
=∑i
µidni
(11.12)
where i runs over, in our specific case, the three components A, B,and C, and nj is all of those n except ni. In the second equality Ihave inserted, as is conventionally done, the chemical potential, µi,which is simply the name for the value of ( ∂G∂ni
)nj ,T,P , which is alsocalled a partial molar free energy (see exercise 11.4.1.1).
To make further progress we need to have a method to determinethe extent of reaction. This is usually done by defining a parametercalled the extent of reaction with the symbol ξ. For clarity, let’sconsider a specific reaction: A + B = C, and let’s define ξ throughthe equation
dξ =1
νidni (11.13)
where νi is the stoichiometric coefficient of reagent i and is a nega-tive quantity for reactants. We can then express the change in anycomponent’s number of moles with dξ by solving equation 11.13 fordni. At this point you should do exercises 11.4.1.2 to 11.4.1.5. Wecan now solve equation 11.13 for dni and insert those values intoequation 11.12 to give us
dG =∑i
µiνidξ
dG
dξ=∑i
µiνi
(11.14)
To solve the second equation we need to express the partial molarfree energies in terms of the pressures of the gases. We simplyargue that since these are free energies, they should obey the sameequation that a free energy obeys, namely, equation 11.10; so we
Chm 118 Problem Solving in Chemistry

11.4 164
can put in for the µi the value µoi + RT ln[PiP oi
]to give us
dG
dξ=∑i
(µoi +RTln
[PiP oi
])νi
=∑i
µoi νi +∑i
(νiRTln
[PiP oi
]) (11.15)
which, if we define∑
i µoi νi as ∆rG
o and move the νi inside thelogarithm term as an exponent gives us our final equation, which iswritten in terms of ∆rG
o or some such symbol–see section 10.10–forthe reaction νAA + νBB = νCC + νDD:
dG
dξ= ∆rG = ∆rG
o +RTln
[P νCC P νDDP νCoC P
νDoD
P νAoAPνBoB
P νAA P νBB
](11.16)
To summarize our discussion above, the pressure dependence of the free energy change fora reaction when the standard state is taken to be one atmosphere (Po = 1) is given by
∆G = ∆Go +RTln
[Pressure of products to stoichiometric power
Pressure of reactants to stoichiometric power
](11.17)
where we have adopted the usual practice and removed the “r” subscripts. It is the sameequation for species whose concentration can vary as it is for species whose pressure varies.This equation is the gives the pressure (concentration) dependence of the free energy. Theargument of the logarithm in equation 11.17 is often called Q for simplicity. The equationthen reads
∆G = ∆Go +RTln(Q) (11.18)
11.4.1 Exercises
11.4.1.1
Look at the definition of µi in equation 11.12 and see if you can justify the following verbaldescription of a partial molar quantity, stated for reasons of concrete appreciation in termsof a partial molar volume: “The partial molar volume of substance i is the change in volume(dV) of the mixture (of which it is a component) when a small number of moles (dni) ofsubstance i is added, holding the other number of moles of other species constant.”
11.4.1.2
As an exercise in the use of ξ, let the reaction be A + B = C and the initial concentrationsof A and B be 1.0M and that of C be zero. At the point of reaction where the concentrationof A is 0.5M, evaluate ξ. Use that value to find the concentration of B and the concentrationof C at that point.
11.4.1.3
Same reaction as the last problem, but the initial concentrations are A = 1.0M, B = 0.7Mand C = 0.0M. Find ξ when the concentration of A is 0.5M and use that value to find theconcentrations of B and C at that point.
Chm 118 Problem Solving in Chemistry

11.4 165
11.4.1.4
For the reaction A + 3B = 2C, show that the extent of reaction varies from zero at thebeginning of the reaction to 1 at the end (no limiting reagent left) if the initial number ofmoles of A, B, and C are respectively 1, 4, and 1M.
11.4.1.5
For the reaction A + 3B = 2C, show that the extent of reaction varies from zero at thebeginning of the reaction to 1
3 at the end (no limiting reagent left) if the initial number ofmoles of A, B, and C are respectively 3, 1, and 2.
11.4.1.6
Find ∆Go for the reaction 2Mg(s) + O2(g) = 2MgO(s).
11.4.1.7
Give an explanation of the spontaneity of the reaction in exercise 11.4.1.6 in terms of theentropy of the universe and its components. Be complete.
11.4.1.8
Find the free energy change for the reaction in exercise 11.4.1.6 if the pressure of O2(g) is1.0 ×10−4 atm. Is the reaction spontaneous under these conditions? HINT: Since the twosolids have rather small change in volume with a change in pressure, their Go values arenearly independent of P and do not appear in equation 11.17 etc.
11.4.1.9
Find ∆Go for the reaction CH3OH(g) = CO(g) + 2H2(g) at 298K.
11.4.1.10
Find ∆G for the reaction in exercise 11.4.1.9 if the pressure of CH3OH(g) is 0.1 atm, thatof CO(g) is 1.0×10−2 atm, and that of H2(g) is 5.0×10−3 atm.
11.4.1.11
Assuming that ∆Ho and ∆So are independent of T, find ∆Go for the reaction in exer-cise 11.4.1.9 at 50oC and 150oC.
11.4.1.12
A long tube is prepared containing SiCl4(g); at one end ot this tube is placed some impureSi(s) and that end of the tube is heated. Over time at the cool end of the tube pure Si(s)forms. Account for this result using thermodynamic arguments. HINT: Think about theconproportionation of Si(IV) and Si(0):
SiCl4 + Si = 2SiCl2
11.4.1.13
This and the next four exercises give a derivation that suggests that equation 11.17 iscorrect. Write the chemical equation that represents benzene liquid vaporing to benzenegas at a pressure of one atmosphere. HINT: We can express the full conditions of a gas, sayX, in a reaction via X(g, P = 0.5 atm), for instance.
Chm 118 Problem Solving in Chemistry

11.5 166
11.4.1.14
Write the chemical equation that represents benzene liquid vaporing to benzene gas at apressure of P atmosphere.
11.4.1.15
We can couple the two equations from the last two exercises by letting benzene liquid atP = 1 atm. go to benzene liquid at P = P atm. What is your estimate of ∆H for thisprocess. HINT: Think about what energy changes take place, especially if we treat the gasas ideal. What is your guess about ∆S for this process? HINT: Is anything happening tothe benzene liquid?
11.4.1.16
To complete the cycle we have been working on, we could let benzene gas at P = 1 atm.go to benzene gas at P = P atm. To be concrete, if P = 0.1, and we work at constanttemperature, we are changing the pressure of benzene gas at constant temperature. Whatdoes this do to the energy under the assumption of ideal gas behavior? HINT: Note if H =E + PV, then ∆Ho = ∆Eo + ∆PV o = ∆Eo + ∆nRT o; what is the consequence if ∆T o
= 0? What does it do to the entropy?
11.4.1.17
Write the complete thermodynamic cycle from the information in the last four exercises.Let the value of ∆G at P = 1 atm. be ∆Go and the value of ∆G at P = P be ∆G. Writeone in terms of the other and additional terms as appropriate. You have just producedwhat we derived more rigorously earlier in this section.
11.4.1.18
Free energies of formation of benzene liquid and benzene gas at 298K are 124.5 and 129.7kJ/mole, respectively. Find the value of ∆G when the pressure of benzene gas is 10 mmHg. HINT: 760 mm Hg is one atmosphere.
11.4.1.19
Use data from the last exercise to find the value of ∆G when the pressure of benzene vaporis 400 mm Hg at 25oC.
11.5 Free Energy and Equilibrium
Equation 11.17 gives us the information we need to know to predict the spontaneity ofa reaction at constant T and P. When the value of ∆G is less than zero, the reaction isspontaneous; when greater, non-spontaneous. What about the other condition, when ∆G= 0. Under that condition, the process is at equilibrium and there is no net reaction ineither direction. The mathematics of this situation are interesting; when ∆G is zero, then
∆Go = −RTln
[P νCCeqP
νDDeq
P νCoC PνDoD
P νAoAPνBoB
P νAAeqPνBBeq
](11.19)
where the terms PCeq is the equilibrium pressure of substance C, etc. This is a ratherbizarre situation. The appropriate ratio of equilibrium pressures is governed by the free
Chm 118 Problem Solving in Chemistry

11.5 167
energy change when all the pressures are 1 atm. If we define an equilibrium constant as wedid in section 4.1, then we can recognize that equation 11.19 can be written as
∆Go = −RTln(K) (11.20)
We can combine equations 11.18 and 11.20 to give a succinct expression for the free energychange:
∆G = RTln
[Q
K
](11.21)
11.5.1 Exercises
11.5.1.1
The reaction
I2(g) + Cl2(g) = 2ICl(g)
has an equilibrium constant of 7.76×108. What is the value of ∆Go?
11.5.1.2
Is the reaction in exercise 11.5.1.1 spontaneous when the pressure of ICl is 0.001 atm andthe other pressures are 1.0 atm? When the pressure of ICl is 10 atm and the other pressuresare 1.0 atm?
11.5.1.3
Offer a rationalization as to why the standard free energy change for the reaction in exer-cise 11.5.1.1 is so small in magnitude.
11.5.1.4
For the reaction
ZnF2(s) = Zn2+(aq) + 2F–(aq)
the value of K is 0.003. Find the value of ∆Go for this reaction. HINT: The role ofconcentrations and pressures are interchangeable in our equations for free energy.
11.5.1.5
If you make 50 mL of a 2 M solution Zn(NO3)2 and 50 mL of a 4.0M solution of CsF, andmix them, what will happen? HINT: Ignore any reactions of nitrate ion and cesium ion,but pay attention to the last exercise; you might consider using equation 11.21.
11.5.1.6
Assuming that ∆Ho and ∆So are independent of temperature, find the boiling point ofCCl4(`).
Chm 118 Problem Solving in Chemistry

11.5 168
11.5.1.7
The normal boiling point of trimethylphosphine is 38.4oC. Its vapor pressure at -45.2oC is0.017 atm. Find ∆Ho, ∆So, and the vapor pressure at 15oC.
11.5.1.8
Imagine you have a chemical reaction with a ∆Go1 and an associated K1. You also havea second chemical reaction with a a ∆Go2 and an associated K2. Show, using the additiveproperty of ∆Go, that the equilibrium constant for the reaction that is the sum of chemicalreactions 1 and 2, K, is K = K1K2.
11.5.1.9
Use equation 11.8 and the relationship between ∆Go and lnK to show if you determine Kat various temperatures and then plot ln[K] versus 1/T that you can obtain ∆Ho and ∆So
from the corresponding line.
Chm 118 Problem Solving in Chemistry

Chapter 12
Equilibrium and Other Uses of theFree Energy
12.1 Equilibrium in Acid Solutions
We learned in the last chapter how to use free energy changes under standard conditions tolearn about the equilibrium constant for a reaction. To express that equation in a differentway, the equilibrium constant is given by
K = e−∆Go
RT (12.1)
Once you have a value of ∆Go you can get an equilibrium constant, and, having that,you can determine the concentrations of the various components of the solution. It is thisdetermination for weak acids that we deal with in this section.
Suppose we have an acid, HA, in an equilibrium HA = H+ + A−, with a Ka = 1.0×10−5,
Ka =[H+][A−]
[HA](12.2)
where the brackets stand for the concentrations of the various ions and molecular speciesin solution. Let us suppose that the solution at hand was made by dissolving 1.0 moles ofHA in a liter of water. Since water itself is an acid, H2O = H+ + OH−, we have two acidspresent, as well as two “existence” equations, one stating that all the moles of “A” that weadded must still be there, in either the form of HA or the form of A−; this is called a “massbalance” equation. The second “existence” equation concerns the charge of the solution;that must be balanced, or, said another way, the count of cations (times their charge) mustequal the count of anions (times their charge); this is the “charge balance” equation. Thesefour equation are:
Ka =[H+][A−]
[HA]
Kw = [H+][OH−]
TA = [HA] + [A−]
[H+] = [A−] + [OH−]
(12.3)
169

12.1 170
The four equations in equations 12.3 have four unknowns in them and hence can be solvedfor those four unknowns. These equations generate a cubic equation to solve, so they arenot much fun to solve (rigorously) by hand. Also, it is of considerable interest to applychemical intuition to the solution as this allows one to grasp what is happening in thesolution. Let’s commence that exercise. We added an acid to water, so the concentrationof a weak base such as OH– is unlikely to be very large. Let us guess it is zero comparedto the concentration of [A−]. That makes the fourth equation of equations 12.3 state that[H+] = [A−] which in turn makes the first equation of the set become
Ka =[H+]2
[HA]= 1.0 x10−5 (12.4)
We can now use the second equation of equations 12.3 and the simplified fourth equationagain to get rid of the [HA] from the denominator of equation 12.4 to give us
Ka =[H+]2
TA − [H+]= 1.0 x10−5 (12.5)
which is has only one variable, [H+], and is a quadratic equation in that variable. This canbe solved more easily than the cubic equation, but is still messy. A further simplification,using intuition, appears if we note that our acid is weak, has a small Ka. That means thatthe concentration of hydrogen ion is not likely to be very large. So let us guess that [H+] issmall compared to the total molarity of acid, TA. Then the equation is
Ka =[H+]2
TA= 1.0 x10−5 (12.6)
and the solution is [H+] = 3.162 x 10−3M. (The rigorous answer is 3.157 x 10−5M, but wewill see how to improve the approximate answer shortly.) We can (and should) now checkour two guesses. Is 0.003162 small compared to 1.00, the second guess we made. Yes. Thenwe use the second equation in equations 12.3 to compute [OH−] (3.16 x 10−12) and ask ifit is small compared to [A−] = [H+] = 3.16 x 10−3? Yes. The guesses are ok.
Let’s consider a couple of changes in order to see how they influence our answers. First,what is the acid is weaker, say Ka = 1.00 x 10−8. Then following the same procedure gives[H+] = 1.000 x 10−4M by the guesses and rigorously the same (with round-off). So theweaker the acid, the better the guesses. The second change worth making is to lower theconcentration of the acid, lower the value of TA. If that value is 0.01 for the acid with aKa of 1.0 x 10−5, then the guesses yield an answer of 3.162 x 10−4 versus a rigorous answerof 3.113 x 10−4. This error is about 5 parts in 300 or slightly over one percent. Too much?Depends on your needs. But clearly the stronger the acid and the more dilute the totalacid added, the more problematic the accuracy. Fortunately, checking your approximationsalways tells you if you are in trouble or not.
There is a fast way to fix calculations of this sort when the guesses are poor. Let’s do acase to see how this is done. Let’s consider an acid with a Ka = 2.00 x 10−3 and a TA of0.01M. Making our two guesses and solving for [H+] as above gives 4.47 x 10−3M. Is thatsmall compared to TA of 0.010M. No. Rather than going back and solving the quadratic, itis faster to simply put the answer for hydrogen ion concentration into the additive portion
Chm 118 Problem Solving in Chemistry

12.1 171
Figure 12.1: A graph showing how the guessed value of the [H+] approaches the true valueas the number of iterations increases for the example in the text. The straight line is therigorous answer.
of the appropriate equation similar to equation 12.5 and solve again:
[H+]2
TA − [H+]=
[H+]2
0.01− 0.00447= 2.0 x10−3 (12.7)
which leads to [H+] = 0.00332M; do it again to get [H+] = 0.00365M; and again for0.00356M; etc. You will zig-zag toward the correct answer, which in this case is 0.00358M,as shown in Figure 12.1. Incidentally, the method of calculation is roughly how moderncomputer programs compute molecular properties using quantum mechanics.
Finally, recall that the results of a calculation of [H+] are often expressed in terms of thenegative logrithm of that number, or in terms of the “pH.”:
pH = −log([H+]) (12.8)
12.1.1 Exercises
12.1.1.1
Find the pH of a solution prepared by dissolving 0.10 moles of HNO2 in a liter of water.The Ka for HNO2 is 5.1×10−4.
12.1.1.2
Find the pH of a solution prepared by dissolving 0.01 moles of HF in a liter of water. TheKa for HF is 6.75×10−4.
Chm 118 Problem Solving in Chemistry

12.2 172
12.1.1.3
Given the definition of pH, what do you imagine a pKa = 6.35 means?
12.1.1.4
The acid H2S has two protons that can be removed. The first has a pKa = 7.00 and thesecond a pKa of 12.92. This diprotic acid hence has five equations necessary to define thesystem. Write those five equations.
12.1.1.5
Show from your equations of the last exercise, good chemical intuition and good guessesthat the solution to the following problem is no more difficult than the previous exercises:Find the pH of a solution prepared by dissolving 0.010 moles of H2S in a liter of water.HINT: S2– is a base.
12.1.1.6
The two pKa of oxalic acid, HOC(O)C(O)OH, are 1.25 and 4.28, a difference of threeunits. The two pKa of adipic acid, HOC(O)CH2CH2CH2CH2C(O)OH are 4.42 and 5.41, adifference of one unit. Comment.
12.2 Mixtures of Acids and Salts: Buffer Solutions
Let’s apply the logic of the last section to a slightly more complex problem; that applicationyields an extraordinary easy solution. We are going to prepare a solution that has 0.50 molesof acid HA and 0.50 moles of NaA dissolved in one liter of water where HA is a weak acid.Also, note that generally speaking, salts of Group I cations like Na+, completely dissociatefrom anions, so the only role for Na+ in these problems is as an additional mass balanceequation and in the charge balance equation.
We then have for this problem the four equations of equation 12.3 with a modification tothe charge balance equation to include sodium ions, and a new mass balance equation forNa+:
Ka =[H+][A−]
[HA]
Kw = [H+][OH−]
TA = [HA] + [A−]
[Na+] + [H+] = [A−] + [OH−]
TNa = [Na+]
(12.9)
To solve this equation lets think, as we did before, about what we added to the solution.We added an acid, so we might guess that [OH−] might be small compared to [A−]. Butwe also added a base (NaA) to the solution, so we might think that [H+] would be smallcompared to [Na+]. If we make both of these guesses in the fourth equation of the set 12.9,then, since the fifth equation fixes the sodium ion concentration, the [A−] is fixed. Andonce that is true, [HA] is fixed by the third equation. Putting this into the first equation
Chm 118 Problem Solving in Chemistry

12.3 173
gives us:
Ka =[H+][Na+]
(TA − [Na+])(12.10)
which in the case of the numbers we are using gives us that [H+] = Ka.
Solutions such as this one, made by mixing an acid and a salt containing its conjugate baseare called buffer solutions. As you will show in an exercise, these resist a change in [H+]when either acid or base is added.
12.2.1 Exercises
12.2.1.1
Here is a variation on the theme of section 12.1. Find the pH of a solution made from100.0 g of Na(CH3COO) dissolved in a liter of water given that the pKa of acetic acid is4.75. HINTS: (1) The equations are those in 12.9. (2) Since you added a base, a logicalapproximation might be what? (3) Concentrated solution, so likely that [A−] is largecompared to what? (4) Rigorous answer is 9.4216.
12.2.1.2
Find the pH when 1.0 moles of acetic acid (pKa = 4.75) and 0.5 moles of sodium acetateare placed in a liter of water.
12.2.1.3
Determine the pH when the solution in the last exercise is treated with 0.10 moles of NaOH;assume no change in volume.
12.2.1.4
Determine the pH of the solution in exercise 12.2.1.2 is treated with 0.10 moles of HClassuming no change in volume. HINT: You need to modify the charge balance equation forthe presence of Cl– and to add another mass balance equation for chloride.
12.3 Solubility Equilibrium, including Common Ion Effect
Another common type of equilibrium that commonly occurs involves the limited dissolvingof certain solids. The classic example is silver chloride, which dissolves very slightly inaqueous solution:
AgCl(s) = Ag+(aq) + Cl–(aq)
The equilibrium constant for this reaction (classically called a Ksp for “solubility product”)is 1.8×10−10. To solve problems of this sort is very easy with our logic of previous sections.We have two unknowns, the concentration of silver ions and the concentration of chlorideions, and we have the Ksp and the charge balance equation. (See the next example to be
Chm 118 Problem Solving in Chemistry

12.4 174
sure you know how to deal with the charge balance equation when the charges on the twoions differ.) Solution is trivial in this case to give [Ag+] = 1.34×10−5.
Of more interest is asking what the solubility of a compound is in a solvent that containsone of the ions of the compound. This kind of problem has what is called the “common ioneffect.” For instance, what is the solubility of ScF3 is in a solution that is 0.01M in NaF?We now have three unknowns, [Sc3+], [F–], and [Na+]; and three equations:
Ksp = [Sc3+][F−]3 = 4.2× 10−18
3[Sc3+] + [Na+] = [F−]
[Na+] = 0.010
(12.11)
Note that the second equation, the charge balance equation, needs a “3” in front of thescandium ion because the equation is counting charge and each scandium ion carries threecharges. Chemical intuition makes the solving of this set of equations very easy. Since ScF3
is not very soluble, one guesses that the [Sc3+] is small compared to that of [Na+]; therefore,[F–] = [Na+] =0.010M and from the first equation, [Sc3+] = 4.2×10−12. Does our guesscheck out?
One final word. The problem just quoted is not correct, although this is the way it is oftendone. The reason is that fluoride ion is a weak base; to solve this problem correctly requiredthat the equilibrium
HF(aq) = H+(aq) + F–(aq)
and then, of course, the equation for water as an acid, must be included. Such problemsbecome difficult very quickly. In this case the rigorous answer is essentially the same as theapproximation above because fluoride is not a very strong base.
12.3.1 Exercises
12.3.1.1
The Ksp for PbSO4 is 1.6×10−8. Find the solubility of PbSO4 in water.
12.3.1.2
Find the solubility of PbSO4 in a solution that is 0.010M in Na2SO4.
12.3.1.3
The Ksp for PbCl2 is 1.6×10−5. Find the solubility of PbCl2 in water.
12.4 Electrochemical Cells
To examine the other crucial area where free energy calculations are important, we need toestablish the concept of the electrochemical cell; a schematic version of a cell is shown in
Chm 118 Problem Solving in Chemistry

12.4 175
Figure 12.2: A schmatic diagram of an electrochemical cell. Note the assumed direction offlow of electrons is indicated by the arrow.
Figure 12.2. In this cell a piece of zinc metal is suspended in a solution of zinc ions and apiece of copper metal is suspended in a solution of cupric ions. (Note in both sides thereis some “innocent” or “spectator” anion, unspecified, to balance charge.) The two piecesof metal are connected by a wire that allow electrons to flow (through a volt meter, whichmeasures the tendency for the electrons to flow). The two beakers are connected by a “saltbridge” that allows ions to migrate from one beaker to another in order to keep chargebalance on the two sides, but does not allow mass transport of material from one beaker tothe other. When we draw a cell this way, or give the abbreviation to be suggested soon, itis always assumed that charge flows from the left hand cell to the right hand cell throughthe external circuit. (If the potential of the cell reads “positive”–you can look at this asthe electrons pushing the meter in a clockwise fashion, then that is the real direction theelectrons would flow.)
In order to get electrons to flow in the assumed direction, definite processes have to happenin each of the two cells. In the left hand cell, electrons must be produced to provide forthose that are leaving from left to right; therefore the chemistry must be:
Zn(s) = Zn2+(aq) + 2e−
where the “2e−” are electrons that flow down the wire from left to right. Likewise, electronsmust be consumed in the right hand cell, so the reaction is:
Cu2+(aq) + 2e− = Cu(s)
Chm 118 Problem Solving in Chemistry

12.4 176
These two reactions are isolated from each other and are called “half-cell reactions.” Eachof them has a certain tendency to consume electrons (the reaction as written in the caseof the copper cell, but the reverse of the reaction given in the zinc cell) and that tendencyis measured by the half-cell reduction potential. This can be abbreviated as Cu2+/Cu andZn2+/Zn. The values of these two are EoCu = 0.340V and EoZn = -0.763V if all concentrationsare 1.0 M (and are relative to an assumed value of 0.00V for the H+(aq)/H2(g)). Since thezinc reaction is going in the opposite direction, the value for its potential must change sign,and then the two can be added to give the net voltage of the cell of 1.103V.
There is a common abbreviation for electrochemical cells. For the cell above it looks likethis:
Zn(s) | Zn2+(aq, 1.0M) || Cu2+(aq, 1.0M) | Cu
where the single vertical line is a phase separation (in this case between solid and solution)and the double vertical lines in the middle represent the salt bridge. Note that our conven-tion about the direction of electron flow remains the same: from left to right in the externalcircuit, which now must be imagined.
Please note that the potential represents the tendency to push electrons and hence is in-dependent of the number of electrons involved in the reaction that creates that potential.Hence in the cell
Zn(s) | Zn2+(aq, 1.0M) || Ag+(aq, 1.0M) | Ag
the cell potential is 0.763V from the Zn side (sign reversed from conventional “reductionpotential” because it is an oxidation) plus 0.800V for the silver side, for a total of 1.563V,even though the number of electrons differ in the two reactions. When you compute a freeenergy change–see the next section–the number of electrons is introduced. The number ofelectrons affects the free energy, not the potential.
12.4.1 Exercises
12.4.1.1
Make a sketch (as, for example, in Figure 12.2) for the electrochemical cell:
Ag(s) | Ag+(aq, 1.0M) || Fe2+(aq, 1.0M),Fe3+(aq, 1.0M) | Pt
where the Pt in the right hand cell is simply there to get the electrons to the reactivecenters, the ferrous and ferric ions.
12.4.1.2
What is the potential of the cell in the last exercise if all concentrations are 1.0M and theEoAg = 0.800V and EoFe3+ is 0.771V?
Chm 118 Problem Solving in Chemistry

12.5 177
12.4.1.3
What is the potential of the cell
Ag(s) | Ag+(aq, 1.0M) || Cl–(aq, 1.0M),Cl2(g, 1.0 atm) | Pt
if the potential for the reaction
Cl2(g) + 2e− = 2Cl–(aq)
is 1.358V.
12.5 Free Energy, Other Work, and the Nernst Equation
The general equation for the free energy, G, is
G = H − TS (12.12)
which, if we take the total differential at constant T and P and insert the first law withthe work term being the total work of all kinds, and demand that our process be reversible,we get (in a similar fashion to equation 11.9)
dG = dE + PdV − TdS
dG = δqrev + δwPV + δwother + PdV − T δqrevT
dG = −PdV + δwother + PdV
dG = δwother
(12.13)
The free energy change is equal to the work other than pressure volume work, that is doneon the system. The work that we are interested in here is electrical work, which is given bythe product of the potential change (equivalent to the force), E , times the charge that flows(equivalent to distance moved), nF , where n is the number of moles of electrons and F isthe Faraday, the charge per mole; and in order to get work done on the system we need anegative sign in front:
δwelect = −nFE (12.14)
This leads us to the relationship between the potential and the free energy
∆G = −nFE (12.15)
with a similar equation with super ‘o’ (see equation 12.16) to reflect the situation when thechemicals are in their standard states.
We can calculate the free energy change for exercise 12.4.1.3 using the equation 12.16:
∆Go = −nFEo (12.16)
where, if we write the “cell reaction” as
Chm 118 Problem Solving in Chemistry

12.6 178
2Ag(s) + Cl2(g) = 2Ag+(aq) + 2Cl–(aq)
the value is - (2)(96,500 kJ/V)(0.558V) = - 107.7 kJ. Note if we wrote the cell reaction as:
Ag(s) + 0.5Cl2(g) = Ag+(aq) + Cl–(aq)
then the number of electrons that “move down the wire” is one mole and the free energychange is - 53.8 kJ, as you would expect.
Finally we manipulate the equation for the concentration dependence of ∆G, equation 11.18,to give us the same information in terms of cell potential:
∆G = ∆Go +RTln(Q)
−nFE = −nFEo +RTln(Q)
E = Eo − RT
nFln(Q)
(12.17)
where the last expression in equation 12.17 is called the Nernst equation.
12.5.1 Exercises
12.5.1.1
Write the cell reaction and find the free energy change for the cell:
Cu(s) | Cu2+(aq, 1.0M) || Fe2+(aq, 1.0M), Fe3+(aq, 1.0M) | Pt
if Eo for Cu2+/Cu is 0.340 and that for Fe3+/Fe2+ is 0.771.
12.5.1.2
Find the free energy change for the cell of the last exercise if the [Cu2+] = 0.01M, [Fe2+] =1.0M, and [Fe3+] = 0.0001M.
12.6 Using Electrochemical Cells to Solve Problems
The Nernst equation, the last expression in equation 12.17, is very useful. Electrochemicalcells can be constructed to learn about process that are hard to determine in other ways.Under these circumstances, the cell potential, E , is measured and some other quantity canthen be evaluated. The exercises give several examples.
Chm 118 Problem Solving in Chemistry

12.6 179
Experiment [H+(aq) EA 1.0 M 2.193VB 0.10 M 2.134VC 0.01M 2.075V
Table 12.1: The observed voltages for cells described in exercise 12.6.1.3.
12.6.1 Exercises
12.6.1.1
Consider the electrochemical cell
Ag(s) | Ag+(aq, 1.0M) || Ag+(aq, unknown concentration) | Ag
which has E of 0.142V at 298K. What is the concentration of silver ion in the right handcompartment? This kind of problem is called a concentration cell and is integral to nervefirings.
12.6.1.2
Consider the electrochemical cell
Cu(s) | Cu2+(aq, 0.010M) || Ag+(aq), IO–3(aq, 0.10M) | Ag
which has E of 0.154V at 298K. What is the value of the equilibrium constant for thedissolving of AgIO3? HINT: This is really a concentration cell in which the unknown is theconcentration of silver ion in equilibrium with solid AgIO3 and a solution of iodate ion of0.10M.
12.6.1.3
Imagine that the form of Cl(V) in solution is not known. We build an electrochemical cellas follows:
Zn(s) | Zn2+(aq, 1.00M) || Cl(V)(aq, 0.010M), H+(aq, C), Cl–(aq, 1.00M) | Pt
where the concentration of the hydrogen ion in the right hand compartment, C, varies.Table 12.1 gives the potential observed as a function of the hydrogen ion concentration.Show that this data is consistent with ClO–
3 as the form of Cl(V), but not with ClO+2 or
ClO3–4 .
Chm 118 Problem Solving in Chemistry

Index of Important Concepts andTerms
δ bond, 110π acceptor ligand, 132π donor ligand, 130σ bond, 110σ donor ligand, 128pi bond, 110
Absorbance, 85Absorbance of light, 79Absorbance peak width, 82Acid, 20Acidity
Effect of charge, 23Effect of resonance, 24Hess’s law for, 21Inductive effects, 25Nature of Element, 22
Allowedness, 85Average field, 81
Balmer series, 52Band theory, 126Beebe, Boniface, 35, 97, 149, 152Beer’s law, 85, 92Boltzmann’s equation, 146Bond energies, 113Bond energy, 15Bond length, 14Bond lengths, 113Boundary conditions, 50Buffer soltuion, 173
Cell reaction, 177Characteristic combination, formula, 42Characteristic numbers
C2v, 37, 121C3v, 38, 123D3h, 42D4h, 39Oh, 129One object, 37Several objects, 39
Characteristic sets in combination,formula, 41
Charge balance equation, 169Chemical potential, 163Color
Average field, 81Separate orbital, 81
Common ion effect, 174Concentration cell, 179Configuration
Definition, 136Nomenclature, 140Predominant, 140Predominant
Adding heat, 144Adding heat to, 148Appearance, 146
Symmetry notation, 75Conjugate acid, 20Conjugte base, 20Correlation diagram, 74Coulomb’s law, 2Counting electrons, 127Crystal field splitting
Low symmetry, 74Octahedral, 72Tetrahedral, 72
180

12.6 181
Crystal field stabilization energy (CFSE),88
Crystal field theory, 70Crystal field theory
Color in low symmetry, 81Color in octahedral compounds, 79Flaw, 77
de Broglie postulate, 51Degenerate orbitals, 70Diamagnetic, 16, 18, 87Diprotic acids, 172Disorder, 136Distribution curves, 27
Eighteen electron rule, 128, 134Electrochemical cell, 174Electron spin, 4Endothermic, 158Energy levels
Electronic, 101Rotational, 100Translational, 100Vibrational, 100
Energy levels in hydrogen atom, 52Enthalpy, 16Enthalpy of formation, 16Enthalpy of reaction, 17Entropy
Absolute, 153Defined, 149Not disorder, 153Reversible expansion, 151State function, 150
Entropy change in reaction, 155Equilibirum
Weak acid, 169Equilibrium, 166Equilibrium
Aqueous, 169Guesses to solve, 170
Equilibrium constant, 20Ethane
Eclipsed, 33Staggered, 33
Exothermic, 158Extensive property, 154Extent of reaction, 163
Factorials, 140
Faraday, 177
First law of thermodynamics, 143
First order, 95
Formal charge, 7
Four equations/four unknowns, 169
Free energy, 161
Free energy
Equilibrium, 166
Of formation, 161
Partial molar, 163
Pressure dependence, 162
g functions, 85
Gap size
Ligand effect, 78
Metal effect, 79
Gauss’ law, 63
Group
C2v, 34
D3h, 35, 42
D4h, 39
Half-cell reactions, 176
Half-cell reduction potential, 176
Half-life, 97
Heat
Definition, 142
Hess’s law, 17
Heteronuclear diatomic, 132
Heteronuclear diatomics, 114
High field complex, 77
High spin complex, 77
Highest occupied molecular orbital, 132
HOMO, 132
Homonuclear diatomics, 113
Hybridization, 118
Hybridization
sp, 118
sp2, 120
sp3, 120
Hydration energies, 22
Hydration energy, 26
Hypervalent compounds, 10, 126
Integrated rate law
First order, 96
Second order, 97
Chm 118 Problem Solving in Chemistry

12.6 182
Intensive property, 154Ionization energy
Definition, 52Distance effect, 65Effect of electron repulsion, 66Effect of penetration, 66Effect of VSNC, 66Four important factors, 65second, 67
IsomerizationLinkage, 92
Isosbestic behavior, 92
LCAO, 103Lewis structure, 7Ligands, 70Linear combination of atomic orbitals,
103Low field complex, 77Low spin complex, 77Lowest unoccupied molecular orbital, 132LUMO, 132
Magnetic momentSpin only, 87
Mass balance equation, 169Microstate
Definition, 136Probability, 138
MO diagram, 113Molar absorptivity, 85Multi-electronic atoms
Configurations, 63
Nernst equation, 178Nomenclature of orbitals
δ, 110π, 110σ, 110
Normalization, 104
Operator, 49Orbital, 63Orbital conservation, 104Orbital interaction
Antibonding, 104bonding, 104Symmetry restriction, 115
Overlap, 104
Paramagnetic, 16, 87
Parity, 85
Parve, 49
Parve on a pole, 50, 100
Pauli Principle, 5
Pauli principle, 63
Penetration, 63
Periodic table, 64
Perturbation theory, 115
pH, 171
pKa, 172
pKa, 20
Planck’s constant, 49
Polarization, 19
POP, 50
Probability, 4
Probability, quantum, 48
Quantum numbers, 53
Radial distribution curve, 53
Radicals, 16
Rate constant, 95
Rate law
First order, 94
Meaning of sum in, 99
Second order, 95
Rate of reaction, 94
Resonance, 12
Reversible expansion, 150
SALCAO, 121
Schrodiner’s equation, 49
Schrodinger’s equation, 103
Screening, 63
Second order, 95
Separate orbital approach, 81
Solids, 125
Solubility equilibrium, 173
Solubility product, 173
Spectrochemical series, 78, 128
Spin, 4
Spin allowed, 85
Spin forbidden, 85
Spin selection rule, 85
Spinel, 90
Spontaneous process, 148
Spontaneous reaction
Chm 118 Problem Solving in Chemistry

12.6 183
Entropy criterion, 149
Standard state, 155
State function, 157
Stern-Gerlach experiment, 45
Stirling’s approximation, 147
Symmetry adaped LCAO, 121
Symmetry operation
Center of inversion, 33
Definition, 30
Group, 34
Identity, 30
Improper, 30
Plane, 32
Proper, 30
Rotation, 30
Rotation-Reflection, 33
System
Definition, 142
Thermodynamics, 16
Third law of thermodynamics, 153
Transition metal compounds
MO theory, 128
Transmittance, 85Two slit experiment, 4
u functions, 85
Valence orbital ionization energy, 68Valence shell electron repulsion theory, 6Valence shell nuclear charge, 66Vibration in metal complexes, 82VOIE, 68VSEPR, 6VSNC, 66
WCalculation of, 140
Wave equation, 47Wave function, 4Wave function
Radial, 53Wave function, angular, 59Work
Definition, 142electrical, 177other, 177
Chm 118 Problem Solving in Chemistry