Embed Size (px)
Citation preview

Ce
MMa
b
c
d
e
f
g
4
a
ARRAA
KHPITE
1
ec2aalrp
CvRS
0d
Toxicology Letters 199 (2010) 144–152
Contents lists available at ScienceDirect
Toxicology Letters
journa l homepage: www.e lsev ier .com/ locate / tox le t
hitosan nanoparticles show rapid extrapulmonary tissue distribution andxcretion with mild pulmonary inflammation to mice
ina Choia, Minjung Choa, Beom Seok Hanb, Jin Hongc, Jayoung Jeongc, Sangjin Parkd,yung-Haing Choe,f, Kwangmeyung Kimd,∗, Wan-Seob Chog,∗
Division of Toxicological Research, National Institute of Food and Drug Safety Evaluation, 231 Jinhoungno Eunpyung-ku, Seoul 122-704, Republic of KoreaDepartment of Toxicopathology, Hoseo Toxicological Research Center, Hoseo University, Chungnam 336-851, Republic of KoreaNutrition and Functional Food Research Team, National Institute of Food and Drug Safety Evaluation, 231 Jinhoungno Eunpyung-ku, Seoul 122-704, Republic of KoreaBiomedical Research Center, Korea Institute of Science and Technology, 39-1 Hawolgok-dong, Seongbuk-gu, Seoul 136-791, Republic of KoreaLaboratory of Toxicology, College of Veterinary Medicine, Seoul National University, Seoul 151-742, Republic of KoreaDepartment of Nanofusion Technology, Graduate School of Convergence Science and Technology, Seoul National University, Seoul 151-742, Republic of KoreaRespiratory Medicine Unit, ELEGI/Colt Laboratory, Centre for Inflammation Research, The Queen’s Medical Research Institute, The University of Edinburgh,7 Little France Crescent, Edinburgh, EH16 4TJ, UK
r t i c l e i n f o
rticle history:eceived 9 July 2010eceived in revised form 24 August 2010ccepted 25 August 2010vailable online 9 September 2010
eywords:
a b s t r a c t
Pulmonary delivery of nanoparticles (NP) conjugated with therapeutic agents has been consideredrecently for both lung disorders and systemic circulation. Hydrophobically modified glycol chitosan(HGC) NP have previously shown excellent deposition to the tumor site and non-destructive intracel-lular release. Here, we evaluated the kinetics and toxicity of HGC NP by intratracheal instillation to mice.HGC NP showed a positive charge and average hydrodynamic size was around 350 nm. The half-life ofNP in the lung was determined as 131.97 ± 50.51 h. NP showed rapid uptake into systemic circulation
ydrophobically modified glycol chitosanulmonary delivery vehiclentratracheal instillationissue distributionlimination
and excretion via urine which was peaked at 6 h after instillation. Although HGC NP were distributed toseveral extrapulmonary organs, the levels were extremely low and transient. HGC NP induced transientneutrophilic pulmonary inflammation from 6 h to day 3 after instillation. Expression of pro-inflammatorycytokines (IL-1�, IL-6, and TNF-�) and chemokine (MIP-1�) in lung showed an increase from 1 h to 24 hafter instillation and recovered thereafter. Our findings suggest that HGC NP can be successful candi-dates for use as pulmonary delivery vehicles, owing to their excellent biocompatibility, transiency, andlow pulmonary toxicity, and property of rapid elimination without accumulation.
Crow
. Introduction
Pulmonary delivery of therapeutic agents has been consid-red an effective and non-invasive method for treatment of lungancer (Tomoda et al., 2009) and inflammation (Cleland et al.,001). In addition, systemic delivery via the pulmonary route isn alternative non-invasive route for intravenous injection such
s anesthetics (Gonda, 2000). Compared to oral route, lung hasarger surface area for absorption and thin vascular epithelial bar-ier for easy permeation (O’Hagan and Illum, 1990), and lowerroteolytic activity (Gonda, 2007). The advantages of pulmonary∗ Corresponding authors. Respiratory Medicine Unit, ELEGI/Colt Laboratory,entre for Inflammation Research, The Queen’s Medical Research Institute, The Uni-ersity of Edinburgh, 47 Little France Crescent, Edinburgh, EH16 4TJ, UK/Biomedicalesearch Center, Korea Institute of Science and Technology, 39-1 Hawolgok-dong,eoul 151-742, Republic of Korea.
E-mail addresses: [email protected] (K. Kim), [email protected] (W.-S. Cho).
378-4274/$ – see front matter. Crown Copyright © 2010 Published by Elsevier Ireland Ltoi:10.1016/j.toxlet.2010.08.016
n Copyright © 2010 Published by Elsevier Ireland Ltd. All rights reserved.
route for systemic circulation are faster onset of action and higherbioavailability than with subcutaneous injection (Heinemann etal., 1997; Laube et al., 1998). These advantages made possible todevelop insulin for pulmonary delivery (Klingler et al., 2009).
Owing to recent advances of nanotechnology, accurate deliveryof nanoparticles (NP) to the lung is now possible for diagnosis andtherapy (Card et al., 2008). NP present many advantages over otherapplications owing to long retention time in the lung, decreasedmuco-ciliary clearance (Niven, 1995; Tsapis et al., 2002), and multi-functionality including diagnosis, imaging, and therapy (Sajja etal., 2009; Sanvicens and Marco, 2008). There are numerous poten-tial therapeutic NP applications in respiratory and systemic disease(Buxton, 2006; Pison et al., 2006; Sung et al., 2007). Aerosol deliveryof EGF-modified gelatin NP–cisplatin complex showed success-
ful targeting to lung cancer (Tseng et al., 2009). Inhalation ofchitosan-programmed cell death 4 complex inhibited pathwaysof cell proliferation and angiogenesis using a mouse lung cancermodel (Jin et al., 2006). Intranasal administration of chitosan–DNAcomplex successfully inhibited respiratory syncytial viral infec-d. All rights reserved.
y Letters 199 (2010) 144–152 145
t(
acGtsMgsgaewthtf
mloHn
2
2
1hfiLpfDw
2
Iaie
FaD
M. Choi et al. / Toxicolog
ion (Kumar et al., 2002) and reduced allergic airway inflammationKumar et al., 2003).
Chitosan has emerged as a useful delivery carrier for diagnosticnd therapeutic applications, due to its unique physicochemicalharacteristics including biocompatibility and biodegradability.lycol chitosan is a commercially available derivative of chitosan
hat possesses enhanced water solubility, and has been used as acaffold for drug delivery or diagnostic imaging (Cho et al., 2007b;artin et al., 2002). We demonstrated in the previous study that
lycol chitosan NP modified with hydrophobic bile acid analogself-assemble into polymers, comprising the hydrophilic shell oflycol chitosan and hydrophobic multicores of bile acid analogs,nd are promising therapeutic agents in the anticancer drug deliv-ry system (Kim et al., 2005; Park et al., 2007). When particle sizeas appropriately controlled, hydrophobically modified glycol chi-
osan (HGC) NP exhibited distinguished in vivo stability with tumoroming properties by way of the enhanced permeability and reten-ion (EPR) effect and can make stable and effective delivery carriersor drug delivery systems (Hyung Park et al., 2006; Kim et al., 2008).
In this study, we synthesized HGC NP for the purpose of pul-onary delivery vehicle. Before practical application of HGC NP
oading therapeutic agents to the lung, kinetics and toxicity studiesf NP will provide invaluable information for future applications.erein, we evaluated acute toxicity, tissue distribution, and elimi-ation of HGC NP.
. Materials and methods
.1. Materials
Glycol chitosan (GC, MW = 250 kDa, degree of deacetylation = 82.7%),-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC), N-ydroxysuccinimide (NHS), 5�-cholanic acid, chlorpromazine hydrochloride,lipin III, and amiloride hydrochloride hydrate, were purchased from Sigma (St.ouis, MO). Solvents were purchased as anhydrous grade and used without furtherurification. The monoreactive hydroxysuccinimide ester of Cy5.5 was obtainedrom Amersham Biosciences (Piscataway, NJ). Fluorescent amine LysoTracker GreenND-26 was obtained from Molecular Probes (Eugene, OR). All other chemicalsere purchased as reagent grade and used without further purification.
.2. Synthesis of GC-5ˇ-cholanic acid conjugates
Glycol chitosan (250 kDa) was hydrophobically modified with 5�-cholanic acid.n brief, glycol chitosan (250 mg, 1 �mol) was dissolved in distilled water (30 ml),nd mixed with 5�-cholanic acid (75 mg, 200 �mol) in methanol (90 ml). The chem-cal reaction was initiated by addition of equal amounts of NHS and EDC (1.5quivalents of 5�-cholanic acid). The solution was stirred at room temperature
ig. 2. Time-course lung distribution profile of HGC NP, as seen using the NIR imaging tecfter intratracheal instillation of 2 mg/kg of HGC NP. Fluorescence intensities of lungs wata were expressed as mean ± S.D. *p < 0.05 versus vehicle control, n = 5 mice per each g
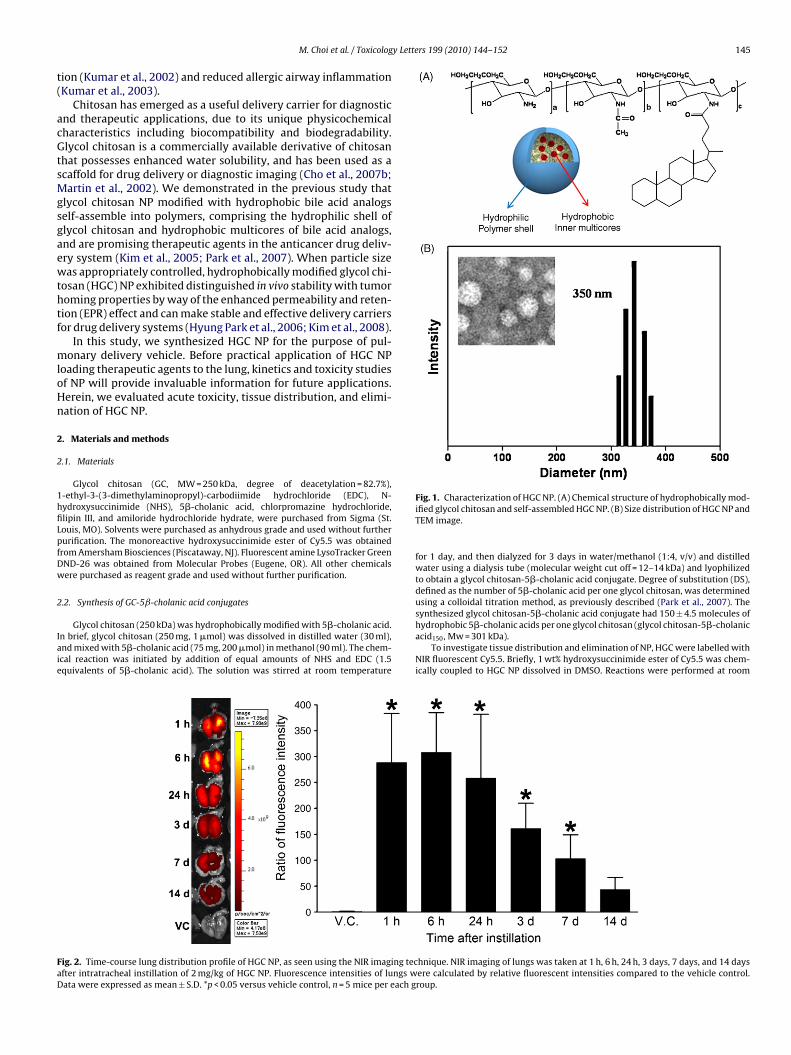
Fig. 1. Characterization of HGC NP. (A) Chemical structure of hydrophobically mod-ified glycol chitosan and self-assembled HGC NP. (B) Size distribution of HGC NP andTEM image.
for 1 day, and then dialyzed for 3 days in water/methanol (1:4, v/v) and distilledwater using a dialysis tube (molecular weight cut off = 12–14 kDa) and lyophilizedto obtain a glycol chitosan-5�-cholanic acid conjugate. Degree of substitution (DS),defined as the number of 5�-cholanic acid per one glycol chitosan, was determinedusing a colloidal titration method, as previously described (Park et al., 2007). Thesynthesized glycol chitosan-5�-cholanic acid conjugate had 150 ± 4.5 molecules of
hydrophobic 5�-cholanic acids per one glycol chitosan (glycol chitosan-5�-cholanicacid150, Mw = 301 kDa).To investigate tissue distribution and elimination of NP, HGC were labelled withNIR fluorescent Cy5.5. Briefly, 1 wt% hydroxysuccinimide ester of Cy5.5 was chem-ically coupled to HGC NP dissolved in DMSO. Reactions were performed at room
hnique. NIR imaging of lungs was taken at 1 h, 6 h, 24 h, 3 days, 7 days, and 14 daysere calculated by relative fluorescent intensities compared to the vehicle control.roup.
146 M. Choi et al. / Toxicology Letters 199 (2010) 144–152
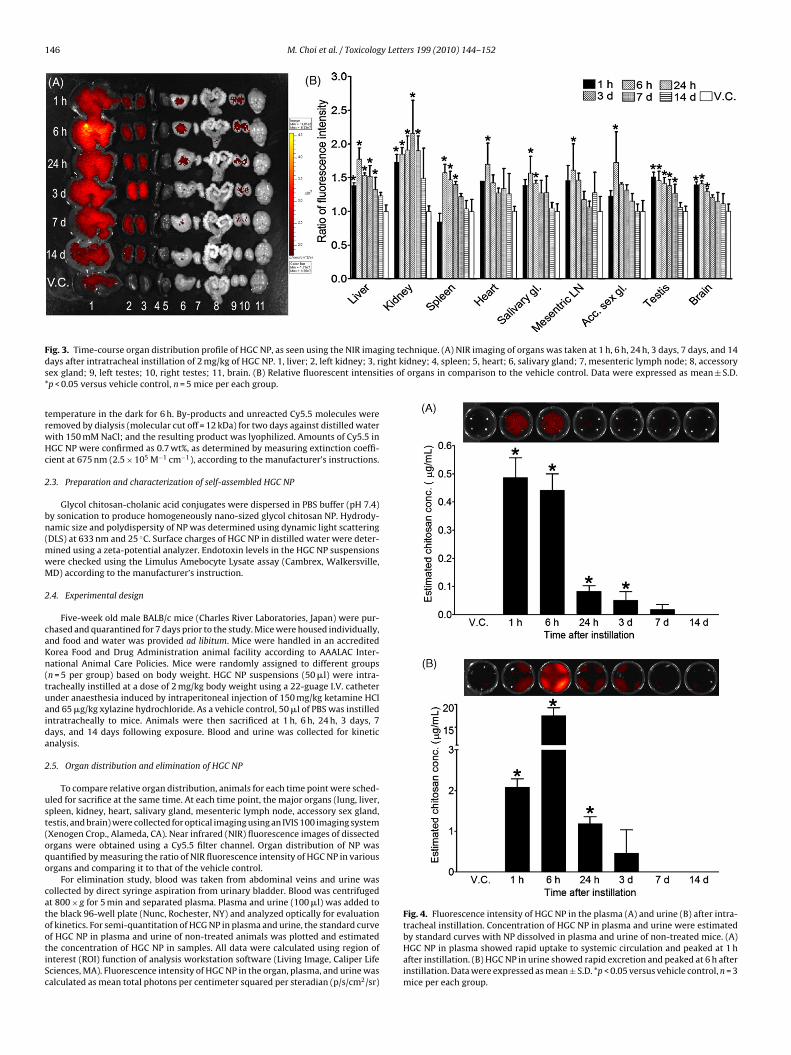
F ing technique. (A) NIR imaging of organs was taken at 1 h, 6 h, 24 h, 3 days, 7 days, and 14d ight kidney; 4, spleen; 5, heart; 6, salivary gland; 7, mesenteric lymph node; 8, accessorys ties of organs in comparison to the vehicle control. Data were expressed as mean ± S.D.*
trwHc
2
bn(mwM
2
caKn(tuaida
2
ust(oqo
catootiSc
Fig. 4. Fluorescence intensity of HGC NP in the plasma (A) and urine (B) after intra-
ig. 3. Time-course organ distribution profile of HGC NP, as seen using the NIR imagays after intratracheal instillation of 2 mg/kg of HGC NP. 1, liver; 2, left kidney; 3, rex gland; 9, left testes; 10, right testes; 11, brain. (B) Relative fluorescent intensip < 0.05 versus vehicle control, n = 5 mice per each group.
emperature in the dark for 6 h. By-products and unreacted Cy5.5 molecules wereemoved by dialysis (molecular cut off = 12 kDa) for two days against distilled waterith 150 mM NaCl; and the resulting product was lyophilized. Amounts of Cy5.5 inGC NP were confirmed as 0.7 wt%, as determined by measuring extinction coeffi-ient at 675 nm (2.5 × 105 M−1 cm−1), according to the manufacturer’s instructions.
.3. Preparation and characterization of self-assembled HGC NP
Glycol chitosan-cholanic acid conjugates were dispersed in PBS buffer (pH 7.4)y sonication to produce homogeneously nano-sized glycol chitosan NP. Hydrody-amic size and polydispersity of NP was determined using dynamic light scatteringDLS) at 633 nm and 25 ◦C. Surface charges of HGC NP in distilled water were deter-
ined using a zeta-potential analyzer. Endotoxin levels in the HGC NP suspensionsere checked using the Limulus Amebocyte Lysate assay (Cambrex, Walkersville,D) according to the manufacturer’s instruction.
.4. Experimental design
Five-week old male BALB/c mice (Charles River Laboratories, Japan) were pur-hased and quarantined for 7 days prior to the study. Mice were housed individually,nd food and water was provided ad libitum. Mice were handled in an accreditedorea Food and Drug Administration animal facility according to AAALAC Inter-ational Animal Care Policies. Mice were randomly assigned to different groupsn = 5 per group) based on body weight. HGC NP suspensions (50 �l) were intra-racheally instilled at a dose of 2 mg/kg body weight using a 22-guage I.V. catheternder anaesthesia induced by intraperitoneal injection of 150 mg/kg ketamine HClnd 65 �g/kg xylazine hydrochloride. As a vehicle control, 50 �l of PBS was instilledntratracheally to mice. Animals were then sacrificed at 1 h, 6 h, 24 h, 3 days, 7ays, and 14 days following exposure. Blood and urine was collected for kineticnalysis.
.5. Organ distribution and elimination of HGC NP
To compare relative organ distribution, animals for each time point were sched-led for sacrifice at the same time. At each time point, the major organs (lung, liver,pleen, kidney, heart, salivary gland, mesenteric lymph node, accessory sex gland,estis, and brain) were collected for optical imaging using an IVIS 100 imaging systemXenogen Crop., Alameda, CA). Near infrared (NIR) fluorescence images of dissectedrgans were obtained using a Cy5.5 filter channel. Organ distribution of NP wasuantified by measuring the ratio of NIR fluorescence intensity of HGC NP in variousrgans and comparing it to that of the vehicle control.
For elimination study, blood was taken from abdominal veins and urine wasollected by direct syringe aspiration from urinary bladder. Blood was centrifugedt 800 × g for 5 min and separated plasma. Plasma and urine (100 �l) was added tohe black 96-well plate (Nunc, Rochester, NY) and analyzed optically for evaluation
f kinetics. For semi-quantitation of HCG NP in plasma and urine, the standard curvef HGC NP in plasma and urine of non-treated animals was plotted and estimatedhe concentration of HGC NP in samples. All data were calculated using region ofnterest (ROI) function of analysis workstation software (Living Image, Caliper Lifeciences, MA). Fluorescence intensity of HGC NP in the organ, plasma, and urine wasalculated as mean total photons per centimeter squared per steradian (p/s/cm2/sr)tracheal instillation. Concentration of HGC NP in plasma and urine were estimatedby standard curves with NP dissolved in plasma and urine of non-treated mice. (A)HGC NP in plasma showed rapid uptake to systemic circulation and peaked at 1 hafter instillation. (B) HGC NP in urine showed rapid excretion and peaked at 6 h afterinstillation. Data were expressed as mean ± S.D. *p < 0.05 versus vehicle control, n = 3mice per each group.
M. Choi et al. / Toxicology Letters 199 (2010) 144–152 147
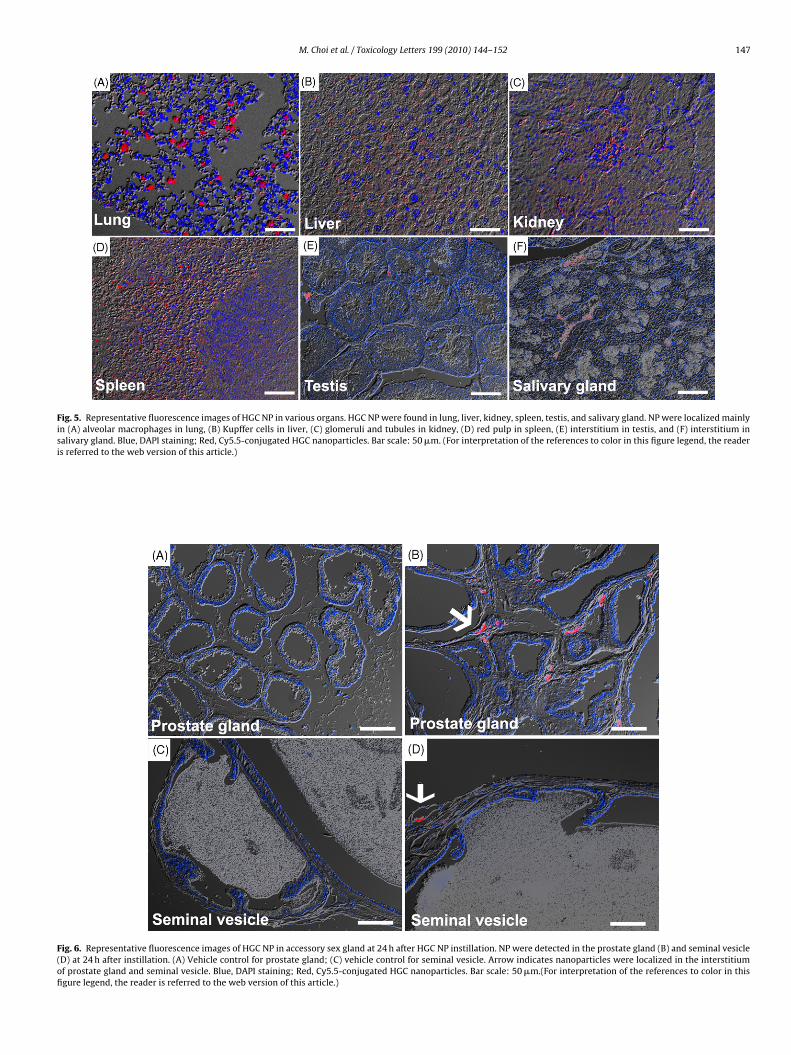
Fig. 5. Representative fluorescence images of HGC NP in various organs. HGC NP were found in lung, liver, kidney, spleen, testis, and salivary gland. NP were localized mainlyin (A) alveolar macrophages in lung, (B) Kupffer cells in liver, (C) glomeruli and tubules in kidney, (D) red pulp in spleen, (E) interstitium in testis, and (F) interstitium insalivary gland. Blue, DAPI staining; Red, Cy5.5-conjugated HGC nanoparticles. Bar scale: 50 �m. (For interpretation of the references to color in this figure legend, the readeris referred to the web version of this article.)
Fig. 6. Representative fluorescence images of HGC NP in accessory sex gland at 24 h after HGC NP instillation. NP were detected in the prostate gland (B) and seminal vesicle(D) at 24 h after instillation. (A) Vehicle control for prostate gland; (C) vehicle control for seminal vesicle. Arrow indicates nanoparticles were localized in the interstitiumof prostate gland and seminal vesicle. Blue, DAPI staining; Red, Cy5.5-conjugated HGC nanoparticles. Bar scale: 50 �m.(For interpretation of the references to color in thisfigure legend, the reader is referred to the web version of this article.)
148 M. Choi et al. / Toxicology Lette
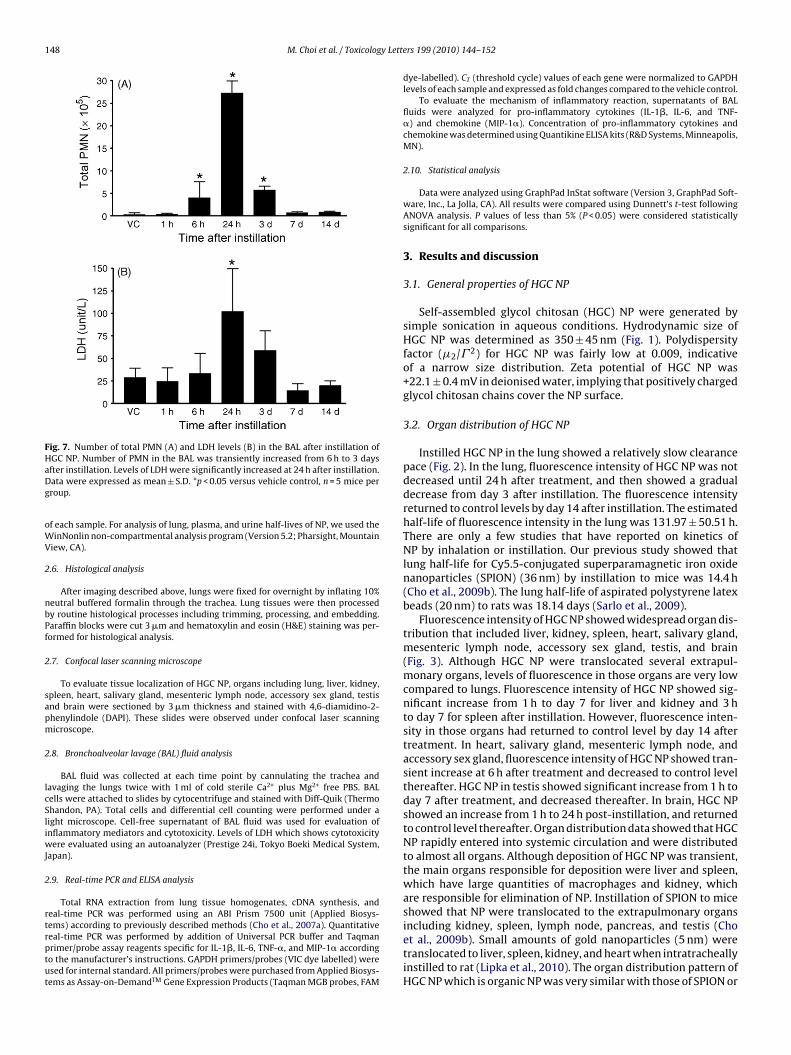
Fig. 7. Number of total PMN (A) and LDH levels (B) in the BAL after instillation ofHaDg
oWV
2
nbPf
2
sapm
2
lcSliwJ
2
rtrptut
GC NP. Number of PMN in the BAL was transiently increased from 6 h to 3 daysfter instillation. Levels of LDH were significantly increased at 24 h after instillation.ata were expressed as mean ± S.D. *p < 0.05 versus vehicle control, n = 5 mice perroup.
f each sample. For analysis of lung, plasma, and urine half-lives of NP, we used theinNonlin non-compartmental analysis program (Version 5.2; Pharsight, Mountain
iew, CA).
.6. Histological analysis
After imaging described above, lungs were fixed for overnight by inflating 10%eutral buffered formalin through the trachea. Lung tissues were then processedy routine histological processes including trimming, processing, and embedding.araffin blocks were cut 3 �m and hematoxylin and eosin (H&E) staining was per-ormed for histological analysis.
.7. Confocal laser scanning microscope
To evaluate tissue localization of HGC NP, organs including lung, liver, kidney,pleen, heart, salivary gland, mesenteric lymph node, accessory sex gland, testisnd brain were sectioned by 3 �m thickness and stained with 4,6-diamidino-2-henylindole (DAPI). These slides were observed under confocal laser scanningicroscope.
.8. Bronchoalveolar lavage (BAL) fluid analysis
BAL fluid was collected at each time point by cannulating the trachea andavaging the lungs twice with 1 ml of cold sterile Ca2+ plus Mg2+ free PBS. BALells were attached to slides by cytocentrifuge and stained with Diff-Quik (Thermohandon, PA). Total cells and differential cell counting were performed under aight microscope. Cell-free supernatant of BAL fluid was used for evaluation ofnflammatory mediators and cytotoxicity. Levels of LDH which shows cytotoxicity
ere evaluated using an autoanalyzer (Prestige 24i, Tokyo Boeki Medical System,apan).
.9. Real-time PCR and ELISA analysis
Total RNA extraction from lung tissue homogenates, cDNA synthesis, andeal-time PCR was performed using an ABI Prism 7500 unit (Applied Biosys-
ems) according to previously described methods (Cho et al., 2007a). Quantitativeeal-time PCR was performed by addition of Universal PCR buffer and Taqmanrimer/probe assay reagents specific for IL-1�, IL-6, TNF-�, and MIP-1� accordingo the manufacturer’s instructions. GAPDH primers/probes (VIC dye labelled) weresed for internal standard. All primers/probes were purchased from Applied Biosys-ems as Assay-on-DemandTM Gene Expression Products (Taqman MGB probes, FAMrs 199 (2010) 144–152
dye-labelled). CT (threshold cycle) values of each gene were normalized to GAPDHlevels of each sample and expressed as fold changes compared to the vehicle control.
To evaluate the mechanism of inflammatory reaction, supernatants of BALfluids were analyzed for pro-inflammatory cytokines (IL-1�, IL-6, and TNF-�) and chemokine (MIP-1�). Concentration of pro-inflammatory cytokines andchemokine was determined using Quantikine ELISA kits (R&D Systems, Minneapolis,MN).
2.10. Statistical analysis
Data were analyzed using GraphPad InStat software (Version 3, GraphPad Soft-ware, Inc., La Jolla, CA). All results were compared using Dunnett’s t-test followingANOVA analysis. P values of less than 5% (P < 0.05) were considered statisticallysignificant for all comparisons.
3. Results and discussion
3.1. General properties of HGC NP
Self-assembled glycol chitosan (HGC) NP were generated bysimple sonication in aqueous conditions. Hydrodynamic size ofHGC NP was determined as 350 ± 45 nm (Fig. 1). Polydispersityfactor (�2/� 2) for HGC NP was fairly low at 0.009, indicativeof a narrow size distribution. Zeta potential of HGC NP was+22.1 ± 0.4 mV in deionised water, implying that positively chargedglycol chitosan chains cover the NP surface.
3.2. Organ distribution of HGC NP
Instilled HGC NP in the lung showed a relatively slow clearancepace (Fig. 2). In the lung, fluorescence intensity of HGC NP was notdecreased until 24 h after treatment, and then showed a gradualdecrease from day 3 after instillation. The fluorescence intensityreturned to control levels by day 14 after instillation. The estimatedhalf-life of fluorescence intensity in the lung was 131.97 ± 50.51 h.There are only a few studies that have reported on kinetics ofNP by inhalation or instillation. Our previous study showed thatlung half-life for Cy5.5-conjugated superparamagnetic iron oxidenanoparticles (SPION) (36 nm) by instillation to mice was 14.4 h(Cho et al., 2009b). The lung half-life of aspirated polystyrene latexbeads (20 nm) to rats was 18.14 days (Sarlo et al., 2009).
Fluorescence intensity of HGC NP showed widespread organ dis-tribution that included liver, kidney, spleen, heart, salivary gland,mesenteric lymph node, accessory sex gland, testis, and brain(Fig. 3). Although HGC NP were translocated several extrapul-monary organs, levels of fluorescence in those organs are very lowcompared to lungs. Fluorescence intensity of HGC NP showed sig-nificant increase from 1 h to day 7 for liver and kidney and 3 hto day 7 for spleen after instillation. However, fluorescence inten-sity in those organs had returned to control level by day 14 aftertreatment. In heart, salivary gland, mesenteric lymph node, andaccessory sex gland, fluorescence intensity of HGC NP showed tran-sient increase at 6 h after treatment and decreased to control levelthereafter. HGC NP in testis showed significant increase from 1 h today 7 after treatment, and decreased thereafter. In brain, HGC NPshowed an increase from 1 h to 24 h post-instillation, and returnedto control level thereafter. Organ distribution data showed that HGCNP rapidly entered into systemic circulation and were distributedto almost all organs. Although deposition of HGC NP was transient,the main organs responsible for deposition were liver and spleen,which have large quantities of macrophages and kidney, whichare responsible for elimination of NP. Instillation of SPION to miceshowed that NP were translocated to the extrapulmonary organs
including kidney, spleen, lymph node, pancreas, and testis (Choet al., 2009b). Small amounts of gold nanoparticles (5 nm) weretranslocated to liver, spleen, kidney, and heart when intratracheallyinstilled to rat (Lipka et al., 2010). The organ distribution pattern ofHGC NP which is organic NP was very similar with those of SPION or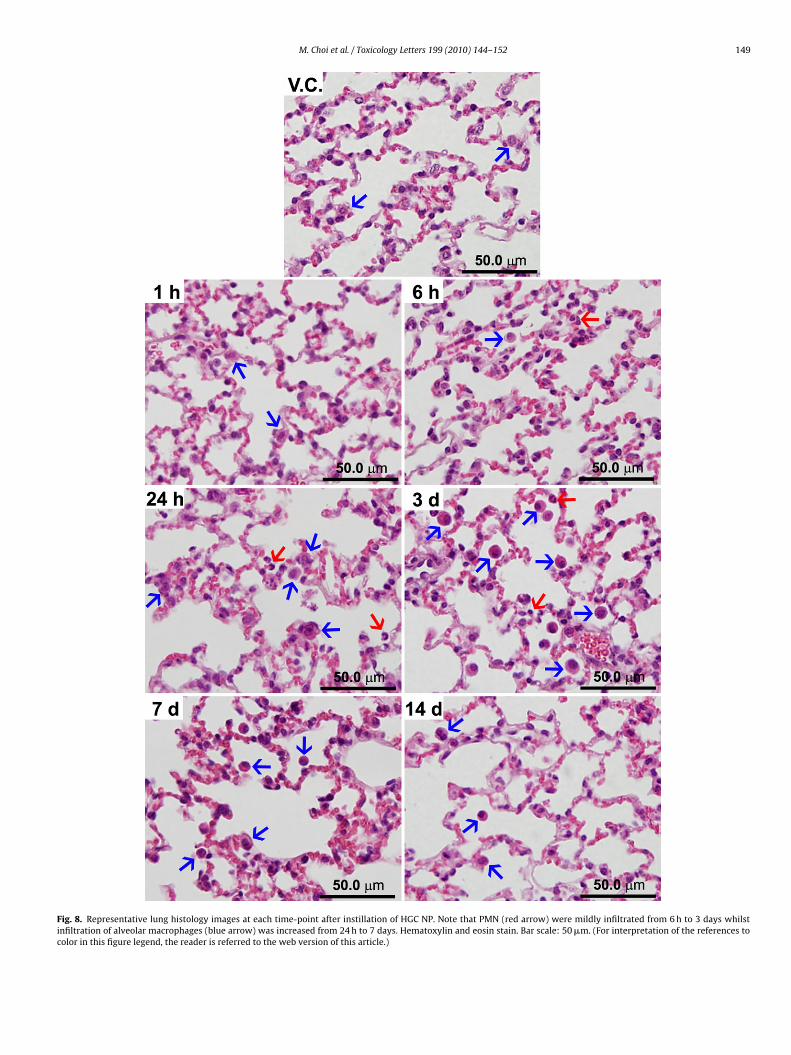
M. Choi et al. / Toxicology Letters 199 (2010) 144–152 149
Fig. 8. Representative lung histology images at each time-point after instillation of HGC NP. Note that PMN (red arrow) were mildly infiltrated from 6 h to 3 days whilstinfiltration of alveolar macrophages (blue arrow) was increased from 24 h to 7 days. Hematoxylin and eosin stain. Bar scale: 50 �m. (For interpretation of the references tocolor in this figure legend, the reader is referred to the web version of this article.)
150 M. Choi et al. / Toxicology Letters 199 (2010) 144–152
F ogenaT to coc
gts
3
bapdmmuto1iNteSilitru
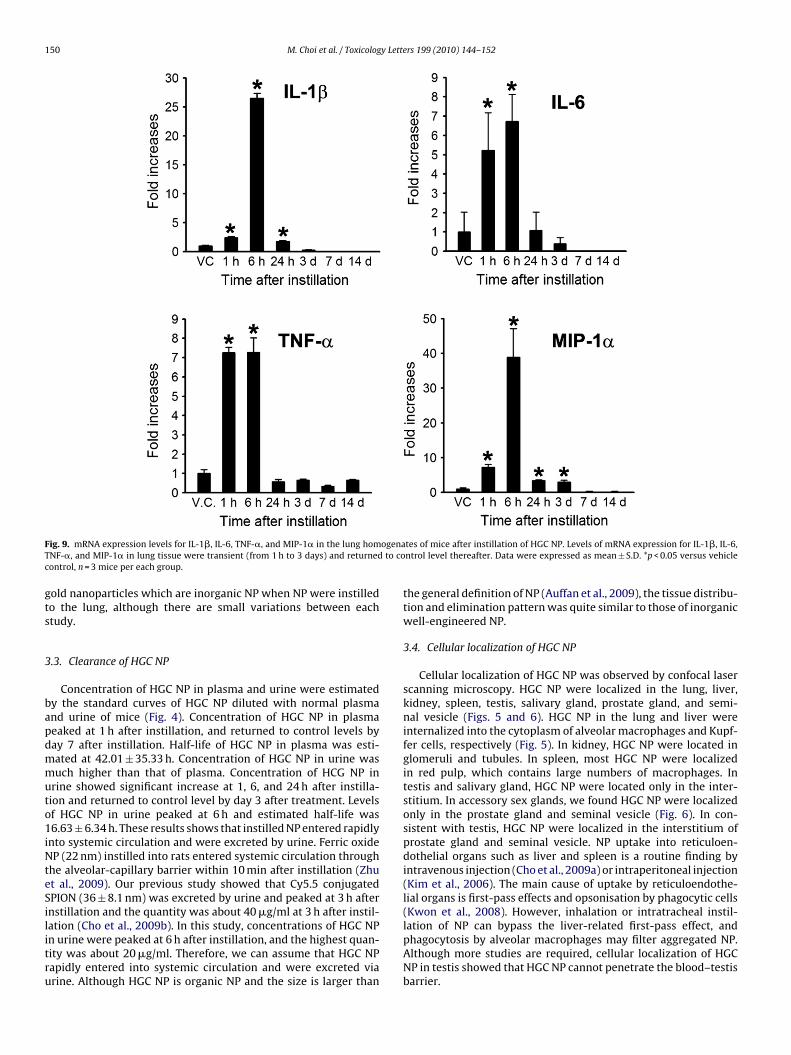
ig. 9. mRNA expression levels for IL-1�, IL-6, TNF-�, and MIP-1� in the lung homNF-�, and MIP-1� in lung tissue were transient (from 1 h to 3 days) and returnedontrol, n = 3 mice per each group.
old nanoparticles which are inorganic NP when NP were instilledo the lung, although there are small variations between eachtudy.
.3. Clearance of HGC NP
Concentration of HGC NP in plasma and urine were estimatedy the standard curves of HGC NP diluted with normal plasmand urine of mice (Fig. 4). Concentration of HGC NP in plasmaeaked at 1 h after instillation, and returned to control levels byay 7 after instillation. Half-life of HGC NP in plasma was esti-ated at 42.01 ± 35.33 h. Concentration of HGC NP in urine wasuch higher than that of plasma. Concentration of HCG NP in
rine showed significant increase at 1, 6, and 24 h after instilla-ion and returned to control level by day 3 after treatment. Levelsf HGC NP in urine peaked at 6 h and estimated half-life was6.63 ± 6.34 h. These results shows that instilled NP entered rapidly
nto systemic circulation and were excreted by urine. Ferric oxideP (22 nm) instilled into rats entered systemic circulation through
he alveolar-capillary barrier within 10 min after instillation (Zhut al., 2009). Our previous study showed that Cy5.5 conjugatedPION (36 ± 8.1 nm) was excreted by urine and peaked at 3 h afternstillation and the quantity was about 40 �g/ml at 3 h after instil-
ation (Cho et al., 2009b). In this study, concentrations of HGC NPn urine were peaked at 6 h after instillation, and the highest quan-ity was about 20 �g/ml. Therefore, we can assume that HGC NPapidly entered into systemic circulation and were excreted viarine. Although HGC NP is organic NP and the size is larger thantes of mice after instillation of HGC NP. Levels of mRNA expression for IL-1�, IL-6,ntrol level thereafter. Data were expressed as mean ± S.D. *p < 0.05 versus vehicle
the general definition of NP (Auffan et al., 2009), the tissue distribu-tion and elimination pattern was quite similar to those of inorganicwell-engineered NP.
3.4. Cellular localization of HGC NP
Cellular localization of HGC NP was observed by confocal laserscanning microscopy. HGC NP were localized in the lung, liver,kidney, spleen, testis, salivary gland, prostate gland, and semi-nal vesicle (Figs. 5 and 6). HGC NP in the lung and liver wereinternalized into the cytoplasm of alveolar macrophages and Kupf-fer cells, respectively (Fig. 5). In kidney, HGC NP were located inglomeruli and tubules. In spleen, most HGC NP were localizedin red pulp, which contains large numbers of macrophages. Intestis and salivary gland, HGC NP were located only in the inter-stitium. In accessory sex glands, we found HGC NP were localizedonly in the prostate gland and seminal vesicle (Fig. 6). In con-sistent with testis, HGC NP were localized in the interstitium ofprostate gland and seminal vesicle. NP uptake into reticuloen-dothelial organs such as liver and spleen is a routine finding byintravenous injection (Cho et al., 2009a) or intraperitoneal injection(Kim et al., 2006). The main cause of uptake by reticuloendothe-lial organs is first-pass effects and opsonisation by phagocytic cells(Kwon et al., 2008). However, inhalation or intratracheal instil-
lation of NP can bypass the liver-related first-pass effect, andphagocytosis by alveolar macrophages may filter aggregated NP.Although more studies are required, cellular localization of HGCNP in testis showed that HGC NP cannot penetrate the blood–testisbarrier.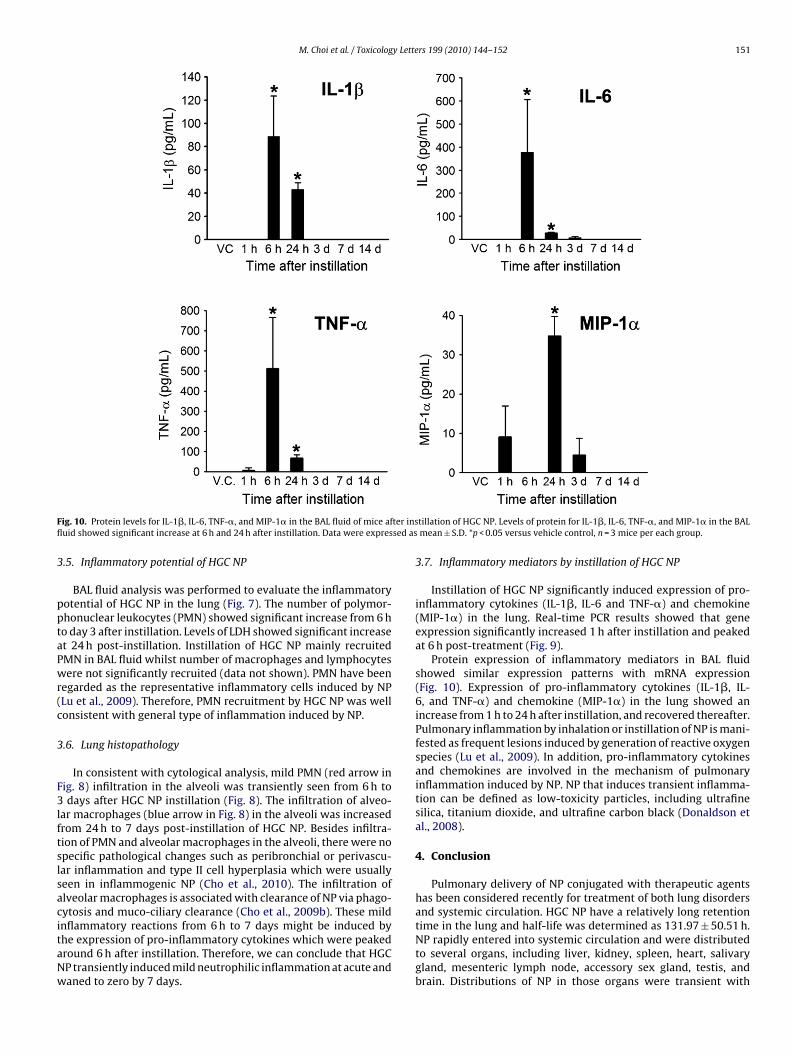
M. Choi et al. / Toxicology Letters 199 (2010) 144–152 151
F ter insfl sed as
3
pptaPwr(c
3
F3lftslsacitaNw
ig. 10. Protein levels for IL-1�, IL-6, TNF-�, and MIP-1� in the BAL fluid of mice afuid showed significant increase at 6 h and 24 h after instillation. Data were expres
.5. Inflammatory potential of HGC NP
BAL fluid analysis was performed to evaluate the inflammatoryotential of HGC NP in the lung (Fig. 7). The number of polymor-honuclear leukocytes (PMN) showed significant increase from 6 ho day 3 after instillation. Levels of LDH showed significant increaset 24 h post-instillation. Instillation of HGC NP mainly recruitedMN in BAL fluid whilst number of macrophages and lymphocytesere not significantly recruited (data not shown). PMN have been
egarded as the representative inflammatory cells induced by NPLu et al., 2009). Therefore, PMN recruitment by HGC NP was wellonsistent with general type of inflammation induced by NP.
.6. Lung histopathology
In consistent with cytological analysis, mild PMN (red arrow inig. 8) infiltration in the alveoli was transiently seen from 6 h todays after HGC NP instillation (Fig. 8). The infiltration of alveo-
ar macrophages (blue arrow in Fig. 8) in the alveoli was increasedrom 24 h to 7 days post-instillation of HGC NP. Besides infiltra-ion of PMN and alveolar macrophages in the alveoli, there were nopecific pathological changes such as peribronchial or perivascu-ar inflammation and type II cell hyperplasia which were usuallyeen in inflammogenic NP (Cho et al., 2010). The infiltration oflveolar macrophages is associated with clearance of NP via phago-ytosis and muco-ciliary clearance (Cho et al., 2009b). These mild
nflammatory reactions from 6 h to 7 days might be induced byhe expression of pro-inflammatory cytokines which were peakedround 6 h after instillation. Therefore, we can conclude that HGCP transiently induced mild neutrophilic inflammation at acute andaned to zero by 7 days.tillation of HGC NP. Levels of protein for IL-1�, IL-6, TNF-�, and MIP-1� in the BALmean ± S.D. *p < 0.05 versus vehicle control, n = 3 mice per each group.
3.7. Inflammatory mediators by instillation of HGC NP
Instillation of HGC NP significantly induced expression of pro-inflammatory cytokines (IL-1�, IL-6 and TNF-�) and chemokine(MIP-1�) in the lung. Real-time PCR results showed that geneexpression significantly increased 1 h after instillation and peakedat 6 h post-treatment (Fig. 9).
Protein expression of inflammatory mediators in BAL fluidshowed similar expression patterns with mRNA expression(Fig. 10). Expression of pro-inflammatory cytokines (IL-1�, IL-6, and TNF-�) and chemokine (MIP-1�) in the lung showed anincrease from 1 h to 24 h after instillation, and recovered thereafter.Pulmonary inflammation by inhalation or instillation of NP is mani-fested as frequent lesions induced by generation of reactive oxygenspecies (Lu et al., 2009). In addition, pro-inflammatory cytokinesand chemokines are involved in the mechanism of pulmonaryinflammation induced by NP. NP that induces transient inflamma-tion can be defined as low-toxicity particles, including ultrafinesilica, titanium dioxide, and ultrafine carbon black (Donaldson etal., 2008).
4. Conclusion
Pulmonary delivery of NP conjugated with therapeutic agentshas been considered recently for treatment of both lung disordersand systemic circulation. HGC NP have a relatively long retention
time in the lung and half-life was determined as 131.97 ± 50.51 h.NP rapidly entered into systemic circulation and were distributedto several organs, including liver, kidney, spleen, heart, salivarygland, mesenteric lymph node, accessory sex gland, testis, andbrain. Distributions of NP in those organs were transient with
1 y Lette
eowNafea
C
A
F(
R
A
B
C
C
C
C
C
C
C
D
GG
H
H
J
52 M. Choi et al. / Toxicolog
xtremely low levels and mainly localized in reticuloendothelialrgans and kidney which are responsible for NP elimination. NPere rapidly excreted via urine and peaked at 6 h after instillation.P induced transient pulmonary inflammation from 6 h to day 3fter instillation. Our findings suggest that HGC NP can be success-ul candidates for use as pulmonary delivery vehicles, owing to theirxcellent biocompatibility, transiency, and low pulmonary toxicity,nd property of rapid elimination without accumulation.
onflict of interest
This paper has no conflict of interest.
cknowledgment
This work was supported by grants from the Koreaood and Drug Administration, the Nanotoxicology Program09151KFDA693).
eferences
uffan, M., Rose, J., Bottero, J.Y., Lowry, G.V., Jolivet, J.P., Wiesner, M.R., 2009. Towardsa definition of inorganic nanoparticles from an environmental, health and safetyperspective. Nat. Nanotechnol. 4, 634–641.
uxton, D., 2006. The promise of nanotechnology for heart, lung and blood diseases.Expert Opin. Drug Deliv. 3, 173–175.
ard, J.W., Zeldin, D.C., Bonner, J.C., Nestmann, E.R., 2008. Pulmonary applicationsand toxicity of engineered nanoparticles. Am. J. Physiol. Lung Cell. Mol. Physiol.295, L400–411.
ho, W.S., Cho, M., Jeong, J., Choi, M., Cho, H.Y., Han, B.S., Kim, S.H., Kim, H.O., Lim,Y.T., Chung, B.H., 2009a. Acute toxicity and pharmacokinetics of 13 nm-sizedPEG-coated gold nanoparticles. Toxicol. Appl. Pharmacol. 236, 16–24.
ho, W.S., Cho, M., Kim, S.R., Choi, M., Lee, J.Y., Han, B.S., Park, S.N., Yu, M.K., Jon,S., Jeong, J., 2009b. Pulmonary toxicity and kinetic study of Cy5.5-conjugatedsuperparamagnetic iron oxide nanoparticles by optical imaging. Toxicol. Appl.Pharmacol. 239, 106–115.
ho, W.S., Choi, M., Han, B.S., Cho, M., Oh, J., Park, K., Kim, S.J., Kim, S.H., Jeong, J.,2007a. Inflammatory mediators induced by intratracheal instillation of ultrafineamorphous silica particles. Toxicol. Lett. 175, 24–33.
ho, W.S., Duffin, R., Poland, C.A., Howie, S.E., Macnee, W., Bradley, M., Megson,I.L., Donaldson, K., 2010. Metal oxide nanoparticles induce unique inflammatoryfootprints in the lung; important implications for nanoparticle testing. Environ.Health Perspect. doi:10.1289/ehp.1002201.
ho, Y.W., Park, S.A., Han, T.H., Son, D.H., Park, J.S., Oh, S.J., Moon, D.H., Cho, K.J.,Ahn, C.H., Byun, Y., Kim, I.S., Kwon, I.C., Kim, S.Y., 2007b. In vivo tumor targetingand radionuclide imaging with self-assembled nanoparticles: mechanisms, keyfactors, and their implications. Biomaterials 28, 1236–1247.
leland, J.L., Daugherty, A., Mrsny, R., 2001. Emerging protein delivery methods.Curr. Opin. Biotechnol. 12, 212–219.
onaldson, K., Borm, P.J., Oberdorster, G., Pinkerton, K.E., Stone, V., Tran, C.L., 2008.Concordance between in vitro and in vivo dosimetry in the proinflammatoryeffects of low-toxicity, low-solubility particles: the key role of the proximalalveolar region. Inhal. Toxicol. 20, 53–62.
onda, I., 2000. The ascent of pulmonary drug delivery. J. Pharm. Sci. 89, 940–945.onda, I., 2007. Peptides and proteins: pulmonary absorption. In: Encyclopedia of
Pharmaceutical Technology. Informa Healthcare, USA.einemann, L., Traut, T., Heise, T., 1997. Time-action profile of inhaled insulin. Dia-
bet. Med. 14, 63–72.yung Park, J., Kwon, S., Lee, M., Chung, H., Kim, J.H., Kim, Y.S., Park, R.W., Kim,
I.S., Bong Seo, S., Kwon, I.C., Young Jeong, S., 2006. Self-assembled nanopar-ticles based on glycol chitosan bearing hydrophobic moieties as carriers fordoxorubicin: in vivo biodistribution and anti-tumor activity. Biomaterials 27,
119–126.in, H., Kim, T.H., Hwang, S.K., Chang, S.H., Kim, H.W., Anderson, H.K., Lee, H.W., Lee,K.H., Colburn, N.H., Yang, H.S., Cho, M.H., Cho, C.S., 2006. Aerosol delivery ofurocanic acid-modified chitosan/programmed cell death 4 complex regulatedapoptosis, cell cycle, and angiogenesis in lungs of K-ras null mice. Mol. CancerTher. 5, 1041–1049.
rs 199 (2010) 144–152
Kim, J.H., Kim, Y.S., Park, K., Lee, S., Nam, H.Y., Min, K.H., Jo, H.G., Park, J.H., Choi, K.,Jeong, S.Y., Park, R.W., Kim, I.S., Kim, K., Kwon, I.C., 2008. Antitumor efficacy ofcisplatin-loaded glycol chitosan nanoparticles in tumor-bearing mice. J. Control.Release 127, 41–49.
Kim, J.S., Yoon, T.J., Yu, K.N., Noh, M.S., Woo, M., Kim, B.G., Lee, K.H., Sohn, B.H.,Park, S.B., Lee, J.K., Cho, M.H., 2006. Cellular uptake of magnetic nanoparticleis mediated through energy-dependent endocytosis in A549 cells. J. Vet. Sci. 7,321–326.
Kim, K., Kwon, S., Park, J.H., Chung, H., Jeong, S.Y., Kwon, I.C., Kim, I.S., 2005.Physicochemical characterizations of self-assembled nanoparticles of glycolchitosan-deoxycholic acid conjugates. Biomacromolecules 6, 1154–1158.
Klingler, C., Muller, B.W., Steckel, H., 2009. Insulin-micro- and nanoparticles forpulmonary delivery. Int. J. Pharm. 377, 173–179.
Kumar, M., Behera, A.K., Lockey, R.F., Zhang, J., Bhullar, G., De La Cruz, C.P., Chen,L.C., Leong, K.W., Huang, S.K., Mohapatra, S.S., 2002. Intranasal gene transferby chitosan–DNA nanospheres protects BALB/c mice against acute respiratorysyncytial virus infection. Hum. Gene Ther. 13, 1415–1425.
Kumar, M., Kong, X., Behera, A.K., Hellermann, G.R., Lockey, R.F., Mohapatra,S.S., 2003. Chitosan IFN-gamma-pDNA nanoparticle (CIN) therapy for allergicasthma. Genet. Vaccin. Ther. 1, 3.
Kwon, J.T., Hwang, S.K., Jin, H., Kim, D.S., Minai-Tehrani, A., Yoon, H.J., Choi, M., Yoon,T.J., Han, D.Y., Kang, Y.W., Yoon, B.I., Lee, J.K., Cho, M.H., 2008. Body distributionof inhaled fluorescent magnetic nanoparticles in the mice. J. Occup. Health 50,1–6.
Laube, B.L., Benedict, G.W., Dobs, A.S., 1998. Time to peak insulin level, relativebioavailability, and effect of site of deposition of nebulized insulin in patientswith noninsulin-dependent diabetes mellitus. J. Aerosol Med. 11, 153–173.
Lipka, J., Semmler-Behnke, M., Sperling, R.A., Wenk, A., Takenaka, S., Schleh, C.,Kissel, T., Parak, W.J., Kreyling, W.G., 2010. Biodistribution of PEG-modified goldnanoparticles following intratracheal instillation and intravenous injection. Bio-materials 31, 6574–6581.
Lu, S., Duffin, R., Poland, C., Daly, P., Murphy, F., Drost, E., Macnee, W., Stone, V.,Donaldson, K., 2009. Efficacy of simple short-term in vitro assays for predictingthe potential of metal oxide nanoparticles to cause pulmonary inflammation.Environ. Health Perspect. 117, 241–247.
Martin, L., Wilson, C.G., Koosha, F., Tetley, L., Gray, A.I., Senel, S., Uchegbu, I.F., 2002.The release of model macromolecules may be controlled by the hydrophobicityof palmitoyl glycol chitosan hydrogels. J. Control. Release 80, 87–100.
Niven, R.W., 1995. Delivery of biotherapeutics by inhalation aerosol. Crit. Rev. Ther.Drug Carrier Syst. 12, 151–231.
O’Hagan, D.T., Illum, L., 1990. Absorption of peptides and proteins from the respi-ratory tract and the potential for development of locally administered vaccine.Crit. Rev. Ther. Drug Carrier Syst. 7, 35–97.
Park, K., Kim, J.H., Nam, Y.S., Lee, S., Nam, H.Y., Kim, K., Park, J.H., Kim, I.S., Choi, K.,Kim, S.Y., Kwon, I.C., 2007. Effect of polymer molecular weight on the tumor tar-geting characteristics of self-assembled glycol chitosan nanoparticles. J. Control.Release 122, 305–314.
Pison, U., Welte, T., Giersig, M., Groneberg, D.A., 2006. Nanomedicine for respiratorydiseases. Eur. J. Pharmacol. 533, 341–350.
Sajja, H.K., East, M.P., Mao, H., Wang, Y.A., Nie, S., Yang, L., 2009. Development of mul-tifunctional nanoparticles for targeted drug delivery and noninvasive imagingof therapeutic effect. Curr. Drug Discov. Technol. 6, 43–51.
Sanvicens, N., Marco, M.P., 2008. Multifunctional nanoparticles—properties andprospects for their use in human medicine. Trends Biotechnol. 26, 425–433.
Sarlo, K., Blackburn, K.L., Clark, E.D., Grothaus, J., Chaney, J., Neu, S., Flood, J.,Abbott, D., Bohne, C., Casey, K., Fryer, C., Kuhn, M., 2009. Tissue distribution of20 nm, 100 nm and 1000 nm fluorescent polystyrene latex nanospheres follow-ing acute systemic or acute and repeat airway exposure in the rat. Toxicology263, 117–126.
Sung, J.C., Pulliam, B.L., Edwards, D.A., 2007. Nanoparticles for drug delivery to thelungs. Trends Biotechnol. 25, 563–570.
Tomoda, K., Ohkoshi, T., Hirota, K., Sonavane, G.S., Nakajima, T., Terada, H., Komuro,M., Kitazato, K., Makino, K., 2009. Preparation and properties of inhalablenanocomposite particles for treatment of lung cancer. Colloids Surf. B: Bioin-terfaces 71, 177–182.
Tsapis, N., Bennett, D., Jackson, B., Weitz, D.A., Edwards, D.A., 2002. Trojan particles:large porous carriers of nanoparticles for drug delivery. Proc. Natl. Acad. Sci.U.S.A. 99, 12001–12005.
Tseng, C.L., Su, W.Y., Yen, K.C., Yang, K.C., Lin, F.H., 2009. The use of biotinylated-
EGF-modified gelatin nanoparticle carrier to enhance cisplatin accumulation incancerous lungs via inhalation. Biomaterials 30, 3476–3485.Zhu, M.T., Feng, W.Y., Wang, Y., Wang, B., Wang, M., Ouyang, H., Zhao, Y.L., Chai,Z.F., 2009. Particokinetics and extrapulmonary translocation of intratracheallyinstilled ferric oxide nanoparticles in rats and the potential health risk assess-ment. Toxicol. Sci. 107, 342–351.

























![Excretion [2015]](https://img.pdfslide.us/doc/110x75/55d39c87bb61eb05278b46dd/excretion-2015-55d47f0693bf7.jpg)



