Embed Size (px)
Citation preview

10914 Phys. Chem. Chem. Phys., 2010, 12, 10914–10918 This journal is c the Owner Societies 2010
Chemisorption-induced gap state at organic–metal interface: Benzenethiol
on Pt(111)
Shigeru Masuda,*a Toyohiro Kamada,a Keita Sasaki,a Masaru Aokiaa and
Yoshitada Morikawab
Received 15th January 2010, Accepted 1st June 2010
DOI: 10.1039/c001016b
Electron emission spectra obtained by thermal collisions of He*(23S) metastable atoms with
benzenethiol (C6H5SH) on Pt(111) were measured to characterize the chemisorption-induced
gap state (CIGS) formed at the organic–metal interface. First-principles calculations using
density functional theory were also performed for an ordered thiolate (C6H5S) monolayer on
Pt(111). Our data exhibit that the CIGS due to the S 3p–Pt 5d mixings appears just below the
Fermi level (EF) of the substrate, where the local density of states decreases drastically from the
S terminal to the benzene ring. Furthermore, strong benzene p(1e1g)–S 3p couplings are
apparently lifted upon the formation of thiolate. These features indicate that thiolate is
not a good mediator of metal wave functions at EF, which is closely related to tunneling
probability (and eventually electric conductance) in the relevant metal–organic–metal
junctions at zero bias.
1. Introduction
Charge transport across the organic–metal interface has
attracted considerable attention for the basic understanding
and potential use in organic-based electronic devices,1–5
e.g., organic thin-film solar cells, organic light emitting diodes,
functional metal–organic–metal junctions. One of the critical
issues in the field is to control the gap states induced at the
organic–metal interface, as in the case of heterojunctions in
semiconductor devices. When the organic molecule is bound
chemically to a metal substrate, the discrete levels of the
molecule are shifted and broadened to some extent, some of
which may be split to form new electronic states in the
HOMO–LUMO gap (HOMO: highest occupied molecular
orbital and LUMO: lowest unoccupied molecular orbital).
The chemisorption-induced gap state (CIGS) thus formed
mediates the extension of metal wave functions to the chemisorbed
species, so that it plays a crucial role in tunneling probability
and eventually electric conductivity in the relevant molecular
junction. The CIGS is also responsible for the formation of a
dipole layer at the organic–metal interface, which directly
affects the charge injection barrier. Thus, the systematic under-
standing and controlling of the CIGS is a key factor for
fabricating novel organic–metal systems. Here, we report a
new characterization of the CIGS based on the asymptotic
behavior of metal wave functions exposed inside the organic
molecule by taking benzenethiolate (C6H5S) on Pt(111) as a
model system.
Metastable atom electron spectroscopy (MAES) used here is
based on the energy analysis of electrons emitted by thermal
collisions of rare gas metastable atoms such as He*(1s2s, 23S)
with a solid surface.6,7 On a metal surface such as Pt(111), the
He*(23S) atom deexcites to the ground state predominantly via
resonance ionization (RI) followed by Auger neutralization
(AN). Since the AN process produces two holes in the valence
bands, the electron emission spectrum shows a broad structure,
reflecting the self-convolution of the local density of states. On
an insulator surface, such as an ordinary organic film, the RI
process is suppressed and Penning ionization (PI) occurs instead,
where an electron in the valence state of the film fills the He* 1s
hole and simultaneously the 2s electron is emitted. The PI
process yields a single-hole spectrum, as in the case of ultraviolet
photoemission spectroscopy (UPS). The He* atoms do not
penetrate into the bulk, and therefore, the local electronic states
at the outermost layer can be selectively probed. This unique
feature has been applied to identify the CIGS emerged in the
organic–metal systems, e.g., C6H6/Pd(110),8 alkanethiolates
(CnH2n+1S, n = 1–3)/Pt(111),9 C60 on Pt(111).10 Furthermore,
it has been used to clarify the relationship between CIGS and the
charge injection barrier, by taking bathocuproine (BCP) on
polycrystalline Au as a model system.11
The purpose of the present study is two-fold. The first
purpose is to identify the CIGS formed at the C6H5S–Pt(111)
interface by MAES and first-principles calculations using
density functional theory (DFT). Our data show that the
local density of CIGS near the Fermi level (EF) significantly
decreases from the terminal S atom to the benzene ring,
indicating that benzenethiolate is not a good mediator of
metal wave functions. The second purpose is to establish that
the asymptotic feature of CIGS is closely related to the charge
transport across the organic–metal interface. The current–voltage
(I–V) characteristics in benzenedithiolate (BDT, SC6H4S)
bridged by a pair of metal electrodes have been exten-
sively studied both from experimental12–15 and theoretical16–22
points of view. The measured conductances at zero bias in
aDepartment of Basic Science, Graduate School of Arts and Sciences,The University of Tokyo, Komaba, Meguro, Tokyo 153-8902, Japan.E-mail: [email protected]
b The Institute of Scientific and Industrial Research, Osaka University,8-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan
PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics
Publ
ishe
d on
26
July
201
0. D
ownl
oade
d by
Uni
vers
ity o
f C
onne
ctic
ut o
n 29
/10/
2014
02:
01:2
2.
View Article Online / Journal Homepage / Table of Contents for this issue

This journal is c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 10914–10918 10915
Au–BDT–Au13,14 and Pt–BDT–Pt15 junctions are much lower
than that in a Pt–C60–Pt junction,23 but is the same order as
that in a Pt–hexanedithiolate–Pt junction.24
2. Experimental
The experimental apparatus and related procedure are reported
elsewhere.25,26 The Pt(111) substrate was cleaned by repeated
Ar+ ion sputtering and heating cycles. The clean substrate
showed a well-ordered 1 � 1 pattern, and no impurities were
detected within the limit of Auger electron spectroscopy (AES).
The chemisorbed overlayers and condensed films of C6H5SH
were prepared by exposing the clean Pt(111) substrate held at
200 and 135 K to the gaseous molecules, respectively. The
thickness of the condensed films was estimated by exposure-
dependent UPS and MAES spectra.
3. Computational details
All calculations based on a generalized gradient approxima-
tion (GGA)27 in DFT were performed using a program package
called simulation tool for atom technology (STATE).28 The
pseudopotentials of H 1s, C 2p, and Pt 5d states are con-
structed by Vanderbilt’s ultrasoft scheme,29 whereas those of
other components are constructed by normconserving
scheme.30,31 The cut-off energies for the wave function and
the augmented charge are 25 Ry and 225 Ry, respectively. A
periodic slab model is used, with each slab composed of six
atomic layers, separated by a vacuum region of 15.9 A.
Benzenethiolate is introduced on one side of the slab. For
the structural optimization, benzenethiolate and the top three
layers of the substrate atoms are allowed to relax. For
comparison, a periodic slab with three atomic layers separated
by a vacuum region of 22.8 A was also used.
4. Results and discussion
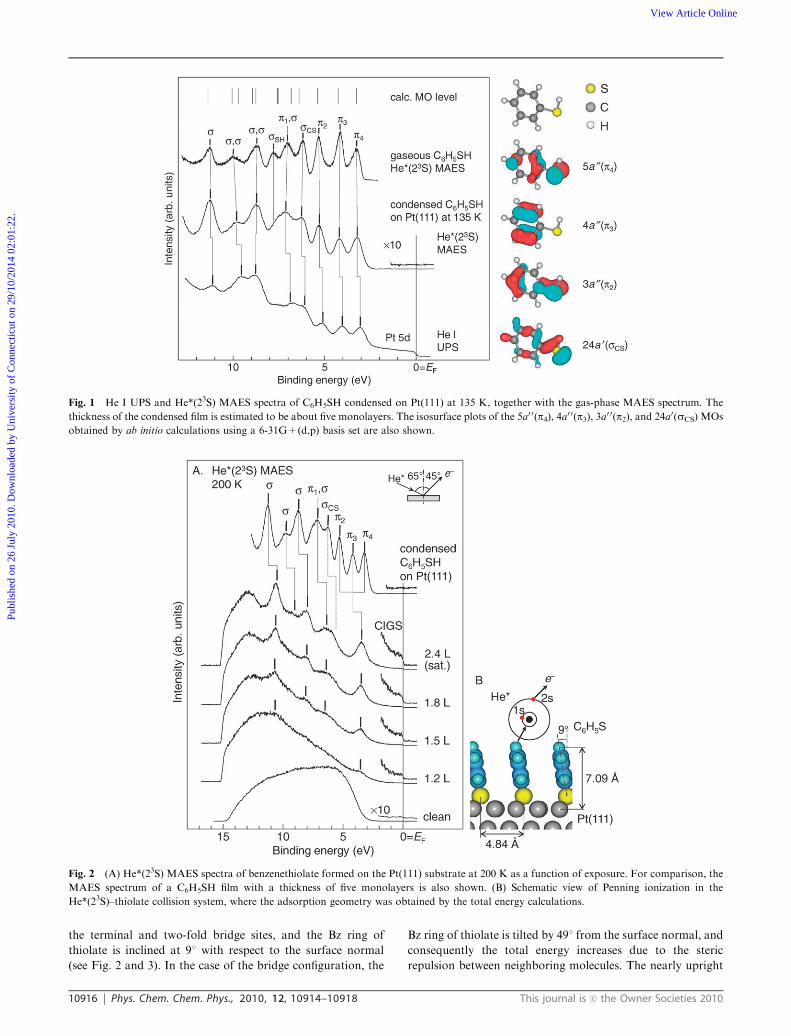
To clarify the electronic states of C6H5SH without direct
contact with the metal substrate, we measured the He I UPS
and He*(23S) MAES spectra of C6H5SH in the gas and con-
densed phases. Fig. 1 shows the typical data for a condensed
film on Pt(111) with a thickness of about five monolayers. The
binding energy is referred to the Fermi level (EF) of the
substrate. As a reference, the gas-phase MAES spectrum and
some molecular orbitals (MOs) of C6H5SH obtained by
ab initio calculations at the B3LYP level are also shown in
the figure, where the notations are based on the Cs symmetry.
The gas-phase spectrum exhibits several bands due to PI from
the relevant MOs.32,33 The 5a0 0(p4) and 3a0 0(p2) MOs are
composed of antibonding and bonding couplings between
doubly-degenerate benzene (Bz) 1e1g and S 3p orbitals,
whereas the 4a0 0(p3) MO is composed of Bz 1e1g with a nodal
plane on the S atom. The ionization energies of the p4, p3 andp2 bands determined by the UPS spectrum (not shown)
are 8.53, 9.53, and 10.63 eV, respectively. The large energy
splittings correspond well to the calculated values (DE(p4–p3) =1.01 eV and DE(p3–p2) = 1.12 eV), indicating a strong Bz p–S3p conjugation. The DFT calculation for free C6H5S also
shows the strong Bz p–S 3p coupling (DE(p4–p3) = 0.98 eV
and DE(p3–p2) = 1.21 eV) (see below). The 2a0 0(p1) MO is
derived mainly from the lower-lying Bz 1a2u orbital.
The 24a0(sCS) and 22a0(sSH) MOs are distributed along the
molecular skeleton with a large amount of the S 3p components.
As is seen in Fig. 1, the corresponding bands appear in the
condensed-phase spectra. The Pt 5d bands are clearly observed
just below EF in the UPS spectrum, but are missing in the
MAES spectrum. Further, the C6H5SH-derived bands in the
MAES are slightly shifted to the higher binding energy side
relative to those in the UPS spectrum. These features are due
to the fact that electron emission upon He*(23S) impact
selectively takes place at the topmost layer.6,34 The absence
of electron emission near EF in the MAES spectrum clearly
indicates that the topmost molecules (without a chemical bond
to the Pt substrate) are insulating in character, with a wide
HOMO–LUMO gap.
Fig. 2 shows the coverage dependence of the He*(23S)
MAES spectra obtained by exposing the Pt(111) substrate at
200 K to gaseous C6H5SH. At 200 K the molecules are known
to chemisorb on the substrate through the S atom with a
cleavage of the S–H bond, forming thiolate (C6H5S–Pt).35
According to scanning tunneling microscopy (STM) and
AES studies,36 the saturation coverage is estimated to be
0.33. At low exposure (below 1.2 L), the He*(23S) atoms
deexcite predominantly via the two-step process (RI + AN),
as in the case of the clean surface. At high exposure, PI occurs
as a competing process, and its contribution peaks at satura-
tion coverage (2.4 L). Three spectral features can be deduced.
(1) The sCS-derived band is strongly suppressed upon
chemisorption, in spite of a clear appearance of the other
s-derived bands. This suggests that the thiolate species is
bound in a tilted manner through the S–Pt bond. As illustrated
in Fig. 2, in such an orientation, the He*(23S) atoms can access
more readily the outer-located benzene ring than the inner-
located S atom and Pt substrate. The tilted orientation is also
supported by comparison with the MAES spectrum of benzene
chemisorbed on Pt(111), where the RI + AN process is
dominant even at saturation coverage, reflecting their flat
orientation.37
(2) The energy splitting among the p4, p3 and p2 bands is
scarcely seen in thiolate, in contrast to the cases of free
C6H5SH, free C6H5S and condensed C6H5SH as mentioned
above. This indicates that the original p MOs in C6H5SH,
particularly the p4 and p2 MOs including the S 3p components,
are drastically altered by the formation of the S–Pt bond.
(3) A weak emission with an edge structure at EF appears
upon chemisorption, and its intensity grows with increasing
coverage. We assigned it to the CIGS formed at the thiolate–
Pt(111) interface, because the corresponding emission is missing
in the cases of bare Pt(111) and multilayer film. A similar
CIGS with metallic character has been observed in the MAES
spectra of CnH2n+1S (n = 1–3)/Pt(111),9 C6H5S/Au(111),38
C6H5Se/Pt(111),38 C60/Pt(111),
10 etc.
To examine the above-mentioned features, we performed
the first-principles DFT calculations for C6H5S on Pt(111).
The periodic slabs with three and six Pt layers were used, and a
(O3 � O3)R301 overlayer was assumed, based on the STM
studies.36 In both cases, the total energy calculations show that
the three-fold hollow site of the (111) plane is more stable than
Publ
ishe
d on
26
July
201
0. D
ownl
oade
d by
Uni
vers
ity o
f C
onne
ctic
ut o
n 29
/10/
2014
02:
01:2
2.
View Article Online
10916 Phys. Chem. Chem. Phys., 2010, 12, 10914–10918 This journal is c the Owner Societies 2010
the terminal and two-fold bridge sites, and the Bz ring of
thiolate is inclined at 91 with respect to the surface normal
(see Fig. 2 and 3). In the case of the bridge configuration, the
Bz ring of thiolate is tilted by 491 from the surface normal, and
consequently the total energy increases due to the steric
repulsion between neighboring molecules. The nearly upright
Fig. 1 He I UPS and He*(23S) MAES spectra of C6H5SH condensed on Pt(111) at 135 K, together with the gas-phase MAES spectrum. The
thickness of the condensed film is estimated to be about five monolayers. The isosurface plots of the 5a0 0(p4), 4a0 0(p3), 3a0 0(p2), and 24a0(sCS) MOs
obtained by ab initio calculations using a 6-31G+(d,p) basis set are also shown.
Fig. 2 (A) He*(23S) MAES spectra of benzenethiolate formed on the Pt(111) substrate at 200 K as a function of exposure. For comparison, the
MAES spectrum of a C6H5SH film with a thickness of five monolayers is also shown. (B) Schematic view of Penning ionization in the
He*(23S)–thiolate collision system, where the adsorption geometry was obtained by the total energy calculations.
Publ
ishe
d on
26
July
201
0. D
ownl
oade
d by
Uni
vers
ity o
f C
onne
ctic
ut o
n 29
/10/
2014
02:
01:2
2.
View Article Online
This journal is c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 10914–10918 10917
configuration of thiolate is consistent with a previous vibrational
analysis.35 A similar DFT calculation has been performed for
Pt(111)(O3 � O3)R301–CnH2n+1S (n = 1–3),9 which show
that alkanethiolates are bound preferentially on the bridge
sites of the (111) plane.
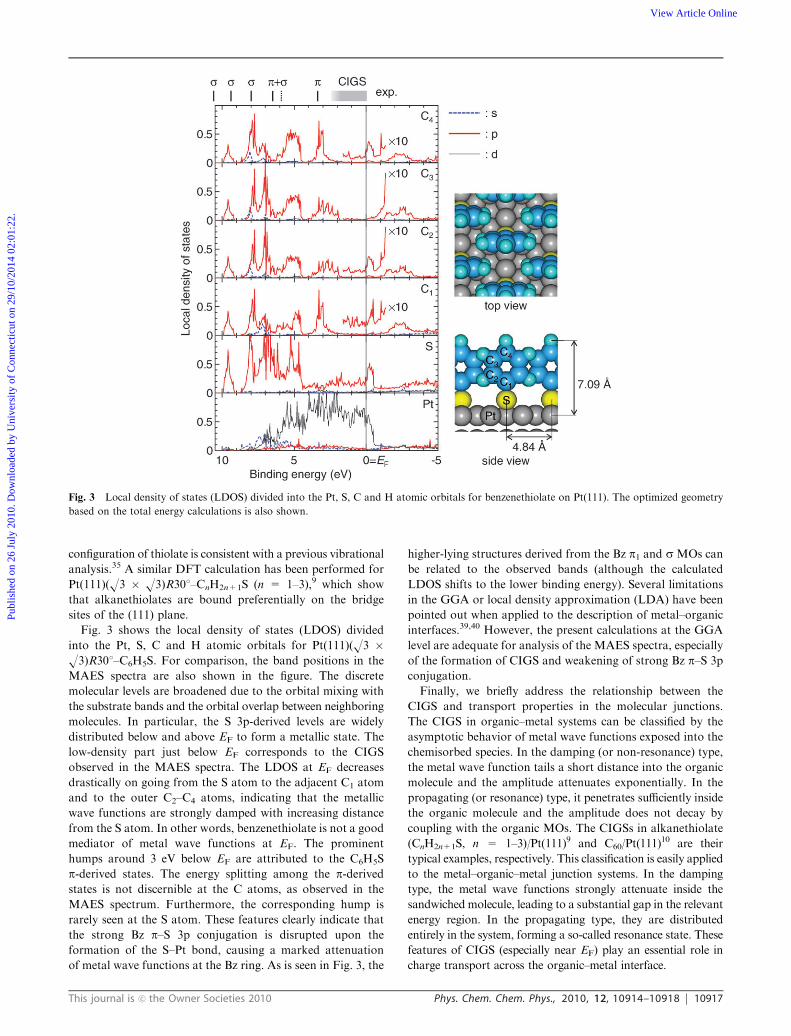
Fig. 3 shows the local density of states (LDOS) divided
into the Pt, S, C and H atomic orbitals for Pt(111)(O3 �O3)R301–C6H5S. For comparison, the band positions in the
MAES spectra are also shown in the figure. The discrete
molecular levels are broadened due to the orbital mixing with
the substrate bands and the orbital overlap between neighboring
molecules. In particular, the S 3p-derived levels are widely
distributed below and above EF to form a metallic state. The
low-density part just below EF corresponds to the CIGS
observed in the MAES spectra. The LDOS at EF decreases
drastically on going from the S atom to the adjacent C1 atom
and to the outer C2–C4 atoms, indicating that the metallic
wave functions are strongly damped with increasing distance
from the S atom. In other words, benzenethiolate is not a good
mediator of metal wave functions at EF. The prominent
humps around 3 eV below EF are attributed to the C6H5S
p-derived states. The energy splitting among the p-derivedstates is not discernible at the C atoms, as observed in the
MAES spectrum. Furthermore, the corresponding hump is
rarely seen at the S atom. These features clearly indicate that
the strong Bz p–S 3p conjugation is disrupted upon the
formation of the S–Pt bond, causing a marked attenuation
of metal wave functions at the Bz ring. As is seen in Fig. 3, the
higher-lying structures derived from the Bz p1 and s MOs can
be related to the observed bands (although the calculated
LDOS shifts to the lower binding energy). Several limitations
in the GGA or local density approximation (LDA) have been
pointed out when applied to the description of metal–organic
interfaces.39,40 However, the present calculations at the GGA
level are adequate for analysis of the MAES spectra, especially
of the formation of CIGS and weakening of strong Bz p–S 3p
conjugation.
Finally, we briefly address the relationship between the
CIGS and transport properties in the molecular junctions.
The CIGS in organic–metal systems can be classified by the
asymptotic behavior of metal wave functions exposed into the
chemisorbed species. In the damping (or non-resonance) type,
the metal wave function tails a short distance into the organic
molecule and the amplitude attenuates exponentially. In the
propagating (or resonance) type, it penetrates sufficiently inside
the organic molecule and the amplitude does not decay by
coupling with the organic MOs. The CIGSs in alkanethiolate
(CnH2n+1S, n = 1–3)/Pt(111)9 and C60/Pt(111)10 are their
typical examples, respectively. This classification is easily applied
to the metal–organic–metal junction systems. In the damping
type, the metal wave functions strongly attenuate inside the
sandwiched molecule, leading to a substantial gap in the relevant
energy region. In the propagating type, they are distributed
entirely in the system, forming a so-called resonance state. These
features of CIGS (especially near EF) play an essential role in
charge transport across the organic–metal interface.
Fig. 3 Local density of states (LDOS) divided into the Pt, S, C and H atomic orbitals for benzenethiolate on Pt(111). The optimized geometry
based on the total energy calculations is also shown.
Publ
ishe
d on
26
July
201
0. D
ownl
oade
d by
Uni
vers
ity o
f C
onne
ctic
ut o
n 29
/10/
2014
02:
01:2
2.
View Article Online

10918 Phys. Chem. Chem. Phys., 2010, 12, 10914–10918 This journal is c the Owner Societies 2010
Recently Kiguchi et al. traced the I–V curves for the
Pt–BDT–Pt system using STM and break junction methods,
and obtained the differential conductance of B3 � 10�2 G0
(where G0 is the quantum unit of conductance) at zero bias.15
This indicates that the transmission coefficient through the
junction is only B3% in the Landauer limit.41 Although this
value is higher than those of ordinary Au–BDT–Au junctions,
i.e., 4� 10�4 G0 (1 V bias),12 4� 10�3 G0,14 or 1.1� 10�2 G0,
13
it is much lower than that in a Pt–C60–Pt junction (B0.7 G0)23
and the same order of magnitude as that of a Pt–1,6-hexane-
dithiolate–Pt junction.24 These results may be strange at first
glance, because free BDT and C60 are typical p-conjugatedsystems while alkanethiolate is a non-conjugated system. As
indicated by the present study for C6H5S/Pt(111), the Bz p–S3p conjugation is heavily destroyed upon chemisorption and
the metal wave functions at EF attenuate rapidly from the
terminal S atom to the Bz ring. This would also be the case for
the Pt–BDT–Pt junction system. As a consequence, the charge
transport through BDT is governed mainly by non-resonant
tunneling rather than resonant tunneling, causing low con-
ductivity at zero bias.
5. Conclusion
We have examined the local electronic states of benzenethiolate
formed on Pt(111) using MAES and first-principles DFT
calculations. The chemisorption-induced gap states are derived
from the S 3p–Pt 5d couplings, yielding a sharp Fermi edge.
The local density of states at EF decreases drastically from the
S terminal to the benzene ring. Furthermore, the strong
benzene p–S 3p conjugation is apparently lifted upon the
formation of thiolate. These features indicate that thiolate is
not a good mediator of metal wave functions at EF, which is
closely related to tunneling probability (and eventually electric
conductance) in the relevant metal–organic–metal junctions at
zero bias.
Acknowledgements
This study is financially supported through Special Coordination
Funds of theMinistry of Education, Culture, Sports, and Science
and Technology of the Japanese Government.
References
1 W. R. Salaneck, S. Stafstrom and J.-L. Bredas, ConjugatedPolymer Surfaces and Interfaces: Electronic and Chemical Structureof Interfaces for Polymer Light Emitting Devices, CambridgeUniversity Press, Cambridge, 1996.
2 H. Ishii, K. Sugiyama, E. Ito and K. Seki, Adv. Mater., 1999, 11,605.
3 In Conjugated Polymer and Molecular Interfaces: Science andTechnology for Photonic and Optoelectronic Applications, ed.W. R. Salaneck, K. Seki, A. Kahn and J.-J. Pireaux, MarcelDekker, New York, 2001.
4 N. Koch, ChemPhysChem, 2007, 8, 1438.
5 F. Flores, J. Ortega and H. Vazquez, Phys. Chem. Chem. Phys.,2009, 11, 8658.
6 Y. Harada, S. Masuda and H. Ozaki, Chem. Rev., 1997, 97, 1897.7 H. Morgner, Adv. At. Mol. Opt. Phys., 2000, 42, 387.8 J. Yoshinobu, M. Kawai, I. Imamura, F. Marumo, R. Suzuki,H. Ozaki, M. Aoki, S. Masuda and M. Aida, Phys. Rev. Lett.,1997, 79, 3942.
9 S. Masuda, Y. Koide, M. Aoki and Y. Morikawa, J. Phys. Chem.C, 2007, 111, 11747.
10 M. Sogo, Y. Sakamoto, M. Aoki, S. Masuda, S. Yanagisawa andY. Morikawa, J. Phys. Chem. C, 2010, 114, 3504.
11 M. Aoki, S. Toyoshima, T. Kamada, M. Sogo, S. Masuda,T. Sakurai and K. Akimoto, J. Appl. Phys., 2009, 106, 043715.
12 M. A. Reed, C. Zhou, C. J. Muller, T. P. Burgin and J. M. Tour,Science, 1997, 278, 252.
13 X. Xiao, B. Xu and N. J. Tao, Nano Lett., 2004, 4, 267.14 M. Kiguchi, S. Miura, K. Hara, M. Sawamura and K. Murakoshi,
Appl. Phys. Lett., 2006, 89, 213104.15 M. Kiguchi, S. Miura, K. Hara, M. Sawamura and K. Murakoshi,
Appl. Phys. Lett., 2007, 91, 053110.16 M. P. Samanta, W. Tian, S. Datta, J. I. Henderson and
C. P. Kubiak, Phys. Rev. B: Condens. Matter, 1996, 53, R7626.17 M. Di Ventra and N. D. Lang, Phys. Rev. B: Condens. Matter
Mater. Phys., 2001, 65, 045402.18 S. N. Yaliraki and M. A. Ratner, J. Chem. Phys., 1998, 109,
5036.19 J. M. Seminario, C. E. De La Cruz and P. A. Derosa, J. Am. Chem.
Soc., 2001, 123, 5616.20 Y. Luo, C.-K. Wang and Y. Fu, J. Chem. Phys., 2002, 117, 10283.21 J. Nara, H. Kino, N. Kobayashi, M. Tsukada and T. Ohno, Thin
Solid Films, 2003, 438–439, 221.22 J. W. Lawson and C. W. Bauschlicher Jr., Phys. Rev. B: Condens.
Matter Mater. Phys., 2006, 74, 125401.23 M. Kiguchi, Appl. Phys. Lett., 2009, 95, 073301.24 V. B. Engelkes, J. M. Beebe and C. D. Frisbie, J. Am. Chem. Soc.,
2004, 126, 14287.25 M. Aoki, Y. Ohashi, S. Masuda, S. Ojima and N. Ueno, J. Chem.
Phys., 2005, 122, 194508.26 M. Aoki, Y. Koide and S. Masuda, J. Electron Spectrosc. Relat.
Phenom., 2007, 156–158, 383.27 J. P. Perdew, K. Bruke and M. Ernzerhof, Phys. Rev. Lett., 1996,
77, 3865.28 Y. Morikawa, Phys. Rev. B: Condens. Matter, 1995, 51, 14802.29 D. Vanderbilt, Phys. Rev. B: Condens. Matter, 1990, 41, 7892.30 G. B. Bachelet, D. R. Hamann and M. Schluter, Phys. Rev. B:
Condens. Matter, 1982, 26, 4199.31 N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter,
1991, 43, 1993.32 A. Abduaini, S. Kera, M. Aoki, K. K. Okudaira, N. Ueno and
Y. Harada, J. Electron Spectrosc. Relat. Phenom., 1998, 88–91,849.
33 N. Kishimoto, M. Furuhashi and K. Ohno, J. Electron Spectrosc.Relat. Phenom., 2000, 113, 35.
34 Y. Harada, H. Ozaki and K. Ohno, Phys. Rev. Lett., 1984, 52,2269.
35 D. A. Stern, E. Wellner, G. N. Salaita, L. Laguren-Davidson,F. Lu, N. Batina, D. G. Frank, D. C. Zapien, N. Walton andA. T. Hubbard, J. Am. Chem. Soc., 1988, 110, 4885.
36 Y.-C. Yang, Y.-P. Yen, L.-Y. O. Yang, S.-L. Yau and K. Itaya,Langmuir, 2004, 20, 10030.
37 M. Sogo, Y. Sakamoto, M. Aoki and S. Masuda, to be published.38 M. Aoki, T. Kamada, K. Sasaki, S. Masuda and Y. Morikawa, to
be published.39 E. Fabiano, M. Piacenza, S. D’Agostino and F. Della Sala,
J. Chem. Phys., 2009, 131, 234101.40 J. M. Garcia-Lastra, C. Rostgaard, A. Rubio and K. S. Thygesen,
Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 245427.41 R. Landauer, Phys. Lett. A, 1981, 85, 91.
Publ
ishe
d on
26
July
201
0. D
ownl
oade
d by
Uni
vers
ity o
f C
onne
ctic
ut o
n 29
/10/
2014
02:
01:2
2.
View Article Online









![MODELLING OF CESIUM CHEMISORPTION UNDER NUCLEAR … 2019 - Final... · chemisorption procesimprovement of s, the Cs chemisorption model Nakajima et alby . [17] is then described](https://img.pdfslide.us/doc/110x75/6079e6b83b443d67370b949a/modelling-of-cesium-chemisorption-under-nuclear-2019-final-chemisorption.jpg)



















