Embed Size (px)
Citation preview

FRONTIERS ARTICLE
Ambiguities in surface nonlinear spectroscopy calculations
Steven J. Byrnes a, Phillip L. Geissler b,c, Y.R. Shen a,!aDepartment of Physics, University of California at Berkeley, Berkeley, CA 94720, United StatesbDepartment of Chemistry, University of California at Berkeley, Berkeley, CA 94720, United StatescChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, United States
a r t i c l e i n f o
Article history:Available online 16 August 2011
a b s t r a c t
Recent molecular simulations of liquid interfaces yield surface sum-frequency vibrational spectrathat can be highly sensitive to the seemingly-arbitrary choice of molecular center in the calculationmethod. We show that the ambiguity arises because widely-used approaches, focusing exclusively onelectric-dipole contributions from the surface, neglect coordinate-dependent quadrupole-order contribu-tions of comparable magnitude from the bulk. The correct calculation includes both surface and bulkresponses. With a judicious choice of molecular center, however, it may be possible to minimize the bulkcontribution, allowing the surface-only calculation to produce a reasonably accurate spectrum. We usewater as an example to elucidate the problem.
! 2011 Elsevier B.V. All rights reserved.
1. Introduction
1.1. Background
Sum-frequency generation (SFG), including its special case ofsecond-harmonic generation (SHG), has been established as a pow-erful technique for studying surfaces and interfaces [1] because ofits ability to provide surface-specific electronic and vibrationalspectra. The technique has already been widely used to probe sys-tems with fundamental importance in physics, chemistry, biology,and geology. In some cases, however, interpretation of the spectracan be difficult. This is particularly true for surface vibrationalspectra of liquids, such as water, where the diversity of moleculararrangements leads to a broad, but featured, spectrum. To under-stand such a spectrum and hence be able to deduce structuralinformation about the surface or interface, we would need a theo-retical calculation that can reproduce the experimental spectrum.Agreement between theory and experiment would lend weightto calculations which can then be further extended to predictnew properties or phenomena about the surface. So far, molecularsimulations have been the only theoretical technique used to cal-culate SF vibrational spectra of liquid interfaces, in particular thewater interfaces because of their importance. However, their suc-cess in reproducing experimental spectra has been limited. Evenin the case of the neat water/vapor interface, the calculated spec-trum in the OH stretch range often does not fully agree with theexperimental one, especially on the low-frequency side [2]
(however, more recent studies claim to have reconciled thelow-frequency discrepancies [3,4]). Different groups have also re-ported somewhat different calculated spectra [2].
Recently, Noah-Vanhoucke et al. found that different seemingly-valid ways to perform the calculation could yield very differentspectra [5]. Therefore it is appropriate to carefully reexamine theapproach and assumptions used in the simulations. In this Letter,we show existing deficiencies in current computational approachesand suggest ways to minimize them. We shall focus on the neatwater/vapor interface as a representative example, but ourdiscussion is generally applicable to all interfaces.
Let us first briefly review the basic theory of reflected SFG froman interface (with more details presented in Section 2). Considertwo input beams at frequencies x1 and x2 overlapping on theinterface that generates a SF output at frequency xSF =x1 +x2 inthe reflected direction (Figure 1). The SF signal can be written as[1]:
S!eSF ; e1; e2" / eSF # v$!2"S;eff : e1e2
!!!!!!2; v
$!2"S;eff $ v
$!2"S % v
$!2"B
&i D~k!!!
!!!; !1"
where v$!2"S and v
$!2"B are defined as the second-order surface and bulk
nonlinear susceptibilities, respectively, êi is the polarization unitvector of the ith field, D~k $~kSF &~k1 &~k2 is the wave vector mis-match between input and output beams, and v
$!2"S;eff is the net ‘‘effec-
tive’’ surface susceptibility. The boundary between ‘‘surface’’ and‘‘bulk’’ in this context is wherever the perturbing effects of the sur-face can no longer be felt (Figure 1), for example a few monolayersfor neat water, or a screening length for ionic solutions. In centro-symmetric media, v
$!2"B vanishes under the electric-dipole approxi-
mation, and is therefore dominated by the electric-quadrupole
0009-2614/$ - see front matter ! 2011 Elsevier B.V. All rights reserved.doi:10.1016/j.cplett.2011.08.027
! Corresponding author.E-mail addresses: [email protected] (P.L. Geissler), [email protected]
(Y.R. Shen)
Chemical Physics Letters 516 (2011) 115–124
Contents lists available at SciVerse ScienceDirect
Chemical Physics Letters
journal homepage: www.elsevier .com/locate /cplet t

and magnetic-dipole response. On the other hand, v$!2"
S is dominatedby the electric-dipole contribution because of the broken inversionsymmetry at the interface. Its resonant spectrum, especially thevibrational one, can provide information about the interfacial struc-ture as well as the orientation of the interfacial molecules. In somecases, the bulk term may be negligible compared with the surfaceterm. In general, however, this is not necessarily true [1], and thereis no simple theory that can be used to predict whether the bulkterm can be neglected or not for a given interface. An order-of-mag-nitude estimate yields that the ratio of the bulk to the surface termis equal to the ratio of the geometric dimension of the induced elec-tric-quadrupole on individual molecules to the average distance be-tween molecules along the surface normal [6]. Thus, roughlyspeaking, if molecules are polar, electric-quadrupoles are well local-ized on the molecules and the surface layer thickness is large com-pared to a chromophore, the bulk term can be significantly smallerthan the surface term. Experimentally, it is generally impossible toseparate surface and bulk contributions [1,7–9]. We usually resortto the observed sensitivity of the spectrum to surface perturbations,for example by adsorbed molecules, to judge whether the surfaceterm dominates or not.
More careful examination of the theory reveals that generallyv$!2"
B =i D~k!!!
!!! consists of three separate terms: one comes from elec-tric-quadrupole (and magnetic-dipole) contribution of interfacialmolecules due to the strong field gradient at the interface betweentwo media of different refractive indices [8]. It would vanish if therefractive indices were matched. Because this contribution is notrelevant to the study here, we shall neglect it in our discussion.The other two come from bulk quadrupole contributions:
v$!2"B !~k1;~k2;~ks"
&i D~k!!!
!!!' v
$!2"BS !~k1;~kf2;~ks" %
v$!2"BB
&i D~k!!!
!!!
v$!2"BB $ v
$!2"B !~k10;~k20;~ks0"
v$!2"
BS !~k1;~k2;~ks" $ v$!2"
B !~k1;~k2;~ks" & v$!2"BB
" #.& i D~k
!!!!!!
where !~k10; ~k20; ~ks0" are nearby ‘‘reference’’ wavevectors that areperfectly phase-matched !D~k0 ' 0", and therefore give a large SFGsignal which can be unambiguously identified with the bulk. (Un-less there is exactly one nearby phase-matched condition, there willbe multiple choices for !~k10; ~k20; ~ks0", and each will give a differentv$!2"BB and v
$!2"BS . If there is no nearby phase-matched condition, the
above still works as a mathematical construction. See Ref. [7] fora similar but not identical construction.) The total effective suscep-tibility is then:
v$!2"S;eff !~k1;~k2;~ks" ' v
$!2"S % v
$!2"BS !~k1;~k2;~ks" %
v$!2"BB
&i D~k!!!
!!!
and it can be shown that v$!2"
S and v$!2"
BS are not separable in measure-ment, while v
$!2"BB can be measured by varying the~ki [7]. Actually, as
we shall show below, v$!2"S and v
$!2"BS are fundamentally inseparable
because the division of electric dipole and electric quadrupole con-tributions from molecules is not unique, but depends on how themultipole expansion is defined [7]. In the remainder of the Letter,for simplicity and clarity, we shall use v
$!2"B in the analysis, but we
note that the ambiguities under discussion are restricted more spe-cifically to its v
$!2"BS component, while the v
$!2"BB component plays no
role in the ambiguities.
1.2. Overview of molecular simulation ambiguities (fundamentalcause)
Consider now the molecular simulation of SFG from surfaces.Conventional approaches were recently discovered to be ambigu-ous and ill-defined [5]. We will show that this problem arises fromthe neglect of electric-quadrupole and magnetic-dipole contribu-tions, i.e., the calculations implicitly assume v
$!2"B ' 0 and
v$!2"S;eff ' v
$!2"S . However, it is deeply problematic to ignore v
$!2"B , not
merely because it may lead to inaccuracies, but more fundamen-tally because it makes the whole calculation ill-defined. Whilev$!2"S;eff of Eq. 1 is well-defined, v
$!2"S on its own and v
$!2"B on its own
are not. Therefore, any attempt to calculate v$!2"
S by ignoring v$!2"B
will necessarily yield an incorrect and ambiguous result. The ambi-guities in defining v
$!2"S and v
$!2"B are the main subject of this Letter,
and in Section 2, we will explain and quantify their origins andtheir consequences.
These ambiguities stem from the usual ambiguity in multipole-expansion: the electromagnetic response of a system can be cor-rectly described using different forms of multipole expansion[7,10–12]. The ambiguity in multipole expansions appears in manyareas of physics—perhaps the most famous example is the fact thatthe dipole moment of a charged molecule is coordinate-system-dependent. Similarly, the electric-quadrupole moment of a polarmolecule is also coordinate-system-dependent. In the case ofSFG, different multipole expansions give different weights to thedipole response v
$!2"S versus the quadrupole response v
$!2"B , but their
combined response v$!2"
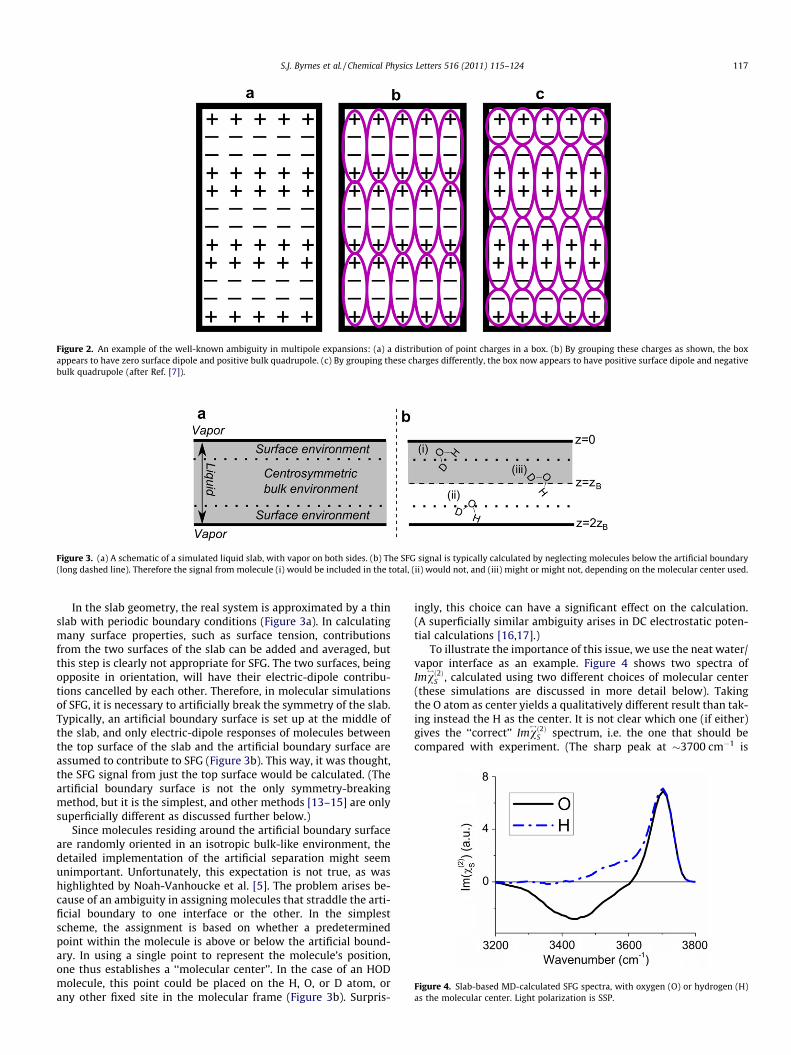
S;eff is constant, as we shall show later.In Figure 2, we illustrate this classical ambiguity in multipole
expansions with a simple example [7]: the same charge-distribu-tion of a charge-neutral system (Figure 2a) can be described ashaving zero surface dipole and positive bulk quadrupole density(Figure 2b), or positive surface dipole and negative bulk quadru-pole density (Figure 2c). These are two equally-valid descriptionsof the same system [10–12].
1.3. Overview of molecular simulation ambiguities (specific cause)
As explained above, from a fundamental and general perspec-tive, previous molecular-simulation calculations of SFG have beenambiguous because they calculated the ill-defined quantity v
$!2"S ,
rather than the unambiguous quantity v$!2"
S;eff . Different multipoleexpansion schemes (e.g., Figure 2) in simulation yield differentvalues of v
$!2"S . Such arbitrariness is often hidden deep within sub-
tle details of the algorithms. We focus on the common case ofmolecular simulations in a slab geometry: here, the arbitrary partof the algorithm was recently discovered by Noah-Vanhouckeet al. [5].
Figure 1. A schematic of the water/vapor interface probed by an SFG experiment.The water is depicted as infinitely deep, which corresponds to an experiment wherelight attenuation or spatial filtering suppresses the signal of the opposite (bottom)interface.
116 S.J. Byrnes et al. / Chemical Physics Letters 516 (2011) 115–124
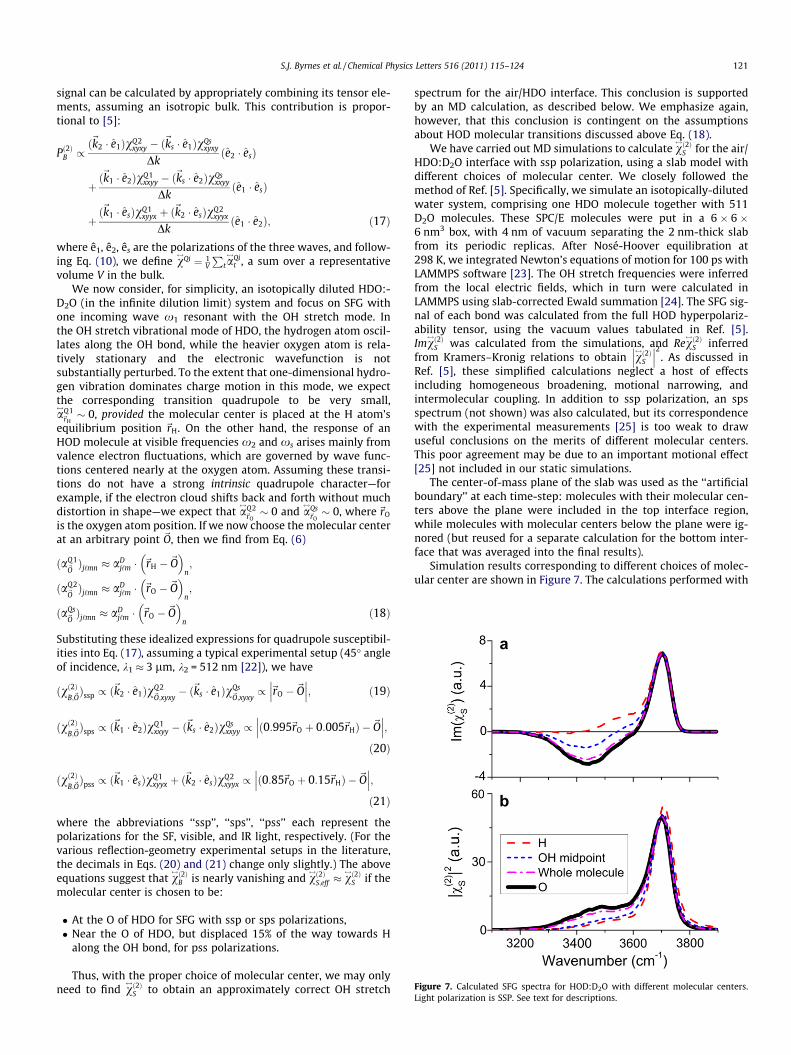
In the slab geometry, the real system is approximated by a thinslab with periodic boundary conditions (Figure 3a). In calculatingmany surface properties, such as surface tension, contributionsfrom the two surfaces of the slab can be added and averaged, butthis step is clearly not appropriate for SFG. The two surfaces, beingopposite in orientation, will have their electric-dipole contribu-tions cancelled by each other. Therefore, in molecular simulationsof SFG, it is necessary to artificially break the symmetry of the slab.Typically, an artificial boundary surface is set up at the middle ofthe slab, and only electric-dipole responses of molecules betweenthe top surface of the slab and the artificial boundary surface areassumed to contribute to SFG (Figure 3b). This way, it was thought,the SFG signal from just the top surface would be calculated. (Theartificial boundary surface is not the only symmetry-breakingmethod, but it is the simplest, and other methods [13–15] are onlysuperficially different as discussed further below.)
Since molecules residing around the artificial boundary surfaceare randomly oriented in an isotropic bulk-like environment, thedetailed implementation of the artificial separation might seemunimportant. Unfortunately, this expectation is not true, as washighlighted by Noah-Vanhoucke et al. [5]. The problem arises be-cause of an ambiguity in assigning molecules that straddle the arti-ficial boundary to one interface or the other. In the simplestscheme, the assignment is based on whether a predeterminedpoint within the molecule is above or below the artificial bound-ary. In using a single point to represent the molecule’s position,one thus establishes a ‘‘molecular center’’. In the case of an HODmolecule, this point could be placed on the H, O, or D atom, orany other fixed site in the molecular frame (Figure 3b). Surpris-
ingly, this choice can have a significant effect on the calculation.(A superficially similar ambiguity arises in DC electrostatic poten-tial calculations [16,17].)
To illustrate the importance of this issue, we use the neat water/vapor interface as an example. Figure 4 shows two spectra ofImv
$!2"S , calculated using two different choices of molecular center
(these simulations are discussed in more detail below). Takingthe O atom as center yields a qualitatively different result than tak-ing instead the H as the center. It is not clear which one (if either)gives the ‘‘correct’’ Imv
$!2"S spectrum, i.e. the one that should be
compared with experiment. (The sharp peak at (3700 cm&1 is
Figure 2. An example of the well-known ambiguity in multipole expansions: (a) a distribution of point charges in a box. (b) By grouping these charges as shown, the boxappears to have zero surface dipole and positive bulk quadrupole. (c) By grouping these charges differently, the box now appears to have positive surface dipole and negativebulk quadrupole (after Ref. [7]).
Figure 3. (a) A schematic of a simulated liquid slab, with vapor on both sides. (b) The SFG signal is typically calculated by neglecting molecules below the artificial boundary(long dashed line). Therefore the signal from molecule (i) would be included in the total, (ii) would not, and (iii) might or might not, depending on the molecular center used.
Figure 4. Slab-based MD-calculated SFG spectra, with oxygen (O) or hydrogen (H)as the molecular center. Light polarization is SSP.
S.J. Byrnes et al. / Chemical Physics Letters 516 (2011) 115–124 117

the same in the two cases, because it is associated with a surfacemode—dangling OH vibration—that does not exist near the artifi-cial boundary surface [2].)
Actually, the correct spectrum must come from calculation ofv$!2"
S;eff , which includes both v$!2"S and v
$!2"B . As we shall show in Section
2, both the v$!2"
S and v$!2"B spectra depend on the choice of the molec-
ular center, but that of v$!2"S;eff does not. With current simulation ap-
proaches, it can be difficult to calculate v$!2"
B . One therefore hopesthat the v
$!2"B contribution can be negligible in comparison with
v$!2"
S . For this to be true, we must have a sufficiently large v$!2"S , such
as in the cases of a surface layer of polar molecules with a signifi-cant net polar orientation, and quite importantly, a choice ofmolecular center that minimizes the absolute value of v
$!2"B . For
our example in Section 4, an SFG reflection measurement of theair/water interface, we shall suggest that the most appropriatemolecular center is at the oxygen atom for SSP polarization (denot-ing S-, S-, and P-polarizations for the fields at xSF, visible x2, andinfrared x1, respectively) and SPS polarization, but slightly dis-placed towards the hydrogen atom for PSS polarization.
The Letter is organized as follows: Section 2 describes the basicsof sum-frequency generation. We first discuss SF response fromindividual molecules in terms of multipole expansion and showthat the division into terms of electric-dipole, electric-quadrupole,and so on is not unique, but depends on the choice of molecularcenter. We then show the same ambiguity arises in describing non-linear susceptibilities, v
$!2"S and v
$!2"B , but that the effective surface
susceptibility, v$!2"
S;eff , which takes into account both surface and bulkcontributions, is independent of the molecular center. Section 3discusses how such ambiguities will lead to different SF vibrationalspectra calculated by molecular simulation using different choicesof molecular center. Section 4 presents molecular dynamics simu-lations of the air/water interface as an example, and shows howthe effect of v
$!2"B can be minimized by a proper choice of molecular
center.
2. Basics of sum-frequency generation
We describe in this section the basics of SF response of an inter-facial system and show that v
$!2"S;eff of Eq. (1) does not depend on the
choice of molecular center, but v$!2"S and v
$!2"B do. We start from SF
responses of individual molecules, and show that the sum of theelectric-dipole and electric-quadrupole responses of an individualmolecule is independent of the choice of molecular center, as anyobservable property should be.
2.1. SF response from individual molecules
The SF response of an individual molecule in the incoming fields~E1!x1;~k1" and ~E2!x2;~k2" can be characterized by its effective elec-tric dipole~p!2"
eff , defined as the electric-dipole moment which, by it-self, would give the same electromagnetic signal in the direction~kSF as the combined effects of its true electric-dipole and higher-or-der moments together. The effective dipole is given in the form of amultipole expansion (i.e., power series in the wavevectors ~k1, ~k2,~kSF" as [1]:
~p!2"eff !xSF ;~kSF" '~p!2"!xSF ;~kSF" & i~kSF # q
$!2"!xSF ;~kSF"
& cxSF
~kSF ) ~l!2"!xSF ;~kSF" % # # #
~p!2" ' a$D :~E1
~E2 % a$Q1;EQ : !r~E1"~E2 % a
$Q2;EQ :~E1!r~E2"
% a$Q1;MD :~B1
~E2 % a$Q2;MD :~E1
~B2
' a$D :~E1
~E2 % ia$Q1 :~E1
~E2~k1 % ia
$Q2 :~E1~E2~k2
q$!2" ' a
$Qs;EQ :~E1~E2
~l!2" ' a$Qs;MD :~E1
~E2 !2"
where ~p!2", q$!2", and ~l!2" are the induced electric dipole, electric
quadrupole, and magnetic dipole at the sum frequency, respec-tively; a
$D, a$Qi;EQ , a
$Qi;MD are corresponding nonlinear electric-dipole,electric-quadrupole, and magnetic-dipole polarizabilities (see Fig-ure 5); and a
$Qi is a linear combination of a$Qi;EQ and a
$Qi;MD (seeAppendix A for details). For simplicity, we shall henceforth usethe term ‘‘quadrupole’’ to refer to both the electric-quadrupoleand magnetic-dipole responses together, as described by the tensora$Qi which combines their effects.
Since (a/k)* 1, where a is a molecular dimension and k is alight wavelength, we are well justified in ignoring the higher-ordermultipoles beyond those shown in Eq. (2). More specifically, for agiven molecule, each higher order in the multipole expansion issuppressed by an additional factor of order (a/k), as usual. On theother hand, when averaged over many molecules in a centrosym-metric bulk, the signal associated with odd-rank susceptibility ten-sors (like a
$D" vanishes, while even-rank contributions (like a$Qi" do
not. Since there are many more molecules in the bulk than the sur-face, the effect of even-rank tensors is boosted by a factor of order(a/k)&1 compared to odd-rank. From these two considerations, a
$D
and a$Qi together comprise the lowest-order term of an (a/k)* 1
expansion, justifying the choice of terms shown in Eq. (2) [1].
Figure 5. The four lowest-order contributions to SFG susceptibility in the multipole expansion. The light-matter interaction is specified by ‘‘ED’’ for ‘‘Electric Dipole’’, or‘‘EQ/MD’’ for ‘‘Electric Quadrupole or Magnetic Dipole’’.
118 S.J. Byrnes et al. / Chemical Physics Letters 516 (2011) 115–124

We can formally rewrite Eq. (2) in a way that distinguishespurely dipolar coupling between the molecule and input fields(yielding a response ~p!2"
D " from those involving a field gradient!~p!2"
Q ". We will soon see, however, that the division is not unique.
~p!2"eff !xSF ;~kSF" '~p!2"
D !xSF ;~kSF" %~p!2"Q !xSF ;~kSF"
~p!2"D ' a
$D :~E1~E2
~p!2"Q ' i+a
$Q1 :~E1~E2~k1 % a
$Q2 :~E1~E2~k2 & a
$Qs :~E1~E2~kSF , !3"
As a physical quantity, the magnitude of ~p!2"eff !xSF ;~kSF" should of
course be independent of the origin for the multipole expansionof the molecular charge oscillations. However, those of ~p!2"
D !xSF ;~kSF" and ~p!2"
Q !xSF ;~kSF" may not be. For explicit discussion of the
problem, we need to define a ‘‘molecular center’’ ~O, which is takenas the origin for the multipole expansion above. (Usually, one wouldchoose ~O to be at a point on the molecule.) With ~O chosen, we can
write ~p!2"eff !xSF ;~kSF" $~p!2"
eff ;~O, ~p!2"
D !xSF ;~kSF" $~p!2"D;~O
and ~p!2"Q !xSF ;~kSF" $
~p!2"Q ;~O
. Here, ~p!2"eff ;~O
is the effective dipole centered at ~O, oscillating at
the sum frequency, that is induced on the molecule by the fields~E1 $~E0
1ei~k1 #~O and ~E2 $~E0
2ei~k1 #~O at ~O, and ~E0
1;~E02 are the amplitudes of
the incoming waves. The SF radiation field at the detector is propor-tional to ~p!2"
eff ;~O$~p!2"
eff ;~Oe&i~kSF #~O; we therefore expect ~p!2"
eff ;~Oto be inde-
pendent of ~O in both phase and magnitude.To confirm that ~p!2"
eff ;~Ois independent of ~O, we rewrite Eq. (2) as
~p!2"eff ;~O
' +~p!2"D;~O
%~p!2"Q ;~O
,e&i~kSF #~O !4"
with
~p!2"D;~O
' a$D~O :~E0
1~E02
" #e&iD~k#~Oei
~kSF #~O
~p!2"Q ;~O
' i a$Q1~O
:~E01~E02~k1 % a
$Q2~O
:~E01~E02~k2 & a
$Qs~O
:~E01~E02~ks
" #e&iD~k#~Oei
~kSF #~O !5"
where D~k $~kSF &~k1 &~k2. We can show that a$D~Ois independent of ~O,
but a$Qi~O
depends on ~O: if ~O and ~O0 $ ~O& D~O are two molecularcenters,
a$Qi~O0 ' a
$Qi~O% a
$DD~O: !6"
The explicit proof is given in Appendix A. The changes of~p!2"D and~p!2"
Qdue to the shift of ~O to ~O0 are given by
~p!2"D;~O0e
&i~kSF #~O0 &~p!2"D;~O
e&i~kSF #~O ' a$D :~E0
1~E02
" #!e&iD~k#~O0 & e&iD~k#~O"
' a$D :~E0
1~E02
" #+iD~k # D~O% # # #,e&iD~k#~O
~p!2"Q ;~O0e
&i~kSF #~O0 &~p!2"Q ;~O
e&i~kSF #~O ' &i a$D :~E0
1~E02
" #!D~k # D~O"e&iD~k#~O % # # # : !7"
We then see, in the limit of neglecting higher-order terms ofD~k # !~O&~O0"!!!
!!! (i.e., in the spirit of multipole expansion, neglectingresponses of higher order than quadrupole), that the molecular-center dependences of ~p!2"
D and ~p!2"Q cancel each other:
~p!2"eff ;~O
' +~p!2"D;~O
%~p!2"Q ;~O
,e&i~kSF #~O ' +~p!2"D;~O0 %~p!2"
Q ;~O0 ,e&i~kSF #~O0 ' ~p!2"
eff ;~O0 !8"
This confirms the earlier statement that ~p!2"eff does not depend on the
choice of the molecular center ~O of the coordinate system, but ~p!2"D
and ~p!2"Q do. The latter often leads to ambiguity in distinguishing
electric-dipole and quadrupole responses of a molecule.
2.2. Surface and bulk SF susceptibilities
The bulk SF susceptibility of a system is usually defined as
v$!2"
B !xSF ;~kSF" $1V
X
j
+~p!2"eff ;j=
~E1j~E2j,; !9"
where the sum is over individual molecules (j) in a mesoscopic bulkvolume V with dimensions large compared to a molecule, and~E1j '~E0
1ei~k1 #~Oj , ~E2j '~E0
2ei~k2 #~Oj . (The vector division in (9) is defined in
the obvious way: for the (i,m,n)th Cartesian component of v$!2"B ,
use the ith component of~p!2"eff ;j, mth of~E1j, and nth of~E2j.) For a med-
ium with inversion symmetry, the electric-dipole part, ~p!2"D , of ~p!2"
effvanishes in summation, but the quadrupole part, ~p!2"
Q , survives; aswe showed in the preceding section, ~p!2"
Q depends on the choice ofmolecular center. Hence, we expect that v
$!2"B must also depend on
the choice of molecular center. From the expression for ~p!2"Q in Eq.
(3), we obtain, for the choice of molecular center of the jth moleculeat ~Oj,
v$!2"B;~O
' 1V
X
j
i aQ1j;~Oj
#~k1 % aQ2j;~Oj
#~k2 & aQsj;~Oj
#~kSF$ %
: !10"
If the molecular center is shifted from ~Oj to ~O0j ' ~Oj & D~Oj, then from
Eq. (6) we have
v$!2"B;~O0 & v
$!2"B;~O
' &iV
X
j
!a$Dj "!D~Oj # D~k"
$ &in a$DD~O
D E# D~k !11"
where n denotes the density of molecules and angular bracketsindicate an average over molecular orientations and arrangementsin the bulk environment.
As mentioned above, v$!2"
B can be split into a v$!2"
BB term which can
be measured by varying ~ki, and a v$!2"
BS term which cannot be mea-sured separately from the surface [7]. The molecular-center depen-
dence in Eq. (11) comes entirely from a change in v$!2"BS , not v
$!2"BB . The
components of v$!2"
BB , such as a$Q2
xyxy & a$Qs
xyxy
" #[7], are independent of
molecular center by Eq. (6). This is consistent with our assertion
that v$!2"
BS and v$!2"
S are fundamentally inseparable, while v$!2"
BB isseparate.
It was already known in the early development of second-har-monic generation and SFG for surface studies that the surfaceand bulk terms in v
$!2"S;eff of Eq. (1) are not separable, either in theory
or in measurement [8,9]. On the other hand, as a measurable phys-ical quantity, v
$!2"S;eff naturally is independent of the choice of molec-
ular center. Thus if v$!2"
B depends on the choice of molecular center,so must v
$!2"S , but the sum of v
$!2"S and v
$!2"B =!&iDk" must not. The ex-
plicit proof is briefly outlined here, with more details given inAppendix C.
Similar to the bulk case, the surface SF susceptibility is generallydefined as
v$!2"S !xSF ;~kSF" $
1A
X
j
+~p!2"eff ;t=
~E1t~E2t , !12"
where the summation is on molecules in a surface layer over a sur-face area A. The surface layer is a thin region at the interface that isstructurally different from the bulk. For second-order nonlinearoptical response of an isotropic liquid like water, for example, thesurface layer is the layer that has broken inversion symmetry. Inpractice, the surface layer is microscopically thin, so that the quad-rupole part,~p!2"
Q , of~p!2"eff can be neglected in the summation restricted
to the surface layer. We then find, for the molecular center of the jthmolecule taken at ~Ot ,
v$!2"S;~O
' 1A
X
j
a$Dj !13"
Here, witha$Dj;~Oj
being independent of ~Oj, the dependence on thechoice of molecular center comes through counting of the set ofmolecules included in the summation: in reality, we should writeEq. (13) as
S.J. Byrnes et al. / Chemical Physics Letters 516 (2011) 115–124 119

v$!2"S;~O
' 1A
X
Oj;z>zB
a$D
j !14"
(see Appendix B) Here, as sketched in Figure 6a, the interface is atz = 0, the semi-infinite bulk medium under discussion is at z < 0,and z = zB (<0) is a plane sufficiently deep to be in the bulk environ-ment, but separated from the interface only by a microscopicallysmall distance. Changing the molecular center from ~Oj to~O0
j ' ~Oj & D~Oj, but keeping zB unchanged for simplicity, will addsome molecules near zB to the sum in Eq. (A.10), and remove others.For illustration, we show in Figure 6b a system of HDO molecules,with the plane z = zB denoted by the dashed line. There are a num-ber of randomly oriented HDO molecules with their oxygen (O) justabove the plane (Figure 6b). If the molecular center of HDO is takento be on O, these molecules will all be counted in the summation ofEq. (14). However, if the molecular center is taken to be on thehydrogen (H), then half of these molecules will no longer becounted in the sum, yielding a different value for v
$!2"S . Since the ef-
fect of changing the molecular center on v$!2"S is roughly from mole-
cules within a layer of D~O!!!
!!! thick, the change of v$!2"
S is expected to be
of the order of aDD~O!!!
!!!. In Appendix C, we derive the net change of
v$!2"S due to the change from ~Oj to ~O0
j precisely as:
v$!2"S;~O0 & v
$!2"S;~O
' &n a$DDOz
D E!15"
(consistent with Ref. [5]). Since a$DDOz
D E–0 in general, this explic-
itly shows that a different choice of ~O will yield a different v$!2"
S .From Eqs. (11) and (15), we see readily
v$!2"S;~O
%v$!2"B;~O
&i D~k!!!
!!!' v
$!2"S;~O0 %
v$!2"B;~O0
&i D~k!!!
!!!: !16"
As a result, v$!2"S;eff is independent of the choice of molecular center ~O.
3. Ambiguities in molecular-dynamics calculations
Discussion in the previous section provides the correct frame-work for calculation to compare with experiment of SFG, empha-sizing the need to calculate v
$!2"S;eff . Unfortunately, this is not the
usual practice reported in the literature. Instead, it is simply as-sumed that v
$!2"B would vanish and just v
$!2"S is calculated, ignoring
the fact that v$!2"S depends on the choice of molecular center. As no-
ticed recently by Noah-Vanhoucke et al. [5], different choices ofmolecular center can yield significantly different SF surface spec-tra. We shall elucidate this by an example in a later section.
In practice, the ambiguity in v$!2"
S is typically resolved by arbi-trarily choosing a molecular center to arrive at a definite answer.However, the arbitrary step (which may be subtle and uninten-
tional) depends on the simulation approach. Therefore we focuson molecular simulations using a slab model, as described in theIntroduction. For surface SFG, these simulations often impose anartificial boundary surface at the middle of the slab to break theinherent inversion symmetry of the slab [5]. We can relate thisprocedure to Eq. (14) by assuming the artificial boundary surfaceis at z = zB. The molecules with molecular centers above the artifi-cial boundary surface are summed up, with the other molecules ig-nored. This procedure is consistent with Eq. (14), so it is alegitimate way to calculate v
$!2"S . However, it also inherits the
pathology that v$!2"
S is an ambiguous quantity whose value dependson the choice of molecular center. Consequently, as noted in Ref.[5], the SF spectrum calculated from v
$!2"S by molecular simulation
depends on the choice of molecular center. Clearly, the correct SFspectrum must come from calculating v
$!2"S;eff $ v
$!2"S % v
$!2"B =!&iDk",
which is independent of the choice of molecular center.The higher-order bulk nonlinear susceptibility, v
$!2"B , given by Eq.
(10) (with further details in Appendix A), is generally more difficultto calculate. Although detailed calculation procedures can be foundin Refs. [18–21], the facts that v
$!2"B is an ambiguous quantity and
that its value depends on the choice of molecular center have notbeen discussed in the literature. As shown in Appendix A, a properquantum–mechanical calculation of v
$!2"B;~O
should use the ‘‘relative’’position operators ~r!~O" $~r &~O in the expressions for the momentoperators ~p, q
$, ~l. Therefore, the result will in general depend on
the choice of ~O.In a molecular-simulation calculation of SFG, one would ideally
determine v$!2"S;eff by calculating both v
$!2"S and v
$!2"B with a consistent
choice of molecular center. The difficulty of computing v$!2"B , how-
ever, may render this approach impractical. A simpler approachis to choose a molecular center that will minimize v
$!2"B , and
therefore maximize v$!2"
S , for the frequency-range and other param-eters under investigation. Then, we may be able to argue that
v$!2"
B;~O=D~k
!!!!!! * v
$!2"S;~O
!!!!!!, and hence v
$!2"S;eff - v
$!2"S;~O, allowing us to obtain a
fairly accurate SF surface spectrum from calculating only v$!2"
S . Thisis likely the case for isotropic liquids composed of small molecules,or small functional groups on larger molecules. In the followingsection, we use water as an example to illustrate these points.
4. Water as an example
The air/neat-water interface has been studied extensively bothin theory and in experiment. There are quite a few molecular sim-ulations of the SF surface spectrum of the system reported in theliterature, but to our knowledge, none of them includes v
$!2"B in
the calculation. Here, we show that v$!2"
B is not negligible if themolecular center is not judiciously chosen. We begin by analyzingv$!2"B more closely. The contribution of v
$!2"B to the experimental
Figure 6. (a) An illustration for Eq. (14). The system is the same as the one described in Figure 1, with the plane z = zB indicated by a dashed line. The notional dividing planez = zB could be placed at any arbitrary depth within the isotropic bulk environment. (b) A close-up view of the group of molecules with oxygen atoms immediately above theplane z = zB. If oxygen is chosen as the molecular center, all of these molecules are included in Eq. (14); if hydrogen is chosen instead, only half are.
120 S.J. Byrnes et al. / Chemical Physics Letters 516 (2011) 115–124
signal can be calculated by appropriately combining its tensor ele-ments, assuming an isotropic bulk. This contribution is propor-tional to [5]:
P!2"B /
!~k2 # e1"vQ2xyxy & !~ks # e1"vQs
xyxy
Dk!e2 # es"
%!~k1 # e2"vQ1
xxyy & !~ks # e2"vQsxxyy
Dk!e1 # es"
%!~k1 # es"vQ1
xyyx % !~k2 # es"vQ2xyyx
Dk!e1 # e2"; !17"
where ê1, ê2, ês are the polarizations of the three waves, and follow-ing Eq. (10), we define v
$Qi ' 1V
Pta$Qit , a sum over a representative
volume V in the bulk.We now consider, for simplicity, an isotopically diluted HDO:-
D2O (in the infinite dilution limit) system and focus on SFG withone incoming wave x1 resonant with the OH stretch mode. Inthe OH stretch vibrational mode of HDO, the hydrogen atom oscil-lates along the OH bond, while the heavier oxygen atom is rela-tively stationary and the electronic wavefunction is notsubstantially perturbed. To the extent that one-dimensional hydro-gen vibration dominates charge motion in this mode, we expectthe corresponding transition quadrupole to be very small,a$Q1~rH
( 0, provided the molecular center is placed at the H atom’sequilibrium position ~rH. On the other hand, the response of anHOD molecule at visible frequencies x2 and xs arises mainly fromvalence electron fluctuations, which are governed by wave func-tions centered nearly at the oxygen atom. Assuming these transi-tions do not have a strong intrinsic quadrupole character—forexample, if the electron cloud shifts back and forth without muchdistortion in shape—we expect that a
$Q2~rO
( 0 and a$Qs~rO
( 0, where~rOis the oxygen atom position. If we now choose the molecular centerat an arbitrary point ~O, then we find from Eq. (6)
!aQ1~O"j‘mn - aD
j‘m # ~rH &~O" #
n;
!aQ2~O"j‘mn - aD
j‘m # ~rO &~O" #
n;
!aQs~O"j‘mn - aD
j‘m # ~rO &~O" #
n!18"
Substituting these idealized expressions for quadrupole susceptibil-ities into Eq. (17), assuming a typical experimental setup (45" angleof incidence, k1 - 3 lm, k2 = 512 nm [22]), we have
!v!2"B;~O
"ssp / !~k2 # e1"vQ2~O;xyxy
& !~ks # e1"vQs~O;xyxy
/ ~rO &~O!!!
!!!; !19"
!v!2"B;~O
"sps / !~k1 # e2"vQ1xxyy & !~ks # e2"vQs
xxyy / 0:995~rO % 0:005~rH! " &~O!!!
!!!;
!20"
!v!2"B;~O
"pss / !~k1 # es"vQ1xyyx % !~k2 # es"vQ2
xyyx / 0:85~rO % 0:15~rH! " &~O!!!
!!!;
!21"
where the abbreviations ‘‘ssp’’, ‘‘sps’’, ‘‘pss’’ each represent thepolarizations for the SF, visible, and IR light, respectively. (For thevarious reflection-geometry experimental setups in the literature,the decimals in Eqs. (20) and (21) change only slightly.) The aboveequations suggest that v
$!2"B is nearly vanishing and v
$!2"S;eff - v
$!2"S if the
molecular center is chosen to be:
. At the O of HDO for SFG with ssp or sps polarizations,
. Near the O of HDO, but displaced 15% of the way towards Halong the OH bond, for pss polarizations.
Thus, with the proper choice of molecular center, we may onlyneed to find v
$!2"S to obtain an approximately correct OH stretch
spectrum for the air/HDO interface. This conclusion is supportedby an MD calculation, as described below. We emphasize again,however, that this conclusion is contingent on the assumptionsabout HOD molecular transitions discussed above Eq. (18).
We have carried out MD simulations to calculate v$!2"
S for the air/HDO:D2O interface with ssp polarization, using a slab model withdifferent choices of molecular center. We closely followed themethod of Ref. [5]. Specifically, we simulate an isotopically-dilutedwater system, comprising one HDO molecule together with 511D2O molecules. These SPC/E molecules were put in a 6 ) 6 )6 nm3 box, with 4 nm of vacuum separating the 2 nm-thick slabfrom its periodic replicas. After Nosé-Hoover equilibration at298 K, we integrated Newton’s equations of motion for 100 ps withLAMMPS software [23]. The OH stretch frequencies were inferredfrom the local electric fields, which in turn were calculated inLAMMPS using slab-corrected Ewald summation [24]. The SFG sig-nal of each bond was calculated from the full HOD hyperpolariz-ability tensor, using the vacuum values tabulated in Ref. [5].Imv
$!2"S was calculated from the simulations, and Rev
$!2"S inferred
from Kramers–Kronig relations to obtain v$!2"S
!!!!!!2. As discussed in
Ref. [5], these simplified calculations neglect a host of effectsincluding homogeneous broadening, motional narrowing, andintermolecular coupling. In addition to ssp polarization, an spsspectrum (not shown) was also calculated, but its correspondencewith the experimental measurements [25] is too weak to drawuseful conclusions on the merits of different molecular centers.This poor agreement may be due to an important motional effect[25] not included in our static simulations.
The center-of-mass plane of the slab was used as the ‘‘artificialboundary’’ at each time-step: molecules with their molecular cen-ters above the plane were included in the top interface region,while molecules with molecular centers below the plane were ig-nored (but reused for a separate calculation for the bottom inter-face that was averaged into the final results).
Simulation results corresponding to different choices of molec-ular center are shown in Figure 7. The calculations performed with
Figure 7. Calculated SFG spectra for HOD:D2O with different molecular centers.Light polarization is SSP. See text for descriptions.
S.J. Byrnes et al. / Chemical Physics Letters 516 (2011) 115–124 121
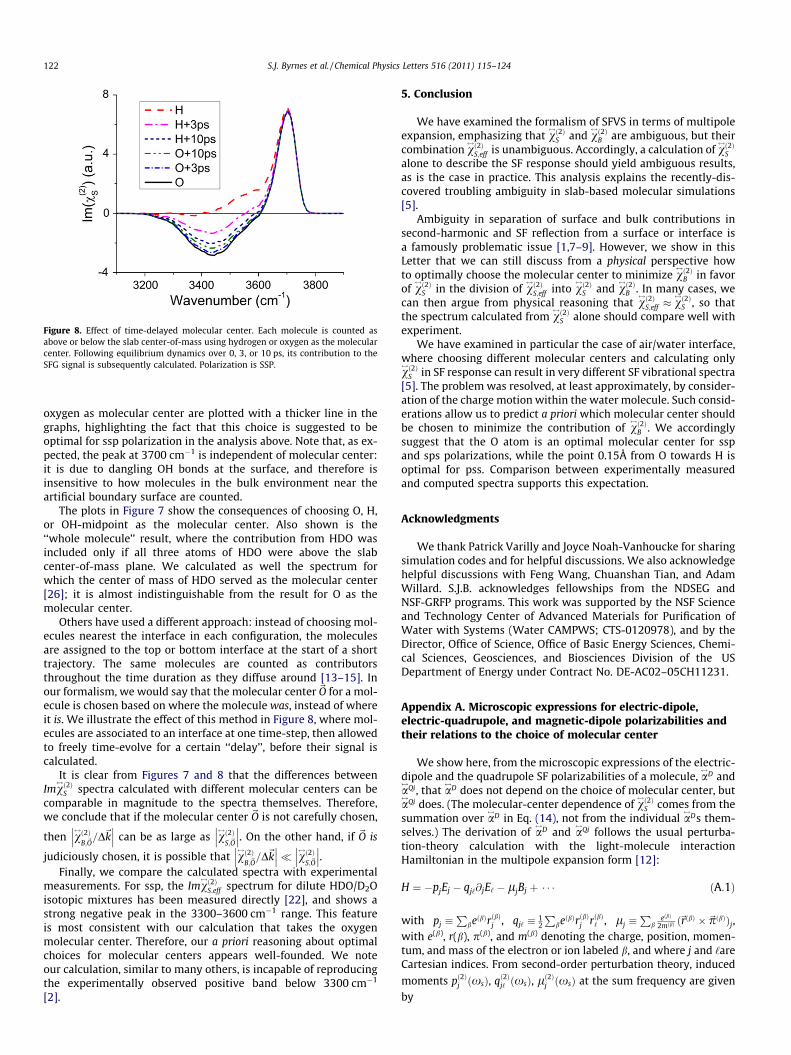
oxygen as molecular center are plotted with a thicker line in thegraphs, highlighting the fact that this choice is suggested to beoptimal for ssp polarization in the analysis above. Note that, as ex-pected, the peak at 3700 cm&1 is independent of molecular center:it is due to dangling OH bonds at the surface, and therefore isinsensitive to how molecules in the bulk environment near theartificial boundary surface are counted.
The plots in Figure 7 show the consequences of choosing O, H,or OH-midpoint as the molecular center. Also shown is the‘‘whole molecule’’ result, where the contribution from HDO wasincluded only if all three atoms of HDO were above the slabcenter-of-mass plane. We calculated as well the spectrum forwhich the center of mass of HDO served as the molecular center[26]; it is almost indistinguishable from the result for O as themolecular center.
Others have used a different approach: instead of choosing mol-ecules nearest the interface in each configuration, the moleculesare assigned to the top or bottom interface at the start of a shorttrajectory. The same molecules are counted as contributorsthroughout the time duration as they diffuse around [13–15]. Inour formalism, we would say that the molecular center ~O for a mol-ecule is chosen based on where the molecule was, instead of whereit is. We illustrate the effect of this method in Figure 8, where mol-ecules are associated to an interface at one time-step, then allowedto freely time-evolve for a certain ‘‘delay’’, before their signal iscalculated.
It is clear from Figures 7 and 8 that the differences betweenImv
$!2"S spectra calculated with different molecular centers can be
comparable in magnitude to the spectra themselves. Therefore,we conclude that if the molecular center ~O is not carefully chosen,
then v$!2"B;~O
=D~k!!!
!!! can be as large as v$!2"
S;~O
!!!!!!. On the other hand, if ~O is
judiciously chosen, it is possible that v$!2"B;~O
=D~k!!!
!!! * v$!2"S;~O
!!!!!!.
Finally, we compare the calculated spectra with experimentalmeasurements. For ssp, the Imv
$!2"S;eff spectrum for dilute HDO/D2O
isotopic mixtures has been measured directly [22], and shows astrong negative peak in the 3300–3600 cm&1 range. This featureis most consistent with our calculation that takes the oxygenmolecular center. Therefore, our a priori reasoning about optimalchoices for molecular centers appears well-founded. We noteour calculation, similar to many others, is incapable of reproducingthe experimentally observed positive band below 3300 cm&1
[2].
5. Conclusion
We have examined the formalism of SFVS in terms of multipoleexpansion, emphasizing that v
$!2"S and v
$!2"B are ambiguous, but their
combination v$!2"S;eff is unambiguous. Accordingly, a calculation of v
$!2"S
alone to describe the SF response should yield ambiguous results,as is the case in practice. This analysis explains the recently-dis-covered troubling ambiguity in slab-based molecular simulations[5].
Ambiguity in separation of surface and bulk contributions insecond-harmonic and SF reflection from a surface or interface isa famously problematic issue [1,7–9]. However, we show in thisLetter that we can still discuss from a physical perspective howto optimally choose the molecular center to minimize v
$!2"B in favor
of v$!2"S in the division of v
$!2"S;eff into v
$!2"S and v
$!2"B . In many cases, we
can then argue from physical reasoning that v$!2"S;eff - v
$!2"S , so that
the spectrum calculated from v$!2"
S alone should compare well withexperiment.
We have examined in particular the case of air/water interface,where choosing different molecular centers and calculating onlyv$!2"S in SF response can result in very different SF vibrational spectra
[5]. The problem was resolved, at least approximately, by consider-ation of the charge motion within the water molecule. Such consid-erations allow us to predict a priori which molecular center shouldbe chosen to minimize the contribution of v
$!2"B . We accordingly
suggest that the O atom is an optimal molecular center for sspand sps polarizations, while the point 0.15Å from O towards H isoptimal for pss. Comparison between experimentally measuredand computed spectra supports this expectation.
Acknowledgments
We thank Patrick Varilly and Joyce Noah-Vanhoucke for sharingsimulation codes and for helpful discussions. We also acknowledgehelpful discussions with Feng Wang, Chuanshan Tian, and AdamWillard. S.J.B. acknowledges fellowships from the NDSEG andNSF-GRFP programs. This work was supported by the NSF Scienceand Technology Center of Advanced Materials for Purification ofWater with Systems (Water CAMPWS; CTS-0120978), and by theDirector, Office of Science, Office of Basic Energy Sciences, Chemi-cal Sciences, Geosciences, and Biosciences Division of the USDepartment of Energy under Contract No. DE-AC02–05CH11231.
Appendix A. Microscopic expressions for electric-dipole,electric-quadrupole, and magnetic-dipole polarizabilities andtheir relations to the choice of molecular center
We show here, from the microscopic expressions of the electric-dipole and the quadrupole SF polarizabilities of a molecule, a
$D anda$Qi, that a
$D does not depend on the choice of molecular center, buta$Qi does. (The molecular-center dependence of v
$!2"S comes from the
summation over a$D in Eq. (14), not from the individual a
$Ds them-selves.) The derivation of a
$D and a$Qi follows the usual perturba-
tion-theory calculation with the light-molecule interactionHamiltonian in the multipole expansion form [12]:
H ' &pjEj & qj‘@ jE‘ & ljBj % # # # !A:1"
with pj $P
be!b"r!b"j , qj‘ $ 1
2
Pbe
!b"r!b"j r!b"‘ , lj $P
be!b"2m!b" !~r!b" )~p!b""j,
with e(b), r(b), p(b), and m(b) denoting the charge, position, momen-tum, and mass of the electron or ion labeled b, and where j and ‘areCartesian indices. From second-order perturbation theory, inducedmoments p!2"
j !xs", q!2"j‘ !xs", l!2"
j !xs" at the sum frequency are givenby
Figure 8. Effect of time-delayed molecular center. Each molecule is counted asabove or below the slab center-of-mass using hydrogen or oxygen as the molecularcenter. Following equilibrium dynamics over 0, 3, or 10 ps, its contribution to theSFG signal is subsequently calculated. Polarization is SSP.
122 S.J. Byrnes et al. / Chemical Physics Letters 516 (2011) 115–124

p!2"j !xs" ' aD
j‘nE1‘E2n % aQ1;EQj‘nt !@tE1‘"E2n % aQ2;EQ
j‘nt E1‘!@tE2n"
% aQ1;MDj‘n B1‘E2n % aQ2;MD
j‘n E1‘B2n
q!2"jt xs! " ' aQs;EQ
j‘nt E1‘E2n
l!2"j xs! " ' aQs;MD
j‘n E1‘E2n !A:2"
with
aDj‘n ' F!pj;p‘;pn"
aQs;EQj‘nt ' F!qjt; p‘;pn"
aQs;MDj‘n ' F!lj;p‘;pn"
aQ1;EQj‘nt ' F!pj; q‘t; pn" !A:3"
aQ1;MDj‘n ' F!pj;l‘;pn"
aQ2;EQj‘nt ' F!pj;p‘; qnt"
aQ2;MDj‘n ' F!pj;p‘;ln"
F!A;B;C" ' &1!h2
X
g;s;t
q!0"gg
!A"gs!B"st!C"tg!x2 &xtg % iCtg"!xs &xsg % iCsg"
% # # #& '
!A:4"
where ‘‘# # #’’ denotes five other similar terms,xsg and Csg are the fre-quency and damping constant of the resonance between states sand g, and A,B, and C represent operators involved in the light-mat-ter interaction (A.1) at frequencies xs,x1, andx2, respectively. (Theconclusions would not be altered if theC’s were replaced with moresophisticated treatments of dephasing.) The effective induced elec-tric-dipole moment at xs can be written as:
p!2"eff ;j!xs ' x1 %x2;~kSF"
' p!2"j & !i~kSF # q
$!2""j &cxSF
!~kSF ) ~l!2""j
' aDj‘n % i aQ1
j‘ntk1t % aQ2j‘ntk2t & aQs
j‘ntkSF;t" #" #
E1‘E2n !A:5"
in which a$Qi includes both electric-quadrupole and magnetic-dipole
parts:
aQsj‘nt ' aQs;EQ
j‘nt % ixSF
aQs;MDb‘n ebtj
aQ1j‘nt ' aQ1;EQ
j‘nt & ix1aQ1;MDjbn ebt‘
aQ2j‘nt ' aQ2;EQ
j‘nt & ix2aQ2;MDj‘b ebtn
!A:6"
where ej‘n is the Levi–Civita tensor. We now show that a$D does not
depend on the choice of molecular center, while a$Qi does. In
calculating the matrix element +f !~r",ab of an operator f !~r" represent-ing a property of the molecule, we implicitly have ~r referred to amolecular center. If the molecular center is at ~O away from theorigin of the coordinate system used in the calculation, then wemust replace f !~r" by f !~r &~O". Thus, the operators pj $
Pbe
!b"r!b"j ,qj‘ $ 1
2
Pbe
!b"r!b"j r!b"‘ , and lj $P
be!b"2m!b" !~r!b" )~p!b""j in the above
equations should be replaced by p!~O"j $
Pbe
!b"!r!b"j &~O", q!~O"j‘ $
12
Pbe
!b"!r!b"j &~O"!r!b"‘ &~O", and l!~O"j $
Pb
e!b"2m!b" +!~r!b" &~O" )~p!b",j,
respectively.Since ~O is a constant vector, we have F(Oj,X,Y) = F(X,Oj,Y) =
F(X,Y,Oj) = 0 for any operators X,Y. It is then readily seen that a$D
is independent of ~O, i.e., independent of the choice of molecularcenter.
However, a$Qi does have molecular-center dependence. If we
change the molecular center from ~O to ~O0, the electric quadrupoleand magnetic dipole operators change from q!~O"
‘t and ~l!~O"j to
q!~O0"‘t ' 1
2
X
b
e!b"!r!b"‘ & O0‘"!r
!b"t & O0
t"
' q!~O"‘t % 1
2!~O&~O0"tp
!~O"‘ % 1
2!~O&~O0"‘p
!~O0"t
~l!~O0" 'X
b
e!b"
2m!b" !~r!b" &~O0" )~p!b"
' ~l!~O0" % !~O&~O0" )X
b
e!b"
2m!b"~p!b": !A:7"
We apply these with the help of the identityP
be!b"m!b" F!pj;
p!b"‘ ;pn" ' ix1F!pj;p‘;pn", which follows from ~p!b" ' mi
!h +H0;~r!b",,where H0 is the unperturbed Hamiltonian, H0 $ V!r!1"; r!2"; . . ."%P
bj~p!b" j2
2m!b" . The result is
aQ1;EQ ;!~O0"j‘nt ' aQ1;EQ ;!~O"
j‘nt % 12aDj‘n!~O&~O0"t %
12aDjtn!~O&~O0"‘
aQ1;MD;!~O0"j‘n ' aQ1;MD;!~O"
j‘n % ix1
2e‘tbaD
jbn!~O&~O0"t
and therefore
aQ1;!~O0"j‘nt & aQ1;!~O"
j‘nt ' 12aDjtn!~O&~O0"‘ %
12aDj‘n!~O&~O0"t
% 12ebt‘ebdfaD
jfn!~O&~O0"t ' aDj‘n!~O&~O0"t :
Likewise for all a$Qi,
aQi;!~O0"j‘nt & aQi;!~O"
j‘nt ' aDj‘n!~O&~O0"t: !A:8"
Thus, a$Qi depends on the choice of molecular center in a way that
incorporates some part of the dipole susceptibility.
Appendix B. Effective surface susceptibility
The SF signals in transmission and reflection from a semi-infi-nite medium are proportional to jv
$!2"S;eff j
2, where the effective sur-
face nonlinear susceptibility v$!2"S;eff is related to the surface and
bulk nonlinear susceptibilities v$!2"
S;~Oand v
$!2"B;~O
by
v$!2"S;eff $ v
$!2"S;~O
%v$!2"B;~O
&iDk!B:1"
Macroscopically, this relation is usually derived from a three-layermodel [27]. Here, we give a microscopic derivation valid for thesimple case we have been considering, where the index of refractionis constant everywhere. The purpose of this is to show explicitlythat (B.1) is consistent with Eqs. (10) and (14), the molecularexpressions for v
$!2"B;~O
and v$!2"
S;~O, respectively.
The SF output field from the surface and the bulk of the mediumcan be considered as generated by an equivalent surface sheet ofnonlinear polarization, ~P!2"
S;eff , which contains contributions fromall molecules:
~P!2"eff ' 1
A
X
t
~p!2"eff ;j;~Oj
e&i~kSF #~Oj
' 1A
Z 0%
zB
X
!~Oj"z'z
~p!2"eff ;j;~Oj
e&i~kSF #~Oj
0
@
1
Adz
% 1A
Z zB
&1
X
!~Oj"z'z
~p!2"eff ;j;~Ot
e&i~kSF #~Oj
0
@
1
Adz !B:2"
The second line of the above equation has the surface and bulk partsseparately presented, each of which depends in general on how themolecular center ~O is chosen within the molecule. Here, the coordi-nate system is defined so that the medium spans z < 0, and jzBj is the
S.J. Byrnes et al. / Chemical Physics Letters 516 (2011) 115–124 123

thickness of the surface layer (Figure 6a), assumed to be much lessthan a wavelength. With the incoming fields given by~E1!~r" '~E0
1ei~k1 #~r
and ~E2!~r" '~E02e
i~k2 #~r , we can write, for the bulk part,
1A
Z zB
&1
X
!~Oj"z'z
~p!2"eff ;j;~Ot
e&i~kSF #~Oj
0
@
1
Adz
' 1A
Z zB
&1
X
!~Oj"z'z
e&iD~k#~Oj i+a$Q1
j;~Oj#~k1 % a
$Q2j;~Oj
#~k2 & a$Qsj;~Oj
#~kSF ,
0
@
1
Adz
0
@
1
A :~E01~E02
' v$!2"
B;~O:~E0
1~E02
" #Z zB
&1e&i Dkj jzdz / v
$!2"B;~O
:~E01~E02
" #Z 0
&1e&i Dkj jzdz
'v$!2"
B;~O
&i Dkj j:~E0
1~E02
0
@
1
A !B:3"
(using Eq. (10)), and for the surface part,
1A
Z 0%
zB
X
!~Oj"z'z
~p!2"eff ;j;~Oj
e&i~kSF #~Oj
0
@
1
Adz
' 1A
Z 0%
zB
X
!~Oj"z'zB
a$Dj e
&iD~k#~Oj
0
@
1
Adz :~E01~E02
/1A
X
!~Oj"z>zB
a$D
j
0
@
1
A :~E01~E02
' v$!2"
S;~O:~E0
1~E02 !B:4"
(using Eq. (14)). With ~P!2"S;eff $ v
$!2"S;eff :
~E01~E02, we obtain Eq. (1).
Appendix C. Dependence of electric-dipole-order susceptibilityon the choice of molecular center
To calculate the dependence of v$!2"
S on ~O, we start with Eq. (14),v$!2"
S;~O' 1
A
P!~Oj"z>zB
a$Dt . We assume that the molecular center~O for each
molecule is fixed in molecular coordinates. Changing the molecularcenter from ~O to ~O0 ' ~O& D~O, but keeping zB unchanged, will re-move from the sum those molecules with Oz > zB but O0
z < zB, i.e.,molecules originally in the region zB < Oz < zB + DOz. Likewise, itwill add to the sum molecules with Oz < zB but O0
z > zB, i.e., mole-cules originally in the region zB > Oz > zB +DOz.
Consider an ensemble of molecules with a given orientation X$.
For this ensemble, DOz has a constant value DO!X$"
z . If DO!X$"
z > 0,some molecules from this ensemble will be eliminated from thesum, namely those in the region zB < Oz < zB % DO!X
$"
z . The volumeof this region is ADO!X
$"
z , so the change in v$!2"
S due to these elimi-
nated molecules is &n!X$" a
$DD E
X$DO!X
$"
z dX$, where n!X
$"dX
$is the differ-
ential number density of molecules with the given orientation X$.
Likewise, if DO!X$"
z < 0, some molecules from this ensemble will beadded into the sum (14); namely those in the region zB > Oz >
zB % DO!X$"
z . The volume of this region is ADO!X$"
z
!!!!
!!!! ' &ADO!X$"
z , so the
change in v$!2"
S due to these molecules is &n!X$" a
$DD E
X$DO!X
$"
z dX$, the
same expression as before.Altogether,
v$!2"S;~O0 & v
$!2"S;~O
'Z
dX$
&n!X$" a
$DD E
X$DO!X
$"
z
$ %' &n a
$DDOz
D E:
where the angle-brackets are a statistical average over molecules’orientations and local environments. This formula is consistent thatderived in Noah-Vanhoucke et al. [5].
References
[1] Y.R. Shen, Appl. Phys. B 68 (1999) 295.[2] C.S. Tian, Y.R. Shen, Chem. Phys. Lett. 470 (2009) 1.[3] T. Ishiyama, A. Morita, J. Chem. Phys. 131 (2009) 244714.[4] P.A. Pieniazek, C.J. Tainter, J.L. Skinner, J. Chem. Phys. 135 (2011) 044701.[5] J. Noah-Vanhoucke, J.D. Smith, P.L. Geissler, J. Phys. Chem. B 113 (2009) 4065.[6] Y.R. Shen, The Principles of Nonlinear Optics, John Wiley & Sons, Inc, Hoboken,
1984.[7] H. Held, A.I. Lvovsky, X. Wei, Y.R. Shen, Phys. Rev. B 66 (2002) 205110.[8] P. Guyot-Sionnest, Y.R. Shen, Phys. Rev. B 38 (1988) 7985.[9] J.E. Sipe, V. Mizrahi, G.I. Stegeman, Phys. Rev. B 35 (1987) 9091.[10] L.D. Landau, E.M. Lifshitz, Electrodynamics of Continuous Media, second edn.,
Butterworth-Heinemann, Oxford, 1960.[11] P.S. Pershan, Phys. Rev. 130 (1963) 919.[12] R.E. Raab, O.L. De Lange, Multipole Theory in Electromagnetism, Clarendon
Press, Oxford, 2005.[13] B.M. Auer, J.L. Skinner, J. Chem. Phys. 129 (2008) 214705.[14] E.C. Brown, M. Mucha, P. Jungwirth, D.J. Tobias, J. Phys. Chem. B 109 (2005)
7934.[15] A. Perry, H. Ahlborn, B. Space, P.B. Moore, J. Chem. Phys. 118 (2003) 8411.[16] L.R. Pratt, J. Phys. Chem. 96 (1992) 25.[17] M.A. Kastenholz, P.H. Huünenberger, J. Chem. Phys. 124 (2006) 124106.[18] A. Morita, Chem. Phys. Lett. 398 (2004) 361.[19] C. Neipert, B. Space, A.B. Roney, J. Phys. Chem. C 111 (2007) 8749.[20] R.W. Munn, Mol. Phys. 89 (1996) 555.[21] X.D. Zhu, A. Wong, Phys. Rev. B 46 (1992) 2540.[22] C.S. Tian, Y.R. Shen, J. Am. Chem. Soc. 131 (2009) 2790.[23] S. Plimpton, J. Comput. Phys. 117 (1995) 1.[24] I.C. Yeh, M.L. Berkowitz, J. Chem. Phys. 111 (1999) 3155.[25] X. Wei, Y.R. Shen, Phys. Rev. Lett. 86 (2001) 4799.[26] A. Morita, J. Phys. Chem. B 110 (2006) 3158.[27] P. Guyot-Sionnest, W. Chen, Y.R. Shen, Phys. Rev. B 33 (1986) 8254.
Steven J. Byrnes was born in Lexington, Massachusettsin 1984. He received a B.A. summa cum laude fromHarvard University in 2007, majoring in physics andmathematics. He is currently a pursuing a Ph.D. inphysics at the University of California, Berkeley, work-ing with Y.R. Shen and Feng Wang on both theoreticaland experimental studies of nonlinear optical spec-troscopy, ultrafast optics, and nanoscience.
Phillip L. Geisslerwas born in Ithaca, New York, in 1974and raised in Virginia, Phillip Geissler returned to hisnatal roots as an undergraduate at Cornell University.He received his Ph.D. at the University of California,Berkeley, in 2000, working with David Chandler. Gei-ssler pursued postdoctoral research at Harvard withEugene Shakhnovich, and at MIT as a Science Fellow. Hereturned to his western roots in 2003, joining the fac-ulty at UC Berkeley as a professor of chemistry. Geiss-ler’s research group develops and applies theoreticaltools of statistical mechanics to explore emergentmolecular behavior in complex chemical and biophysi-
cal systems.
Y.R. Shen is a Professor of Physics at the University ofCalifornia at Berkeley. He received his Ph.D. in solid-state physics from Harvard University. He is the authorof the well-received book The Principles of NonlinearOptics and more than 500 publications in fields rangingfrom nonlinear optics to laser spectroscopy, surfacescience, chemical physics, and condensed matter phys-ics. He and his group have developed optical second-harmonic and sum-frequency generation as surface-specific probes for studies of surfaces and interfaces.
124 S.J. Byrnes et al. / Chemical Physics Letters 516 (2011) 115–124













![[C. J. LeVine] Mechanics Solutions Manual(Berkeley Physics Course,(BookFi.org)](https://img.pdfslide.us/doc/110x75/55cf8eb1550346703b949e6d/c-j-levine-mechanics-solutions-manualberkeley-physics-coursebookfiorg.jpg)















