Embed Size (px)
Citation preview

This journal is c the Owner Societies 2011 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 1925
Cite this: Phys. Chem. Chem. Phys., 2011, 13, 1925–1938
Characterization and reactivity of oxygen-centred radicals over transition
metal oxide clusters
Yan-Xia Zhao,ab
Xiao-Nan Wu,ab
Jia-Bi Ma,ab
Sheng-Gui He*aand
Xun-Lei Ding*a
Received 14th July 2010, Accepted 24th November 2010
DOI: 10.1039/c0cp01171a
We introduce chemical structures and reactivity of oxygen-centred radicals (O��) over transition
metal oxide (TMO) clusters based on mass spectrometric and density functional theory studies.
Two main issues will be discussed: (1) the compositions of TMO clusters that have the bonding
characteristics of (or contain) the O�� radicals; and (2) the dependences (cluster structures, sizes,
charge states, metal types, etc.) of the chemical reactivity and selectivity for the O�� radicals over
TMO clusters. One of the goals of cluster chemistry is to understand the elementary reactions
involved with complex heterogeneous catalysis. The study of the O�� containing TMO clusters
permits rather detailed descriptions for how mono-nuclear oxygen-centred radicals may exist and
react with small molecules over TMO based catalysts.
1. Introduction
Transition metal oxides (TMOs) are widely used as both
catalysts and catalytic support materials.1–7 The catalytic role
of the oxide surface is in terms of forming or providing oxygen
in an activated state.8 In a general scheme of O2 dissociation: O2
(molecular oxygen)-O2�� (superoxide)-O2
2� (peroxide)-
2O�� (mono-nuclear oxygen-centred radical) - 2O2� (lattice
oxygen), the O2��, O2
2�, and O�� are considered as reactive
oxygen species (ROS).9–11 Note that O2�� is usually denoted as
O2� while O�� is often simplified as O� or O� in the literature.
In condensed-phase studies, spectroscopy-based methods
including infrared, Raman, electron spin resonance, etc. have
been widely used to characterize the role of the ROS in surface
reactions and the achievements have been frequently
reviewed.8,9,11–14 It has been generally considered that
oxidations or oxidative transformations of very stable
molecules (such as CO12,15–17 and CH4)18–23 over TMO
surfaces at low temperature often must involve ROS.24–26
However, in some cases, ROS may not be generated with
sufficient concentrations or their lifetimes may be too short9,27
for condensed phase studies. As a result, it is very useful to
adopt alternative ways to investigate the chemistry of the ROS
involved with the condensed-phase systems.
One such alternative way is to study the TMO clusters, in
order to understand the elementary steps involved in the ROS
a Beijing National Laboratory for Molecular Science (BNLMS), StateKey Laboratory for Structural Chemistry of Unstable and StableSpecies, Institute of Chemistry, Chinese Academy of Sciences, Beijing100190, People’s Republic of China. E-mail: [email protected],[email protected]; Fax: +86-10-62559373; Tel: +86-10-62536990
bGraduate School of Chinese Academy of Sciences, Beijing 100039,People’s Republic of China
Yan-Xia Zhao
Yan-Xia Zhao received her BSdegree in chemistry fromShanxi Normal University in2005 and her MS degreein chemistry from BeijingNormal University in 2008.She is now a PhD candidateat the Institute of Chemistry,Chinese Academy of Science.Her research interests mainlyfocus on the chemistry oftransition metal oxide speciesin the gas phase.
Xiao-Nan Wu
Xiao-Nan Wu received aBS degree in chemistryfrom Beijing University ofChemical Technology in 2007.He is now a PhD student at theInstitute of Chemistry, ChineseAcademy of Sciences. Hisresearch activities are on thestudy of chemical reactions oftransition metal oxide clusters,especially lanthanide metaloxide clusters by using time offlight mass spectrometry.
PCCP Dynamic Article Links
www.rsc.org/pccp PERSPECTIVE
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online / Journal Homepage / Table of Contents for this issue

1926 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 This journal is c the Owner Societies 2011
over the TMO catalysts. The TMO materials, especially those
with metals being in the highest oxidation states (such as TiO2
and V2O5) are usually insulators or semiconductors in which
the motion of the majority of the electrons is localized in
nature. As a result, small TMO clusters may well be effective
models of real surface species or active sites over the bulk TMO
materials.28–30 This article is to review the research progress
from the cluster chemistry to understand one of the ROS, the
mono-nuclear oxygen-centred radical species O��, over the
TMO surfaces. It is noticeable that in addition to the O2
(or other molecules such as N2O)9,12 dissociation, the O��
species may also be generated through photon irradiation in
photocatalytic processes because the lattice oxygen O2� may
be converted to O�� directly (O2� + hn - O�� + e�)
or through recombination with pre-generated holes (O2� +
h+ - O��).9,14,31,32 Meanwhile, the dehydroxylation of the
surface hydroxyl groups by evacuation as a pretreatment of
materials at high temperature may also generate O��
centres.33–35
Fig. 1a shows the electron configuration of the simplest O��
species, the free O� anion system, of which the unpaired
electron or the spin densities (SDs) are located in one of the
O 2p orbitals. Density functional theory (DFT) calculations36
predicted (Fig. 1b–d) that the oxide clusters of metals, such as
the first d-block transition metal Sc, can have essentially the
same characteristics for the SD distributions as the free
O� does: the unpaired electron or SD values of about 1 mB in
each of Sc2O4�, Sc3O5, and Sc4O6
+ clusters are also
distributed in the 2p orbital of one of the oxygen atoms. As
a result, one can consider that each of these clusters contains
one unit of O��.
The O�� radicals can be terminally bonded (denoted as Ot��,
Fig. 1b and c) or bridgingly bonded (denoted as Ob��, Fig. 1d)
over the TMO clusters. The O�� containing clusters can be
positively (Fig. 1d) or negatively (Fig. 1b) charged, or overall
neutral (Fig. 1c), which may correspond to the surface O��
species in different local charge environments.37–39 One can
also imagine that the sizes of the O�� containing clusters can
vary from several to dozens of atoms. The strength of the O��
binding onto the clusters may depend on cluster sizes,
structures, charge states, and the types of the transition
metals. All of these issues can ultimately influence the
chemical reactivity and selectivity of the O�� radicals over
the TMO clusters. The O�� containing TMO clusters are thus
ideal models to investigate a detailed and possibly rich
chemistry of the surface O�� species, under isolated,
controlled, and reproducible conditions.
Combined experimental (mostly mass spectrometry based)
and theoretical (such as DFT based) methods can be used to
study the bonding and reactivity of the TMO clusters in
different charge states. The mass spectrometric study of
neutral TMO cluster reactivity is difficult and progress has
been made recently by Prof. Elliot R. Bernstein’s group40–46 by
using single-photon (soft) ionisation which prevents severe
cluster fragmentation that would occur in the process of
electron-impact or multi-photon (hard) ionisation.47 It
should be noted that spectroscopic method is often very
useful for studying the structures and reactivity of neutral
TMO clusters.48 For example, important mechanistic details
involved in MO + CH4 2 M + CH3OH reactions can been
obtained with matrix isolation infrared absorption
spectroscopy.49 The transition metal species are challengingJia-Bi Ma
Jia-Bi Ma received her BSdegree in chemistry from JilinUniversity in 2008. She is nowa PhD student at the Instituteof Chemistry, Chinese Academyof Sciences. Her researchactivities focus on experimentaland theoretical investigationsof the reactions of thetransition metal oxide clusterswith small molecules.
Sheng-Gui He
Sheng-Gui He received his BSdegree in physics and PhDdegree in chemistry from theUniversity of Science andTechnology of China in 1997and 2002, respectively. Afterpostdoctoral stays at theUniversity of Kentucky(September 2002–February2005) with Prof. Dennis J.Clouthier and at ColoradoState University (February2005–January 2007) withProf. Elliot R. Bernstein,he joined the Institute ofChemistry, Chinese Academy
of Science in January 2007. His research interests are onexperimental and theoretical studies of reactive intermediatesincluding free radicals and atomic clusters.
Xun-Lei Ding
Xun-Lei Ding received his BSdegree in physics in 1999 fromthe University of Science andTechnology of China (USTC)and PhD degree in chemistryin 2004 under the supervisionof Professor Jin-Long Yang.After a one-year postdoctoralstay at USTC with ProfessorJian-Guo Hou and a two-yearpostdoctoral stay at theTheory@Elettra group ofCNR-INFM DEMOCRITOS(Italy) with Professor MariaPeressi, he joined the Instituteof Chemistry, Chinese Academy
of Sciences in 2007. His research interests include first principlestudies on structural and reactivity properties of clusters andsurfaces.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online

This journal is c the Owner Societies 2011 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 1927
systems for state-of-the-art quantum chemistry calculations.50
The experimental data, such as bond and ionisation energies,
electron affinities, vibrational spectra, and reaction channels
and rate constants may be used to gauge the validity of chosen
theoretical methods (functionals of DFT, basis sets, etc.).51–54
We do not attempt to go into further details of the
experimental and theoretical methods for studying the TMO
clusters and readers may refer to the original references. The
study of bonding and reactivity of atomic clusters including
many of those without oxygen-centred radicals has broad
scientific significance55,56 and several aspects of recent
achievements have been reviewed29,30,48,49,57–68 and selected
individual contributions may be found in refs. 69–85. This
review article focuses on the research progress in bonding and
reactivity of the O�� radicals over TMO clusters based on our
own works and other closely related investigations,
particularly those of the groups headed by Prof. Helmut
Schwarz, Prof. A. Welford Castleman, Jr., and their
collaborators.
2. homonuclear transition metal oxide clusters
2.1 d-block transition metal oxide clusters
2.1.1 Compositions of O�� containing TMO clusters.
To consider possible compositions of the TMO clusters
(MxOyq, M is a metal atom and q is the charge number,
q = 0, �1) with the oxygen-centred radical O��, one may
quickly find the stoichiometric metal oxide cluster cations such
as Sc2kO3k+, TikO2k
+, V2kO5k+, CrkO3k
+, and Mn2kO7k+
because these species can be generated by exciting one electron
from each of the stoichiometric neutral clusters into vacuum.
The unpaired electron left in each of the cations can be
localized on oxygen atom(s) to form an oxygen-centred
radical under the conditions that all of the metal atoms are
in the maximum oxidation state and electron excitation does
not result in significant structure relaxation. One may also
think that the above predicted O�� containing clusters are
oxygen-rich by (averagely) a half oxygen atom if one counts
the total valence numbers of metal and oxygen atoms and takes
into account the net charge. To follow a rule that O��
containing clusters are oxygen-rich by a half oxygen atom,
one finds thatMxOyq clusters with O�� may satisfy the general
equation:36
D R 2y � nx + q = 1, (1)
in which n is the number of valence electrons of elementM that
can be oxidized by oxygen to the +n oxidation state. The
D=1 anions are Sc2kO3k+1�, TikO2k+1
�, V2kO5k+1�, etc. and
the D = 1 neutrals are Sc2kO3kScO2, V2kO5kVO3, etc. (note
that there is no D = 1 neutral oxide cluster for groups 4 and 6
metals). The D value for a general MxOyq cluster can be
used to judge whether the cluster is oxygen-rich (D > 0) or
oxygen-poor (D o 0) and the extent of the richness and
Fig. 1 DFT calculated profiles of spin density distributions for free
O� anion (a), and Sc2O4� (b), Sc3O5 (c), and Sc4O6
+ (d) clusters. The
electron configuration of O� is also shown. The structures of the
clusters are adapted from ref. 36 and some of the Sc–O bond lengths
are given in pm.
Fig. 2 DFT calculated profiles of SOMOs forM3Oyq (D=1) clusters. TheMulliken spin density values greater than 0.5 mB over oxygen atoms are
given in parentheses. Adapted from ref. 36.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online

1928 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 This journal is c the Owner Societies 2011
poorness. The D value may be called extent of cluster over-
oxidation.
A DFT study with a hybrid functional B3LYP86,87
predicted that for early transition metals (M = groups 3–7
and 3d–5d metals), except for M = Cr and Mn, all of the
investigated oxide clusters MxOyq with D = 1 and x = 1–3
(x = 1–6 for M = Sc) have the character of oxygen-centred
radicals.36 Fig. 2 shows the profiles of singly occupied
molecular orbitals (SOMOs) for the D = 1 clusters with
three metal atoms. Like in the free O� anion, the unpaired
electrons in these clusters are mainly located over the O 2p
orbitals. Note that in the D = 1 oxide clusters (such as V3O8
and W3O9+) of groups 5–7 metals, the unpaired electron can
be distributed over two oxygen atoms in each cluster. This
is reasonable if one considers that adsorption of a free O�
onto a cluster (such as onto V3O7+ to form V3O8) can result
in re-distribution of the SDs. Each of these clusters in Fig. 2
can be considered to contain one (or equivalently one) unit of
O�� radical.
The DFT study suggested that the D = 1 clusters of CrxOyq
[(CrO3)1–3+ and (CrO3)2–3O
�] and MnxOyq (MnO4, Mn2O7
+,
Mn2O8�, and Mn3O11) have unbroken O–O bonds and these
clusters do not have O�� radicals.36 This means that chromium
(Cr) and manganese (Mn) of the 3d metals do not belong to the
type of transition metals of which all of the oxide clusters
MxOyqwith D=1 contain O�� radicals. This is in parallel with
the smallest bond energies of CrO (4.78 eV) and MnO
(3.83 eV)88 among MO (M = groups 3–7 and 3d–5d), as
well as quite large bond energy of O2 (5.12 eV).89
An analysis of the geometrical parameters (such as in
Fig. 1b–c, see details in ref. 36) indicates that the M–Ot�� or
M–Ob�� bond in each cluster is significantly longer than the
normal MQOt double or M–Ob single bond, respectively. For
example, the Sc–Ot�� bond length in Sc2O4
� (Fig. 1b) is
206 pm in contrast to the much shorter length (176 pm) of
the other Sc–O terminal bond in the same cluster. The
elongation of the M–Ot�� bonds in the D = 1 clusters is also
consistent with the result of Wiberg bond order analysis for
selected clusters: the bond order values are 1.14, 1.06, 0.80,
0.75 for Ta–Ot�� in TaO3, Mo–Ot
�� in MoO4�, Sc–Ot
�� in
ScO2, and Zr–Ot�� in ZrO3
�, respectively.36 This suggests that
the Ot�� (as well as Ob
��) atoms with spin density values close
to one unit mB are singly bonded with the transition metal
centres, in agreement with the picture that one of the 2p
orbitals of Ot�� or Ob
�� atom is singly occupied and such
electronic structure (or electron configuration) is the same as
that for the free O� anion (Fig. 1a).
The Ot�� (or Ob
��) atom with elongated M–Ot��
(or M–Ob��) bond and a locally open shell electronic
structure is expected to be the active site of a D = 1 cluster
in the reaction with molecules such as CO, CH4, C2H4, and so
on. Oxygen atom transfer is an important type of chemical
reaction, so it is useful for predicting the M–O bond
dissociation energies [energy cost for O atom loss: MxOyq
(D = 1) - MxOy–1q (D = �1)+O, M = groups 3–7 metals
and xr 3] in order to study the reactivity of the D=1 clusters
with O�� centres. The computedM–O dissociation energies by
B3LYP cover a broad energy range from 1.90 eV (Tc3O10) to
5.06 eV (Ta2O6�, see ref. 36 for details), suggesting a possibly
rich chemistry for the early transition metal oxide clusters with
oxygen-centred radicals.
2.1.2 Reactivity of O�� radicals over cationic TMO
clusters. A systematic verification of the theoretical results36
came from an experimental study of the reactions of early
transition metal oxide cluster cations (MxOy+,M= Sc, Y, La;
Ti, Zr, Hf; V, Nb, Ta; Cr, Mo, W; Mn, Re) with methane
(CH4) under near room-temperature conditions.90 The CH4
molecule is chemically very stable21,65,67,91 and not too many
oxide clusters (see a list in Table 1) are able to activate CH4
at low- or room-temperature.92–105 By using a time of flight
mass spectrometer that is coupled with a laser ablation
cluster source and a fast flow reactor,106 several series of
stoichiometric metal oxide cluster cations (D = 1 clusters)
(TiO2)1–5+, (ZrO2)1–4
+, (HfO2)1–2+, (V2O5)1–5
+,
(Nb2O5)1–3+, (Ta2O5)1–2
+, (MoO3)1–2+, (WO3)1–3
+, and
Re2O7+ have been identified to be able to abstract a
hydrogen atom from CH4 to produce CH3 under near room-
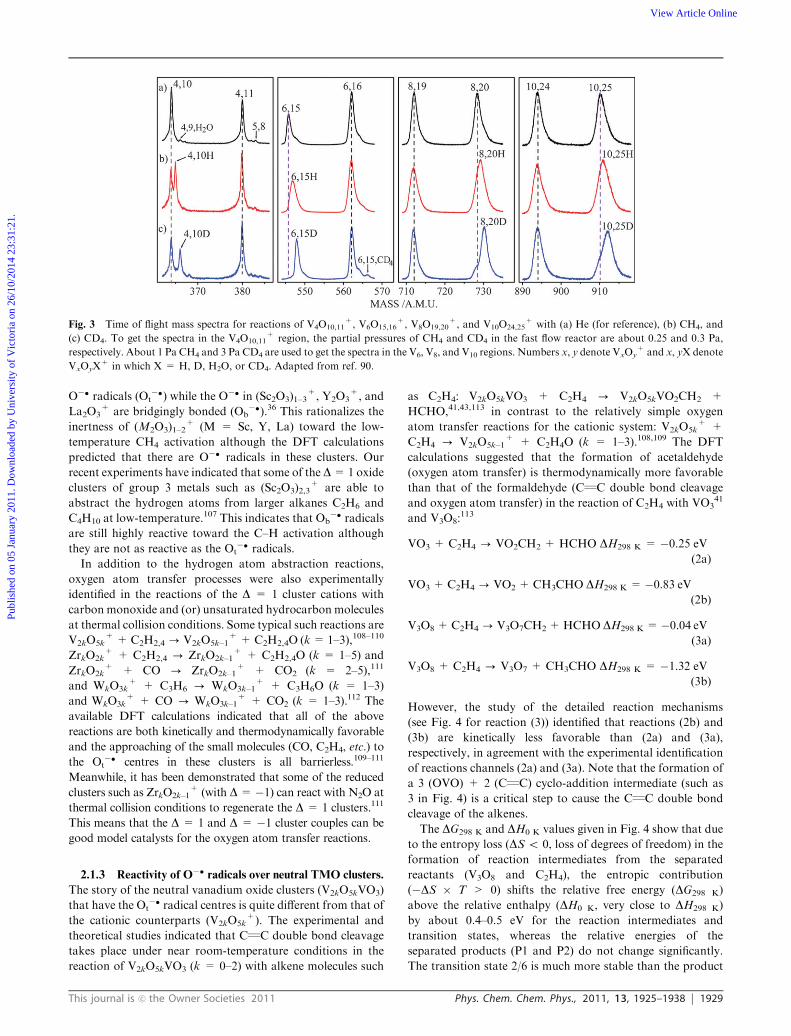
temperature conditions.90 The mass spectra that suggest
reactions of (V2O5)2–5+ + CH4 - (V2O5)2–5H
+ + CH3
are given in Fig. 3. Note that a lot of other cluster cations
with D = �1, 0, 2, etc. were also generated in the clusters
source and reacted/collided with the CH4 in the fast flow
reactor. However, there was no evidence of the CH4
activation by any of these D a 1 clusters.
The experiments also showed that the D = 1 clusters of
(M2O3)1–2+ (M = Sc, Y, La), (CrO3)1–2
+, and Mn2O7+ are
inert or react very slowly with CH4 under near room-
temperature conditions. The inertness of (CrO3)1–2+ and
Mn2O7+ [in contrast to high reactivity of (MoO3)1–2
+,
(WO3)1–3+, and Re2O7
+] toward the CH4 activation is in
agreement with the B3LYP predictions36 that there are no
O�� radicals in these chromium or manganese species. A few
DFT studies indicated that the homolytic C–H bond activation
of CH4 by the O�� radicals over the cluster cations [such as
V4O10+,98 (Al2O3)3–5
+,99 VAlO4+,103 (V2O5)(SiO2)1–4
+,104
and so on] is (overall) barrierless. Except for MnO+, the
available theoretical calculations36,98–105 suggested that all
of the clusters listed in Table 1 contain terminally bonded
Table 1 Oxide cations that are able to activate methane under nearroom-temperature conditions
Year Homonuclear oxide clusters References
1989 OsO4+ 92
1992 FeO+ 931995 MnO+ 941997 MoO3
+ 951999 TiO2
+, ZrO2+ 96
2006 MgO+ 97V4O10
+ 982008 (Al2O3)3–5
+ 992009 SO2
+ 100P4O10
+ 1012010 (TiO2)1–5
+, (V2O5)1–5+, 90
(ZrO2)1–4+, (Nb2O5)1–3
+, (MoO3)1–2+
(HfO2)1–2+, (Ta2O5)1–2
+, (WO3)1–3+, Re2O7
+
(CeO2)2–4+ 102
Heteronuclear oxide clusters2010 VAlO4
+ 103V2O5(SiO2)1–4
+, (V2O5)2SiO2+ 104
V3PO10+ 105
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online
This journal is c the Owner Societies 2011 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 1929
O�� radicals (Ot��) while the O�� in (Sc2O3)1–3
+, Y2O3+, and
La2O3+ are bridgingly bonded (Ob
��).36 This rationalizes the
inertness of (M2O3)1–2+ (M = Sc, Y, La) toward the low-
temperature CH4 activation although the DFT calculations
predicted that there are O�� radicals in these clusters. Our
recent experiments have indicated that some of the D=1 oxide
clusters of group 3 metals such as (Sc2O3)2,3+ are able to
abstract the hydrogen atoms from larger alkanes C2H6 and
C4H10 at low-temperature.107 This indicates that Ob�� radicals
are still highly reactive toward the C–H activation although
they are not as reactive as the Ot�� radicals.
In addition to the hydrogen atom abstraction reactions,
oxygen atom transfer processes were also experimentally
identified in the reactions of the D = 1 cluster cations with
carbon monoxide and (or) unsaturated hydrocarbon molecules
at thermal collision conditions. Some typical such reactions are
V2kO5k++C2H2,4-V2kO5k–1
++C2H2,4O (k=1–3),108–110
ZrkO2k+ + C2H2,4 - ZrkO2k–1
+ + C2H2,4O (k = 1–5) and
ZrkO2k+ + CO - ZrkO2k–1
+ + CO2 (k = 2–5),111
and WkO3k+ + C3H6 - WkO3k–1
+ + C3H6O (k = 1–3)
and WkO3k+ + CO - WkO3k–1
+ + CO2 (k = 1–3).112 The
available DFT calculations indicated that all of the above
reactions are both kinetically and thermodynamically favorable
and the approaching of the small molecules (CO, C2H4, etc.) to
the Ot�� centres in these clusters is all barrierless.109–111
Meanwhile, it has been demonstrated that some of the reduced
clusters such as ZrkO2k–1+ (with D= �1) can react with N2O at
thermal collision conditions to regenerate the D = 1 clusters.111
This means that the D = 1 and D = �1 cluster couples can be
good model catalysts for the oxygen atom transfer reactions.
2.1.3 Reactivity of O�� radicals over neutral TMO clusters.
The story of the neutral vanadium oxide clusters (V2kO5kVO3)
that have the Ot�� radical centres is quite different from that of
the cationic counterparts (V2kO5k+). The experimental and
theoretical studies indicated that CQC double bond cleavage
takes place under near room-temperature conditions in the
reaction of V2kO5kVO3 (k = 0–2) with alkene molecules such
as C2H4: V2kO5kVO3 + C2H4 - V2kO5kVO2CH2 +
HCHO,41,43,113 in contrast to the relatively simple oxygen
atom transfer reactions for the cationic system: V2kO5k+ +
C2H4 - V2kO5k–1+ + C2H4O (k = 1–3).108,109 The DFT
calculations suggested that the formation of acetaldehyde
(oxygen atom transfer) is thermodynamically more favorable
than that of the formaldehyde (CQC double bond cleavage
and oxygen atom transfer) in the reaction of C2H4 with VO341
and V3O8:113
VO3 + C2H4 - VO2CH2 + HCHO DH298 K = �0.25 eV
(2a)
VO3 +C2H4 - VO2 + CH3CHO DH298 K =�0.83 eV(2b)
V3O8+C2H4-V3O7CH2+HCHO DH298 K=�0.04 eV(3a)
V3O8 + C2H4 - V3O7 + CH3CHO DH298 K = �1.32 eV
(3b)
However, the study of the detailed reaction mechanisms
(see Fig. 4 for reaction (3)) identified that reactions (2b) and
(3b) are kinetically less favorable than (2a) and (3a),
respectively, in agreement with the experimental identification
of reactions channels (2a) and (3a). Note that the formation of
a 3 (OVO) + 2 (CQC) cyclo-addition intermediate (such as
3 in Fig. 4) is a critical step to cause the CQC double bond
cleavage of the alkenes.
The DG298 K and DH0 K values given in Fig. 4 show that due
to the entropy loss (DS o 0, loss of degrees of freedom) in the
formation of reaction intermediates from the separated
reactants (V3O8 and C2H4), the entropic contribution
(�DS � T > 0) shifts the relative free energy (DG298 K)
above the relative enthalpy (DH0 K, very close to DH298 K)
by about 0.4–0.5 eV for the reaction intermediates and
transition states, whereas the relative energies of the
separated products (P1 and P2) do not change significantly.
The transition state 2/6 is much more stable than the product
Fig. 3 Time of flight mass spectra for reactions of V4O10,11+, V6O15,16
+, V8O19,20+, and V10O24,25
+ with (a) He (for reference), (b) CH4, and
(c) CD4. To get the spectra in the V4O10,11+ region, the partial pressures of CH4 and CD4 in the fast flow reactor are about 0.25 and 0.3 Pa,
respectively. About 1 Pa CH4 and 3 Pa CD4 are used to get the spectra in the V6, V8, and V10 regions. Numbers x, y denote VxOy+ and x, yX denote
VxOyX+ in which X = H, D, H2O, or CD4. Adapted from ref. 90.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online

1930 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 This journal is c the Owner Societies 2011
P1 (V3O7CH2+HCHO) in terms of DH0 K (�0.43 eV versus
�0.03 eV) while 2/6 is less stable than P1 in terms of DG298 K
(+0.03 eV versus�0.05 eV). The experimental identification of
V3O7CH2 (reaction channel 3a)41 suggests that the entropic
contribution to the free energies possibly plays an important
role. Otherwise, a much faster conversion 2 - 2/6 - 6 - P2
(reaction channel 3b) would make the reaction channel 3a
incompatible and not observable.
Partial oxidation of propylene (C3H6) catalyzed by the
simplest O�� containing vanadium oxide cluster VO3 was
also studied by DFT calculations.114 The overall barrierless
catalytic cycles at room-temperature are summarized in Fig. 5.
The calculations predicted that, in the model catalytic cycles,
the most favorable products are acetaldehyde (CH3CHO) and
CO2, which are also the major products in propylene selective
oxidation over V2O5/SiO2 catalyst.115 The match of the simple
gas phase model catalysis with the complex condensed phase
heterogeneous reactions may not be a coincidence. The match
can be rationalized by similar chemistry of the O�� radicals
over condensed phase system and the gas phase clusters. The
electron paramagnetic resonance (EPR) spectroscopy
characterized that the interaction of N2O with VO2 centres in
zeolite BEA (Si/Al = 30) generates vanadium(V) bound O��
radicals with gx = 2.0202, gy = 2.0173, and gz = 2.0284
(anisotropic g-factors); and |Ax| = 1.65, |Ay| = 1.58, and
|Az| = 1.49 mT (hyperfine structure constants).116 In the case
Fig. 4 DFT calculated potential energy profiles (a) and structures of reaction intermediates and transition states (b) for V3O8 + C2H4. An integer
n is used to denote reaction intermediate and two integer combination n1/n2 is used to denote transition state that connects reaction intermediates n1and n2. The relative Gibbs free energies (DG298 K in eV) and zero-point vibration corrected energies (DH0 K in eV) with respect to V3O8 + C2H4
are given in parentheses as (DG298 K/DH0 K). The bond lengths are given in 0.1 nm. Adapted from ref. 113.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online

This journal is c the Owner Societies 2011 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 1931
of propylene adsorption onto the zeolite with the V–O��
radicals, single oxygen transfer leading to the rupture of the
CQC double bond was observed. The mechanism of the
propylene CQC bond rupture rather than the formation of
C3H6O (acetone, propanal, etc.) by the V–O�� radicals in the
condensed phase system116 can be well interpreted by the gas
phase studies of the reactions of C3H6 with VO3(V2O5)0–2(=VO3, V3O8, and V5O13) clusters that are exactly the
V–O�� radical species.41,43,113
2.1.4 Reactivity of O�� radicals over anionic TMO clusters.
The reactivity of the O�� radicals over negatively charged
TMO clusters is generally much lower than that of the O��
centres over the positively charged clusters in the reaction with
hydrocarbon molecules. The O�� containing anions V2O6�
and V4O11�,108,117,118 and ZrkO2k+1
� (k = 1–4)119 display
association or minor oxygen atom transfer channels with
unsaturated hydrocarbon molecules such as C2H2 and C2H4,
in contrast to the efficient oxygen atom transfer processes in
the reactions of (V2O5)1–3+ as well as (ZrO2)1–5
+ with these
molecules.108–111 In addition, the activation of the most
stable alkane molecule CH4 by (V2O5)1–5+, (Nb2O5)1–3
+,
and (Ta2O5)1–2+ is very fast (the first order rate constants
k1 B 10�10 cm3 molecule�1 s�1)90,98 while it was considered
that the group 5 metal oxide cluster anions are inert toward
saturated hydrocarbon molecules such as C4H10.108,120
The DFT results121–125 in Fig. 6a show that the reactivity
(both thermodynamically and kinetically) of the O��
containing clusters toward CH4 activation depends
significantly on the charge states. The cationic (V2O5+) and
anionic (V2O6�) clusters have the highest and lowest reactivity,
respectively, while the reactivity of the neutral one (V3O8) is in
the middle. The C–H activation barrier (0.26 eV) in V2O6�+
CH4 is still very small as compared with the large barriers
(1.7–3.8 eV)126,127 of CH4 activation by the MQO double
bonds in TMO cluster species V3O6Cl3, Cr3O9, Mo3O9, and
W3O9 without the O�� centres. The barriers for the reactions of
larger alkanes (C2H6, n-C4H10, etc.) with O�� containing
clusters (such as V2O6�, see Fig. 6b) can be small enough to
provide a chance to observe the C–H activation by TMO
anions at low-temperature. Note that the examples for the
activation of alkanes by anions are scarce.68
With the above considerations in mind and by increasing the
pressures of alkanes in the fast flow reactor, we were able to
observe evidences for the C–H activation of C2H6 and n-C4H10
by V2O6� and V4O11
� 118 as well as by Zr2O5� and Zr3O7
� 125
(see Fig. 7 for one example). The estimated rate constants (k1)
for V2O6� + C4H10 and Zr2O5
� + C4H10 are 1 � 10�12 and
2 � 10�11 cm3 molecule�1 s�1, respectively. Since the methane
activation is a holy grail in alkane chemistry, it is interesting to
consider the rate of CH4 activation by these O�� containing
cluster anions. The k1(V2O6�+CH4) value may be scaled from
k1(V2O6�+C4H10) by a factor of exp(�DEa/kBT), in which
DEa is the difference of the C–H activation barrier (0.245 eV,
Fig. 6b), kB is the Boltzmann constant, and T is the
temperature. For T = 298 K, k1(V2O6� + CH4) is estimated
to be 7.2 � 10�17 cm3 molecule�1 s�1. Similarly, with the DFT
calculated DEa = 0.206 eV for the Zr2O5� reaction systems,
the k1(Zr2O5�+CH4) at room temperature can be estimated to
be 6.6 � 10�15 cm3 molecule�1 s�1, which is close to
k1(OH+CH4) = 7.89 � 10�15 cm3 molecule�1 s�1.128,129
This means that some of the O�� containing cluster anions
may be as reactive as the free OH radical toward the C–H
activation. The higher reactivity of Zr2O5� versus V2O6
� in the
C–H s bond activation is in parallel with the localized SDs
(over one O atom) in the former and delocalized (over two
O atoms) SDs in the latter.36 The influence of the SD
localization and delocalization on the cluster reactivity will
be discussed in more detail in the text below. It should be
pointed out that the reactivity of the O�� containing TMO
anions (ZrkO2k+1�, k = 1–4) and cations (ZrkO2k
+, k = 2–5)
toward the CO oxidation can be very similar and the
rate constants were measured to be on the order of
10�12 B 10�13 cm3 molecule�1 s�1 in refs. 111 and 119.
2.2 Cerium oxide cluster cations
Despite the fact that there are comprehensive data available on
the chemistry of d-block transition metal oxide clusters, the
investigations on oxide clusters of f-block metals (lanthanides
and actinides) are very limited.130–137 Cerium [Xe]4f15d16s2 is
the first 4f-element and its oxides (such as ceria, CeO2) are
widely used as catalysts and as promoters of heterogeneous
catalytic reactions,5,138 so the understanding of structure-
reactivity relationship of cerium–oxygen systems is very
important.139,140 Based on the chemistry of d-block TMO
clusters with O�� radicals, one would expect that the
stoichiometric cerium oxide cluster cations CekO2k+ contain
oxygen-centred radicals that are very reactive toward CH4,
C2H4, CO, etc. Meanwhile, the bulk CeO2 and ZrO2 can have
the same cubic fluorite (CaF2) structure141 while zirconium
([Kr]4d25s2) has one more d and one less f electrons than
cerium does. Is the behavior of f electron in cerium very similar
to that of the additional d electron in zirconium in terms of the
oxide cluster reactivity?
The experimental study of the reactions between cerium
oxide cluster cations and a series of small molecules under
near room-temperature conditions indicated that the CekO2k+
cluster can transfer one oxygen atom to C2H4 (see the spectra
Fig. 5 Schematic diagram showing barrierless reaction channels that
form catalytic cycles for propylene (C3H6) partial oxidation over the
VO3 cluster under gas phase, room-temperature conditions. Adapted
from ref. 114.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online

1932 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 This journal is c the Owner Societies 2011
in Fig. 8) or CO for k = 2–6 and abstract one hydrogen atom
from CH4 or C2H6 for k = 2–4 (the mass resolution is limited
for k = 5 and 6 clusters).102 However, CekO2k+ clusters have
high reactivity toward C2H4 and relatively much lower
reactivity toward CO, CH4, and C2H6. This is in sharp
contrast with the fact that ZrkO2k+ is highly reactive toward
all of the small molecules. Table 2 lists the rate constants for
reactions of Ce2O4+ as well as Zr2O4
+ with a few small
molecules in the same fast flow reactor and the uncertainties
of the relative rates are about 30–50%.102
The DFT study indicated that the SDs in CekO2k+ clusters
with k=2–5 (see Fig. 9, the large k=6 cluster was not studied
by the DFT) are mainly distributed over one Ce and two
O atoms, which forms (OCeO)� heteroatom radicals.
The terminal oxygen atom with a fraction of the SDs
(B0.1–0.6 mB) in CekO2k+ still has radical character and may
be denoted as Ot��f. Note that the SDs are primarily localized
over a single O atom (Ot�� radical) in ZrkO2k
+ (k = 2–4).111
The C–H activation of alkanes by a Ot�� or Ot
��f containing
cluster involves interactions of the cluster SOMO with the
C–H s orbital that may be written as hSOMO|H|si, in
which H is the interacting Hamiltonian.102 The SD
(or SOMO) localization (Ot��) leads to larger values
of oSOMO|H|s> than the SD delocalization (Ot��f) does
for a given distance between the C–H bond and the reacting
Ot atom. As a result, Ot�� is more reactive than Ot
��f in terms
of C–H s bond activation which rationalizes the experimental
results of k1(Zr2O4++CH4) c k1(Ce2O4
++CH4) in Table 2
as well as k1(Zr2O5� + C4H10) c k1(V2O6
�+C4H10)
discussed in subsection 2.1.4. The oxidation of CO molecule
by Ot�� or Ot
��f involves participation of carbon (2s) lone pair
orbital which has s character. It is thus expected that the
extent of SD localization also significantly affects/controls the
rate constants for CO oxidation, in agreement with a much
faster reaction of Zr2O4+ + CO - Zr2O3
+ + CO2 than
Ce2O4+ + CO - Ce2O3
+ + CO2 although the latter is more
exothermic than the former.102,111
As to the oxidation of C2H4 with a CQC p bond, the DFT
study indicated that approaching C2H4 with Ot��f in Ce2O4
+
to form a C–O bond is barrierless (Fig. 10) and the subsequent
steps to produce acetaldehyde are driven by the highly
favorable thermodynamics.102 One may conclude that for the
relatively reactive p system (alkenes), the extent of SD
localization or delocalization does not significantly affect the
rates of oxidation by Ot��f or Ot
�� containing clusters
(CekO2k+ and ZrkO2k
+). The SD values over Ot��f
(see Fig. 9) of CekO2k+ increases as the cluster size (k)
increases and the largest SD change occurs when k = 1
(CeO2+, 0.183 mB) goes to k = 2 (Ce2O4
+, 0.333 mB). Suchchange results in significantly enhanced reactivity for the C–H
s bond activation since CH4 and C2H6 activation by Ce2O4+
was observed while CeO2+ was not reactive with both CH4
and C2H6.135 In contrast, the rate constant of CeO2
+ + C2H4 -
CeO++C2H4O ison theorderof 1� 10�10 cm3molecule�1 s�1,135
which is still close to the rate of Ce2O4+ + C2H4 - Ce2O3
+ +
C2H4O.102
The nature of the electronic ground state of the bulk CeO2
has been controversial for a long time.141,142 The first scenario
features a formally fully occupied O 2p band and empty 4f
states (Ce 4f0) in the band gap,143,144 and the second one
involves a mixture of Ce 4f0 and Ce 4f1 O 2p hole
states.145,146 The unique SD distribution over CekO2k+
Fig. 6 DFT calculated energy profiles for hydrogen abstraction
reactions: (a) VxOyq + CH4 - VxOyH
q + CH3 in which
VxOyq = V2O5
+, V3O8, and V2O6�; and (b) V2O6
� + CnH2n+2 -
V2O6H�+ CnH2n+1 in which n = 1, 2, and 4 (n-butane). The relative
energies of the reactants, products, and the transition states for C–H
activation are shown.
Fig. 7 Time of flight mass spectra for reactions of Zr2Oy� with (a)
pure He, (b) 30 Pa C2H6, (c) 70 Pa C2H6, (d) 5 Pa C4H10, and (e) 70 Pa
C4H10 in a fast flow reactor. The relative signal intensities from 271 to
305 amu (for Zr2O6� and Zr2O7
�) are scaled by a factor of 1/4.
Adapted from ref. 125.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online

This journal is c the Owner Societies 2011 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 1933
(Fig. 9) disagrees with the first scenario, i.e. if the bulk CeO2
has the electronic ground state featured with the fully occupied
O 2p valence band (like ZrO2), one would expect that the
SOMOs of CekO2k+ have the character of pure oxygen 2p
orbitals (like ZrkO2k+), which is not true from the cluster
reactivity studies. Note that the electronic structures of a few
cerium containing compounds Ce(C8H8)2,147 Ce(C5H5)3
+,148
and Ce(C14H18)2149 are also unusual due to the special 4f
electron in cerium atom. The cluster study102 indicated that
cerium can be used to tune (by cluster sizes) the amount of SD
distributions over the Ot��f radicals that are generally reactive
with p while it can be much less reactive with s bonds of
hydrocarbon molecules. This clearly provides a clue
with which to enhance the selectivity (by decreasing the
byproducts H2O/CO2 formation that involves C–H bond
activation) without losing the reactivity of working catalysts
for partial oxidation of alkenes such as selective oxidation of
propylene that is very important in industry.150,151
3. Heteronuclear oxide clusters
The practical metal oxide catalysts are usually supported on or
mixed with other materials in heterogeneous catalytic
reactions. For example, V2O5 catalyst (active phase) may be
supported on Al2O3,152 SiO2,
153 TiO2,154 ZrO2,
155 CeO2,156
and so on. The mechanistic behavior of the support materials
in the surface reactions remains poorly understood and in
some cases, the usually considered inactive components (such
as CeO2 in VOx/CeO2 material) are finally characterized to be
of primary importance.157 The study of the O�� radicals
over heteronuclear oxide clusters (such as Vx1Six2Oyq for
V2O5/SiO2) may provide an opportunity to understand the
possible roles of both the active (such as V2O5) and inactive
(such as SiO2) phases in surface reactions.
The D value (extent of cluster over-oxidation) defined for
homonuclear TMO clusters in eqn (1) may be extended as,
D R 2y � Sinixi + q = 1 (4)
for the compositions of O�� containing heteronuclear oxide
clusters M1x1M2x2M3x3. . .Oyq, in which ni is the number of
valence electrons of elementMi that can be oxidized by oxygen
to +ni oxidation state. The DFT studies have indicated that a
Fig. 8 TOF mass spectra for reactions of CexOy+ (x= 2–7) with (a) 0% (for reference), (b) 0.5%, (c) 2.0%, and (d) 4.0% C2H4 seeded in helium
carrier gas. The numbers x,y and x,y,z denote CexOy+ and CexOy(C2H4)z
+, respectively. Adapted from ref. 102.
Table 2 Estimated absolute (k1) and relative (k1rel) rate constants for
reactions of Ce2O4+ and Zr2O4
+ clusters with small molecules
X
Ce2O4+ + X Zr2O4
+ + X
k1 k1rel k1 k1
rel
C2H4 4.6 � 10�10 1.0 3.2 � 10�10 0.7C2H6 1.1 � 10�11 2.5 � 10�2 3.2 � 10�10 0.7CO 1.9 � 10�12 4.1 � 10�3 1.8 � 10�10 0.4CH4 2.2 � 10�13 5.0 � 10�4 9.1 � 10�11 0.2
a k1 is in unit of cm3 molecule�1 s�1 and k1rel is relative to
k1(Ce2O4+ + C2H4). Adapted from ref. 102.
Fig. 9 DFT calculated profiles of SOMOs for CekO2k+ (k = 1–5)
clusters. The Mulliken spin density values in mB over the relevant
terminal oxygen atoms are shown. Adapted from ref. 102.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online

1934 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 This journal is c the Owner Societies 2011
few selected bimetallic oxide clusters with D = 1 (such as
V2Ti2O10�,158 TiVO5 and CrVO6,
36 and ZrScO4 and
ZrNbO5)159 contain O�� radicals.
Our recent experiments have indicated that under near room-
temperature conditions, the D = 1 heteronuclear oxide clusters
cations VAlO4+,103 V2O5(SiO2)1–4
+ and (V2O5)2SiO2+,104 and
V3PO10+105 can activate methane with rate constants (k1) on the
order of 10�10B10�11 cm3 molecule�1 s�1, while the D = 1
anions V2O5(SiO2)1–4O� and (V2O5)2SiO2O
� are able to
activate C–H bonds of n-butane with k1 B 10�11 cm3
molecule�1 s�1.160 The experiments thus suggest that the
above D = 1 clusters contain Ot�� radicals. However, the
DFT studies of the cluster structures (see Fig. 11 for
examples) provided an interesting result: the Ot�� radicals are
bonded with Al, Si, or P atoms rather than the vanadium atoms
in most of the clusters including VAlO4+, V2O5(SiO2)1–4
+,
V3PO10+, V2O5(SiO2)1–4O
�, and (V2O5)2SiO2O�. This is in
contrast with the traditional concept that transition metal
oxides (such as V2O5) are active phase of catalysts (such as
V2O5/SiO2) while the main group metal or non-metal oxides
(such as SiO2) are support materials and do not participate
directly in surface reactions.
The O�� radical species were suggested to be important in the
selective oxidation of methane, ethane, and benzene.18,22,161–166
It was usually considered that the O�� species are involved
directly with transition metal atoms. For example, the study of
methane conversion to methanol and formaldehyde over
vanadium oxide and molybdenum oxide supported on
mesoporous silica suggested a mechanism that the reactions
are initiated by the formation of O�� coordinated with V
and Mo at the surface.18,22 The heteronuclear oxides
VAlO4+, V2O5(SiO2)1–4
+, V3PO10+, V2O5(SiO2)1–4O
�, and
(V2O5)2SiO2O� are with diameters ranging from 0.6 to
0.8 nm. The cluster study thus implies that for catalysts such
as V2O5/SiO2 and V2O5/Al2O3 in nano-size and further smaller
regions, or for related surface defect sites, one should take into
account that the usually considered support materials (Si–O and
Al–O) may well play an important role in surface reactions such
as in the process of C–H activation. The vanadium loaded SiO2
was used as photocatalyst for the partial oxidation of methane.
It was confirmed that the high photo-activity for formaldehyde
formation (1.8–2.6% yield) with high selectivity (80–92%) was
obtained when the loading amount of V was very low, ca.
0.01–0.1 mol%. In contrast, higher loading of V resulted in
lower activity and selectivity to formaldehyde.35 We speculate
that the O�� centres for the methane activation in the
photocatalytic conversion of methane at very low vanadium
loading can be bounded with Si rather than with V.
The study of the O�� containing heteronuclear oxide clusters
also resulted in some new mechanistic details that had not been
realized in the study of related homonuclear oxide systems.
Fig. 11 shows that in V2O5(SiO2)2O� and V2O5(SiO2)4O
�
clusters, the SDs are delocalized over two oxygen atoms
(Ot��f), which is also the case for V2O6
� cluster.36,64
However, the V2O5(SiO2)2O� and V2O5(SiO2)4O
� clusters
react much faster than V2O6� does in the reaction with
C4H10, in contrast with the idea that Ot��f radicals have
relatively slow reactivity toward the C–H activation.102 The
DFT study indicated that the SDs can be localized if one of the
two Si–Ot��f or V–Ot
��f bonds in each cluster is lengthened
and the other is shortened. However, the energy cost for
such SD localization process is significantly smaller in
V2O5(SiO2)2O� [and possibly in V2O5(SiO2)4O
�] than in
V2O6�, leading to higher reactivity of the former than the
latter toward C–H activation, in agreement with the
experiments.160 It indicates that the reactivity of Ot��f
toward C–H activation depends not only on the amount of
SDs but also on the energy cost for localizing the SDs.
The comparative study of methane activation by V3PO10+
and V4O10+ clusters105 suggested that intra-cluster SD
transfer in high symmetry V4O10+ and P4O10
+ clusters:
O1��–M–Ob–M=O2 - O1=M–Ob–M–O2
�� (M = V or P,
and O1 and O2 are different terminal oxygen atoms), is fast
(B109–1012 s�1) and the reacting CH4 molecule further
Fig. 10 DFT calculated reaction pathway for Ce2O4+ (2A) + C2H4 (
1Ag)- Ce2O3+ (2A0) + CH3CHO (1A0). The zero-point vibration corrected
energy (DH0 K in eV) and Gibbs free energy at 298 K (DG298 K in eV) are given in parentheses as (DH0 K/DG298 K). Some bond lengths in 0.1 nm are
given. Adapted from ref. 102.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online

This journal is c the Owner Societies 2011 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 1935
enhances the speed of the SD transfer by lowering down
the transfer barrier. In contrast, the SD transfer in the
heteronuclear cluster V3PO10+ is impossible due to
unfavorable thermodynamics. This means that the number of
effective Ot�� centres in V4O10
+ or P4O10+ is larger than that
in V3PO10+, which rationalizes the experimental result stating
that the rate constant of V4O10+ + CH4 (almost the same as
that of P4O10+ + CH4)
98,101 is larger than that of V3PO10+ +
CH4 by a factor of 2.5 � 0.2. The model cluster study also
suggested that the hole-centres (O��) that can be photo-
generated in bulk materials may transfer preferentially
toward pre-adsorbed small molecules over the surfaces.105
4. Additional remarks
It has been demonstrated that for groups 3–7 metals (except Cr
and Mn) and related heteronuclear systems, the O�� radicals
generally exist over the D = 1 oxide clusters (eqn (1) and (4)).
We have tried to identify O�� radicals over later transition
metal oxide clusters including FexOy� (such as Fe2kO3k
+167
and Fe2kO3k+1�) and CoxOy
� (x Z 2) by searching for
the cluster reactivity toward C–H activation at near room-
temperature conditions. However, none of the studied clusters
is able to activate CH4, C2H6, or C4H10. This implies that the
later transition metal oxides such as Fe2O3 and Co3O4 are
fundamentally different from the early transition metal oxides
such as Sc2O3, TiO2, etc. in terms of electron excitation and
hole (O��) formation.168–169 The later transition metal oxide
clusters (even for those with metal in the normally highest
oxidation state, such as Fe2kO3k)170 are also very difficult for
the quantum chemistry studies because of the additional
valence d electrons (3d5 for Fe3+) and their possible partial
participations in the bonding with oxygen.
For early transition metals and some main group metals, the
oxygen-more-rich (D = 2, 3,. . .) oxide clusters can be
superoxide (O2��) or peroxide (O2
2�) complexes.110,171–174
However, our experimental investigation on the early
transition metal oxide cluster cations indicated that the
D > 1 cluster systems are inert with CH4 while many of
the D = 1 systems can activate CH4 with rate constants on
the order of 10�10 cm3 molecule�1 s�1 under near-room-
temperature conditions.90 The chemical reactivity and
selectivity of the D > 1 TMO clusters remain poorly
understood although it is important to study these clusters
for interpretation of the chemistry of the surface O2�� and
O22� species.8,175 Zhou and co-authors have recently identified
that the neutral cluster TaO4 (D = 3) can spontaneously
activate dihydrogen at cryogenic temperatures85 while we
have recently characterized that the oxygen-very-rich cluster
Zr2O8� (D = 7) can activate C–H bonds of n-butane under
mild conditions.176 It is noticeable that for some of the D = 2
TMO clusters, such as WO4177 and VTi3O10
�,158 the DFT
studies suggested that there are two O�� centres (bi-radicals) in
each cluster. The reactivity study of these bi-radical species
may be important to interpret the chemistry of coupled O��
dimer species (O��� � �O��) over the metal surfaces. It has been
pointed out that it is difficult to characterize such adsorbed
dimer species due to complications such as heterogeneity of
isolated O�� sites and transition of a pair of O�� to peroxide
ion O22� in the condensed phase study,9 while a gas phase
cluster study may provide mechanistic details for such species.
5. Conclusions
The mono-nuclear oxygen-centred radicals (O��) generally
exist over oxide clusters of groups 3–7 metals (except Cr
and Mn) and related heteronuclear systems of which the
compositions satisfy eqn (1) or (4). In terms of the C–H
activation of alkanes such as CH4, the charge state of the
TMO clusters and the extent of spin density localization largely
influence the reactivity of O�� radicals: (1) the anions are
generally much less reactive than the corresponding cations
while the neutrals may be in the middle; (2) the O�� containing
cluster anions may still be as reactive as the OH free radical;
and (3) the localized spin densities over a single O atom in the
cluster lead to relatively high reactivity. The cerium element
may be used to tune the amount of spin densities over the O��
radicals. The oxygen atom transfer usually occurs in the
reactions of the O�� containing clusters with CO, alkenes
(sometimes with CQC bond cleavage), etc. The intra-cluster
spin density transfer in some of the O�� containing clusters can
be fast and the speed of the transfer can be enhanced by the
interacting small molecules. The O�� radicals are bonded with
the Al, Si, or P rather than the V atoms in most of the studied
V–Al, V–Si, and V–P heteronuclear oxide clusters. The
chemistry of O�� containing TMO clusters can be closely
related with that of the surface O�� species in terms of C–H
activation, oxygen atom transfer, alkene CQC double bond
cleavage, hole transfer, support effect, etc. in catalysis or
Fig. 11 DFT calculated structures and profiles of SOMOs for
V2O5(SiO2)1–4+ (a) and V2O5(SiO2)1–4O
� (b) clusters. The Mulliken
spin density values in mB over the relevant oxygen atoms are given in
parentheses. Some bond lengths are given in 0.1 nm. Adapted from
refs. 104 and 160.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online

1936 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 This journal is c the Owner Societies 2011
photocatalysis. In general, the investigation of O�� containing
TMO clusters provides a good chance to learn the chemistry of
the reactive O�� species in a bulk system as well as that of the
metal valence d and f electrons.
Acknowledgements
This work was supported by the Chinese Academy of Sciences
(Hundred Talents Fund), the National Natural Science
Foundation of China (Nos. 20703048, 20803083, and
20933008), CMS Foundation of the ICCAS (No. CMS-
LX200902), and the 973 Program (No. 2011CB932302).
Notes and references
1 G. Ertl, H. Knozinger and J. Weikamp, Handbook ofHeterogeneous Catalysis, Wiley-VCH,Weinheim, Germany, 1997.
2 J. L. G. Fierro, Metal Oxides, Taylor & Francis Group, BocaRaton, FL, 2006.
3 R. A. van Santen and M. Neurock, Molecular HeterogeneousCatalysis, Wiley-VCH, Weinheim, Germany, 2006.
4 M. P. Rosynek, Catal. Rev. Sci. Eng., 1977, 16, 111.5 A. Trovarelli, Catal. Rev. Sci. Eng., 1996, 38, 439.6 M. A. Barteau, Chem. Rev., 1996, 96, 1413.7 B. M. Weckhuysen and D. E. Keller, Catal. Today, 2003, 78, 25.8 M. Che and A. J. Tench, Adv. Catal., 1983, 32, 1.9 M. Che and A. J. Tench, Adv. Catal., 1982, 31, 77.10 C. Li, K. Domen, K. Maruya and T. Onishi, J. Am. Chem. Soc.,
1989, 111, 7683.11 G. I. Panov, K. A. Dubkov and E. V. Starokon, Catal. Today,
2006, 117, 148.12 J. H. Lunsford, Catal. Rev. Sci. Eng., 1974, 8, 135.13 K. Dyrek and M. Che, Chem. Rev., 1997, 97, 305.14 M. Chiesa, E. Giamello andM. Che, Chem. Rev., 2010, 110, 1320.15 Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science,
2003, 301, 935.16 J. Guzman, S. Carrettin and A. Corma, J. Am. Chem. Soc., 2005,
127, 3286.17 J. Guzman, S. Carrettin, J. C. Fierro-Gonzalez, Y. Hao,
B. C. Gates and A. Corma,Angew. Chem., Int. Ed., 2005, 44, 4778.18 H. F. Liu, R. S. Liu, K. Y. Liew, R. E. Johnson and
J. H. Lunsford, J. Am. Chem. Soc., 1984, 106, 4117.19 J. H. Lunsford, Angew. Chem., Int. Ed. Engl., 1995, 34, 970.20 M. M. Khan and G. A. Somorjai, J. Catal., 1985, 91, 263.21 R. H. Crabtree, Chem. Rev., 1995, 95, 987.22 H. Launay, S. Loridant, D. L. Nguyen, A. M. Volodin,
J. L. Dubois and J. M. M. Millet, Catal. Today, 2007, 128, 176.23 M. S. Palmer, M. Neurock and M. M. Olken, J. Am. Chem. Soc.,
2002, 124, 8452.24 The involvement of ROS or radical species for the transformation
of very stable molecules is also often necessary for biologicalprocesses and low-temperature organic synthesis.8,25–26 The low-temperature or relatively-lower-temperature chemical processesare reliable basis for solving some of the current-day environmentand energy problems. We hope this article falls into the scope ofICCAS (Institute of Chemistry, Chinese Academy of Sciences)Special Issue: Processes in Chemical Reactions Related toEnvironment, Energy and Life Sciences.
25 F. Thomas, Eur. J. Inorg. Chem., 2007, 2379.26 W. Buckel, Angew. Chem., Int. Ed., 2009, 48, 6779.27 C. Zhao and I. E. Wachs, Catal. Today, 2006, 118, 332.28 This does not necessarily mean that, in terms of catalysis, it is
unimportant to study the pure metal clusters because the motion ofa fair amount of valance electrons in metals is delocalized in nature(metals are usually good conductors). The unique reactivity andproperties obtained for metal clusters at certain size may be used togenerate new, efficient, and selective catalysts.29–30.
29 P. B. Armentrout, Annu. Rev. Phys. Chem., 2001, 52, 423.30 R. A. J. O’Hair and G. N. Khairallah, J. Cluster Sci., 2004, 15,
331.
31 A. L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95,735.
32 M. Sterrer, T. Berger, O. Diwald and E. Knozinger, J. Am. Chem.Soc., 2003, 125, 195.
33 For example, the high-temperature treatment of mesoporoussilica (RSi–OH + RSi–OH - RSi–O�� + RSi� + H2O)can produce a kind of surface defect on silica, non-bridgingoxygen hole centre (RSi–O��) accompanied with E0 centre(RSi�) that was clarified to be the photoactive sites for thenon-oxidative coupling of methane.34–35.
34 L. Yuliati, M. Tsubota, A. Satsuma, H. Itoh and H. Yoshida,J. Catal., 2006, 238, 214.
35 L. Yuliatiw and H. Yoshida, Chem. Soc. Rev., 2008, 37, 1592.36 Y.-X. Zhao, X.-L. Ding, Y.-P. Ma, Z.-C. Wang and S.-G. He,
Theor. Chem. Acc., 2010, 127, 449.37 The local charges over TMO catalytic surface may be generated
through charge transfers and tuned by supporting and supportedmaterials.3,38,39 As a result, it is necessary to study the O��
containing TMO clusters in different charge states.38 M. Sterrer, T. Risse, U. M. Pozzoni, L. Giordano, M. Heyde,
H.-P. Rust, G. Pacchioni andH.-J. Freund,Phys. Rev. Lett., 2007,98, 096107.
39 N. Nilius, M. V. Ganduglia-Pirovano, V. Brazdova, M. Kulawik,J. Sauer and H.-J. Freund, Phys. Rev. Lett., 2008, 100, 096802.
40 S.-G. He, Y. Xie, F. Dong, S. Heinbuch, E. Jakubikova,J. J. Rocca and E. R. Bernstein, J. Phys. Chem. A, 2008, 112,11067.
41 F. Dong, S. Heinbuch, Y. Xie, J. J. Rocca, E. R. Bernstein,Z.-C. Wang, K. Deng and S.-G. He, J. Am. Chem. Soc., 2008, 130,1932.
42 W. Xue, Z.-C.Wang, S.-G. He, Y. Xie and E. R. Bernstein, J. Am.Chem. Soc., 2008, 130, 15879.
43 F. Dong, S. Heinbuch, Y. Xie, E. R. Bernstein, J. J. Rocca,Z.-C. Wang, X.-L. Ding and S.-G. He, J. Am. Chem. Soc., 2009,131, 1057.
44 Y. Xie, F. Dong, S. Heinbuch, J. J. Rocca and E. R. Bernstein,J. Chem. Phys., 2009, 130, 114306.
45 F. Dong, S. Heinbuch, Y. Xie, J. J. Rocca and E. R. Bernstein,J. Phys. Chem. A, 2009, 113, 3029.
46 Y. Xie, F. Dong, S. Heinbuch, J. J. Rocca and E. R. Bernstein,Phys. Chem. Chem. Phys., 2010, 12, 947.
47 F. Dong, S. Heinbuch, S.-G. He, Y. Xie, J. J. Rocca andE. R. Bernstein, J. Chem. Phys., 2006, 125, 164318.
48 Y. Gong and M.-F. Zhou, Chem. Rev., 2009, 109, 6765.49 G.-J. Wang and M.-F. Zhou, Int. Rev. Phys. Chem., 2008, 27, 1.50 C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009,
11, 10757.51 S.-G. He, Y. Xie, Y.-G. Guo and E. R. Bernstein, J. Chem. Phys.,
2007, 126, 194315.52 Y. Xie, S.-G. He, F. Dong and E. R. Bernstein, J. Chem. Phys.,
2008, 128, 044306.53 Y.-P. Ma, W. Xue, Z.-C. Wang, M.-F. Ge and S.-G. He, J. Phys.
Chem. A, 2008, 112, 3731.54 W. Xue, S. Yin, X.-L. Ding, S.-G. He and M.-F. Ge, J. Phys.
Chem. A, 2009, 113, 5302.55 A.W. Castleman, Jr. and K. H. Bowen, J. Phys. Chem., 1996, 100,
12911.56 P. Jena and A. W. Castleman, Jr., Proc. Natl. Acad. Sci. U. S. A.,
2006, 103, 10560.57 D. Schroder and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1995,
34, 1973.58 M. B. Knickelbein, Annu. Rev. Phys. Chem., 1999, 50, 79.59 V. E. Bondybey and M. K. Beyer, J. Phys. Chem. A, 2001, 105,
951.60 K. A. Zemski, D. R. Justes and A. W. Castleman, Jr., J. Phys.
Chem. B, 2002, 106, 6136.61 D. K. Bohme and H. Schwarz, Angew. Chem., Int. Ed., 2005, 44,
2336.62 T. M. Bernhardt, Int. J. Mass Spectrom., 2005, 243, 1.63 D. Schroder and H. Schwarz, Top. Organomet. Chem., 2007, 22, 1.64 K. R. Asmis and J. Sauer, Mass Spectrom. Rev., 2007, 26, 542.65 D. Schroder and H. Schwarz,Proc. Natl. Acad. Sci. U. S. A., 2008,
105, 18114.66 L.-F. Cui and L.-S. Wang, Int. Rev. Phys. Chem., 2008, 27, 139.67 M. Schlangen and H. Schwarz, Dalton Trans., 2009, 10155.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online

This journal is c the Owner Societies 2011 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 1937
68 J. Roithova and D. Schroder, Chem. Rev., 2010, 110, 1170.69 S. F. Vyboishchikov and J. Sauer, J. Phys. Chem. A, 2001, 105,
8588.70 M. Calatayud, J. Andres and A. Beltran, J. Phys. Chem. A, 2001,
105, 9760.71 H. Hakkinen and U. Landman, J. Am. Chem. Soc., 2001, 123,
9704.72 H.-Y. Wang, R.-B. Huang, H. Chen, M.-H. Lin and L.-S. Zheng,
J. Phys. Chem. A, 2001, 105, 4653.73 W. T. Wallace and R. L. Whetten, J. Am. Chem. Soc., 2002, 124,
7499.74 M. Andersson and A. Rosen, J. Chem. Phys., 2002, 117, 7051.75 A. Fielicke, R. Mitric, G. Meijer, V. Bonacic-Koutecky and
G. von Helden, J. Am. Chem. Soc., 2003, 125, 15716.76 A. S. Worz, K. Judai, S. Abbet and U. Heiz, J. Am. Chem. Soc.,
2003, 125, 7964.77 J. M. Lightstone, H. A. Mann, M. Wu, P. M. Johnson and
M. G. White, J. Phys. Chem. B, 2003, 107, 10359.78 J. M. Parnis, E. Escobar-Cabrera, M. G. K. Thompson,
J. P. Jacula, R. D. Lafleur, A. Guevara-Garcıa, A. Martınezand D. M. Rayner, J. Phys. Chem. A, 2005, 109, 7046.
79 R. B. Wyrwas and C. C. Jarrold, J. Am. Chem. Soc., 2006, 128,13688.
80 M. Ichihashi, T. Hanmura and T. Kondow, J. Chem. Phys., 2006,125, 133404.
81 R. Burgert, H. Schnockel, A. Grubisic, X. Li, S. T. Stokes,K. H. Bowen, G. F. Gantefor, B. Kiran and P. Jena, Science,2008, 319, 438.
82 Z. D. Reed andM. A. Duncan, J. Phys. Chem. A, 2008, 112, 5354.83 S. H. Li, A. Mirabal, J. Demuth, L. Woste and T. Siebert, J. Am.
Chem. Soc., 2008, 130, 16832.84 H.-B. Li, S.-X. Tian and J.-L. Yang, Chem.–Eur. J., 2009, 15,
10747.85 M.-F. Zhou, C.-X. Wang, Z.-H. Li, J. Zhuang, Y.-Y. Zhao,
X.-M. Zhen and K.-N. Fan, Angew. Chem., Int. Ed., 2010, 49,7757.
86 A. D. Becke, J. Chem. Phys., 1993, 98, 5648.87 C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens.
Matter, 1988, 37, 785.88 G. L. Gutsev, L. Andrews and C. W. Bauschlicher, Jr., Theor.
Chem. Acc., 2003, 109, 298.89 J. B. Pedley and E. M. Marshall, J. Phys. Chem. Ref. Data, 1983,
12, 967.90 Y.-X. Zhao, X.-N. Wu, Z.-C. Wang, S.-G. He and X.-L. Ding,
Chem. Commun., 2010, 46, 1736.91 R. A. Periana, O. Mironov, D. Taube, G. Bhalla and C. J. Jones,
Science, 2003, 301, 814.92 K. K. Irikura and J. L. Beauchamp, J. Am. Chem. Soc., 1989, 111,
75.93 D. Schroder, A. Fiedler, J. Hrusak and H. Schwarz, J. Am. Chem.
Soc., 1992, 114, 1215.94 M. F. Ryan, A. Fiedler, D. Schroder and H. Schwarz, J. Am.
Chem. Soc., 1995, 117, 2033.95 I. Kretzschmar, A. Fiedler, J. N. Harvey, D. Schroder and
H. Schwarz, J. Phys. Chem. A, 1997, 101, 6252.96 J. N. Harvey, M. Diefenbach, D. Schroder and H. Schwarz, Int. J.
Mass Spectrom., 1999, 182–183, 85.97 D. Schroder and J. Roithova, Angew. Chem., Int. Ed., 2006, 45,
5705.98 S. Feyel, J. Dobler, D. Schroder, J. Sauer and H. Schwarz,Angew.
Chem., Int. Ed., 2006, 45, 4681.99 S. Feyel, J. Dobler, R. Hockendorf, M. K. Beyer, J. Sauer and
H. Schwarz, Angew. Chem., Int. Ed., 2008, 47, 1946.100 G. de Petris, A. Troiani, M. Rosi, G. Angelini and O. Ursini,
Chem.–Eur. J., 2009, 15, 4248.101 N. Dietl, M. Engeser and H. Schwarz, Angew. Chem., Int. Ed.,
2009, 48, 4861.102 X.-N. Wu, Y.-X. Zhao, W. Xue, Z.-C. Wang, S.-G. He and
X.-L. Ding, Phys. Chem. Chem. Phys., 2010, 12, 3984.103 Z.-C. Wang, X.-N. Wu, Y.-X. Zhao, J.-B. Ma, X.-L. Ding and
S.-G. He, Chem. Phys. Lett., 2010, 489, 25.104 X.-L. Ding, Y.-X. Zhao, X.-N. Wu, Z.-C. Wang, J.-B. Ma and
S.-G. He, Chem.–Eur. J., 2010, 16, 11463.105 J.-B. Ma, X.-N. Wu, Y.-X. Zhao, X.-L. Ding and S.-G. He, Phys.
Chem. Chem. Phys., 2010, 12, 12223.
106 M. E. Geusic, M. D. Morse, S. C. Obrien and R. E. Smalley, Rev.Sci. Instrum., 1985, 56, 2123.
107 X.-N. Wu, Y.-X. Zhao, J.-Bi. Ma, X.-L. Ding and S.-G. He,unpublished work.
108 K. A. Zemski, D. R. Justes and A. W. Castleman, Jr., J. Phys.Chem. A, 2001, 105, 10237.
109 D. R. Justes, R. Mitric, N. A. Moore, V. Bonacic-Koutecky andA. W. Castleman, Jr., J. Am. Chem. Soc., 2003, 125, 6289.
110 S. Yin, Y.-P. Ma, L. Du, S.-G. He and M.-F. Ge, Chin. Sci. Bull.,2008, 53, 3829.
111 G. E. Johnson, R. Mitric, E. C. Tyo, V. Bonacic-Koutecky andA. W. Castleman, Jr., J. Am. Chem. Soc., 2008, 130, 13912.
112 G. E. Johnson, E. C. Tyo and A. W. Castleman, Jr., Proc. Natl.Acad. Sci. U. S. A., 2008, 105, 18108.
113 Y.-P. Ma, X.-L. Ding, Y.-X. Zhao and S.-G. He,ChemPhysChem., 2010, 11, 1718.
114 Z.-C. Wang, W. Xue, Y.-P. Ma, X.-L. Ding, S.-G. He, F. Dong,S. Heinbuch, J. J. Rocca and E. R. Bernstein, J. Phys. Chem. A,2008, 112, 5984.
115 M. Ruszel, B. Grzybowska, M. Gasior, K. Samson, I. Gressel andJ. Stoch, Catal. Today, 2005, 99, 151.
116 P. Pietrzyk, Z. Sojka, S. Dzwigaj and M. Che, J. Am. Chem. Soc.,2007, 129, 14174.
117 R. C. Bell and A. W. Castleman, Jr., J. Phys. Chem. A, 2002, 106,9893.
118 J.-B. Ma, X.-N. Wu, Y.-X. Zhao, S.-G. He and X.-L. Ding, ActaPhys. Chim. Sin., 2010, 26, 1761.
119 G. E. Johnson, R. Mitric, M. Nossler, E. C. Tyo, V. Bonacic-Koutecky andA.W. Castleman, Jr., J. Am. Chem. Soc., 2009, 131,5460.
120 K. A. Zemski, D. R. Justes, R. C. Bell and A. W. Castleman, Jr.,J. Phys. Chem. A, 2001, 105, 4410.
121 The DFT results in Fig. 6 were obtained with B3LYPfunctional86,87 and TZVP basis sets122 for H, C, O, and V andSDD123 for Zr. The Gaussian03 program124 was used. Thestructures of V2O5
+, V2O6�, V3O8, and Zr2O5
� can be found inliterature.36,64,109,113,119 See refs. 118 and 125 for more details.
122 A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100,5829.
123 D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss,Theor. Chim. Acta, 1990, 77, 123.
124 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria,M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr.,T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam,S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi,G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada,M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida,T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene,X. Li,J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken,C. Adamo,J. Jaramillo, R. Gomperts, R. E. Stratmann,O. Yazyev,A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski,P. Y. Ayala,K. Morokuma, G. A. Voth, P. Salvador,J. J. Dannenberg,V. G. Zakrzewski, S. Dapprich, A. D. Daniels,M. C. Strain,O. Farkas, D. K. Malick, A. D. Rabuck,K. Raghavachari,J. B. Foresman, J. V. Ortiz, Q. Cui,A. G. Baboul, S. Clifford,J. Cioslowski, B. B. Stefanov, G. Liu,A. Liashenko, P. Piskorz,I. Komaromi, R. L. Martin, D. J. Fox,T. Keith, M. A. Al-Laham,C. Y. Peng, A. Nanayakkara,M. Challacombe, P. M. W. Gill,B. G. Johnson, W. Chen,M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03(Revision C.02), Gaussian, Inc.,Wallingford, CT, 2004.
125 J.-B. Ma, X.-N. Wu, Y.-X. Zhao, X.-L. Ding and S.-G. He, Chin.J. Chem. Phys., 2010, 23, 133.
126 G. Fu, X. Xu, X. Lu and H.-L. Wan, J. Am. Chem. Soc., 2005,127, 3989.
127 G. Fu, X. Xu and H.-L. Wan, Catal. Today, 2006, 117, 133.128 K.-M. Jeong and F. Kaufman, J. Phys. Chem., 1982, 86,
1808.129 A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551.130 J. K. Gibson, J. Phys. Chem., 1994, 98, 11321.131 J. K. Gibson, J. Phys. Chem., 1996, 100, 507.132 S.-F. Li, H.-Q. Lu, P.-L. Li, Z.-X. Yang and Z.-X. Guo, J. Chem.
Phys., 2008, 128, 164718.133 C. Chen, H.-L. Chen, M.-H. Weng, S.-P. Ju, J.-G. Chang and
C.-S. Chang, Chin. J. Catal., 2008, 29, 1117.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online

1938 Phys. Chem. Chem. Phys., 2011, 13, 1925–1938 This journal is c the Owner Societies 2011
134 F. Aubriet, J. J. Gaumet, W. A. de Jong, G. S. Groenewold,A. K. Gianotto, M. E. McIlwain, M. J. Van Stipdonk andC. M. Leavitt, J. Phys. Chem. A, 2009, 113, 6239.
135 C. Heinemann, H. H. Cornehl, D. Schroder, M. Dolg andH. Schwarz, Inorg. Chem., 1996, 35, 2463.
136 S. P. Willson and L. Andrews, J. Phys. Chem. A, 1999, 103, 3171.137 Z.-J. Wu, W. Guan, J. Meng and Z.-M. Su, J. Cluster Sci., 2007,
18, 444.138 G. A. Deluga, J. R. Salge, L. D. Schmidt and X. E. Verykios,
Science, 2004, 303, 993.139 C. T. Campbell and C. H. F. Peden, Science, 2005, 309, 713.140 F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero,
G. Comelli and R. Rosei, Science, 2005, 309, 752.141 M. V. Ganduglia-Pirovano, A. Hofmann and J. Sauer, Surf. Sci.
Rep., 2007, 62, 219.142 F. Marabelli and P. Wachter, Phys. Rev. B: Condens. Matter,
1987, 36, 1238.143 E. Wuilloud, B. Delley, W. D. Schneider and Y. Baer, Phys. Rev.
Lett., 1984, 53, 202.144 P. Wachter, Phys. B, 2001, 300, 105.145 A. Fujimori, Phys. Rev. Lett., 1984, 53, 2518.146 M. Matsumoto, K. Soda, K. Ichikawa, S. Tanaka, Y. Taguchi,
K. Jouda, O. Aita, Y. Tezuka and S. Shin, Phys. Rev. B: Condens.Matter, 1994, 50, 11340.
147 M. D. Walter, C. H. Booth, W. W. Lukens and R. A. Andersen,Organometallics, 2009, 28, 698.
148 M. Coreno, M. de Simone, J. C. Green, N. Kaltsoyannis,N. Narband and A. Sella, Chem. Phys. Lett., 2006, 432, 17.
149 A. Ashley, G. Balazs, A. Cowley, J. Green, C. H. Booth andD. O’Hare, Chem. Commun., 2007, 1515.
150 M. Li and J. Shen, J. Catal., 2002, 205, 248.151 Z. Su, N. Borho and Y. J. Xu, J. Am. Chem. Soc., 2006, 128, 17126.152 J. M. L. Nieto, Top. Catal., 2006, 41, 3.153 M. Cavalleri, K. Hermann, A. Knop-Gericke, M. Havecker,
R. Herbertb, C. Hess, A. Oestereich, J. Dobler and R. Schlogl,J. Catal., 2009, 262, 215.
154 U. Bentrup, A. Bruckner, C. Rudinger and H. J. Eberle, Appl.Catal., A, 2004, 269, 237.
155 P. R. Shah, I. Baldychev, J. M. Vohs and R. J. Gorte,Appl. Catal.,A, 2009, 361, 13.
156 M. Baron, H. Abbott, O. Bondarchuk, D. Stacchiola, A. Uhl,S. Shaikhutdinov, H.-J. Freund, C. Popa, M. V. Ganduglia-Pirovano and J. Sauer, Angew. Chem., Int. Ed., 2009, 48, 8006.
157 M. V. Ganduglia-Pirovano, C. Popa, J. Sauer, H. Abbott, A. Uhl,M. Baron, D. Stacchiola, O. Bondarchuk, S. Shaikhutdinov andH.-J. Freund, J. Am. Chem. Soc., 2010, 132, 2345.
158 E. Janssens, G. Santambrogio, M. Brummer, L. Woste,P. Lievens, J. Sauer, G. Meijer and K. R. Asmis, Phys. Rev.Lett., 2006, 96, 233401.
159 M. Nobler, R. Mitric, V. Bonacic-Koutecky, G. E. Johnson,E. C. Tyo and A. W. Castleman, Jr., Angew. Chem. Int. Ed.,2010, 49, 407.
160 Y.-X. Zhao, X.-N. Wu, J.-B. Ma, S.-G. He and X.-L. Ding,J. Phys. Chem. C, 2010, 114, 12271.
161 V. A. Shvets and V. B. Kazansky, J. Catal., 1972, 25, 123.162 T. Y. Ben and J. H. Lunsford, Chem. Phys. Lett., 1973,
19, 348.163 M. B. Ward, M. J. Lin and J. H. Lunsford, J. Catal., 1977,
50, 306.164 N. I. Lipatkina, V. A. Shvets and V. B. Kazansky, Kinet. Katal.,
1978, 19, 979.165 R. S. Liu, M. Iwamoto and J. H. Lunsford, J. Chem. Soc., Chem.
Commun., 1982, 78.166 M. V. Parfenov, E. V. Starokon, S. V. Semikolenov and
G. I. Panov, J. Catal., 2009, 263, 173.167 S. Yin,W. Xue, X.-L. Ding,W.-G.Wang, S.-G. He andM.-F. Ge,
Int. J. Mass Spectrom., 2009, 281, 72.168 A. Kay, I. Cesar and M. Grtzel, J. Am. Chem. Soc., 2006, 128,
15714.169 S. V. Yanina and K. M. Rosso, Science, 2008, 320, 218.170 X.-L. Ding, W. Xue, Y.-P. Ma, Z.-C. Wang and S.-G. He,
J. Chem. Phys., 2009, 130, 014303.171 S. R. Desai, H. B. Wu, C. M. Rohlfing and L.-S. Wang, J. Chem.
Phys., 1997, 106, 1309.172 Y. Gong, C.-F. Ding and M.-F. Zhou, J. Phys. Chem. A, 2007,
111, 11572.173 Z.-C. Wang, X.-L. Ding, Y.-P. Ma, H. Cao, X.-N. Wu,
Y.-X. Zhao and S.-G. He, Chin. Sci. Bull., 2009, 54, 2814.174 B. Wang, W.-J. Chen, B.-C. Zhao, Y.-F. Zhang and X. Huang,
J. Phys. Chem. A, 2010, 114, 1964.175 X. Huang, H.-J. Zhai, T. Waters, J. Li and L.-S. Wang, Angew.
Chem., Int. Ed., 2006, 45, 657.176 J.-B. Ma, X.-N. Wu, Y.-X. Zhao, X.-L. Ding and S.-G. He,
J. Phys. Chem. A, 2010, 114, 10024.177 H.-J. Zhai, B. Kiran, L.-F. Cui, X. Li, D. A. Dixon and
L.-S. Wang, J. Am. Chem. Soc., 2004, 126, 16134.
Publ
ishe
d on
05
Janu
ary
2011
. Dow
nloa
ded
by U
nive
rsity
of
Vic
tori
a on
26/
10/2
014
23:3
1:21
. View Article Online





























