Embed Size (px)
Citation preview

122
CHAPTER- VII
FT-IR and FT-Raman spectroscopic investigation on 2-Ethyl Pyridine using
HF and DFT (B3LYP and B3PW91) calculations
7.1. Introduction
Heterocyclic nitrogen containing compounds such as pyridine and its derivatives
are commonly present in synthetic and natural products [1-2]. The study of the
vibrational spectra of substituted pyridine mainly amino pyridine attracts the attention of
many spectroscopists due to their wide application in pharmacology and agro- chemistry.
Pyridine heterocyclic compounds and its derivatives are repeated moiety in many large
molecules with interesting photo physical, electrochemical and catalytic applications [3-
10]. They serve as good anesthetic agent and hence are used in the preparation of drugs
for certain brain disease. These pharmaceutically acceptable salts and the pre drugs are
used for the treatment (or) prevention of diabetic neuropathy [11-12]. The presence of
ethyl group in the molecule shows some difference in photo physical properties relative
to the pyridine. 2-Ethyl pyridine constitutes an important class of heterocyclic organic
compound. Investigations on the structure of these organic molecules have been a subject
of interest because of their peculiar photo physical properties and pharmaceutical
importance [13-15].
Adnan Saglam et al [16] has recorded the Fourier transform infrared and laser
Raman spectra of 4-Pyridine acid in the regions of 100- 4000 cm-1
. The optimized
molecular structures, vibrational frequencies and corresponding vibrational assignments
of the cis and trans conformers of 2-, 3- and 4-pyridine carboxaldehydes have been

123
calculated using ab initio Hartree–Fock (HF) and density functional theory (B3LYP)
methods with 6-311++G(d, p) basis set. The calculations were adapted to the CS
symmetries of all the molecules. The mean vibrational deviations between the vibrational
frequency values of the two conformers of all the compounds have been seen to increase
while the relative energies increase and it was concluded the more different the molecular
structure of the two conformers is the higher the relative energy between them, which
gives a bigger mean vibrational deviation.
Michalski et al [17] has studied the Synthesis of 2-phenylazo-5-nitro-6-methyl-
pyridine. Synthesis of 2-phenylazo-5-nitro-6-methyl-pyridine was described. Its X-ray
structure was reported and discussed in terms of the molecular conformation of the
compound. The crystal is triclinic, space group P-1, with the unit cell parameters
aZ6.372(1), bZ7.522(2), cZ12.495(2) A° , and aZ6.372(1), bZ89.62(3)8 and
gZ101.57(3)8. The pyridine and phenyl rings were planar deflected by torsional angle
JZ4.8 (3)8. The crystal structure was stabilized by non-classical hydrogen interaction of
the C–H/O type with C/O distance 3.307(5) A ° , H/O distance 2.481(3) A ° and C–H/O
angle equal to 147.8(3)8. These interactions in the crystal structure couple pairs of the
molecules related by an inversion centre. FT-IR, Raman and NMR spectra of this
compound have also been measured. The 6-31G (d,p) basis set with the B3LYP
functional has been used to discuss the structure and dynamics of the compound studied.
Literature survey reveals that no ab initio HF or DFT with 6-311G (d, p) basis
sets calculations have been reported 2-Ethyl Pyridine (2-EP) so far. Hence the present

124
work has been undertaken to carry out a complete vibrational analysis on this molecule,
based on both experimental and theoretical study.
7.2. Computational details
In the present work, the HF and some of the hybrid methods such as B3LYP and
B3PW91 are carried out using the basis sets 6-31G (d, p), 6-31+G (d, p), 6-311G(d, p)
and 6-311++G (d, p). All these calculations were performed using GAUSSIAN 03W
program package on Pentium IV processor in personal computer [18-21].
The calculated frequencies are scaled down by suitable factors in comparison with
the experimental frequencies. The scaling factors are 0.903 and 0.904 for HF and in
agreement with the literature [22-23]. In the case of B3LYP with 6-31G (d, p)
calculation, the scaling factors are 0.955, 0.971, 0.959, 0.939, 0.738 and 0.795; for 6-
311++G (d, p) basis sets, the scaling factors are 0.961, 0.969, 0.979, 0.939, 0.789 and
0.845. In the case of B3PW91/6-311G (d, p) calculation, the scaling factors are 0.954,
0.988, 0.849, 0.788, 0.709 and 1.04 and in good agreement with the literature [24].
7. 3 Results and Discussion
7.3.1 Molecular Geometry:
The most optimized geometry performed by HF and DFT of 2-Ethyl pyridine
molecule with atoms numbering is shown in Figure 7.1. The molecule consists of ethyl
group connected to a pyridine ring. The zero point vibrational energy as predicted by
HF/6-31+G (d, p), B3LYP/6-31G (d, p), B3LYP/6-311++G (d, p) and B3PW91/6-311G
(d, p) of the molecule is 70.90, 70.54, 66.18, 65.94 and 66.17 Kcal/mol respectively.

125
The structural parameters; bond lengths, bond angles and dihedral angles calculated using
different basis sets are presented in Table 7.1.
In comparison with the experimental values, it is observed that most of the
calculated bond length values are slightly larger than the experimental values. This may
be due to the fact that the calculations are performed for the isolated molecules (gaseous
phase) while the experimental spectra are recorded in solid phase. This is in accordance
with the earlier work [25]. Comparing bond angles and lengths of B3LYP/B3PW91 with
those of HF, as a whole the values of former are bigger than later. The calculated values
from B3LYP/B3PW91 correlate well with the experimental data. Although the
differences, calculated geometrical parameters represent a good approximation and they
are the bases for calculating other parameters, such as vibrational frequencies. Optimized
geometrical parameters, namely, bond lengths and bond angles at HF/6-31+G (d, p),
B3LYP/6-31+G (d, p), B3LYP/6-311++G (d, p) and B3PW91/6-311G (d, p) levels are
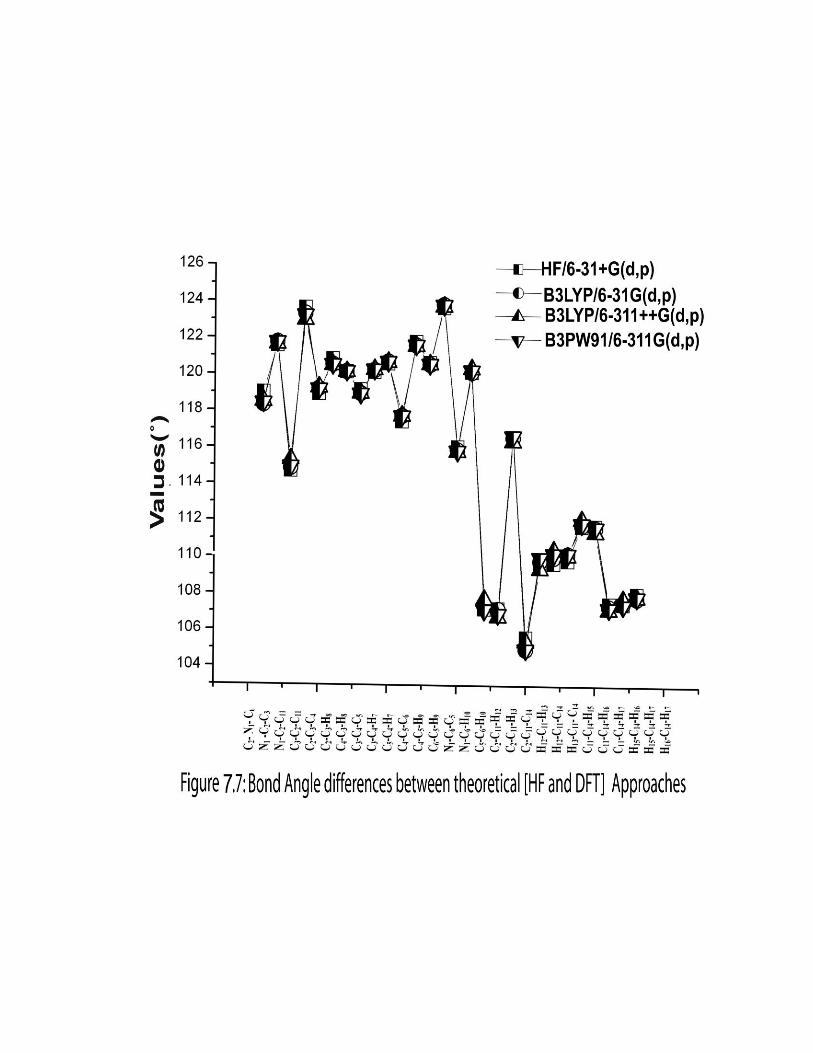
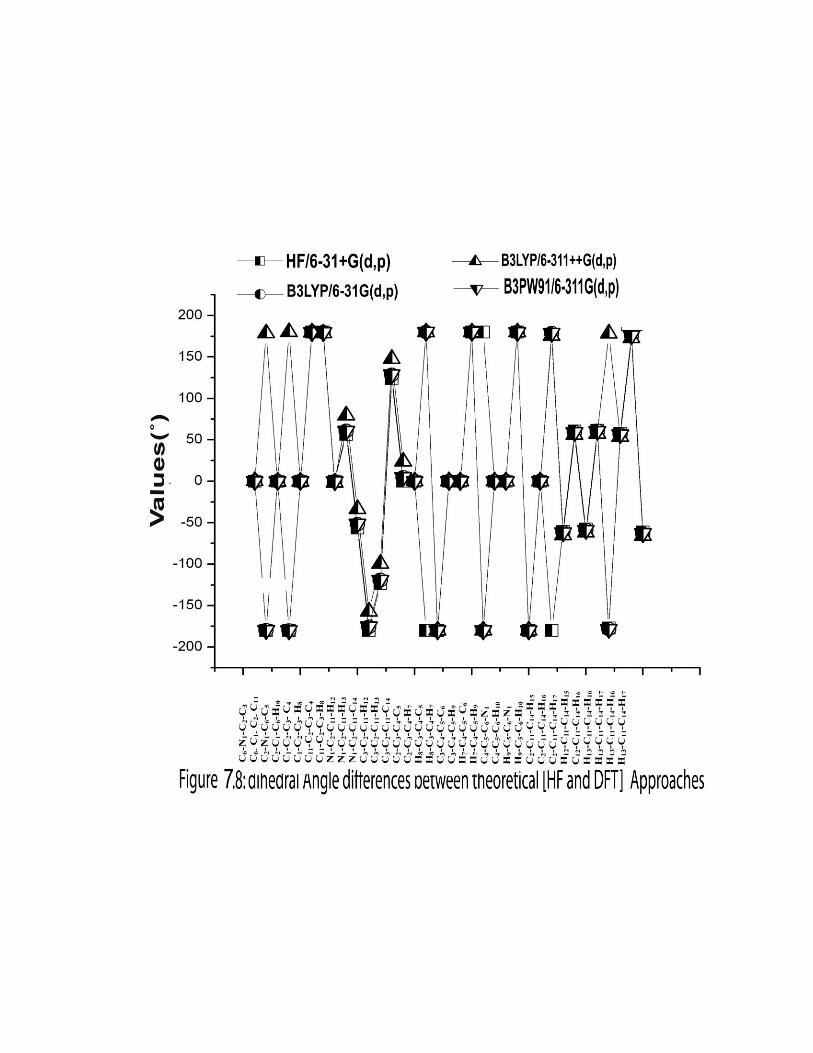
given in the Table 7.1. The comparative graphs of bond lengths, bond angles and dihedral
angles for three sets are presented in the Figures 7.6, 7.7 & 7.8.
Optimized structure yields fairly accurate bond length pairs for the bonds N1-C2
and N1-C6, C2-C3, C3-H8 and C4-H7 at all five levels of calculations. Bond lengths are
found decreasing in going from HF/6-31+G (d, p) to B3PW91/6-311G (d, p) to
B3LYP/6-311++G (d, p) to B3LYP/6-31G (d, p). According to the observed values [26],
the bond lengths N1-C2 and N1-C6 are almost equal (~1.340 Å) whereas in DFT
calculation, the bond length N1-C2 is 0.008 Å at B3LYP/6-311++G (d, p) level and

126
0.011 Å at B3PW91/6-311++G (d, p) level is greater than bond length N1-C6. This
increase in bond length is due to the substitutions and single (C-N) and double (C=N)
bonds in the ring. In accordance with the observed values, the bond lengths C2-C3 and
C5-C6 are equal (expt. 1.395 Å). The bond length C2-C3 (1.400 Å) calculated by
B3LYP/6-31G (d, p)) level is 0.05 Å lesser and C5-C6 (1.396Å) is 0.001 Å greater than
the experimental values [27-29]. This may be due to the substitution of CH2CH3 instead
of H. The bond lengths computed by B3LYP/6-311G (d, p) for the bonds C14-H15, C14-
H16 and C14-H17 are fairly accurate with experimental values [30]. The observed bond
length C11-C14 (~1.483 Å) [31] is 0.047 Å less than computed value (1.530 Å) by
B3LYP/6-311G (d, p).
Analogues to the bond length, order of the bond angle lie as C4-C5-C6<C2-N1-
C6<C3-C4-C5<C2-C3-C4<N1-C2-C3<N1-C6-C5. The pyridine ring appears to be a
little distorted with the decrease in bond angle N1-C2-C3 (121.65˚) than N1-C6-C5
(123.78˚) which is due to the substitution of ethyl group. The observed bottom ring angle
C2-N1-C6 (117.30˚) is 2.10 ˚ less than the top ring angle C3-C4-C5 (119.40˚) due to the
replacement of C by N in the ring. Also the ring carbon atom exerts a large attraction on
valence electron cloud of nitrogen which results in an increase in C-N force constant and
decrease in the corresponding bond length [32].
7.3.2. Vibrational assignments:
The title molecule belongs to CS point group of symmetry. The present molecule
has 17 atoms; hence there can be 45 normal modes of vibrations, of which 32 are in–

127
plane vibrations (A′ species) and 13 out–of–plane vibrations (A″ species). They can be
distributed as:
ΓVib = 32 A′ + 13A
″.
All the 45 fundamental vibrations are active in both Raman scattering and IR
absorption. The calculated and experimental frequency values, for different methods and
basis sets and the corresponding assignments are presented in the table 7.2. The
comparative graph of IR and Raman spectra are given in the figure 7.2 and 7.3.
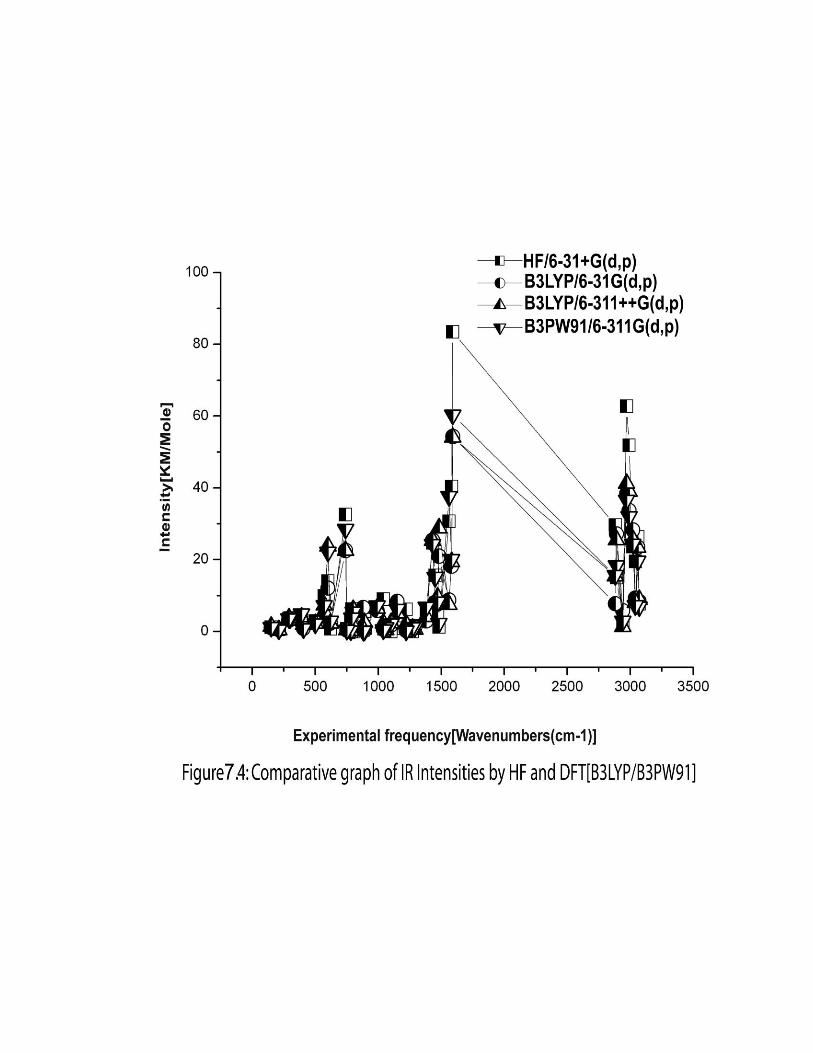
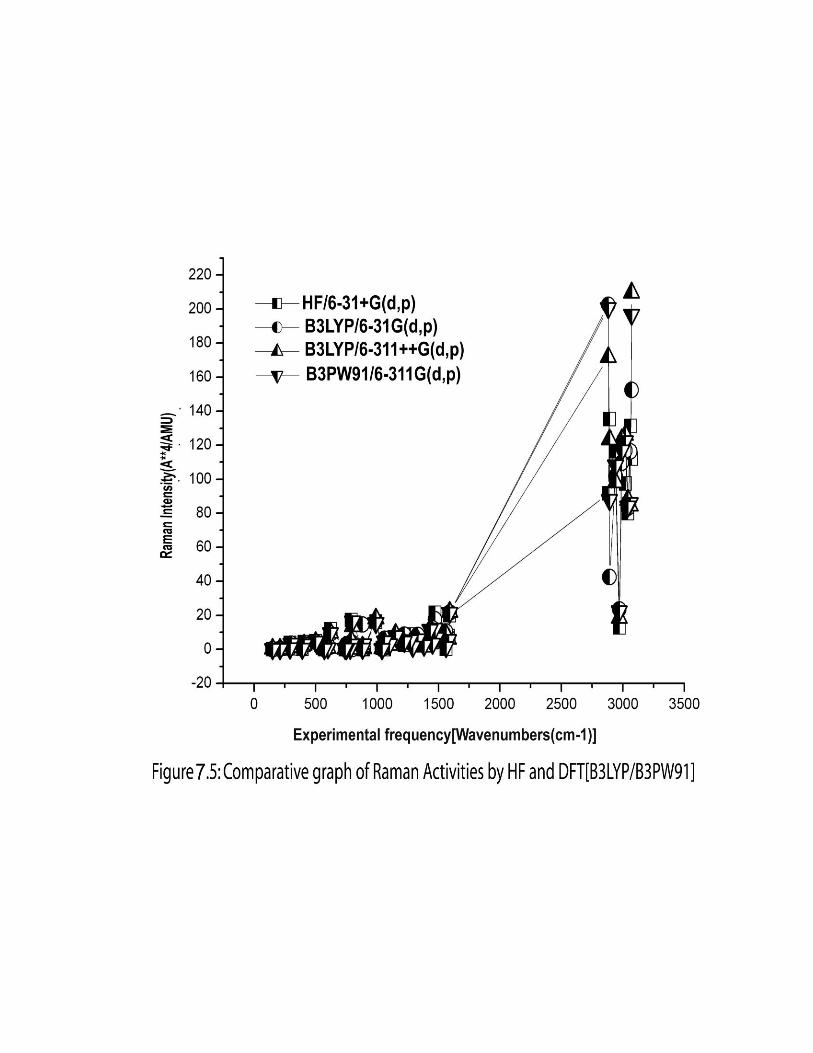
7.3.3. Computed IR intensity and Raman activity analysis:
The computed IR intensities and Raman activities of the 2-EP for different modes
of vibrations with corresponding frequencies, calculated by different methods and basis
sets are given in the Table 7.3. The IR intensity values predicted by HF methods are
found to be larger when compared to hybrid methods whereas the Raman activity values
predicted by hybrid methods are found to be larger when compared to HF. The similar
effect was also observed in the earlier work [33].The comparison of IR intensity and
Raman activity among different methods and basis sets are graphically shown in figures
7.4 and 7.5 respectively.
7.3.4. Computed vibrational frequency analysis:
The standard deviation (SD) calculation made between experimental and
computed values (HF/DFT) for the title molecule is presented in the Table 7.4. According
to the SD, the frequency deviation decrease going from HF/6-31+G (d, p) to B3LYP/6-
31G (d, p) to B3LYP/6-311++G (d, p) to B3PW91/6-311G (d, p). The deviation ratio of
HF/6-31+G (d, p) to B3LYP/6-31G (d, p) is 1.90, HF/6-31+G (d, p) to B3LYP/6-311++G

128
(d, p) is 2.03 and HF/6-31+G (d, p) to B3PW91/6-311G (d, p) is 1.97. The comparative
graph of calculated vibrational frequencies by HF and DFT methods for the title molecule
are given in the Figure 7.9 and it is found that the frequencies are calculated by B3LYP
with 6-311++G (d, p) basis set is much closer to the experimental values than HF
method.
7.3.5 C-H Vibrations
The Carbon – Hydrogen stretching vibrations give rise to bands in the region
3000 – 3100 cm-1
in all aromatic compounds [35-36]. As 2-EP is a mono substituted
hetero aromatic molecule, it has four C-H moieties. The expected four stretching
vibrations are observed at 3070, 3060, 3040 and 3020 cm-1
. These assigned values are in
good agreement with B3LYP method as well as literature data.
The bands corresponding to the C-H in-plane and out-of-plane bending vibrations
normally occur in the region 1000 – 1300 cm-1
and 750 – 1000 cm-1
respectively [37-41].
The sharp bands are observed at 1380, 1330, 1290 and 1220 cm-1
for C-H in-plane
bending vibrations. The bands with medium intensity are found at 890, 880, 800 and 790
cm-1
are assigned to C-H out-of-plane bending vibrations. Except for the first two bands
in C-H in-plane bending vibrations all the assigned values are in well within the expected
range. The deviation of the bands is due to the interaction between the ring C-H and
substituted ethyl C-H group. The assigned frequencies are found to be well above the
expected range. This shows that, though the out-of-plane bending vibrations are not

129
influenced, the in-plane bending vibrations are clearly found influenced. This is naturally
due to the presence of ethyl group in the place of methyl group in the ring.
7.3.6. Ethyl group vibrations
Vibrational spectra studies on methyl pyridine show that asymmetric and
symmetric C-H stretching vibrations are observed between 2846 and 2960 cm-1
. However
in this case, the ethyl group is attached with the ring. This give rise to five bands
associated with ethyl C-H stretching vibrations. In 2-EP, the C-H stretching vibrations are
observed at 2990, 2970, 2940, 2890 and 2880 cm-1
sequentially. The C-H in-plane
bending vibrations are found at 1150, 1100 and 1060, 1040 and 990 cm-1
. Similarly, the
C-H out-of-plane bending vibrations for ethyl group are found at 780, 750, 740, 620 and
600 cm-1
. Except for two bands in the stretching mode, two in plane bending and one in
out of plane bending the above assignments are coherent with the literature data [42-
43].This may be naturally due to the presence of n in the ring. Last two bands of out of
plane bending are moved down to the expected range. This view indicates that the ethyl
group is slightly affected. The computed values by B3LYP/6-311++G (d, p) are in perfect
match with the experimental values.
7.3.7 C-C vibrations:
The ring carbon–carbon stretching vibrations occur between the regions 1430 –
1625 cm-1
with variable intensity [44]. The bands between 1590 - 1650 cm-1
in pyridine
derivatives are usually assigned to C=C stretching modes [45]. In the present work, the
C=C stretching vibrations are observed at 1590 and 1580 cm-1
. The ring C-C stretching

130
vibrations normally occur in the region 1590-1430 cm-1
[46]. The C-C vibrations are
observed at 1480 and 1470 cm-1
sequentially. One band of C=C stretching vibration is
moved down to the expected range which is due to the substitution of ethyl group.
The C-C bonds present in the ethyl group give rise to two C-C stretching
vibrations and are observed at 1450 and 1430 cm-1
. When compared with ring C-C
vibrations, these vibrations are found to be well within the expected range. Though this
bond C-C is out of the ring, the corresponding vibrations are observed within the
literature range. The C-C-C in-plane and out-of-plane bending vibrations are observed
with the strong intensity at 570, 500 cm-1
and 390, 300 cm-1
respectively. According to
the literature [47-49], these vibrations are found to be shifted too below the expected
range. This is due to the suppression of ethyl group vibrations. The C-C in plane and out
of plane bending vibrations associated with ethyl group are observed at 290 and 150 cm-1
respectively which are also found to be deviated abruptly. This is mainly due to the C-C
bonds connected between the ring and ethyl group.
7.3.8. C-N vibrations
The existence of N in the ring give rise to a strong C=N stretching and C-N
stretching vibrations. The C=N and C-N stretching vibrations normally occur in the
region 1500 – 1600 cm-1
and 1266 – 1382 cm-1
respectively [50-53]. A strong band for
C=N stretching vibration is observed at 1560 cm-1
and C-N stretching vibration observed
at 1270 cm-1
for 2-EP. The C-N in plane and out of plane bending vibrations are observed
at 405 and 210 cm-1
. These assigned vibrational modes are apparently moved down due
to the ethyl group.

131
7.4. Conclusion:
A complete vibrational investigation on 2-Ethyl pyridine is performed by HF and
DFT (B3LYP and B3PW91) levels of theory. The observed and stimulated spectra have
shown a good frequency fit. The difference between theoretical and experimental wave
numbers within 10 cm–1
is confirmed by the qualitative agreement between the calculated
and observed frequencies. The global minimum energy between the different methods
shows the difference in optimizations between the same and the different sets. Various
quantum chemical calculations help us to identify the structural and symmetry properties
of the titled molecule. From the vibrational investigation, the following observations are
made;
1. In molecular geometry it is observed that, the bond lengths of all pairs
decrease in going from HF/6-31+G (d, p) to B3PW91/6-311G (d, p) to
B3LYP/6-311++G (d, p) to B3LYP/6-31G (d, p).
2. The pyridine ring appears to be a little distorted with the decrease in bond
angle N1-C2-C3 than N1-C6-C5 which is due to the substitution of ethyl
and methyl coupling.
3. The IR intensity values predicted by HF methods are found to be larger
when compared to hybrid methods whereas the Raman activity values
predicted by hybrid methods are found to be larger when compared to HF.
4. In computed frequency analysis, it is found that the frequencies are
calculated by B3LYP with 6-311++G (d, p) basis set is much closer to the
experimental values than HF method.

132
5. In C-H vibrations, except for the two bands in C-H in-plane bending
vibrations all the assigned values are well within the expected range. The
deviation of the bands is due to the interaction between the ring C-H and
substituted ethyl C-H group.
6. As the present molecule is mono substituted pyridine, the C-H vibrations
for ethyl group are not much affected. The computed values by B3LYP/6-
311++G (d, p) are in perfectly match with the experimental values.
7. One band of C=C stretching vibration is moved down to the expected
range which is due to the substitution of ethyl group.
8. In ethyl C-C vibrations, though this bond C-C is out of the ring, the
corresponding vibrations are observed within the literature range.

FIGURES




1
CHAPTER- IV
FT-IR and FT-Raman spectroscopic investigation on 2- Ethyl Pyridine using HF
and DFT (B3LYP and B3PW91) Calculations
4.1. Introduction
Heterocyclic nitrogen containing compounds, such as pyridine and its derivatives are
commonly present in synthetic and natural products [1-2]. The study of the vibrational spectra of
substituted pyridine mainly amino pyridine attracts the attention of many spectroscopists due to
their wide application in pharmacology and agro- chemistry. Pyridine heterocyclic compounds
and its derivatives are a repeated moiety in many large molecules with interesting photo
physical, electrochemical and catalytic applications [3-10]. They serve as good anesthetic agent
and hence are used in the preparation of drugs for certain brain disease. These pharmaceutically
acceptable sults and the pre drugs are used for the treatment (or) prevention of diabetic
neuropathy [11-12]. The methyl substitution on the CH2 group of title molecule shows some
difference in photo physical properties relative to the pyridine. 2-Ethyl pyridine constitutes an
important class of heterocyclic organic compound. Investigations on the structure of these
organic molecules have been a subject of great interest because of their peculiar photo physical
properties and pharmaceutical importance [13-15].
Adnan Sa˘glam et.al [16] have recorded the Fourier transform infrared and laser Raman
spectra of 4-Pyridine acid in the regions of 100- 4000 cm-1
, respectively. The optimized
molecular structures, vibrational frequencies and corresponding vibrational assignments of the
cis and trans conformers of 2-, 3- and 4-pyridine carboxaldehydes have been calculated using ab
initio Hartree–Fock (HF) and density functional theory (B3LYP) methods with 6-311++G(d, p)
basis set. The calculations were adapted to the CS symmetries of all the molecules. The mean
vibrational deviations between the vibrational frequency values of the two conformers of all the

2
compounds have been seen to increase while the relative energies increase and it was concluded
the more different the molecular structure of the two conformers is the higher the relative energy
is between them, and thus a bigger mean vibrational deviation.
J. Michalski et.al [17] have studied the Synthesis of 2-phenylazo-5-nitro-6-methyl-
pyridine. Synthesis of 2-phenylazo-5-nitro-6-methyl-pyridine was described. Its X-ray structure
was reported and discussed in terms of the molecular conformation of the compound. The crystal
is triclinic, space group P-1, with the unit cell parameters aZ6.372(1), bZ7.522(2), cZ12.495(2)
A° , and aZ6.372(1), bZ89.62(3)8 and gZ101.57(3)8. The pyridine and phenyl rings were planar
deflected by torsional angle JZ4.8(3)8. The crystal structure was stabilised by non-classical
hydrogen interaction of the C–H/O type with C/O distance 3.307(5) A ° , H/O distance 2.481(3)
A ° and C–H/O angle equal to 147.8(3)8. These interactions in the crystal structure couple pairs
of the molecules related by an inversion centre. FT-IR, Raman and NMR spectra of this
compound have also been measured. The 6-31G(d,p) basis set with the B3LYP functional has
been used to discuss the structure and dynamics of the compound studied.
Literature survey reveals that to the best of our knowledge no ab initio HF/DFT with 6-
311G (d, p) basis sets calculations of 2-EP have been reported so far. Hence the present work has
been undertaken to carry out a complete vibrational analysis on these molecules, based on both
experimental and theoretical study.

3
4.3. Computational details
In the present work, the HF and some of the hybrid methods such as B3LYP and
B3PW91 were carried out using the basis sets 6-31G (d, p), 6-31+G (d, p), 6-311G(d, p) and 6-
311++G (d, p). All these calculations were performed using GAUSSIAN 03W program package
on Pentium IV processor in personal computer [18-21].
The calculated frequencies are scaled down by suitable factors in comparison with the
experimental frequencies. The scaling factors are 0.903 and 0.904 for HF and in agreement with
the literature [22-23]. In the case of B3LYP with 6-31G (d, p) calculation, the scaling factors
are0.955, 0.971, 0.959, 0.939, 0.738 and 0.795; for 6-311++G (d, p)basis sets, the scaling factors
are0.961, 0.969, 0.979, 0.939, 0.789 and 0.845. In the case of B3PW91/6-311G (d, p)
calculation, the scaling factors are 0.954, 0.988, 0.849, 0.788, 0.709 and 1.04 and in good
agreement with the literature [24].
4. Results and Discussion
4.1 Molecular Geometry:
The most optimized geometries are performed by HF and DFT of 2-Ethyl pyridine
molecule with atoms numbering are shown in figure 4.1. The molecule consists of amino group
connected to a pyridine ring. The zero point vibrational energy of the molecule is 70.90, 70.54,
66.18, 65.94 and 66.17 Kcal/mol respectively as predicted by HF/6-31+G (d, p), B3LYP/6-31G
(d, p), B3LYP/6-311++G (d, p) and B3PW91/6-311G (d, p). The structural parameters; bond
lengths, bond angles and dihedral angles calculated using different basis sets are presented in
Table 4.1.

4
In comparison with the experimental values, it is observed that most of the calculated
bond length values are slightly larger than the experimental values. This may be due to the fact
that the calculations are performed for the isolated molecules (gaseous phase) while the
experimental spectra are recorded in solid phase. This is in accordance with the earlier work
[25]. Comparing bond angles and lengths of B3LYP/B3PW91 with those of HF, as a whole the
formers are bigger than later and the B3LYP/B3PW91 calculated values correlates well
compared with the experimental data. Although the differences, calculated geometrical
parameters represent a good approximation and they are the bases for calculating other
parameters, such as vibrational frequencies. Optimized geometrical parameters, namely, bond
lengths and bond angles at HF/6-31+G (d, p), B3LYP/6-31+G (d, p), B3LYP/6-311++G (d, p)
and B3PW91/6-311G (d, p) levels are given in the Table 4.1. The comparative graphs of bond
lengths, bond angles and dihedral angles for three sets are presented in the Figures 6, 7 & 8.
Optimized structure yields fairly accurate bond length pairs for the bonds N1-C2 and N1-
C6, C2-C3, C3-H8 and C4-H7 at all five levels of calculations. Bond lengths of all pairs decrease
in going from HF/6-31+G (d, p) to B3PW91/6-311G (d, p) to B3LYP/6-311++G (d, p) to
B3LYP/6-31G (d, p). According to the observed values [26], the bond lengths N1-C2 and N1-C6
are almost equal (~1.340 Å) whereas in DFT calculation, the bond length N1-C2 is 0.008 Å at
B3LYP/6-311++G (d, p) level and 0.011 Å at B3PW91/6-311++G (d, p) level is greater than
bond length N1-C6. This increase in bond length is due to the substitutions and single (C-N) and
double (C=N) bonds in the ring. In accordance with the observed values, the bond lengths C2-
C3 and C5-C6 are equal (expt. 1.395 Å). The bond length C2-C3 (1.400 Å) calculated by
B3LYP/6-31G (d, p)) level is 0.05 Å lesser and C5-C6 (1.396Å) is 0.001 Å greater than the

5
experimental values [27-29]. This may be due to the substitution of CH2CH3 instead of H. The
bond lengths computed by B3LYP/6-311G (d, p) for the bonds C14-H15, C14-H16 and C14-
H17 are fairly accurate with experimental values [30]. The observed bond length C11-C14
(~1.483 Å) [31] is 0.047 Å less than computed value (1.530 Å) by B3LYP/6-311G (d, p).
Analogues to the bond length, order of the bond angle lie as C4-C5-C6<C2-N1-C6<C3-
C4-C5<C2-C3-C4<N1-C2-C3<N1-C6-C5. The pyridine ring appears to be a little distorted with
the decrease in bond angle N1-C2-C3 (121.65˚) than N1-C6-C5 (123.78˚) which is due to the
substitution of ethyl and methyl coupling. The observed bottom ring angle C2-N1-C6 (117.30˚)
is 2.10 ˚ less than the top ring angle C3-C4-C5 (119.40˚) due to the replacement of C by N in the
ring. Also the ring carbon atom exerts a large attraction on valence electron cloud of nitrogen
resulting in an increase in C-N force constant and decrease in the corresponding bond length
[32].
4.2. Vibrational assignments:
The title molecule belongs to CS point group of symmetry and the optimized geometrical
parameters are calculated according to labeling of atoms. The present molecule has 17 atoms;
hence there can be 45 normal modes of vibrations, of which 32 are in–plane vibrations (A′
species) and 13 out–of–plane vibrations (A″ species). They can be distributed as:
ΓVib = 32 A′ + 13A
″.
All the 45 fundamental vibrations are active in both Raman scattering and IR absorption.
The calculated and experimental frequency values, for different methods and basis sets and the

6
corresponding assignments are presented in the table 4.2.The comparative graph of IR and
Raman spectra are given in the figure 4.2 and 4.3.
4.2.1. Computed IR Intensity and Raman Activity Analysis:
The computed IR intensities and Raman activities of the 2-EP for different modes of
vibrations with corresponding frequencies, calculated by different methods and basis sets are
given in the Table 4.3. The IR intensity values predicted by HF methods are found to be larger
when compared to hybrid methods whereas the Raman activity values predicted by hybrid
methods are found to be larger when compared to HF. The similar effect was also observed in
the earlier work [33].The comparison of IR intensity and Raman activity among different
methods and basis sets are graphically shown in figures 4.4 and 4.5 respectively.
4.2.2. Computed Vibrational frequency Analysis:
The standard deviation (SD) calculation made between experimental and computed
(HF/DFT) for the title molecule is presented in the Table 4.4. According to the SD, the frequency
deviation decrease is going from HF/6-31+G (d, p) to B3LYP/6-31G (d, p) to B3LYP/6-311++G
(d, p) to B3PW91/6-311G (d, p). The deviation ratio of HF/6-31+G (d, p) to B3LYP/6-31G (d, p)
is 1.90, HF/6-31+G (d, p) to B3LYP/6-311++G (d, p) is 2.03 and HF/6-31+G (d, p) to
B3PW91/6-311G (d, p) is 1.97. The comparative graph of calculated vibrational frequencies by
HF and DFT methods for the title molecule are given in the Figure 4.9 and it is found that the
frequencies are calculated by B3LYP with 6-311++G (d, p) basis set is much closer to the
experimental values than HF method.

7
4.2.3. C-H Vibrations
The Carbon – Hydrogen stretching vibrations give rise to bands in the region 3000 –
3100 cm-1
in all aromatic compounds [35-36]. As 2-EP is a mono substituted hetero aromatic
molecule, it has four C-H moieties. The expected four stretching vibrations are observed at 3070,
3060, 3040 and 3020 cm-1
. These assigned values are in good agreement with B3LYP method as
well as literature data. The bands corresponding to the C-H in-plane and out-of-plane bending
vibrations normally occur in the region 1000 – 1300 cm-1
and 750 – 1000 cm-1
respectively [37-
41]. The sharp bands are observed at 1330, 1290, 1270 and 1230 cm-1
for C-H in-plane bending
vibrations. The bands with medium intensity are found at 890, 880, 800 and 780 cm-1
are
assigned to C-H out-of-plane bending vibrations. Except for the first band in C-H in-plane
bending vibrations all the assigned values are in well within the expected range. The deviation of
first band is due to the interaction between the ring C-H and substituted ethyl C-H group. The
assigned frequencies are found to be well above the expected range. This shows that, though the
out-of-plane bending vibrations are not influenced, the in-plane bending vibrations are clearly
found influenced. This is naturally due to the interaction of ring C-H and substituted ethyl C-H
group vibrations as expected.
4.2.4. Methyl and ethyl group vibrations
Vibrational spectra studies on Methyl pyridine shows that asymmetric and symmetric C-
H stretching vibrations are observed between 2846 and 2960 cm-1
. However in this case, the
methyl group is attached with CH2 in the ring. This give rise to five bands associated with ethyl
and methyl C-H stretching vibrations. In 2-EP, the C-H stretching vibrations corresponding to
methyl group are observed at 2990, 2970 and 2940 cm-1
and corresponding to ethyl group are

8
found at 2890 and 2880 cm-1
sequentially. The C-H in-plane bending vibrations of methyl group
are found at 1150, 1100 and 1060 cm-1
and of ethyl group are found at 1040 and 990 cm-1
. The
C-H out-of-plane bending vibrations for methyl group are assigned at 780, 750 and 740 cm-1
.
The above assignments are coherent with the literature data [42-43]. As the molecule is mono
substituted pyridine, the C-H vibrations for ethyl and methyl groups are not much affected. The
computed values by B3LYP/6-311++G (d, p) are in perfectly match with the experimental
values.
4.2.5. C-C vibrations:
The ring carbon–carbon stretching vibrations occur between the regions 1430 – 1625
cm-1
with variable intensity [44]. In the present work, the C=C stretching vibrations are observed
at 1590 and 1580 cm-1
and C-C vibrations are observed at 1480 and 1470 cm-1
sequentially.
Though the 2-EP is a pyridine ring, the C-C stretching vibrations are not shifted from the
expected range [45]. The C-C-C in-plane and out-of-plane bending vibrations are observed with
the strong intensity at 620 and 500 cm-1
respectively. These assignments are also supported by
the literature [46-49]. The observed values of C=C vibrations are well within the expected range,
however the C-C vibrations are shifted down slightly which is due to the interruption of C=N
vibrations.
4.2.6. C-N vibrations
The existence of N in the ring give rise to a strong C=N stretching and C-N stretching
vibrations. The C=N and C-N stretching vibrations normally occur in the region 1500 – 1600
cm-1
and 1266 – 1382 cm-1
respectively [50-52]. A strong band for C=N stretching vibration is
observed at 1560 cm-1
and C-N stretching vibration observed at 1270 cm-1
for 2-EP. The

9
associated C=N in-plane bending vibration is shifted up by 30 cm-1
from C-N (570 cm-1
) in-plane
bending vibration. These assigned vibrational values also supported by the literature [53].
4.2.7. C- CH3 vibrations:
The asymmetric deformation of CH3 group is usually observed at around 1450 cm-1
for
methyl substituted molecules [54]. In the present compound, the CH3 deformations are observed
at 1450 and 1430 cm-1
. These assigned values are well within the expected range. Though it is
connected next to ethyl group with the ring, the C-CH2CH3 stretching vibration is observed at
1380 cm-1
. According to the literature [55-56], this absorption overlaps with C-C ring stretching
vibrations. The C-CH2 in- plane bending vibrations and C-CH2 out-of-plane bending vibrations
observed at 405 and 300 cm-1
. The CH3 and CH2 twisting vibrations are observed at 210 and 150
cm-1
which are also supported by the literature values [57-59]. Though the methyl group is
attached with the CH2, the vibrations of methyl group remain unaffected.
5. Conclusion:
A complete vibrational investigation on 2-Ethyl pyridine is performed by HF and DFT
(B3LYP and B3PW91) levels of theory. The observed and stimulated spectra have shown a good
frequency fit. The difference between theoretical and experimental wave numbers within 10
cm–1
is confirmed by the qualitative agreement between the calculated and observed frequencies.
The global minimum energy between the different methods shows the difference in
optimizations between the same and the different sets. Various quantum chemical calculations
help us to identify the structural and symmetry properties of the titled molecule. From the
vibrational investigation, the following observations are made;

10
1. In molecular geometry it is observed that, the bond lengths of all pairs decrease in
going from HF/6-31+G (d, p) to B3PW91/6-311G (d, p) to B3LYP/6-311++G (d,
p) to B3LYP/6-31G (d, p).
2. The pyridine ring appears to be a little distorted with the decrease in bond angle
N1-C2-C3 than N1-C6-C5 which is due to the substitution of ethyl and methyl
coupling.
3. The IR intensity values predicted by HF methods are found to be larger when
compared to hybrid methods whereas the Raman activity values predicted by
hybrid methods are found to be larger when compared to HF.
4. In computed frequency analysis, it is found that the frequencies are calculated by
B3LYP with 6-311++G (d, p) basis set is much closer to the experimental values
than HF method.
5. In C-H vibrations, except for the first band in C-H in-plane bending vibrations all
the assigned values are in well within the expected range. The deviation of first
band is due to the interaction between the ring C-H and substituted ethyl C-H
group.
6. As the present molecule is mono substituted pyridine, the C-H vibrations for ethyl
and methyl groups are not much affected. The computed values by B3LYP/6-
311++G (d, p) are in perfectly match with the experimental values.
7. The observed values of C=C vibrations are well within the expected range,
however the C-C vibrations are shifted down slightly which is due to the
interruption of C=N vibrations.

11
8. In C-CH3 vibrations, though the methyl group is attached with the CH2, the
vibrations of methyl group remain unaffected.
References
[1] Gilchrist .T.L, Heterocylic Chemistry, John Wiley & Sons, New York, 1988.
[2] Fallas . J.A, Gonzalez .L, Corral .I, Journal of Tetrahedron letters, 65, 2009, 232-236.
[3] Zucchi .F, Trabanelli .G, Gonzalez .N.A, Journal of Archaeological Modern Chemistry,
132, 1995, 4579.
[4] Khan .B.T, Khan . S.R.A, Annapoorna .K, Indian Journal of Chemical Society, 34 1995,
11878.
[5] Lizarraga .M.E, Navarro .R, Urriolabeitia .E.P, Journal of Organo metallic Chemistry,
542, 1997, 51-55.
[6] Georgopoulou .A.S, Ulvenlund .S, Mingos . D.M.P, Baxter .I, Williams .D.J, Journal of
Chemical Society, 4, 1999, 547-551.
[7] Liaw .W, Lee .N, Chen .C, Lee .C, Lee .G, Peng .S, Journal of American Chemical
Society, 122, 2000, 488-492.
[8] Trotter .P.J, White .P.A, Journal of Applied Spectroscopy, 32, 1978, 323-327.

12
[9] Rajpure .K.Y, Bhosale .C.H, Journal of Materials Chemistry and Physics, 64 2000, 70-
76.
[10] Licht .S, Journal of Solar Energy Materials and Solar Cells 38, 1995, 305-310.
[11] Altenburger .J.M, Lassalle .G.Y, Matrougui .M, Galtier .D, Jetha .J.C, Bocskei .Z,
Berry .C.N, Lunven .C, Lorrain .J, Herault .J.P, Schaeffer .P, O’Connor .S.E,
Herbert .J.M, Bioorganic and Medicinal Chemistry letters, 12, 2004, 1713.
[12] Camp .H, Perk .J, Hand book of American chemical society, 2000, 31-32.
[13] Mukherjee .A.K, Kumar .P, Journal of Hetrocycles, 16, 1981, 1995-1998.
[14] Mukherjee .A.K, Journal of Hetrocycles, 26, 1987, 1077-1083.
[15] Rao .Y.S, Filler .R, Chemistry of Heterocyclic Compounds, vol. 45, John Wiley and Sons
Inc., 1988, 361-365.
[16] Adnan Sa˘glam, Fatih Ucun and Vesile Guclu, Spectrochimica Acta Part A 67 2007,
465–471.
[17] J. Michalski, E. Kucharska, M. Wandas, Hanuza .J, Was´kowska .A, Ma˛czka .M,
Talik .Z, Olejniczak .S and Potrzebowski .M.J, Journal of Molecular Structure 744–
747, 2005, 377–392.
[18] Mehmet Karabacak, Dilek Karagoz, Mustafa Kurt, Spectrochimica Acta A 72 2009,
1076-1083.
[19] Schlegel .H.B, Journal of Computational Chemistry, 3, 1982, 214 -219.
[20] Gaussian 03 program, (Gaussian Inc., Wallingford CT), 2000.
[21] Frisch .M.J, Nielsen .M.B, Holder .A.J, Gauss view Users Manual, Gaussian Inc., Pitts-
burgh, PA, 2000.

13
[22] Young .D.C, Computational Chemistry: A Practical guide for applying Techniques to
Real world Problems (Electronic), John Wiley & Sons Inc., New York, 2001.
[23] Sekerci .M, Atalay .Y, Yakuphanoglu .F, Avci .D, Basoglu .A, Spectrochimica Acta Part
A 67, 2007, 503-508.
[24] Sundaraganesan .N, IIlakiamani .S, Saleem .H, Wojiciechowski .P.M, Michalska .D,
Spectrochimica Acta A 61 2005, 2995-3001.
[25] Sundaraganesan .N, Kalaichelvan .S, Meganathan .C, Dominic Joshua .D, Cornard
.J, Spectrochimica Acta Part A 71 2008, 898-906.
[26] Ming Chao, Ellory Schempp, Acta Crystallography, B33, 1977, 1557.
[27] Sverdlav .L.M, Kovner .M.A, Krainov .E.P, Vibrational Spectra of Polyatomic
Molecules, Nauka, Moscow, 1970.
[28] Sharma .S.D, Doraiswamy .S, Chemical Physics Letter, 41 1976, 192.
[29] Wei .P.G, Mak .T.C.W, Journal of Chemical Crystallography, 26 1996, 133-135.
[30] Tzeng .W.B, Narayan .K, Lin .J.L, Tung .C.C, Spectrochimica Acta 55 A 1999, 153-159.
[31] Sajan .D, Joe .H, Jayakumar .V.S, Zeleski .J, Journal of Molecular Structure, 785 2006,
43.
[32] Sundaraganesan .N, Karpagam .J, Sabastian .S, Cornard .J, Spectrochimica Acta Part A
71 2009, 11-19.
[33] Ramalingam .S, Periandy .S, Govindarajan .M, Mohan .S, Spectrochimica Acta 75A
2010, 1308-1314.

14
[34] Ramalingam .S, Periandy .S, Narayanan .B, Mohan .S, Spectrochimica Acta 76A 2010,
84-92.
[35] Varsanyi .G, vibrational spectra of Benzene Derivatives, Academic Press, NewYork,
1969.
[36] Socrates .G, Infrared Characteristic Group Frequencies, Wiley , New York, 1980.
[37] Pagannone .M, Formari .B, Mattel .G, Spectrochimica Acta A, 43 1986, 621.
[38] Prabakaran .A.R, Mohan .S, Indian Journal of Physics, 63B (4) 1989, 468 – 473.
[39] Jag Mohan, Organic Spectroscopy – Principle and Applications, second ed., Narosa
Publishing House, New Delhi, pp.30-32
[40] Kalsi .P.S, Spectroscopy of organic compounds, wiley Eastern Limited, New Delhi,
1993, p. 117.
[41] Ngabalasubramanian .P.B, Periandy .S, Mohan .S Spectrochimica Acta Part A 74, 2009,
1280 – 1287
[42] Colthup .N.B, Paly L.H, Wiberley .S.E, Introduction to Infrared and Raman
spectroscopy, Academic Press, New York, 1990
[43] Dollish .F.R, Fateley .W.G, Bentely .F.F, Characteristic Raman Frequencies on Organic
compounds, Wiley, New York, 1997.
[44] Mohan .S, SundaraGanesan .N, Mink .J, Spectrochimica Acta A 47, 1991, 1111.
[45] Krishnakumar .V, Prabavathi .N, Spectrochimica Acta Part A 72, 2009, 743 – 747.
[46] Sundaraganesan .N, Kalaichelvan .S, Meganathan .C, Dominic Joshua .B, Cornard .G
Spectrochimica Acta Part A 71, 2008, 898-906.
[47] Sathyanarayana .D.N, vibrational spectroscopy theory and application, New Age
International publishers, New Delhi, 2004.

15
[48] Periandy .S and Mohan .S, Proceedings National Academic Science India, 68 (A), III
1998.
[49] Anjaneyulu .A, Ramana Rao .G, Spectrochimica Acta A 55, 1999, 749.
[50] Singh .D.N, Singh .I.D, Indian Journal of Physics, 58B (6), 1984, 556.
[51] Colthup .N.B, Daly .L.H, Wiberley .S.E, Introduction to Infrared and Raman
Spectroscopy, Academic Press Inc., London, 1964.
[52] Bellamy .L.J, The Infrared Spectra of Complex Molecules, Chapman and Hall, London,
1975.
[53] Socrates .G, Infrared and Raman characteristics group frequencies Tables and charts, 3rd
ed, wiley, chichoster, 2001.
[54] Sundaraganesan .N, Meganathan .C, Mustafa Kurt, Journal of Molecular Structure 891,
2008, 284-291.
[55] Shanmugam .R, Sathayanarayana .D, Spectrochimica Acta A 40, 1984.
[56] Krishnakumar .V, John Xavier .R, Spectrochimica Acta Part A 60, 2004, 709-714.
[57] Green .J.H.S, Harison D.J, Kynoston .W, Spectrochimica Acta Part A 27, 1971, 807.
[58] Singh .R.N, Prasad .S.C, Spectrochimi. Acta A 34, 1974, 39.
[59] Silverstein .M, Clayton Basseler .G, Morill .C, Spectrometric Identification of Organic
Compounds, Wiley New York, 1981.

Table 7.1: Optimized geometrical parameters for 2-Ethyl pyridine computed at HF/6-31+G (d, p), B3LYP/6-31G (d, p) and 6-311++G (d,
p) and B3PW91/6-311G (d, p) basis sets
Geometrical
Parameters
Methods
HF/6-31+G(d, p) B3lyp/6-
31G(d, p)
B3lyp/6-
311++G(d, p)
B3PW91/6-
311G(d, p)
Experimental
Value
Bong length(Å)
N1 - C2 1.328 1.346 1.342 1.340 1.341
N1 - C6 1.316 1.335 1.334 1.329 1.340
C2 – C3 1.388 1.400 1.398 1.395 1.395
C2 – C11 1.514 1.517 1.515 1.509 -
C3 - C4 1.385 1.394 1.391 1.390 1.374
C3 – H8 1.072 1.084 1.082 1.083 1.081
C4 - C5 1.380 1.391 1.390 1.387 1.394
C4 – H7 1.076 1.086 1.084 1.085 1.081
C5 - C6 1.385 1.396 1.392 1.391 1.395
C5 – H9 1.074 1.085 1.083 1.084 1.081
C6 – H10 1.077 1.089 1.087 1.088 1.081
C11 –H12 1.086 1.097 1.097 1.096 1.071
C11 - H13 1.086 1.096 1.093 1.095 -
C11 - C14 1.526 1.529 1.530 1.522 1.483
C14 - H15 1.084 1.093 1.092 1.092 1.090
C14 - H16 1.085 1.095 1.093 1.094 1.090
C14 - H17 1.087 1.095 1.093 1.094 1.090
Bong Angle(˚)
C2- N1- C6 119.02 118.34 118.50 118.44 117.30
N1 -C2-C3 121.60 121.77 121.65 121.77 123.60
N1-C2-C11 114.72 114.87 115.33 114.95 -
C3-C2-C11 123.66 123.34 123.00 123.26 -
C2-C3-C4 118.96 119.23 119.29 119.21 118.50
C2-C3-H8 120.86 120.57 120.53 120.57 -
C4-C3-H8 120.17 120.18 120.16 120.20 -
C3-C4-C5 119.18 118.98 118.99 118.96 119.40
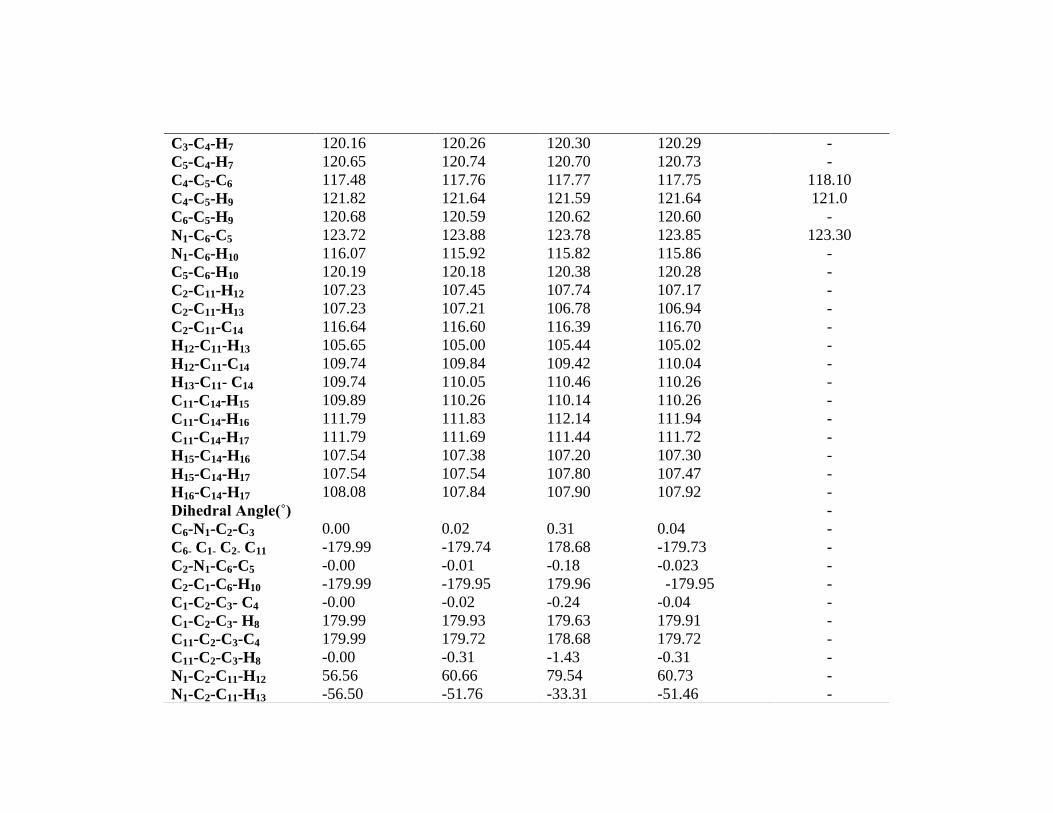
C3-C4-H7 120.16 120.26 120.30 120.29 -
C5-C4-H7 120.65 120.74 120.70 120.73 -
C4-C5-C6 117.48 117.76 117.77 117.75 118.10
C4-C5-H9 121.82 121.64 121.59 121.64 121.0
C6-C5-H9 120.68 120.59 120.62 120.60 -
N1-C6-C5 123.72 123.88 123.78 123.85 123.30
N1-C6-H10 116.07 115.92 115.82 115.86 -
C5-C6-H10 120.19 120.18 120.38 120.28 -
C2-C11-H12 107.23 107.45 107.74 107.17 -
C2-C11-H13 107.23 107.21 106.78 106.94 -
C2-C11-C14 116.64 116.60 116.39 116.70 -
H12-C11-H13 105.65 105.00 105.44 105.02 -
H12-C11-C14 109.74 109.84 109.42 110.04 -
H13-C11- C14 109.74 110.05 110.46 110.26 -
C11-C14-H15 109.89 110.26 110.14 110.26 -
C11-C14-H16 111.79 111.83 112.14 111.94 -
C11-C14-H17 111.79 111.69 111.44 111.72 -
H15-C14-H16 107.54 107.38 107.20 107.30 -
H15-C14-H17 107.54 107.54 107.80 107.47 -
H16-C14-H17 108.08 107.84 107.90 107.92 -
Dihedral Angle(˚) -
C6-N1-C2-C3 0.00 0.02 0.31 0.04 -
C6- C1- C2- C11 -179.99 -179.74 178.68 -179.73 -
C2-N1-C6-C5 -0.00 -0.01 -0.18 -0.023 -
C2-C1-C6-H10 -179.99 -179.95 179.96 -179.95 -
C1-C2-C3- C4 -0.00 -0.02 -0.24 -0.04 -
C1-C2-C3- H8 179.99 179.93 179.63 179.91 -
C11-C2-C3-C4 179.99 179.72 178.68 179.72 -
C11-C2-C3-H8 -0.00 -0.31 -1.43 -0.31 -
N1-C2-C11-H12 56.56 60.66 79.54 60.73 -
N1-C2-C11-H13 -56.50 -51.76 -33.31 -51.46 -
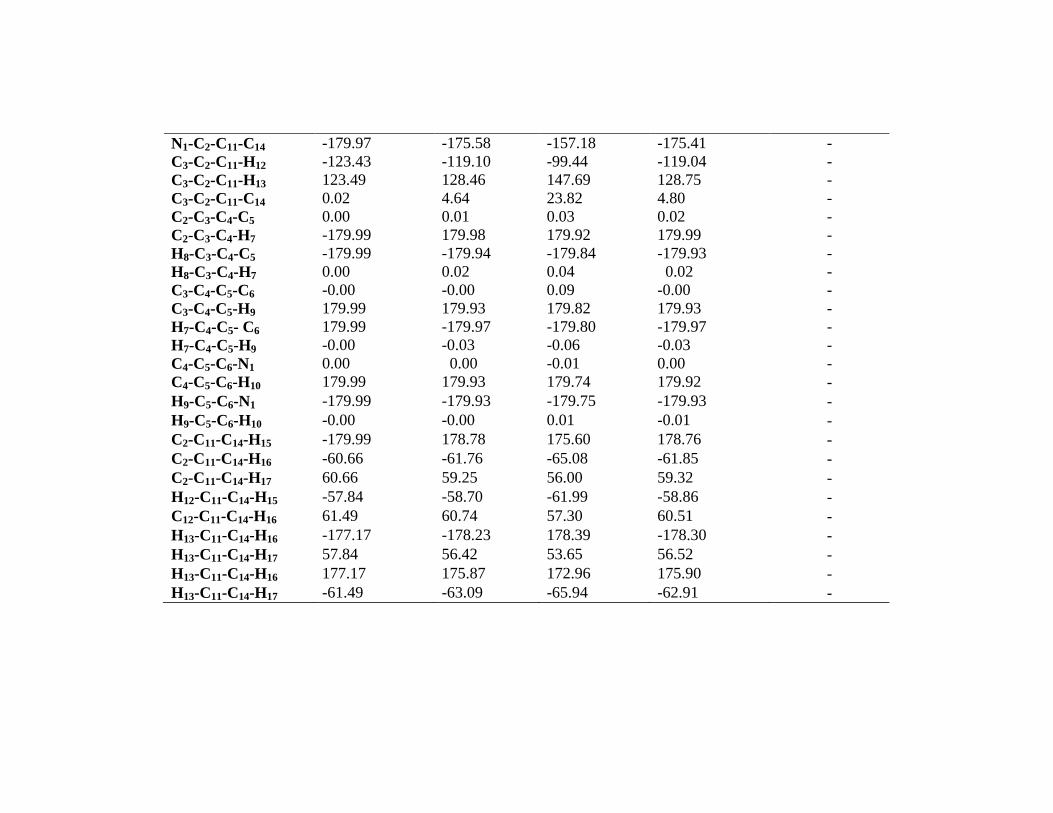
N1-C2-C11-C14 -179.97 -175.58 -157.18 -175.41 -
C3-C2-C11-H12 -123.43 -119.10 -99.44 -119.04 -
C3-C2-C11-H13 123.49 128.46 147.69 128.75 -
C3-C2-C11-C14 0.02 4.64 23.82 4.80 -
C2-C3-C4-C5 0.00 0.01 0.03 0.02 -
C2-C3-C4-H7 -179.99 179.98 179.92 179.99 -
H8-C3-C4-C5 -179.99 -179.94 -179.84 -179.93 -
H8-C3-C4-H7 0.00 0.02 0.04 0.02 -
C3-C4-C5-C6 -0.00 -0.00 0.09 -0.00 -
C3-C4-C5-H9 179.99 179.93 179.82 179.93 -
H7-C4-C5- C6 179.99 -179.97 -179.80 -179.97 -
H7-C4-C5-H9 -0.00 -0.03 -0.06 -0.03 -
C4-C5-C6-N1 0.00 0.00 -0.01 0.00 -
C4-C5-C6-H10 179.99 179.93 179.74 179.92 -
H9-C5-C6-N1 -179.99 -179.93 -179.75 -179.93 -
H9-C5-C6-H10 -0.00 -0.00 0.01 -0.01 -
C2-C11-C14-H15 -179.99 178.78 175.60 178.76 -
C2-C11-C14-H16 -60.66 -61.76 -65.08 -61.85 -
C2-C11-C14-H17 60.66 59.25 56.00 59.32 -
H12-C11-C14-H15 -57.84 -58.70 -61.99 -58.86 -
C12-C11-C14-H16 61.49 60.74 57.30 60.51 -
H13-C11-C14-H16 -177.17 -178.23 178.39 -178.30 -
H13-C11-C14-H17 57.84 56.42 53.65 56.52 -
H13-C11-C14-H16 177.17 175.87 172.96 175.90 -
H13-C11-C14-H17 -61.49 -63.09 -65.94 -62.91 -

Table 7.2: Observed and HF /6-31+G (d, p), B3LYP/6-31G (d, p), B3LYP/6-311+G (d, p) and B3PW91/6-311++G (d, p) level
calculated vibrational frequencies of 2-Ethyl Pyridine
Sl.
No.
Symmetry
species
CS
Observed
fundamentals
(cm-1
)
Calculated Frequencies (cm-1
)
Vibrational
assignments FTIR
FT
Raman
HF/6-31+G(d, p) B3LYP/6-31G
(d, p)
B3LYP/6-311++
G(d, p)
B3PW91/6-311G(d,
p)
Unscaled Scaled Unscaled Scaled Unscaled Scaled Unscaled Scaled
1 A´ - 3070 3392 3066 3217 3075 3195 3073 3208 3063 (C-H) υ
2 A´ 3060 3060 3377 3053 3208 3066 3188 3066 3201 3056 (C-H) υ
3 A´ - 3040 3348 3027 3185 3044 3165 3044 3179 3035 (C-H) υ
4 A´ 3020 3020 3335 3015 3156 3017 3137 3017 3149 3007 (C-H) υ
5 A´ 2990 2990 3254 2941 3119 2981 3092 2974 3113 2972 (C-H) υ
6 A´ 2970 2970 3253 2941 3112 2975 3090 2972 3106 2966 (C-H) υ
7 A´ 2940 2940 3224 2915 3070 2934 3065 2948 3068 2929 (C-H) υ
8 A´ 2890 2890 3197 2890 3044 2891 3025 2910 3039 2902 (C-H) υ
9 A´ 2880 2880 3183 2877 3041 2888 3011 2896 3035 2898 (C-H) υ
10 A´ 1590 1590 1804 1631 1646 1600 1632 1583 1646 1571 (C=C) υ
11 A´ 1580 1580 1780 1609 1629 1583 1614 1565 1628 1554 (C=C) υ
12 A´ 1560 - 1654 1495 1522 1479 1510 1555 1509 1580 (C=N) υ
13 A″ 1480 - 1636 1479 1516 1473 1507 1476 1501 1484 (C-C) υ
14 A″ 1470 - 1629 1473 1514 1471 1500 1470 1496 1479 (C-C) υ
15 A″ 1450 1450 1607 1453 1487 1445 1481 1451 1472 1455 (C-C) υ
16 A´ 1430 - 1596 1443 1470 1428 1462 1432 1456 1439 (C-C) υ
17 A´ 1380 - 1551 1402 1427 1387 1415 1386 1407 1391 (C-H) δ
18 A´ - 1330 1498 1354 1377 1338 1371 1343 1368 1325 (C-H) δ
19 A´ 1290 1290 1428 1291 1320 1285 1315 1288 1320 1279 (C-H) δ
20 A´ 1270 - 1402 1267 1312 1259 1297 1271 1307 1266 (C-N) υ
21 A´ 1220 1220 1355 1225 1290 1238 1278 1229 1282 1283 (C-H) δ

υ- Stretching; α– Deformation; δ-In plane bending; γ- Out plane bending; τ- Twisting:
22 A´ 1150 1150 1313 1187 1241 1166 1237 1162 1241 1149 (C-H) δ
23 A´ 1100 1100 1217 1100 1182 1111 1177 1106 1174 1087 (C-H) δ
24 A´ 1060 - 1210 1094 1131 1063 1128 1060 1126 1042 (C-H) δ
25 A´ 1040 1040 1192 1077 1109 1042 1102 1035 1100 101 (C-H) ) δ
26 A´ 990 990 1155 1044 1079 992 1072 1007 1078 998 (C-H) ) δ
27 A″ 890 1141 1031 1072 986 1065 894 1071 910 (C-H) γ
28 A″ 880 880 1129 1021 1008 802 1011 850 1011 859 (C-H) γ
29 A″ 800 - 1100 994 1006 800 1010 797 1010 797 (C-H) γ
30 A″ 790 - 1090 985 995 792 991 782 995 786 (C-H) γ
31 A″ 780 780 1058 957 976 776 980 774 980 774 (C-H) γ
32 A″ 750 - 996 900 905 720 906 766 905 769 (C-H) γ
33 A″ 740 - 884 799 813 647 815 706 812 755 (C-H) γ
34 A″ 620 - 845 764 791 629 789 623 792 625 (C-H) γ
35 A´ 600 - 844 763 765 608 764 603 764 603 (C-H) γ
36 A´ 570 570 779 704 721 573 733 579 721 569 (CCC) δ
37 A´ 500 500 685 619 638 507 640 505 635 501 (CCC) δ
38 A´ 405 405 595 538 555 410 563 444 555 394 (C-N) δ
39 A´ - 390 528 477 480 355 486 383 479 378 (CCC) γ
40 A´ - 300 460 416 425 314 417 329 426 302 (CCC) γ
41 A″ 290 290 453 410 416 307 415 327 412 292 (C-C) δ
42 A″ 210 - 311 281 281 207 267 224 276 218 (C-N) τ
43 A″ 150 150 239 216 221 163 235 185 217 171 (C-C) γ
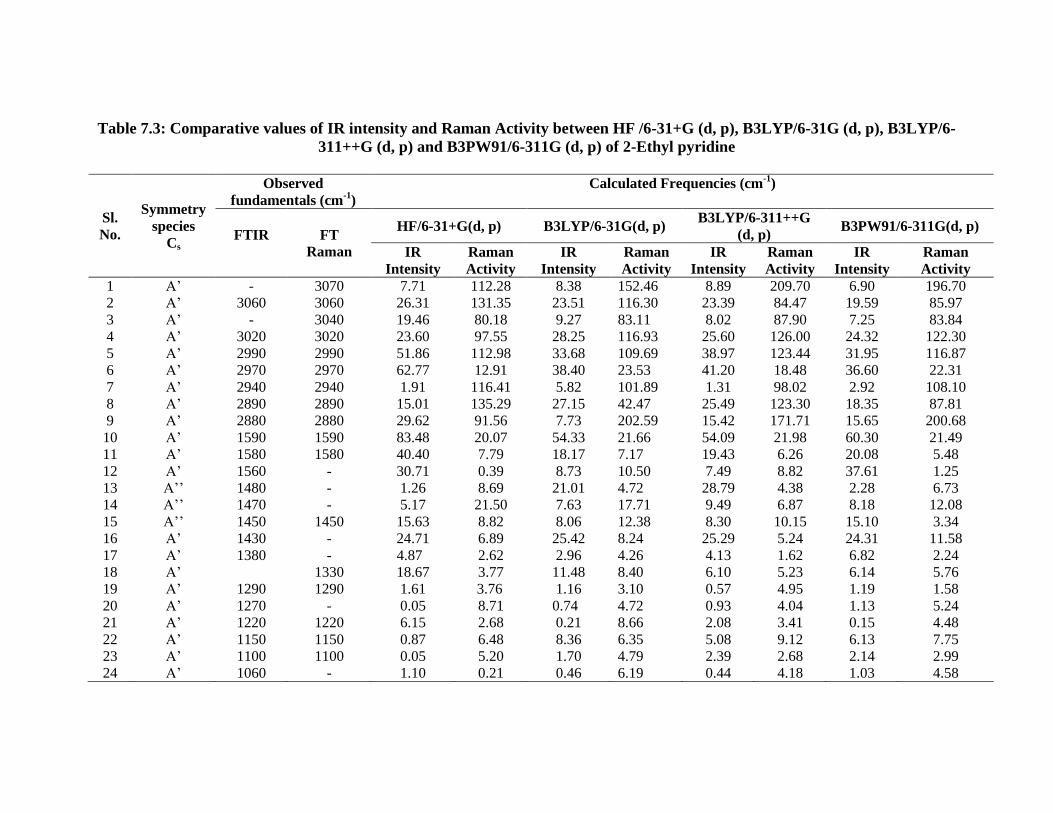
Table 7.3: Comparative values of IR intensity and Raman Activity between HF /6-31+G (d, p), B3LYP/6-31G (d, p), B3LYP/6-
311++G (d, p) and B3PW91/6-311G (d, p) of 2-Ethyl pyridine
Sl.
No.
Symmetry
species
Cs
Observed
fundamentals (cm-1
)
Calculated Frequencies (cm-1
)
FTIR
FT
Raman
HF/6-31+G(d, p) B3LYP/6-31G(d, p) B3LYP/6-311++G
(d, p) B3PW91/6-311G(d, p)
IR
Intensity
Raman
Activity
IR
Intensity
Raman
Activity
IR
Intensity
Raman
Activity
IR
Intensity
Raman
Activity
1 A’ - 3070 7.71 112.28 8.38 152.46 8.89 209.70 6.90 196.70
2 A’ 3060 3060 26.31 131.35 23.51 116.30 23.39 84.47 19.59 85.97
3 A’ - 3040 19.46 80.18 9.27 83.11 8.02 87.90 7.25 83.84
4 A’ 3020 3020 23.60 97.55 28.25 116.93 25.60 126.00 24.32 122.30
5 A’ 2990 2990 51.86 112.98 33.68 109.69 38.97 123.44 31.95 116.87
6 A’ 2970 2970 62.77 12.91 38.40 23.53 41.20 18.48 36.60 22.31
7 A’ 2940 2940 1.91 116.41 5.82 101.89 1.31 98.02 2.92 108.10
8 A’ 2890 2890 15.01 135.29 27.15 42.47 25.49 123.30 18.35 87.81
9 A’ 2880 2880 29.62 91.56 7.73 202.59 15.42 171.71 15.65 200.68
10 A’ 1590 1590 83.48 20.07 54.33 21.66 54.09 21.98 60.30 21.49
11 A’ 1580 1580 40.40 7.79 18.17 7.17 19.43 6.26 20.08 5.48
12 A’ 1560 - 30.71 0.39 8.73 10.50 7.49 8.82 37.61 1.25
13 A’’ 1480 - 1.26 8.69 21.01 4.72 28.79 4.38 2.28 6.73
14 A’’ 1470 - 5.17 21.50 7.63 17.71 9.49 6.87 8.18 12.08
15 A’’ 1450 1450 15.63 8.82 8.06 12.38 8.30 10.15 15.10 3.34
16 A’ 1430 - 24.71 6.89 25.42 8.24 25.29 5.24 24.31 11.58
17 A’ 1380 - 4.87 2.62 2.96 4.26 4.13 1.62 6.82 2.24
18 A’ 1330 18.67 3.77 11.48 8.40 6.10 5.23 6.14 5.76
19 A’ 1290 1290 1.61 3.76 1.16 3.10 0.57 4.95 1.19 1.58
20 A’ 1270 - 0.05 8.71 0.74 4.72 0.93 4.04 1.13 5.24
21 A’ 1220 1220 6.15 2.68 0.21 8.66 2.08 3.41 0.15 4.48
22 A’ 1150 1150 0.87 6.48 8.36 6.35 5.08 9.12 6.13 7.75
23 A’ 1100 1100 0.05 5.20 1.70 4.79 2.39 2.68 2.14 2.99
24 A’ 1060 - 1.10 0.21 0.46 6.19 0.44 4.18 1.03 4.58
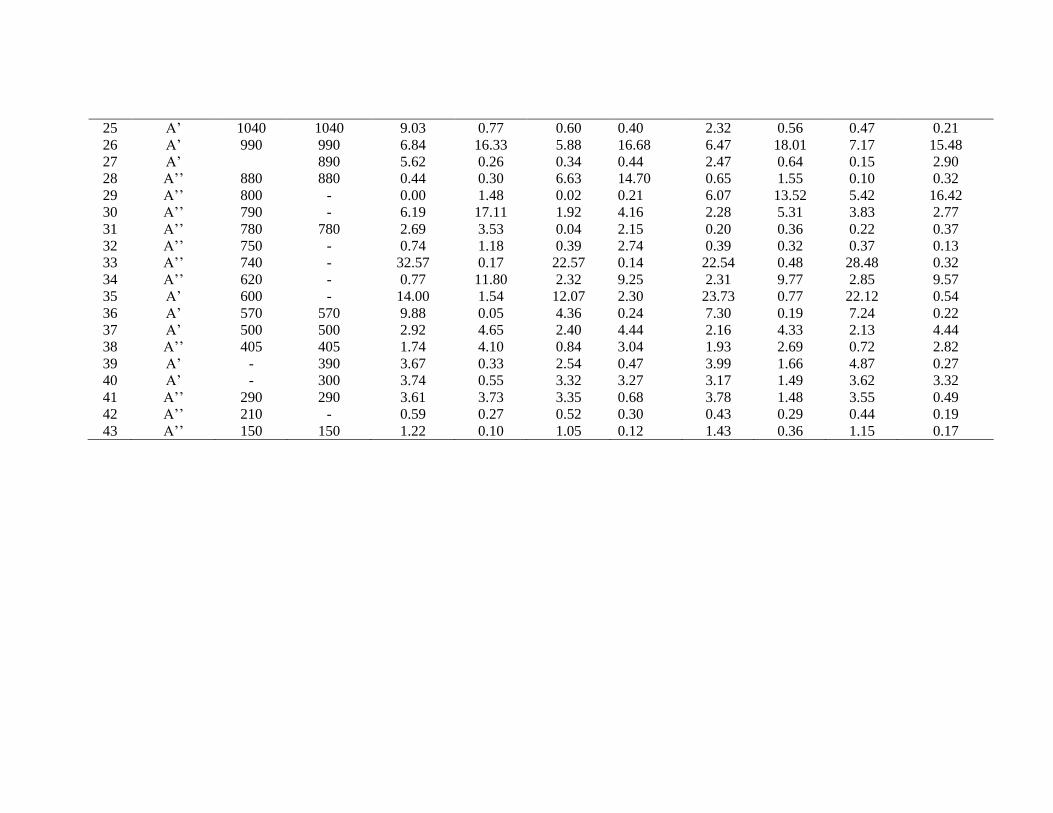
25 A’ 1040 1040 9.03 0.77 0.60 0.40 2.32 0.56 0.47 0.21
26 A’ 990 990 6.84 16.33 5.88 16.68 6.47 18.01 7.17 15.48
27 A’ 890 5.62 0.26 0.34 0.44 2.47 0.64 0.15 2.90
28 A’’ 880 880 0.44 0.30 6.63 14.70 0.65 1.55 0.10 0.32
29 A’’ 800 - 0.00 1.48 0.02 0.21 6.07 13.52 5.42 16.42
30 A’’ 790 - 6.19 17.11 1.92 4.16 2.28 5.31 3.83 2.77
31 A’’ 780 780 2.69 3.53 0.04 2.15 0.20 0.36 0.22 0.37
32 A’’ 750 - 0.74 1.18 0.39 2.74 0.39 0.32 0.37 0.13
33 A’’ 740 - 32.57 0.17 22.57 0.14 22.54 0.48 28.48 0.32
34 A’’ 620 - 0.77 11.80 2.32 9.25 2.31 9.77 2.85 9.57
35 A’ 600 - 14.00 1.54 12.07 2.30 23.73 0.77 22.12 0.54
36 A’ 570 570 9.88 0.05 4.36 0.24 7.30 0.19 7.24 0.22
37 A’ 500 500 2.92 4.65 2.40 4.44 2.16 4.33 2.13 4.44
38 A’’ 405 405 1.74 4.10 0.84 3.04 1.93 2.69 0.72 2.82
39 A’ - 390 3.67 0.33 2.54 0.47 3.99 1.66 4.87 0.27
40 A’ - 300 3.74 0.55 3.32 3.27 3.17 1.49 3.62 3.32
41 A’’ 290 290 3.61 3.73 3.35 0.68 3.78 1.48 3.55 0.49
42 A’’ 210 - 0.59 0.27 0.52 0.30 0.43 0.29 0.44 0.19
43 A’’ 150 150 1.22 0.10 1.05 0.12 1.43 0.36 1.15 0.17

Table 7.4: Standard Deviation of frequencies by HF/DFT (B3LYP/B3PW91) at 6-31G (d, p), 6-31+G (d, p), 6-31++G (d, p), 6-
311G (d, p) and 6-311++G (d, p) basis sets
S.No. Basic set levels Total
values Average
Standard
Deviation
Deviation
ratio
Experimental 58915 1370.1
1 HF/6-31+(d, p) 67727 1575.0 144.90 1.90
2 B3LYP/6-31(d, p) 63472 1476.0 76.18
3 B3LYP/6-311++(d, p) 63153 1468.6 71.33 2.03
4 B3PW91/6-311(d, p) 63270 1471.3 73.29 1.97








![Spectroscopic investigation (FT-IR, FT-Raman, UV, NMR ... · study, we report a detailed spectroscopic investigation of 2-[(acetyloxy)methyl]-4-(2-amino-9H-purin-9-yl)butyl acetate](https://img.pdfslide.us/doc/110x75/60df22b7b968a35a227444f1/spectroscopic-investigation-ft-ir-ft-raman-uv-nmr-study-we-report-a-detailed.jpg)




















