Embed Size (px)
Citation preview

TABLE OF CONTENTS8.1 Introduction8.2 The Theory of Temperature-gradient Gel
Electrophoresis8.2.1 Melting Behavior of Short Double-stranded
DNA Fragments8.2.2 Electrophoretic Mobility and the Melting State
of DNA Fragments8.2.3 Mutations Are Detectable Only in the Lowest
Melting Domain(s)8.2.4 GC- and Psoralen Clamps Extend the
Usefulness of TGGE8.3 The Practice of Temperature-gradient Gel
Electrophoresis8.3.1 Primer Design for TGGE/DGGE8.3.2 Perpendicular TGGE for the Determination of
the Tm
8.3.3 Travel Schedule Experiments8.3.4 Bipolar Clamping
8.4 Denaturing Gradient Gel Electrophoresis (DGGE)8.4.1 Optimization of Gel Running Conditions
8.5 The Use of TGGE/DGGE for Mutation Detection8.6 Detection Rate and Sensitivity8.7 Related Techniques and Variants8.8 Technical Equipment for TGGE/DGGE8.9 Applications of TGGE/DGGE and Related Methods8.10 ConclusionsAcknowledgmentsReferences
8.1 INTRODUCTION
Temperature gradient gel electrophoresis (TGGE) and the related method denaturing-gradient gel electrophoresis(DGGE) are both based on the principle that the elec-trophoretic mobility of double-stranded DNA fragments issignificantly reduced by their partial denaturation. Owing tothe sequence dependence of the melting properties of DNAfragments, sequence variations can be detected. Althoughthe sensitivity of TGGE and DGGE in detecting point
mutations in genetic disorders and other settings has beenreported to be close to 100%, these methods have neverbecome as popular as other mutation detection methods suchas SSCP (see Chapter 6), which may be related to the per-ception that it is difficult to design adequate PCR primersand set up the assays.
In this chapter, the basic principles of TGGE/DGGE willbe discussed and procedures for setting up assays will bedescribed, including how to design and test PCR primerssuitable for TGGE/DGGE analysis. Furthermore, studies onthe sensitivity of TGGE/DGGE for mutation analysis ofgenetic disorders will be reviewed and an overview of vari-ations on the basic TGGE/DGGE method will be provided.TGGE and DGGE are robust and highly sensitive methodsfor mutation screening of genetic disorders that have manyadvantages that counterbalance the extra effort required inestablishing the method.
8.2 THE THEORY OF TEMPERATURE-GRADIENT GEL ELECTROPHORESIS
8.2.1 Melting Behavior of Short Double-stranded DNA Fragments
Myers and colleagues (1985b) originally developed amethod of separating DNA fragments differing by singlenucleotide substitutions in denaturing gradient gels. Themethod was based on the notion that the denaturation(melting) of DNA fragments can be regarded as an equilib-rium for each base pair (bp) between two distinct states: 1)double helical, and 2) a more random state in which basesare neither paired nor stacked on adjacent bases in anyorderly way (Myers et al., 1987). The change from the firstto the second state is caused by increasing temperature orincreasing concentration of denaturing agents.
In the case of single-nucleotide substitutions, the replace-ment of an A :Tbp (two hydrogen bonds) by a G :C pair(three hydrogen bonds) generally will be expected toincrease the temperature at which the corresponding DNAsequence melts. The context of the nucleotide substitutionalso plays a role, and substitutions of A :T by T :A pairs, or
CHAPTER 8
Temperature and Denaturing GradientGel Electrophoresis
HARTMUT PETERS AND PETER N. ROBINSONInstitute of Medical Genetics, Charité University Hospital, Berlin, Germany
83Copyright © 2005 by Elsevier, Inc.
All rights reserved.Molecular DiagnosticsPatrinos and Ansorge

84 Molecular Diagnostics
G:C by C :G pairs, also can affect the temperature at whicha DNA sequence dissociates.
Furthermore, a DNA fragment dissociates in a stepwisefashion as the temperature is gradually increased. Dissocia-tion occurs nearly simultaneously in distinct, approximately50 to 300 nucleotide long regions, termed “meltingdomains.” All nucleotides in a given melting domain disso-ciate in an all-or-nothing manner within a narrow tempera-ture interval.
The melting temperature (Tm) indicates the temperatureat which 50% of the individual molecules are dissociated inthe given melting domain, and 50% are double helical. Asindicated earlier, the Tm is strongly dependent on the individual DNA sequence and can be altered significantly by small changes in the DNA sequence including singlenucleotide substitutions.
8.2.2 Electrophoretic Mobility and the MeltingState of DNA Fragments
TGGE is based on detecting differences in the elec-trophoretic mobility between molecules that may differ onlyat a single position. DNA fragments produced by the poly-merase chain reaction (PCR) are subjected to electrophore-sis through a linearly increasing gradient of temperature (orconcentration gradient of denaturing agents such as urea andformamide for DGGE). Nucleotide substitutions and othersmall changes in the DNA sequence are associated withadditional bands following TGGE.
The electrophoretic mobility of DNA fragments differsaccording to whether the fragment is completely doublehelical, if one or more melting domains has dissociated, orif complete dissociation to two single-stranded moleculeshas occurred. Each of these states can be visualized using aperpendicular TGGE experiment, as will be discussedfurther in Section 8.3.2.
The electrophoretic mobility of a double helical (nonde-natured) DNA fragment is not significantly altered by singlenucleotide substitutions within it, but is primarily dependenton the length and perhaps the curvature of the fragment(Haran et al., 1994). Therefore, assuming that PCR productscontain a mixture of two DNA fragments that differ at asingle position, as would be the case for a heterozygouspoint mutation, both fragments initially will progressthrough the gel at the same speed.
When the molecules reach that point in the gel where thetemperature equals their Tm, the molecules will experiencea decrease in mobility owing to a transition from a com-pletely duplex (double helical) conformation to a partiallydenatured one. Dissociation of the first or first few meltingdomains generally results in a dramatic reduction in themobility of the DNA fragment, because the fragment takeson a complex, branched conformation.
Due to the strong sequence dependence of the meltingtemperature, branching (dissociation) and consequent retar-
dation of electrophoretic mobility occurs at different levelsof the temperature gradient associated with bands at differ-ent positions in the gel (Myers et al., 1987). In addition to the two homoduplex molecules (wt/wt and mt/mt), twodifferent heteroduplex molecules (wt/mt and mt/wt) can beformed by dissociating and reannealing DNA fragmentscontaining a heterozygous mutation prior to performingTGGE (see Fig. 8.1). In practice, it is also possible toperform 40 cycles of PCR; the activity of the Taq poly-merase is exhausted in the final cycles of PCR, such that heteroduplices are formed as efficiently as is one performeddenaturation and reannealing following PCR. Heteroduplexfragments then contain unpaired bases or “bulges” in theotherwise double helical DNA, resulting in a significantreduction in the Tm of the affected melting domain (Ke andWartell, 1995). The melting temperatures of the two heteroduplex molecules are generally different from oneanother, so that each heteroduplex is separately visible in thegel. A heterozygous point mutation will thus be visualizedby the appearance of four bands: a band representing thenormal allele (homoduplex), a band representing the mutanthomoduplex that will lie above or underneath the wild-typehomoduplex band, depending on the effect of the mutationon the Tm, and two heteroduplex bands that are always abovethe homoduplex bands (see Fig. 8.2; Myers et al. 1987).Mutant and wild-type homoduplex bands generally are sep-arated by 2–10mm, and the heteroduplex bands are oftenthree or more cm above the homoduplex bands.
8.2.3 Mutations Are Detectable Only in the Lowest Melting Domain(s)
In the preceding discussion, a significant issue is that muta-tions are detectable only in the melting domain(s) with thelowest melting temperature. If, however, a DNA moleculecontains several melting domains with different melting
FIGURE 8.1 Mechanism of heteroduplex formation. In the caseof heterozygous point mutations in genetic disorders, PCR pro-duces two alleles differing only at the position of the point muta-tion. A wild-type (AA) and a mutant (aa) molecule are present atan approximately 1 :1 ratio. Denaturation followed by reannealingof these molecules produces both wild-type (AA) and mutant (aa)homoduplex molecules, as well as two heteroduplex molecules,consisting of a wild-type and a mutant strand (Aa and aA).

temperatures, it is generally not possible to visualize muta-tions located elsewhere than in the melting domain with thelowest Tm. Once the DNA fragments reach the temperatureat which the first melting domain dissociates, the mobilityof the fragment is greatly reduced so that it may not reachtemperatures relevant for the higher Tm domains under the conditions of the experiment. Also, dissociation of thehighest Tm domain results in complete dissociation of theDNA fragment into two single-stranded DNA molecules.Single-stranded DNA, like completely double helical DNA,does not demonstrate differences in electrophoretic mobil-ity, owing to small sequence changes, and hence there is no
possibility of distinguishing two sequences once completedissociation has occurred.
The consequence of these observations is that only muta-tions in the lowest Tm domain can be detected reliably byTGGE or DGGE (Myers et al., 1987).
8.2.4 GC- and Psoralen Clamps Extend the Usefulness of TGGE
Myers and colleagues (1985a) presented an extension of theoriginal DGGE protocol that allowed mutations in everyregion of the DNA fragment under analysis to be detected.These researchers attached a 135bp, GC-rich sequence,known as GC-clamp, to the b-globin promoter region inwhich mutations were being sought. The b-globin promoterregion was found to contain two melting domains; withoutthe GC-clamp, only mutations in the domain with the lowerTm could be visualized in the gel. Owing to its high GCcontent, the GC-clamp has a significantly higher meltingtemperature than most naturally occurring sequences. Theattachment of the GC-clamp was found to significantly alterthe melting properties of the b-globin sequence and muta-tions in the entire b-globin sequence could be experimen-tally detected (Myers et al., 1985a). By adding a 40nt G+Crich sequence to one of the two PCR primers, a GC-clampcan conveniently be added to any DNA fragment producedby PCR (Sheffield et al., 1989). It is also possible to use auniversal GC-clamp that is incorporated into amplified DNAfragments during PCR, thereby avoiding the expense of syn-thesizing long primers (Top, 1992).
Psoralen-modified PCR primers are an alternative to GC-clamps. One of the two PCR primers is 5¢ modified by 5-(w-hexyloxy)-psoralen. The 5¢ terminus of the primer shouldhave two adenosine residues; if the natural sequence doesnot have AA, this sequence should be appended to the spe-cific DNA sequence of the primer. Psoralens are bifunctionalphotoreagents that can form covalent bonds with pyrimidinebases (especially thymidine). If intercalated at 5¢-TpT indouble helical DNA (this will be the complementarysequence of the 3¢ terminus of the other strand followingPCR), psoralen forms a covalent bond with thymidine afterphotoinduction (Costes et al., 1993b). Photoinduction canbe performed by exposing to the PCR products to a sourceof UV light (365) for 5 to 15 minutes, which can be doneconveniently in the original PCR tubes or 96-well plates.
In general, psoralen clamping provides comparableresults to GC clamping, except that cross-linking of the PCRfragments is only approximately 85% efficient, so that one observes single-stranded, denatured DNA fragmentsrunning below the main bands in the TGGE. Psoralenclamping sometimes is preferred over GC-clamping becausethe PCR is often easier to optimize, and bipolar clamping ispossible if necessary (see section 8.3.4). Psoralen modifica-tion of primers is available from many commercial oligonu-cleotide sources.
CHAPTER 8 • Temperature and Denaturing Gradient Gel Electrophoresis 85
FIGURE 8.2 Parallel TGGE/DGGE. Mutation screening gener-ally is performed with the temperature or denaturing gradient par-allel to the direction of electrophoresis. In this example, results ofelectrophoresis from top to bottom for a hypothetical family seg-regating an autosomal recessive disorder are shown. Cases 3 and4 are normal, carrying only the wild-type allele (d). Cases 2, 5, and6 are heterozygous for a point mutation resulting in the appearanceof an additional homoduplex band (c), as well as two additionalheteroduplex bands (a and b). Case 1, who is homozygous for themutation, shows just the mutant homoduplex band (c).
86 Molecular Diagnostics
8.3 THE PRACTICE OF TEMPERATURE-GRADIENT GEL ELECTROPHORESIS
Detailed protocols for TGGE and DGGE are available else-where (Kang et al., 1995; Murdaugh and Lerman, 1996). Inthe following sections, the most important issues concern-ing how to set up TGGE or DGGE assays successfully arediscussed, including especially the issues related to primerdesign and optimization procedures. Several points thatapply only to DGGE are discussed in Section 8.4.
8.3.1 Primer Design for TGGE/DGGE
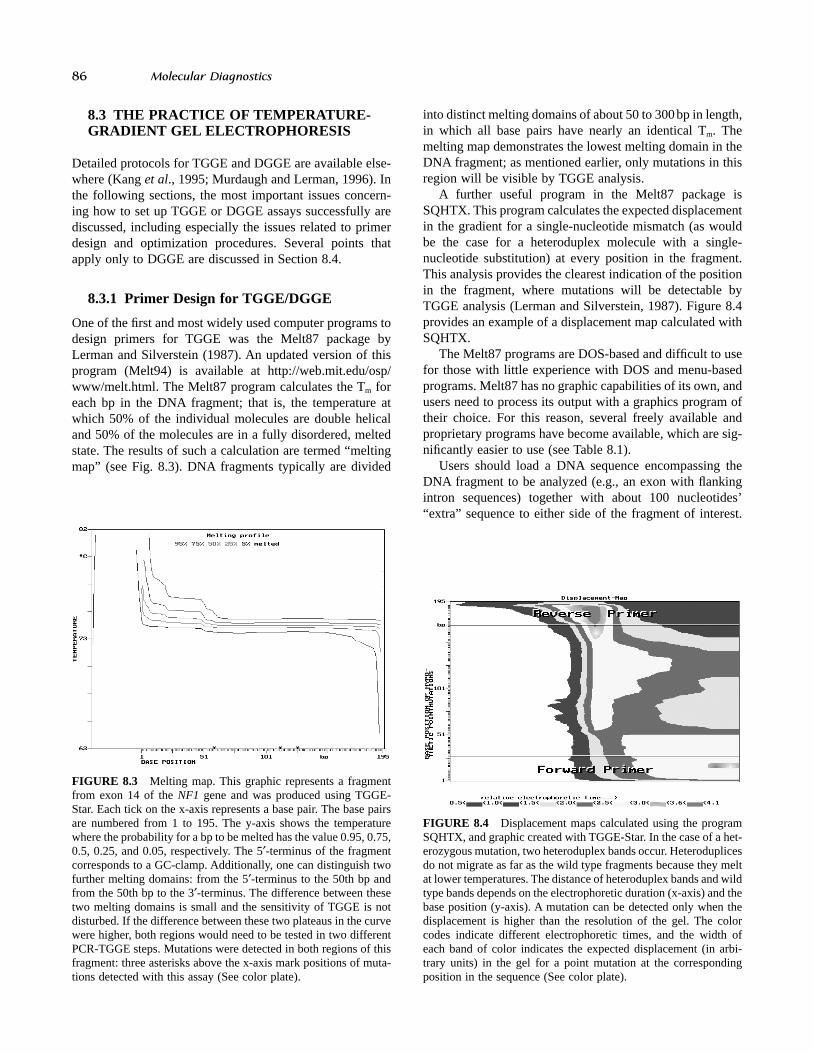
One of the first and most widely used computer programs todesign primers for TGGE was the Melt87 package byLerman and Silverstein (1987). An updated version of thisprogram (Melt94) is available at http://web.mit.edu/osp/www/melt.html. The Melt87 program calculates the Tm foreach bp in the DNA fragment; that is, the temperature atwhich 50% of the individual molecules are double helicaland 50% of the molecules are in a fully disordered, meltedstate. The results of such a calculation are termed “meltingmap” (see Fig. 8.3). DNA fragments typically are divided
into distinct melting domains of about 50 to 300bp in length,in which all base pairs have nearly an identical Tm. Themelting map demonstrates the lowest melting domain in theDNA fragment; as mentioned earlier, only mutations in thisregion will be visible by TGGE analysis.
A further useful program in the Melt87 package isSQHTX. This program calculates the expected displacementin the gradient for a single-nucleotide mismatch (as wouldbe the case for a heteroduplex molecule with a single-nucleotide substitution) at every position in the fragment.This analysis provides the clearest indication of the positionin the fragment, where mutations will be detectable byTGGE analysis (Lerman and Silverstein, 1987). Figure 8.4provides an example of a displacement map calculated withSQHTX.
The Melt87 programs are DOS-based and difficult to usefor those with little experience with DOS and menu-basedprograms. Melt87 has no graphic capabilities of its own, andusers need to process its output with a graphics program oftheir choice. For this reason, several freely available andproprietary programs have become available, which are sig-nificantly easier to use (see Table 8.1).
Users should load a DNA sequence encompassing theDNA fragment to be analyzed (e.g., an exon with flankingintron sequences) together with about 100 nucleotides’“extra” sequence to either side of the fragment of interest.
FIGURE 8.3 Melting map. This graphic represents a fragmentfrom exon 14 of the NF1 gene and was produced using TGGE-Star. Each tick on the x-axis represents a base pair. The base pairsare numbered from 1 to 195. The y-axis shows the temperaturewhere the probability for a bp to be melted has the value 0.95, 0.75,0.5, 0.25, and 0.05, respectively. The 5¢-terminus of the fragmentcorresponds to a GC-clamp. Additionally, one can distinguish twofurther melting domains: from the 5¢-terminus to the 50th bp andfrom the 50th bp to the 3¢-terminus. The difference between thesetwo melting domains is small and the sensitivity of TGGE is notdisturbed. If the difference between these two plateaus in the curvewere higher, both regions would need to be tested in two differentPCR-TGGE steps. Mutations were detected in both regions of thisfragment: three asterisks above the x-axis mark positions of muta-tions detected with this assay (See color plate).
FIGURE 8.4 Displacement maps calculated using the programSQHTX, and graphic created with TGGE-Star. In the case of a het-erozygous mutation, two heteroduplex bands occur. Heteroduplicesdo not migrate as far as the wild type fragments because they meltat lower temperatures. The distance of heteroduplex bands and wildtype bands depends on the electrophoretic duration (x-axis) and thebase position (y-axis). A mutation can be detected only when thedisplacement is higher than the resolution of the gel. The colorcodes indicate different electrophoretic times, and the width ofeach band of color indicates the expected displacement (in arbi-trary units) in the gel for a point mutation at the correspondingposition in the sequence (See color plate).

The previously mentioned programs can be used to findprimers that result in a DNA fragment with melting proper-ties adequate for TGGE or DGGE. In general, some amountof trial and error is needed to find optimal primers for anygiven sequence. Users need to decide both the position ofthe forward and reverse primers as well as whether the GC-clamp is to be placed on the 5¢ or 3¢ PCR primer or both (seelater for a discussion of bipolar clamping). Programs suchas TGGE-Star and MELTingeny facilitate this process byallowing users to easily shift primer positions and recalcu-late the melting maps. It should be mentioned that a 40-nucleotide GC-clamp can be substituted for a psoralenclamp in the computer analysis.
8.3.2 Perpendicular TGGE forthe Determination of the Tm
In most cases in which TGGE is used for mutation analysis,parallel electrophoresis with simultaneous analysis of mul-tiple samples will be performed. For each such assay, theoptimal temperature gradient and run time must be deter-mined experimentally. The procedures used for this purposeare described in this and the following section.
The optimization process begins with a perpendicularTGGE experiment, in which electrophoresis is performedperpendicularly to the temperature gradient (see Fig. 8.5).Perpendicular TGGE is used to verify the reversible meltingbehavior of the DNA fragment and to determine its Tm underthe experimental conditions. Perpendicular TGGE is runwith a gradient of 20°C–60°C, which will be adequate forthe vast majority of PCR fragments. Electrophoresis is ini-tially performed at room temperature for 10–15 minutes torun the sample into the gel. Then, electrophoresis is stoppedwhile a temperature gradient of 20°C–60°C is established,after which electrophoresis should be continued for 90–120minutes. Figure 8.5 demonstrates the use of this analysis todetermine the Tm of the DNA fragment being analyzed.
8.3.3 Travel Schedule Experiments
Up to three novel bands are observed upon TGGE/DGGEanalysis of a heterozygous mutation or polymorphism. The
separation will begin to become apparent when the het-eroduplex molecules have reached their Tm, as their mobil-ity will be retarded by partial denaturation. Separation of thehomoduplex molecules will occur in a region of the gradi-ent surrounding the Tm of the lowest melting domain of theDNA fragment. Therefore, TGGE assays are set up to avoida long running time before the samples reach the effectiverange of separation. One should choose the temperature gra-dient such that the effective range of separation is approxi-mately in the middle or somewhat above the middle of thegel, and that the upper and lower temperature ranges are sep-arated by about 15°C from the Tm of the DNA fragment.
Once an appropriate temperature gradient has beenchosen, the optimal running time can be determined by atravel-schedule experiment—a parallel TGGE experiment inwhich samples are applied every 30 minutes for three hours(or longer), such that the last sample to be loaded has run30 minutes, and the first sample, three hours. Usually, onewill see a reduction on electrophoretic mobility of samplesafter a certain period of time (generally 60 to 90 minutes ifthe temperature gradient was chosen correctly). Samplesoften do not continue to wander in the gel with any signifi-cant velocity once their melting temperature has beenreached. These gels generally are run for about 30 minuteslonger than the time determined in this manner (see Fig.8.6). Different choices of the range and starting point of thetemperature gradient affect both the range in the gel at whichmutations will be visible as well as the optimal running time(see Fig. 8.7).
8.3.4 Bipolar Clamping
Occasionally, TGGE analysis will result in fuzzy bands thatare difficult to evaluate, despite apparently adequate meltingbehavior, as predicted by Melt94 or other programs. Bipolarclamping of PCR products, by means of attaching a psoralenclamp to each of the two PCR primers rather than just one,is an efficacious method to improve melting characteristicsof PCR fragments that are otherwise not amenable toTGGE/DGGE analysis (Gille et al., 1998). Bipolar clamp-ing is a simple procedure that can significantly improve
CHAPTER 8 • Temperature and Denaturing Gradient Gel Electrophoresis 87
TABLE 8.1 Programs for the design of PCR primers for use in TGGE/DGGE
Name Comment URL
Melt94 DOS-based http://web.mit.edu/osp/www/melt.html
TGGE-Star DOS-based, freely available user-friendly wrapper for http://www.charite.de/bioinf/tgge/Melt87 (Gille and Gille, 2002)
Poland Server-based implementation of Poland’s algorithm http://www.biophys.uni-(Steger, 1994) duesseldorf.de/local/POLAND/poland.html
MELTingeny A commercial, Java-based GUI program with flexible http://www.ingeny.comroutines for designing DGGE/TGGE primers
WinMelt, MacMelt Commercial GUI programs for melting profile analysis http://www.medprobe.com/uk/melt.html

88 Molecular Diagnostics
results of TGGE analysis in cases where analysis with onlyone clamp has yielded suboptimal results. Programs such asTGGE-Star (Gille and Gille, 2002) offer the possibility ofcomputer analysis with two clamps, and may suggest the useof bipolar clamping for amplicons whose predicted meltingproperties are otherwise not satisfactory.
8.4 DENATURING GRADIENT GELELECTROPHORESIS (DGGE)
The theory of DGGE/TGGE is described in detail in the firstpart of this chapter. Parallel DGGE is a form of polyacry-
lamide gel electrophoresis in which a double-stranded DNAfragment migrates into a gradient of linearly increasingdenaturing conditions. The denaturing gradient is function-ally equivalent to the temperature gradient of TGGE. Thedenaturants used are heat (a constant temperature of gener-ally 60°C) and a fixed ratio of formamide (ranging from0–40%) and urea (ranging from 0–7M). The temperature of60°C was empirically chosen to exceed the melting temper-ature of an AT-rich DNA fragment in the absence of a denat-urant. For extremely GC-rich DNA sequences, highertemperatures (e.g., 75°C) can be used. To achieve a uniformtemperature distribution the electrophoresis unit is attachedto a circulating water bath.
FIGURE 8.5 a) Schematic drawing of a perpendicular TGGE/DGGE gel. A temperature gradient from t1 (e.g., 20°C) to t2 (e.g., 60°C)is established perpendicularly to the direction of electrophoresis (indicated by - and +). Fragments at lower temperatures remain completelydouble helical and have a relatively high electrophoretic mobility (a). Once the melting temperature of the lowest-temperature meltingdomain is reached, partial denaturation of the DNA fragment (b) causes a significant reduction of electrophoretic mobility. The tempera-ture at which 50% of individual molecules are melted is denoted as the melting temperature (Tm), and is indicated by the arrow in the figure(b). A reversible denaturation step is observed as a continuous transition (curve). Once the temperature of the highest-melting domain isreached, irreversible melting occurs, causing a discontinuous transition in the melting curve (c). b) Perpendicular TGGE gel. In this example,a PCR fragment corresponding to NF1 gene exon 14 was analyzed. PCR product was applied and run into the gel at 10°C for 15 minutes.Then, a temperature gradient from 20°C to 60°C was established perpendicularly to the direction of electrophoresis, which was then per-formed for an additional 60 minutes. One observes a high electrophoretic mobility in portions of the gel with temperatures below the Tm
of the fragment. The gradual decrease in mobility around the middle of the gel indicates reversible melting of the lowest-temperature meltingdomain. In portions of the gel with temperatures above the Tm of the fragment, partial denaturation of the fragment leads to a significantlyreduced electrophoretic mobility. The arrow at the midway point of the curve indicates the Tm of the fragment under the experimental con-ditions (approximately 39°C).
(a)
(b)

8.4.1 Optimization of Gel Running Conditions
The computer programs (e.g., Melt94) described earlierreduce the number of preliminary experiments required foroptimization of the gel running conditions. However, it isstill necessary to run some preliminary gels to determine theoptimal electrophoresis conditions and running times and toconfirm that the optimal denaturing gradient has beenchosen. The aim of these travel schedule gels is to have well-separated bands (normal and mutation positive control aresimultaneously loaded on the gels) that are focused by thegradient. PCR products with two low-melting domainsrequire different gel conditions for the analysis of eachdomain.
The choice of the denaturant concentration range can bedetermined as follows. The differences in gradient depth (thedisplacement) between a fragment and the same fragmentwith a change at a specified bp are calculated by the program
SQHTX (Lerman and Silverstein, 1987) as described inSection 8.3.1. SQHTX calculates the displacement as thedifference in temperature at which the wild-type homoduplexand the heteroduplex molecules partially melt (see Fig. 8.4).To convert between the temperature values and the denatu-rant concentration, a difference of 1°C is converted to a dif-ference of 3% denaturant concentration (approximatelyequivalent to 1cm distance within a 20% urea gradient gel).An experimental determination of gradient behavior can be achieved by perpendicular gel electrophoresis. Data from the perpendicular gels help to estimate the denaturantconcentration range to use in parallel gel electrophoresis.For parallel gels, the gradient initially should be chosen witha 25% to 30% difference in denaturant concentration cen-tered around the melting temperature of the domain (Myerset al., 1987). Once optimized gel running conditions havebeen established, the method can be used for mutationscreening.
CHAPTER 8 • Temperature and Denaturing Gradient Gel Electrophoresis 89
FIGURE 8.6 Travel schedule experiment. This experiment is used to determine the optimal running time of a TGGE experiment. Frag-ments, corresponding to exon 19a of the NF1 gene, in which one of the primers was modified with psoralen (see Section 8.2.4), were appliedat intervals of 30 minutes, such that the first fragments had a total running time of 5 hours, and the last fragments to be applied had a runningtime of 60 minutes. Lanes labeled U contain PCR fragments that were not UV-irradiated to effect psoralen-mediated crosslinking, and laneslabeled N (two lanes were loaded for each timepoint) contain irradiated PCR fragments. One sees that the fragments initially are completelydouble-helical (1h), such that irradiated and nonirradiated fragments display the same band pattern. Starting at the Tm of this fragment(40°C), the nonirradiated fragments (U) undergo complete dissociation so that only a single-strand band running well below the main bandof the irradiated (cross-linked) fragments is visible (compare the time points at 2.5 and 3 hours. Additionally, the irradiated fragments (N)show a large reduction in electrophoretic mobility following partial denaturation at about 40°C. Under these conditions, an optimal runningtime would be 3 hours, although the running time could be reduced by adjusting the temperature gradient (see also Fig. 8.7). (See colorplate)

90 Molecular Diagnostics
specificity of TGGE/DGGE is exquisitely high. In otherwords, a false-positive four-band pattern occurs rarely if atall.
8.6 DETECTION RATE AND SENSITIVITY
By using DGGE Myers and colleagues (1985b) detected anestimated 40% of the sequence variants in a DNA fragmentup to 500bp in their initial study. The use of GC-clamps,psoralen clamps, or bipolar clamping, which aid the forma-tion of uniform low melting domains, significantly improvedthe detection rate of TGGE/DGGE, which in many casesapproaches nearly 100%.
The sensitivity of TGGE/DGGE for detecting knownmutations is generally reported to be nearly 100%, gener-ally performing as well as or better than other mutationdetection methods (Abrams et al., 1990; Ferec et al., 1992;Gelfi et al., 1997; Gejman et al., 1998; Tchernitchko et al.,1999; Zschocke et al., 2000; Breton et al., 2003). In onestudy with a panel of known mutations, DGGE detected 201of 201 known mutations in the CFTR gene (Macek et al.,1997). The reasons for lower reported detection rates ofunknown mutations in some studies has been speculated tobe due to genetic heterogeneity (Ferec et al., 1999), clinicaloverdiagnosis (Katzke et al., 2002), or location of mutationsin intronic or promoter regions that were not included in thescreening program. Optimization of the TGGE/DGGE assayconditions and primers, perhaps including the use of bipolarclamping (Gille and Gille, 2002), may increase sensitivity.In summary, the sensitivity of TGGE/DGGE, when properlyused, is close to 100%.
TGGE/DGGE also has been shown to be very sensitivein the detection of mutations in situations where the muta-tion sequence is present in proportions less than 50% (as isgenerally the case when heterozygous mutations are soughtin genomic DNA). This has proved useful in detection ofheteroplasmy in mitochondrial disorders with heteroplasmicproportions as low as 1% (Tully et al., 2000), as well as intesting for residual disease in cancer (Ahnhudt et al., 2001;Alkan et al., 2001).
FIGURE 8.7 The effect of different temperature gradients onseparation of samples. A relatively wide gradient (a) leads to a rel-atively small separation of homoduplex and heteroduplex bands inthe case of a heterozygous mutation, while a shallower gradient (b)increases the separation. The lower temperature of the gradient canalso be adjusted to reduce the amount of time needed before theTm of the lowest melting domain is reached.
FIGURE 8.8 A heterozygous point mutation in exon 31 of theNF1 gene detected by TGGE. The third and fifth lanes display a classical four-band appearance due to the presence of the heterozygous mutation 5839CÆT.
8.5 THE USE OF TGGE/DGGE FORMUTATION DETECTION
TGGE and DGGE have been used to investigate a largenumber of disease genes, some of which are listed in the fol-lowing sections. Due to the relative ease of detecting het-erozygous mutations owing to the occurrence of up to threenovel bands, TGGE and DGGE have been particularlyuseful for disorders characterized by heterozygous muta-tions or frequent de novo mutations (reviewed in Fodde andLosekoot, 1994). In light of the effort involved in designingprimers and optimizing conditions, TGGE or DGGE gener-ally is reserved for situations when large numbers of samplesare to be screened for mutations.
Most mutation screening protocols involve the simulta-neous analysis of 24 or more samples on one parallelTGGE/DGGE. In general, altered band patterns are easy tospot. The classic appearance of heterozygous mutations (seeFigs. 8.2 and 8.8) is due to the appearance of three addi-tional bands. With some mutations, only one or two addi-tional bands are seen. In the authors’ experience, the

8.7 RELATED TECHNIQUES AND VARIANTS
A wide range of improvements and further developments ofthe principles underlying DGGE and TGGE have appearedin the last decade, the most important of which are brieflysummarized here.
Broad range DGGE. A single gel and a single set of conditions is used to screen all the exons of one gene(Guldberg and Guttler, 1994; Hayes et al., 1999).
Multiplex DGGE. Several exons are simultaneously ana-lyzed in one DGGE gel (Costes et al., 1993a).
Genomic DGGE (gDGGE). Genomic DNA is digestedwith a restriction enzyme, electrophoresed by DGGE,transferred to nylon membrane, and hybridized to aunique DNA probe (Borresen et al., 1988).
Constant DGGE (cDGGE). Gels contain constant con-centrations of denaturants. This allows an increased res-olution of mutant fragments since they will constantlymigrate with a different electrophoretic mobility throughthe whole length of the gel (Hovig et al., 1991).
Constant denaturant capillary electrophoresis (CDCE).DNA migrates through a 30cm quartz capillary of 75 mminner diameter, filled with a viscous polyacrylamide solu-tion. A 10cm part of the capillary, prior to the detector,is heated to a temperature permitting partial melting (seealso previous chapter). Usually the DNA is fluorescein-labeled and detected by laser-induced fluorescence(Khrapko et al., 1994). Separation of DNA fragments is achieved by the differential velocity of partly melted DNA in a medium with uniform denaturant concentration.
Temporal temperature gradient gel electrophoresis(TTGE). A constant concentration of urea or formamideis used as in cDGGE, but the temperature during the runis increased gradually (Yoshino et al., 1991; Wiese et al.,1995). The denaturant concentration (usually 6–8% urea)used in TTGE can be determined either from the theo-retical melting curve or experimentally from a perpen-dicular DGGE.
Microtemperature-gradient gel electrophoresis (mTGGE).A minimized gel (20 ¥ 20 ¥ 0.5mm) leads to the reduc-tion of the amount of DNA required and to shorterrunning times (approximately 12min at 100V, 10mA).The method was used in microbial ecology and epidemi-ology (Biyani and Nishigaki, 2001).
Double-gradient, denaturing gradient gel electrophore-sis (DG-DGGE). In addition to the chemical denaturinggradient (formamide and urea) a second sieving gradient(e.g., 6%–12% polyacrylamide gradient) is used (Cremonesi et al., 1997).
Two-dimensional DNA fingerprinting/two-dimensionalgene scanning (TDGS, 2D-DNA typing). Combinessize fractionation of DNA fragments in the first dimen-sion with their sequence-specific separation throughDGGE in the second dimension (see also next chapter).
Denaturing HPLC (dHPLC). Uses an ion-pair chro-matography separation principle, combined with aprecise control of the column temperature and optimizedmobile phase gradient for separation of mutant DNAmolecules (reviewed in Xiao and Oefner, 2001).
Cycling gradient capillary electrophoresis (CGCE).DNA sequence variants are detected based on their dif-ferential migration in a polymer-filled capillary system.A cycling (oscillating) temporal temperature gradient
CHAPTER 8 • Temperature and Denaturing Gradient Gel Electrophoresis 91
FIGURE 8.9 Mutation detection using DGGE analysis. a) Screening for the Gg -158 CÆT polymorphism in the promoter region of thehuman Gg-globin gene. Lanes 3, 4, and 5 correspond to homozygous samples for that polymorphism; lanes 2 and 7 correspond to het-erozygous samples; and lanes 1 and 6 correspond to samples that do not carry this polymorphism to either of the two alleles (photo cour-tesy of Dr. George P. Patrinos). b) DGGE analysis of the promoter region of the human Ag-globin gene. Lanes 2 and 4 correspond toheterozygous cases for the Ag -117 GÆA mutation, leading to the Greek type of nondeletional Hereditary Persistence of Fetal Hemoglo-bin (nd-HPFH); lane 3 corresponds to a homozygous case for the same mutation; lane 1 corresponds to a wild-type control (photo courtesyof Dr. George P. Patrinos). c) Mutation analysis of exons 11 and 13 of the CFTR gene. Lanes 1, 2, 4, and 5 correspond to wild-type cases;lane 3 corresponds to a heterozygous case for the E822X nonsense mutation; lane 6 corresponds to a heterozygous case for the G542X non-sense mutation, leading to cystic fibrosis (photo courtesy of Dr. Angeliki Balassopoulou, Athens, Greece). The gradient of denaturing agentsis depicted at the left side of each gel.

92 Molecular Diagnostics
is applied. This improvement enables utilization of a multiple injection technique, in which multiple samplesare injected into the same capillary (or set of capillaries)separated by predefined time intervals of partial electrophoresis. A 96-capillary system is able to screenover 15,000 samples in 24h (Minarik et al., 2003).
8.8 TECHNICAL EQUIPMENTFOR TGGE/DGGE
In general, for DGGE, preexisting vertical electrophoresisequipment with buffer-tank and combined heater/stirrerthermostat can be adapted. For TGGE, special equipment toachieve a constant temperature gradient is necessary.
The Biometra TGGE (Goettingen, Germany; www.biometra.de) system uses a temperature block powered byPeltier technology, which enables a strictly linear gradientthat may allow more reproducible conditions than with con-ventional chemical gradients or temperature gradients usingwater baths. The Biometra TGGE system is available in twoformats: A TGGE “mini” system operates small gels and istherefore suitable for fast, serial experiments. A TGGE maxisystem provides a large separation distance and allows highparallel sample throughput.
The DCode mutation detection system (Bio-Rad Lab-oratories, Hercules USA) can be used to screen mutationsby DGGE, TGGE, CDGE, TTGE, and other techniques. Thesystem performs TTGE by controlling the buffer tempera-ture during the electrophoresis run. A temperature controlmodule regulates the rate of temperature increase in auniform and linear fashion.
Sooner Scientific (Garvin, USA, www.soonersci.com)offers five different-sized DGGE Systems variants (for 2, 4,or 8 smaller gels or one large gel).
The INGENYphorU system (Ingeny International, GPGoes, The Netherlands; www.ingeny.com) is suitable forDGGE, TGGE, CDGE, and other techniques.
8.9 APPLICATIONS OF TGGE/DGGE ANDRELATED METHODS
TGGE/DGGE has been applied in an increasing number ofstudies. A recent search in PubMed database found over1,100 citations. The following applications have beendescribed:
• Screening for polymorphisms in human genes; forexample, COL1A2 gene (Borresen et al., 1988), alpha-1-antitrypsin (Hayes, 2003), human g-globin genes (Patrinoset al., 1998; 2001; Fig. 8.9a,b)
• Mutation detection in human genes; for example, p53(Pignon et al., 1994), FBN1 (Tiecke et al., 2001; Katzkeet al., 2002; Robinson et al., 2002), NF1 (Peters et al.,
1999; Fahsold et al., 2000), dystrophin gene (Hofstra etal., 2004), d-globin (Papadakis et al., 1997) and b-globingenes (Losekoot et al., 1990), CFTR gene (Fig. 8.9c), andso on
• Mutation and polymorphism detection in mitochondrialDNA (Hanekamp et al., 1996; Chen et al., 1999)
• Analysis in microbial ecology, determination of biodiver-sity of bacterial populations in soil, fresh, or salt water(Muyzer and Smalla, 1998; van Elsas et al., 2002)
• Genome profiling and provisional microbial species identification on the basis of random PCR and TGGE(Watanabe et al., 2002)
• Determination of biodiversity in fecal or intestinalmicroflora (Tannock, 2002)
• HLA typing (Uhrberg et al., 1994)• Analysis of proteins and antibody binding (Riesner et al.,
1991; Arakawa et al., 1993)• Clonality analysis of T-cell or T-cell receptors (Plonquet
et al., 2002; Lukowsky, 2003)• Mutation detection and detection of variation between
genomes of viral strains (Lu et al., 2002; Motta et al.,2002)
• Analysis of biodiversity and polymorphisms in plants(Gomes et al., 2003; Nikolcheva et al., 2003)
• Examination of the fidelity of DNA polymerases (Keohavong and Thilly, 1989)
8.10 CONCLUSIONS
TGGE/DGGE and related methods provide a very high sen-sitivity and are relatively easy and cheap to perform oncethe assays have been designed and optimized. The mainadvantages are in the high detection rate and specificity and improved heterozygote detection. The methodology issimple, nonradioactive, and relatively nontoxic. The disad-vantages of TGGE and DGGE include mainly the limitationof PCR fragment length to about 500 nucleotides, the diffi-culties of analyzing GC-rich fragments, and the need forcomputer analysis of potential PCR fragments (which on theother hand can save time and money by eliminating the useof inadequate primers). However, once primers and condi-tions have been chosen, TGGE/DGGE is a robust and easy-to-perform mutation screening method. It is particularly wellsuited for the detection of known and unknown mutationsin large genes, where high sensitivity is required and whenlarge numbers of samples are to be tested.
Acknowledgments
The authors would like to thank Christoph Gille, Anja Klose,Angelika Pletschacher, and Horst Schlechte.

References
Abrams, E. S., Murdaugh, S. E., and Lerman, L. S. (1990). Com-prehensive detection of single base changes in human genomicDNA using denaturing gradient gel electrophoresis and a GCclamp. Genomics 7, 463–475.
Ahnhudt, C., Muche, J. M., Dijkstal, K., Sterry, W., and Lukowsky,A. (2001). An approach to the sensitivity of temperature-gradient gel electrophoresis in the detection of clonally expandedT-cells in cutaneous T-cell lymphoma. Electrophoresis 22, 33–38.
Alkan, S., Cosar, E., Ergin, M., and Hsi, E. (2001). Detection of T-cell receptor-gamma gene rearrangement in lymphoprolifera-tive disorders by temperature gradient gel electrophoresis. Arch.Pathol. Lab. Med. 125, 202–207.
Arakawa, T., Hung, L., Pan, V., Horan, T. P., Kolvenbach, C. G.,and Narhi, L. O. (1993). Analysis of the heat-induced denatu-ration of proteins using temperature gradient gel electrophore-sis. Anal. Biochem. 208, 255–259.
Biyani, M., and Nishigaki, K. (2001). Hundredfold productivity ofgenome analysis by introduction of microtemperature-gradientgel electrophoresis. Electrophoresis 22, 23–28.
Borresen, A. L., Hovig, E., and Brogger, A. (1988). Detection ofbase mutations in genomic DNA using denaturing gradient gelelectrophoresis (DGGE) followed by transfer and hybridizationwith gene-specific probes. Mutat. Res. 202, 77–83.
Breton, J., Sichel, F., Abbas, A., Marnay, J., Arsene, D., andLechevrel, M. (2003). Simultaneous use of DGGE and DHPLCto screen TP53 mutations in cancers of the esophagus and cardiafrom a European high incidence area (Lower Normandy,France). Mutagenesis 18, 299–306.
Chen, T. J., Boles, R. G., and Wong, L. J. (1999). Detection of mito-chondrial DNA mutations by temporal temperature gradient gelelectrophoresis. Clin. Chem. 45, 1162–1167.
Costes, B., Fanen, P., Goossens, M., and Ghanem, N. (1993a). Arapid, efficient, and sensitive assay for simultaneous detec-tion of multiple cystic fibrosis mutations. Hum. Mutat. 2, 185–191.
Costes, B., Girodon, E., Ghanem, N., Chassignol, M., Thuong, N. T., Dupret, D., and Goossens, M. (1993b). Psoralen-modifiedoligonucleotide primers improve detection of mutations bydenaturing gradient gel electrophoresis and provide an alterna-tive to GC-clamping. Hum. Mol. Genet. 2, 393–397.
Cremonesi, L., Firpo, S., Ferrari, M., Righetti, P. G., and Gelfi, C.(1997). Double-gradient DGGE for optimized detection ofDNA point mutations. Biotechniques 22, 326–330.
Fahsold, R., Hoffmeyer, S., Mischung, C., Gille, C., Ehlers, C.,Kucukceylan, N., Abdel-Nour, M., Gewies, A., Peters, H.,Kaufmann, D., Buske, A., Tinschert, S., and Nurnberg, P.(2000). Minor lesion mutational spectrum of the entire NF1gene does not explain its high mutability but points to a func-tional domain upstream of the GAP-related domain. Am. J.Hum. Genet. 66, 790–818.
Ferec, C., Audrezet, M. P., Mercier, B., Guillermit, H., Moullier,P., Quere, I., and Verlingue, C. (1992). Detection of over 98%cystic fibrosis mutations in a Celtic population. Nat. Genet. 1,188–191.
Ferec, C., Raguenes, O., Salomon, R., Roche, C., Bernard, J. P.,Guillot, M., Quere, I., Faure, C., Mercier, B., Audrezet, M. P.,Guillausseau, P. J., Dupont, C., Munnich, A., Bignon, J. D., andLe Bodic, L. (1999). Mutations in the cationic trypsinogen gene
and evidence for genetic heterogeneity in hereditary pancreati-tis. J. Med. Genet. 36, 228–232.
Fodde, R., and Losekoot, M. (1994). Mutation detection by dena-turing gradient gel electrophoresis (DGGE). Hum. Mutat. 3,83–94.
Gejman P. V., Cao Q., Guedj F., and Sommer S. (1998). The sen-sitivity of denaturing gradient gel electrophoresis: a blindedanalysis. Mutat. Res. 382, 109–114.
Gelfi, C., Righetti, S. C., Zunino, F., Della Torre, G., Pierotti, M. A.,and Righetti, P. G. (1997). Detection of p53 point mutations by double-gradient, denaturing gradient gel electrophoresis.Electrophoresis 18, 2921–2927.
Gille, C., Gille, A., Booms, P., Robinson, P. N., and Nurnberg, P.(1998). Bipolar clamping improves the sensitivity of mutationdetection by temperature gradient gel electrophoresis. Elec-trophoresis 19, 1347–1350.
Gille, C., and Gille, A. (2002). TGGE-STAR: primer design for melting analysis using PCR gradient gel electrophoresis.Biotechniques 32, 264, 266, 268.
Gomes, N. C., Fagbola, O., Costa, R., Rumjanek, N. G., Buchner,A., Mendona-Hagler, L., and Smalla, K. (2003). Dynamics offungal communities in bulk and maize rhizosphere soil in thetropics. Appl. Environ. Microbiol. 69, 3758–3766.
Guldberg, P., and Guttler, F. (1994). “Broad-range” DGGE forsingle-step mutation scanning of entire genes: application tohuman phenylalanine hydroxylase gene. Nucleic Acids Res. 22,880–881.
Hanekamp, J. S., Thilly, W. G., and Chaudhry, M. A. (1996).Screening for human mitochondrial DNA polymorphisms withdenaturing gradient gel electrophoresis. Hum. Genet. 98,243–245.
Haran, T. E., Kahn, J. D., and Crothers, D. M. (1994). Sequenceelements responsible for DNA curvature. J. Mol. Biol. 244,135–143.
Hayes, V. M. (2003). Genetic diversity of the alpha-1-antitrypsingene in Africans identified using a novel genotyping assay.Hum. Mutat. 22, 59–66.
Hayes, V. M., Wu, Y., Osinga, J., Mulder, I. M., van der Vlies, P.,Elfferich, P., Buys, C. H., and Hofstra, R. M. (1999). Improve-ments in gel composition and electrophoretic conditions forbroad-range mutation analysis by denaturing gradient gel elec-trophoresis. Nucleic Acids Res. 27, e29.
Hofstra, R. M., Mulder, I. M., Vossen, R., de Koning-Gans, P. A.,Kraak, M., Ginjaar, I. B., van der Hout, A. H., Bakker, E., Buys,C. H., van Ommen, G. J., van Essen, A. J., and den Dunnen, J. T. (2004). DGGE-based whole-gene mutation scanning of thedystrophin gene in Duchenne and Becker muscular dystrophypatients. Hum. Mutat. 23, 57–66.
Hovig, E., Smith-Sorensen, B., Brogger, A., and Borresen, A. L.(1991). Constant denaturant gel electrophoresis, a modificationof denaturing gradient gel electrophoresis, in mutation detec-tion. Mutat. Res. 262, 63–71.
Kang, J., Kühn, J. E., Schäfer, P., Immelmann, A., and Henco, K.(1995) Quantification of DNA and RNA by PCR. In A Practi-cal Approach, M. J. McPherson, B. D. Hames, G. R. Taylor,eds. PCR 2, pp. 119–133. IRL Press, Oxford.
Katzke, S., Booms, P., Tiecke, F., Palz, M., Pletschacher, A.,Turkmen, S., Neumann, L. M., Pregla, R., Leitner, C.,Schramm, C., Lorenz, P., Hagemeier, C., Fuchs, J., Skovby, F.,Rosenberg, T., and Robinson, P. N. (2002). TGGE screening of
CHAPTER 8 • Temperature and Denaturing Gradient Gel Electrophoresis 93

94 Molecular Diagnostics
the entire FBN1 coding sequence in 126 individuals withmarfan syndrome and related fibrillinopathies. Hum. Mutat. 20,197–208.
Ke, S. H., and Wartell, R. M. (1995). Influence of neighboring basepairs on the stability of single base bulges and base pairs in aDNA fragment. Biochemistry 34, 4593–4600.
Keohavong, P., and Thilly, W. G. (1989). Fidelity of DNA poly-merases in DNA amplification. Proc. Natl. Acad. Sci. USA 86,9253–9257.
Khrapko, K., Hanekamp, J. S., Thilly, W. G., Belenkii, A., Foret,F., and Karger, B. L. (1994). Constant denaturant capillary elec-trophoresis (CDCE): a high resolution approach to mutationalanalysis. Nucleic Acids Res. 22, 364–369.
Lerman, L. S., and Silverstein, K. (1987). Computational simula-tion of DNA melting and its application to denaturing gradientgel electrophoresis. Methods Enzymol. 155, 482–501.
Losekoot, M., Fodde, R., Harteveld, C. L., van Heeren, H., Giordano, P. C., and Bernini, L. F. (1990). Denaturing gradientgel electrophoresis and direct sequencing of PCR amplifiedgenomic DNA: a rapid and reliable diagnostic approach to betathalassaemia. Br. J. Haematol. 76, 269–274.
Lu, Q., Hwang, Y. T., and Hwang, C. B. (2002). Mutation spectraof herpes simplex virus type 1 thymidine kinase mutants. J.Virol. 76, 5822–5828.
Lukowsky, A. (2003). Clonality analysis by T-cell receptor gammaPCR and high-resolution electrophoresis in the diagnosis ofcutaneous T-cell lymphoma (CTCL). Methods Mol. Biol. 218,303–320.
Macek, M., Jr., Mercier, B., Mackova, A., Miller, P. W., Hamosh,A., Ferec, C., and Cutting, G. R. (1997). Sensitivity of the dena-turing gradient gel electrophoresis technique in detection ofknown mutations and novel Asian mutations in the CFTR gene.Hum. Mutat. 9, 136–147.
Minarik, M., Minarikova, L., Bjorheim, J., and Ekstrom, P. O.(2003). Cycling gradient capillary electrophoresis: a low-costtool for high-throughput analysis of genetic variations. Elec-trophoresis 24, 1716–1722.
Motta, F. C., Rosado, A. S., and Couceiro, J. N. (2002). Standard-ization of denaturing gradient gel electrophoresis for mutantscreening of influenza A (H3N2) virus samples. J. Virol.Methods 101, 105–115.
Murdaugh, S. E., and Lerman, L. S. (1996) DGGE. DenaturingGradient Gel Electrophoresis and Related Techniques. In Lab-oratory Protocols for Mutation Detection, U. Landegren, ed.,pp. 33–37, Oxford University Press, Oxford.
Muyzer, G., and Smalla, K. (1998). Application of denaturing gra-dient gel electrophoresis (DGGE) and temperature gradient gelelectrophoresis (TGGE) in microbial ecology. Antonie VanLeeuwenhoek 73, 127–141.
Myers, R. M., Fischer, S. G., Lerman, L. S., and Maniatis, T.(1985a). Nearly all single base substitutions in DNA frag-ments joined to a GC-clamp can be detected by denaturing gradient gel electrophoresis. Nucleic Acids Res. 13, 3131–3145.
Myers, R. M., Lumelsky, N., Lerman, L. S., and Maniatis, T.(1985b). Detection of single base substitutions in total genomicDNA. Nature 313, 495–498.
Myers, R. M., Maniatis, T., and Lerman, L. S. (1987). Detectionand localization of single base changes by denaturing gradientgel electrophoresis. Methods Enzymol. 155, 501–527.
Nikolcheva, L. G., Cockshutt, A. M., and Barlocher, F. (2003).Determining diversity of freshwater fungi on decaying leaves:comparison of traditional and molecular approaches. Appl.Environ. Microbiol. 69, 2548–2554.
Papadakis, M., Papapanagiotou, E., and Loutradi-Anagnostou, A.(1997). Scanning method to identify the molecular heterogene-ity of delta-globin gene especially in delta-thalassemias: detec-tion of three novel substitutions in the promoter region of thegene. Hum. Mutat. 9, 465–472.
Patrinos, G. P., Kollia, P., Loutradi-Anagnostou, A., Loukopoulos,D., and Papadakis, M. N. (1998). The Cretan type of non-deletional hereditary persistence of fetal hemoglobin [Agamma-158CÆT] results from two independent gene conversionevents. Hum. Genet. 102, 629–634.
Patrinos, G. P., Kollia, P., Papapanagiotou, E., Loutradi-Anagnos-tou, A., Loukopoulos, D., and Papadakis, M. N. (2001).Agamma-haplotypes: a new group of genetic markers for tha-lassemic mutations inside the 5¢ regulatory region of the humanAgamma-globin gene. Am. J. Hematol. 66, 99–104.
Peters, H., Hess, D., Fahsold, R., and Schulke, M. (1999). A novelmutation L1425P in the GAP-region of the NF1 gene detectedby temperature gradient gel electrophoresis (TGGE). Mutationin brief no. 230. Online. Hum. Mutat. 13, 337.
Pignon, J. M., Vinatier, I., Fanen, P., Jonveaux, P., Tournilhac, O.,Imbert, M., Rochant, H., and Goossens, M. (1994). Exhaustiveanalysis of the P53 gene coding sequence by denaturing gradi-ent gel electrophoresis: application to the detection of pointmutations in acute leukemias. Hum. Mutat. 3, 126–132.
Plonquet, A., Gherardi, R. K., Creange, A., Antoine, J. C.,Benyahia, B., Grisold, W., Drlicek, M., Dreyfus, P., Honnorat,J., Khouatra, C., Rouard, H., Authier, F. J., Farcet, J. P., Delattre, J. Y., and Delfau-Larue, M. H. (2002). Oligoclonal T-cells in blood and target tissues of patients with anti-Hu syndrome. J. Neuroimmunol. 122, 100–105.
Riesner, D., Henco, K., and Steger, G. (1991) Temperature-gradient gel electrophoresis: A method for the analysis of conformational transitions and mutations in nucleic acids andproteins. In Advances in electrophoresis, A. Chrambach, M. J.Dunn, B. J. Radola, eds., Vol. 4. VCH, pp. 169–250. Verlags-gesellschaft, Weinhein.
Robinson, P. N., Booms, P., Katzke, S., Ladewig, M., Neumann,L., Palz, M., Pregla, R., Tiecke, F., and Rosenberg, T. (2002).Mutations of FBN1 and genotype-phenotype correlations inMarfan syndrome and related fibrillinopathies. Hum. Mutat. 20,153–161.
Sheffield, V. C., Cox, D. R., Lerman, L. S., and Myers, R. M.(1989). Attachment of a 40-base-pair G + C-rich sequence (GC-clamp) to genomic DNA fragments by the polymerase chainreaction results in improved detection of single-base changes.Proc. Natl. Acad. Sci. USA 86, 232–236.
Steger, G. (1994). Thermal denaturation of double-stranded nucleicacids: prediction of temperatures critical for gradient gel elec-trophoresis and polymerase chain reaction. Nucleic Acids Res.22, 2760–2768.
Tannock, G. W. (2002). Analysis of the intestinal microflora using molecular methods. Eur. J. Clin. Nutr. 56(Suppl. 4),S44–49.
Tchernitchko, D., Lamoril, J., Puy, H., Robreau, A. M., Bogard, C.,Rosipal, R., Gouya, L., Deybach, J. C., and Nordmann, Y.(1999). Evaluation of mutation screening by heteroduplex

analysis in acute intermittent porphyria: comparison with denaturing gradient gel electrophoresis. Clin. Chim. Acta. 279,133–143.
Tiecke, F., Katzke, S., Booms, P., Robinson, P. N., Neumann, L.,Godfrey, M., Mathews, K. R., Scheuner, M., Hinkel, G. K.,Brenner, R. E., Hovels-Gurich, H. H., Hagemeier, C., Fuchs, J.,Skovby, F., and Rosenberg, T. (2001). Classic, atypically severeand neonatal Marfan syndrome: twelve mutations and genotype-phenotype correlations in FBN1 exons 24–40. Eur. J.Hum. Genet. 9, 13–21.
Top, B. (1992). A simple method to attach a universal 50-bp GC-clamp to PCR fragments used for mutation analysis by DGGE.PCR Methods Appl. 2, 83–85.
Tully, L. A., Parsons, T. J., Steighner, R. J., Holland, M. M.,Marino, M. A., and Prenger, V. L. (2000). A sensitive denatur-ing gradient-Gel electrophoresis assay reveals a high frequencyof heteroplasmy in hypervariable region 1 of the humanmtDNA control region. Am. J. Hum. Genet. 67, 432–443.
Uhrberg, M., Hinney, A., Enczmann, J., and Wernet, P. (1994).Analysis of the HLA-DR gene locus by temperature gradientgel electrophoresis and its application for the rapid selection of
unrelated bone marrow donors. Electrophoresis 15, 1044–1050.van Elsas, J. D., Garbeva, P., and Salles, J. (2002). Effects of agro-
nomical measures on the microbial diversity of soils as relatedto the suppression of soil-borne plant pathogens. Biodegrada-tion 13, 29–40.
Watanabe, T., Saito, A., Takeuchi, Y., Naimuddin, M., and Nishigaki, K. (2002). A database for the provisional identifi-cation of species using only genotypes: web-based genome profiling. Genome Biol. 3, RESEARCH0010.
Wiese, U., Wulfert, M., Prusiner, S. B., and Riesner, D. (1995).Scanning for mutations in the human prion protein open readingframe by temporal temperature gradient gel electrophoresis.Electrophoresis 16, 1851–1860.
Xiao, W., and Oefner, P. J. (2001). Denaturing high-performanceliquid chromatography: A review. Hum. Mutat. 17, 439–474.
Yoshino, K., Nishigaki, K., and Husimi, Y. (1991). Temperaturesweep gel electrophoresis: a simple method to detect pointmutations. Nucleic Acids Res. 19, 3153.
Zschocke, J., Quak, E., Guldberg, P., and Hoffmann, G. F. (2000).Mutation analysis in glutaric aciduria type I. J. Med. Genet. 37,177–181.
CHAPTER 8 • Temperature and Denaturing Gradient Gel Electrophoresis 95




























