Embed Size (px)
Citation preview

131
CHAPTER-3
STUDIES TOWARDS NEW AND IMPROVED
SYNTHESIS OF VALACYCLOVIR

132
3.1 INTRODUCTION
Viruses [95a] are almost as versatile as bacteria in the range of
diseases they can cause. A virus is defined as various numbers of
submicroscopic parasites that can infect any animal. All viruses replicate
only within living cells and cannot usually survive for long time outside
the host cell. Most prominent mechanism of viruses entry to human body
is by the absorption to superficial cells. [95b-c].
Out of several viral families across the globe, one of the most
commonly existing viral families is represented by Herpes viruses [96, 97
a-c]. Table-3.1 gives a glimpse of the herpes viruses that infect humans.
Table 3.1: Herpes viruses that infect humans
Type Synonym
Human herpesvirus 1 (HHV-1) Herpes simplex virus 1 (HSV-1)
Human herpesvirus 2 (HHV-2) Herpes simplex virus 2 (HSV-2)
Human herpesvirus 3 (HHV-3) Varicella-Zoster virus (VZV)
Human herpesvirus 4 (HHV-4) Epstein-Barr virus (EBV),
Human herpesvirus 5 (HHV-5) Cytomegalovirus (CMV)
Human herpesvirus 6 (HHV-6) Roseolovirus
Human herpesvirus 7 (HHV-7) Roseolovirus
Human herpesvirus 8 (HHV-8) Kaposi's sarcoma-associated
herpesvirus (KSHV),
Significant progress has been achieved in the development of antiviral
compounds [98] and an increasing number of antiviral agents are now in
regular use (Table-3.2). Many are nucleoside analogues that interfere
with viral replication, but other targets have also been successfully
133
exploited [99]. Much research emphasis has been concentrated on agents
that act to inhibit the replication of HIV [100].
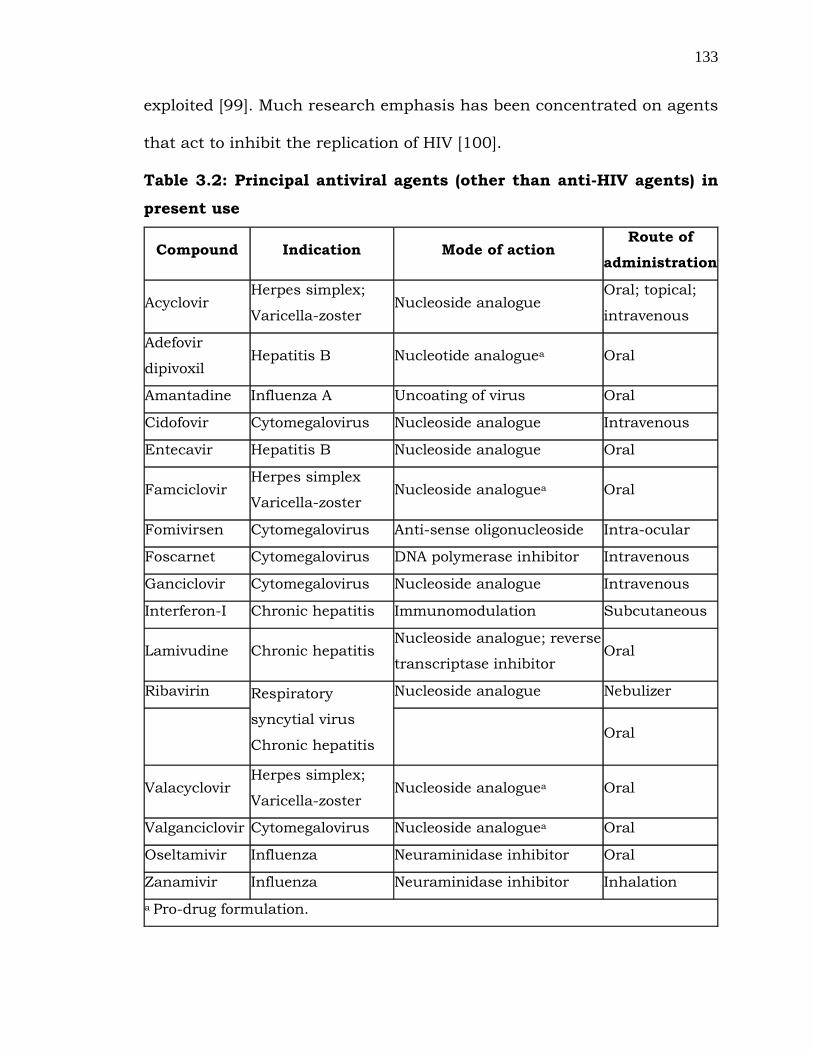
Table 3.2: Principal antiviral agents (other than anti-HIV agents) in
present use
Compound Indication Mode of action Route of
administration
Acyclovir Herpes simplex;
Varicella-zoster Nucleoside analogue
Oral; topical;
intravenous
Adefovir
dipivoxil Hepatitis B Nucleotide analoguea Oral
Amantadine Influenza A Uncoating of virus Oral
Cidofovir Cytomegalovirus Nucleoside analogue Intravenous
Entecavir Hepatitis B Nucleoside analogue Oral
Famciclovir Herpes simplex
Varicella-zoster Nucleoside analoguea Oral
Fomivirsen Cytomegalovirus Anti-sense oligonucleoside Intra-ocular
Foscarnet Cytomegalovirus DNA polymerase inhibitor Intravenous
Ganciclovir Cytomegalovirus Nucleoside analogue Intravenous
Interferon-I Chronic hepatitis Immunomodulation Subcutaneous
Lamivudine Chronic hepatitis Nucleoside analogue; reverse
transcriptase inhibitor Oral
Ribavirin Respiratory
syncytial virus
Chronic hepatitis
Nucleoside analogue Nebulizer
Oral
Valacyclovir Herpes simplex;
Varicella-zoster Nucleoside analoguea Oral
Valganciclovir Cytomegalovirus Nucleoside analoguea Oral
Oseltamivir Influenza Neuraminidase inhibitor Oral
Zanamivir Influenza Neuraminidase inhibitor Inhalation
a Pro-drug formulation.

134
By and large, an antiviral drug exerts its effect by inhibiting the Virus
by any of the following methods; (a) binding with the receptor molecule
(b) avoid the replication and (c) stopping the virus from releasing its
proteins [101].(d) by strengthening the immune system of the body
[102a].
Valacyclovir, 49 a most promising second generation nucleoside
analogue DNA polymerase inhibitor which rapidly converted to acyclovir,
45 which has demonstrated antiviral activity against HSV types 1 (HSV-
1) and 2 (HSV-2) and VZV both in cell culture and in vivo [102b]. The
chemical name of 49 is L-valine, 2-[(2-amino-1,6-dihydro-6-oxo-9H-
purin-9-yl)methoxy]ethylester mono hydrochloride salt. Valacyclovir (49)
is indicated for the treatment of; herpes zoster (shingles), suppression of
genital herpes in immunocompetent individuals, suppression of
recurrent genital herpes in HIV-infected [102c] individuals and is also
indicated for the treatment of cold sores (herpes labialis).
This chapter is dedicated to discuss the process research and
development activity of valacyclovir (49) carried out in our laboratory.
Before discussing the process development activity, a small section to
introduce the history of valacyclovir, its mechanism of action, and
detailed review on available synthetic processes from its birth to till date
is illustrated.

135
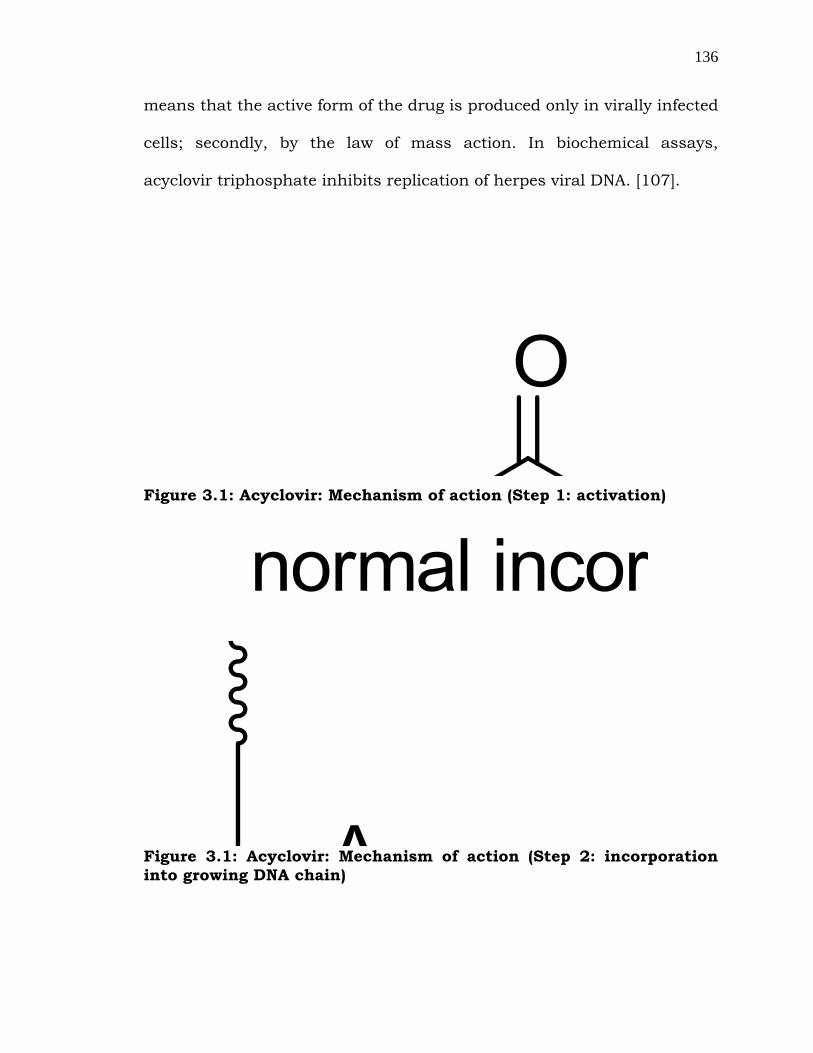
3.1.1 Mechanism of action
Valacyclovir (49) is well absorbed to release 45 into the bloodstream
[103]. Uptake of acyclovir is enhanced in herpes virus-infected cells,
presumably because of its rapid activation to the monophosphate form,
46 catalyzed by a herpes virus-encoded thymidine kinase (TK) [104].
(Step-1, Figure-3.1) Subsequent conversion of the monophosphate to the
active form, acyclovir triphosphate, 48 is accomplished by host-cell
enzymes. Compound 48 functions as a substrate for viral, but not
cellular, DNA polymerase, competing with deoxyguanosine triphosphate
for incorporation into the elongating chain [105]. The incorporation of
acyclovir triphosphate into the growing chain of viral DNA results in
chain termination, as acyclovir lacks the 3'-hydroxyl group necessary for
subsequent elongation (Step-2, Figure-3.1)
Structurally, acyclovir is acycloguanosine, an analogue of the purine
nucleoside, guanosine, in which the deoxyribose moiety has lost its cyclic
configuration. Acyclovir itself [106a] is inactive, in order to achieve its
antiviral effect, it must first be phosphorylated to the triphosphate form
within the infected cell [106b]. Although the second and third phosphate
groups are added by cellular enzymes, the initial phosphorylation step is
accomplished by a viral thymidine kinase, specified by herpes simplex
and varicella zoster viruses. The cellular form of this enzyme is much
less efficient in producing acyclovir monophosphate. This unique feature
is the basis for the selective toxicity of acyclovir, for two reasons: first, it
136
means that the active form of the drug is produced only in virally infected
cells; secondly, by the law of mass action. In biochemical assays,
acyclovir triphosphate inhibits replication of herpes viral DNA. [107].
Figure 3.1: Acyclovir: Mechanism of action (Step 1: activation)
Figure 3.1: Acyclovir: Mechanism of action (Step 2: incorporation
into growing DNA chain)

137
3.2 REVIEW OF LITERATURE
3.2.1 Development history of Valacyclovir and its pharmaceutical
properties
Among the several structural analogues of acyclovir [108-110],
valacyclovir, 49 is one of the successful candidates to be enjoyed as a
drug. The later is metabolized into the former after oral administration
similar to famciclovir [111], 51 a pro-drug of penciclovir, 50 [112]
(Figure-3.2). These two drugs are subsequently approved by the U.S Food
and Drug Administration (FDA) for the treatment of acute herpes zoster
and herpes simplex genitalis [113].
Figure 3.2: Second generation antiherpes drugs
Since, we are primarily interested in valacyclovir (49), systematic
review of literature on various synthetic methodologies adopted by
various researchers has been detailed here.

138
3.2.2 Review of reported synthetic approaches
The synthesis of valacyclovir (49) reported till date, mainly starts from
acyclovir (45) by its condensation with protected valine derivatives. The
philosophy of synthetic routes to 49 is briefly described in the following
section.
3.2.2.1 Beauchamp et al. approach: The first synthesis [114] for 49
involves condensation of acyclovir (45) with N-Cbz-L-valine (52) in the
presence of 1,3-Dicyclohexylcarbodiimide (DCC) in the presence of
catalytic amount of dimethylaminopyridine (DMAP) in DMF followed by
hydrogenolysis of resulting N-Cbz protected valacyclovir (53) using 5%
Pd/C catalyst as depicted in Scheme-3.1.
Scheme 3.1: Beauchamp et al. synthetic approach to Valacyclovir.
Similar kind of work reported by Wang and co-workers [114b] involves
condensation of methoxy (54a) or nitro (54b) substituted N-Cbz-L-valine
as protected aminoacid as depicted in Scheme-3.2. Once the
condensation is achieved, hydrogenolysis of the resulting intermediates
using Pd/C in formic acid resulted 49.

139
Scheme 3.2: Synthesis of Valacyclovir by Wang and co-workers.
3.2.2.2 Jackson et al. approach: Jackson et al. [115] described an
improved process for the preparation of 49 by condensing acyclovir (45)
with N-carboxyanhydride of Z-valine (56) in the presence of DMAP in a
single step with 97.7% yield and 95% purity (Scheme-3.3).
Scheme 3.3: Jackson’s synthetic approach.
The process may be performed in a single step without the need for
protection and subsequent deprotection and is thus much simpler and
quicker to perform than the previously reported procedures.
3.2.2.3 Montoro et al. approach: Montoro et al. [116] developed a new
route by sequentially converting the hydroxy group in the side chain of
acyclovir (45) to a good leaving group Z (Z= tosylate, or mesylate, or
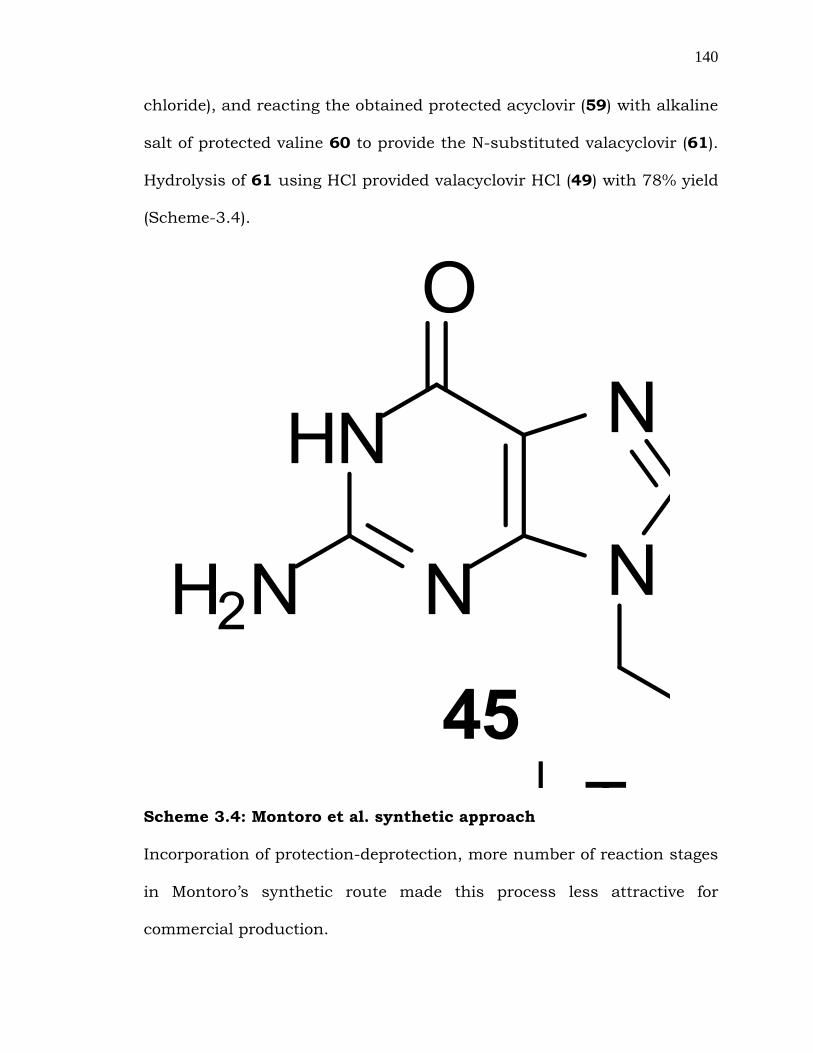
140
chloride), and reacting the obtained protected acyclovir (59) with alkaline
salt of protected valine 60 to provide the N-substituted valacyclovir (61).
Hydrolysis of 61 using HCl provided valacyclovir HCl (49) with 78% yield
(Scheme-3.4).
Scheme 3.4: Montoro et al. synthetic approach
Incorporation of protection-deprotection, more number of reaction stages
in Montoro‟s synthetic route made this process less attractive for
commercial production.

141
3.2.2.4 Etinger Marina Yu et al. approach: The process reported by
Etinger et al. [117] involves the condensation of acyclovir (45) with N-
Boc-L-valine (62) in the presence of N-(3-dimethylaminopropyl)-N-
ethylcarbodiimide as a coupling agent to get the N-BOC protected
valacyclovir (63) with 97% yield and 98% purity. The N-BOC group was
then removed by hydrolysis using 12N HCl furnishing Valacyclovir
hydrochloride with 92% yield and 98% purity by HPLC. (Scheme-3.5)
Scheme 3.5: Etinger et al. synthetic approach
3.2.3 Summary of literature review
Careful examination of the reported approaches reveal that they follow
common approach in designing the synthesis, by protecting amino group
in L-valine with different protecting groups followed by removing the
protected groups at the end. Beauchamp‟s first synthetic approach to
valacyclovir appears to be promising, but covers insufficient information
on control of process related impurities specifically the enantiomer (D-
isomer).D-Isomer impurity is possible due to the carryover from L-Valine
and or racemisation during reaction conditions. Montoro‟s synthetic

142
route though economical compared to Beauchamp‟s route but involves
multiple stages. Etinger et al approach also involves protection-
deprotection stages though simple compared to both Beauchamp‟s and
Montoro‟s synthetic approaches. Jackson‟s approach is a simple, safe,
quick and economical process for the preparation of 49 and is
appreciable compared to the other approaches. But, it is silent on the
formation of isomeric impurity; moreover, the bulky protecting groups
used in almost all the known synthetic routes may cause steric
hindrance and resulting to incomplete formation of the required L-valine
ester. The protection-deprotection steps further increase the risk of
racemization leading to formation of other isomer in the product. In view
of above limitations associated with reported procedures, we felt a need
for not only a simple, impurity free, cost effective and scalable process,
but also to have an IP advantage by means of freedom to operate to enter
into the market.
3.3. OBJECTIVE OF THE PRESENT WORK
With the above background, we adopted two approaches to achieve
the goal. First approach is the identification of alternative route of
synthesis and second one being the improvement of the original route to
achieve 49 by circumventing the said disadvantages. Both the
approaches have been described separately in two sections as below.

143
Section 1. Development of an alternative, facile, and efficient synthesis to
49
Section 2. To provide an improved process for 49 by circumventing the
challenges associated with the reported process.
3.4 RESULTS & DISCUSSION
3.4.1 Section-1: Studies towards new synthesis of Valacyclovir
To circumvent the problem of steric hindrance and racemization, we
thought to use a small size versatile synthetic precursor of amino group
such as azide functionality. The azido group being an excellent masked
amino equivalent is relatively stable and can be readily reduced to the
desired amino function when required. This protocol led to the synthesis
of valacyclovir, 49 from acyclovir, 45 and the results are presented here.
Our synthesis of valacyclovir [118] started with reaction of imidazole,
64 with sodium azide and sulfuryl chloride to furnish hydrochloride salt
of Imidazole-1-sulfonyl azide (65), which is then treated with L-valine
(57) to give α-azido acid derivative of L-valine (66) as shown in Scheme-
3.6. Compound 65 is proved to be equal to triflylazide in its ability to act
as diazo-donor in the conversion of α-amino acid to α-azido acid. The
azido acid (66) was characterized by proton NMR which showed the
presence of acidic proton at 11.5 ppm and by the absence of amino
protons of valine moiety. Further, the structure of 66 was confirmed by

144
the presence of IR absorption frequency peak at 2110 cm-1 corresponds
to azide moiety.
Scheme 3.6: Preparation of 2(S)-azido-3-methylbutyric acid (66).
The 2(S)-azido-3-methylbutyric acid (66) obtained is then condensed
with acyclovir (45) in presence of DCC and DMAP to obtain a novel
intermediate, 9-[(2-(2S-Azido-3-methylbutyroxy) ethoxy) methyl] guanine
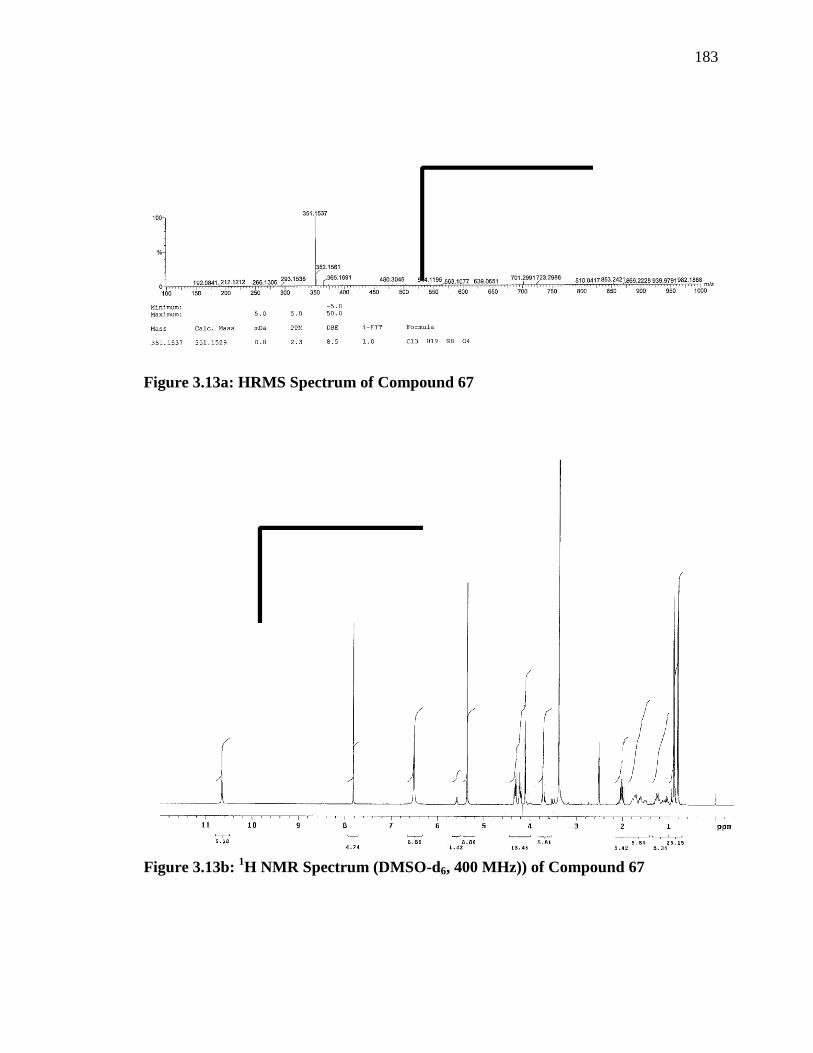
(67) as shown in Scheme 3.7. The structure of 67 was confirmed by
HRMS, which shown the mass value of 325 against the calculated value
of 325 that corresponds to mass value of 67. The IR frequencies at 2099
cm-1 and 1732 cm-1 indicated the formation of desired azido ester (67).
Further, the presence of azomethane proton as a singlet at δ value of 7.8
and protons of two methyl groups of isopropyl moiety as two doublets at
δ values at 0.80 and 0.89 respectively and the absence of alcohol proton
of acyclovir moiety and acid protons of azido acid moiety in the proton
NMR confirmed the structure of 67 (Figure-3.13b). Finally, the catalytic
reduction of 67 with raney nickel yielded the valacyclovir freebase. Mass
spectrum showed protonated molecular ion at m/z 325. IR Spectrum

145
displayed absence of azide stretching frequency. Further, carbonyl
stretching at 1732 cm-1 along with significant maxima at 3327, 3110 and
1H NMR displayed singlet for azomethane proton at δ 7.84, amido proton
at δ 10.81 and isopropyl protons in the range of δ 0.80-0.90. The
freebase is then treated with aqueous hydrochloric acid to form
Valacyclovir hydrochloride (49) with an overall yield of 67.3% as depicted
in Scheme-3.7.
Scheme 3.7: New synthesis for Valacyclovir HCl (49) using azido L-
valine (66).
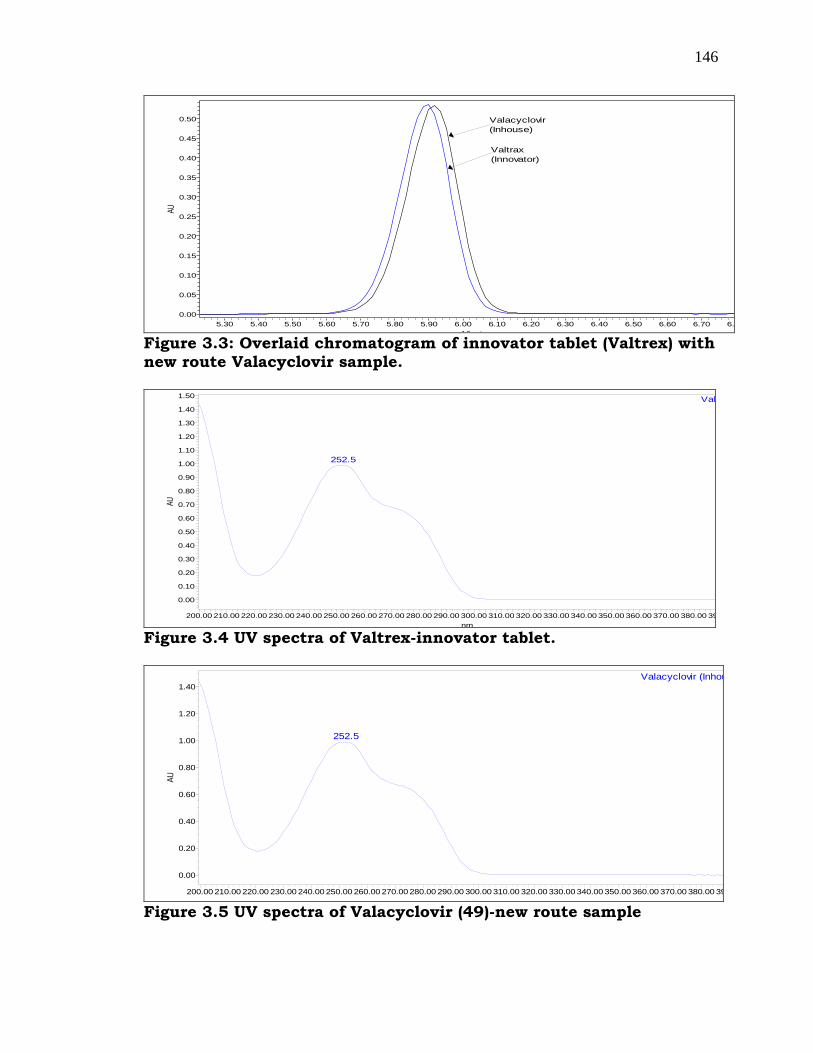
Comparison of obtained sample with Innovator sample [113] by HPLC
revealed that the principle peak obtained using Innovator product
matches with the product obtained by our route as described below. The
analytical results by HPLC [119] were depicted in Figure-3.3, 3.4 & 3.5
which revealed that the product obtained is matching with the innovator
samples and confirms the feasibility of the product as per the synthetic
methodology followed as depicted in Scheme-3.7.
146
AU
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
Minutes
5.30 5.40 5.50 5.60 5.70 5.80 5.90 6.00 6.10 6.20 6.30 6.40 6.50 6.60 6.70 6.80
Valtrax
(Innovator)
Valacyclovir
(Inhouse)
Figure 3.3: Overlaid chromatogram of innovator tablet (Valtrex) with new route Valacyclovir sample.
AU
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
0.80
0.90
1.00
1.10
1.20
1.30
1.40
1.50
nm
200.00 210.00 220.00 230.00 240.00 250.00 260.00 270.00 280.00 290.00 300.00 310.00 320.00 330.00 340.00 350.00 360.00 370.00 380.00 390.00
252.5
Valtrax
Figure 3.4 UV spectra of Valtrex-innovator tablet.
AU
0.00
0.20
0.40
0.60
0.80
1.00
1.20
1.40
nm
200.00 210.00 220.00 230.00 240.00 250.00 260.00 270.00 280.00 290.00 300.00 310.00 320.00 330.00 340.00 350.00 360.00 370.00 380.00 390.00
252.5
Valacyclovir (Inhouse)
Figure 3.5 UV spectra of Valacyclovir (49)-new route sample

147
Having established the feasibility of the route, selected at research
phase, we have taken up optimization of each step to establish the
robustness and ruggedness for process for making 49. The stage wise
optimization details of the route are discussed below in detail.
3.4.1.1 Optimization studies for coupling reaction:
Coupling experiment performed using DCC [120] in DMF for the
synthesis of 67 during feasibility was discouraging because of tedious
process of removing byproduct DCU from the product and also low yield.
During optimization we have explored the coupling reaction using
various acid activating agents such as acid chlorides, mixed anhydride
and using various dehydrating agents such as, EDC [121], MeSO2Cl-Et3N
and N,N‟-carbonyldiimidazole [122] (CDI). To our surprise, reaction did
not proceed with acid chloride prepared using thionylchloride or
oxalylchloride. Reaction using methane sulfonyl chloride as acid
activating agent gave poor yield and quality. Though reaction is going for
completion using EDC, CDI, and DEAD, yields and quality obtained were
not encouraging as shown in Table-3.3
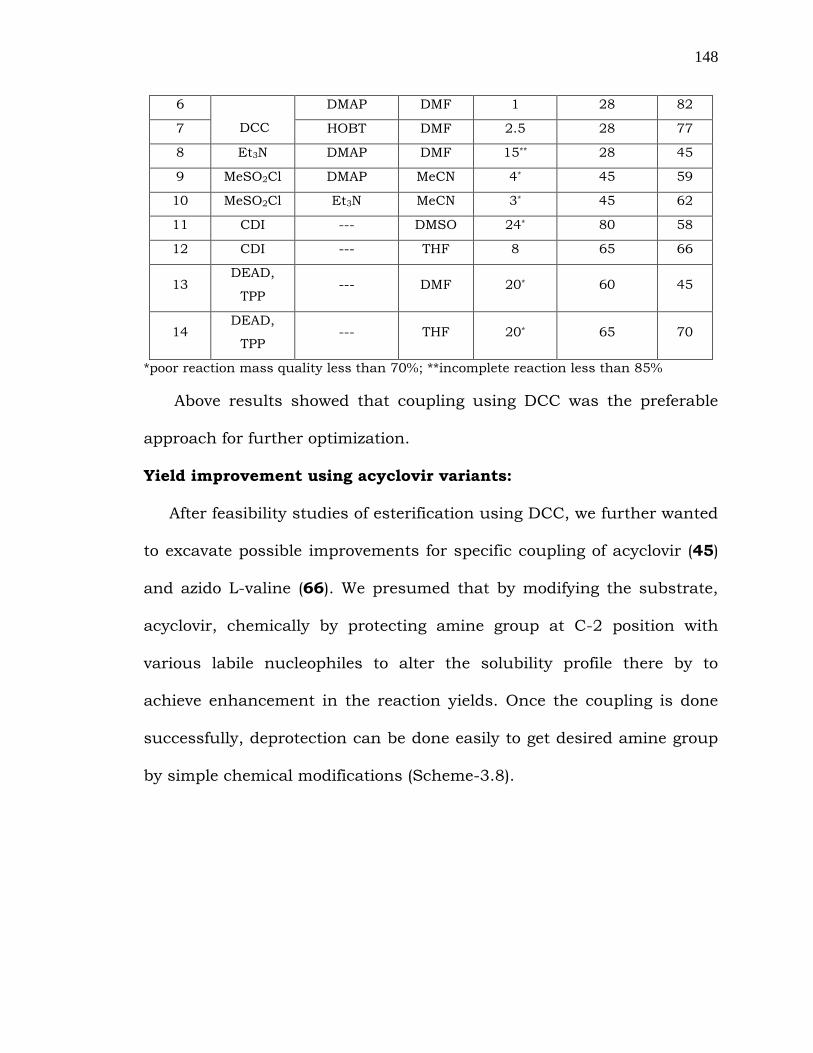
Table 3.3: Selected esterification attempts for the synthesis of 67.
entry reagent catalyst solvent reaction
time (h)
reaction
temp (°C)
yield
(%)
1
EDC
DMAP DCM 6 28 62
2 HOBT DMSO 5* 28 55
3 Et3N,DMAP DMF 4* 28 59
4
DCC
DMAP DCM 4* 28 69
5 DMAP DMSO 2 28 72
148
6
DCC
DMAP DMF 1 28 82
7 HOBT DMF 2.5 28 77
8 Et3N DMAP DMF 15** 28 45
9 MeSO2Cl DMAP MeCN 4* 45 59
10 MeSO2Cl Et3N MeCN 3* 45 62
11 CDI --- DMSO 24* 80 58
12 CDI --- THF 8 65 66
13 DEAD,
TPP --- DMF 20* 60 45
14 DEAD,
TPP --- THF 20* 65 70
*poor reaction mass quality less than 70%; **incomplete reaction less than 85%
Above results showed that coupling using DCC was the preferable
approach for further optimization.
Yield improvement using acyclovir variants:
After feasibility studies of esterification using DCC, we further wanted
to excavate possible improvements for specific coupling of acyclovir (45)
and azido L-valine (66). We presumed that by modifying the substrate,
acyclovir, chemically by protecting amine group at C-2 position with
various labile nucleophiles to alter the solubility profile there by to
achieve enhancement in the reaction yields. Once the coupling is done
successfully, deprotection can be done easily to get desired amine group
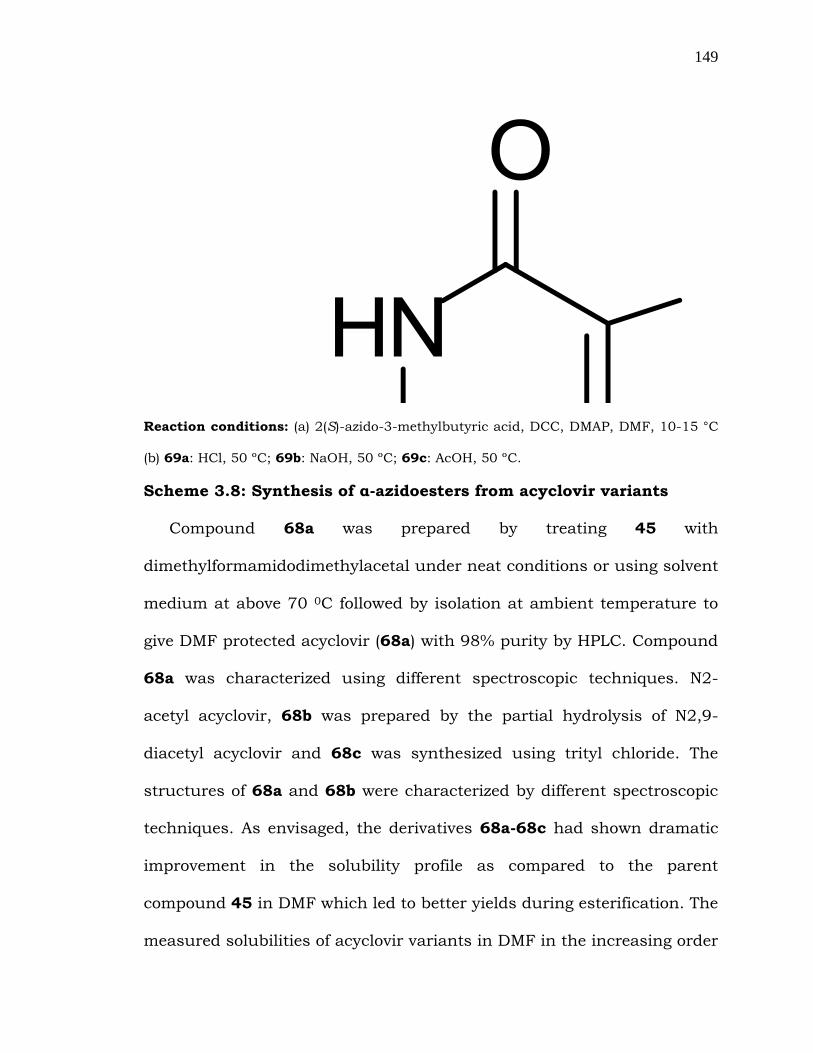
by simple chemical modifications (Scheme-3.8).
149
Reaction conditions: (a) 2(S)-azido-3-methylbutyric acid, DCC, DMAP, DMF, 10-15 °C
(b) 69a: HCl, 50 ºC; 69b: NaOH, 50 ºC; 69c: AcOH, 50 ºC.
Scheme 3.8: Synthesis of α-azidoesters from acyclovir variants
Compound 68a was prepared by treating 45 with
dimethylformamidodimethylacetal under neat conditions or using solvent
medium at above 70 0C followed by isolation at ambient temperature to
give DMF protected acyclovir (68a) with 98% purity by HPLC. Compound
68a was characterized using different spectroscopic techniques. N2-
acetyl acyclovir, 68b was prepared by the partial hydrolysis of N2,9-
diacetyl acyclovir and 68c was synthesized using trityl chloride. The
structures of 68a and 68b were characterized by different spectroscopic
techniques. As envisaged, the derivatives 68a-68c had shown dramatic
improvement in the solubility profile as compared to the parent
compound 45 in DMF which led to better yields during esterification. The
measured solubilities of acyclovir variants in DMF in the increasing order

150
are Acyclovir (150 volumes), Acetyl acyclovir, 68b (80 volumes), DMF
acyclovir, 68a (75 volumes) and trityl acyclovir, 68c (5 volumes) as
captured in Figure-3.6.
8285
92 95
150
80 75
5
0
50
100
150
200
250
Acy Ac-Acy DMF-Acy Tr-Acy
Comparison of solubilities and yields
% yield Solubility in DMF (No. of times)
Figure 3.6: Comparison of solubility of 45, 68a-c and yields during
condensation
It was observed that trityl protected acyclovir resulted in enhanced
yield (95%) over other protections or without protection when same
procedure was employed for esterification. Even though protected
acyclovir derivatives showed improvement in yield due to increased
solubility, cycle time and number of stage will increase for the synthesis
of Valacyclovir HCl. Further, trityl protected derivative (68c) generates
huge amount of by-product, trityl alcohol, on deprotection and adds to
cost and generates huge effluent made this option relatively less
attractive. Thus, we focused our attention on the optimization of DCC
catalyzed esterification using 45 and Azido acid, 66 [123].
151
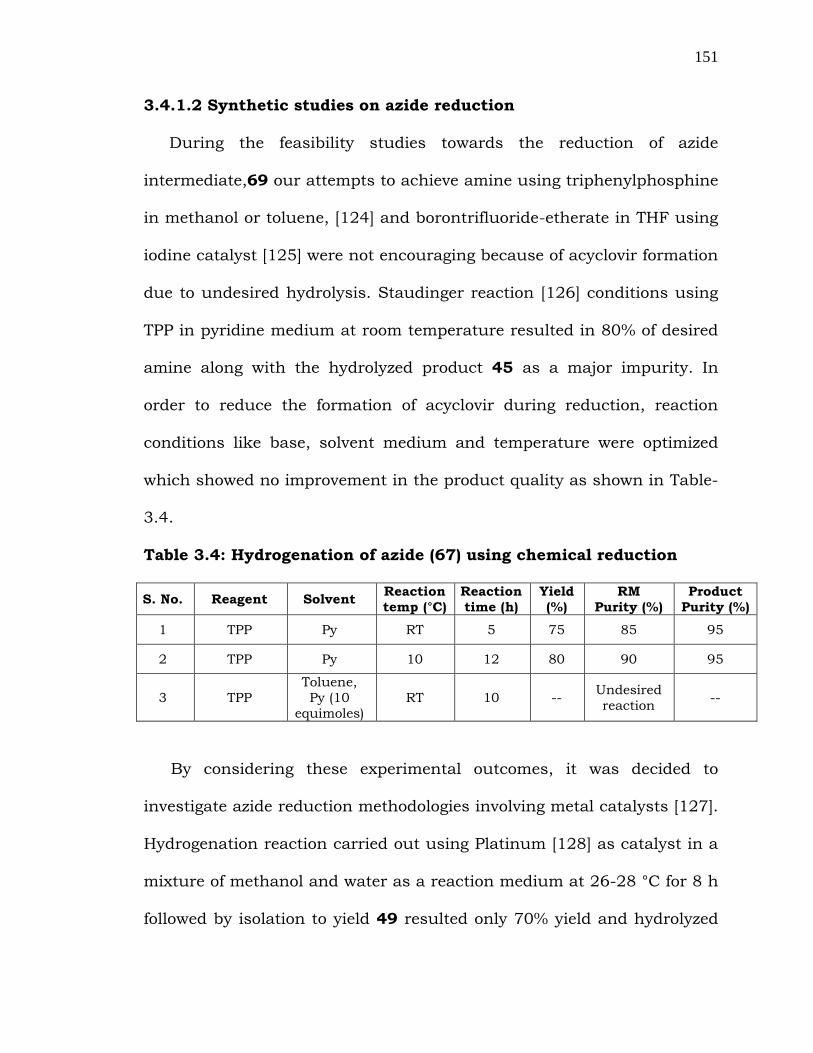
3.4.1.2 Synthetic studies on azide reduction
During the feasibility studies towards the reduction of azide
intermediate,69 our attempts to achieve amine using triphenylphosphine
in methanol or toluene, [124] and borontrifluoride-etherate in THF using
iodine catalyst [125] were not encouraging because of acyclovir formation
due to undesired hydrolysis. Staudinger reaction [126] conditions using
TPP in pyridine medium at room temperature resulted in 80% of desired
amine along with the hydrolyzed product 45 as a major impurity. In
order to reduce the formation of acyclovir during reduction, reaction
conditions like base, solvent medium and temperature were optimized
which showed no improvement in the product quality as shown in Table-
3.4.
Table 3.4: Hydrogenation of azide (67) using chemical reduction
S. No. Reagent Solvent Reaction
temp (°C)
Reaction
time (h)
Yield
(%)
RM
Purity (%)
Product
Purity (%)
1 TPP Py RT 5 75 85 95
2 TPP Py 10 12 80 90 95
3 TPP
Toluene,
Py (10
equimoles)
RT 10 -- Undesired
reaction --
By considering these experimental outcomes, it was decided to
investigate azide reduction methodologies involving metal catalysts [127].
Hydrogenation reaction carried out using Platinum [128] as catalyst in a
mixture of methanol and water as a reaction medium at 26-28 °C for 8 h
followed by isolation to yield 49 resulted only 70% yield and hydrolyzed
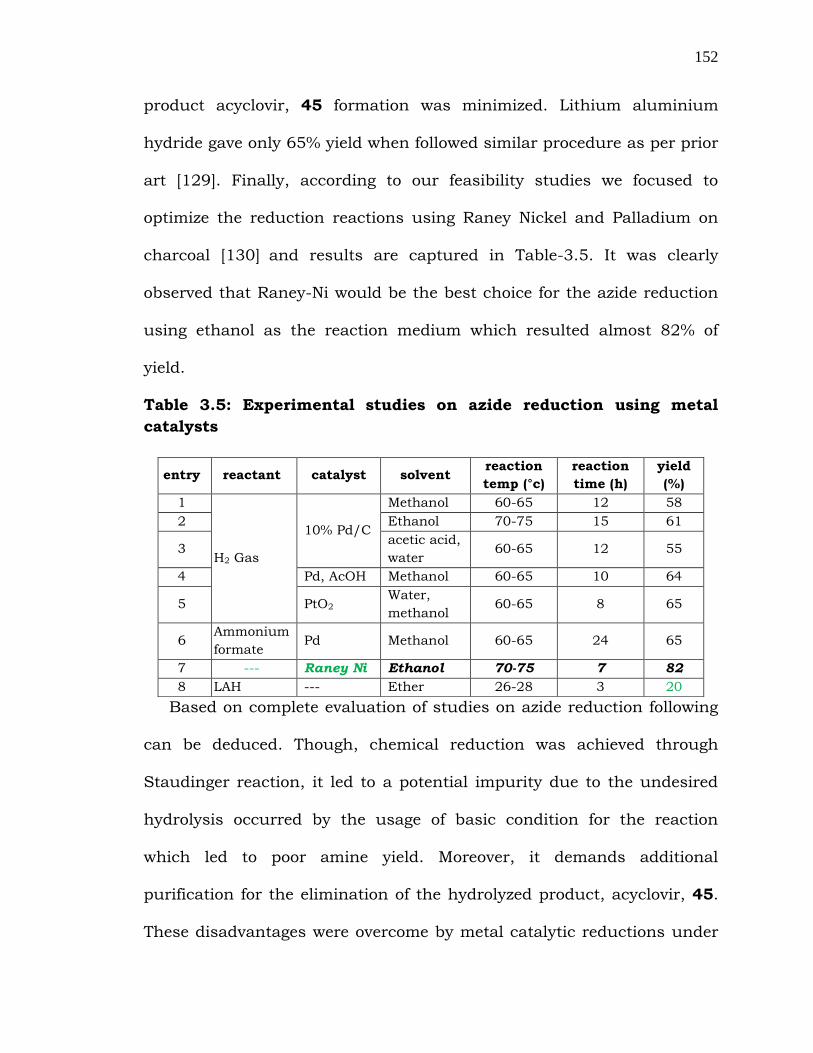
152
product acyclovir, 45 formation was minimized. Lithium aluminium
hydride gave only 65% yield when followed similar procedure as per prior
art [129]. Finally, according to our feasibility studies we focused to
optimize the reduction reactions using Raney Nickel and Palladium on
charcoal [130] and results are captured in Table-3.5. It was clearly
observed that Raney-Ni would be the best choice for the azide reduction
using ethanol as the reaction medium which resulted almost 82% of
yield.
Table 3.5: Experimental studies on azide reduction using metal
catalysts
Based on complete evaluation of studies on azide reduction following
can be deduced. Though, chemical reduction was achieved through
Staudinger reaction, it led to a potential impurity due to the undesired
hydrolysis occurred by the usage of basic condition for the reaction
which led to poor amine yield. Moreover, it demands additional
purification for the elimination of the hydrolyzed product, acyclovir, 45.
These disadvantages were overcome by metal catalytic reductions under
entry reactant catalyst solvent reaction
temp (°c)
reaction
time (h)
yield
(%)
1
H2 Gas
10% Pd/C
Methanol 60-65 12 58
2 Ethanol 70-75 15 61
3 acetic acid,
water 60-65 12 55
4 Pd, AcOH Methanol 60-65 10 64
5 PtO2 Water,
methanol 60-65 8 65
6 Ammonium
formate Pd Methanol 60-65 24 65
7 --- Raney Ni Ethanol 70-75 7 82
8 LAH --- Ether 26-28 3 20

153
mild conditions. Therefore, in the current context, catalytic reduction
using Raney Nickel is more preferable over chemical reduction to achieve
optimum yield and quality of final product, 49.
Finally, valacyclovir salt was prepared using aqueous hydrochloric
acid at pH 2.5-3.0. The reaction mixture was treated with charcoal to
remove color impurities and the purity of the product is found to be
98.33% by HPLC and chiral purity is 97.06%.
3.4.1.3 Process safety assessment
Having completed the feasibility studies for the new route, our next
aim was to ensure safe scale up of the process. With the scale up there
is always an increased liability from accidents, culminate to injury of
personal, loss of equipment and delay in key deliverables. One of the
anticipated key challenges for our route of synthesis is handling of sodium
azide and azide intermediates. Hitherto there is no literature precedence
for safety studies during preparation of 66. Therefore it is imperative to
perform reaction safety assessment using reaction calorimeter (RC) [131],
to understand the thermal events associated with each step. The
preliminary dust explosion studies to find out an engineering solution
during scale up were also helpful in concluding the safety assessment.
The key outcomes are to be essentially used for designing of plant
equipment, services and operations to impart scalability for the optimized
process.

154
Investigations using reaction calorimeter: We have investigated the
thermal behavior during preparation of Azido acid (66) from imidazole,
64 using reaction calorimeter and summary of observations is outlined
in this section. Reaction Calorimeter (Mettler make, Model- SV 01 800m1
capacity, all glass reactor equipped with a propeller type up flow agitator,
jacketed with controlled temperature circulator, addition funnel,
condenser and the probes) was used for performing the safety studies.
A suspension of sodium azide (26 g) in toluene (260 ml) was cooled to 0 °C
and charged sulfuryl chloride (32.4mL) to it. The temperature was raised to 25
°C and maintained mass for l6 hrs at 25°C- 30°C. The above solution was
charged into RC reactor, cooled to 0 °C and added imidazole (49 g) to the
reactor in portion wise. Temperature of the mass was raised to 25 °C and
continued stirring at 25 °C for 3hrs. Reaction mass was then cooled to 0°C, 5%
Na2CO3 solution (26O mL) was added, raised the temperature to 25 °C and
continued stirring for 15 min. Toluene and aqueous layer were separated,
toluene layer was washed with water (200 mL X 2), further with 5% brine
solution (200 mL) and again charged to the RC reactor. Ice-cooled HCI solution
(25mL conc. HCl in l00mL of water) was added to the solution and maintained
the temperature at 0 °C for 15mins before separation of layers. Meanwhile
water (275mL) was taken in a separate RB flask, cooled to 0 °C and added
K2C03 (70.8 g) followed by L-Valine (27.4 g) and CuSO4. 5H20 (50mg) at 0 °C.
This Valine complex was then charged to the empty RC reactor and stirred at 0
°C. To the above solution charged the aqueous layer (collected after HCI
addition) and maintained reaction temperature at 0 °C and continued stirring

155
for 1 hr before the reaction mixture was drained-off. The brief details are
summarized in Figure-3.7.
Figure 3.7: Step wise observations of reaction calorimetric studies
Summary of investigations: Our investigations with Reaction calorimeter
studies have revealed that, out of all the reaction steps “Imidazole addition” is
one of the first key step which can lead to an adiabatic temperature rise of
about 32 °C with an overall evolved heat of 261.2KJ/Kg of Imidazole. Second
key step is aqueous layer containing Imidazole-1-sulfonyl azide as
hydrochloride salt (65) addition to L-Valine complex which can lead to an

156
adiabatic temperature rise of about 10°C with an overall evolved heat of 236.7
KJ/Kg of imidazole.
Precussion Studies: Physical operations like drying, milling, sieving and
packing are commonly employed operations post isolation of an
intermediate and/or an active pharmaceutical ingredient in
manufacturing. The impact sensitivity of a product is easily measured to
understand the sensitivity of a product to shock and therefore exposable,
if it disintegrates with a hammer upon its contact to shock energy under
given test conditions. The process of this measurement is known to be
precussion test or fall hammer test [132].
General test procedure involves; Place the test sample (100mg
sample) enclosed in aluminum foil between the upper and lower parts of
the stamp and let the drop (Drop weight 5Kgs) hammer fall ( drop height
80-100cms). Generally, this test is performed in presence of two persons
in a dark room, to observe the reactions keenly. When a test sample
burns or produces sparks or smoke, it is construed as positive [133].
Three specimen samples of 67 from various experiments were subjected
to “fall hammer test” and found that it is “negative”. It implies that α-
azido ester (67) is not shock sensitive and can be handled in a
pharmaceutical manufacturing environment with normal powder
handling conditions without any safety hazard.

157
Based on safety assessment studies, we are pleased to find that we
could achieve our goal by maintaining sufficient cooling requirements
and we safely were able to test the process robustness under pilot plant
and successfully prepared multi kilogram amounts of 66 and challenges
anticipated in handling azide intermediates have been well addressed
during our safety assessment studies. Therefore, reaction calorimeter
proved to be an invaluable tool in the development of a safer procedure to
prepare 66.
3.4.2 Section-2: Studies towards an improved synthesis of
Valacyclovir (49)
Though we have a new route as described above, the synthesis of
valacyclovir (49) reported by Beauchamp and co-workers involving
condensation of acyclovir (45) and N-carbobenzyloxy-L-valine (52, N-Cbz-
L-valine) followed by de-protection of resulted N-Cbz protected
valacyclovir (53) using palladium catalyst with 55% yield (Scheme-3.9)
was taken for improvement. We have developed an improved process
[134] making use of the foresaid reported process.

158
Scheme 3.9: Synthetic scheme of Valacyclovir (49)
After critical evaluation of the scheme, following criticalities were
identified while practicing Beauchamp‟s approach and are listed below;
1. 3-4% of enantiomer (D-Isomer, 70) was formed [135] along with N-Cbz
valacyclovir (53).
2. Metal impurities palladium and aluminium carried from
palladium/alumina reagent and could not be removed to the
desirable levels by the conventional purifications.
3. Column chromatography and/or repeated crystallizations were
required to meet the desired purity.
4. Process was not economic to practice at industrial scale

159
Having understood the limitations of the reported process, we
attempted to improve the same by considering some of parameters to
address the issues and the process was described in detail below.
When condensation of 45 and 52 was performed in DMF using DCC
in the presence of catalytic amount of DMAP at about 60 °C, 3-4% of
unwanted D-isomer, 70 was observed along with the product, 53. The
formation of D-isomer is a function of reaction temperature. The results
of the controlled experiments were shown in Table-3.6. As the reaction
temperature is raised, impurity was gradually raised and reached to the
level of 3.3% at 25-30°C (Table-3.6, entry 8). Thus, the optimized reaction
temperature was fixed to -5 to 0 °C, wherein 70 was controlled to a level
of 1% in the reaction mass, and hence the temperature is one of the
critical parameter to control this impurity.
Table 3.6: Effect of reaction temperature on racemisaton
entry reaction
temperature (°C)
compound
70 (%)
01 -10-5 1.0
02 -5-0 1.0
03 0-5 1.1
04 5-10 1.0
05 10-15 1.5
06 15-20 1.8
07 20-25 2.7
08 25-30 3.3

160
However, the content of this impurity was increased significantly
during the scale up at the above set temperature. For example, in 50 kg
batch this impurity (70) was raised to 2.6-3% (Table-3.7).
Table 3.7: Variation in 70 content at different scales at -5 to 0 °C
entry scale compound
70 (%)
01 lab scale (upto 100 g) 1.0-1.8
02 kilo lab scale (upto 25 kg) 1.5-2.2
03 plant scale (upto 50 kg) 2.6-3.0
Further examination of the experiment revealed that compound 53
underwent racemization while distilling the DMF at 85 °C over a period of
10-12 h during the work-up of the reaction as shown in Table-3.8. From
this experiment it is understood that, though it is controlled in the
reaction part, distillation of DMF at higher temperature found to be the
culprit for the same at scale.
Table 3.8: Study on thermal sensitivity of N-Cbz Valacyclovir, 53
entry distillation condition compound
70 (%)
01 before distillation 1.1
02 distillation at 85 °C for 12 h 2.6
03 distillation at 85 °C for 30 h 3.2
Thus, the appropriate solvent system was sought to purify the
compound 53 in order to reduce the level of 70. Among the several
solvents explored, acetone-water and methanol-water solvent
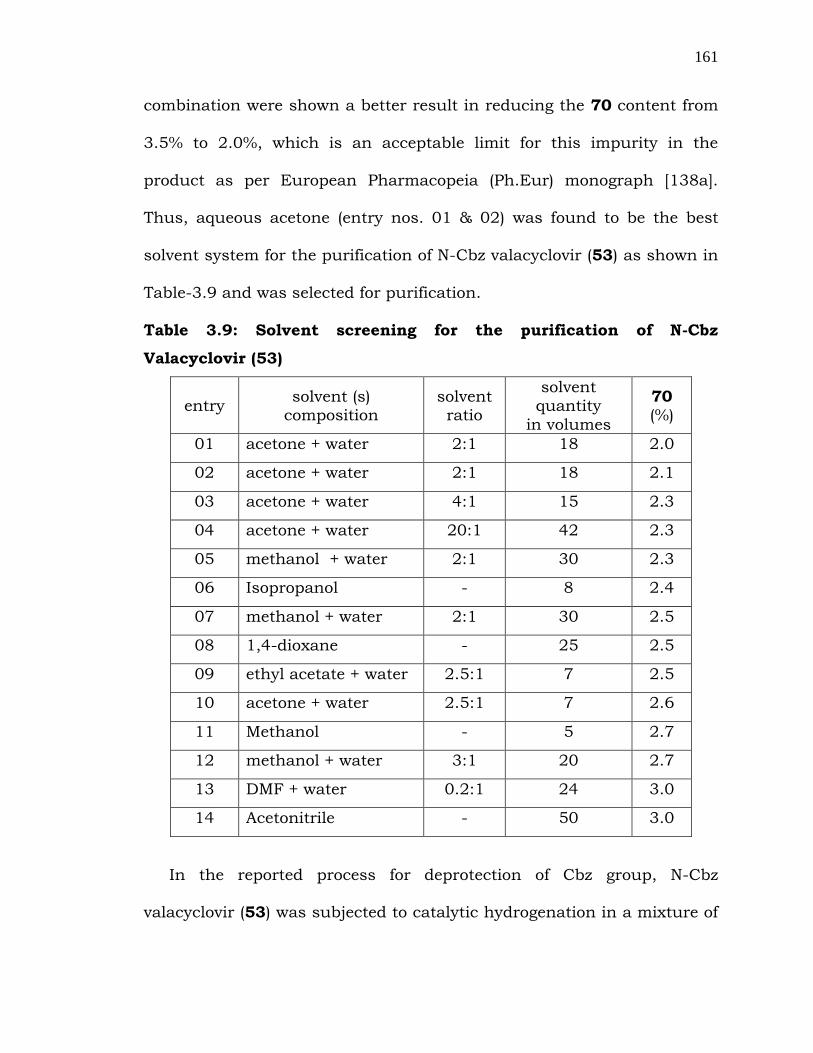
161
combination were shown a better result in reducing the 70 content from
3.5% to 2.0%, which is an acceptable limit for this impurity in the
product as per European Pharmacopeia (Ph.Eur) monograph [138a].
Thus, aqueous acetone (entry nos. 01 & 02) was found to be the best
solvent system for the purification of N-Cbz valacyclovir (53) as shown in
Table-3.9 and was selected for purification.
Table 3.9: Solvent screening for the purification of N-Cbz
Valacyclovir (53)
entry solvent (s)
composition solvent ratio
solvent quantity
in volumes
70 (%)
01 acetone + water 2:1 18 2.0
02 acetone + water 2:1 18 2.1
03 acetone + water 4:1 15 2.3
04 acetone + water 20:1 42 2.3
05 methanol + water 2:1 30 2.3
06 Isopropanol - 8 2.4
07 methanol + water 2:1 30 2.5
08 1,4-dioxane - 25 2.5
09 ethyl acetate + water 2.5:1 7 2.5
10 acetone + water 2.5:1 7 2.6
11 Methanol - 5 2.7
12 methanol + water 3:1 20 2.7
13 DMF + water 0.2:1 24 3.0
14 Acetonitrile - 50 3.0
In the reported process for deprotection of Cbz group, N-Cbz
valacyclovir (53) was subjected to catalytic hydrogenation in a mixture of

162
methanol, THF and aqueous HCl. In our process for the deprotection,
catalytic hydrogenation reaction was conducted in DMF using palladium
supported on alumina. Filtration of the catalyst through celite followed
by usual work-up yielded crude valacyclovir (49) in 92% yield with 98.5%
purity. However, this isolated product contains corresponding
enantiomer (70) to an extent of 3.5-4.5%. As aqueous acetone solvent
used for the washability of 70 could not eliminate/reduce the content of
D-isomer of Valacyclovir, 71 it was decided to study the purification
procedure of Valacyclovir, 49 to achieve more enatiomeric purity. Hence,
various solvents were screened to remove D-isomer (71) from the
compound 49. Acetonitrile-water combination (entries 13 and 14) was
found to be ideal in reducing the 71 content from 4.2% to 2.7% as shown
in Table-3.10.
Table 3.10: Solvent screening for the purification of Valacyclovir
(49)
entry solvent (s)
composition
solvent
ratio
solvent qty. in
vol.
71
(%)
01 Methanol -- 10 4.57
02 Methanol -- 10 4.16
03 DMF -- 5 3.59
04 1,4-dioxane -- 10 3.47
05 tert-butanol -- 10 3.44
06 Dichloromethane -- 10 3.44
07 THF -- 10 3.44
08 methanol + water 12:3 12.5 3.22
09 methanol + water 11.25:3 19 3.00
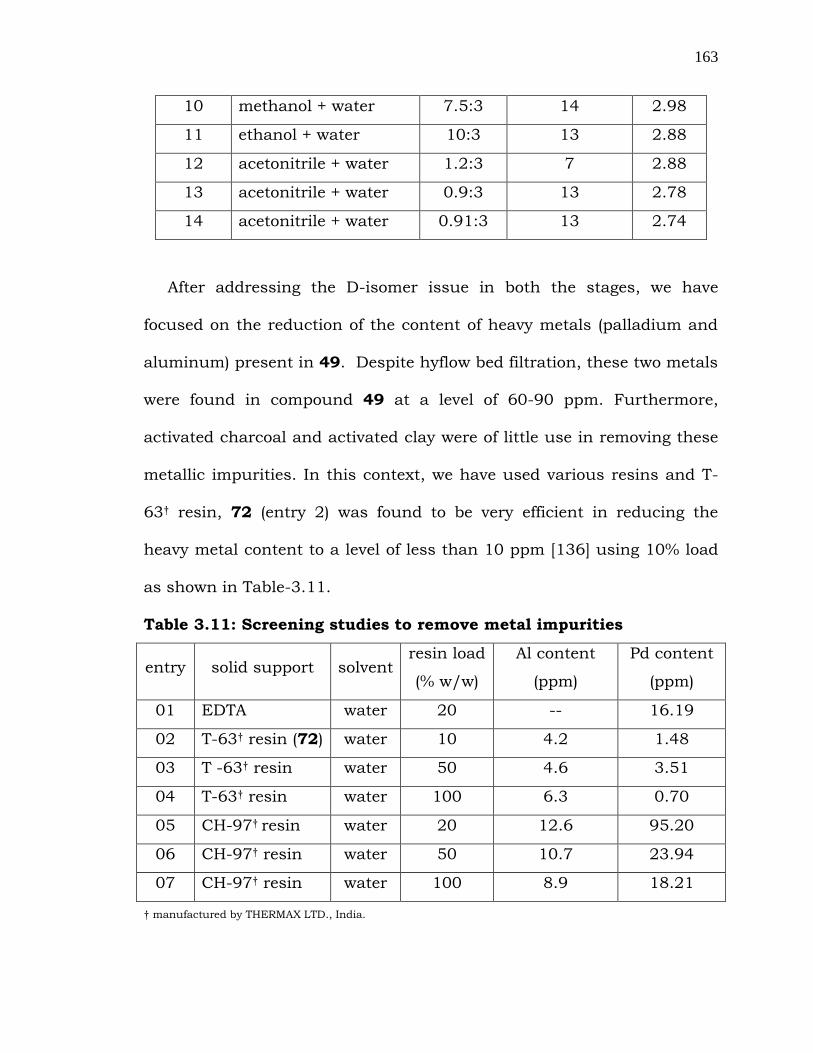
163
10 methanol + water 7.5:3 14 2.98
11 ethanol + water 10:3 13 2.88
12 acetonitrile + water 1.2:3 7 2.88
13 acetonitrile + water 0.9:3 13 2.78
14 acetonitrile + water 0.91:3 13 2.74
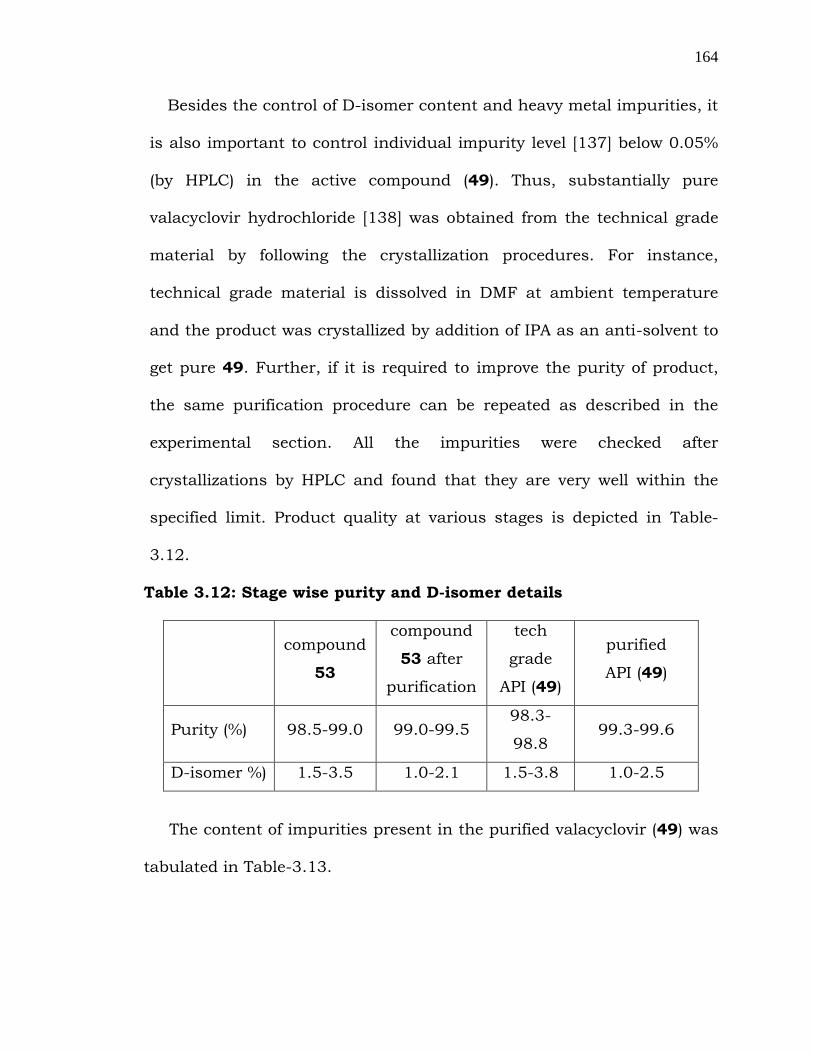
After addressing the D-isomer issue in both the stages, we have
focused on the reduction of the content of heavy metals (palladium and
aluminum) present in 49. Despite hyflow bed filtration, these two metals
were found in compound 49 at a level of 60-90 ppm. Furthermore,
activated charcoal and activated clay were of little use in removing these
metallic impurities. In this context, we have used various resins and T-
63† resin, 72 (entry 2) was found to be very efficient in reducing the
heavy metal content to a level of less than 10 ppm [136] using 10% load
as shown in Table-3.11.
Table 3.11: Screening studies to remove metal impurities
entry solid support solvent resin load
(% w/w)
Al content
(ppm)
Pd content
(ppm)
01 EDTA water 20 -- 16.19
02 T-63† resin (72) water 10 4.2 1.48
03 T -63† resin water 50 4.6 3.51
04 T-63† resin water 100 6.3 0.70
05 CH-97† resin water 20 12.6 95.20
06 CH-97† resin water 50 10.7 23.94
07 CH-97† resin water 100 8.9 18.21
† manufactured by THERMAX LTD., India.
164
Besides the control of D-isomer content and heavy metal impurities, it
is also important to control individual impurity level [137] below 0.05%
(by HPLC) in the active compound (49). Thus, substantially pure
valacyclovir hydrochloride [138] was obtained from the technical grade
material by following the crystallization procedures. For instance,
technical grade material is dissolved in DMF at ambient temperature
and the product was crystallized by addition of IPA as an anti-solvent to
get pure 49. Further, if it is required to improve the purity of product,
the same purification procedure can be repeated as described in the
experimental section. All the impurities were checked after
crystallizations by HPLC and found that they are very well within the
specified limit. Product quality at various stages is depicted in Table-
3.12.
Table 3.12: Stage wise purity and D-isomer details
compound
53
compound
53 after
purification
tech
grade
API (49)
purified
API (49)
Purity (%) 98.5-99.0 99.0-99.5 98.3-
98.8 99.3-99.6
D-isomer %) 1.5-3.5 1.0-2.1 1.5-3.8 1.0-2.5
The content of impurities present in the purified valacyclovir (49) was
tabulated in Table-3.13.
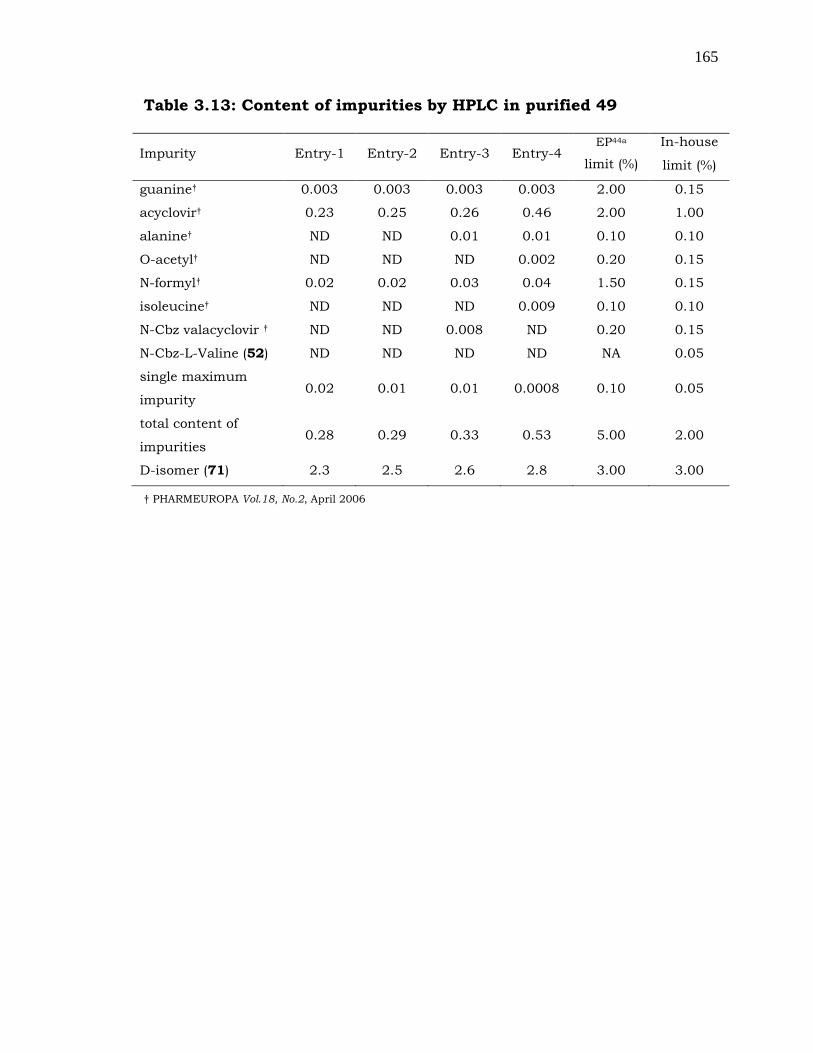
165
Table 3.13: Content of impurities by HPLC in purified 49
Impurity Entry-1 Entry-2 Entry-3 Entry-4 EP44a
limit (%)
In-house
limit (%)
guanine† 0.003 0.003 0.003 0.003 2.00 0.15
acyclovir† 0.23 0.25 0.26 0.46 2.00 1.00
alanine† ND ND 0.01 0.01 0.10 0.10
O-acetyl† ND ND ND 0.002 0.20 0.15
N-formyl† 0.02 0.02 0.03 0.04 1.50 0.15
isoleucine† ND ND ND 0.009 0.10 0.10
N-Cbz valacyclovir † ND ND 0.008 ND 0.20 0.15
N-Cbz-L-Valine (52) ND ND ND ND NA 0.05
single maximum
impurity 0.02 0.01 0.01 0.0008 0.10 0.05
total content of
impurities 0.28 0.29 0.33 0.53 5.00 2.00
D-isomer (71) 2.3 2.5 2.6 2.8 3.00 3.00
† PHARMEUROPA Vol.18, No.2, April 2006

166
3.5 CONCLUSIONS
We achieved our goal in two aspects. Firstly, a new and concise
synthesis for Valacyclovir, 49 was developed by the azide precursor. The
azide chemistry is explored for the first time in the synthesis of
valacyclovir. Challenges anticipated in handling azide intermediates have
been well addressed during our safety assessment studies. We have seen
how the reaction calorimeter is a common tool to investigate chemical
reaction kinetics, to determine required data for chemical process safety
and to access fundamental information about phase changes. We have
presented that the integrated calorimetric approach substantiates that
the process parameters can be envisaged to ensure successful scale up of
optimized process at industrial level. The approach is extremely
attractive and also opens a new pathway to prepare other guanine
containing acyclic nucleosides.
Secondly, a facile and efficient process was developed for Valacyclovir
by controlling the related substances including chiral impurity following
the well known process by Beckump. Furthermore, process related heavy
metal impurities were effectively controlled to acceptable limits using
inexpensive and commercially available resin.

167
3.6 EXPERIMENTAL SECTION
General Procedures: All starting materials were commercial products.
The solvents and reagents were used without any purification. Melting
points (mp) were recorded with Buchi melting point B-540 instrument
and are uncorrected. IR spectra were recorded in the solid state as a KBr
dispersion using a Perkin- Elmer FT-IR spectrophotometer and only
diagnostic and/or intense peaks are reported. PNMR spectra were
recorded in CDCl3 and DMSO-d6 with Varian Mercury Plus 400 MHz
instrument. The chemical shifts are reported in δ ppm relative to TMS.
Multiplicity is indicated by one or more of the following: s (singlet), d
(doublet), t (triplet), q (quartet), m (multiplet), br (broad); Analysis of
metals was done by the use of inductively coupled plasma-optical
emission spectroscopy (ICP-OES). ICP-OES analysis is accurate to parts
per billion (ppb) to parts per trillion (ppt) range and applicable to most of
the metals in pharmaceuticals.
3.6.1 Preparation of N2-DMF acyclovir 68a: A mixture of acyclovir (50
g, 0.222 mol) and N,N-dimethylformamido dimethylacetal (500 mL, 5 vol.)
was maintained at reflux for 90 minutes. The reaction mixture was
cooled to 25 °C and water was charged for solid separation. The
separated solid was filtered and dried at 50 °C to give the desired product
68a (45 g, 98 % by HPLC).

168
3.6.2 Preparation of N2-Acetyl acyclovir 68b: N2,N9-Diacetylacyclovir
(25 g, 0.081 mol) was taken in methanol (125 mL) at ambient
temperature. TMS chloride (25 g, 0.231 mol) was then added (for
selective deprotection) to the mixture at 25 °C and maintained at the
same temperature for an hour. After completion of the reaction by TLC,
the separated solid was filtered followed by methanol washings. The solid
was dried at 60 °C to get the desired product 68b (20 g, 96.4% by HPLC).
3.6.3 Preparation of N2-Trityl acyclovir 68c: Acyclovir (25 g, 0.111
mol) was taken in DMF (250 mL) along with TEA (59.4 g, 0.588 mol). The
mixture was heated to 50 °C followed by drop wise addition of trityl
chloride solution in DMF (250 mL) at 50 °C (80.5 g, 0.289 mol). Reaction
mixture was maintained for completion of the reaction at 50 °C. Reaction
mass was cooled to room temperature and unwanted solids were filtered.
Add water (500 mL) to the filtrate and the separated solid was filtered.
The obtained filtrate was triturated with water (1000 mL). The reaction
mixture was aged for 1 hr and the solid was filtered and dried at 65 °C
under reduced pressure to get the desired mono N-trityl acyclovir 68c
(22 g, 98% by HPLC); IR (νmax, cm-1) 3468, 3101, 2931, 1686, 1632,
1579, 1269, 1120, 790; 1H NMR (DMSO-d6, 400 MHz) δ: 10.64 (s, 1 H),
7.73 (s, 1 H), 7.67 (s, 1 H), 7.3 (m, 15 H), 4.83 (s, 2 H), 4.51 (t, J = 5.6
Hz, 1 H), 3.20 (q, J = 5.6 Hz, 2 H), 2.92 (t, J = 4.8 Hz, 2 H).

169
3.6.4 Preparation of 69a: L-Valine azide (60%, 13.5 g, 0.056 mol) was
taken along with DMF (150 mL) and cooled to 10-15 °C. DCC (15.4 g,
0.074 mol) was charged under nitrogen and the mixture was aged for 30
minutes at 10-15 °C. DMF-acyclovir, 68a (10 g, 0.037 mol) was charged
followed by DMAP (0.686 g, 0.0056 mol) at the same temperature.
Reaction mass was aged for 2 h and solid was separated from the
reaction mass by filtration. Clear filtrate was subjected to cooling to 10-
15 °C followed by drop wise addition of water (750 mL) for separation of
solid. Separated solid was filtered and dried at 60 °C to give desired
product 69a (13.6 g, 90% by HPLC); IR (νmax, cm-1) 3085, 3044, 2102,
1741, 1663, 1633, 1113; 1H NMR (DMSO-d6, 400 MHz) δ: 11.33 (s, 1 H),
8.57 (s, 1 H), 7.93 (s, 1 H), 5.45 (s, 2 H), 4.32 (m, 1 H), 4.23 (m, 1 H),
4.10 (d, J = 4.8 Hz, 1 H), 3.16 (s, 3 H), 3.03 (s, 3 H), 2.03 (m, J = 6.8 Hz,
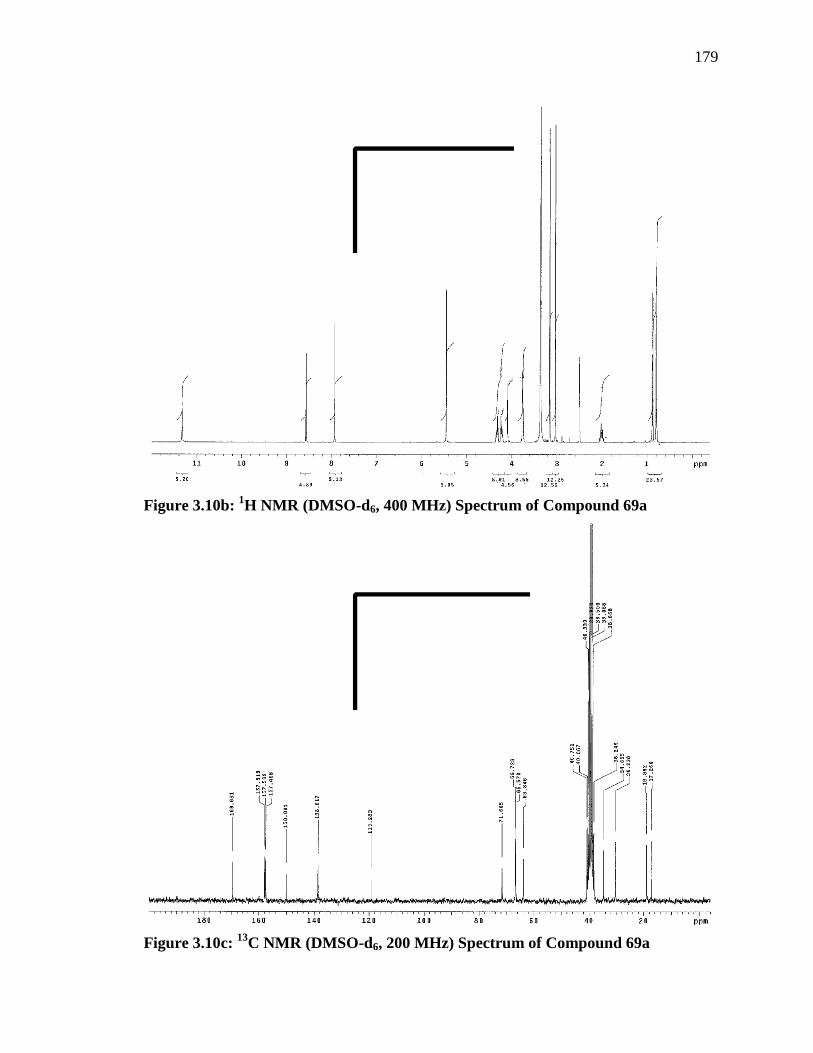
1 H), 0.88 (d, J = 7.2 Hz, 3 H), 0.80 (d, J = 6.8 Hz, 3 H).
3.6.5 Preparation of 69b: L-Valine azide (60%, 6.7 g, 0.028 mol) was
taken along with DMF (50 mL) under nitrogen atmosphere and cooled to
10-15 °C. DCC (11.25 g, 0.054 mol) was charged and the mixture was
aged for 30 minutes at 10-15 °C. A pre cooled (10-15 °C) mixture of N2-
acetyl acyclovir, 68b (5 g, 0.019 mol), DMAP (2.5 g, 0.020 mol) and DMF
(75 mL) was added to the azide solution at 10-15 °C. After completion of
the reaction by TLC, by-product was removed by filtration and the filtrate
was triturated with water (625 mL) at 10-15 °C. Separated solid was

170
filtered and dried at 70 °C to get the desired compound 69b (3 g, 97.2%
by HPLC); IR (νmax, cm-1) 3238, 3167, 3085, 2107, 1743, 1688, 1667,
1264, 1102; 1H NMR (DMSO-d6, 400 MHz) δ: 12.0 (s, 1 H), 11.78 (s, 1
H), 8.13 (s, 1 H), 5.48 (s, 2 H), 4.31 (m, 1 H), 4.21 (m, 1 H), 4.08 (d, J =
5.2 Hz, 1 H), 2.01 (m, J = 5.2 Hz, 1 H), 2.18 (s, 3 H), 0.88 (d, J = 6.8 Hz,
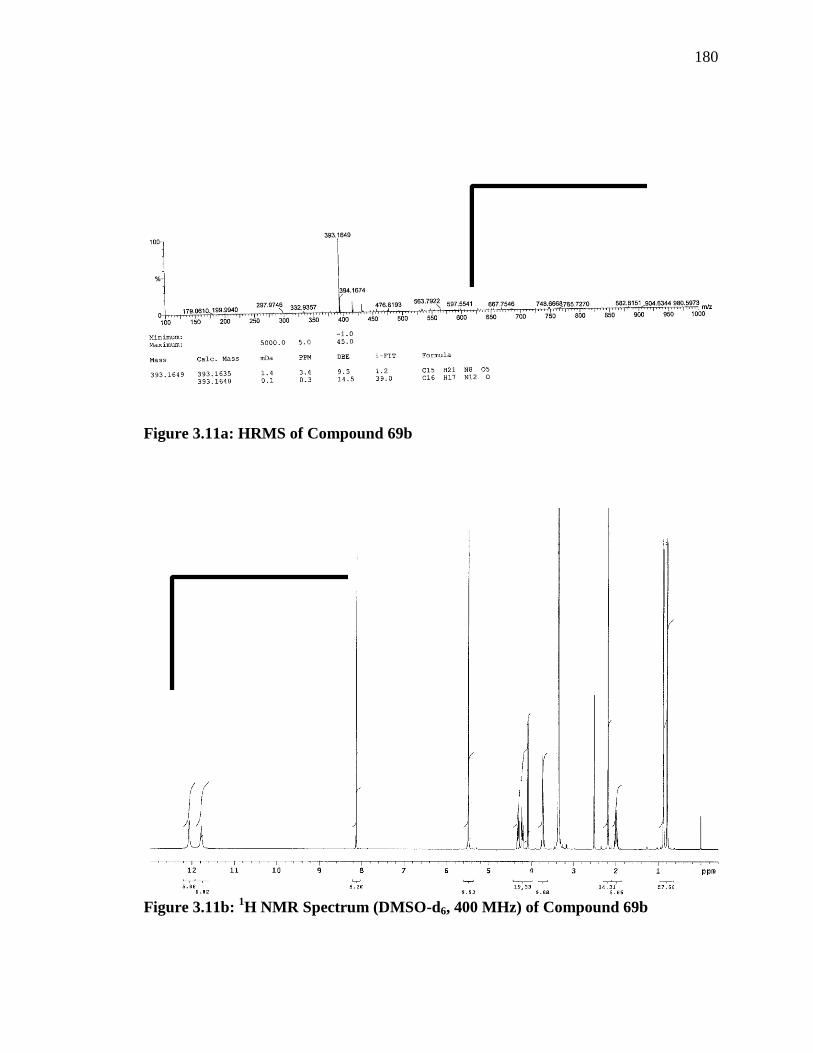
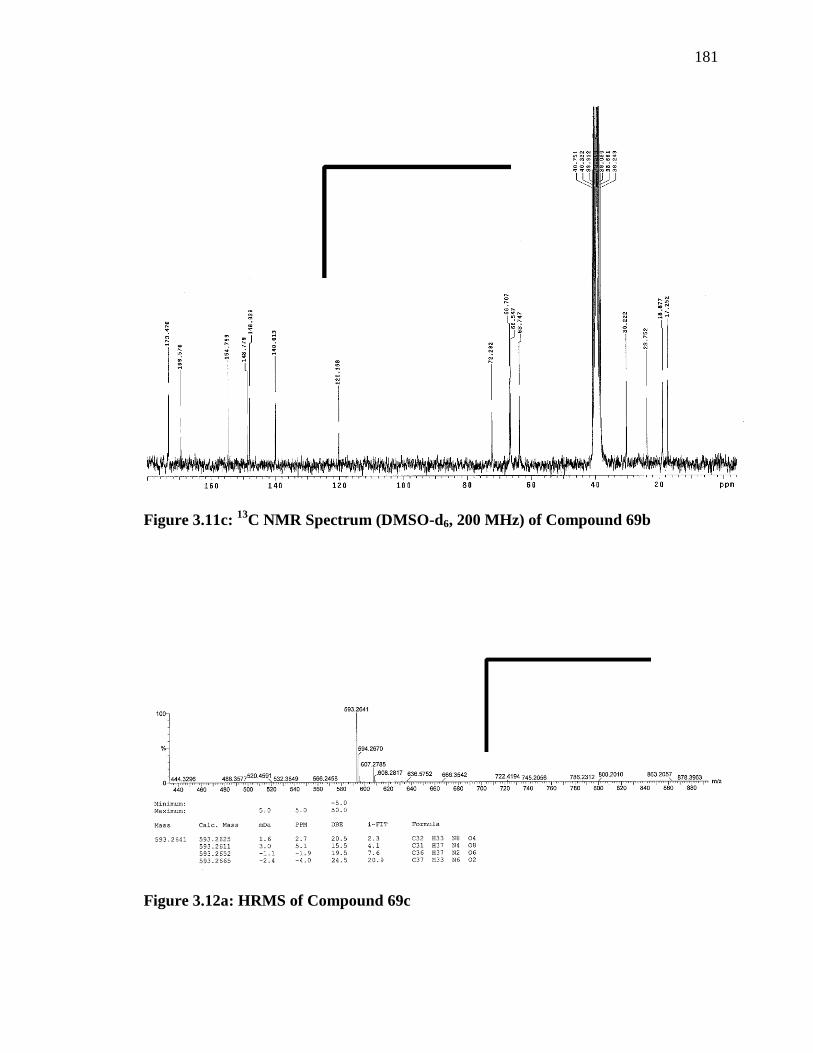
3 H), 0.79 (d, J = 6.8 Hz, 3 H).
3.6.6 Preparation of 69c: L-Valine azide (60%, 7.67 g, 0.032 mol) was
taken along with DMF (150 mL) and cooled to 10-15 °C. DCC (8.8 g,
0.042 mol) was charged under nitrogen and the mixture was aged for 30
minutes at 10-15 °C. N-Trityl-acyclovir, 68c (10 g, 0.021 mol) was
charged followed by DMAP (0.4 g, 0.0033 mol) at the same temperature.
Reaction mass was aged for 1 h and solid was separated from the
reaction mass by filtration. Clear filtrate was subjected to cooling to 10-
15 °C followed by drop wise addition of water (750 mL) for separation of
solid. Separated solid was filtered and dried at 60 °C to give desired
product, 69c (16.5 g, 90% by HPLC); IR (νmax, cm-1) 3326, 3117, 3050,
2928, 2102, 1743, 1706, 1625, 1090, 737, 699; 1H NMR (DMSO-d6, 400
MHz) δ: 10.66 (s, 1 H), 7.7 (s, 1H), 7.65 (s, 1 H), 7.21 (m, 15 H), 4.84 (s, 2
H), 4.05 (d, J = 5.2 Hz, 1 H), 4.01 (m, 1 H), 3.86 (m, 1 H), 3.77 (m, 2 H),
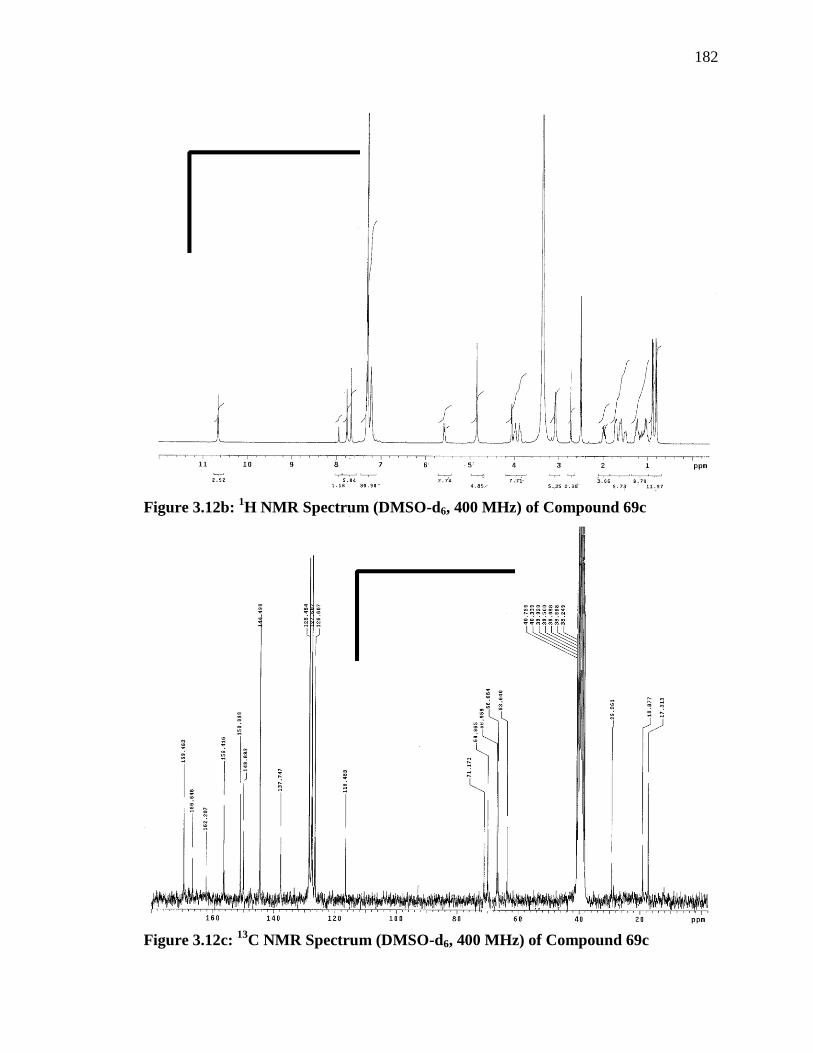
2.0 (m, 1 H), 0.88 (m, J = 6.8, 2 H), 0.81 (m, J = 6.8, 2 H).

171
3.6.7 Preparation of 9-[(2-(2S-Azido-3-methylbutyroxy) ethoxy)
methyl] guanine, 67: L-Valine azide (66, 90%, 21.3 g, 0.13 moles) was
taken along with DMF (400 mL) under nitrogen atmosphere and cooled to
10-15 °C. DCC was charged and the mixture was aged for 30 minutes at
10-15 °C. Acyclovir (20 g, 0.089 moles) was charged followed by DMAP
(1.62 g, 0.013 moles) at the same temperature. Reaction mass was aged
for 45 minutes and solid was separated from the reaction mass by
filtration. Clear filtrate was subjected to cooling to 0-5°C followed by drop
wise addition of water (1350 mL) for separation of solid. Separated solid
was filtered and dried at 60 °C to give desired product, 67 (25.5 g, 82%)
which was confirmed by spectral data and purity by HPLC was found to
be 92.81%.
3.6.8 Preparation of valacyclovir hydrochloride 49: Compound 67 (20
g) was taken in ethanol (300 mL) along with raney nickel (4 g) and
maintained at 50-60 °C for about 7 hours. After reaction was completed,
reaction mass was cooled to ambient temperature and unwanted solid
was separated by filtration. Filtrate pH was adjusted to 2.5-3.0 using
aqueous hydrochloric acid (10%, 35 mL). The homogeneous reaction was
treated with charcoal at 50-60 °C and then charcoal was separated by
hyflow filtration. The filtrate was concentrated under reduced pressure at
50°C. The obtained residue was treated with acetone (100 mL) and the

172
product, 49 (16.9 g, 82%) was separated by filtration followed by drying
at 50 °C and 49 is confirmed by spectral data.
3.6.9 Preparation of Valacyclovir (49)
(a) Preparation of Cbz-protected Valacyclovir (53): Cbz-L-Valine (52,
83.6 g, 0.332 mol) was dissolved in DMF (350 mL) and the solution was
cooled to -5 °C. A solution of DCC (68.6 g, 0.333 mol) in DMF (150 mL)
was added below 0 °C. After aging for 20 minutes, acyclovir (45, 50 g,
0.222 mol) and DMAP (4 g, 0.032 mol) were charged and reaction
mixture was stirred at -5-0 °C for about six hours. Dicyclohexylurea was
filtered, 80% of the solvent was removed by distillation and the remaining
solution was diluted with water (300 mL). Precipitated compound 53 was
filtered at ambient temperature and recrystallised from methanol (88.5 g,
87%). Purity by HPLC 99.3%, mp 157-159 °C; IR (KBr) cm-1 3311, 2931,
2855, 1726, 1630, 1606, 1536, 1390, 1183, 1128, 1101, 1041; 1H NMR
(CDCl3, 200 MHz) δ: 10.8 (br, 1H), 7.9 (br, 1H), 7.7 (s, 1H), 7.22 (m, 5H),
5.38 (s, 2H), 5.1 (s, 2H), 4.25 (m, 1Ha), 4.35 (m, 1Hb), 3.75 (d, 1H), 3.7
(m, 2H), 2.15 (m, 1H), 0.91 (d, 3H), 0.88 (d, 3H).
Purification of Cbz-protected Valacyclovir (53): Cbz Protected
valacyclovir (53, 25 g) having 3.1% of D-isomer, 70 was dissolved in a
mixture of acetone (300 mL) and water (75 mL) at reflux temperature and
cooled to ambient temperature. The mixture was stirred for few hours

173
after diluting with additional water (75 mL) and filtered to provide
compound 53 (21.2 g, 84.8%, 2.1% 70 by HPLC).
(b) Deprotection of Cbz group of 53: Cbz Protected valacyclovir (53, 5
g, 0.011 mol) and dry 5% Pd on alumina mixture (0.5 g) were taken in
DMF (50 mL) in a hydrogenator vessel. Hydrogen pressure of 4 kg/cm2
was applied at about 30 °C for the completion of the reaction. 70% of the
solvent was removed by distillation under vacuum below 80 °C and the
resultant concentrated solution was cooled to 10 °C. pH was adjusted to
3.0-4.0 using aqueous HCl at 10 °C, diluted with water (12.5 mL) and
catalyst was removed by filtration through celite at ambient temperature.
Filtrate was saturated with acetone (225 mL), precipitated valacyclovir
HCl (49) was filtered and dried under suction (4.7 g, 98.5% pure by
HPLC).
3.6.10 Purification of Valacyclovir HCl (49) to remove 71:
Valacyclovir HCl (49, 25 g) having 3.5% of 71 was dissolved in 25%
aqueous acetonitrile (250 mL) at 70 °C. The mixture was cooled to 30 °C
and diluted slowly with acetonitrile (75 mL) at 30 °C under stirring. The
separated solid was filtered and dried under vacuum to yield compound
49 (19.5 g, 75%, 2.6% 71 by HPLC).
3.6.11 Procedure for removal of heavy metal impurities: A
suspension of valacyclovir hydrochloride (49, 25 g) in water (50 mL) was

174
heated to 65 °C with simultaneous stirring. To the obtained solution, T-
63 resin (procured from Thermax Ltd.) was added and the mixture was
stirred for 20 minutes. The suspension was filtered through 0.45 m
filter paper and washed with water (25 mL). To the filtrate, acetone (500
mL) was added slowly. The obtained suspension was stirred for 1 h and
the precipitate was filtered and dried to yield compound 49 (24 g, 96%,
both Pd and Al content less than 10 ppm).
3.6.12 Purification of Valacyclovir HCl (49): Valacyclovir HCl (49, 50
g) wet material obtained from the above procedure was taken in DMF
(230 mL) and maintained for 7 h at 30 °C. Isopropanol (115 mL) was
added to the above mixture over a period of an hour and isolated product
was filtered at ambient temperature, washed with isopropanol and dried
under vacuum at 60 °C. Dried material was subjected to same
purification process to provide pure 49 which was further converted into
dihydrate (49, 20.7 g, 99.66% purity by HPLC) having individual
impurity below 0.05%, mp decomposed with foaming at 178 °C (lit.
Valacyclovir HCl monohydrate mp 150 °C, decomposes with foaming at
195 °C); IR (KBr) cm-1 3327, 3196, 2967, 1732, 1631, 1101, 1037; 1H
NMR (DMSO-d6, 200 MHz) δ: 10.9 (br, 1H), 8.5 (br, 3H), 7.83 (s,1H), 6.6
(s, 2H), 5.38 (s, 2H), 4.40 (m, 1Hb), 4.28 (m, 1Ha), 3.82 (d, 1H), 3.74 (m,
2H), 2.13 (m, 1H), 0.91 (d, 3H), 0.88 (d, 3H).
175
3.7 APPENDIX
Spectral data of active pharmaceutical ingredient, novel intermediates
and impurities are mentioned as follows.
Figure 3.8a: Mass Spectrum of Compound 49
Figure 3.8b:
1H NMR Spectrum (DMSO-d6, 400 MHz) of Compound 49
176
Figure 3.8c: D2O- NMR Spectrum (DMSO-d6, 400 MHz) of Compound 49
Figure 3.8d:
13C NMR Spectrum (DMSO-d6, 400 MHz) of Compound 49
177
Figure 3.9a: HRMS of Compound 68c
Figure 3.9b:
1H NMR spectrum (DMSO-d6, 400 MHz) of Compound 68c
178
Figure 3.9c:
13C NMR spectrum (DMSO-d6, 200 MHz) of Compound 68c
Figure 3.10a: HRMS of Compound 69a
179
Figure 3.10b:
1H NMR (DMSO-d6, 400 MHz) Spectrum of Compound 69a
Figure 3.10c:
13C NMR (DMSO-d6, 200 MHz) Spectrum of Compound 69a
180
Figure 3.11a: HRMS of Compound 69b
Figure 3.11b:
1H NMR Spectrum (DMSO-d6, 400 MHz) of Compound 69b
181
Figure 3.11c: 13
C NMR Spectrum (DMSO-d6, 200 MHz) of Compound 69b
Figure 3.12a: HRMS of Compound 69c
182
Figure 3.12b: 1H NMR Spectrum (DMSO-d6, 400 MHz) of Compound 69c
Figure 3.12c: 13
C NMR Spectrum (DMSO-d6, 400 MHz) of Compound 69c
183
Figure 3.13a: HRMS Spectrum of Compound 67
Figure 3.13b:
1H NMR Spectrum (DMSO-d6, 400 MHz)) of Compound 67

184
Figure 3.13c: 13
C NMR Spectrum (DMSO-d6, 200 MHz) of Compound 67








![INDEX 0269 [globalgenealogy.com]globalgenealogy.com/countries/canada/ontario/eastern-ontario/... · BEAUCHAMP, Yvon Gerard 0216 BERTRAND, Rose Delima 0093 BEAUCHAMP, Yvonne 0084A](https://img.pdfslide.us/doc/110x75/5b9b79ef09d3f2d06f8cf723/index-0269-beauchamp-yvon-gerard-0216-bertrand-rose-delima-0093-beauchamp.jpg)



















