Embed Size (px)
Citation preview

Pergolide (Permax)
Oral: 0.05, 0.25, 1.0 mg tablets
Protirelin (Thypinone, Relefact TRH, Thyrel TRH)
Parenteral: 500 mg/mL for injection
Sermorelin (Geref)
Parenteral: 0.5, 1.0 mg for subcutaneous injection; 50 g powder to reconstitute for intravenous injection
Somatrem (Protropin)
Parenteral: 5, 10 mg/vial with diluent for subcutaneous or IM injection
Somatropin (Genotropin, Humatrope, Nutropin, Nutropin AQ, Norditropin, Serostim, Saizen)
Parenteral: 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.5, 1.6, 1.8, 2, 4, 5, 5.8, 6, 8, 10, 12, 13.5, 13.8, 15, 18, 22.5, 24 mg/vial with diluent for subcutaneous or IM injection
Thyrotropin alpha (Thyrogen)
Parenteral: 1.1 mg (> 4 IU)/vial with diluent for IM injection
Triptorelin (Trelstar)
Parenteral: 3.75, 11.25 mg for IM injection
Urofollitropin (Fertinex, Bravelle)
Parenteral: powder to reconstitute for injection, 75, 150 IU FSH activity per ampule
Vasopressin (generic, Pitressin)
Parenteral: 20 pressor units/mL for IM or subcutaneous administration Chapter 38. Thyroid & Antithyroid Drugs Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 38. Thyroid & Antithyroid Drugs >
Thyroid & Antithyroid Drugs: Introduction
Because of its anatomic prominence, the thyroid was one of the first of the endocrine glands to be associated with the clinical conditions caused by its malfunction.
Thyroid Physiology

The normal thyroid gland secretes sufficient amounts of the thyroid hormones—triiodothyronine (T3) and tetraiodothyronine (T4, thyroxine)—to normalize growth and development, body temperature, and energy levels. These hormones contain 59% and 65% (respectively) of iodine as an essential part of the molecule. Calcitonin, the second type of thyroid hormone, is important in the regulation of calcium metabolism and is discussed in Chapter 42: Agents That Affect Bone Mineral Homeostasis.
Iodide Metabolism
The recommended daily adult iodide (I–)* intake is 150 g (200 g during pregnancy).
* In this chapter, the term "iodine" denotes all forms of the element; the term "iodide" denotes only the ionic form, I–.
Iodide, ingested from food, water, or medication, is rapidly absorbed and enters an extracellular fluid pool. The thyroid gland removes about 75 g a day from this pool for hormone secretion, and the balance is excreted in the urine. If iodide intake is increased, the fractional iodine uptake by the thyroid is diminished.
Biosynthesis of Thyroid Hormones
Once taken up by the thyroid gland, iodide undergoes a series of enzymatic reactions that convert it into active thyroid hormone (Figure 38–1). The first step is the transport of iodide into the thyroid gland by an intrinsic follicle cell basement membrane protein called the sodium/iodide symporter (NIS). This can be inhibited by such anions as SCN–, TcO4
–, and ClO4–. Iodide is then oxidized by
thyroidal peroxidase to iodine, in which form it rapidly iodinates tyrosine residues within the thyroglobulin molecule to form monoiodotyrosine (MIT) and diiodotyrosine (DIT). This process is called iodide organification. Thyroidal peroxidase is transiently blocked by high levels of intrathyroidal iodide and blocked more persistently by thioamide drugs. Two molecules of DIT combine within the thyroglobulin molecule to form L-thyroxine (T4). One molecule of MIT and one molecule of DIT combine to form T3. In addition to thyroglobulin, other proteins within the gland may be iodinated, but these iodoproteins do not have hormonal activity. Thyroxine, T3, MIT, and DIT are released from thyroglobulin by exocytosis and proteolysis of thyroglobulin at the apical colloid border. The MIT and DIT are deiodinated within the gland, and the iodine is reutilized. This process of proteolysis is also blocked by high levels of intrathyroidal iodide. The ratio of T4 to T3 within thyroglobulin is approximately 5:1, so that most of the hormone released is thyroxine. Most of the T3 circulating in the blood is derived from peripheral metabolism of thyroxine (see below).
Figure 38–1.
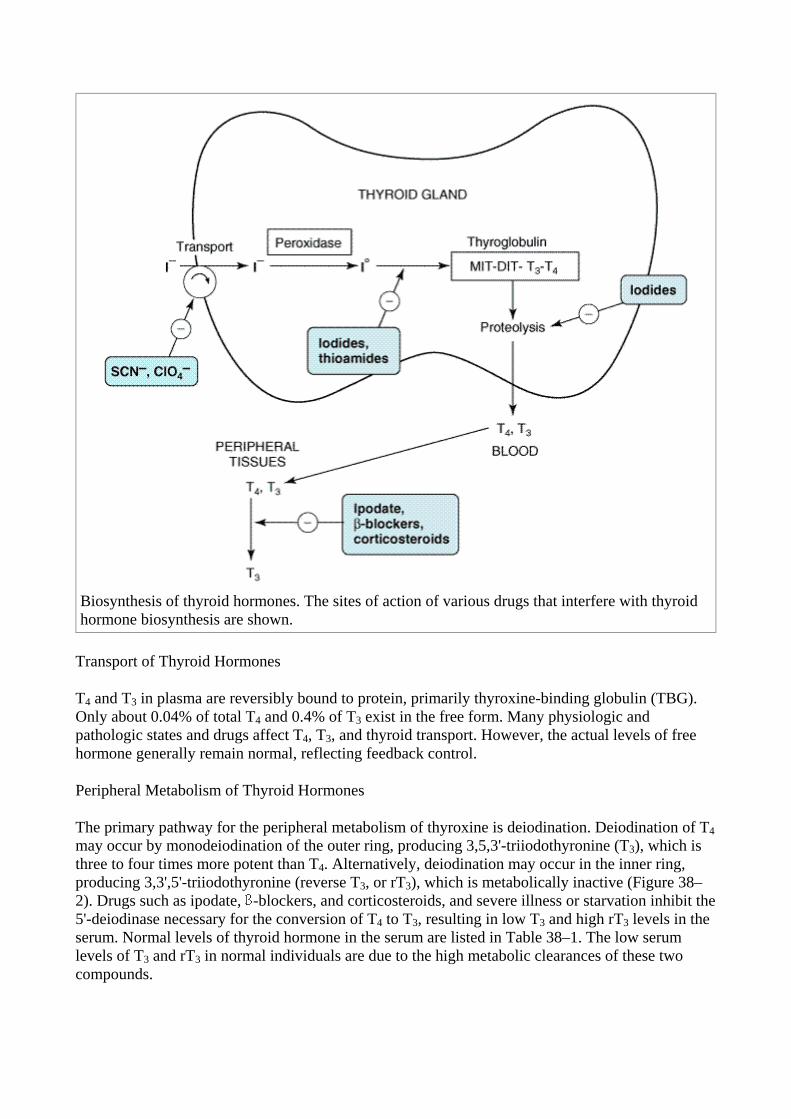
Biosynthesis of thyroid hormones. The sites of action of various drugs that interfere with thyroid hormone biosynthesis are shown.
Transport of Thyroid Hormones
T4 and T3 in plasma are reversibly bound to protein, primarily thyroxine-binding globulin (TBG). Only about 0.04% of total T4 and 0.4% of T3 exist in the free form. Many physiologic and pathologic states and drugs affect T4, T3, and thyroid transport. However, the actual levels of free hormone generally remain normal, reflecting feedback control.
Peripheral Metabolism of Thyroid Hormones
The primary pathway for the peripheral metabolism of thyroxine is deiodination. Deiodination of T4may occur by monodeiodination of the outer ring, producing 3,5,3'-triiodothyronine (T3), which is three to four times more potent than T4. Alternatively, deiodination may occur in the inner ring, producing 3,3',5'-triiodothyronine (reverse T3, or rT3), which is metabolically inactive (Figure 38–2). Drugs such as ipodate, -blockers, and corticosteroids, and severe illness or starvation inhibit the 5'-deiodinase necessary for the conversion of T4 to T3, resulting in low T3 and high rT3 levels in the serum. Normal levels of thyroid hormone in the serum are listed in Table 38–1. The low serum levels of T3 and rT3 in normal individuals are due to the high metabolic clearances of these two compounds.
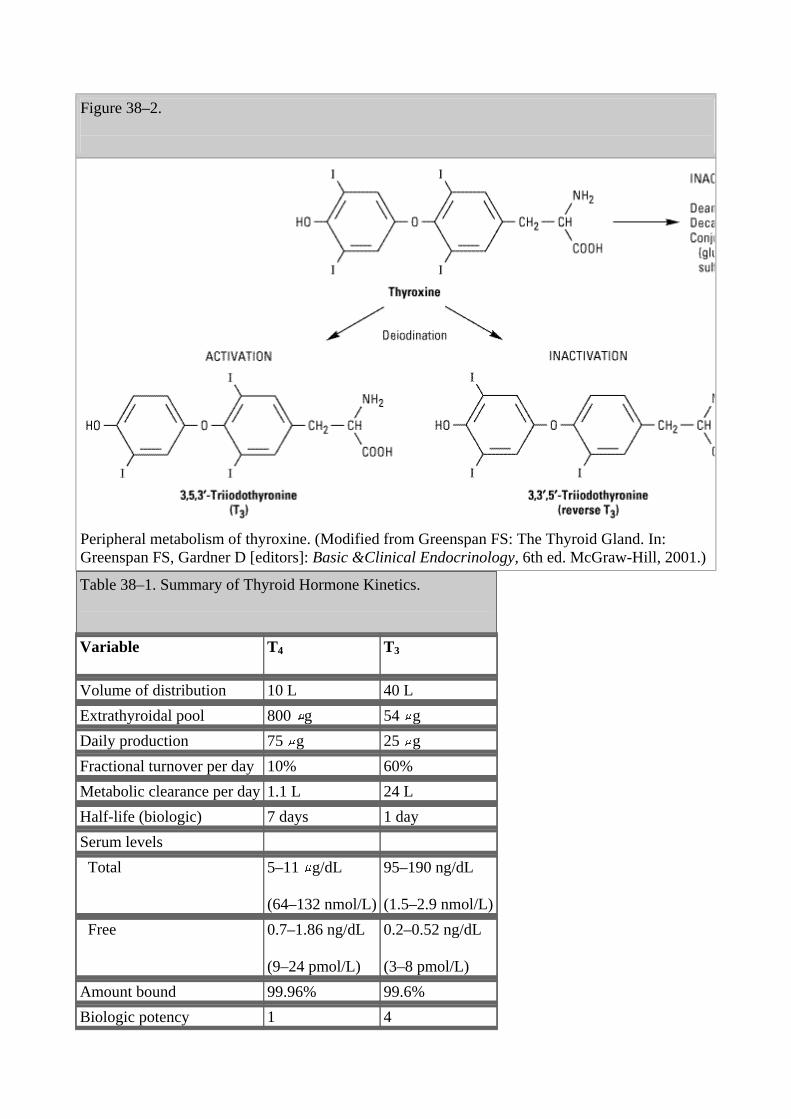
Figure 38–2.
Peripheral metabolism of thyroxine. (Modified from Greenspan FS: The Thyroid Gland. In: Greenspan FS, Gardner D [editors]: Basic &Clinical Endocrinology, 6th ed. McGraw-Hill, 2001.) Table 38–1. Summary of Thyroid Hormone Kinetics.
Variable T4
T3
Volume of distribution 10 L 40 L Extrathyroidal pool 800 g 54 g Daily production 75 g 25 g Fractional turnover per day 10% 60% Metabolic clearance per day 1.1 L 24 L Half-life (biologic) 7 days 1 day Serum levels Total 5–11 g/dL
(64–132 nmol/L)
95–190 ng/dL
(1.5–2.9 nmol/L) Free 0.7–1.86 ng/dL
(9–24 pmol/L)
0.2–0.52 ng/dL
(3–8 pmol/L) Amount bound 99.96% 99.6% Biologic potency 1 4
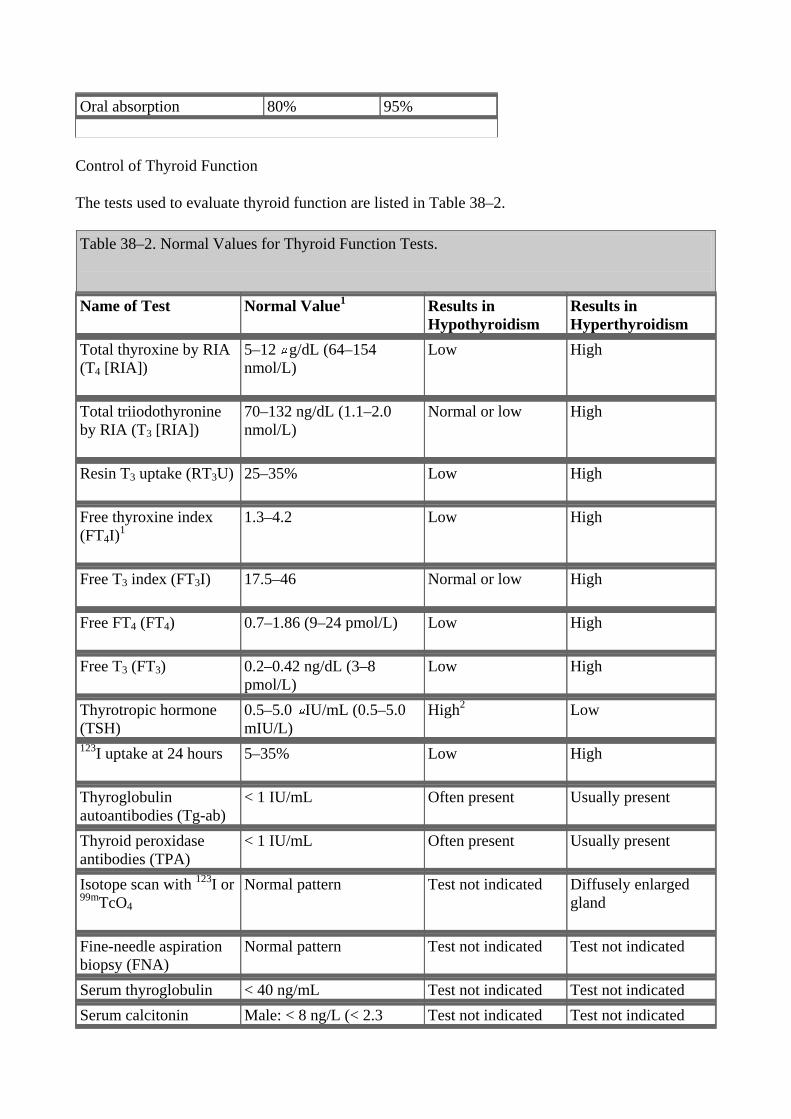
Oral absorption 80% 95%
Control of Thyroid Function
The tests used to evaluate thyroid function are listed in Table 38–2.
Table 38–2. Normal Values for Thyroid Function Tests.
Name of Test Normal Value1
Results in Hypothyroidism
Results in Hyperthyroidism
Total thyroxine by RIA (T4 [RIA])
5–12 g/dL (64–154 nmol/L)
Low High
Total triiodothyronine by RIA (T3 [RIA])
70–132 ng/dL (1.1–2.0 nmol/L)
Normal or low High
Resin T3 uptake (RT3U)
25–35% Low High
Free thyroxine index (FT4I)1
1.3–4.2 Low High
Free T3 index (FT3I)
17.5–46 Normal or low High
Free FT4 (FT4)
0.7–1.86 (9–24 pmol/L) Low High
Free T3 (FT3)
0.2–0.42 ng/dL (3–8 pmol/L)
Low High
Thyrotropic hormone (TSH)
0.5–5.0 IU/mL (0.5–5.0 mIU/L)
High2
Low
123I uptake at 24 hours
5–35% Low High
Thyroglobulin autoantibodies (Tg-ab)
< 1 IU/mL Often present Usually present
Thyroid peroxidase antibodies (TPA)
< 1 IU/mL Often present Usually present
Isotope scan with 123I or 99mTcO4
Normal pattern Test not indicated Diffusely enlarged gland
Fine-needle aspiration biopsy (FNA)
Normal pattern Test not indicated Test not indicated
Serum thyroglobulin < 40 ng/mL Test not indicated Test not indicated Serum calcitonin Male: < 8 ng/L (< 2.3 Test not indicated Test not indicated
pmol/L); female: < 4 ng/L (< 1.17 pmol/L)
1Results may vary with different laboratories.
2Exception is central hypothyroidism
Thyroid-Pituitary Relationships
Control of thyroid function via thyroid-pituitary feedback is also discussed in Chapter 37: Hypothalamic & Pituitary Hormones. Briefly, hypothalamic cells secrete thyrotropin-releasing hormone (TRH) (Figure 38–3). TRH is secreted into capillaries of the pituitary portal venous system, and in the pituitary gland, TRH stimulates the synthesis and release of thyroid-stimulating hormone (TSH). TSH in turn stimulates an adenylyl cyclase–mediated mechanism in the thyroid cell to increase the synthesis and release of T4 and T3. These thyroid hormones act in a negative feedback fashion in the pituitary to block the action of TRH and in the hypothalamus to inhibit the synthesis and secretion of TRH. Other hormones or drugs may also affect the release of TRH or TSH.
Figure 38–3.

The hypothalamic-pituitary-thyroid axis. Acute psychosis or prolonged exposure to cold may activate the axis. Hypothalamic TRH stimulates pituitary TSH release, while somatostatin and dopamine inhibit it. TSH stimulates T4 and T3 synthesis and release from the thyroid, and they in turn inhibit both TRH and TSH synthesis and release. Small amounts of iodide are necessary for hormone production, but large amounts inhibit T3 and T4 production and release. (Solid arrows, stimulatory influence; dashed arrows, inhibitory influence. H, hypothalamus, HP, anterior pituitary.)
Autoregulation of the Thyroid Gland
The thyroid gland also regulates its uptake of iodide and thyroid hormone synthesis by intrathyroidal mechanisms that are independent of TSH. These mechanisms are primarily related to the level of iodine in the blood. Large doses of iodine inhibit iodide organification (Figure 38–1). In certain disease states (eg, Hashimoto's thyroiditis), this can result in inhibition of thyroid hormone synthesis and hypothyroidism.
Abnormal Thyroid Stimulators
In Graves' disease (see below), lymphocytes secrete a TSH receptor-stimulating antibody (TSH-R Ab [stim]), also known as thyroid-stimulating immunoglobulin (TSI). This immunoglobulin binds to the TSH receptor and turns on the gland in the same fashion as TSH itself. The duration of its effect, however, is much longer than that of TSH. TSH receptors are also found in orbital fibrocytes, which may be stimulated by high levels of TSH-R Ab [stim]. Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 38. Thyroid & Antithyroid Drugs >
Basic Pharmacology of Thyroid & Antithyroid Drugs
Thyroid Hormones
Chemistry
The structural formulas of thyroxine and triiodothyronine as well as reverse triiodothyronine (rT3) are shown in Figure 38–2. All of these naturally occurring molecules are levo (L) isomers. The synthetic dextro (D) isomer of thyroxine, dextrothyroxine, has approximately 4% of the biologic activity of the L isomer as evidenced by its lesser ability to suppress TSH secretion and correct hypothyroidism.
Pharmacokinetics
Thyroxine is absorbed best in the duodenum and ileum; absorption is modified by intraluminal factors such as food, drugs, and intestinal flora. Oral bioavailability of current preparations of L-thyroxine averages 80% (Table 38–1). In contrast, T3 is almost completely absorbed (95%). T4 and T3 absorption appears not to be affected by mild hypothyroidism but may be impaired in severe myxedema with ileus. These factors are important in switching from oral to parenteral therapy. For parenteral use, the intravenous route is preferred for both hormones.
In patients with hyperthyroidism, the metabolic clearances of T4 and T3 are increased and the half-lives decreased; the opposite is true in patients with hypothyroidism. Drugs that induce hepatic
microsomal enzymes (eg, rifampin, phenobarbital, carbamazepine, phenytoin) increase the metabolism of both T4 and T3 (Table 38–3). Despite this change in clearance, the normal hormone concentration is maintained in euthyroid patients as a result of compensatory hyperfunction of the thyroid. However, patients receiving T4 replacement medication may require increased dosages to maintain clinical effectiveness. A similar compensation occurs if binding sites are altered. If TBG sites are increased by pregnancy, estrogens, or oral contraceptives, there is an initial shift of hormone from the free to the bound state and a decrease in its rate of elimination until the normal hormone concentration is restored. Thus, the concentration of total and bound hormone will increase, but the concentration of free hormone and the steady state elimination will remain normal. The reverse occurs when thyroid binding sites are decreased.
Table 38–3. Drug Effects and Thyroid Function.
Drug Effect Drugs Change in thyroid hormone synthesis Inhibition of TRH or TSH secretion without induction of hypothyroidism
Dopamine, levodopa, corticosteroids, somatostatin
Inhibition of thyroid hormone synthesis or release with the induction of hypothyroidism (or occasionally hyperthyroidism)
Iodides (including amiodarone), lithium, aminoglutethimide
Alteration of thyroid hormone transport and serum total T3 and T4 levels, but usually no modification of FT4 or TSH
Increased TBG Estrogens, tamoxifen, heroin, methadone, mitotane
Decreased TBG Androgens, glucocorticoids Displacement of T3 and T4 from TBG with transient hyperthyroxinemia
Salicylates, fenclofenac, mefenamic acid, furosemide
Alteration of T4 and T3 metabolism with modified serum T3 and T4 levels but not FT4 or TSH levels
Induction of increased hepatic enzyme activity Phenytoin, carbamazepine, phenobarbital, rifampin, rifabutin
Inhibition of 5'-deiodinase with decreased T3, increased rT3
Iopanoic acid, ipodate, amiodarone, -blockers, corticosteroids, propylthiouracil
Other interactions Interference with T4 absorption
Cholestyramine, colestipol, aluminum hydroxide, sucralfate, reloxifene, ferrous sulfate, some calcium preparations, bran
Induction of autoimmune thyroid disease with hypothyroidism or hyperthyroidism
Interferon- , interleukin-2
Mechanism of Action
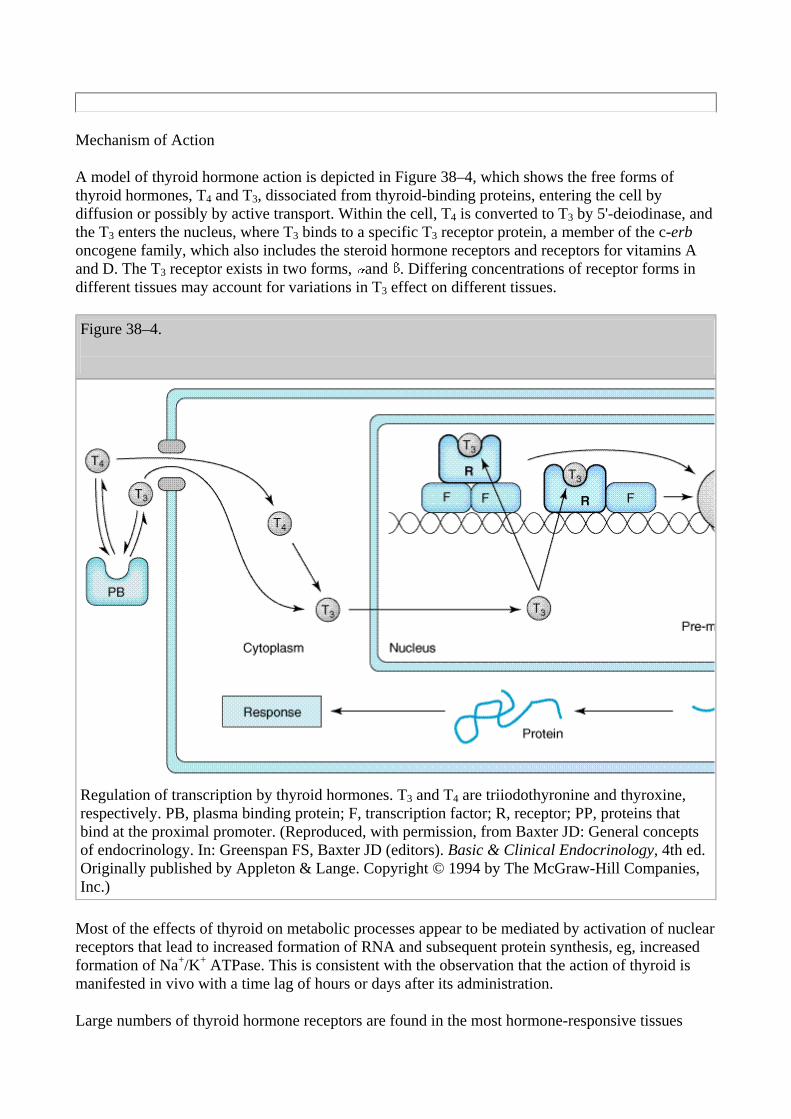
A model of thyroid hormone action is depicted in Figure 38–4, which shows the free forms of thyroid hormones, T4 and T3, dissociated from thyroid-binding proteins, entering the cell by diffusion or possibly by active transport. Within the cell, T4 is converted to T3 by 5'-deiodinase, and the T3 enters the nucleus, where T3 binds to a specific T3 receptor protein, a member of the c-erb oncogene family, which also includes the steroid hormone receptors and receptors for vitamins A and D. The T3 receptor exists in two forms, and . Differing concentrations of receptor forms in different tissues may account for variations in T3 effect on different tissues.
Figure 38–4.
Regulation of transcription by thyroid hormones. T3 and T4 are triiodothyronine and thyroxine, respectively. PB, plasma binding protein; F, transcription factor; R, receptor; PP, proteins that bind at the proximal promoter. (Reproduced, with permission, from Baxter JD: General concepts of endocrinology. In: Greenspan FS, Baxter JD (editors). Basic & Clinical Endocrinology, 4th ed. Originally published by Appleton & Lange. Copyright © 1994 by The McGraw-Hill Companies, Inc.)
Most of the effects of thyroid on metabolic processes appear to be mediated by activation of nuclear receptors that lead to increased formation of RNA and subsequent protein synthesis, eg, increased formation of Na+/K+ ATPase. This is consistent with the observation that the action of thyroid is manifested in vivo with a time lag of hours or days after its administration.
Large numbers of thyroid hormone receptors are found in the most hormone-responsive tissues
(pituitary, liver, kidney, heart, skeletal muscle, lung, and intestine), while few receptor sites occur in hormone-unresponsive tissues (spleen, testes). The brain, which lacks an anabolic response to T3, contains an intermediate number of receptors. In congruence with their biologic potencies, the affinity of the receptor site for T4 is about ten times lower than that for T3. The number of nuclear receptors may be altered to preserve body homeostasis. For example, starvation lowers both circulating T3 hormone and cellular T3 receptors.
Effects of Thyroid Hormones
The thyroid hormones are responsible for optimal growth, development, function, and maintenance of all body tissues. Excess or inadequate amounts result in the signs and symptoms of thyrotoxicosis or hypothyroidism (Table 38–4). Since T3 and T4 are qualitatively similar, they may be considered as one hormone in the discussion that follows.
Table 38–4. Manifestations of Thyrotoxicosis and Hypothyroidism.
System Thyrotoxicosis Hypothyroidism Skin and appendages
Warm, moist skin; sweating; heat intolerance; fine, thin hair; Plummer's nails; pretibial dermopathy (Graves' disease)
Pale, cool, puffy skin; dry and brittle hair; brittle nails
Eyes, face Retraction of upper lid with wide stare; periorbital edema; exophthalmos; diplopia (Graves' disease)
Drooping of eyelids; periorbital edema; loss of temporal aspects of eyebrows; puffy, nonpitting facies; large tongue
Cardiovascular system
Decreased peripheral vascular resistance, increased heart rate, stroke volume, cardiac output, pulse pressure; high-output heart failure; increased inotropic and chronotropic effects; arrhythmias; angina
Increased peripheral vascular resistance; decreased heart rate, stroke volume, cardiac output, pulse pressure; low-output heart failure; ECG: bradycardia, prolonged PR interval, flat T wave, low voltage; pericardial effusion
Respiratory system
Dyspnea; decreased vital capacity Pleural effusions; hypoventilation and CO2 retention
Gastrointestinal system
Increased appetite; increased frequency of bowel movements; hypoproteinemia
Decreased appetite; decreased frequency of bowel movements; ascites
Central nervous system
Nervousness; hyperkinesia; emotional lability
Lethargy; general slowing of mental processes; neuropathies
Musculoskeletal system
Weakness and muscle fatigue; increased deep tendon reflexes; hypercalcemia; osteoporosis
Stiffness and muscle fatigue; decreased deep tendon reflexes; increased alkaline phosphatase, LDH, AST
Renal system Mild polyuria; increased renal blood flow; increased glomerular filtration rate
Impaired water excretion; decreased renal blood flow; decreased glomerular filtration rate
Hematopoietic Increased erythropoiesis; anemia1 Decreased erythropoiesis; anemia1
system Reproductive system
Menstrual irregularities; decreased fertility; increased gonadal steroid metabolism
Hypermenorrhea; infertility; decreased libido; impotence; oligospermia; decreased gonadal steroid metabolism
Metabolic system
Increased basal metabolic rate; negative nitrogen balance; hyperglycemia; increased free fatty acids; decreased cholesterol and triglycerides; increased hormone degradation; increased requirements for fat- and water-soluble vitamins; increased drug metabolism
Decreased basal metabolic rate; slight positive nitrogen balance; delayed degradation of insulin, with increased sensitivity; increased cholesterol and triglycerides; decreased hormone degradation; decreased requirements for fat- and water-soluble vitamins; decreased drug metabolism
1The anemia of hyperthyroidism is usually normochromic and caused by increased red blood cell turnover. The anemia of hypothyroidism may be normochromic, hyperchromic, or hypochromic and may be due to decreased production rate, decreased iron absorption, decreased folic acid absorption, or to autoimmune pernicious anemia.
Thyroid hormone is critical for nervous, skeletal, and reproductive tissues. Its effects depend on protein synthesis as well as potentiation of the secretion and action of growth hormone. Thyroid deprivation in early life results in irreversible mental retardation and dwarfism—symptoms typical of congenital cretinism.
Effects on growth and calorigenesis are accompanied by a pervasive influence on metabolism of drugs as well as carbohydrates, fats, proteins, and vitamins. Many of these changes are dependent upon or modified by activity of other hormones. Conversely, the secretion and degradation rates of virtually all other hormones, including catecholamines, cortisol, estrogens, testosterone, and insulin, are affected by thyroid status.
Many of the manifestations of thyroid hyperactivity resemble sympathetic nervous system overactivity (especially in the cardiovascular system), although catecholamine levels are not increased. Changes in catecholamine-stimulated adenylyl cyclase activity as measured by cAMP are found with changes in thyroid activity. Possible explanations include increased numbers of receptors or enhanced amplification of the receptor signal. Other clinical symptoms reminiscent of excessive epinephrine activity (and partially alleviated by adrenoceptor antagonists) include lid lag and retraction, tremor, excessive sweating, anxiety, and nervousness. The opposite constellation of symptoms is seen in hypothyroidism (Table 38–4).
Thyroid Preparations
See the Preparations Available section at the end of this chapter for a list of available preparations. These preparations may be synthetic (levothyroxine, liothyronine, liotrix) or of animal origin (desiccated thyroid).
Synthetic levothyroxine is the preparation of choice for thyroid replacement and suppression therapy because of its stability, content uniformity, low cost, lack of allergenic foreign protein, easy laboratory measurement of serum levels, and long half-life (7 days), which permits once-daily administration. In addition, T4 is converted to T3 intracellularly; thus, administration of T4 produces both hormones. Generic levothyroxine preparations can be used because they provide comparable

efficacy and are more cost-effective than branded preparations.
Although liothyronine is three to four times more potent than levothyroxine, it is not recommended for routine replacement therapy because of its shorter half-life (24 hours), which requires multiple daily doses; its higher cost; and the greater difficulty of monitoring its adequacy of replacement by conventional laboratory tests. Furthermore, because of its greater hormone activity and consequent greater risk of cardiotoxicity, T3 should be avoided in patients with cardiac disease. It is best used for short-term suppression of TSH. Because oral administration of T3 is unnecessary, use of the more expensive mixture of thyroxine and liothyronine (liotrix) instead of levothyroxine is never required.
The use of desiccated thyroid rather than synthetic preparations is never justified, since the disadvantages of protein antigenicity, product instability, variable hormone concentrations, and difficulty in laboratory monitoring far outweigh the advantage of low cost. Significant amounts of T3 found in some thyroid extracts and liotrix may produce significant elevations in T3 levels and toxicity. Equivalent doses are 100 mg (1.5 g) of desiccated thyroid, 100 g of levothyroxine, and 37.5 g of liothyronine.
The shelf life of synthetic hormone preparations is about 2 years, particularly if they are stored in dark bottles to minimize spontaneous deiodination. The shelf life of desiccated thyroid is not certainly known, but its potency is better preserved if it is kept dry.
Antithyroid Agents
Reduction of thyroid activity and hormone effects can be accomplished by agents that interfere with the production of thyroid hormones; by agents that modify the tissue response to thyroid hormones; or by glandular destruction with radiation or surgery. "Goitrogens" are agents that suppress secretion of T3 and T4 to subnormal levels and thereby increase TSH, which in turn produces glandular enlargement (goiter). The antithyroid compounds used clinically include the thioamides, iodides, and radioactive iodine.
Thioamides
The thioamides methimazole and propylthiouracil are major drugs for treatment of thyrotoxicosis. In the United Kingdom, carbimazole, which is converted to methimazole in vivo, is widely used. Methimazole is about ten times more potent than propylthiouracil.
The chemical structures of these compounds are shown in Figure 38–5. The thiocarbamide group is essential for antithyroid activity.
Figure 38–5.

Structure of thioamides. The thiocarbamide moiety is shown in color.
Pharmacokinetics
Propylthiouracil is rapidly absorbed, reaching peak serum levels after 1 hour. The bioavailability of 50–80% may be due to incomplete absorption or a large first-pass effect in the liver. The volume of distribution approximates total body water with accumulation in the thyroid gland. Most of an ingested dose of propylthiouracil is excreted by the kidney as the inactive glucuronide within 24 hours.
In contrast, methimazole is completely absorbed but at variable rates. It is readily accumulated by the thyroid gland and has a volume of distribution similar to that of propylthiouracil. Excretion is slower than with propylthiouracil; 65–70% of a dose is recovered in the urine in 48 hours.
The short plasma half-life of these agents (1.5 hours for propylthiouracil and 6 hours for methimazole) has little influence on the duration of the antithyroid action or the dosing interval because both agents are accumulated by the thyroid gland. For propylthiouracil, giving the drug every 6–8 hours is reasonable since a single 100 mg dose can inhibit 60% of iodine organification for 7 hours. Since a single 30 mg dose of methimazole exerts an antithyroid effect for longer than 24 hours, a single daily dose is effective in the management of mild to moderate hyperthyroidism.
Both thioamides cross the placental barrier and are concentrated by the fetal thyroid, so that caution must be employed when using these drugs in pregnancy. Of the two, propylthiouracil is preferable in pregnancy because it is more strongly protein-bound and therefore crosses the placenta less readily. In addition, it is not secreted in sufficient quantity in breast milk to preclude breast-feeding.
Pharmacodynamics
The thioamides act by multiple mechanisms. The major action is to prevent hormone synthesis by

inhibiting the thyroid peroxidase-catalyzed reactions and blocking iodine organification. In addition, they block coupling of the iodotyrosines. They do not block uptake of iodide by the gland. Propylthiouracil and (to a much lesser extent) methimazole inhibit the peripheral deiodination of T4 and T3 (Figure 38–1). Since the synthesis rather than the release of hormones is affected, the onset of these agents is slow, often requiring 3–4 weeks before stores of T4 are depleted.
Toxicity
Adverse reactions to the thioamides occur in 3–12% of treated patients. Most reactions occur early. The most common adverse effect is a maculopapular pruritic rash, at times accompanied by systemic signs such as fever. Rare adverse effects include an urticarial rash, vasculitis, arthralgia, a lupus-like reaction, cholestatic jaundice, hepatitis, lymphadenopathy, hypoprothrombinemia, exfoliative dermatitis, polyserositis, and acute arthralgia.
The most dangerous complication is agranulocytosis, an infrequent but potentially fatal adverse reaction. It occurs in 0.3–0.6% of patients taking thioamides, but the risk may be increased in older patients and in those receiving high-dose methimazole therapy (over 40 mg/d). The reaction is usually rapidly reversible when the drug is discontinued, but antibiotic therapy may be necessary for complicating infections. Colony-stimulating factors (eg, G-CSF; see Chapter 33: Agents Used in Anemias; Hematopoietic Growth Factors) may hasten recovery of the granulocytes. The cross-sensitivity between propylthiouracil and methimazole is about 50%; therefore, switching drugs in patients with severe reactions is not recommended.
Anion Inhibitors
Monovalent anions such as perchlorate (ClO4–), pertechnetate (TcO4
–), and thiocyanate (SCN–) can block uptake of iodide by the gland through competitive inhibition of the iodide transport mechanism. Since these effects can be overcome by large doses of iodides, their effectiveness is somewhat unpredictable.
The major clinical use for potassium perchlorate is to block thyroidal reuptake of I– in patients with iodide-induced hyperthyroidism (eg, amiodarone-induced hyperthyroidism). However, potassium perchlorate is rarely used clinically because it has been shown to cause aplastic anemia.
Iodides
Prior to the introduction of the thioamides in the 1940s, iodides were the major antithyroid agents; today they are rarely used as sole therapy.
Pharmacodynamics
Iodides have several actions on the thyroid. They inhibit organification and hormone release and decrease the size and vascularity of the hyperplastic gland. In susceptible individuals, iodides can induce hyperthyroidism (jodbasedow phenomenon) or precipitate hypothyroidism.
In pharmacologic doses (> 6 mg daily), the major action of iodides is to inhibit hormone release, possibly through inhibition of thyroglobulin proteolysis. Rapid improvement in thyrotoxic symptoms occurs within 2–7 days—hence the value of iodide therapy in thyroid storm. In addition, iodides decrease the vascularity, size, and fragility of a hyperplastic gland, making the drugs valuable as preoperative preparation for surgery.

Clinical Use of Iodide
Disadvantages of iodide therapy include an increase in intraglandular stores of iodine, which may delay onset of thioamide therapy or prevent use of radioactive iodine therapy for several weeks. Thus, iodides should be initiated after onset of thioamide therapy and avoided if treatment with radioactive iodine seems likely. Iodide should not be used alone, because the gland will escape from the iodide block in 2–8 weeks, and its withdrawal may produce severe exacerbation of thyrotoxicosis in an iodine-enriched gland. Chronic use of iodides in pregnancy should be avoided, since they cross the placenta and can cause fetal goiter. In radiation emergencies, the thyroid-blocking effects of potassium iodide can protect the gland from subsequent damage if administered before radiation exposure.
Toxicity
Adverse reactions to iodine (iodism) are uncommon and in most cases reversible upon discontinuance. They include acneiform rash (similar to that of bromism), swollen salivary glands, mucous membrane ulcerations, conjunctivitis, rhinorrhea, drug fever, metallic taste, bleeding disorders and, rarely, anaphylactoid reactions.
Iodinated Contrast Media
The iodinated contrast agents—ipodate and iopanoic acid by mouth, or diatrizoate intravenously—are valuable in the treatment of hyperthyroidism, although they are not labeled for this indication. These drugs rapidly inhibit the conversion of T4 to T3 in the liver, kidney, pituitary gland, and brain. This accounts for the dramatic improvement in both subjective and objective parameters. For example, a decrease in heart rate is seen after only 3 days of oral administration of 0.5–1 g/d. T3 levels often return to normal during this time. The prolonged effect of suppressing T4 as well as T3 suggests that inhibition of hormone release due to the iodine released may be an additional mechanism of action. Fortunately, these agents are relatively nontoxic. They provide useful adjunctive therapy in the treatment of thyroid storm and offer valuable alternatives when iodides or thioamides are contraindicated. Surprisingly, these agents may not interfere with 131I retention as much as iodides despite their large iodine content. Their toxicity is similar to that of the iodides, and their safety in pregnancy is undocumented.
Radioactive Iodine
131I is the only isotope used for treatment of thyrotoxicosis (others are used in diagnosis). Administered orally in solution as sodium 131I, it is rapidly absorbed, concentrated by the thyroid, and incorporated into storage follicles. Its therapeutic effect depends on emission of rays with an effective half-life of 5 days and a penetration range of 400–2000 m. Within a few weeks after administration, destruction of the thyroid parenchyma is evidenced by epithelial swelling and necrosis, follicular disruption, edema, and leukocyte infiltration. Advantages of radioiodine include easy administration, effectiveness, low expense, and absence of pain. Fears of radiation-induced genetic damage, leukemia, and neoplasia have not been realized after more than 30 years of clinical experience with radioiodine. Radioactive iodine should not be administered to pregnant women or nursing mothers, since it crosses the placenta and is excreted in breast milk.
Adrenoceptor-Blocking Agents
Beta blockers without intrinsic sympathomimetic activity are effective therapeutic adjuncts in the management of thyrotoxicosis since many of these symptoms mimic those associated with
sympathetic stimulation. Propranolol has been the -blocker most widely studied and used in the therapy of thyrotoxicosis. Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 38. Thyroid & Antithyroid Drugs >
Clinical Pharmacology of Thyroid & Antithyroid Drugs
Hypothyroidism
Hypothyroidism is a syndrome resulting from deficiency of thyroid hormones and is manifested largely by a reversible slowing down of all body functions (Table 38–4). In infants and children, there is striking retardation of growth and development that results in dwarfism and irreversible mental retardation.
The etiology and pathogenesis of hypothyroidism are outlined in Table 38–5. Hypothyroidism can occur with or without thyroid enlargement (goiter). The laboratory diagnosis of hypothyroidism in the adult is easily made by the combination of a low free thyroxine (or low free thyroxine index) and elevated serum TSH (Table 38–2).
Table 38–5. Etiology and Pathogenesis of Hypothyroidism.
Cause Pathogenesis Goiter Degree of
Hypothyroidism Hashimoto's thyroiditis Autoimmune destruction of thyroid Present
early, absent later
Mild to severe
Drug-induced1
Blocked hormone formation Present Mild to moderate
Dyshormonogenesis Impaired synthesis of T4 due to enzyme deficiency
Present Mild to severe
Radiation, 131I, x-ray, thyroidectomy
Destruction or removal of gland Absent Severe
Congenital (cretinism) Athyreosis or ectopic thyroid, iodine deficiency; TSH receptor-blocking antibodies
Absent or present
Severe
Secondary (TSH deficit) Pituitary or hypothalamic disease Absent Mild
1Iodides, lithium, fluoride, thioamides, aminosalicylic acid, phenylbutazone, amiodarone, etc.
The most common cause of hypothyroidism in the USA at this time is probably Hashimoto's thyroiditis, an immunologic disorder in genetically predisposed individuals. In this condition, there is evidence of humoral immunity in the presence of antithyroid antibodies and lymphocyte sensitization to thyroid antigens.

Management of Hypothyroidism
Except for hypothyroidism caused by drugs (Table 38–5), which can be treated by simply removing the depressant agent, the general strategy of replacement therapy is appropriate. The most satisfactory preparation is levothyroxine. Infants and children require more T4 per kilogram of body weight than adults. The average dosage for an infant 1–6 months of age is 10–15 g/kg/d, whereas the average dosage for an adult is about 1.7 g/kg/d. There is some variability in the absorption of thyroxine, so this dosage may vary from patient to patient. Because of the long half-life of thyroxine, the dose can be given once daily. Children should be monitored for normal growth and development. Serum TSH and free thyroxine should be measured at regular intervals and maintained within the normal range. It takes 6–8 weeks after starting a given dose of thyroxine to reach steady state levels in the bloodstream. Thus, dosage changes should be made slowly.
In long-standing hypothyroidism, in older patients, and in patients with underlying cardiac disease, it is imperative to start treatment with reduced dosage. In such adult patients, levothyroxine is given in a dosage of 12.5–25 g/d for 2 weeks, increasing the daily dose by 25 g every 2 weeks until euthyroidism or drug toxicity is observed. In older patients, the heart is very sensitive to the level of circulating thyroxine, and if angina pectoris or cardiac arrhythmia develops, it is essential to stop or reduce the dose of thyroxine immediately. In younger patients or those with very mild disease, full replacement therapy may be started immediately.
The toxicity of thyroxine is directly related to the hormone level. In children, restlessness, insomnia, and accelerated bone maturation and growth may be signs of thyroxine toxicity. In adults, increased nervousness, heat intolerance, episodes of palpitation and tachycardia, or unexplained weight loss may be the presenting symptoms. If these symptoms are present, it is important to monitor serum TSH (Table 38–2), which will determine whether the symptoms are due to excess thyroxine blood levels. Chronic overtreatment with T4, particularly in elderly patients, can increase the risk of atrial fibrillation and accelerated osteoporosis.
Special Problems in Management of Hypothyroidism
Myxedema and Coronary Artery Disease
Since myxedema frequently occurs in older persons, it is often associated with underlying coronary artery disease. In this situation, the low levels of circulating thyroid hormone actually protect the heart against increasing demands that could result in angina pectoris or myocardial infarction. Correction of myxedema must be done cautiously to avoid provoking arrhythmia, angina, or acute myocardial infarction.
Myxedema Coma
Myxedema coma is an end state of untreated hypothyroidism. It is associated with progressive weakness, stupor, hypothermia, hypoventilation, hypoglycemia, hyponatremia, water intoxication, shock, and death.
Management of myxedema coma is a medical emergency. The patient should be treated in the intensive care unit, since tracheal intubation and mechanical ventilation may be required. Associated illnesses such as infection or heart failure must be treated by appropriate therapy. It is important to give all preparations intravenously, because patients with myxedema coma absorb drugs poorly from other routes. Intravenous fluids should be administered with caution to avoid excessive water intake. These patients have large pools of empty T3 and T4 binding sites that must

be filled before there is adequate free thyroxine to affect tissue metabolism. Accordingly, the treatment of choice in myxedema coma is to give a loading dose of levothyroxine intravenously—usually 300–400 g initially, followed by 50 g daily. Intravenous T3 can also be used but may be more cardiotoxic and more difficult to monitor. Intravenous hydrocortisone is indicated if the patient has associated adrenal or pituitary insufficiency but is probably not necessary in most patients with primary myxedema. Opioids and sedatives must be used with extreme caution.
Hypothyroidism and Pregnancy
Hypothyroid women frequently have anovulatory cycles and are therefore relatively infertile until restoration of the euthyroid state. This has led to the widespread use of thyroid hormone for infertility, although there is no evidence for its usefulness in infertile euthyroid patients. In a pregnant hypothyroid patient receiving thyroxine, it is extremely important that the daily dose of thyroxine be adequate because early development of the fetal brain depends on maternal thyroxine. In many hypothyroid patients, a modest increase in the thyroxine dose (about 20–30%) is required to normalize the serum TSH level during pregnancy. Because of the elevated maternal TBG, the free thyroxine index (FT4I) or free thyroxine (FT4) and TSH (Table 38–2) must be used to monitor maternal thyroxine dosages.
Hyperthyroidism
Hyperthyroidism (thyrotoxicosis) is the clinical syndrome that results when tissues are exposed to high levels of thyroid hormone (Table 38–4).
Graves' Disease
The most common form of hyperthyroidism is Graves' disease, or diffuse toxic goiter. The presenting signs and symptoms of Graves' disease are set forth in Table 38–4.
Pathophysiology
Graves' disease is considered to be an autoimmune disorder in which there is a genetic defect in suppressor T lymphocytes, and helper T lymphocytes stimulate B lymphocytes to synthesize antibodies to thyroidal antigens. The antibody described previously (TSH-R Ab [stim]) is directed against the TSH receptor site in the thyroid cell membrane and has the capacity to stimulate the thyroid cell. Spontaneous remission occurs but may require 1 to 15 years.
Laboratory Diagnosis
In most patients with hyperthyroidism, T3, T4, RT3U, FT4, and FT4I will all be elevated and TSH is suppressed (Table 38–2). Radioiodine uptake is usually markedly elevated as well. Antithyroglobulin antibodies, thyroid peroxidase, and TSH-R Ab [stim] are often present.
Management of Graves' Disease
The three primary methods for controlling hyperthyroidism are antithyroid drug therapy, surgical thyroidectomy, and destruction of the gland with radioactive iodine.
Antithyroid Drug Therapy
Drug therapy is most useful in young patients with small glands and mild disease. Methimazole or

propylthiouracil is administered until the disease undergoes spontaneous remission. This is the only therapy that leaves an intact thyroid gland, but it does require a long period of treatment and observation (1–2 years), and there is a 60–70% incidence of relapse.
Antithyroid drug therapy is usually begun with large divided doses, shifting to maintenance therapy with single daily doses when the patient becomes clinically euthyroid. However, mild to moderately severe thyrotoxicosis can often be controlled with methimazole given in a single morning dose of 30–40 mg; once-daily dosing may enhance adherence. Maintenance therapy requires 5–15 mg once daily. Alternatively, therapy is started with propylthiouracil, 100–150 mg every 6 or 8 hours, followed after 4–8 weeks by gradual reduction of the dose to the maintenance level of 50–150 mg once daily. In addition to inhibiting iodine organification, propylthiouracil also inhibits the conversion of T4 to T3, so it brings the level of activated thyroid hormone down more quickly than does methimazole. The best clinical guide to remission is reduction in the size of the goiter. Laboratory tests most useful in monitoring the course of therapy are serum T3 by RIA, FT4 or FT4I, and serum TSH.
Reactivation of the autoimmune process may occur when the dosage of antithyroid drug is lowered during maintenance therapy and TSH begins to drive the gland. TSH release can be prevented by the daily administration of 50–150 g of levothyroxine with 5–15 mg of methimazole or 50–150 mg of propylthiouracil for the second year of therapy. The relapse rate with this program is probably comparable to the rate with antithyroid therapy alone, but the risk of hypothyroidism and overtreatment is avoided.
Reactions to antithyroid drugs have been described above. A minor rash can often be controlled by antihistamine therapy. Because the more severe reaction of agranulocytosis is often heralded by sore throat or high fever, patients receiving antithyroid drugs must be instructed to discontinue the drug and seek immediate medical attention if these symptoms develop. White cell and differential counts and a throat culture are indicated in such cases, followed by appropriate antibiotic therapy.
Thyroidectomy
A near-total thyroidectomy is the treatment of choice for patients with very large glands or multinodular goiters. Patients are treated with antithyroid drugs until euthyroid (about 6 weeks). In addition, for 2 weeks prior to surgery, they receive saturated solution of potassium iodide, 5 drops twice daily, to diminish vascularity of the gland and simplify surgery. About 80–90% of patients will require thyroid supplementation following near-total thyroidectomy.
Radioactive Iodine
Radioiodine therapy utilizing 131I is the preferred treatment for most patients over 21 years of age. In patients without heart disease, the therapeutic dose may be given immediately in a range of 80–120 Ci/g of estimated thyroid weight corrected for uptake. In patients with underlying heart disease or severe thyrotoxicosis and in elderly patients, it is desirable to treat with antithyroid drugs (preferably methimazole) until the patient is euthyroid. The medication is then stopped for 5–7 days before the appropriate dose of 131I is administered. Iodides should be avoided to ensure maximal 131I uptake. Six to 12 weeks following the administration of radioiodine, the gland will shrink in size and the patient will usually become euthyroid or hypothyroid. A second dose may be required in some patients. Hypothyroidism occurs in about 80% of patients following radioiodine therapy. Serum FT4 and TSH levels should be monitored. When hypothyroidism develops, prompt replacement with oral levothyroxine, 50–150 g daily, should be instituted.

Adjuncts to Antithyroid Therapy
During the acute phase of thyrotoxicosis, -adrenoceptor-blocking agents without intrinsic sympathomimetic activity are extremely helpful. Propranolol, 20–40 mg orally every 6 hours, will control tachycardia, hypertension, and atrial fibrillation. Propranolol is gradually withdrawn as serum thyroxine levels return to normal. Diltiazem, 90–120 mg three or four times daily, can be used to control tachycardia in patients in whom -blockers are contraindicated, eg, those with asthma. Other calcium channel blockers may not be as effective as diltiazem. Adequate nutrition and vitamin supplements are essential. Barbiturates accelerate T4 breakdown (by hepatic enzyme induction) and may be helpful both as sedatives and to lower T4 levels.
Toxic Uninodular Goiter & Toxic Multinodular Goiter
These forms of hyperthyroidism occur often in older women with nodular goiters. FT4 is moderately elevated or occasionally normal, but T3 by RIA is strikingly elevated. Single toxic adenomas can be managed with either surgical excision of the adenoma or with radioiodine therapy. Toxic multinodular goiter is usually associated with a large goiter and is best treated by preparation with methimazole or propylthiouracil followed by subtotal thyroidectomy.
Subacute Thyroiditis
During the acute phase of a viral infection of the thyroid gland, there is destruction of thyroid parenchyma with transient release of stored thyroid hormones. A similar state may occur in patients with Hashimoto's thyroiditis. These episodes of transient thyrotoxicosis have been termed "spontaneously resolving hyperthyroidism." Supportive therapy is usually all that is necessary, such as propranolol for tachycardia and aspirin or nonsteroidal anti-inflammatory drugs to control local pain and fever. Corticosteroids may be necessary in severe cases to control the inflammation.
Special Problems
Thyroid Storm
Thyroid storm, or thyrotoxic crisis, is sudden acute exacerbation of all of the symptoms of thyrotoxicosis, presenting as a life-threatening syndrome. Vigorous management is mandatory. Propranolol, 1–2 mg slowly intravenously or 40–80 mg orally every 6 hours, is helpful to control the severe cardiovascular manifestations. If propranolol is contraindicated by the presence of severe heart failure or asthma, hypertension and tachycardia may be controlled with diltiazem, 90–120 mg orally three or four times daily or 5–10 mg/h by intravenous infusion (asthmatic patients only). Release of thyroid hormones from the gland is retarded by the administration of saturated solution of potassium iodide, 10 drops orally daily, or iodinated contrast media (eg, sodium ipodate, 1 g orally daily). The latter medication will also block peripheral conversion of T4 to T3. Hormone synthesis is blocked by the administration of propylthiouracil, 250 mg orally every 6 hours. If the patient is unable to take propylthiouracil by mouth, a rectal formulation can be prepared and administered in a dosage of 400 mg every 6 hours as a retention enema. Methimazole may also be prepared for rectal administration in a dose of 60 mg daily. Hydrocortisone, 50 mg intravenously every 6 hours, will protect the patient against shock and will block the conversion of T4 to T3, rapidly bringing down the level of thyroactive material in the blood.
Supportive therapy is essential to control fever, heart failure, and any underlying disease process that may have precipitated the acute storm. In rare situations, where the above methods are not adequate to control the problem, plasmapheresis or peritoneal dialysis has been used to lower the

levels of circulating thyroxine.
Ophthalmopathy
Although severe ophthalmopathy is rare, it is difficult to treat. Management requires effective treatment of the thyroid disease, usually by total surgical excision or 131I ablation of the gland plus oral prednisone therapy (see below). In addition, local therapy may be necessary, eg, elevation of the head to diminish periorbital edema and artificial tears to relieve corneal drying. Smoking cessation should be advised to prevent progression of the ophthalmopathy. For the severe, acute inflammatory reaction, a short course of prednisone, 60–100 mg orally daily for about a week and then 60–100 mg every other day, tapering the dose over a period of 6–12 weeks, may be effective. If steroid therapy fails or is contraindicated, irradiation of the posterior orbit, using well-collimated high-energy x-ray therapy, will frequently result in marked improvement of the acute process. Threatened loss of vision is an indication for surgical decompression of the orbit. Eyelid or eye muscle surgery may be necessary to correct residual problems after the acute process has subsided.
Dermopathy
Dermopathy or pretibial myxedema will often respond to topical corticosteroids applied to the involved area and covered with an occlusive dressing.
Thyrotoxicosis during Pregnancy
Ideally, women in the childbearing period with severe disease should have definitive therapy with 131I or subtotal thyroidectomy prior to pregnancy in order to avoid an acute exacerbation of the disease during pregnancy or following delivery. If thyrotoxicosis does develop during pregnancy, radioiodine is contraindicated because it crosses the placenta and may injure the fetal thyroid. In the first trimester, the patient can be prepared with propylthiouracil and a subtotal thyroidectomy performed safely during the mid trimester. It is essential to give the patient a thyroid supplement during the balance of the pregnancy. However, most patients are treated with propylthiouracil during the pregnancy, and the decision regarding long-term management can be made after delivery. The dosage of propylthiouracil must be kept to the minimum necessary for control of the disease (ie, < 300 mg daily), because it may affect the function of the fetal thyroid gland. Methimazole is a potential alternative, although there is concern about a possible risk of fetal scalp defects.
Neonatal Graves' Disease
Graves' disease may occur in the newborn infant, either due to passage of TSH-R Ab [stim] through the placenta, stimulating the thyroid gland of the neonate, or to genetic transmission of the trait to the fetus. Laboratory studies reveal an elevated free thyroxine, a markedly elevated T3, and a low TSH—in contrast to the normal infant, in whom TSH is elevated at birth. TSH-R Ab [stim] is usually found in the serum of both the child and the mother.
If caused by maternal TSH-R Ab [stim], the disease is usually self-limited and subsides over a period of 4–12 weeks, coinciding with the fall in the infant's TSH-R Ab [stim] level. However, treatment is necessary because of the severe metabolic stress the infant experiences. Therapy includes propylthiouracil in a dose of 5–10 mg/kg/d in divided doses at 8-hour intervals; Lugol's solution (8 mg of iodide per drop), 1 drop every 8 hours; and propranolol, 2 mg/kg/d in divided doses. Careful supportive therapy is essential. If the infant is very ill, oral prednisone, 2 mg/kg/d in divided doses, will help block conversion of T4 to T3. These medications are gradually reduced as

the clinical picture improves and can be discontinued by 6–12 weeks.
Nontoxic Goiter
Nontoxic goiter is a syndrome of thyroid enlargement without excessive thyroid hormone production. Enlargement of the thyroid gland is usually due to TSH stimulation from inadequate thyroid hormone synthesis. The most common cause of nontoxic goiter worldwide is iodide deficiency, but in the USA, it is Hashimoto's thyroiditis. Less common causes include dietary goitrogens, dyshormonogenesis, and neoplasms (see below).
Goiter due to iodide deficiency is best managed by prophylactic administration of iodide. The optimal daily iodide intake is 150–200 g. Iodized salt and iodate used as preservatives in flour and bread are excellent sources of iodine in the diet. In areas where it is difficult to introduce iodized salt or iodate preservatives, a solution of iodized poppyseed oil has been administered intramuscularly to provide a long-term source of inorganic iodine.
Goiter due to ingestion of goitrogens in the diet is managed by elimination of the goitrogen or by adding sufficient thyroxine to shut off TSH stimulation. Similarly, in Hashimoto's thyroiditis and dyshormonogenesis, adequate thyroxine therapy—150–200 g/d orally—will suppress pituitary TSH and result in slow regression of the goiter as well as correction of hypothyroidism.
Thyroid Neoplasms
Neoplasms of the thyroid gland may be benign (adenomas) or malignant. Some adenomas will regress following thyroxine therapy; those that do not should be rebiopsied or surgically removed. Management of thyroid carcinoma requires a total thyroidectomy, postoperative radioiodine therapy in selected instances, and lifetime replacement with levothyroxine. The evaluation for recurrence of some thyroid malignancies requires withdrawal of thyroxine replacement for 4–6 weeks—accompanied by the development of hypothyroidism. Tumor recurrence is likely if there is a rise in serum thyroglobulin (ie, a tumor marker) or a positive 131I scan when TSH is elevated. Alternatively, administration of recombinant human TSH (Thyrogen) can produce comparable TSH elevations without discontinuing thyroxine and avoiding hypothyroidism. Recombinant human TSH is administered intramuscularly once daily for 2 days. A rise in serum thyroglobulin or a positive 131I scan will indicate a recurrence of the thyroid cancer. Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 38. Thyroid & Antithyroid Drugs >
Preparations Available
Thyroid Agents
Levothyroxine [T4] (generic, Levoxyl, Levo-T, Synthroid, Unithroid)
Oral: 0.025, 0.05, 0.075, 0.088, 0.1, 0.112, 0.125, 0.137, 0.15, 0.175, 0.2, 0.3 mg tablets
Parenteral: 200, 500 g per vial (100 g/mL when reconstituted) for injection
Liothyronine [T3] (generic, Cytomel, Triostat)

Oral: 5, 25, 50 g tablets
Parenteral: 10 g/mL
Liotrix [a 4:1 ratio of T4:T3] (Thyrolar)
Oral: tablets containing 12.5, 25, 30, 50, 60, 100, 120, 150, 180 g T4 and one fourth as much T3
Thyroid desiccated [USP] (generic, Armour Thyroid, Thyroid Strong, Thyrar, S-P-T)
Oral: tablets containing 15, 30, 60, 90, 120, 180, 240, 300 mg; capsules (S-P-T) containing 120, 180, 300 mg
Antithyroid Agents
Diatrizoate sodium (Hypaque)
Parenteral: 25% (150 mg iodine/mL); 50% (300 mg iodine/mL) (unlabeled use)
Iodide (131I) sodium (Iodotope, Sodium Iodide I 131 Therapeutic)
Oral: available as capsules and solution
Iopanoic acid (Telepaque)
Oral: 500 mg tablets (unlabeled use)
Ipodate sodium (Oragrafin Sodium, Bilivist)
Oral: 500 mg capsules (unlabeled use)
Methimazole (Tapazole)
Oral: 5, 10 mg tablets
Potassium iodide
Oral solution (generic, SSKI): 1 g/mL
Oral solution (Lugol's solution): 100 mg/mL potassium iodide plus 50 mg/mL iodine
Oral syrup (Pima): 325 mg/5 mL
Oral controlled action tablets (Iodo-Niacin): 135 mg potassium iodide plus 25 mg niacinamide hydroiodide
Oral potassium iodide tablets (generic, IOSAT, RAD-Block, Thyro-Block): 65, 130 mg
Propylthiouracil [PTU] (generic)

Oral: 50 mg tablets
Thyrotropin; recombinant human TSH (Thyrogen)
Parenteral: 0.9 mg per vial Chapter 39. Adrenocorticosteroids & Adrenocortical Antagonists Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 39. Adrenocorticosteroids & Adrenocortical Antagonists >
Adrenocorticosteroids & Adrenocortical Antagonists: Introduction
The natural adrenocortical hormones are steroid molecules produced and released by the adrenal cortex. Both natural and synthetic corticosteroids are used for diagnosis and treatment of disorders of adrenal function. They are also used—more often and in much larger doses—for treatment of a variety of inflammatory and immunologic disorders.
Secretion of adrenocortical steroids is controlled by the pituitary release of corticotropin (ACTH). Secretion of the salt-retaining hormone aldosterone is primarily under the influence of angiotensin. Corticotropin has some actions that do not depend upon its effect on adrenocortical secretion. However, its pharmacologic value as an anti-inflammatory agent and its use in testing adrenal function depend on its secretory action. Its pharmacology is discussed in Chapter 37: Hypothalamic & Pituitary Hormones and will be reviewed only briefly here.
Inhibitors of the synthesis or antagonists of the action of the adrenocortical steroids are important in the treatment of several conditions. These agents are described at the end of this chapter. Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 39. Adrenocorticosteroids & Adrenocortical Antagonists >
Adrenocorticosteroids
The adrenal cortex releases a large number of steroids into the circulation. Some have minimal biologic activity and function primarily as precursors, and there are some for which no function has been established. The hormonal steroids may be classified as those having important effects on intermediary metabolism (glucocorticoids), those having principally salt-retaining activity (mineralocorticoids), and those having androgenic or estrogenic activity (see Chapter 40: The Gonadal Hormones & Inhibitors). In humans, the major glucocorticoid is cortisol and the most important mineralocorticoid is aldosterone. Quantitatively, dehydroepiandrosterone (DHEA) in its sulfated form (DHEAS) is the major adrenal androgen, since about 20 mg is secreted daily. However, DHEA and two other adrenal androgens, androstenediol and androstenedione, are weak androgens or estrogens, mostly by peripheral conversion to testosterone and dehydrotestosterone or estradiol and estrone. Adrenal androgens constitute the major endogenous precursors of estrogen in women after menopause and in younger patients in whom ovarian function is deficient or absent.
The Naturally Occurring Glucocorticoids; Cortisol (Hydrocortisone)

Mifepristone (Mifeprex)
Oral: 200 mg tablets
Nilutamide (Nilandron)
Oral: 50, 150 mg tablets
Raloxifene (Evista)
Oral: 60 mg tablets
Tamoxifen (generic, Nolvadex)
Oral: 10, 20 mg tablets
Toremifene (Fareston)
Oral: 60 mg tablets
1 Oral contraceptives are listed in Table 40–3. Chapter 41. Pancreatic Hormones & Antidiabetic Drugs Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 41. Pancreatic Hormones & Antidiabetic Drugs >
The Endocrine Pancreas
* Deceased.
The endocrine pancreas in the adult human consists of approximately 1 million islets of Langerhans interspersed throughout the pancreatic gland. Within the islets, at least four hormone-producing cells are present (Table 41–1). Their hormone products include insulin, the storage and anabolic hormone of the body; islet amyloid polypeptide (IAPP, or amylin), whose metabolic function remains undefined; glucagon, the hyperglycemic factor that mobilizes glycogen stores; somatostatin, a universal inhibitor of secretory cells; and pancreatic peptide, a small protein that facilitates digestive processes by a mechanism not yet clarified.
Table 41–1. Pancreatic Islet Cells and Their Secretory Products.
Cell Types Approximate Percent of Islet
Mass Secretory Products
A cell (alpha)
20 Glucagon, proglucagon
B cell (beta) 75 Insulin, C-peptide, proinsulin, islet amyloid polypeptide (IAPP)

D cell (delta) 3–5 Somatostatin F cell (PP cell)1
< 2 Pancreatic polypeptide (PP)
1Within pancreatic polypeptide-rich lobules of adult islets, located only in the posterior portion of the head of the human pancreas, glucagon cells are scarce (< 0.5%) and F cells make up as much as 80% of the cells.
The elevated blood glucose associated with diabetes mellitus results from absent or inadequate pancreatic insulin secretion, with or without concurrent impairment of insulin action. The disease states underlying the diagnosis of diabetes mellitus are now classified into four categories: type 1, "insulin-dependent diabetes," type 2, "noninsulin-dependent diabetes," type 3, "other," and type 4, "gestational diabetes mellitus" (Expert Committee 2002, Mayfield, 1998).
Type 1 Diabetes Mellitus
The hallmark of type 1 diabetes is selective B cell destruction and severe or absolute insulin deficiency. Administration of insulin is essential in patients with type 1 diabetes. Type 1 diabetes is further subdivided into immune and idiopathic causes. The immune form is the most common form of type 1 diabetes. In the USA, this form is diagnosed in approximately 1,500,000 individuals. Although most patients are younger than 30 years of age at the time of diagnosis, the onset can occur at any age. Type 1 diabetes is found in all ethnic groups, but the highest incidence is in people from northern Europe and from Sardinia. Susceptibility appears to involve a multifactorial genetic linkage but only 15–20% of patients have a positive family history.
Type 2 Diabetes Mellitus
Type 2 diabetes is characterized by tissue resistance to the action of insulin combined with a relative deficiency in insulin secretion. A given individual may have more resistance or more B cell deficiency, and the abnormalities may be mild or severe. Although insulin is produced by the B cells in these patients, it is inadequate to overcome the resistance, and the blood glucose rises. The impaired insulin action also affects fat metabolism, resulting in increased free fatty acid flux and triglyceride levels, and reciprocally low high-density lipoprotein (HDL) levels.
Individuals with type 2 diabetes may not require insulin to survive, but 30% or more will benefit from insulin therapy to control the blood glucose. It is likely that 10–20% of individuals in whom type 2 diabetes was initially diagnosed actually have both type 1 and type 2, or have a slowly progressing type 1, and ultimately will require full insulin replacement. Although persons with type 2 diabetes ordinarily will not develop ketosis, ketoacidosis may occur as the result of stress such as infection or use of medication that enhances resistance, eg, corticosteroids. Dehydration in untreated and poorly controlled individuals with type 2 diabetes can lead to a life-threatening condition called "non-ketotic hyperosmolar coma". In this condition, the blood glucose may rise to 6–20 times the normal range and an altered mental state develops or the person loses consciousness. Urgent medical care and rehydration is required.
Type 3 Diabetes Mellitus
The type 3 designation refers to multiple other specific causes of an elevated blood glucose:

nonpancreatic diseases, drug therapy, etc. For a complete, detailed list the reader is referred to Expert Committee, 2002 or Mayfield, 1998.
Type 4 Diabetes Mellitus
Gestational Diabetes (GDM) is defined as any abnormality in glucose levels noted for the first time during pregnancy. Gestational diabetes is diagnosed in approximately 4% of all pregnancies in the USA. During pregnancy, the placenta and placental hormones create an insulin resistance that is most pronounced in the last trimester. Risk assessment for diabetes is suggested starting at the first prenatal visit. High risk individuals should be screened immediately. Screening may be deferred in lower risk women until the 24th to 28th week of gestation. Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 41. Pancreatic Hormones & Antidiabetic Drugs >
Insulin
Chemistry
Insulin is a small protein with a molecular weight in humans of 5808. It contains 51 amino acids arranged in two chains (A and B) linked by disulfide bridges; there are species differences in the amino acids of both chains. Proinsulin, a long single-chain protein molecule, is processed within the Golgi apparatus and packaged into granules, where it is hydrolyzed into insulin and a residual connecting segment called C-peptide by removal of four amino acids (shown in dashed circles in Figure 41–1).
Figure 41–1.

Structure of human proinsulin and commercially available insulin analogs. Insulin is shown as the shaded (darker color) peptide chains, A and B. Species differences in the A and B chains and amino acid modifications for insulin aspart, lispro, and glargine are noted.
Insulin and C-peptide are secreted in equimolar amounts in response to all insulin secretagogues; a small quantity of unprocessed or partially hydrolyzed proinsulin is released as well. While proinsulin may have some mild hypoglycemic action, C-peptide has no known physiologic function. Granules within the B cells store the insulin in the form of crystals consisting of two atoms of zinc and six molecules of insulin. The entire human pancreas contains up to 8 mg of insulin, representing approximately 200 biologic units. Originally, the unit was defined on the basis of the hypoglycemic activity of insulin in rabbits. With improved purification techniques, the unit is presently defined on the basis of weight, and present insulin standards used for assay purposes contain 28 units per milligram.
Insulin Secretion
Insulin is released from pancreatic B cells at a low basal rate and at a much higher stimulated rate in response to a variety of stimuli, especially glucose. Other stimulants such as other sugars (eg, mannose), certain amino acids (eg, leucine, arginine), and vagal activity are recognized. One mechanism of stimulated insulin release is diagrammed in Figure 41–2. As shown in the figure, hyperglycemia results in increased intracellular ATP levels, which close the ATP-dependent potassium channels. Decreased outward potassium efflux results in depolarization of the B cell and opening of voltage-gated calcium channels. The resulting increased intracellular calcium triggers secretion of the hormone. As noted below, the insulin secretagogue drug group (sulfonylureas, meglitinides, and D-phenylalanine) exploits parts of this mechanism.
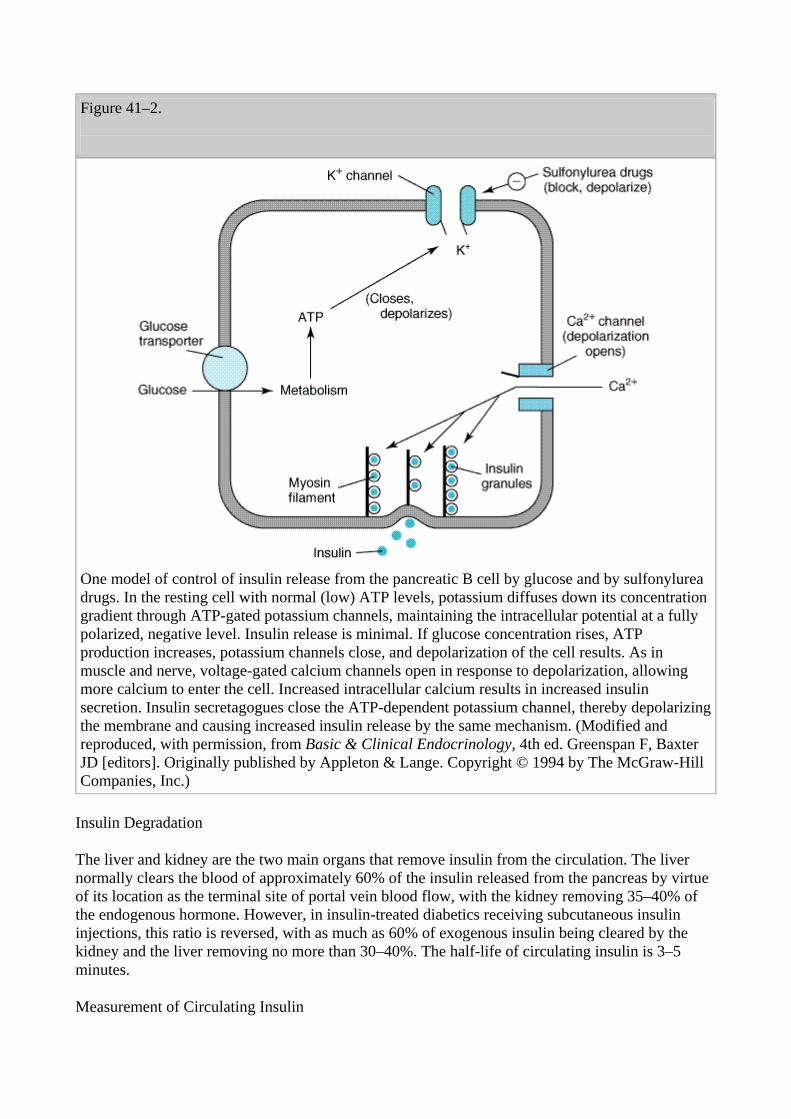
Figure 41–2.
One model of control of insulin release from the pancreatic B cell by glucose and by sulfonylurea drugs. In the resting cell with normal (low) ATP levels, potassium diffuses down its concentration gradient through ATP-gated potassium channels, maintaining the intracellular potential at a fully polarized, negative level. Insulin release is minimal. If glucose concentration rises, ATP production increases, potassium channels close, and depolarization of the cell results. As in muscle and nerve, voltage-gated calcium channels open in response to depolarization, allowing more calcium to enter the cell. Increased intracellular calcium results in increased insulin secretion. Insulin secretagogues close the ATP-dependent potassium channel, thereby depolarizing the membrane and causing increased insulin release by the same mechanism. (Modified and reproduced, with permission, from Basic & Clinical Endocrinology, 4th ed. Greenspan F, Baxter JD [editors]. Originally published by Appleton & Lange. Copyright © 1994 by The McGraw-Hill Companies, Inc.)
Insulin Degradation
The liver and kidney are the two main organs that remove insulin from the circulation. The liver normally clears the blood of approximately 60% of the insulin released from the pancreas by virtue of its location as the terminal site of portal vein blood flow, with the kidney removing 35–40% of the endogenous hormone. However, in insulin-treated diabetics receiving subcutaneous insulin injections, this ratio is reversed, with as much as 60% of exogenous insulin being cleared by the kidney and the liver removing no more than 30–40%. The half-life of circulating insulin is 3–5 minutes.
Measurement of Circulating Insulin

The radioimmunoassay of insulin permits detection of insulin in picomolar quantities. The assay is based on antibodies developed in guinea pigs against bovine or pork insulin. Because of the similarities between these two insulins and human insulin, the assay successfully measures the human hormone as well.
With this assay, basal insulin values of 5–15 U/mL (30–90 pmol/L) are found in normal humans, with a peak rise to 60–90 U/mL (360–540 pmol/L) during meals. Similar assays for measuring all of the known hormones of the endocrine pancreas (including C-peptide and proinsulin) have been developed.
The Insulin Receptor
Once insulin has entered the circulation, it is bound by specialized receptors that are found on the membranes of most tissues. The biologic responses promoted by these insulin-receptor complexes have been identified in the primary target tissues, ie, liver, muscle, and adipose tissue. The receptors bind insulin with high specificity and affinity in the picomolar range. The full insulin receptor consists of two covalently linked heterodimers, each containing an subunit, which is entirely extracellular and constitutes the recognition site, and a subunit that spans the membrane (Figure 41–3). The subunit contains a tyrosine kinase. The binding of an insulin molecule to the subunits at the outside surface of the cell activates the receptor and through a conformational change brings the catalytic loops of the opposing cytoplasmic subunits into closer proximity thereby facilitating phosphorylation of tyrosine residues and tyrosine kinase activity.
Figure 41–3.
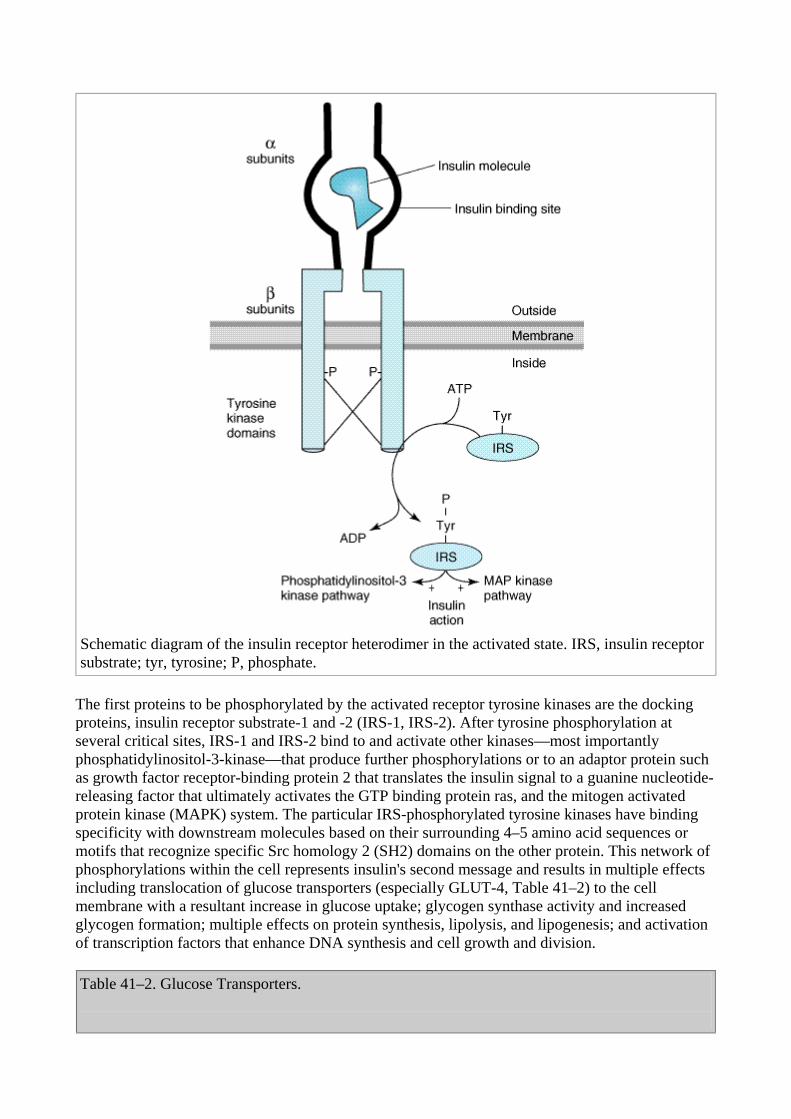
Schematic diagram of the insulin receptor heterodimer in the activated state. IRS, insulin receptor substrate; tyr, tyrosine; P, phosphate.
The first proteins to be phosphorylated by the activated receptor tyrosine kinases are the docking proteins, insulin receptor substrate-1 and -2 (IRS-1, IRS-2). After tyrosine phosphorylation at several critical sites, IRS-1 and IRS-2 bind to and activate other kinases—most importantly phosphatidylinositol-3-kinase—that produce further phosphorylations or to an adaptor protein such as growth factor receptor-binding protein 2 that translates the insulin signal to a guanine nucleotide-releasing factor that ultimately activates the GTP binding protein ras, and the mitogen activated protein kinase (MAPK) system. The particular IRS-phosphorylated tyrosine kinases have binding specificity with downstream molecules based on their surrounding 4–5 amino acid sequences or motifs that recognize specific Src homology 2 (SH2) domains on the other protein. This network of phosphorylations within the cell represents insulin's second message and results in multiple effects including translocation of glucose transporters (especially GLUT-4, Table 41–2) to the cell membrane with a resultant increase in glucose uptake; glycogen synthase activity and increased glycogen formation; multiple effects on protein synthesis, lipolysis, and lipogenesis; and activation of transcription factors that enhance DNA synthesis and cell growth and division.
Table 41–2. Glucose Transporters.
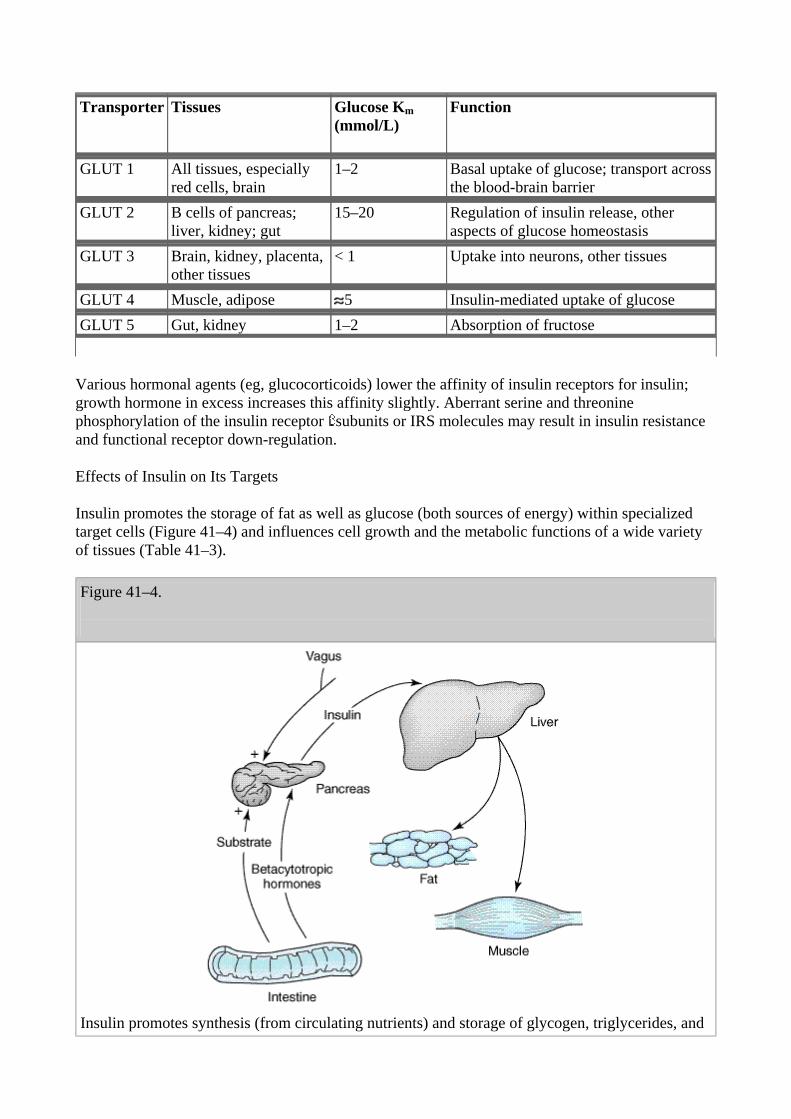
Transporter Tissues Glucose Km (mmol/L)
Function
GLUT 1 All tissues, especially red cells, brain
1–2 Basal uptake of glucose; transport across the blood-brain barrier
GLUT 2 B cells of pancreas; liver, kidney; gut
15–20 Regulation of insulin release, other aspects of glucose homeostasis
GLUT 3 Brain, kidney, placenta, other tissues
< 1 Uptake into neurons, other tissues
GLUT 4 Muscle, adipose 5 Insulin-mediated uptake of glucose GLUT 5 Gut, kidney 1–2 Absorption of fructose
Various hormonal agents (eg, glucocorticoids) lower the affinity of insulin receptors for insulin; growth hormone in excess increases this affinity slightly. Aberrant serine and threonine phosphorylation of the insulin receptor subunits or IRS molecules may result in insulin resistance and functional receptor down-regulation.
Effects of Insulin on Its Targets
Insulin promotes the storage of fat as well as glucose (both sources of energy) within specialized target cells (Figure 41–4) and influences cell growth and the metabolic functions of a wide variety of tissues (Table 41–3).
Figure 41–4.
Insulin promotes synthesis (from circulating nutrients) and storage of glycogen, triglycerides, and
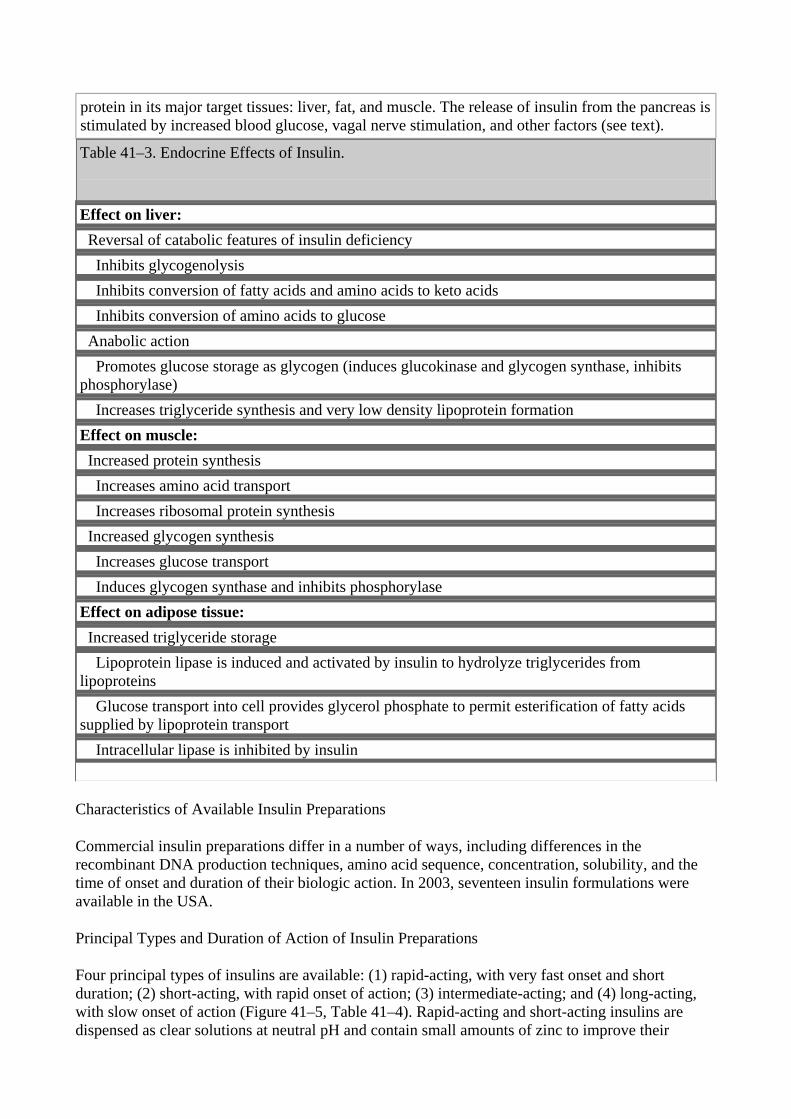
protein in its major target tissues: liver, fat, and muscle. The release of insulin from the pancreas is stimulated by increased blood glucose, vagal nerve stimulation, and other factors (see text). Table 41–3. Endocrine Effects of Insulin.
Effect on liver: Reversal of catabolic features of insulin deficiency Inhibits glycogenolysis Inhibits conversion of fatty acids and amino acids to keto acids Inhibits conversion of amino acids to glucose Anabolic action Promotes glucose storage as glycogen (induces glucokinase and glycogen synthase, inhibits phosphorylase) Increases triglyceride synthesis and very low density lipoprotein formation Effect on muscle: Increased protein synthesis Increases amino acid transport Increases ribosomal protein synthesis Increased glycogen synthesis Increases glucose transport Induces glycogen synthase and inhibits phosphorylase Effect on adipose tissue: Increased triglyceride storage Lipoprotein lipase is induced and activated by insulin to hydrolyze triglycerides from lipoproteins Glucose transport into cell provides glycerol phosphate to permit esterification of fatty acids supplied by lipoprotein transport Intracellular lipase is inhibited by insulin
Characteristics of Available Insulin Preparations
Commercial insulin preparations differ in a number of ways, including differences in the recombinant DNA production techniques, amino acid sequence, concentration, solubility, and the time of onset and duration of their biologic action. In 2003, seventeen insulin formulations were available in the USA.
Principal Types and Duration of Action of Insulin Preparations
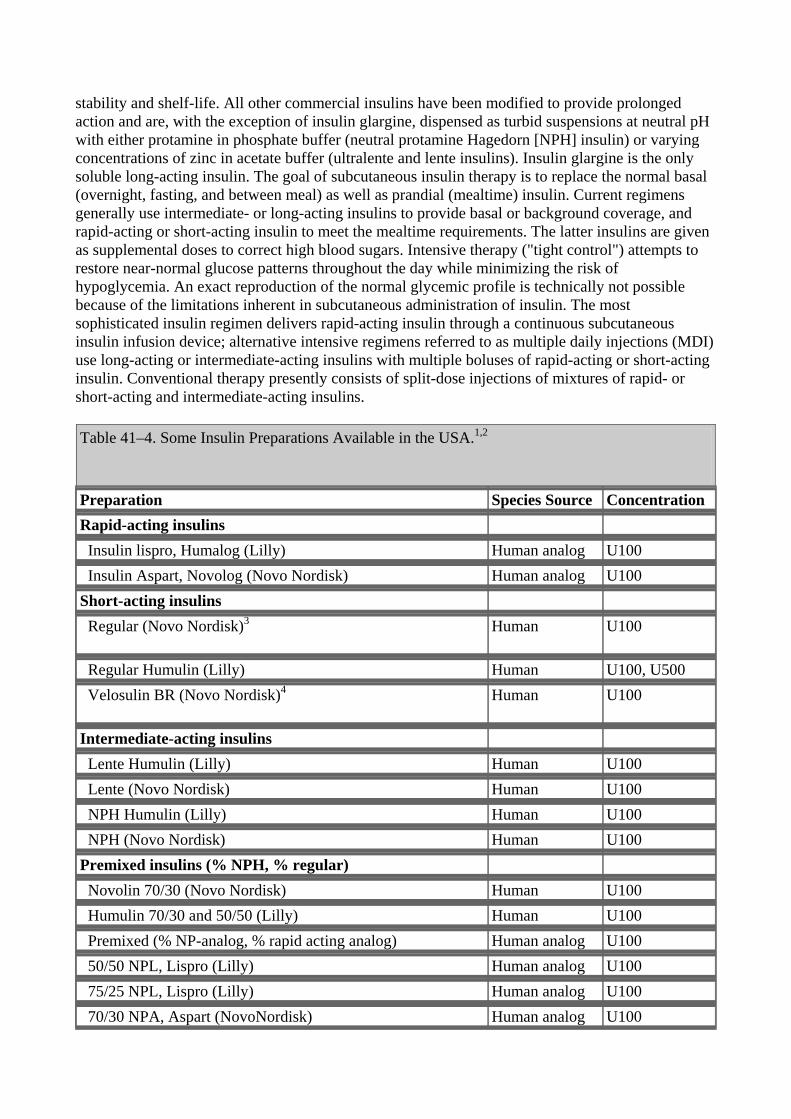
Four principal types of insulins are available: (1) rapid-acting, with very fast onset and short duration; (2) short-acting, with rapid onset of action; (3) intermediate-acting; and (4) long-acting, with slow onset of action (Figure 41–5, Table 41–4). Rapid-acting and short-acting insulins are dispensed as clear solutions at neutral pH and contain small amounts of zinc to improve their
stability and shelf-life. All other commercial insulins have been modified to provide prolonged action and are, with the exception of insulin glargine, dispensed as turbid suspensions at neutral pH with either protamine in phosphate buffer (neutral protamine Hagedorn [NPH] insulin) or varying concentrations of zinc in acetate buffer (ultralente and lente insulins). Insulin glargine is the only soluble long-acting insulin. The goal of subcutaneous insulin therapy is to replace the normal basal (overnight, fasting, and between meal) as well as prandial (mealtime) insulin. Current regimens generally use intermediate- or long-acting insulins to provide basal or background coverage, and rapid-acting or short-acting insulin to meet the mealtime requirements. The latter insulins are given as supplemental doses to correct high blood sugars. Intensive therapy ("tight control") attempts to restore near-normal glucose patterns throughout the day while minimizing the risk of hypoglycemia. An exact reproduction of the normal glycemic profile is technically not possible because of the limitations inherent in subcutaneous administration of insulin. The most sophisticated insulin regimen delivers rapid-acting insulin through a continuous subcutaneous insulin infusion device; alternative intensive regimens referred to as multiple daily injections (MDI) use long-acting or intermediate-acting insulins with multiple boluses of rapid-acting or short-acting insulin. Conventional therapy presently consists of split-dose injections of mixtures of rapid- or short-acting and intermediate-acting insulins.
Table 41–4. Some Insulin Preparations Available in the USA.1,2
Preparation Species Source Concentration Rapid-acting insulins Insulin lispro, Humalog (Lilly) Human analog U100 Insulin Aspart, Novolog (Novo Nordisk) Human analog U100 Short-acting insulins Regular (Novo Nordisk)3
Human U100
Regular Humulin (Lilly) Human U100, U500 Velosulin BR (Novo Nordisk)4
Human U100
Intermediate-acting insulins Lente Humulin (Lilly) Human U100 Lente (Novo Nordisk) Human U100 NPH Humulin (Lilly) Human U100 NPH (Novo Nordisk) Human U100 Premixed insulins (% NPH, % regular) Novolin 70/30 (Novo Nordisk) Human U100 Humulin 70/30 and 50/50 (Lilly) Human U100 Premixed (% NP-analog, % rapid acting analog) Human analog U100 50/50 NPL, Lispro (Lilly) Human analog U100 75/25 NPL, Lispro (Lilly) Human analog U100 70/30 NPA, Aspart (NovoNordisk) Human analog U100

Long-acting insulins Ultralente Humulin U (Lilly) Human U100 Insulin glargine-lantus (Aventis/Hoechst Marion Roussel) Human U100
1 Modified and reproduced, with permission, from Katzung BG (editor): Drug Therapy, 2nd ed. Originally published by Appleton & Lange. Copyright © 1991 by The McGraw-Hill Companies, Inc.
2 All of these agents (except insulin lispro, insulin aspart, insulin glargine, and U500 regular Humulin) are available without a prescription. All insulins should be refrigerated and brought to room temperature just before injection.
3 Novo Nordisk human insulins are termed Novolin R, L, and N.
4Velosulin contains phosphate buffer, which favors its use to prevent insulin aggregation in pump tubing but precludes its being mixed with lente insulin.
Rapid-Acting Insulin
Two rapid-acting insulin analogs are commercially available: insulin lispro and insulin aspart. The rapid-acting insulins permit more physiologic prandial insulin replacement because their rapid onset and early peak action more closely mimics normal endogenous prandial insulin secretion than does regular insulin, and they have the additional benefit of allowing insulin to be taken immediately before the meal without sacrificing glucose control. Their duration of action is rarely more than 3–5 hours, which decreases the risk of late postmeal hypoglycemia. They have the lowest variability of absorption of all available insulin formulations.
Insulin lispro, the first monomeric insulin analog to be marketed, is produced by recombinant technology wherein two amino acids near the carboxyl terminal of the B chain have been reversed in position: proline at position B28 has been moved to B29 and lysine at position B29 has been moved to B28 (Figure 41–1). Reversing these two amino acids does not interfere in any way with insulin lispro's binding to the insulin receptor, its circulating half-life, or with its immunogenicity, all of which are identical with those of human regular insulin. However, the advantage of this analog is its very low propensity—in contrast to human insulin—to self-associate in antiparallel fashion and form dimers. To enhance the shelf-life of insulin in vials, insulin lispro is stabilized into hexamers by a cresol preservative. When injected subcutaneously, the drug quickly dissociates into monomers and is rapidly absorbed with onset of action within 5–15 minutes, and reaching peak activity as early as 1 hour. The time to peak action is relatively constant, regardless of the dose. Its duration is seldom more than 3–5 hours.
Insulin lispro has a low variability of absorption (5%) of all the commercial insulin preparations—compared with 25% for regular insulin and 25–50% or more for intermediate-acting and long-acting insulins. Although not specifically approved for use in continuous subcutaneous insulin infusion (CSII) pumps, when used in these devices or in intensive insulin regimens, insulin lispro is associated with significantly improved glycemic control compared with regular insulin, without increased incidence of hypoglycemia.
Insulin aspart is created by the substitution of the B28 proline with a negatively charged aspartic acid (Figure 41–1). This modification reduces the normal ProB28 and GlyB23 monomer-monomer

interaction, thereby inhibiting insulin self-aggregation. Insulin aspart rapidly breaks into monomers after subcutaneous injection, displays an onset of action within 10–20 minutes, and exerts a peak effect within 1 hour, with an average duration of action of no longer than 3–5 hours. Its absorption and activity profile is similar to insulin lispro and more reproducible than regular insulin, but it has similar binding, activity, and mitogenicity characteristics to regular insulin and equivalent immunogenicity. Insulin aspart is approved for subcutaneous administration by injection as well as through CSII devices.
Short-Acting Insulin
Regular insulin is a short-acting soluble crystalline zinc insulin made by recombinant DNA techniques to produce a molecule identical to human insulin. Its effect appears within 30 minutes and peaks between 2 and 3 hours after subcutaneous injection and generally lasts 5–8 hours. In high concentrations, eg, in the vial, regular insulin molecules self-aggregate in antiparallel fashion to form dimers that stabilize around zinc ions to create insulin hexamers. The hexameric nature of regular insulin causes a delayed onset and prolongs the time to peak action. After subcutaneous injection, the insulin hexamers are too large and bulky to be transported across the vascular endothelium into the bloodstream. As the insulin depot is diluted by interstitial fluid and the concentration begins to fall, the hexamers break down into dimers and finally monomers. This results in three different rates of absorption of the injected insulin, with the final monomeric phase having the fastest uptake out of the injection site. As with all older insulin formulations, the duration of action as well as the time of onset and the intensity of peak action increase with the size of the dose. Clinically, this is a critical issue because the pharmacokinetics and pharmacodynamics of small doses of regular, NPH, lente, and ultralente, insulins differ greatly from those of large doses. Short-acting soluble insulin is the only type that should be administered intravenously as the dilution causes the hexameric insulin to immediately dissociate into monomers. It is particularly useful for intravenous therapy in the management of diabetic ketoacidosis and when the insulin requirement is changing rapidly, such as after surgery or during acute infections.
Intermediate-Acting and Long-Acting Insulins
Lente Insulin
Lente insulin is a mixture of 30% semilente (an amorphous precipitate of insulin with zinc ions in acetate buffer that has a relatively rapid onset of action) with 70% ultralente insulin (a poorly soluble crystal of zinc insulin that has a delayed onset and prolonged duration of action). These two components provide a combination of relatively rapid absorption with sustained long action, making lente insulin a useful therapeutic agent. As with regular insulin, the time of onset, time to peak, and duration of action are dose-dependent.
NPH (Neutral Protamine Hagedorn, or Isophane) Insulin
NPH insulin is an intermediate-acting insulin wherein absorption and the onset of action is delayed by combining appropriate amounts of insulin and protamine so that neither is present in an uncomplexed form ("isophane"). Protamine is a mixture of six major and some minor compounds of similar structure isolated from the sperm of rainbow trout. They appear to be basic, arginine-rich peptides with an average molecular weight of approximately 4400. To form an isophane complex (one in which neither component retains any free binding sites), approximately a 1:10 ratio by weight of protamine to insulin is required, representing approximately six molecules of insulin per molecule of protamine. After subcutaneous injection, proteolytic tissue enzymes degrade the protamine to permit absorption of insulin.
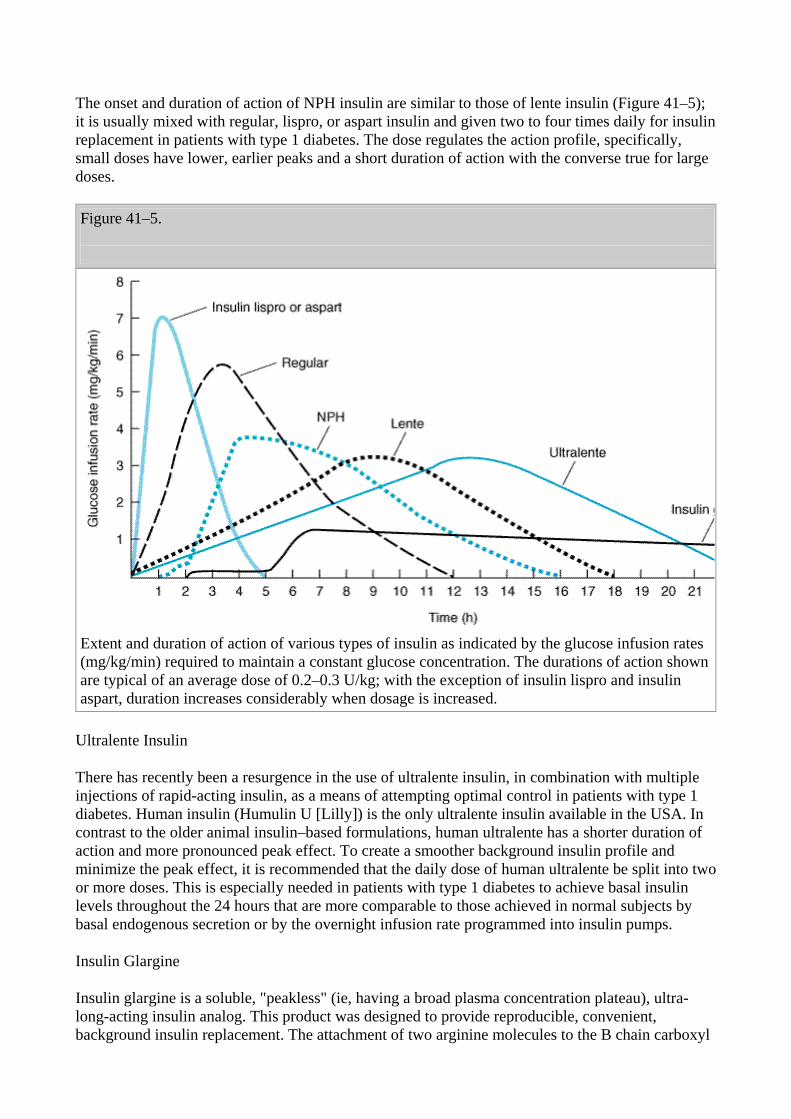
The onset and duration of action of NPH insulin are similar to those of lente insulin (Figure 41–5); it is usually mixed with regular, lispro, or aspart insulin and given two to four times daily for insulin replacement in patients with type 1 diabetes. The dose regulates the action profile, specifically, small doses have lower, earlier peaks and a short duration of action with the converse true for large doses.
Figure 41–5.
Extent and duration of action of various types of insulin as indicated by the glucose infusion rates (mg/kg/min) required to maintain a constant glucose concentration. The durations of action shown are typical of an average dose of 0.2–0.3 U/kg; with the exception of insulin lispro and insulin aspart, duration increases considerably when dosage is increased.
Ultralente Insulin
There has recently been a resurgence in the use of ultralente insulin, in combination with multiple injections of rapid-acting insulin, as a means of attempting optimal control in patients with type 1 diabetes. Human insulin (Humulin U [Lilly]) is the only ultralente insulin available in the USA. In contrast to the older animal insulin–based formulations, human ultralente has a shorter duration of action and more pronounced peak effect. To create a smoother background insulin profile and minimize the peak effect, it is recommended that the daily dose of human ultralente be split into two or more doses. This is especially needed in patients with type 1 diabetes to achieve basal insulin levels throughout the 24 hours that are more comparable to those achieved in normal subjects by basal endogenous secretion or by the overnight infusion rate programmed into insulin pumps.
Insulin Glargine
Insulin glargine is a soluble, "peakless" (ie, having a broad plasma concentration plateau), ultra-long-acting insulin analog. This product was designed to provide reproducible, convenient, background insulin replacement. The attachment of two arginine molecules to the B chain carboxyl

terminal and substitution of a glycine for asparagine at the A21 position created an analog that is soluble in solution but precipitates in the more neutral body pH after subcutaneous injection. Individual insulin molecules slowly dissolve away from the crystalline depot and provide a low, continuous level of circulating insulin. Insulin glargine has a slow onset of action (1–1.5 hours) and achieves a maximum effect after 4–5 hours. This maximum activity is maintained for 11–24 hours or longer. Glargine is usually given once daily, although some very insulin-sensitive individuals will benefit from split (twice a day) dosing. To maintain solubility, the formulation is unusually acidic (pH 4.0) and insulin glargine should not be mixed with other insulin. Separate syringes must be used to minimize the risk of contamination and subsequent loss of efficacy. The absorption pattern of insulin glargine appears to be independent of the anatomic site of injection, and this drug is associated with less immunogenicity than human insulin in animal studies. Glargine's interaction with the insulin receptor is similar to that of native insulin and shows no increase in mitogenic activity in vitro. It has sixfold to sevenfold greater binding than native insulin to the IGF1 receptor, but the clinical significance of this is unclear.
Mixtures of Insulins
Since intermediate-acting insulins require several hours to reach adequate therapeutic levels, their use in type 1 diabetic patients requires supplements of lispro, aspart, or regular insulin before meals. For convenience, these are often mixed together in the same syringe before injection. When regular insulin is used, NPH is preferred to lente insulin as the intermediate-acting component in these mixtures because increased proportions of lente to regular insulin may retard the rapid action of admixed regular insulin, particularly if not injected immediately after mixing. This is due to precipitation of the regular insulin by excess zinc. Premixed formulations of 70%/30% NPH and regular and 50%/50% NPH and regular are available in the USA. Insulin lispro and aspart can be acutely mixed (ie, just before injection) with either NPH, lente, or ultralente insulin without affecting their rapid absorption. However, premixed preparations have thus far been unstable. To remedy this, intermediate insulins composed of isophane complexes of protamine with insulin lispro and insulin aspart have been developed. These intermediate insulins have been designated as "NPL" (neutral protamine lispro) and "NPA" (neutral protamine aspart) and have the same duration of action as NPH insulin. They have the advantage of permitting formulation as premixed combinations of NPL and insulin lispro, and NPA and insulin aspart and they have been shown to be safe and effective in clinical trials. The FDA has approved 50%/50% and 75%/30% NPL/insulin lispro and a 70%/30% NPA/insulin aspart premixed formulations. Additional ratios are available abroad. Insulin glargine must be given as a separate injection. It is not miscible acutely or in a premixed preparation with any other insulin formulation.
Species of Insulin
Beef and Pork Insulins
Historically, commercial insulin in the USA contained beef or pork insulin. Beef insulin differs by three amino acids from human insulin, whereas only a single amino acid distinguishes pork and human insulins (Figure 41–1). The beef hormone is slightly more antigenic than pork insulin in humans. Of the insulins manufactured from animal sources, only purified pork insulin is still available and it requires special ordering.
Human insulin, which is now less expensive than monospecies pork insulin and is also less immunogenic, has supplanted purified pork insulins.
Human Insulins

Mass production of human insulin by recombinant DNA techniques is now carried out by inserting the human proinsulin gene into Escherichia coli or yeast and treating the extracted proinsulin to form the human insulin molecule.
Human insulin from E coli is available for clinical use as Humulin (Lilly) and dispensed as either regular, NPH, lente, or ultralente Humulin. Human insulin prepared biosynthetically in yeast is marketed by Novo Nordisk as human insulin injection in regular, lente, and NPH forms: Novolin R, Monotard Human Insulin (Novolin L), and Novolin N. The same company also produces a human insulin marketed as Velosulin (regular) that contains a phosphate buffer. This reduces aggregation of regular insulin molecules when used in infusion pumps. However, because of the tendency of phosphate to precipitate zinc ions, Velosulin should not be mixed with any of the lente insulins.
Human insulins appear to be as effective as—and considerably less immunogenic in diabetic patients than—beef-pork insulin mixtures and slightly less immunogenic than pork insulin.
Concentration
Currently, all insulins in the USA and Canada are available in a concentration of 100 U/mL (U100) and are dispensed in 10 mL vials. A limited supply of U500 regular human insulin is available for use in rare cases of severe insulin resistance in which larger doses of insulin are required.
Insulin Delivery Systems
The standard mode of insulin therapy is subcutaneous injection using conventional disposable needles and syringes. During the last 3 decades, much effort has gone into exploration of other means of administration.
Portable Pen Injectors
To facilitate multiple subcutaneous injections of insulin, particularly during intensive insulin therapy, portable pen-sized injectors have been developed. These contain cartridges of insulin and replaceable needles. Disposable insulin pens are also available for selected formulations. These include regular insulin, insulin lispro, insulin aspart, NPH insulin, and premixed 70%/30% and 50%/50% NPH/regular, 75% NPL/25% lispro, 50% NPL/50% lispro, and 70% NPA/30% aspart insulin. They have been well accepted by patients because they eliminate the need to carry syringes and bottles of insulin to the workplace and while traveling.
Continuous Subcutaneous Insulin Infusion Devices (Csii, Insulin Pumps)
Continuous subcutaneous insulin infusion devices are external open-loop pumps for insulin delivery. The devices have a user-programmable pump that delivers individualized basal and bolus insulin replacement doses based on blood glucose self-monitoring results. Normally, the 24-hour background basal rates are relatively constant from day to day, although temporarily altered rates can be superimposed to adjust for a short-term change in requirement. For example, the basal delivery rate might need to be decreased for several hours because of the increased insulin sensitivity associated with strenuous activity. In contrast, the bolus amounts frequently vary and are used to correct high blood glucose levels and to cover mealtime insulin requirements based on the carbohydrate content of the food and concurrent activity. The pump—which contains an insulin reservoir, the program chip, the keypad, and the display screen—is about the size of a pager. It is usually placed on a belt or in a pocket, and the insulin is infused through thin plastic tubing that is connected to the subcutaneously inserted infusion set. The abdomen is the favored site for the

infusion set, although flanks and thighs are also used. The insulin reservoir, tubing, and infusion set need to be changed using sterile techniques every 2 or 3 days. CSII delivery is regarded as the most physiologic method of insulin replacement.
The use of these devices is encouraged for individuals who are unable to obtain target control while on multiple injection regimens and in circumstances where excellent glycemic control is desired, such as during pregnancy. Their optimal use requires responsible involvement and commitment by the patient. Velosulin (a regular insulin) and insulin aspart are the only insulins specifically approved for pump use. Although not formally approved for pump use, insulin lispro has been successfully delivered through CSII devices since it became commercially available. Insulins aspart and lispro are preferred pump insulins because their favorable pharmacokinetic attributes allow glycemic control without increasing the risk of hypoglycemia.
Inhaled Insulin
Clinical trials are in progress to evaluate the safety and efficacy of finely powdered and aerosolized insulin formulations delivered by inhalation. Insulin is readily absorbed into the bloodstream through alveolar walls, but the challenge has been to create particles that are small enough to pass through the bronchial tree without being trapped and still enter the alveoli in sufficient amounts to have a clinical effect. Insulin delivered by the inhaled route should have a rapid onset and a relatively short duration of action and could be used to cover mealtime insulin requirements or to correct high glucose levels, but not to provide background or basal insulin coverage. Safety concerns regarding pulmonary fibrosis or hypertension and excessive antibody formation may preclude or delay approval.
Treatment with Insulin
The current classification of diabetes mellitus identifies a group of patients who have virtually no insulin secretion and whose survival depends on administration of exogenous insulin. This insulin-dependent group (type 1) represents 5–10% of the diabetic population in the USA. Most type 2 diabetics do not require exogenous insulin for survival, but many need exogenous supplementation of their endogenous secretion to achieve optimum health. It is estimated that as many as 20% of type 2 diabetics in the USA (2–2.5 million people) are presently taking insulin.
Benefit of Glycemic Control in Diabetes Mellitus
The consensus of the American Diabetes Association is that intensive insulin therapy associated with comprehensive self-management training should become standard therapy in most type 1 patients after puberty (see Benefits of Tight Glycemic Control in Type 1 Diabetes). Exceptions include patients with advanced renal disease and the elderly, since the risks of hypoglycemia outweigh the benefit of tight glycemic control in these groups. In children under the age of 7 years, the extreme susceptibility of the developing brain to damage from hypoglycemia contraindicates attempts at intensive glycemic control, particularly since diabetic complications do not seem to occur until some years after the onset of puberty. A similar conclusion regarding the benefits of tight control in type 2 diabetes was reached as the result of a large study in the United Kingdom.
Complications of Insulin Therapy
Hypoglycemia
Mechanisms and Diagnosis

Hypoglycemic reactions are the most common complication of insulin therapy. They may result from a delay in taking a meal, inadequate carbohydrate consumed, unusual physical exertion, or a dose of insulin that is too large for immediate needs.
Rapid development of hypoglycemia in individuals with intact hypoglycemic awareness causes signs of autonomic hyperactivity, both sympathetic (tachycardia, palpitations, sweating, tremulousness) and parasympathetic (nausea, hunger) and may progress to convulsions and coma if untreated.
In individuals exposed to frequent hypoglycemic episodes during tight glycemic control, autonomic warning signals of hypoglycemia are less frequent or even absent. This dangerous acquired condition is termed "hypoglycemic unawareness." When patients lack the early warning signs of low blood glucose, they may not take corrective measures in time. In patients with persistent, untreated hypoglycemia, the manifestations of insulin excess may develop—confusion, weakness, bizarre behavior, coma, seizures—at which point they may not be able to procure or safely swallow glucose-containing foods. Hypoglycemic awareness may be restored by preventing frequent hypoglycemic episodes. An identification bracelet, necklace, or card in the wallet or purse, as well as some form of rapidly absorbed glucose, should be carried by every diabetic who is receiving hypoglycemic drug therapy.
Treatment of Hypoglycemia
All of the manifestations of hypoglycemia are relieved by glucose administration. To expedite absorption, simple sugar or glucose should be given, preferably in a liquid form. To treat mild hypoglycemia in a patient who is conscious and able to swallow, orange juice, glucose gel, or any sugar-containing beverage or food may be given. If more severe hypoglycemia has produced unconsciousness or stupor, the treatment of choice is to give 20–50 mL of 50% glucose solution by intravenous infusion over a period of 2–3 minutes. If intravenous therapy is not available, 1 mg of glucagon injected either subcutaneously or intramuscularly will usually restore consciousness within 15 minutes to permit ingestion of sugar. If the patient is stuporous and glucagon is not available, small amounts of honey or syrup can be inserted into the buccal pouch. In general, however, oral feeding is contraindicated in unconscious patients. Emergency medical services should be called for all episodes of severely impaired consciousness.
Immunopathology of Insulin Therapy
At least five molecular classes of insulin antibodies may be produced during the course of insulin therapy in diabetes: IgA, IgD, IgE, IgG, and IgM. There are two major types of immune disorders in these patients:
Insulin Allergy
Insulin allergy, an immediate type hypersensitivity, is a rare condition in which local or systemic urticaria results from histamine release from tissue mast cells sensitized by anti-insulin IgE antibodies. In severe cases, anaphylaxis results. Because sensitivity is often to noninsulin protein contaminants, the highly purified and human insulins have markedly reduced the incidence of insulin allergy, especially local reactions.
Immune Insulin Resistance
A low titer of circulating IgG anti-insulin antibodies that neutralize the action of insulin to a

negligable extent develops in most insulin-treated patients. Rarely, the titer of insulin antibodies will lead to insulin resistance and may be associated with other systemic autoimmune processes such as lupus erythematosus.
Lipodystrophy at Injection Sites
Injection of older insulin preparations sometimes led to atrophy of subcutaneous fatty tissue at the site of injection. This type of immune complication is almost never seen since the development of human insulin preparations of neutral pH. Injection of these newer preparations directly into the atrophic area often results in restoration of normal contours. Hypertrophy of subcutaneous fatty tissue remains a problem, even with the purified insulins, if injected repeatedly at the same site. However, this may be corrected by avoidance of that specific injection site or with liposuction. Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 41. Pancreatic Hormones & Antidiabetic Drugs >
Benefits of Tight Glycemic Control in Diabetes
A long-term randomized prospective study involving 1441 type 1 patients in 29 medical centers reported in 1993 that "near normalization" of blood glucose resulted in a delay in onset and a major slowing of progression of microvascular and neuropathic complications of diabetes during follow-up periods of up to 10 years (Diabetes Control and Complications Trial [DCCT] Research Group, 1993).
In the intensively treated group a mean glycated hemoglobin of 7.2% (normal, < 6%) and a mean blood glucose of 155 mg/dL were achieved, while in the conventionally treated group, glycated hemoglobin averaged 8.9% with an average blood glucose of 225 mg/dL. Over the study period, which averaged 7 years, there was an approximately 60% reduction in risk in the tight control group compared with the standard control group with regard to diabetic retinopathy, nephropathy, and neuropathy.
The United Kingdom Prospective Diabetes Study (UKPDS) was a very large randomized prospective study carried out to study the effects of intensive glycemic control with several types of therapies and the effects of blood pressure control in type 2 diabetic patients. A total of 3867 newly diagnosed type 2 diabetic patients were studied over 10 years. A significant fraction of these were overweight and hypertensive. Patients were given dietary treatment alone or intensive therapy with insulin, chlorpropamide, glyburide, or glipizide. Metformin was an option for patients with inadequate response to other therapies. Tight control of blood pressure was added as a variable, with an angiotensin-converting enzyme inhibitor, -blocker or, in some cases, a calcium channel blocker available for this purpose.
Tight control of diabetes, with reduction of HbA1c from 9.1% to 7%, was shown to reduce the risk of microvascular complications overall compared with conventional therapy (mostly diet alone, which decreased HbA1c to 7.9%). Cardiovascular complications were not noted for any particular therapy; metformin treatment alone reduced the risk of macrovascular disease (myocardial infarction, stroke).
Tight control of hypertension also had a surprisingly significant effect on microvascular disease (as well as more conventional hypertension-related sequela) in these diabetic patients. These studies show that tight glycemic control benefits both type 1 and type 2 patients.
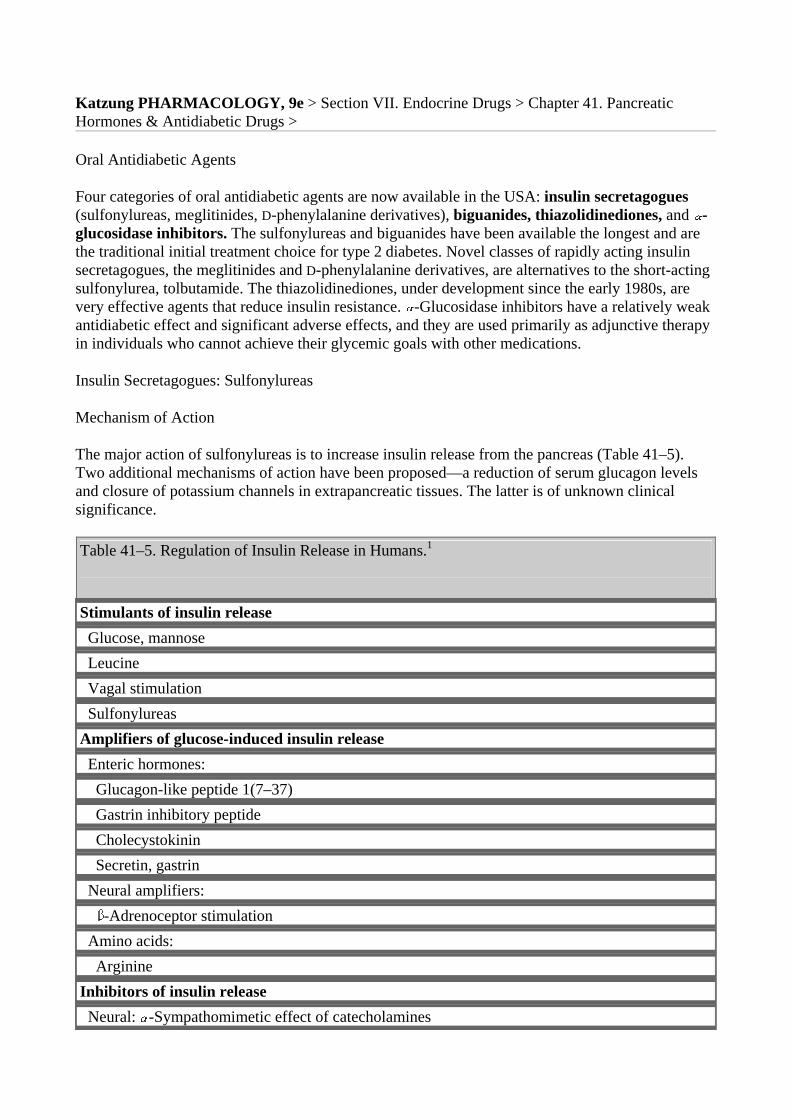
Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 41. Pancreatic Hormones & Antidiabetic Drugs >
Oral Antidiabetic Agents
Four categories of oral antidiabetic agents are now available in the USA: insulin secretagogues (sulfonylureas, meglitinides, D-phenylalanine derivatives), biguanides, thiazolidinediones, and -glucosidase inhibitors. The sulfonylureas and biguanides have been available the longest and are the traditional initial treatment choice for type 2 diabetes. Novel classes of rapidly acting insulin secretagogues, the meglitinides and D-phenylalanine derivatives, are alternatives to the short-acting sulfonylurea, tolbutamide. The thiazolidinediones, under development since the early 1980s, are very effective agents that reduce insulin resistance. -Glucosidase inhibitors have a relatively weak antidiabetic effect and significant adverse effects, and they are used primarily as adjunctive therapy in individuals who cannot achieve their glycemic goals with other medications.
Insulin Secretagogues: Sulfonylureas
Mechanism of Action
The major action of sulfonylureas is to increase insulin release from the pancreas (Table 41–5). Two additional mechanisms of action have been proposed—a reduction of serum glucagon levels and closure of potassium channels in extrapancreatic tissues. The latter is of unknown clinical significance.
Table 41–5. Regulation of Insulin Release in Humans.1
Stimulants of insulin release Glucose, mannose Leucine Vagal stimulation Sulfonylureas Amplifiers of glucose-induced insulin release Enteric hormones: Glucagon-like peptide 1(7–37) Gastrin inhibitory peptide Cholecystokinin Secretin, gastrin Neural amplifiers: -Adrenoceptor stimulation Amino acids: Arginine Inhibitors of insulin release Neural: -Sympathomimetic effect of catecholamines

Humoral: Somatostatin Drugs: Diazoxide, phenytoin, vinblastine, colchicine
1Modified and reproduced, with permission, from Greenspan FS, Strewler GJ (editors): Basic & Clinical Endocrinology, 5th ed. Originally published by Appleton & Lange. Copyright © 1997 by The McGraw-Hill Companies, Inc.
Insulin Release from Pancreatic B Cells
Sulfonylureas bind to a 140 kDa high-affinity sulfonyl-urea receptor that is associated with a B cell inward rectifier ATP-sensitive potassium channel. Binding of a sulfonylurea inhibits the efflux of potassium ions through the channel (Figure 41–2) and results in depolarization. Depolarization, in turn, opens a voltage-gated calcium channel and results in calcium influx and the release of preformed insulin.
Reduction of Serum Glucagon Concentrations
Chronic administration of sulfonylureas to type 2 diabetics reduces serum glucagon levels, which may contribute to the hypoglycemic effect of the drugs. The mechanism for this suppressive effect of sulfonylureas on glucagon levels is unclear but appears to involve indirect inhibition due to enhanced release of both insulin and somatostatin, which inhibit A cell secretion.
Potassium Channel Closure in Extrapancreatic Tissues
Insulin secretagogues bind to sulfonylurea receptors in potassium channels in extrapancreatic tissues but the binding affinity varies among the drug classes and is much less avid than for the B cell receptors. The clinical significance of extrapancreatic binding is not known.
Efficacy & Safety of the Sulfonylureas
In 1970, the University Group Diabetes Program (UGDP) in the USA reported that the number of deaths due to cardiovascular disease in diabetic patients treated with tolbutamide was excessive compared with either insulin-treated patients or those receiving placebos. Due to design flaws, this study and its conclusions were not generally accepted. A study in the United Kingdom, the UKPDS, did not find an untoward cardiovascular effect of sulfonylurea usage in their large, long-term study.
The sulfonylureas continue to be widely prescribed and six are available in the USA (Table 41–6). They are conventionally divided into first-generation and second-generation agents, which differ primarily in their potency and adverse effects. The first-generation sulfonylureas are increasingly difficult to procure, and as the second-generation agents become generic and less expensive, the earlier compounds probably will be discontinued.
Table 41–6. Sulfonureas.
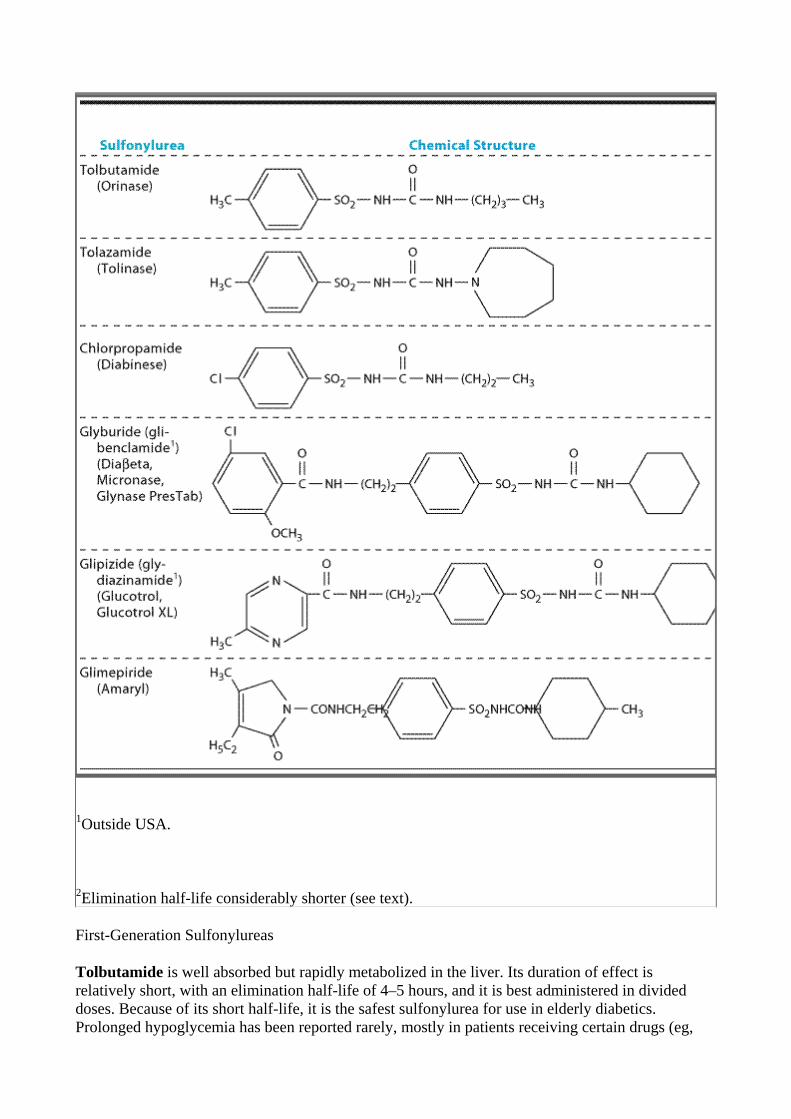
1Outside USA.
2Elimination half-life considerably shorter (see text).
First-Generation Sulfonylureas
Tolbutamide is well absorbed but rapidly metabolized in the liver. Its duration of effect is relatively short, with an elimination half-life of 4–5 hours, and it is best administered in divided doses. Because of its short half-life, it is the safest sulfonylurea for use in elderly diabetics. Prolonged hypoglycemia has been reported rarely, mostly in patients receiving certain drugs (eg,

dicumarol, phenylbutazone, some sulfonamides) that inhibit the metabolism of tolbutamide.
Chlorpropamide has a half-life of 32 hours and is slowly metabolized in the liver to products that retain some biologic activity; approximately 20–30% is excreted unchanged in the urine. Chlorpropamide also interacts with the drugs mentioned above that depend on hepatic oxidative catabolism, and it is contraindicated in patients with hepatic or renal insufficiency. Dosages in excess of 500 mg daily increase the risk of jaundice. The average maintenance dosage is 250 mg daily, given as a single dose in the morning. Prolonged hypoglycemic reactions are more common in elderly patients, and the drug is contraindicated in this group. Other side effects include a hyperemic flush after alcohol ingestion in genetically predisposed patients and dilutional hyponatremia. Hematologic toxicity (transient leukopenia, thrombocytopenia) occurs in less than 1% of patients.
Tolazamide is comparable to chlorpropamide in potency but has a shorter duration of action. Tolazamide is more slowly absorbed than the other sulfonylureas, and its effect on blood glucose does not appear for several hours. Its half-life is about 7 hours. Tolazamide is metabolized to several compounds that retain hypoglycemic effects. If more than 500 mg/d is required, the dose should be divided and given twice daily. Dosages larger than 1000 mg daily do not further improve the degree of blood glucose control.
Second-Generation Sulfonylureas
The second-generation sulfonylureas are more frequently prescribed in the USA than the first-generation agents because they have fewer adverse effects and drug interactions. These potent sulfonylurea compounds—glyburide, glipizide, and glimepiride—should be used with caution in patients with cardiovascular disease or in elderly patients, in whom hypoglycemia would be especially dangerous.
Glyburide is metabolized in the liver into products with very low hypoglycemic activity. The usual starting dosage is 2.5 mg/d or less, and the average maintenance dosage is 5–10 mg/d given as a single morning dose; maintenance dosages higher than 20 mg/d are not recommended. A formulation of "micronized" glyburide (Glynase PresTab) is available in a variety of tablet sizes. However, there is some question as to its bioequivalence with nonmicronized formulations, and the FDA recommends careful monitoring to retitrate dosage when switching from standard glyburide doses or from other sulfonylurea drugs.
Glyburide has few adverse effects other than its potential for causing hypoglycemia. Flushing has rarely been reported after ethanol ingestion and the compound slightly enhances free water clearance. Glyburide is contraindicated in the presence of hepatic impairment and in patients with renal insufficiency.
Glipizide has the shortest half-life (2–4 hours) of the more potent agents. For maximum effect in reducing postprandial hyperglycemia, this agent should be ingested 30 minutes before breakfast, since absorption is delayed when the drug is taken with food. The recommended starting dosage is 5 mg/d, with up to 15 mg/d given as a single dose. When higher daily dosages are required, they should be divided and given before meals. The maximum total daily dosage recommended by the manufacturer is 40 mg/d, although some studies indicate that the maximum therapeutic effect is achieved by 15–20 mg of the drug. An extended-release preparation (Glucotrol XL) provides 24-hour action after a once-daily morning dose (maximum of 20 mg/d). However, this formulation appears to have sacrificed its lower propensity for severe hypoglycemia compared with longer-acting glyburide without showing any demonstrable therapeutic advantages over the latter (which

can be obtained as a generic drug).
Because of its shorter half-life, glipizide is much less likely than glyburide to produce serious hypoglycemia. At least 90% of glipizide is metabolized in the liver to inactive products, and 10% is excreted unchanged in the urine. Glipizide therapy is therefore contraindicated in patients with significant hepatic or renal impairment, who would therefore be at high risk for hypoglycemia.
Glimepiride is approved for once-daily use as monotherapy or in combination with insulin. Glimepiride achieves blood glucose lowering with the lowest dose of any sulfonylurea compound. A single daily dose of 1 mg has been shown to be effective, and the recommended maximal daily dose is 8 mg. It has a long duration of effect with a half-life of 5 hours, allowing once-daily dosing and thereby improving compliance. It is completely metabolized by the liver to inactive products.
Secondary Failure & Tachyphylaxis to Sulfonylureas
Secondary failure, ie, failure to maintain a good response to sulfonylurea therapy over the long term—remains a disconcerting problem in the management of type 2 diabetes. A progressive decrease in B cell mass, reduction in physical activity, decline in lean body mass, or increase in ectopic fat deposition in chronic type 2 diabetes also may contribute to secondary failure.
Insulin Secretagogues: Meglitinides
The meglitinides are a relatively new class of insulin secretagogues. Repaglinide, the first member of the group, was approved for clinical use in 1998 (Table 41–7). These drugs modulate B cell insulin release by regulating potassium efflux through the potassium channels previously discussed. There is overlap with the sulfonylureas in their molecular sites of action since the meglitinides have two binding sites in common with the sulfonylureas and one unique binding site. Unlike the sulfonylureas, they have no direct effect on insulin exocytosis.
Table 41–7. Other Insulin Secretagogues.
Drug Chemical Structure Oral
Dose t1/2
Duration of Action (hours)
Repaglinide (Prandin)
0.25–4 mg before meals
1 hour
4–5

Nateglinide (Starlix)
60–120 mg before meals
1 hour
4
Repaglinide has a very fast onset of action, with a peak concentration and peak effect within approximately 1 hour after ingestion, but the duration of action is 5–8 hours. It is hepatically cleared by CYP3A4 with a plasma half-life of 1 hour. Because of its rapid onset, repaglinide is indicated for use in controlling postprandial glucose excursions. The drug should be taken just before each meal in doses of 0.25–4 mg (maximum, 16 mg/d); hypoglycemia is a risk if the meal is delayed or skipped or contains inadequate carbohydrate. This drug should be used cautiously in individuals with renal and hepatic impairment. Repaglinide is approved as monotherapy or in combination with biguanides. There is no sulfur in its structure, so repaglinide may be used in type 2 diabetic individuals with sulfur or sulfonylurea allergy.
Insulin Secretagogue: D-Phenylalanine Derivative
Nateglinide, a D-phenylalanine derivative, is the latest insulin secretagogue to become clinically available. Nateglinide stimulates very rapid and transient release of insulin from B cells through closure of the ATP-sensitive K+ channel. It also partially restores initial insulin release in response to an intravenous glucose tolerance test. This may be a significant advantage of the drug because type 2 diabetes is associated with loss of this initial insulin response. The restoration of more normal insulin secretion may suppress glucagon release early in the meal and result in less endogenous or hepatic glucose production. Nateglinide may have a special role in the treatment of individuals with isolated postprandial hyperglycemia, but it has minimal effect on overnight or fasting glucose levels. It is efficacious when given alone or in combination with nonsecretagogue oral agents (such as metformin). In contrast to other insulin secretagogues, dose titration is not required.
Nateglinide is ingested just prior to meals. It is absorbed within 20 minutes after oral administration with a time to peak concentration of less than 1 hour and is hepatically metabolized by CYP2C9 and CYP3A4 with a half-life of 1.5 hours. The overall duration of action is less than 4 hours.
Nateglinide amplifies the insulin secretory response to a glucose load but has a markedly diminished effect in the presence of normoglycemia. The incidence of hypoglycemia may be the lowest of all the secretagogues, and it has the advantage of being safe in individuals with very reduced renal function.
Biguanides
The structure of metformin is shown below. Phenformin (an older biguanide) was discontinued in the USA because of its association with lactic acidosis and because there was no documentation of any long-term benefit from its use.

Mechanisms of Action
A full explanation of the biguanides' mechanism of action remains elusive. Their blood glucose-lowering action does not depend on the presence of functioning pancreatic B cells. Patients with type 2 diabetes have considerably less fasting hyperglycemia as well as lower postprandial hyperglycemia after biguanides; however, hypoglycemia during biguanide therapy is essentially unknown. These agents are therefore more appropriately termed "euglycemic" agents. Currently proposed mechanisms of action include (1) direct stimulation of glycolysis in tissues, with increasedglucose removal from blood; (2) reduced hepatic and renal gluconeogenesis; (3) slowing of glucose absorption from the gastrointestinal tract, with increased glucose to lactate conversion by enterocytes; and (4) reduction of plasma glucagon levels.
Metabolism & Excretion
Metformin has a half-life of 1.5–3 hours, is not bound to plasma proteins, is not metabolized, and is excreted by the kidneys as the active compound. As a consequence of metformin's blockade of gluconeogenesis, the drug may impair the hepatic metabolism of lactic acid. In patients with renal insufficiency, biguanides accumulate and thereby increase the risk of lactic acidosis, which appears to be a dose-related complication.
Clinical Use
Biguanides have been most often prescribed for patients whose hyperglycemia is due to ineffective insulin action, ie, insulin resistance syndrome. Because metformin is an insulin-sparing agent and does not increase weight or provoke hypoglycemia, it offers obvious advantages over insulin or sulfonylureas in treating hyperglycemia in such individuals. The UKPDS reported that metformin therapy decreases the risk of macrovascular as well as microvascular disease; this is in contrast to the other therapies, which only modified microvascular morbidity. Biguanides are also indicated for use in combination with insulin secretagogues or thiazolidinediones in type 2 diabetics in whom oral monotherapy is inadequate. Metformin is useful in the prevention of type 2 diabetes; the landmark Diabetes Prevention Program concluded that metformin is efficacious in preventing the new onset of type 2 diabetes in middle-aged, obese individuals with impaired glucose tolerance and fasting hyperglycemia. Interestingly, metformin did not prevent diabetes in older, leaner pre-diabetics.
The dosage of metformin is from 500 mg to a maximum of 2.55 g daily, with the lowest effective dose being recommended. A common schedule would be to begin with a single 500 mg tablet given with breakfast for several days. If this is tolerated without gastrointestinal discomfort and hyperglycemia persists, a second 500 mg tablet may be added with the evening meal. If further dose increases are required after 1 week, an additional 500 mg tablet can be added to be taken with the midday meal, or the larger (850 mg) tablet can be prescribed twice daily or even three times daily (the maximum recommended dosage) if needed. Dosage should always be divided, since ingestion of more than 1000 mg at any one time usually provokes significant gastrointestinal side effects.
Toxicities

The most frequent toxic effects of metformin are gastrointestinal (anorexia, nausea, vomiting, abdominal discomfort, diarrhea) and occur in up to 20% of patients. They are dose related, tend to occur at the onset of therapy, and are often transient. However, metformin may have to be discontinued in 3–5% of patients because of persistent diarrhea. Absorption of vitamin B12 appears to be reduced during long-term metformin therapy, and annual screening of serum vitamin B12 levels and red blood cell parameters has been encouraged by the manufacturer to determine the need for vitamin B12 injections. In the absence of hypoxia or renal or hepatic insufficiency, lactic acidosis is less common with metformin therapy than with phenformin therapy.
Biguanides are contraindicated in patients with renal disease, alcoholism, hepatic disease, or conditions predisposing to tissue anoxia (eg, chronic cardiopulmonary dysfunction), because of an increased risk of lactic acidosis induced by biguanide drugs in the presence of these diseases.
Thiazolidinediones
Thiazolidinediones (Tzds) act to decrease insulin resistance. Their primary action is the nuclear regulation of genes involved in glucose and lipid metabolism and adipocyte differentiation. Tzds areligands of peroxisome proliferator-activated receptor-gamma (PPAR- ), part of the steroid and thyroid superfamily of nuclear receptors. These PPAR receptors are found in muscle, fat, and liver. PPAR- receptors are complex and modulate the expression of the genes involved in lipid and glucose metabolism, insulin signal transduction, and adipocyte and other tissue differentiation. The available Tzds do not have identical clinical effects and new drug development will focus on defining PPAR effects and designing ligands that have selective action—much like the selective estrogen receptor ligands (see Chapter 40: The Gonadal Hormones & Inhibitors).
In addition to targeting adipocytes, myocytes, and hepatocytes, Tzds also have significant effects on vascular endothelium, the immune system, the ovaries, and tumor cells. Some of these responses may be independent of the PPAR- pathway.
In persons with diabetes, a major site of Tzd action is adipose tissue, where the drug promotes glucose uptake and utilization and modulates synthesis of lipid hormones or cytokines and other proteins involved in energy regulation. Tzds also regulate adipocyte apoptosis and differentiation. Numerous other effects have been documented in animal studies but applicability to human tissues has yet to be determined.
Two thiazolidinediones are currently available: pioglitazone and rosiglitazone (Table 41–8). Their distinct side chains create differences in therapeutic action, metabolism, metabolite profile, and adverse effects. A third compound, troglitazone, was withdrawn from the market because of hepatic toxicity thought to be related to its side chain.
Table 41–8. Thiazolidinediones.
Thiazolidinedione Chemical Structure Oral
Dose

Pioglitazone (Actos)
15–45 mg once daily
Rosiglitazone (Avandia)
2–8 mg once daily
Pioglitazone may have PPAR- as well as PPAR- activity. It is absorbed within 2 hours of ingestion; although food may delay uptake, total bioavailability is not affected. Pioglitazone is metabolized by CYP2C8 and CYP3A4 to active metabolites. The bioavailability of numerous other drugs also degraded by these enzymes may be affected by pioglitazone therapy, including estrogen-containing oral contraceptives; additional methods of contraception are advised. Pioglitazone may be taken once daily; the usual starting dose is 15–30 mg. The triglyceride lowering effect is more significant than that observed with rosiglitazone. Pioglitazone is approved as a monotherapy and in combination with metformin, sulfonylureas, and insulin for the treatment of type 2 diabetes.
Rosiglitazone is rapidly absorbed and highly protein bound. It is metabolized in the liver to minimally active metabolites, predominantly by CYP2C8 and to a lesser extent by CYP2C9. It is administered once or twice daily; 4–8 mg is the usual total dose. Rosiglitazone shares the common Tzd adverse effects but does not seem to have any significant drug interactions. The drug is approved for use in type 2 diabetes as monotherapy or in combination with a biguanide or sulfonylurea.
Tzds are considered "euglycemics" and are efficacious in about 70% of new users. The overall response is similar to sulfonylurea and biguanide monotherapy. Individuals experiencing secondary failure to other oral agents should benefit from the addition (rather than substitution) of a Tzd. Because their mechanism of action involves gene regulation, the Tzds have a slow onset and offset of activity over weeks or even months. Combination therapy with sulfonylureas and insulin can lead to hypoglycemia and may require dosage adjustment. Long-term therapy is associated with a drop in triglyceride levels and a slight rise in HDL and low-density lipoprotein (LDL) cholesterol values. An adverse effect common to both Tzds is fluid retention, which presents as a mild anemia and peripheral edema especially when used in combination with insulin or insulin secretagogues. Many users have a dose-related weight gain (average 1–3 kg), which may be fluid-related. These agents should not be used during pregnancy, in the presence of significant liver disease, or if there is a concurrent diagnosis of heart failure. Anovulatory women may resume ovulation and should be counseled on the increased risk of pregnancy. Because of the hepatotoxicity observed with troglitazone, the FDA continues to require regular monitoring of liver function tests for the first year after initiation of Tzd therapy. To date, hepatotoxicity has not been associated with rosiglitazone or pioglitazone.
Thiazolidinediones have a theoretical benefit in the prevention of type 2 diabetes. One study
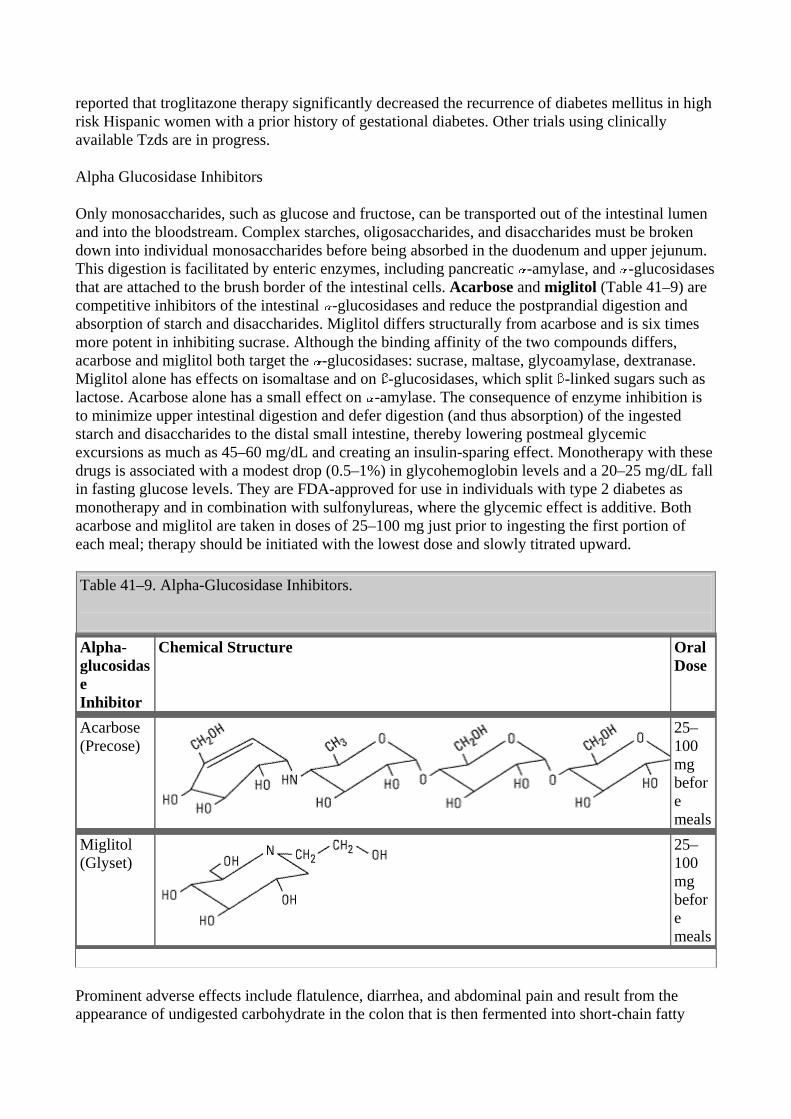
reported that troglitazone therapy significantly decreased the recurrence of diabetes mellitus in high risk Hispanic women with a prior history of gestational diabetes. Other trials using clinically available Tzds are in progress.
Alpha Glucosidase Inhibitors
Only monosaccharides, such as glucose and fructose, can be transported out of the intestinal lumen and into the bloodstream. Complex starches, oligosaccharides, and disaccharides must be broken down into individual monosaccharides before being absorbed in the duodenum and upper jejunum. This digestion is facilitated by enteric enzymes, including pancreatic -amylase, and -glucosidases that are attached to the brush border of the intestinal cells. Acarbose and miglitol (Table 41–9) are competitive inhibitors of the intestinal -glucosidases and reduce the postprandial digestion and absorption of starch and disaccharides. Miglitol differs structurally from acarbose and is six times more potent in inhibiting sucrase. Although the binding affinity of the two compounds differs, acarbose and miglitol both target the -glucosidases: sucrase, maltase, glycoamylase, dextranase. Miglitol alone has effects on isomaltase and on -glucosidases, which split -linked sugars such as lactose. Acarbose alone has a small effect on -amylase. The consequence of enzyme inhibition is to minimize upper intestinal digestion and defer digestion (and thus absorption) of the ingested starch and disaccharides to the distal small intestine, thereby lowering postmeal glycemic excursions as much as 45–60 mg/dL and creating an insulin-sparing effect. Monotherapy with these drugs is associated with a modest drop (0.5–1%) in glycohemoglobin levels and a 20–25 mg/dL fall in fasting glucose levels. They are FDA-approved for use in individuals with type 2 diabetes as monotherapy and in combination with sulfonylureas, where the glycemic effect is additive. Both acarbose and miglitol are taken in doses of 25–100 mg just prior to ingesting the first portion of each meal; therapy should be initiated with the lowest dose and slowly titrated upward.
Table 41–9. Alpha-Glucosidase Inhibitors.
Alpha-glucosidase Inhibitor
Chemical Structure Oral Dose
Acarbose (Precose)
25–100 mg before meals
Miglitol (Glyset)
25–100 mg before meals
Prominent adverse effects include flatulence, diarrhea, and abdominal pain and result from the appearance of undigested carbohydrate in the colon that is then fermented into short-chain fatty

to carbohydrate induces the expression of -glucosidase in the jejunum and ileum, increasing distal small intestine glucose absorption and minimizing the passage of carbohydrate into the colon. Although not a problem with monotherapy or combination therapy with a biguanide, hypoglycemia may occur with concurrent sulfonylurea treatment. Hypoglycemia should be treated with glucose (dextrose) and not sucrose, whose breakdown may be blocked. These drugs are contraindicated in patients with inflammatory bowel disease or any intestinal condition that could be worsened by gas and distention. Because both miglitol and acarbose are absorbed from the gut, these medications should not be prescribed in individuals with renal impairment. Acarbose has been associated with reversible hepatic enzyme elevation and should be used with caution in the presence of hepatic disease.
The STOP-NIDDM trial demonstrated that -glucosidase therapy in prediabetic individuals successfully prevented a significant number of new cases of type 2 diabetes and helped restore cell function. Diabetes prevention may become a further indication for this class of medications.
Combination Therapy with Oral Antidiabetic Agents & Insulin
Combination Therapy in Type 2 Diabetes Mellitus
Bedtime insulin has been suggested as an adjunct to oral antidiabetic therapy in patients with type 2 diabetes patients who have not responded to maximal oral therapy. Clinical practice has evolved to include sulfonylureas, meglitinides, D-phenylalanine derivatives, biguanides, thiazolidinediones, or
-glucosidase inhibitors given in conjunction with insulin.
Individuals unable to achieve glycemic control with bedtime insulin as described above generally require full insulin replacement and multiple daily injections of insulin. Insulin secretagogues are redundant when an individual is receiving multiple daily insulin injections, but cases of severe insulin resistance may benefit from the addition of one of the biguanides, thiazolidinediones, or -glucosidase inhibitors. In some cases, multiple oral agents have been required together with insulin. When oral agents are added to the regimen of someone already taking insulin, the blood glucose should be closely monitored and the insulin dosage decreased as needed to avoid hypoglycemia.
Combination Therapy in Type 1 Diabetes Mellitus
There is no indication for combining insulin with insulin secretagogues (sulfonylureas, meglitinides, or D-phenylalanine derivatives) in individuals with type 1 diabetes. Type 1 diabetics with diets very high in starch may benefit from the addition of -glucosidase inhibitors, but this is not typically practiced in the USA. Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 41. Pancreatic Hormones & Antidiabetic Drugs >
Glucagon
Chemistry & Metabolism
Glucagon is synthesized in the A cells of the pancreatic islets of Langerhans (see Table 41–1). Glucagon is a peptide—identical in all mammals—consisting of a single chain of 29 amino acids, with a molecular weight of 3485. Selective proteolytic cleavage converts a large precursor molecule of approximately 18,000 MW to glucagon. One of the precursor intermediates consists of a 69-amino-acid peptide called glicentin, which contains the glucagon sequence interposed between

peptide extensions.
Glucagon is extensively degraded in the liver and kidney as well as in plasma, and at its tissue receptor sites. Because of its rapid inactivation by plasma, chilling of the collecting tubes and addition of inhibitors of proteolytic enzymes are necessary when samples of blood are collected for immunoassay of circulating glucagon. Its half-life in plasma is between 3 and 6 minutes, which is similar to that of insulin.
"Gut Glucagon"
Glicentin immunoreactivity has been found in cells of the small intestine as well as in pancreatic A cells and in effluents of perfused pancreas. The intestinal cells secrete enteroglucagon, a family of glucagon-like peptides, of which glicentin is a member, along with glucagon-like peptides 1 and 2 (GLP-1 and GLP-2). Unlike the pancreatic A cell, these intestinal cells lack the enzymes to convert glucagon precursors to true glucagon by removing the carboxyl terminal extension from the molecule.
The function of the enteroglucagons has not been clarified, although smaller peptides can bind hepatic glucagon receptors where they exert partial activity. A derivative of the 37-amino-acid form of GLP-1 that lacks the first six amino acids (GLP-1[7–37]) is a potent stimulant of insulin release. It represents the predominant form of GLP in the human intestine and has been termed "insulinotropin." It has been considered as a potential therapeutic agent in type 2 diabetes. However, it requires continuous subcutaneous infusion to produce a sustained lowering of both fasting and postprandial hyperglycemia in type 2 diabetic patients; therefore, its clinical usefulness is limited.
Pharmacologic Effects of Glucagon
Metabolic Effects
The first six amino acids at the amino terminal of the glucagon molecule bind to specific receptors on liver cells. This leads to a Gs protein-linked increase in adeny- lyl cyclase activity and the production of cAMP, which facilitates catabolism of stored glycogen and increases gluconeogenesis and ketogenesis. The immediate pharmacologic result of glucagon infusion is to raise blood glucose at the expense of stored hepatic glycogen. There is no effect on skeletal muscle glycogen, presumably because of the lack of glucagon receptors on skeletal muscle. Pharmacologic amounts of glucagon cause release of insulin from normal pancreatic B cells, catecholamines from pheochromocytoma, and calcitonin from medullary carcinoma cells.
Cardiac Effects
Glucagon has a potent inotropic and chronotropic effect on the heart, mediated by the cAMP mechanism described above. Thus, it produces an effect very similar to that of -adrenoceptor agonists without requiring functioning -receptors.
Effects on Smooth Muscle
Large doses of glucagon produce profound relaxation of the intestine. In contrast to the above effects of the peptide, this action on the intestine may be due to mechanisms other than adenylyl cyclase activation.

Clinical Uses
Severe Hypoglycemia
The major use of glucagon is for emergency treatment of severe hypoglycemic reactions in patients with type 1 diabetes when unconsciousness precludes oral feedings and use of intravenous glucose is not possible. Recombinant glucagon is currently available in 1 mg vials for parenteral use (Glucagon Emergency Kit). Nasal sprays have been developed for this purpose but have not yet received FDA approval.
Endocrine Diagnosis
Several tests use glucagon to diagnose endocrine disorders. In patients with type 1 diabetes mellitus,a standard test of pancreatic B cell secretory reserve utilizes 1 mg of glucagon administered as an intravenous bolus. Since insulin-treated patients develop circulating anti-insulin antibodies that interfere with radioimmunoassays of insulin, measurements of C-peptide are used to indicate B cell secretion.
Beta-Blocker Poisoning
Glucagon is sometimes useful for reversing the cardiac effects of an overdose of -blocking agents because of its ability to increase cAMP production in the heart. However, it is not clinically useful in the treatment of cardiac failure.
Radiology of the Bowel
Glucagon has been used extensively in radiology as an aid to x-ray visualization of the bowel because of its ability to relax the intestine.
Adverse Reactions
Transient nausea and occasional vomiting can result from glucagon administration. These are generally mild, and glucagon is relatively free of severe adverse reactions. Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 41. Pancreatic Hormones & Antidiabetic Drugs >
Islet Amyloid Polypeptide (Iapp, Amylin)
IAPP is a 37-amino-acid peptide originally derived from islet amyloid deposits in pancreas material from patients with long-standing type 2 diabetes or insulinomas. It is produced by pancreatic B cells, packaged within B cell granules in a concentration 1–2% that of insulin, and secreted in response to B cell secretagogues. Approximately one molecule of IAPP is released for every ten molecules of insulin. A physiologic effect has not been established; however, pharmacologic doses inhibit the action of insulin to promote muscle uptake of glucose. IAPP appears to be a member of the superfamily of neuroregulatory peptides, with 46% homology with the calcitonin gene-related peptide CGRP (see Chapter 17: Vasoactive Peptides). Whereas CGRP inhibits insulin secretion, this has not been demonstrated at physiologic concentrations of IAPP. Clinical trials have begun to evaluate IAPP and its analogs, eg, pramlintide, as adjuncts to insulin therapy in type 1 diabetic patients with recurrent episodes of severe insulin-induced hypoglycemia—episodes that are generally refractory to usual preventive measures.

Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 41. Pancreatic Hormones & Antidiabetic Drugs >
Preparations Available1
Sulfonylureas
Acetohexamide (Dymelor) (rarely used)
Oral: 250, 500 mg tablets
Chlorpropamide (generic, Diabinese)
Oral: 100, 250 mg tablets
Glimepiride (Amaryl)
Oral: 1, 2, 4 mg tablets
Glipizide (generic, Glucotrol, Glucotrol XL)
Oral: 5, 10 mg tablets; 5, 10 mg extended release tablets
Glyburide (generic, Dia eta, Micronase, Glynase PresTab)
Oral: 1.25, 2.5, 5 mg tablets; 1.5, 3, 4.5, 6 mg Glynase PresTab, micronized tablets
Tolazamide (generic, Tolinase)
Oral: 100, 250, 500 mg tablets
Tolbutamide (generic, Orinase)
Oral: 500 mg tablets
Meglitinide & Related Drugs
Repaglinide (Prandin)
Oral: 0.5, 1, 2 mg tablets
Nateglinide (Starlix)
Oral: 60, 120 mg tablets
Biguanide & Biguanide Combinations
Metformin (Glucophage, Glucophage XR)

Oral: 500, 850, 1000 mg tablets; extended-release (XR): 500 mg tablets
Metformin Combinations
Glipizide plus metformin (Metaglip)
Oral: 2.5/250, 2.5/500, 5/500 mg tablets
Glyburide plus metformin (Glucovance)
Oral: 1.25/250, 2.5/500, 5/500 mg tablets
Rosiglitazone plus metformin (Avandamet)
Oral: 1/500, 2/500, 4/500 mg tablets
Thiazolidinedione Derivatives
Pioglitazone (Actos)
Oral: 15, 30, 45 mg tablets
Rosiglitazone (Avandia)
Oral: 2, 4, 8 mg tablets
Alpha Glucosidase Inhibitors
Acarbose (Precose)
Oral: 50, 100 mg tablets
Miglitol (Glyset)
Oral: 25, 50, 100 mg tablets
Glucagon
Glucagon (generic)
Parenteral: 1 mg lyophilized powder to reconstitute for injection
1 See Table 41–4 for Insulin Preparations. Chapter 42. Agents That Affect Bone Mineral Homeostasis Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 42. Agents That Affect Bone Mineral Homeostasis >





























