Embed Size (px)
Citation preview

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Phylogenetic Reconstruction: Parsimony
Anders Gorm PedersenAnders Gorm Pedersen
[email protected]@cbs.dtu.dk
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
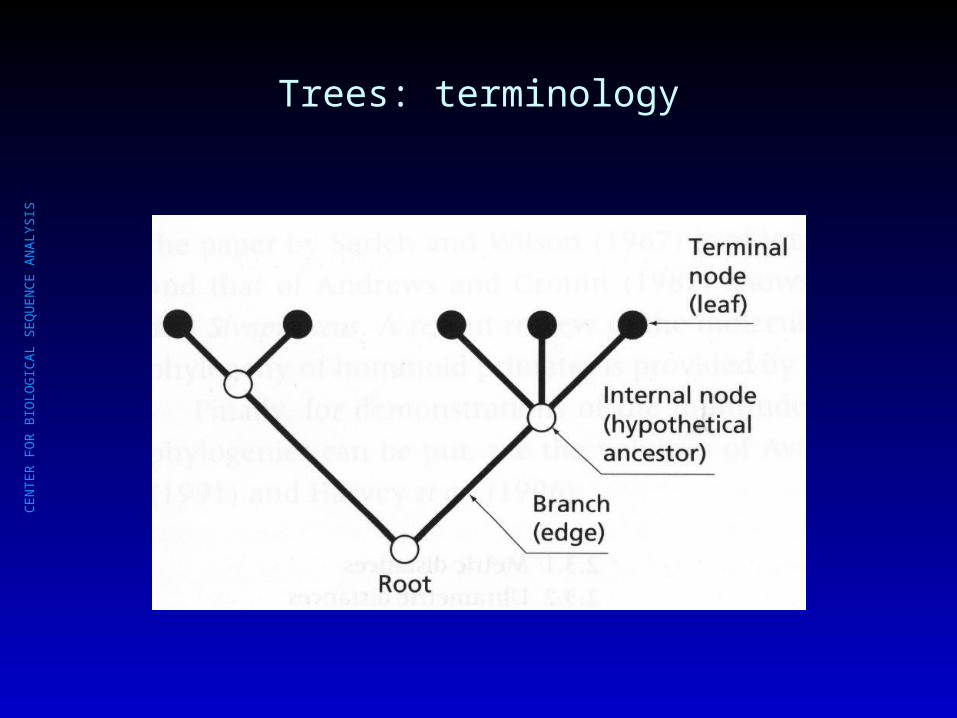
Trees: terminology
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
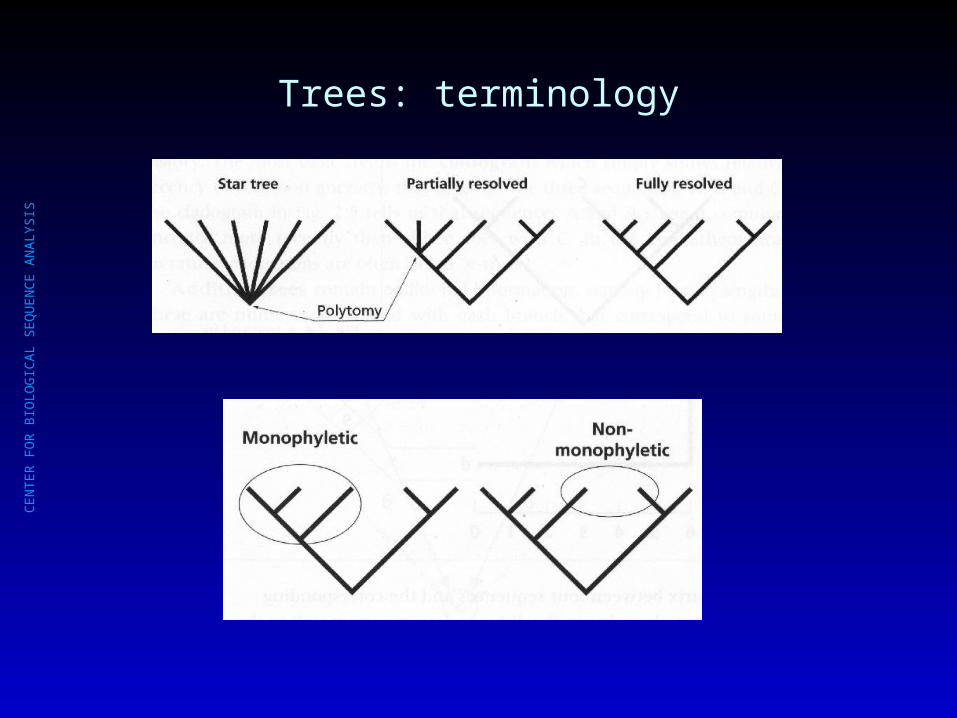
Trees: terminology
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
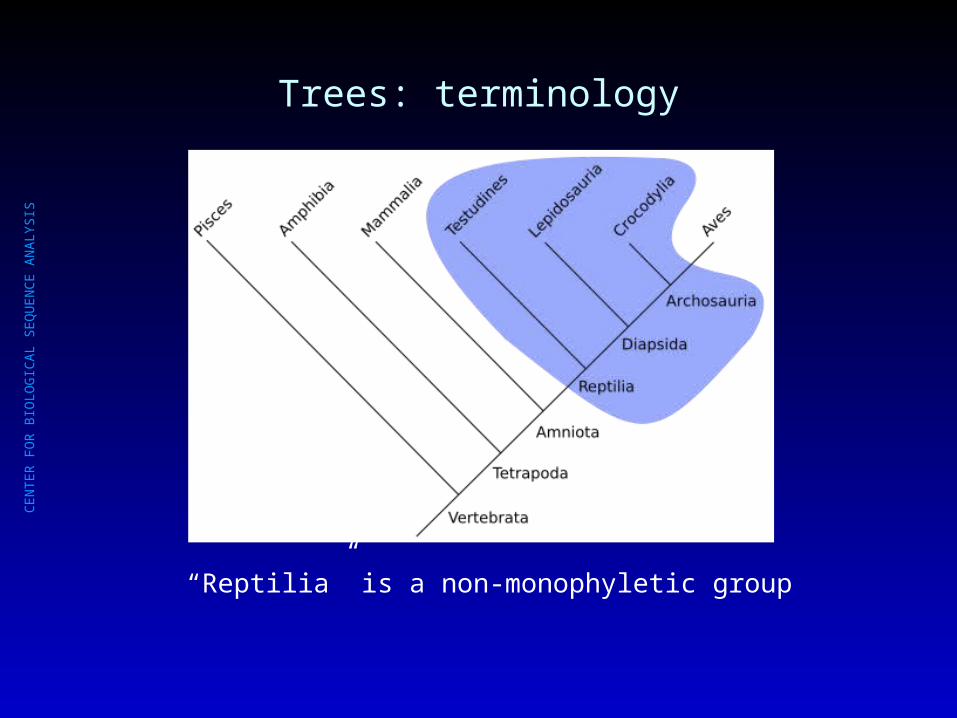
Trees: terminology
“Reptilia” is a non-monophyletic group

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Trees: representations
Three different representations of the same tree-topology
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
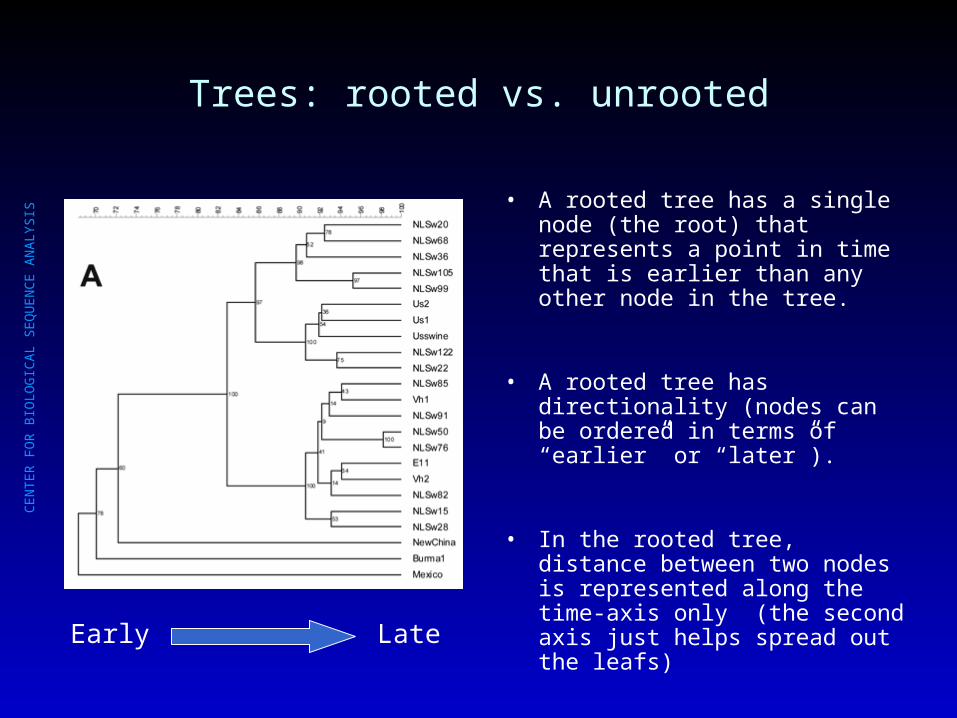
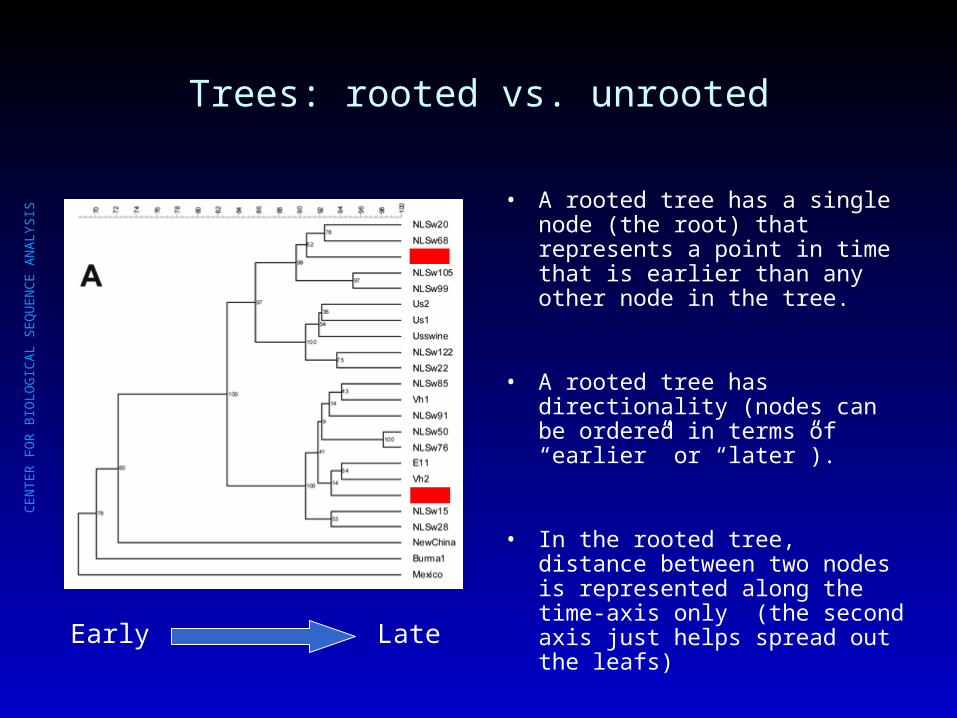
Trees: rooted vs. unrooted
• A rooted tree has a single node (the root) that represents a point in time that is earlier than any other node in the tree.
• A rooted tree has directionality (nodes can be ordered in terms of “earlier” or “later”).
• In the rooted tree, distance between two nodes is represented along the time-axis only (the second axis just helps spread out the leafs)
Early Late
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
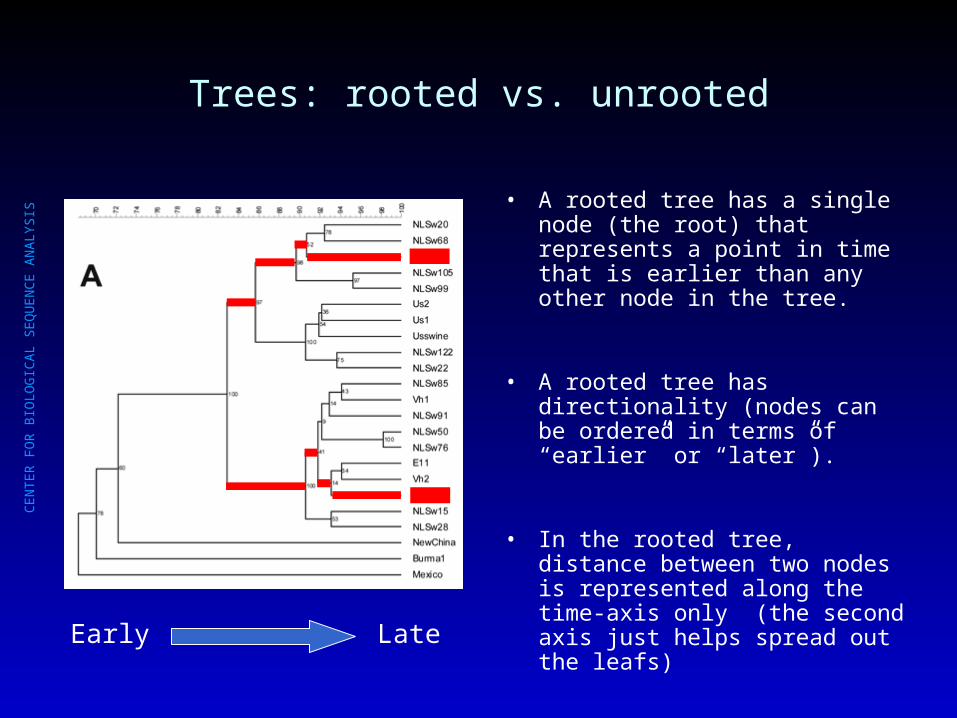
Trees: rooted vs. unrooted
Early Late
• A rooted tree has a single node (the root) that represents a point in time that is earlier than any other node in the tree.
• A rooted tree has directionality (nodes can be ordered in terms of “earlier” or “later”).
• In the rooted tree, distance between two nodes is represented along the time-axis only (the second axis just helps spread out the leafs)
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Trees: rooted vs. unrooted
Early Late
• A rooted tree has a single node (the root) that represents a point in time that is earlier than any other node in the tree.
• A rooted tree has directionality (nodes can be ordered in terms of “earlier” or “later”).
• In the rooted tree, distance between two nodes is represented along the time-axis only (the second axis just helps spread out the leafs)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Trees: rooted vs. unrooted
• In unrooted trees there is no directionality: we do not know if a node is earlier or later In unrooted trees there is no directionality: we do not know if a node is earlier or later than another nodethan another node
• Distance along branches directly represents node distanceDistance along branches directly represents node distance

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Trees: rooted vs. unrooted
• In unrooted trees there is no directionality: we do not know if a node is earlier or later In unrooted trees there is no directionality: we do not know if a node is earlier or later than another nodethan another node
• Distance along branches directly represents node distanceDistance along branches directly represents node distance

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Reconstructing a tree using non-contemporaneous data
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
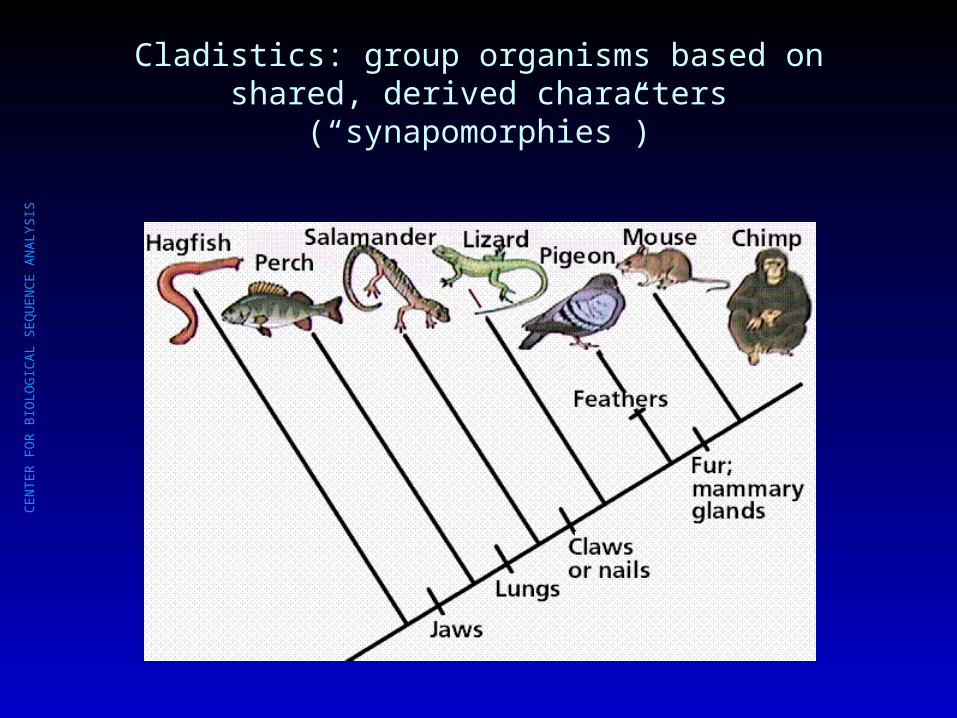
Cladistics: group organisms based on shared, derived characters (“synapomorphies”)
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
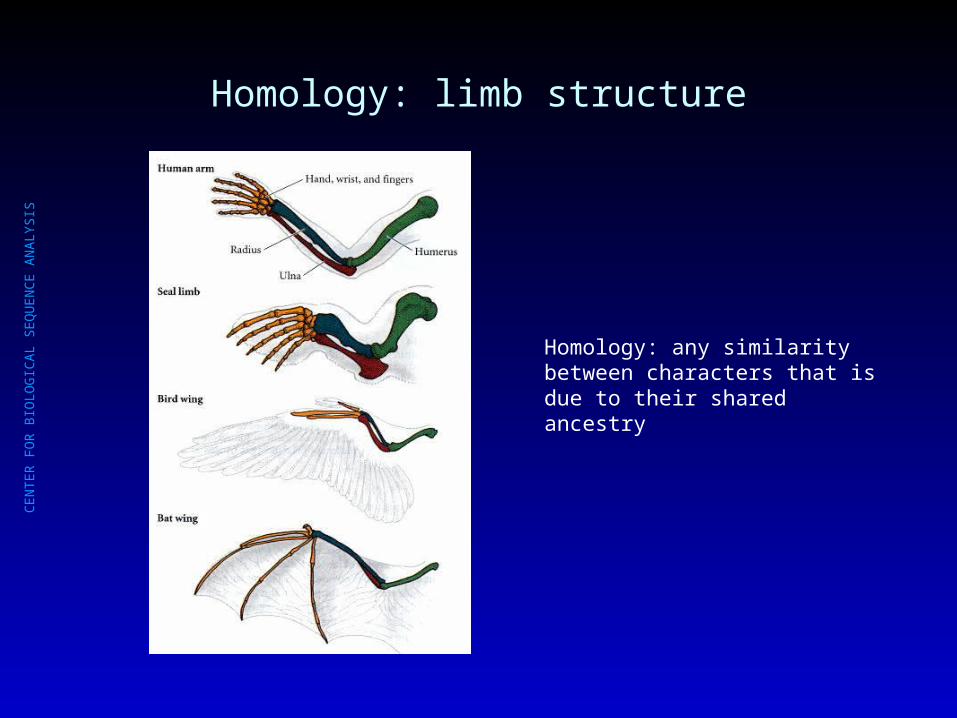
Homology: limb structure
Homology: any similarity between characters that is due to their shared ancestry

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
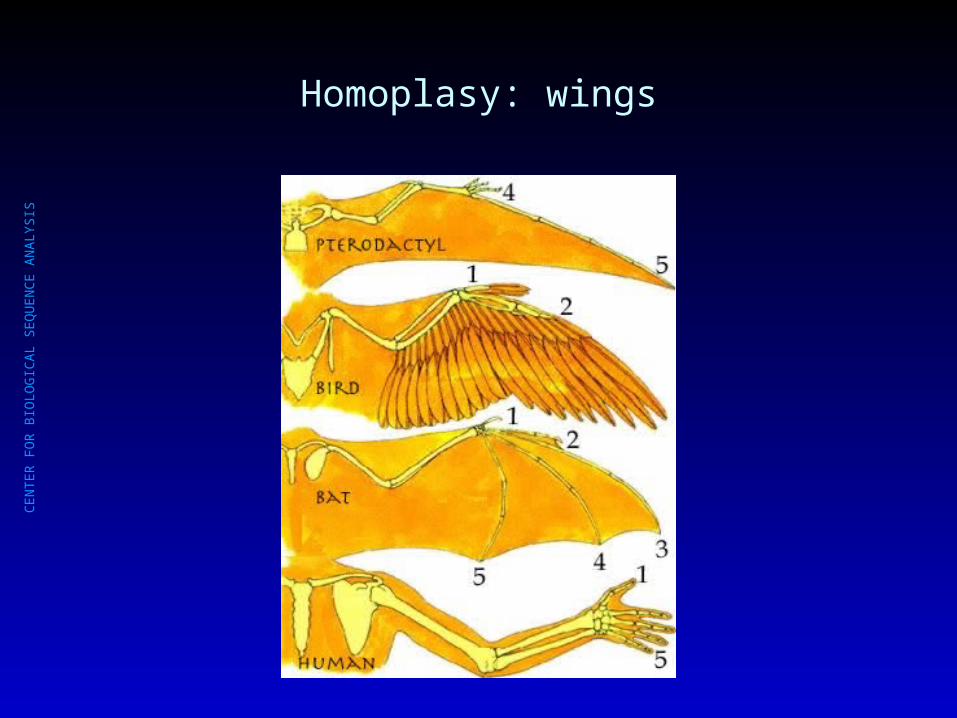
Homology vs. Homoplasy
HomologyHomology: similar traits : similar traits inherited from a common inherited from a common ancestorancestor
HomoplasyHomoplasy: similar traits are : similar traits are not directly caused by common not directly caused by common ancestry (convergent evolution).ancestry (convergent evolution).
XX X X
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Homoplasy: wings

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Molecular phylogeny
AA A G C G T T G G G C A A A G C G T T G G G C A A
BB A G C G T T T G G C A A A G C G T T T G G C A A
CC A G C T T T G T G C A A A G C T T T G T G C A A
DD A G C T T T T T G C A A A G C T T T T T G C A A
1 2 3 1 2 3
• DNA and protein sequences DNA and protein sequences
• Homologous characters inferred Homologous characters inferred from alignment.from alignment.
• Other molecular data: Other molecular data: absence/presence of restriction absence/presence of restriction sites, DNA hybridization data, sites, DNA hybridization data, antibody cross-reactivity, etc. (but antibody cross-reactivity, etc. (but losing importance due to cheap, losing importance due to cheap, efficient sequencing).efficient sequencing).

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Morphology vs. molecular data
African white-backed vulture(old world vulture)
Andean condor (new world vulture)
New and old world vultures seem to be closely related based on morphology.
Molecular data indicates that old world vultures are related to birds of prey (falcons, hawks, etc.) while new world vultures are more closely related to storks
Similar features presumably the result of convergent evolution

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Phylogenetic reconstruction
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Phylogenetic reconstruction
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
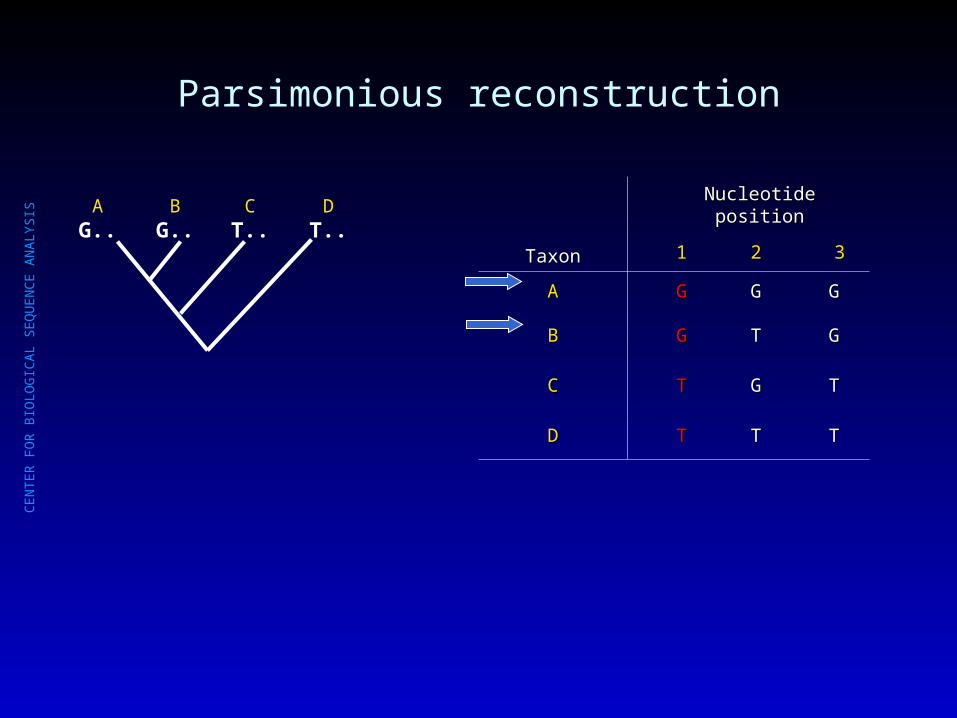
Parsimony criterion: choose simplest hypothesis
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
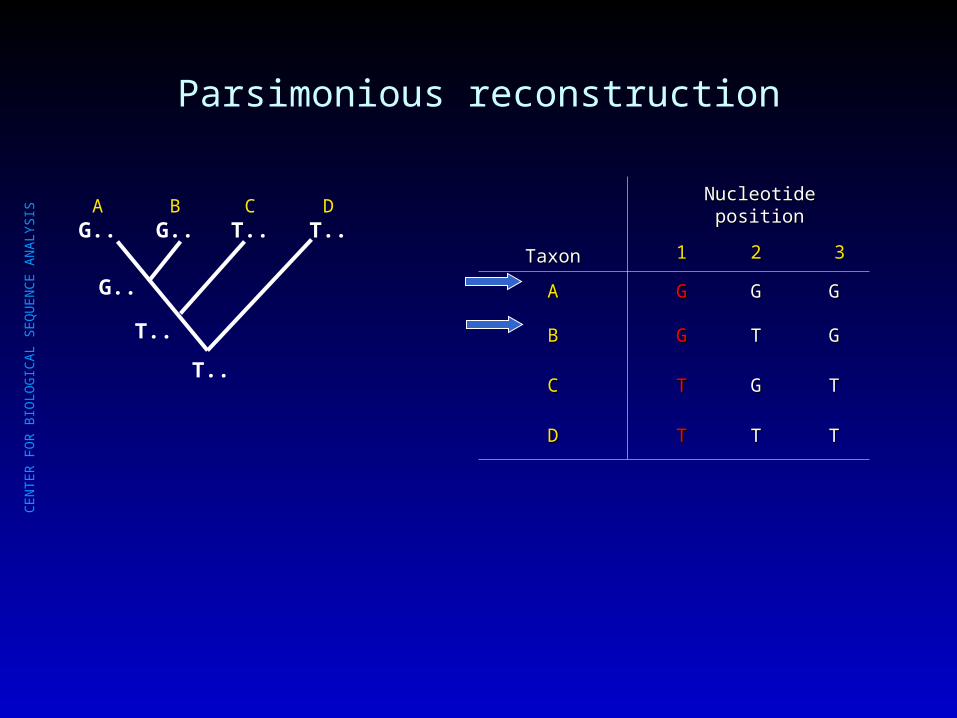
Parsimonious reconstruction
AG..
BG..
CT..
DT..
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
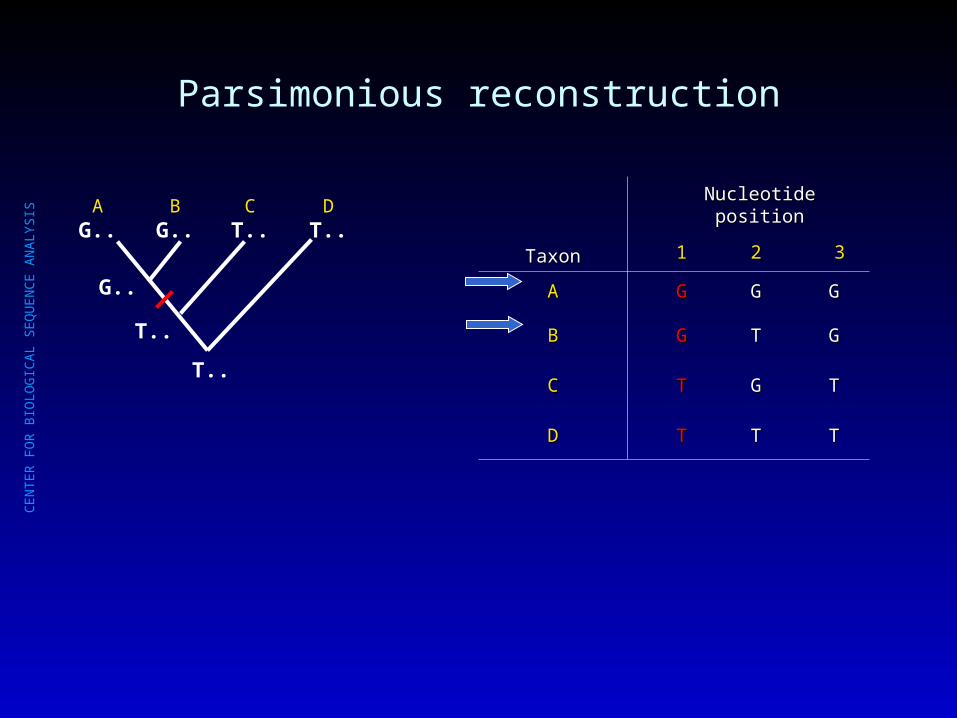
Parsimonious reconstruction
AG..
BG..
CT..
DT..
T..
T..
G..
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Parsimonious reconstruction
AG..
BG..
CT..
DT..
T..
T..
G..
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
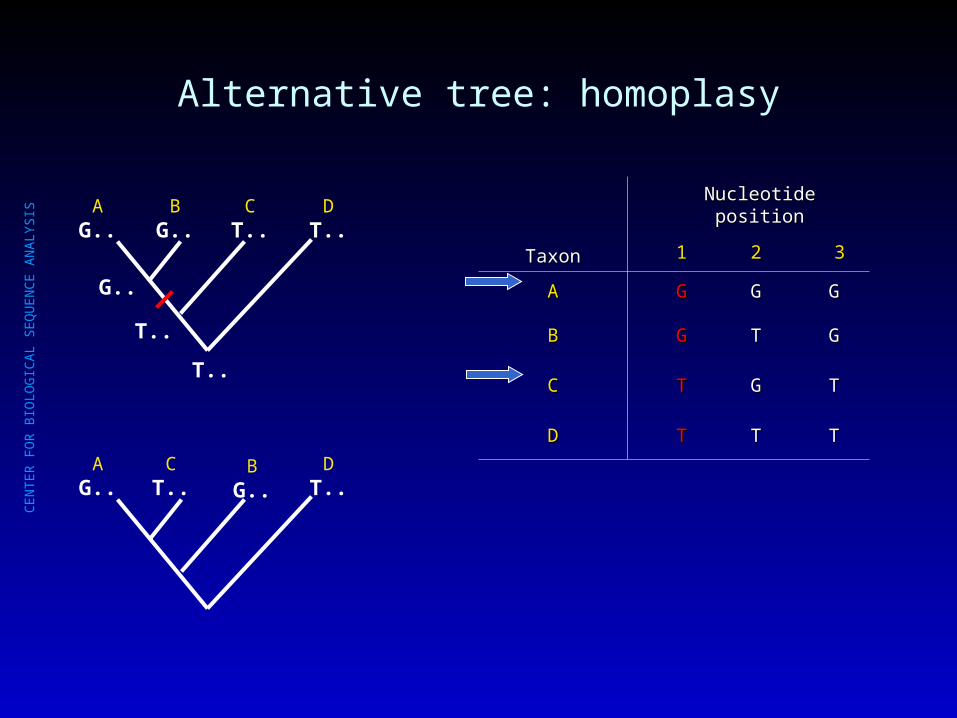
Alternative tree: homoplasy
AG..
BG..
CT..
DT..
AG..
BG..
CT..
DT..
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
T..
T..
G..
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
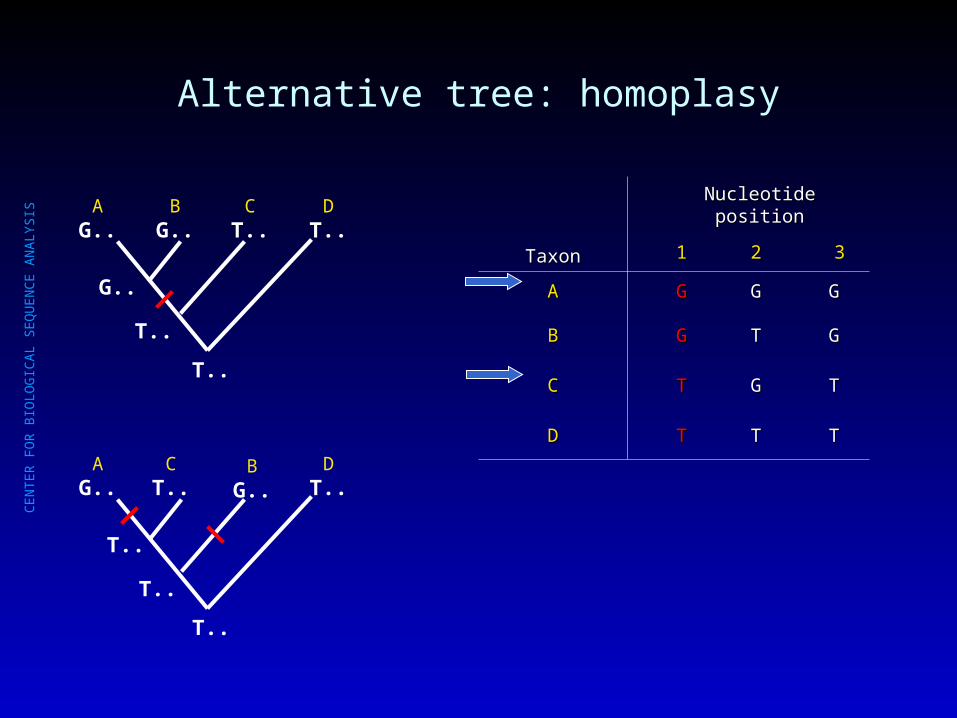
Alternative tree: homoplasy
AG..
BG..
CT..
DT..
AG..
BG..
CT..
DT..
T..
T..
T..
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
T..
T..
G..
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
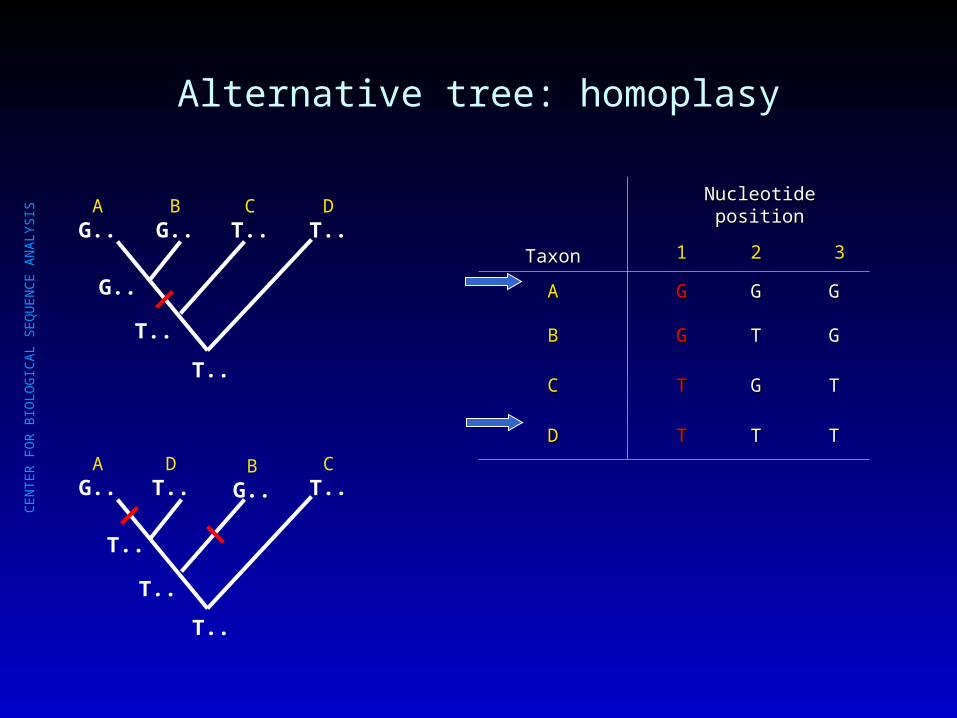
Alternative tree: homoplasy
AG..
BG..
CT..
DT..
AG..
BG..
DT..
CT..
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
T..
T..
G..
T..
T..
T..
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
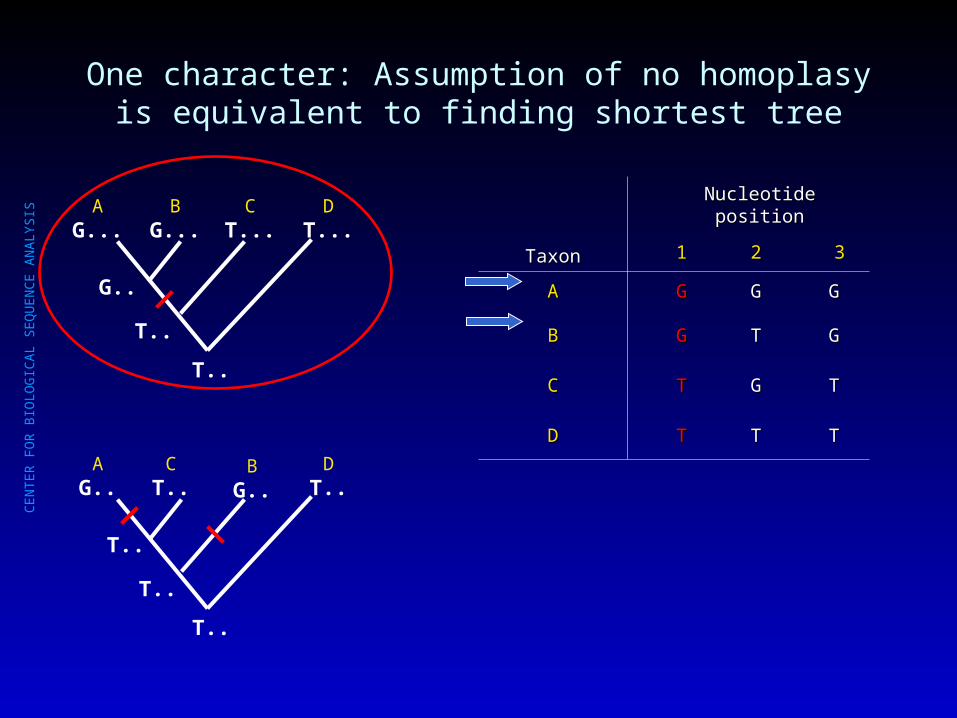
One character: Assumption of no homoplasy is equivalent to finding shortest tree
AG...
BG...
CT...
DT...
AG..
BG..
CT..
DT..
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
T..
T..
G..
T..
T..
T..
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
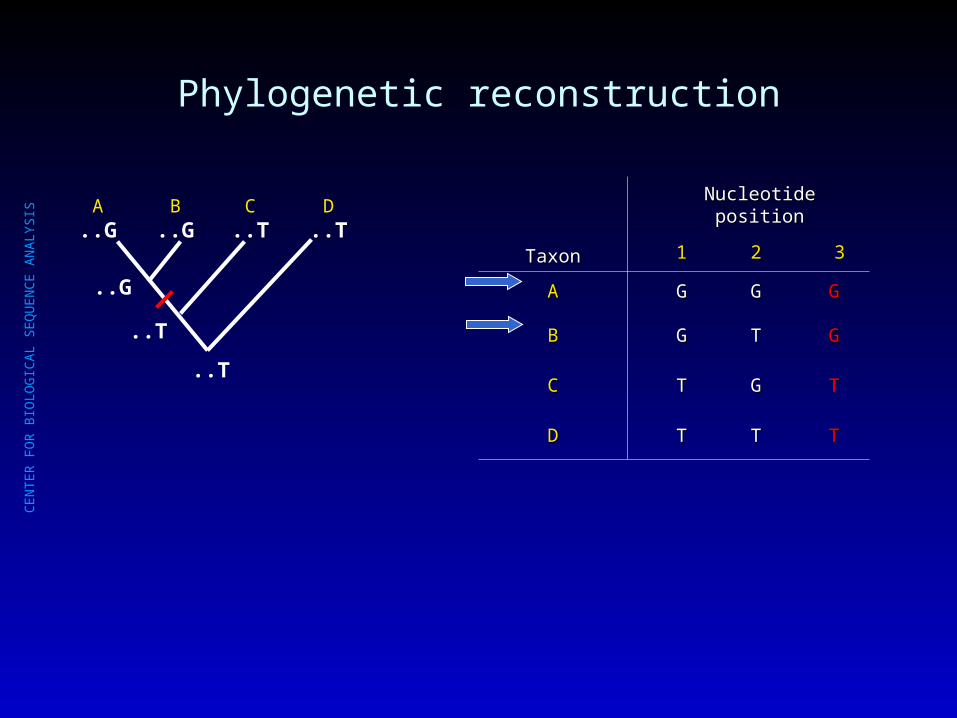
Phylogenetic reconstruction
A..G
B..G
C..T
D..T
..T
..T
..G
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
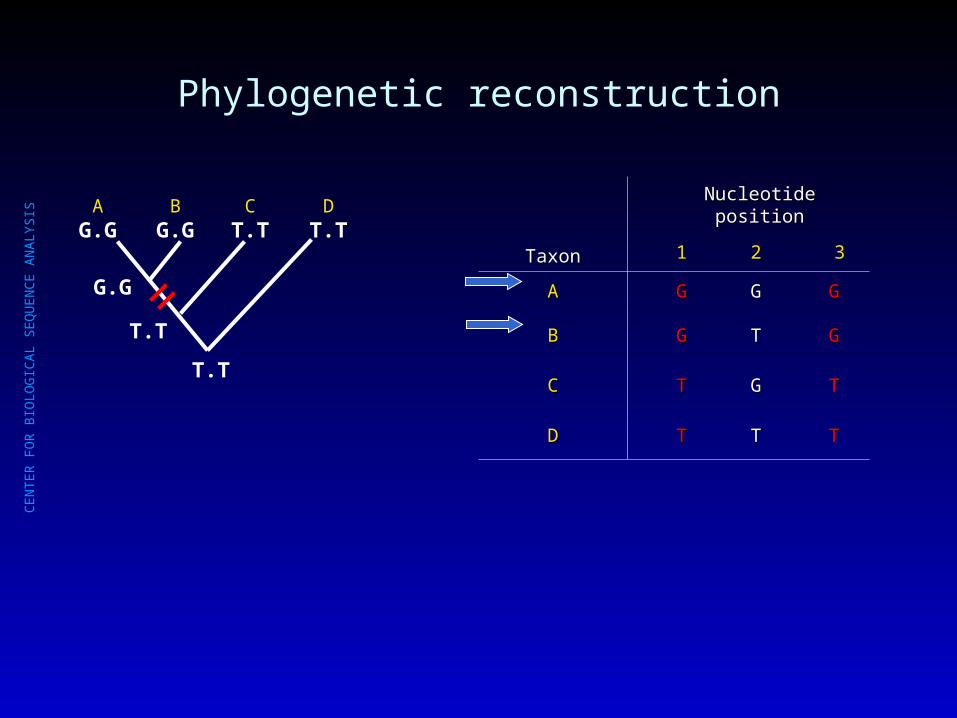
Phylogenetic reconstruction
AG.G
BG.G
CT.T
DT.T
T.T
T.T
G.G
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
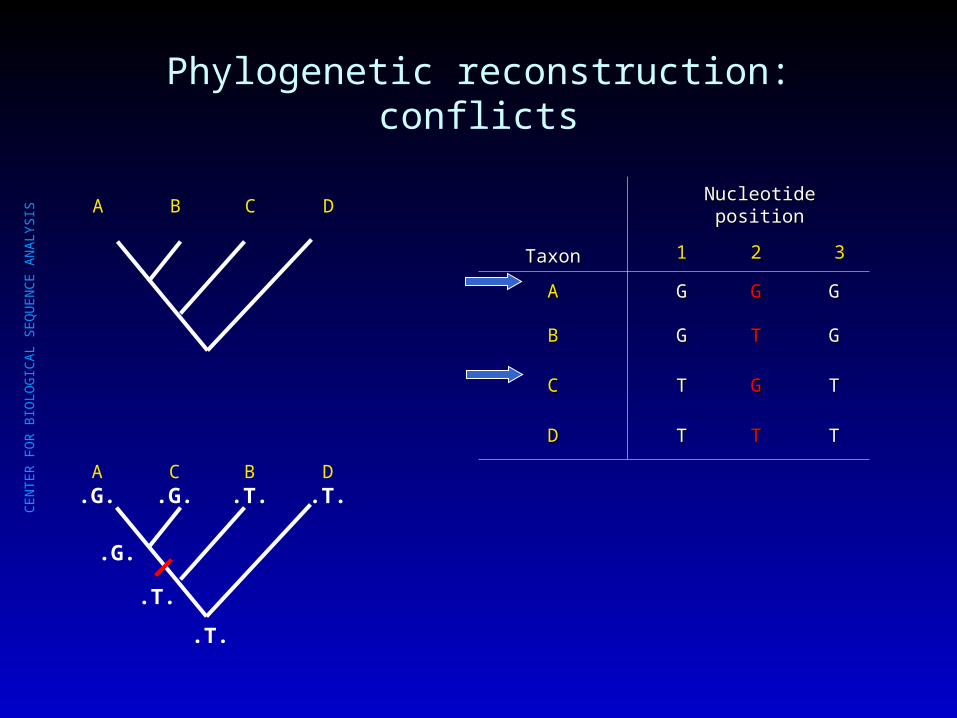
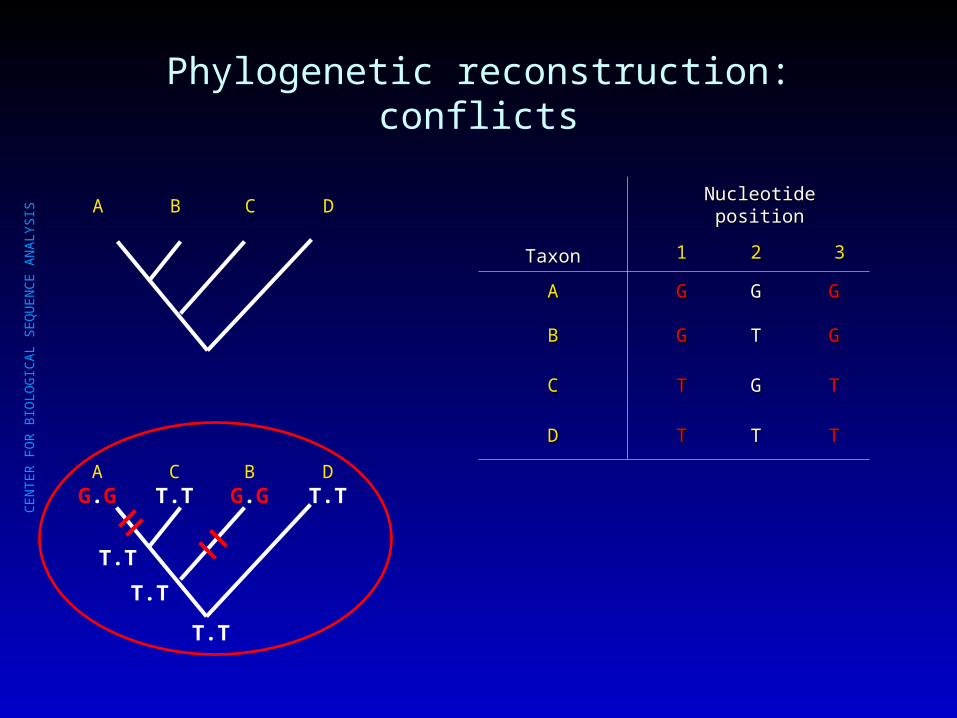
Phylogenetic reconstruction: conflicts
A B C D
A.G.
C.G.
B.T.
D.T.
.T.
.T.
.G.
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
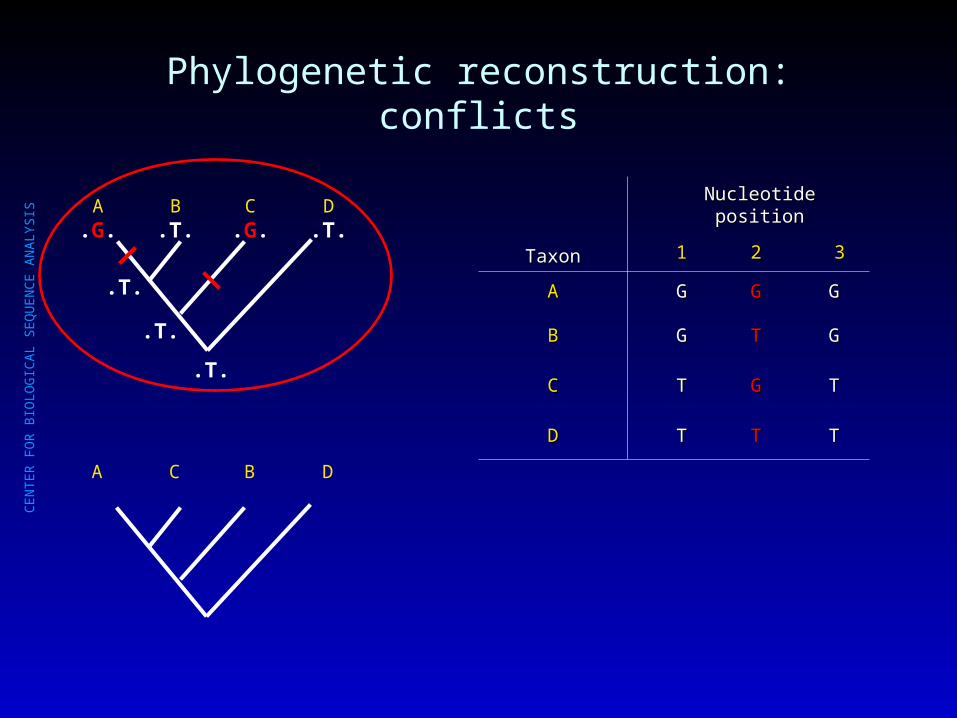
Phylogenetic reconstruction: conflicts
A.G.
B.T.
C.G.
D.T.
.T.
.T.
.T.
A C B D
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Phylogenetic reconstruction: conflicts
A B C D
AG.G
CT.T
BG.G
DT.T
T.T
T.T
T.T
TaxonTaxon
Nucleotide positionNucleotide position
11 22 33
AA GG GG GG
BB GG TT GG
CC TT GG TT
DD TT TT TT
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
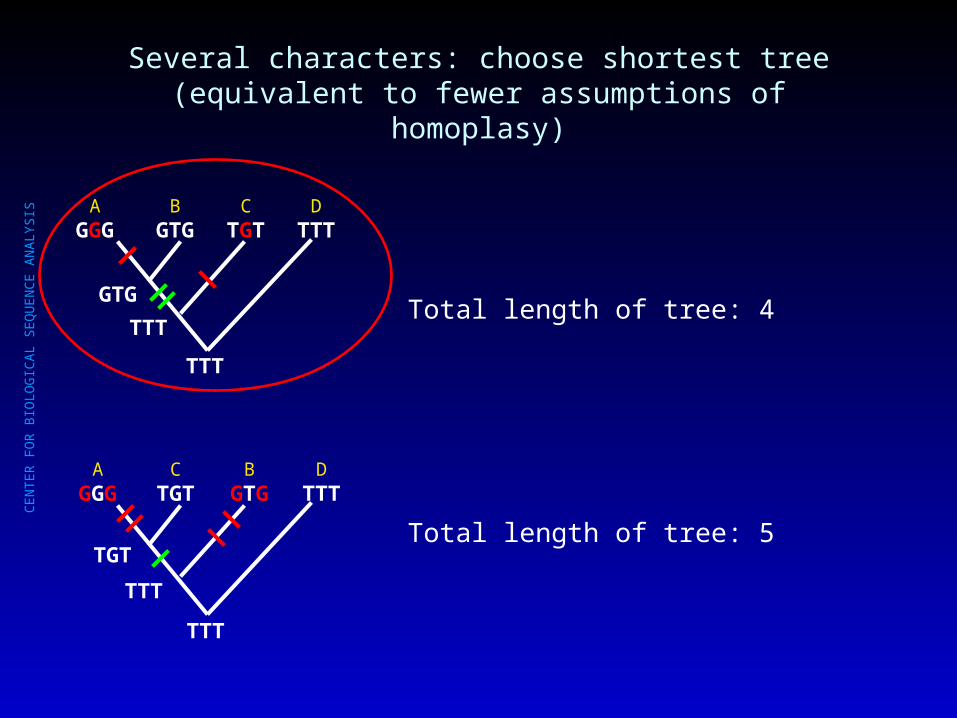
Several characters: choose shortest tree(equivalent to fewer assumptions of homoplasy)
AGGG
BGTG
CTGT
DTTT
AGGG
BGTG
CTGT
DTTT
TTT
TTT
GTG
TTT
TTT
TGT
Total length of tree: 4
Total length of tree: 5

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Maximum Parsimony
• Maximum parsimony: the best tree is the shortest tree (the tree Maximum parsimony: the best tree is the shortest tree (the tree requiring the smallest number of mutational events)requiring the smallest number of mutational events)
• This corresponds to the tree that implies the least amount of homoplasy This corresponds to the tree that implies the least amount of homoplasy (convergent evolution, reversals)(convergent evolution, reversals)
• How do we find the best tree for a given data set?How do we find the best tree for a given data set?

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Maximum Parsimony: first approach
1.1. Construct list of all possible trees for data setConstruct list of all possible trees for data set
2.2. For each tree: determine length, add to list of lengthsFor each tree: determine length, add to list of lengths
3.3. When finished: select shortest tree from listWhen finished: select shortest tree from list
4.4. If several trees have the same length, then they are equally good If several trees have the same length, then they are equally good (equally parsimonious)(equally parsimonious)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Maximum Parsimony: problems
• We need algorithm for constructing list of all possible treesWe need algorithm for constructing list of all possible trees
• We need algorithm for determining length of given treeWe need algorithm for determining length of given tree
• Should all mutational events have same cost?Should all mutational events have same cost?

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Constructing list of all possible unrooted trees
1.1. Construct unrooted tree from first three Construct unrooted tree from first three taxa. There is only one way of doing taxa. There is only one way of doing thisthis
2.2. Starting from (1), construct the three Starting from (1), construct the three possible derived trees by adding taxon possible derived trees by adding taxon 4 to each internal branch4 to each internal branch
3.3. From each of the trees constructed in From each of the trees constructed in step (2), construct the five possible step (2), construct the five possible derived trees by adding taxon 5 to derived trees by adding taxon 5 to each internal branch. each internal branch.
4.4. Continue until all taxa have been Continue until all taxa have been added in all possible locationsadded in all possible locations

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Maximum Parsimony: problems
• We need algorithm for constructing list of all possible trees We need algorithm for constructing list of all possible trees
• We need algorithm for determining length of given treeWe need algorithm for determining length of given tree
• Should all mutational events have same cost?Should all mutational events have same cost?
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
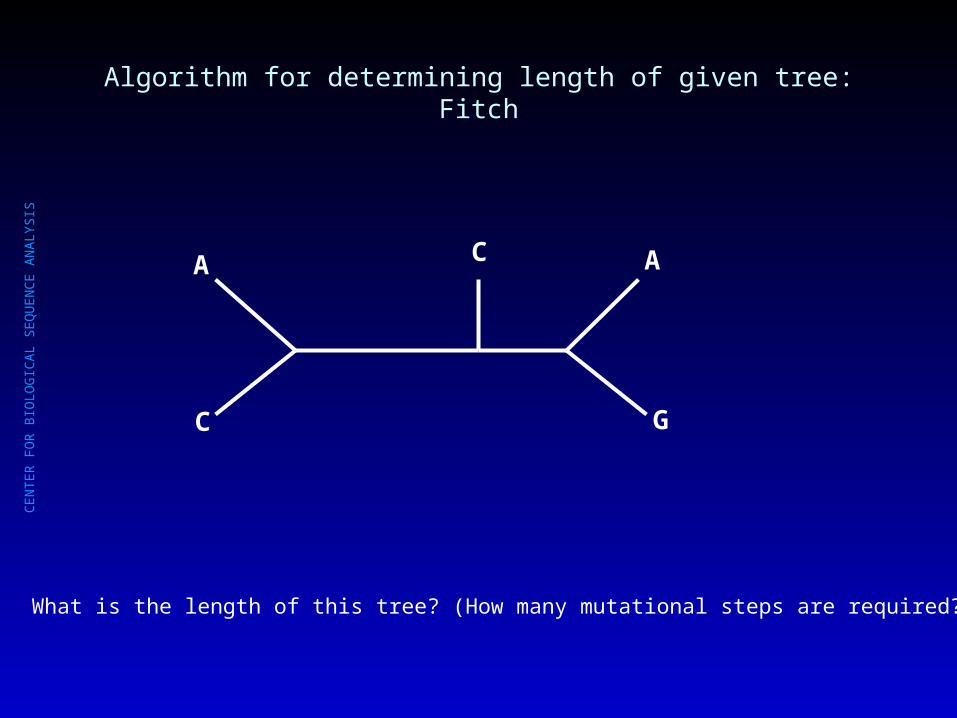
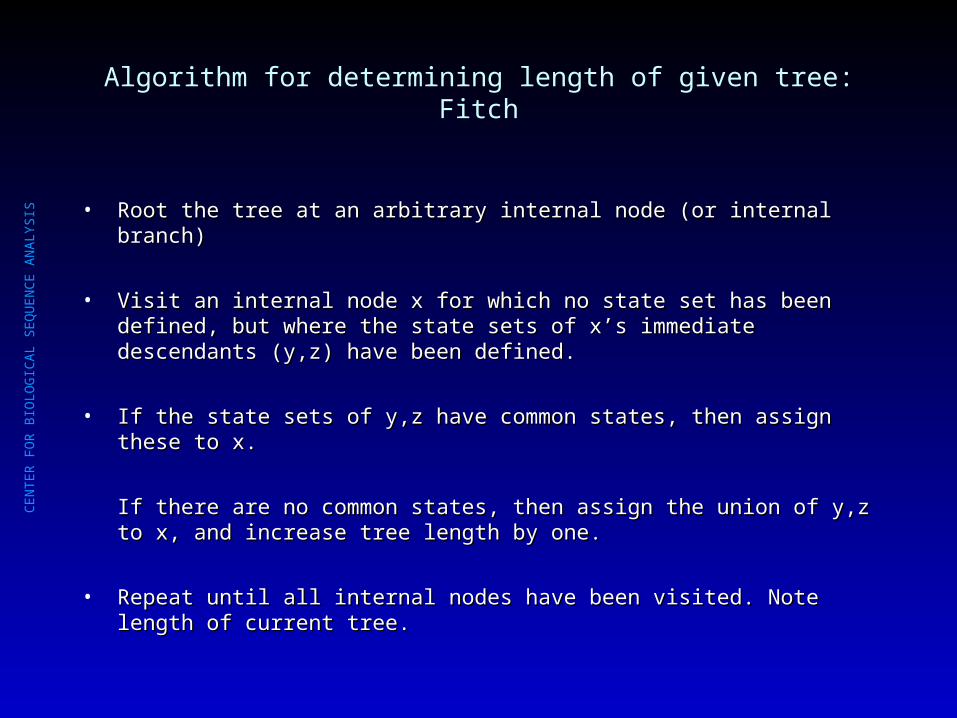
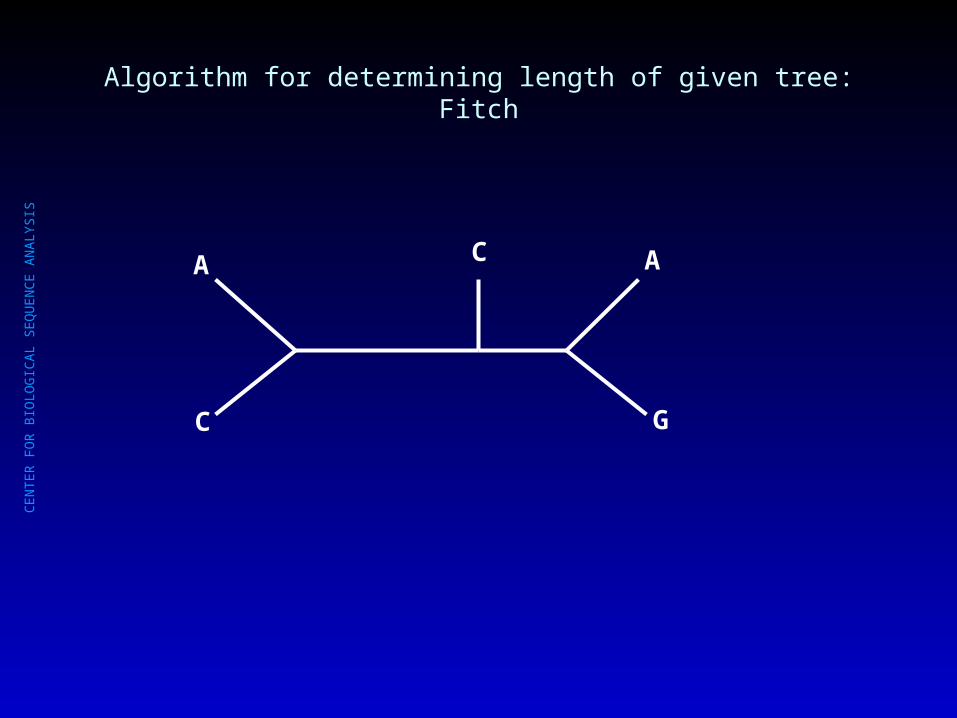
Algorithm for determining length of given tree: Fitch
What is the length of this tree? (How many mutational steps are required?)
C
A C A
G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
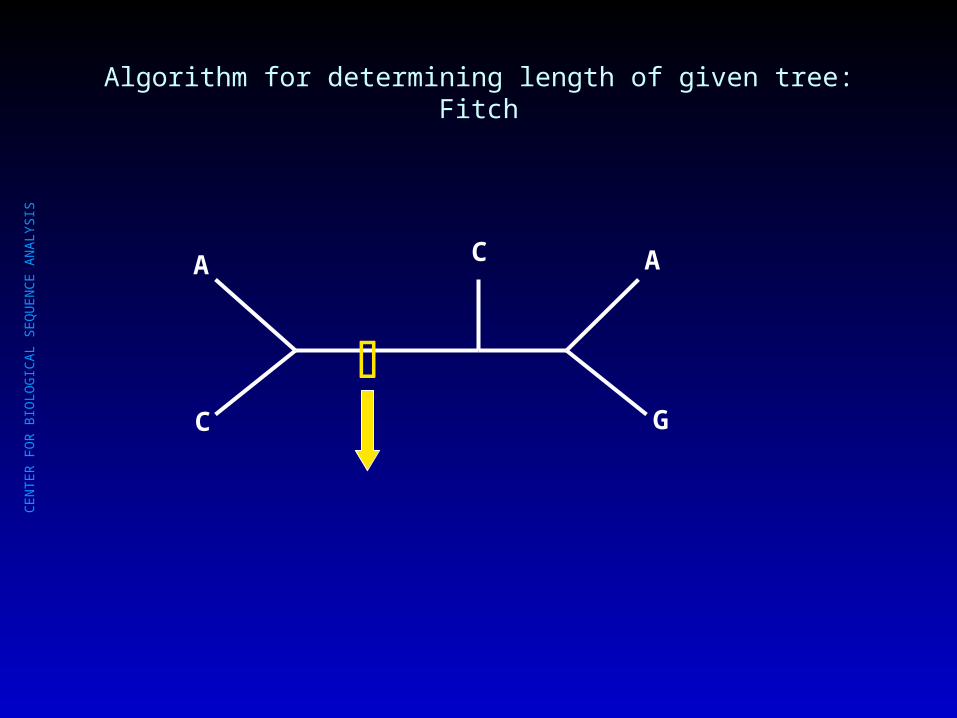
SIS • Root the tree at an arbitrary internal node (or internal branch)Root the tree at an arbitrary internal node (or internal branch)
• Visit an internal node x for which no state set has been defined, but where the Visit an internal node x for which no state set has been defined, but where the state sets of x’s immediate descendants (y,z) have been defined.state sets of x’s immediate descendants (y,z) have been defined.
• If the state sets of y,z have common states, then assign these to x.If the state sets of y,z have common states, then assign these to x.
If there are no common states, then assign the union of y,z to x, and increase If there are no common states, then assign the union of y,z to x, and increase tree length by one.tree length by one.
• Repeat until all internal nodes have been visited. Note length of current tree.Repeat until all internal nodes have been visited. Note length of current tree.
Algorithm for determining length of given tree: Fitch
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
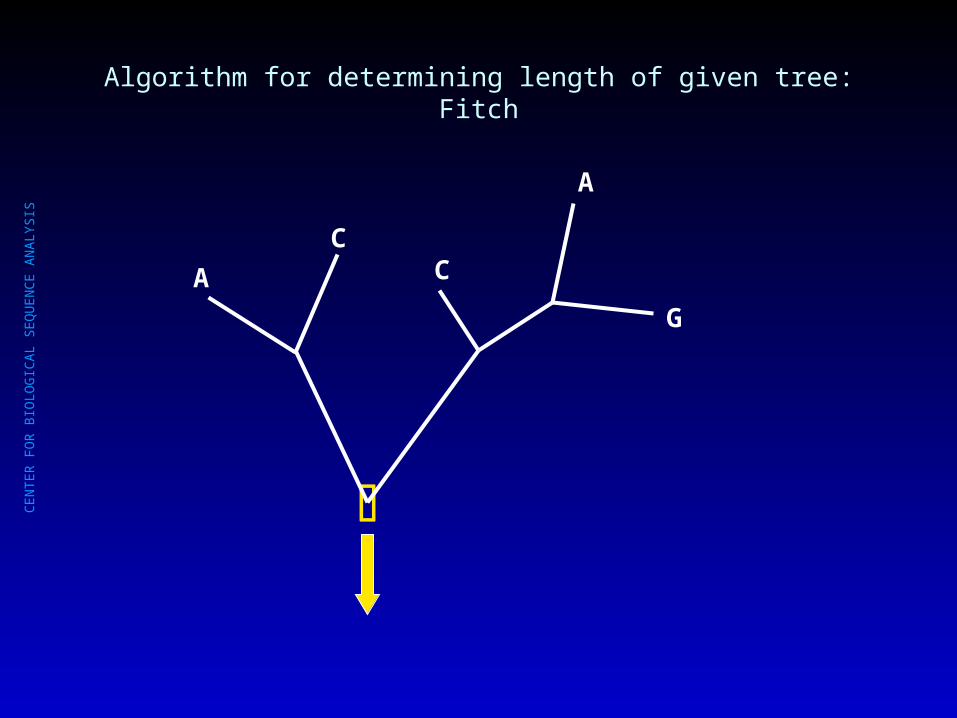
Algorithm for determining length of given tree: Fitch
C
A C A
G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
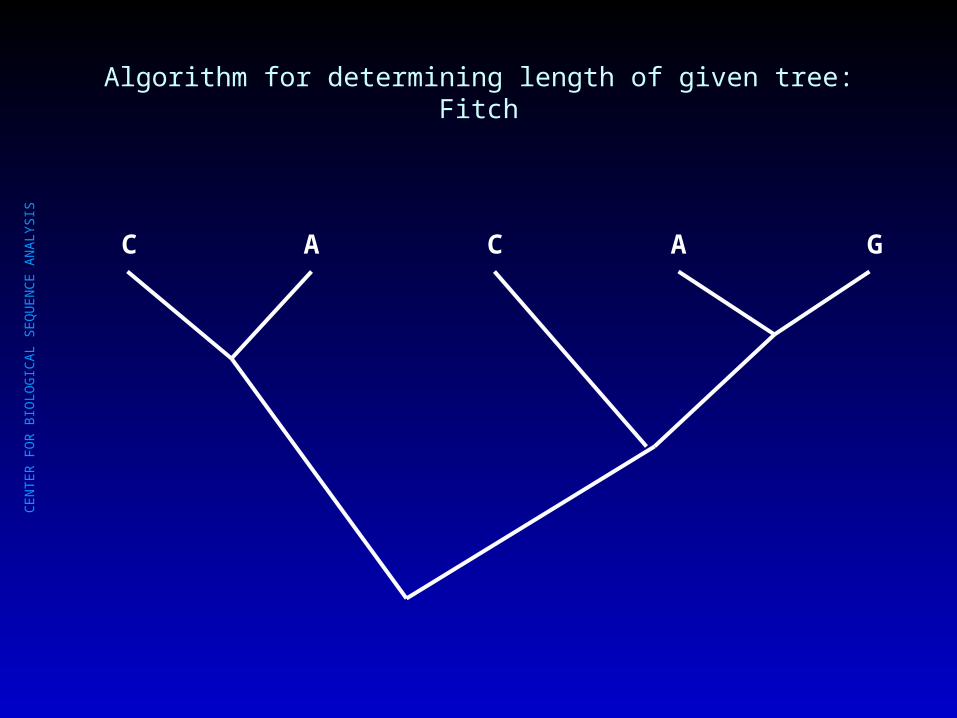
Algorithm for determining length of given tree: Fitch
C
A C A
G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Algorithm for determining length of given tree: Fitch
C
A C
A
G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Algorithm for determining length of given tree: Fitch
C A C A G
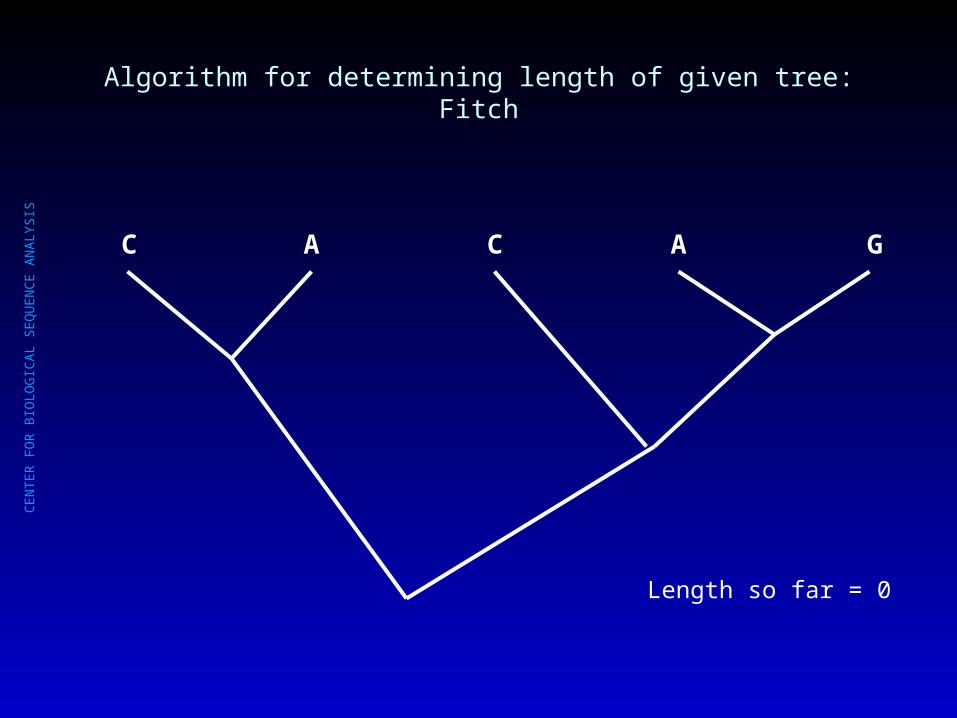
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Algorithm for determining length of given tree: Fitch
C A C A G
Length so far = 0
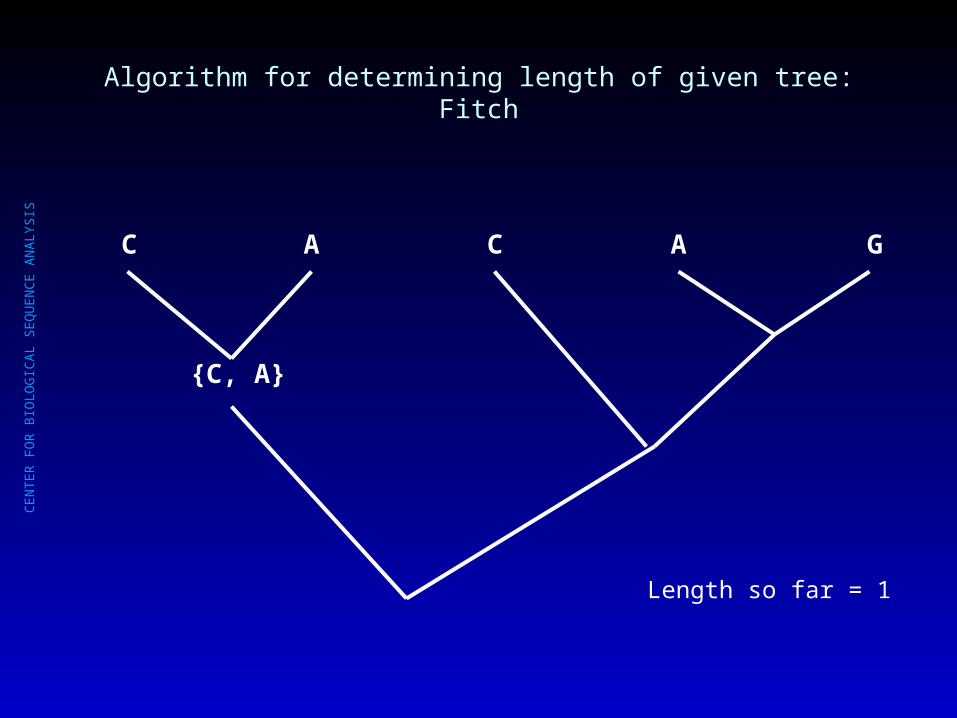
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Algorithm for determining length of given tree: Fitch
Length so far = 1
{C, A}
C A C A G
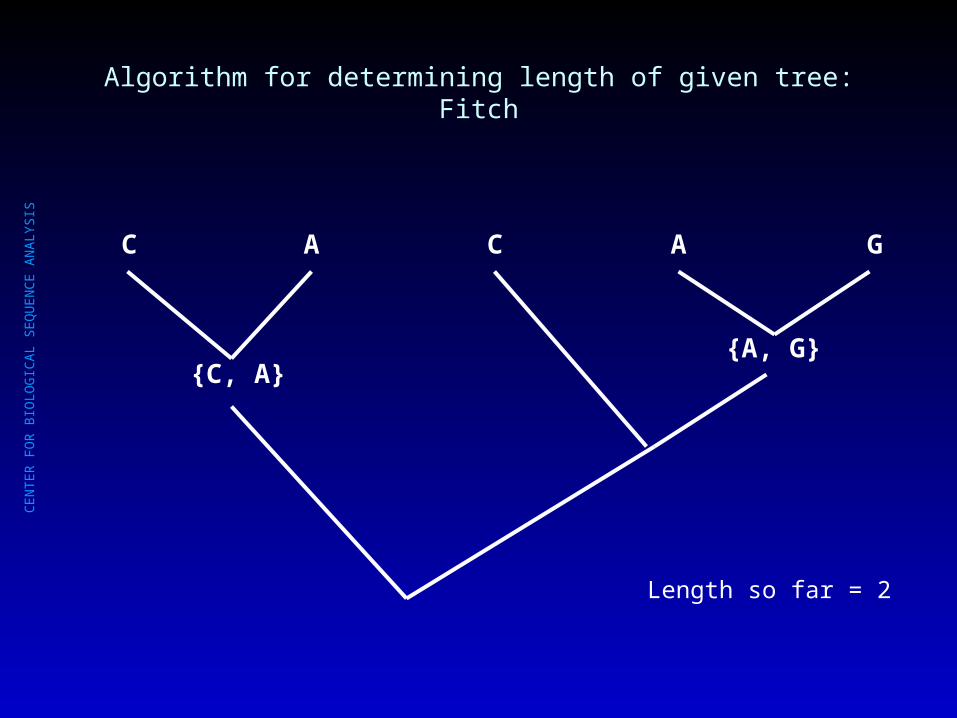
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Algorithm for determining length of given tree: Fitch
Length so far = 2
{C, A}{A, G}
C A C A G
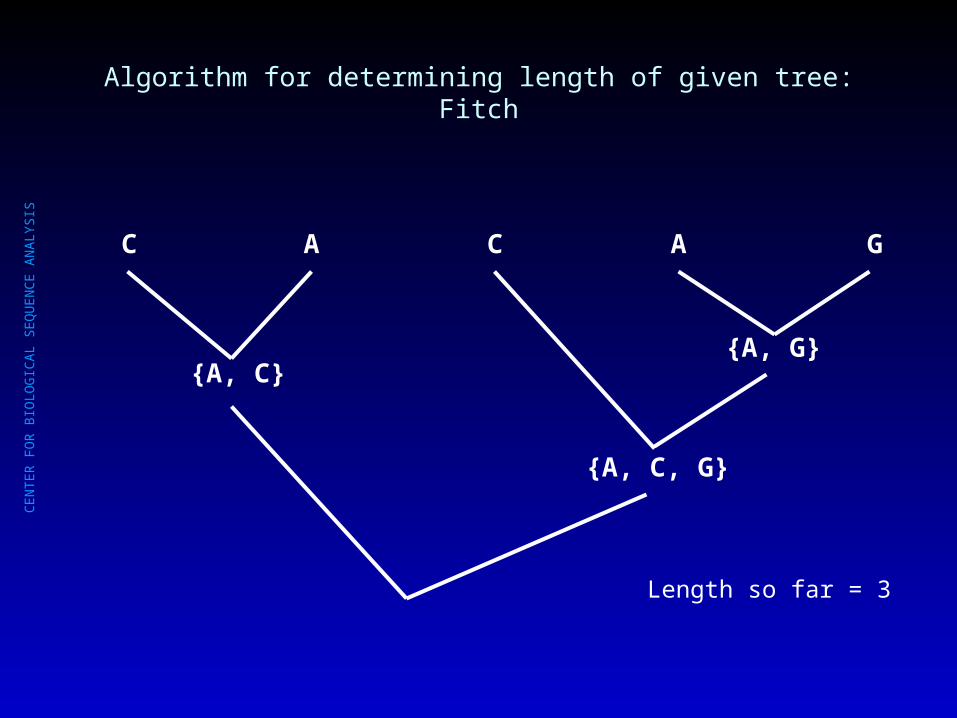
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Algorithm for determining length of given tree: Fitch
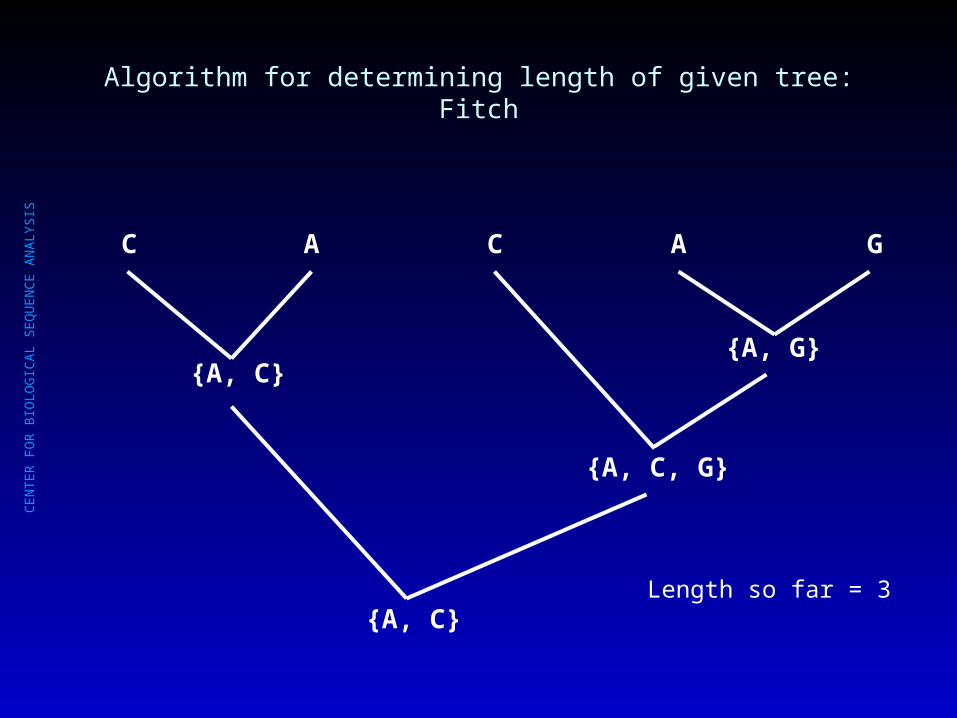
Length so far = 3
{A, C}{A, G}
{A, C, G}
C A C A G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
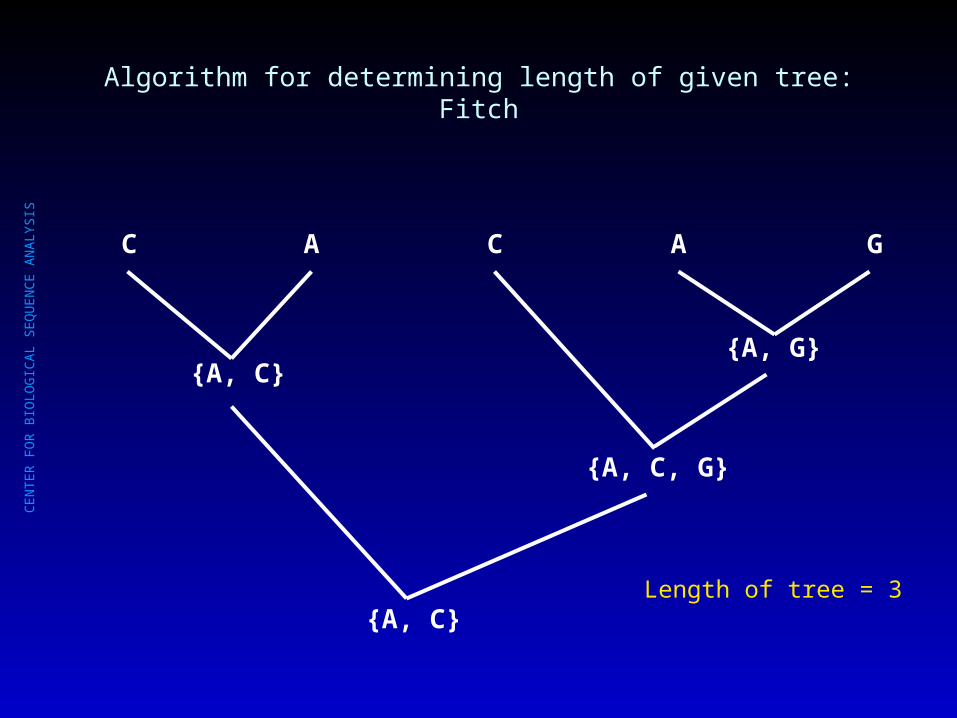
Algorithm for determining length of given tree: Fitch
{A, C}{A, G}
{A, C, G}
{A, C}Length so far = 3
C A C A G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Algorithm for determining length of given tree: Fitch
Length of tree = 3
{A, C}{A, G}
{A, C, G}
{A, C}
C A C A G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
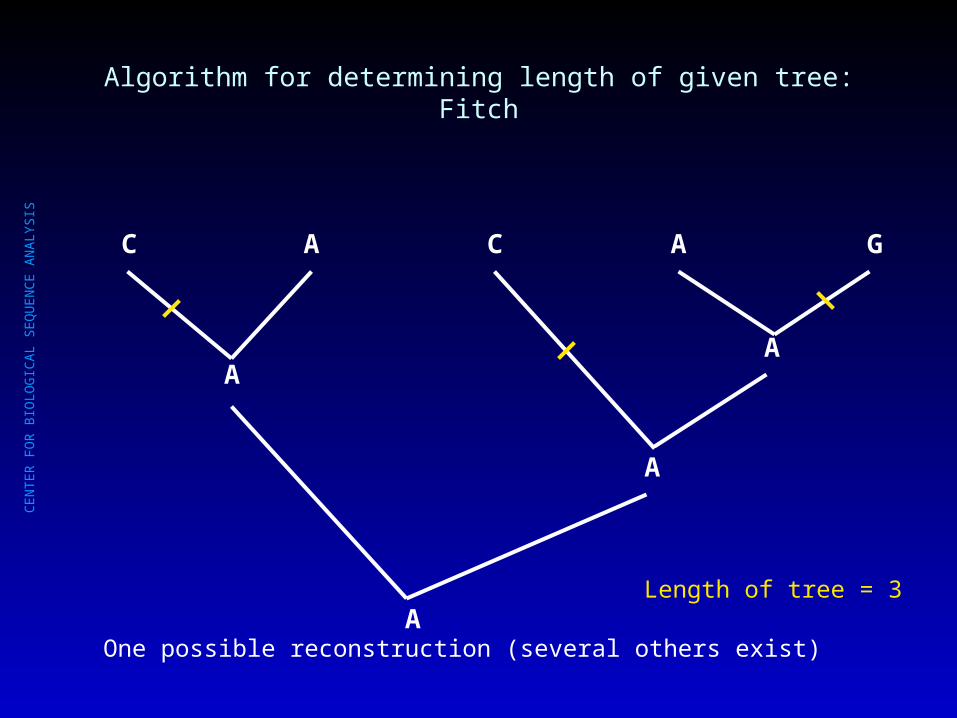
Algorithm for determining length of given tree: Fitch
C A C A G
Length of tree = 3
AA
A
AOne possible reconstruction (several others exist)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Maximum Parsimony: problems
• We need algorithm for constructing list of all possible trees We need algorithm for constructing list of all possible trees
• We need algorithm for determining length of given tree We need algorithm for determining length of given tree
• Should all mutational events have same cost?Should all mutational events have same cost?

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Mutational events need not have the same cost
A C G TA 0 3 1 3C 3 0 3 1G 1 3 0 3T 3 1 3 0
C A C A G
Sankoff algorithm

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Maximum Parsimony: problems
• We need algorithm for constructing list of all possible trees We need algorithm for constructing list of all possible trees
• We need algorithm for determining length of given tree We need algorithm for determining length of given tree
• Should all mutational events have same cost? Should all mutational events have same cost?
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
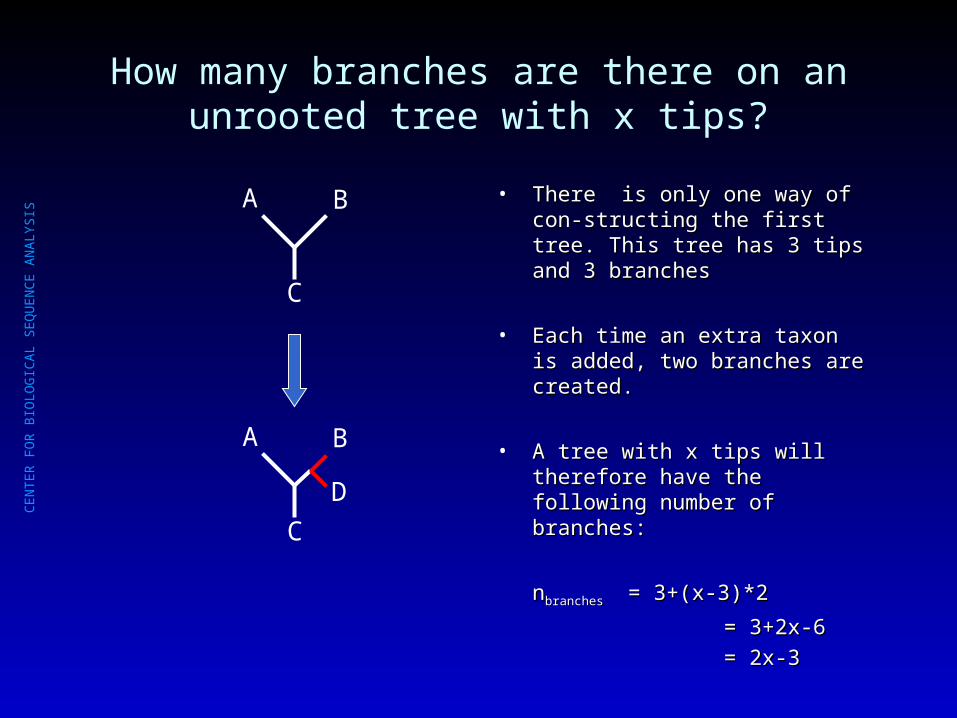
How many branches are there on an unrooted tree with x tips?
• There is only one way of con-There is only one way of con-structing the first tree. This tree has structing the first tree. This tree has 3 tips and 3 branches3 tips and 3 branches
• Each time an extra taxon is added, Each time an extra taxon is added, two branches are created.two branches are created.
• A tree with x tips will therefore have A tree with x tips will therefore have the following number of branches:the following number of branches:
nnbranchesbranches = 3+(x-3)*2= 3+(x-3)*2
= 3+2x-6= 3+2x-6
= 2x-3= 2x-3
A B
C
A B
C
D
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
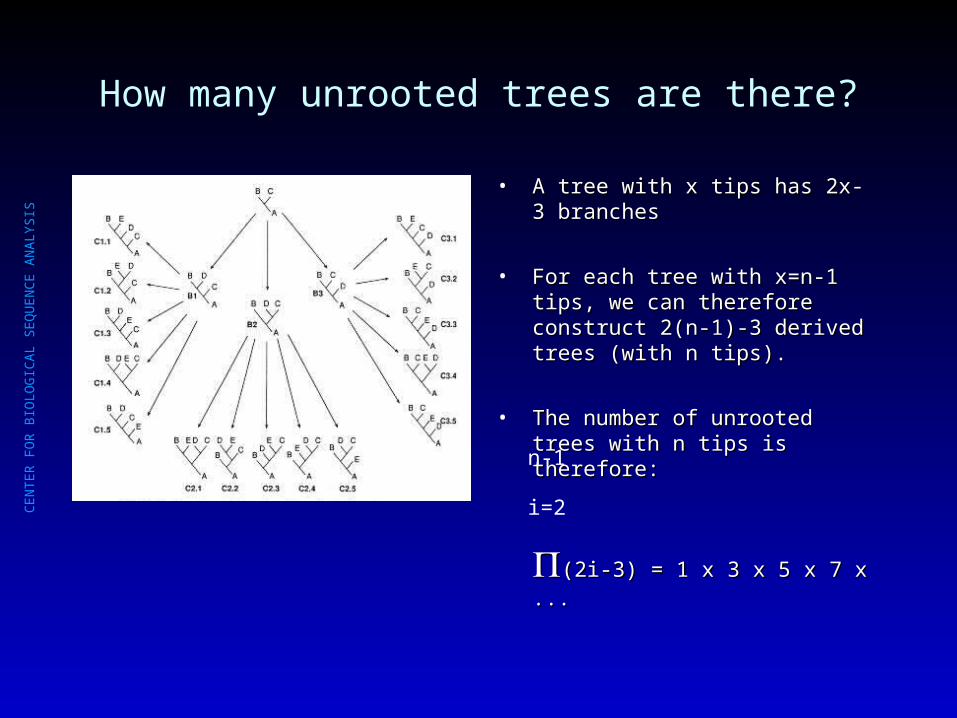
How many unrooted trees are there?
• A tree with x tips has 2x-3 branchesA tree with x tips has 2x-3 branches
• For each tree with x=n-1 tips, we For each tree with x=n-1 tips, we can therefore construct 2(n-1)-3 can therefore construct 2(n-1)-3 derived trees (with n tips). derived trees (with n tips).
• The number of unrooted trees with The number of unrooted trees with n tips is therefore:n tips is therefore:
(2i-3) = 1 x 3 x 5 x 7 x ...(2i-3) = 1 x 3 x 5 x 7 x ...i=2
n-1
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
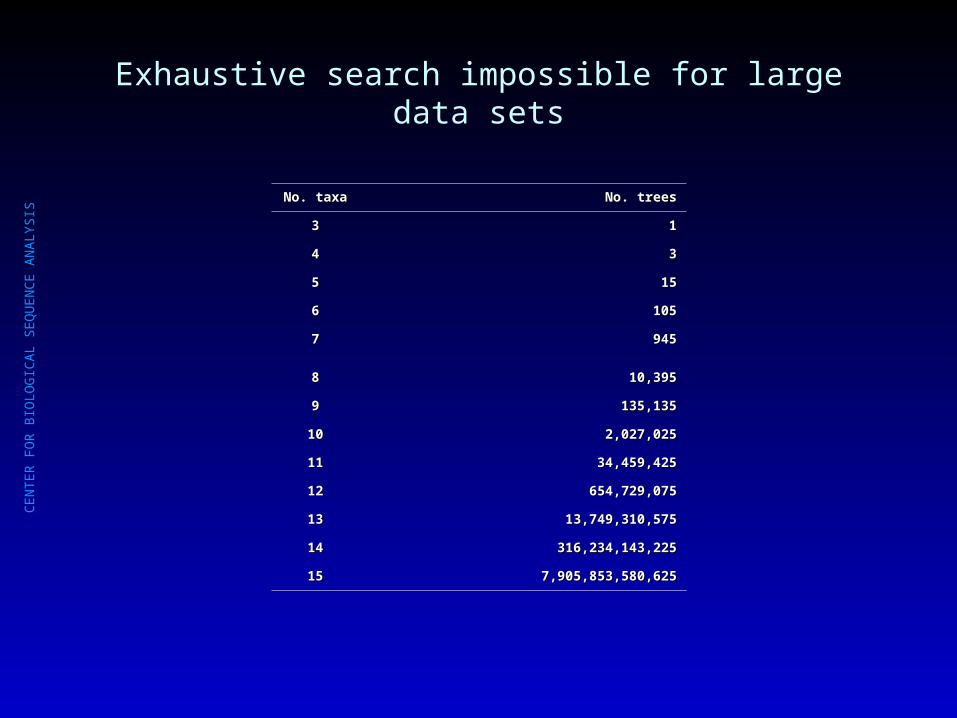
Exhaustive search impossible for large data sets
No. taxaNo. taxa No. treesNo. trees
33 11
44 33
55 1515
66 105105
77 945945
88 10,39510,395
99 135,135135,135
1010 2,027,0252,027,025
1111 34,459,42534,459,425
1212 654,729,075654,729,075
1313 13,749,310,57513,749,310,575
1414 316,234,143,225316,234,143,225
1515 7,905,853,580,6257,905,853,580,625
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
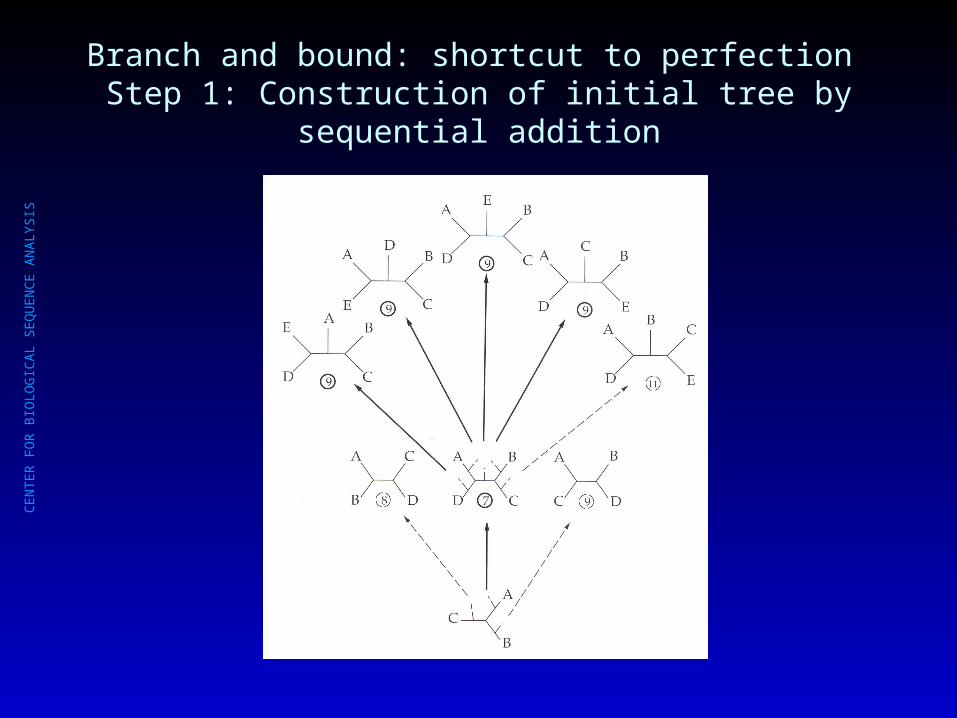
Branch and bound: shortcut to perfection Step 1: Construction of initial tree by sequential addition
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
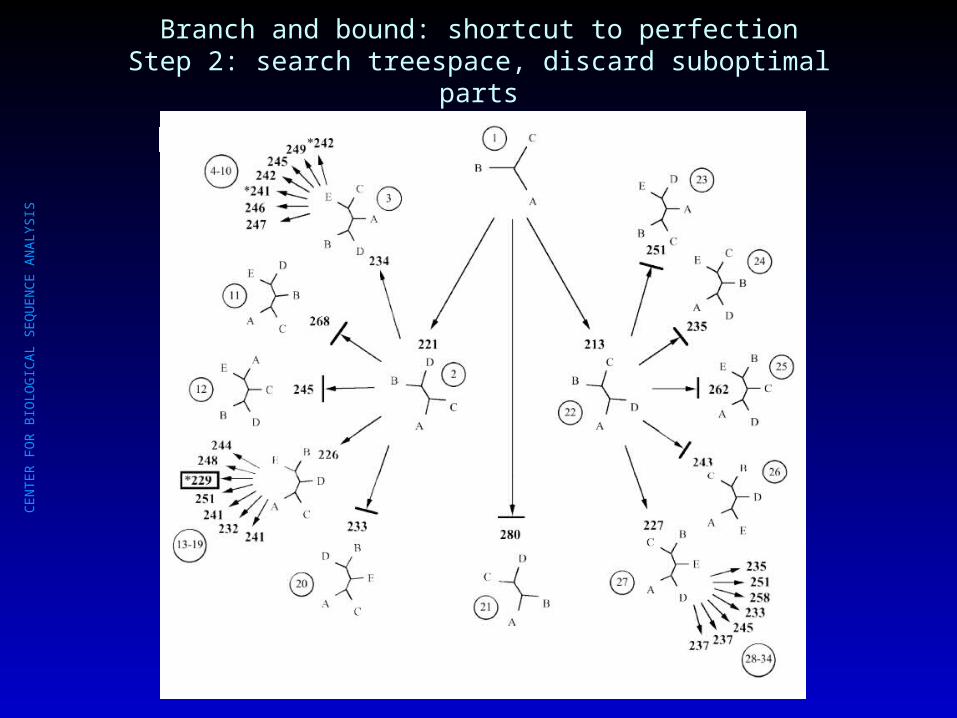
Branch and bound: shortcut to perfectionStep 2: search treespace, discard suboptimal parts
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
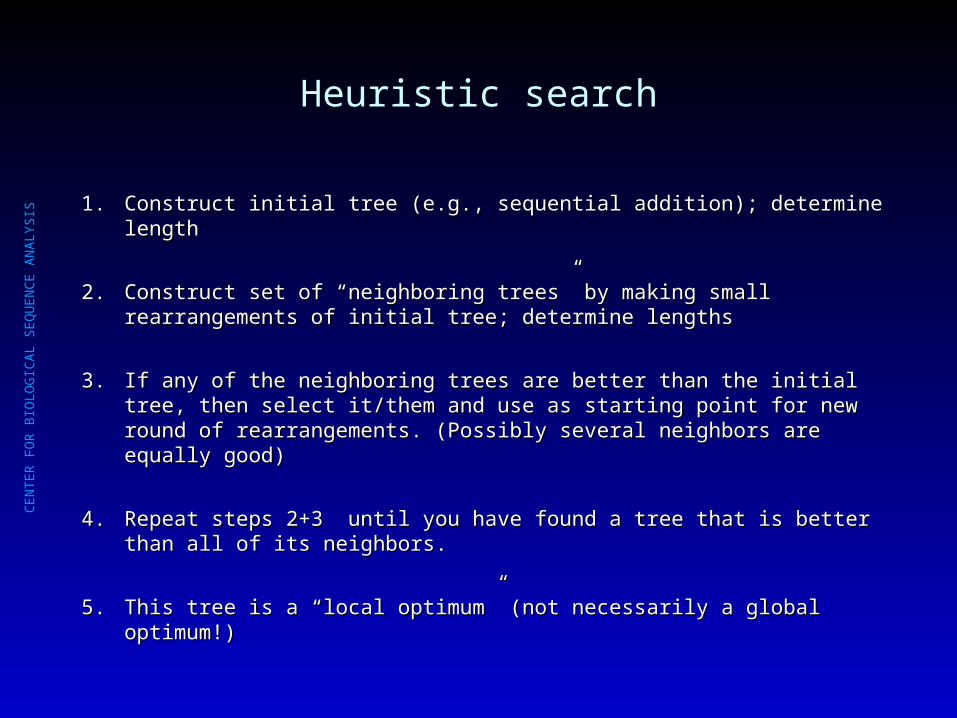
Heuristic search
1.1. Construct initial tree (e.g., sequential addition); determine lengthConstruct initial tree (e.g., sequential addition); determine length
2.2. Construct set of “neighboring trees” by making small rearrangements of initial Construct set of “neighboring trees” by making small rearrangements of initial tree; determine lengthstree; determine lengths
3.3. If any of the neighboring trees are better than the initial tree, then select it/them If any of the neighboring trees are better than the initial tree, then select it/them and use as starting point for new round of rearrangements. (Possibly several and use as starting point for new round of rearrangements. (Possibly several neighbors are equally good)neighbors are equally good)
4.4. Repeat steps 2+3 until you have found a tree that is better than all of its Repeat steps 2+3 until you have found a tree that is better than all of its neighbors.neighbors.
5.5. This tree is a “local optimum” (not necessarily a global optimum!) This tree is a “local optimum” (not necessarily a global optimum!)
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
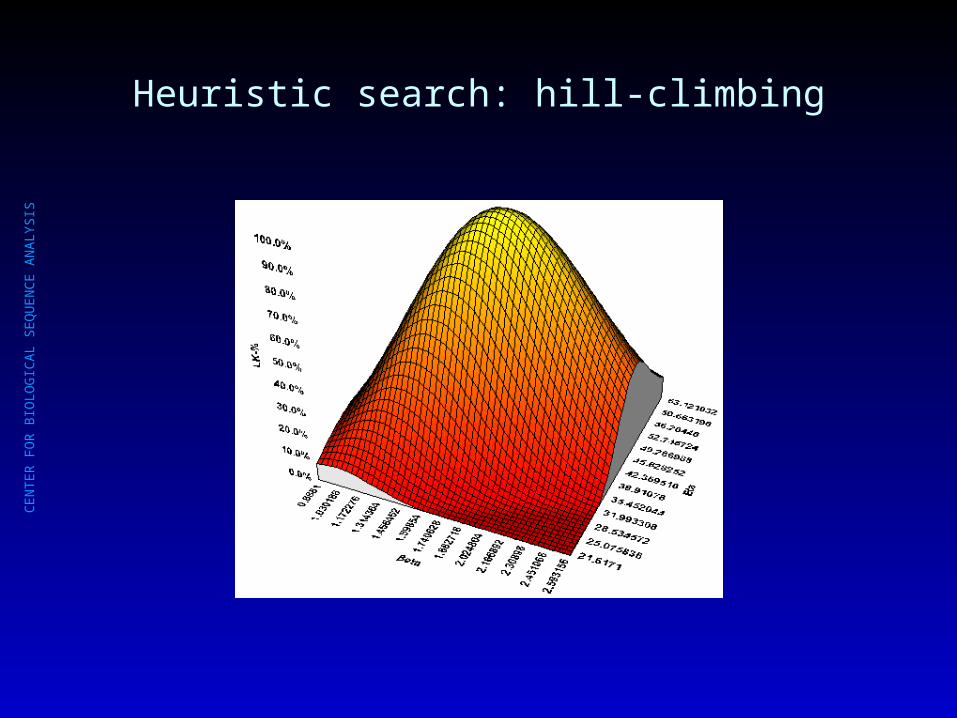
Heuristic search: hill-climbing

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Heuristic search: local vs. global optimum
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
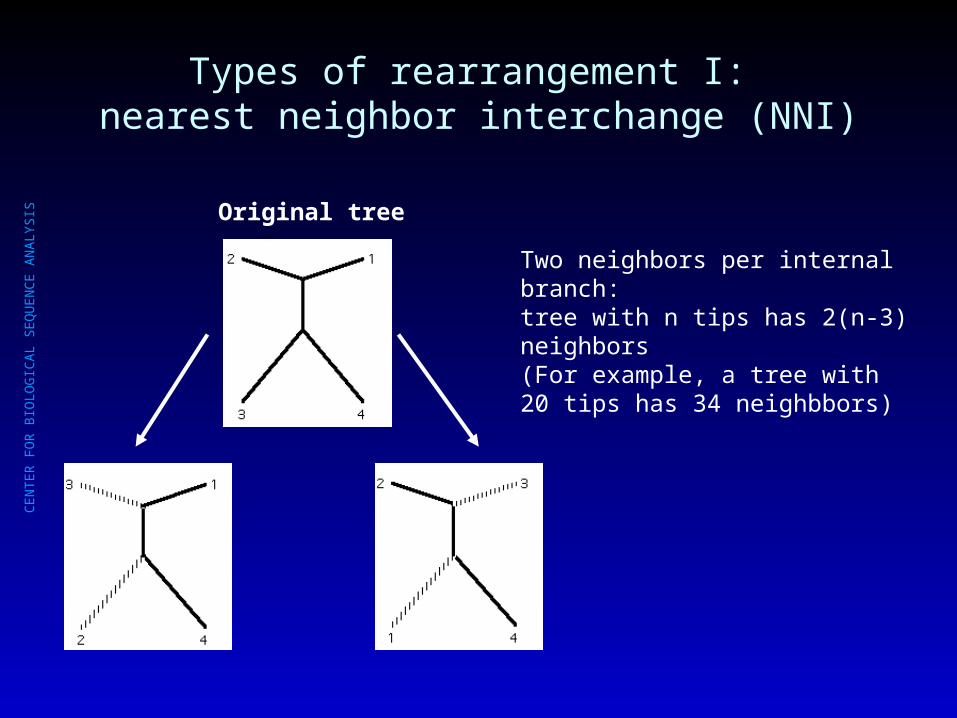
Types of rearrangement I: nearest neighbor interchange (NNI)
Original tree
Two neighbors per internal branch: tree with n tips has 2(n-3) neighbors(For example, a tree with 20 tips has 34 neighbbors)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Types of rearrangement II: subtree pruning and regrafting (SPR)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Types of rearrangement III: tree bisection and reconnection (TBR)
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
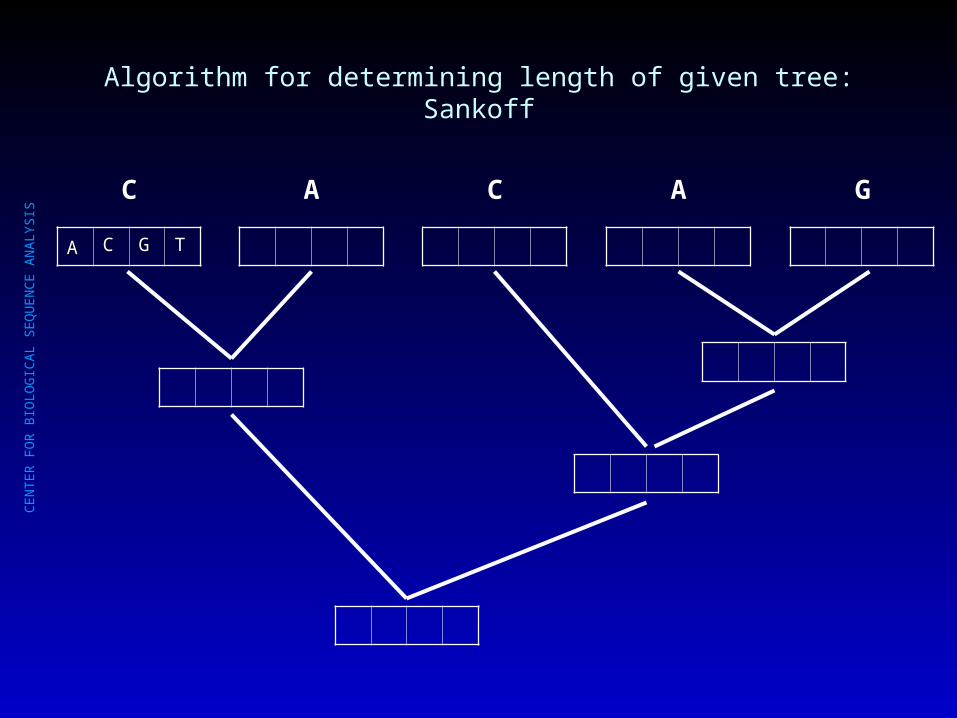
Algorithm for determining length of given tree: Sankoff
AA CC GG TT
C A C A G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
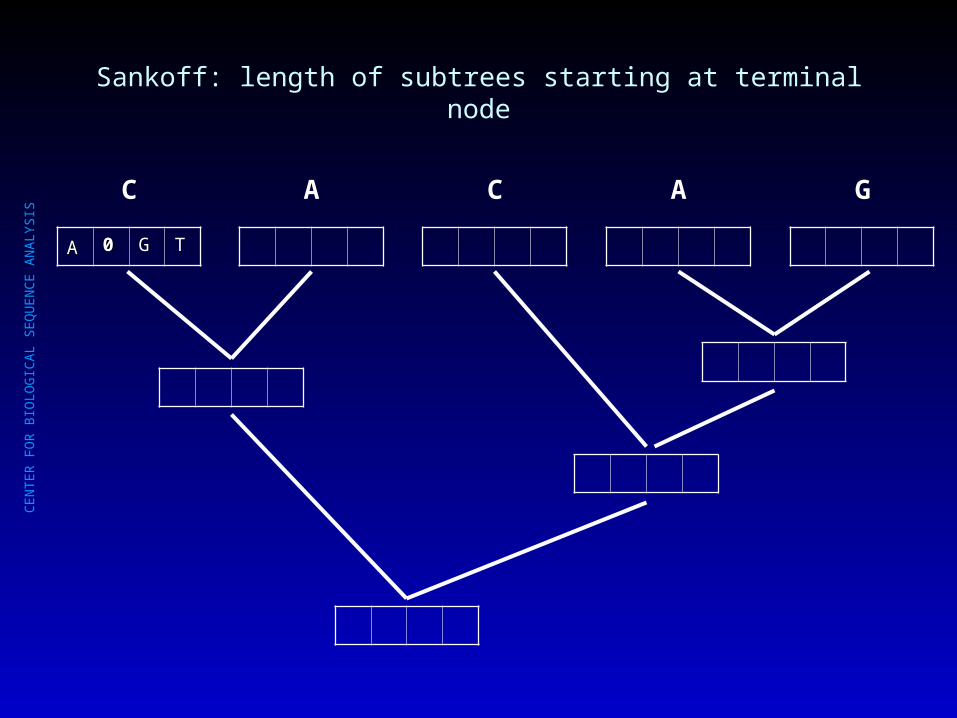
Sankoff: length of subtrees starting at terminal node
AA 00 GG TT
C A C A G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Sankoff: length of subtrees starting at terminal node
00
C A C A G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
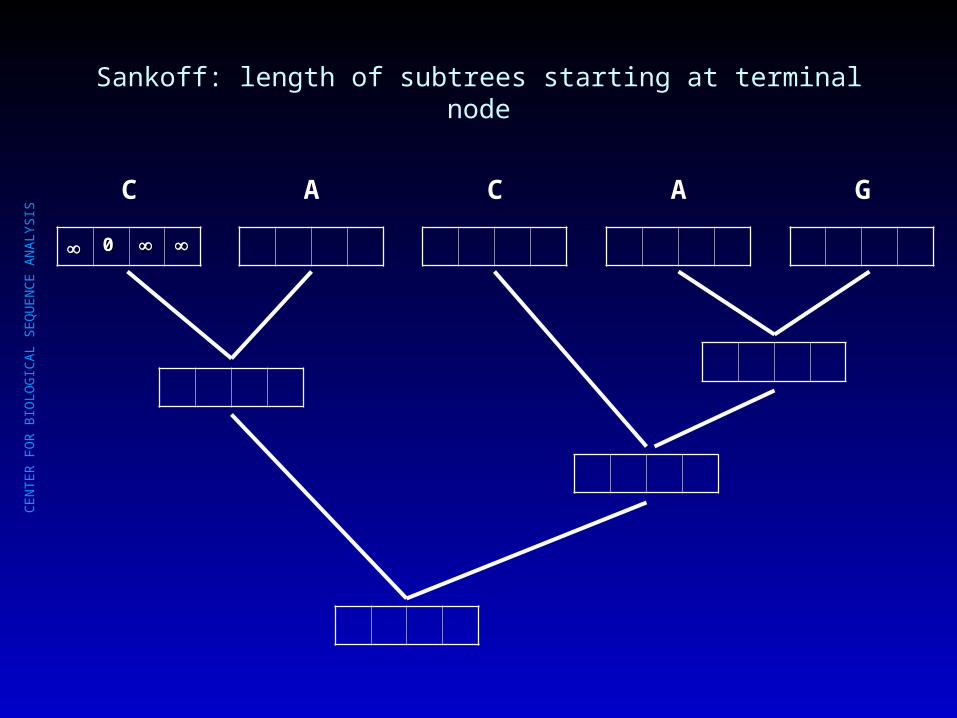
Sankoff: length of subtrees starting at terminal node
00
C
00
A C A G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
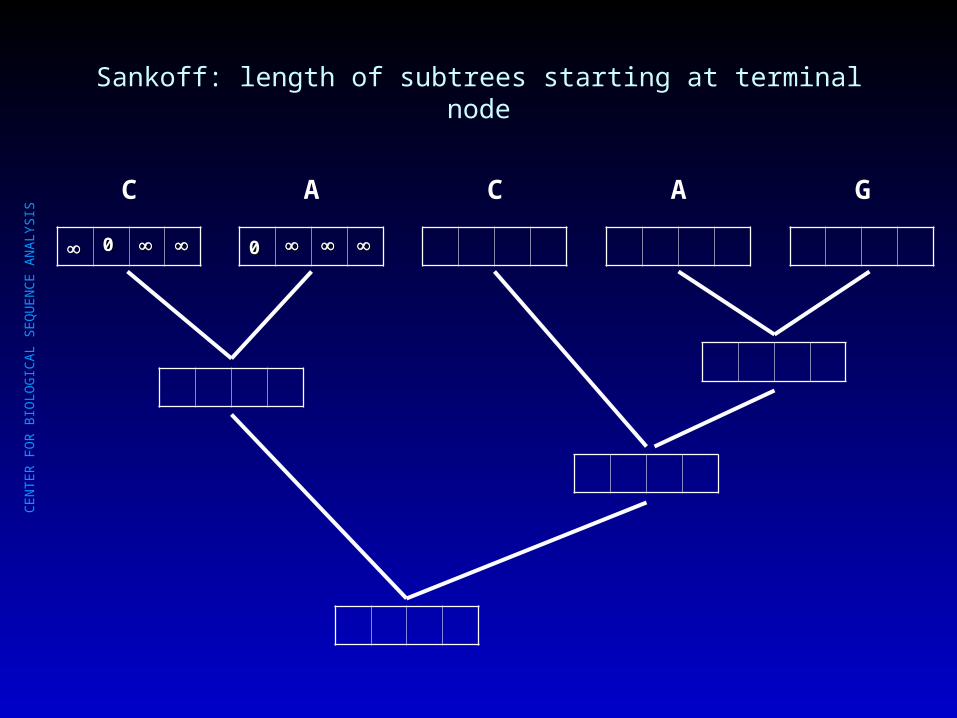
Sankoff: length of subtrees starting at terminal node
00
C
00
A
00
C A G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
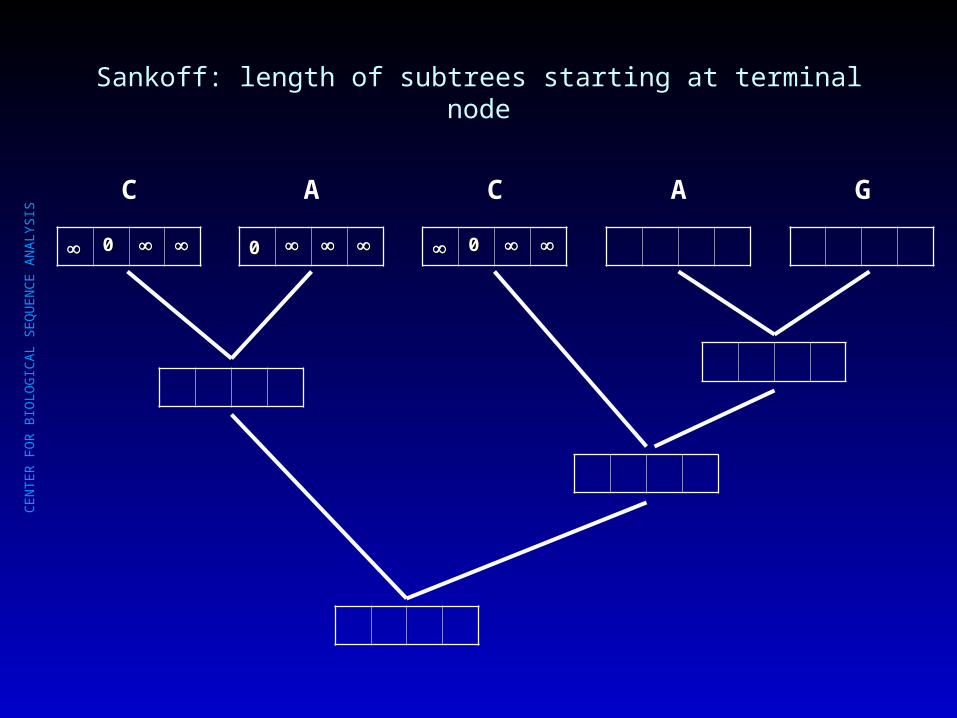
Sankoff: length of subtrees starting at terminal node
00
C
00
A
00
C
00
A G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
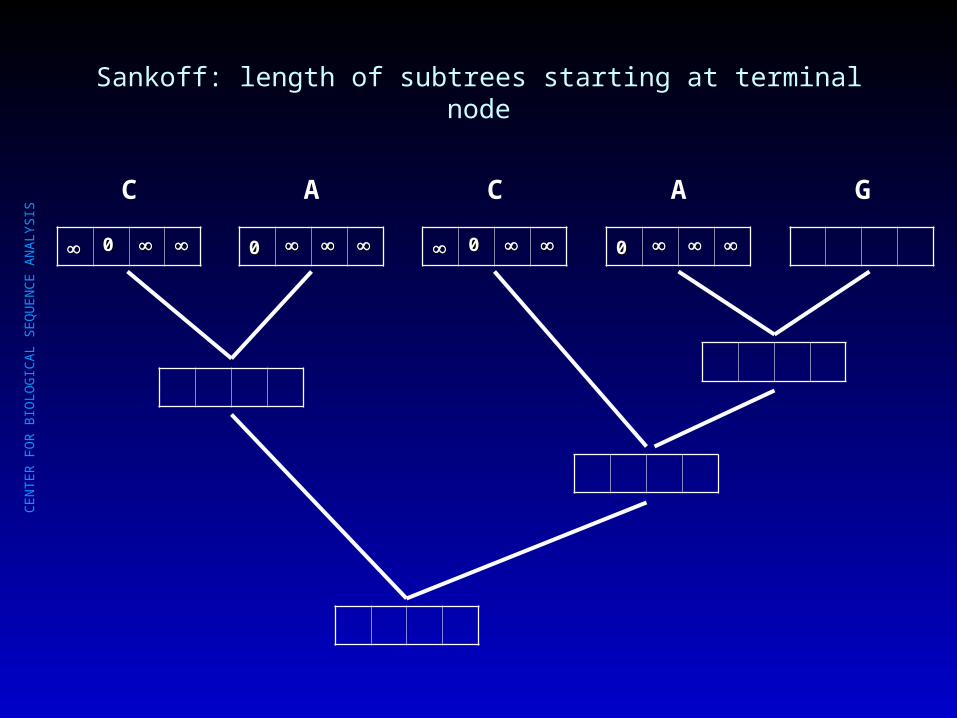
Sankoff: length of subtrees starting at terminal node
00
C
00
A
00
C
00
A
00
G
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
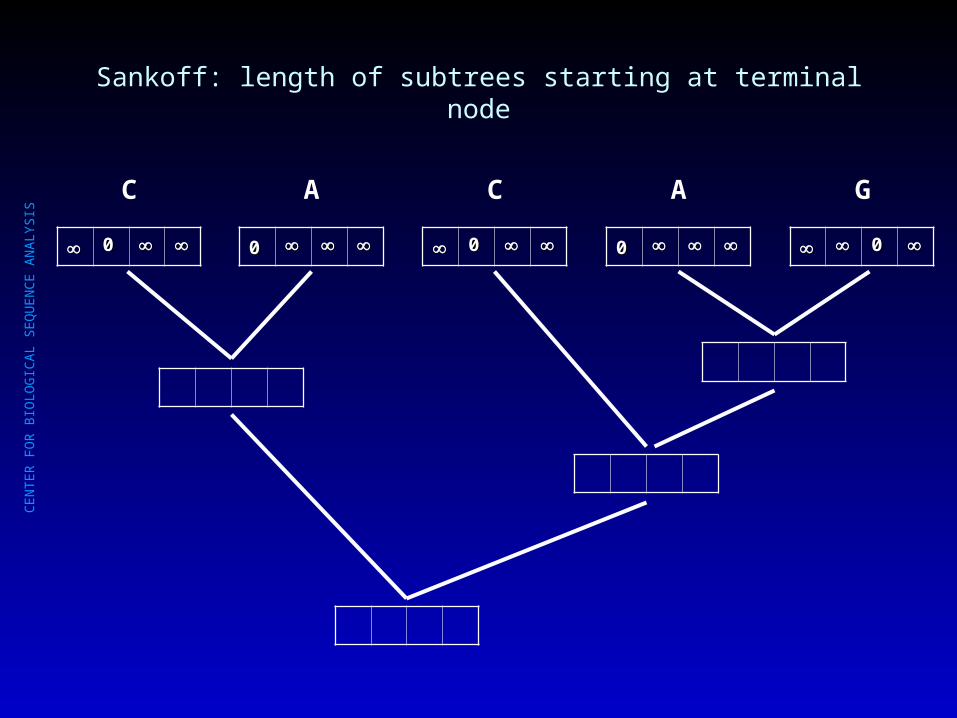
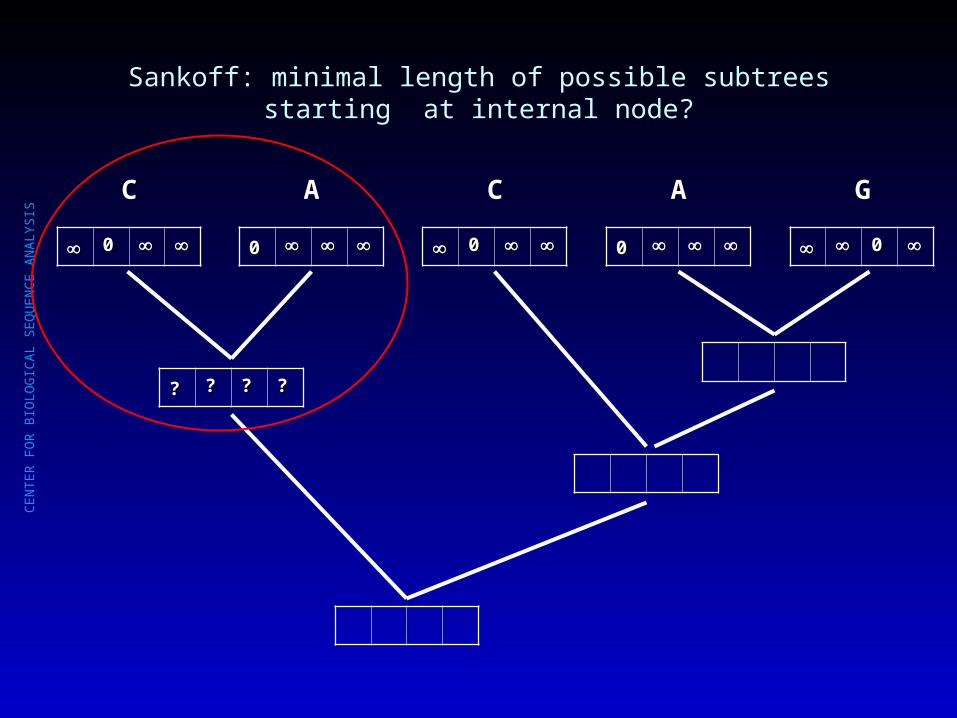
Sankoff: minimal length of possible subtrees starting at internal node?
00
C
00
A
00
C
00
A
00
G
?? ?? ?? ??
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
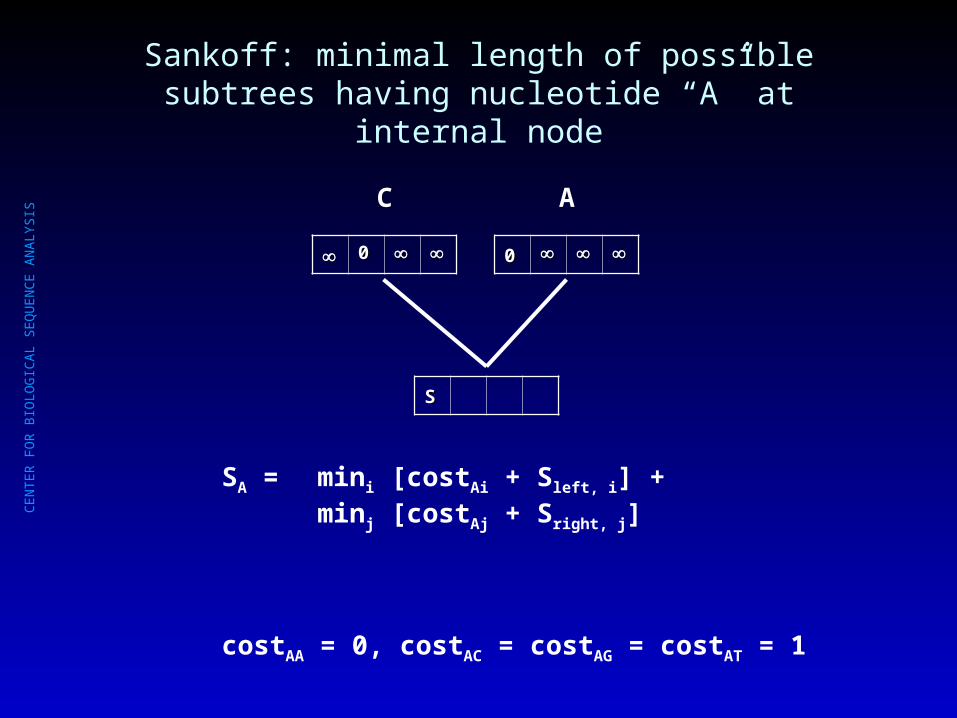
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
00
C
00
A
SS
SA = mini [costAi + Sleft, i] + minj [costAj + Sright, j]
costAA = 0, costAC = costAG = costAT = 1

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
00
C
??
0
Nt on left branchNt on left branch CostCost
AA 00
CC
GG
TT
SA = mini [costAi + Sleft, i] + minj [costAj + Sright, j]

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
00
C
??
0
Nt on left branchNt on left branch CostCost
AA 0 0 + +
CC
GG
TT
SA = mini [costAi + Sleft, i] + minj [costAj + Sright, j]

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
00
C
??
0
Nt on left branchNt on left branch CostCost
AA 0 + 0 + = =
CC
GG
TT
SA = mini [costAi + Sleft, i] + minj [costAj + Sright, j]

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
00
C
??
Nt on left branchNt on left branch CostCost
AA 0 + 0 + = =
CC 1 + 0 = 11 + 0 = 1
GG
TT
1
SA = mini [costAi + Sleft, i] + minj [costAj + Sright, j]

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
00
C
??
Nt on left branchNt on left branch CostCost
AA 0 + 0 + = =
CC 1 + 0 = 11 + 0 = 1
GG 1 + 1 + = =
TT
1
SA = mini [costAi + Sleft, i] + minj [costAj + Sright, j]

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
00
C
??
Nt on left branchNt on left branch CostCost
AA 0 + 0 + = =
CC 1 + 0 = 11 + 0 = 1
GG 1 + 1 + = =
TT 1 + 1 + = =
1
SA = mini [costAi + Sleft, i] + minj [costAj + Sright, j]

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
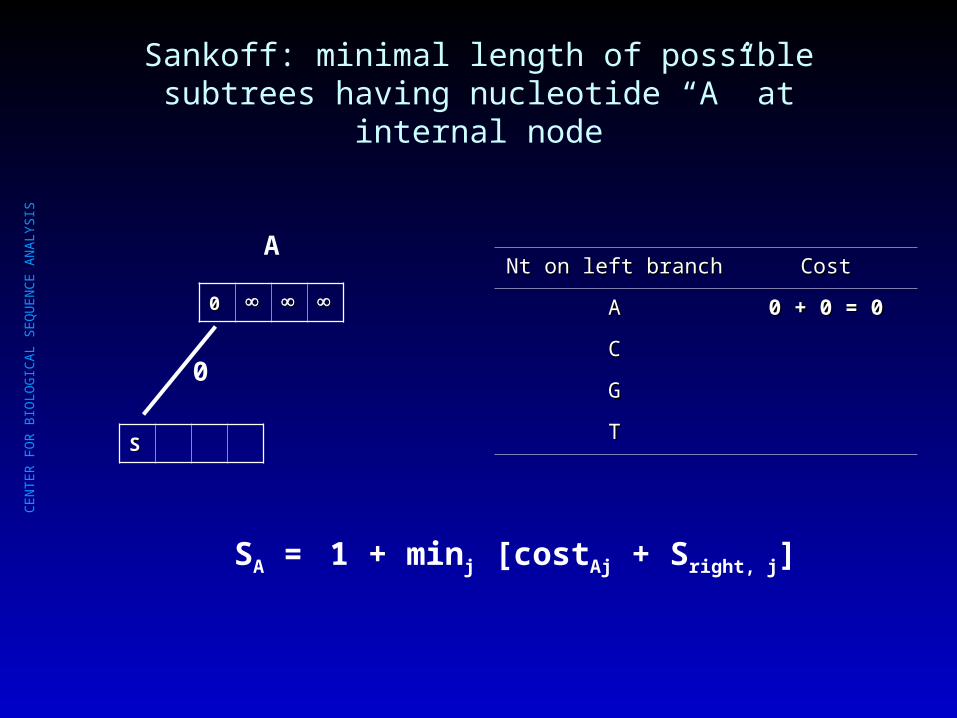
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
00
C
??
Nt on left branchNt on left branch CostCost
AA 0 + 0 + = =
CC 1 + 0 =1 + 0 = 1 1
GG 1 + 1 + = =
TT 1 + 1 + = =
1
SA = mini [costAi + Sleft, i] + minj [costAj + Sright, j]
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
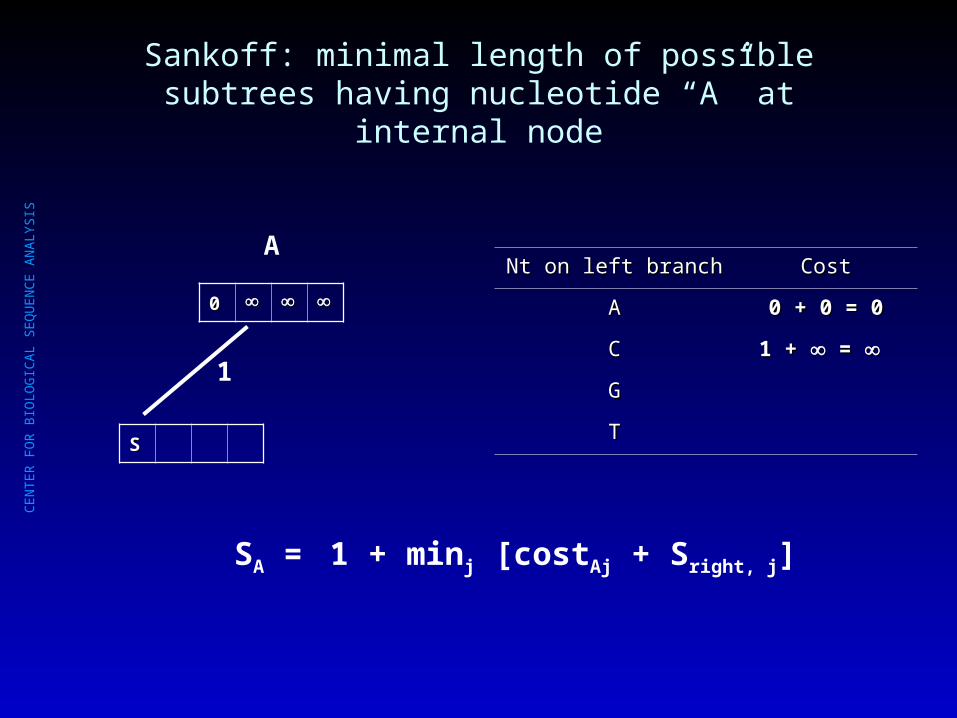
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
Nt on left branchNt on left branch CostCost
AA 0 + 0 = 00 + 0 = 0
CC
GG
TT
SA = 1 + minj [costAj + Sright, j]
00
A
SS
0
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
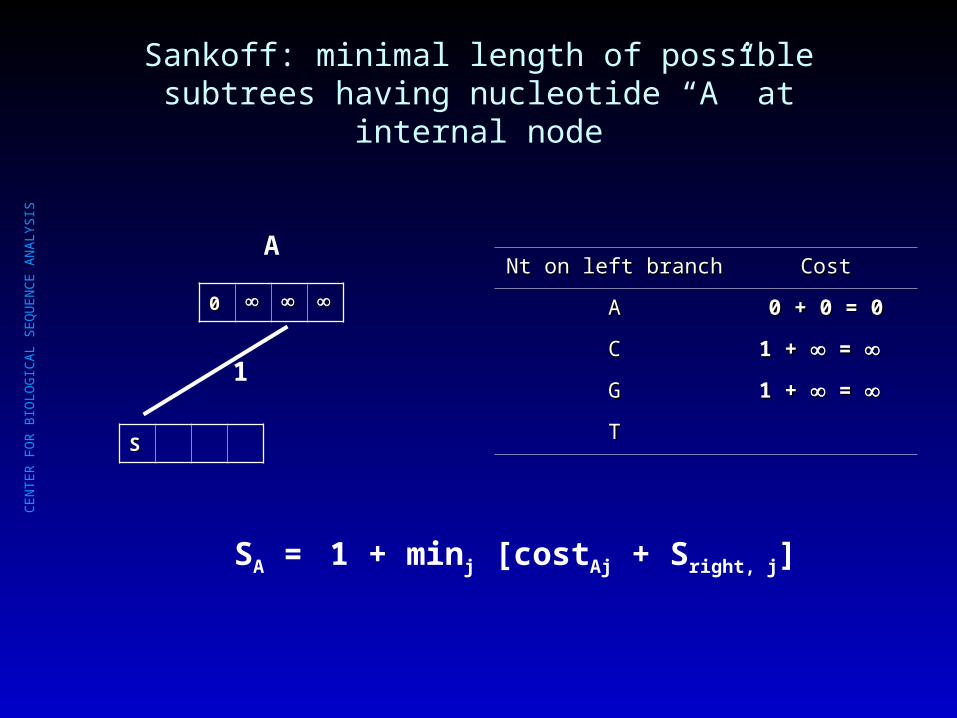
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
Nt on left branchNt on left branch CostCost
AA 0 + 0 = 00 + 0 = 0
CC 1 + 1 + = =
GG
TT
SA = 1 + minj [costAj + Sright, j]
00
A
SS
1
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
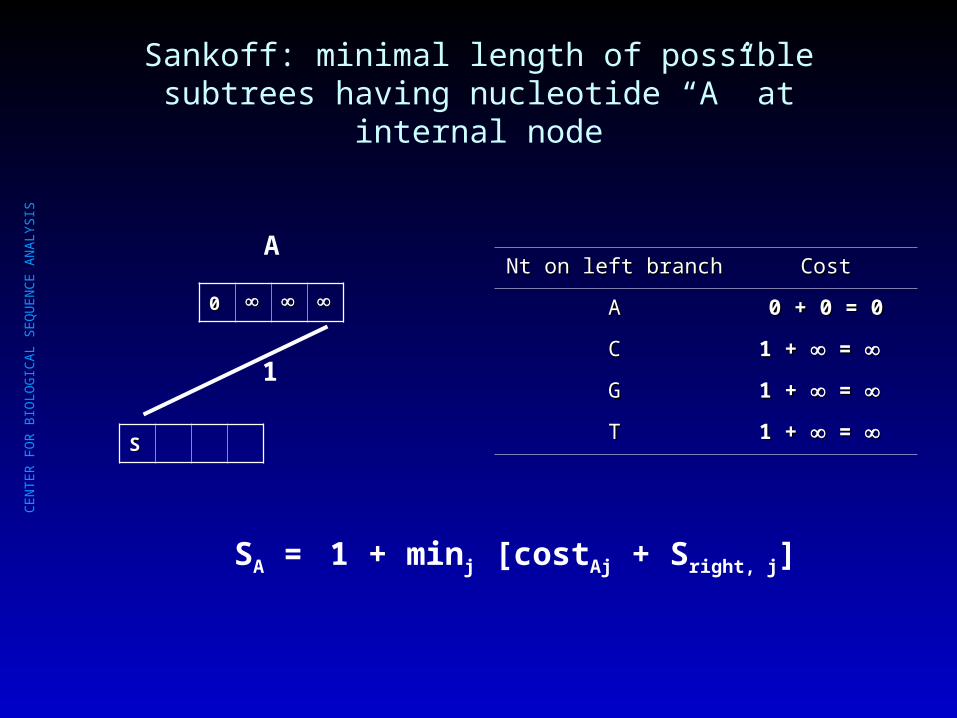
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
Nt on left branchNt on left branch CostCost
AA 0 + 0 = 00 + 0 = 0
CC 1 + 1 + = =
GG 1 + 1 + = =
TT
SA = 1 + minj [costAj + Sright, j]
00
A
SS
1
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
Nt on left branchNt on left branch CostCost
AA 0 + 0 = 00 + 0 = 0
CC 1 + 1 + = =
GG 1 + 1 + = =
TT 1 + 1 + = =
SA = 1 + minj [costAj + Sright, j]
00
A
SS
1

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
Nt on left branchNt on left branch CostCost
AA 0 + 0 = 00 + 0 = 0
CC 1 + 1 + = =
GG 1 + 1 + = =
TT 1 + 1 + = =
SA = 1 + minj [costAj + Sright, j]
00
A
SS
0
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
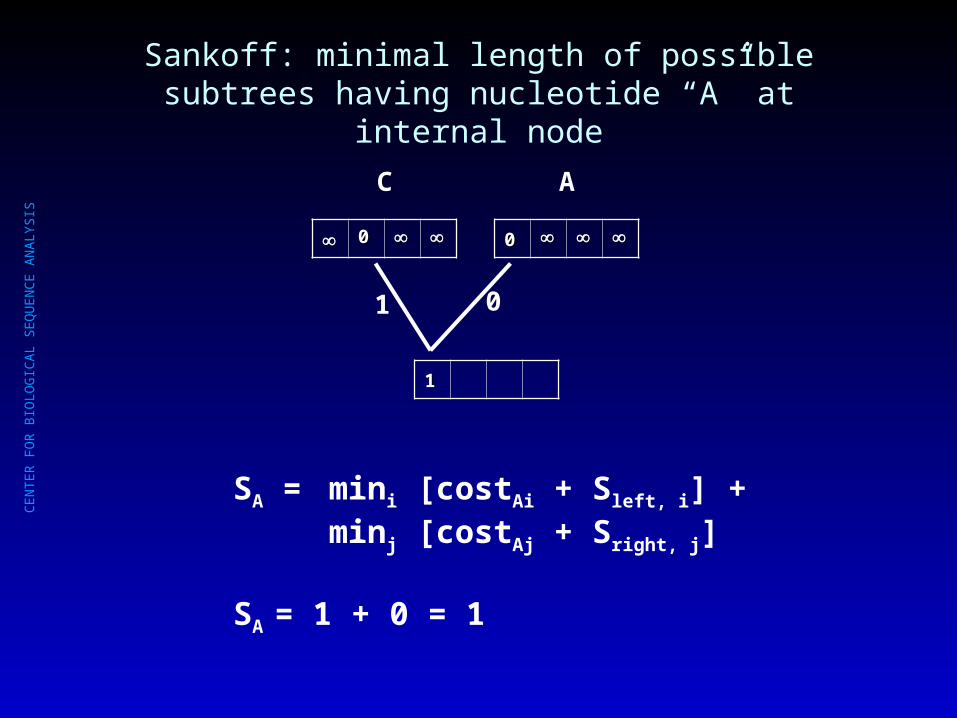
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
SA = mini [costAi + Sleft, i] + minj [costAj + Sright, j]
SA = 1 + 0 = 1
00
C
00
A
11
1 0
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Sankoff: minimal length of possible subtrees having nucleotide “A” at internal node
00
C
00
A
00
C
00
A
00
G
11
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
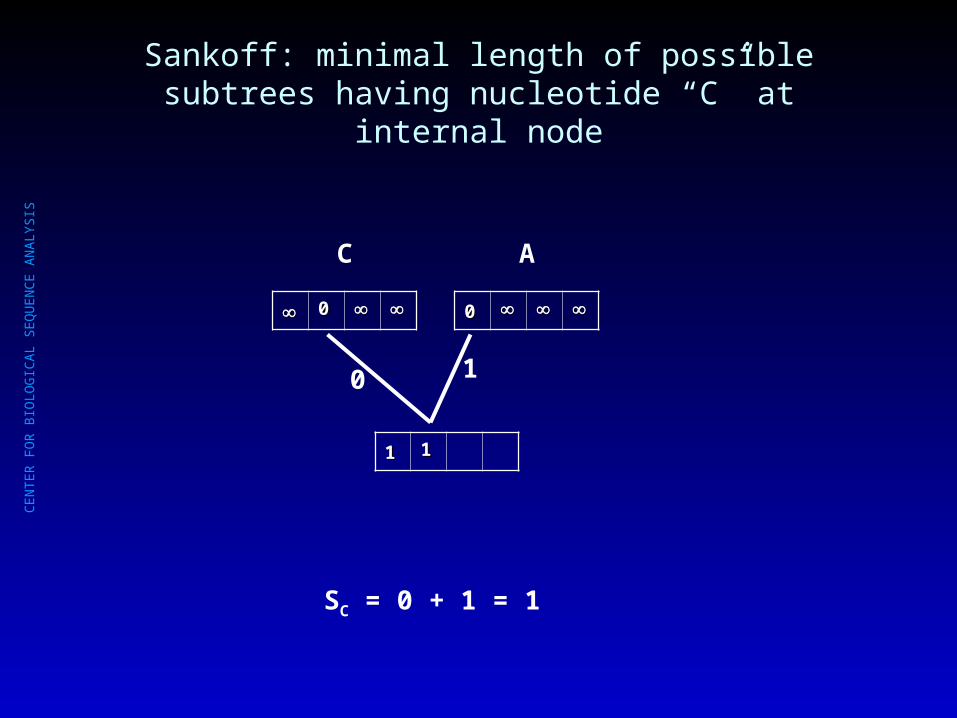
Sankoff: minimal length of possible subtrees having nucleotide “C” at internal node
00
C
00
A
11 11
0 1
SC = 0 + 1 = 1
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
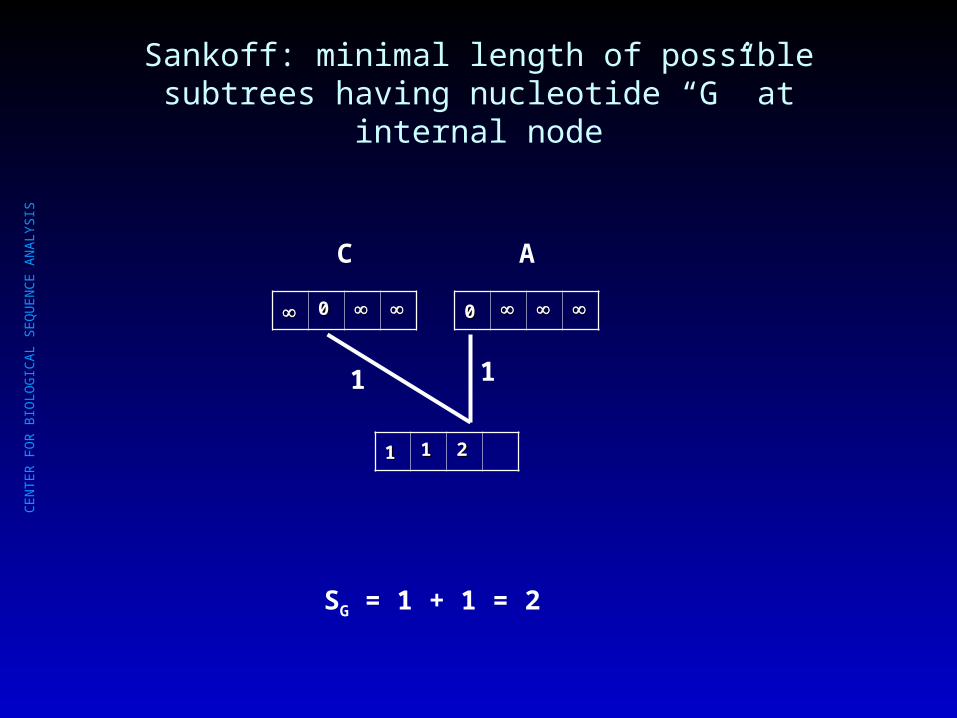
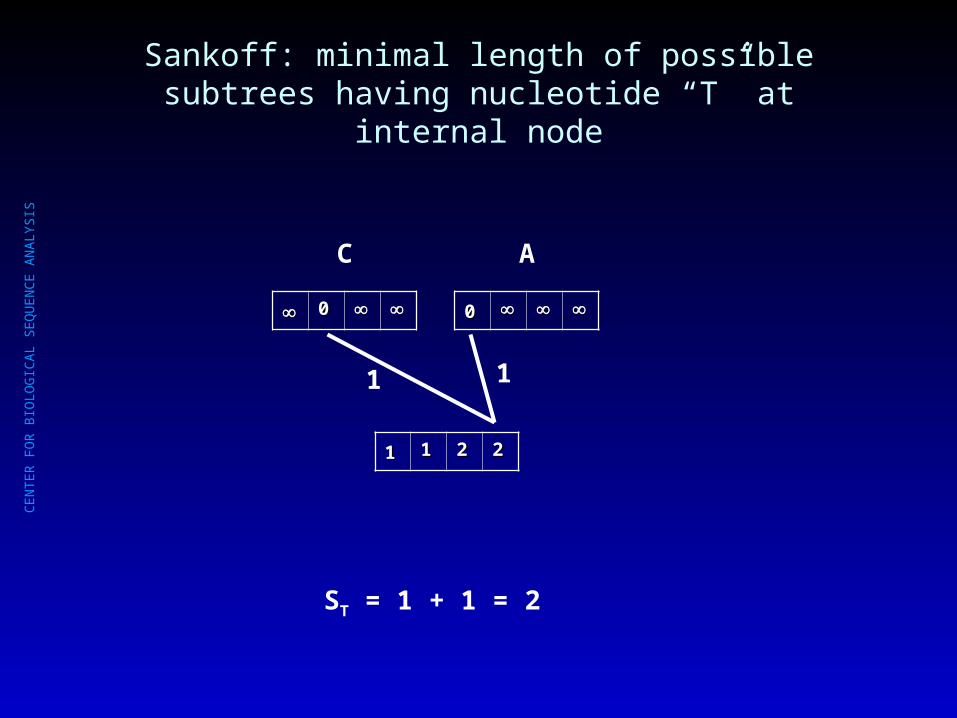
Sankoff: minimal length of possible subtrees having nucleotide “G” at internal node
00
C
00
A
11 11 22
1 1
SG = 1 + 1 = 2
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Sankoff: minimal length of possible subtrees having nucleotide “T” at internal node
00
C
00
A
11 11 22 22
1 1
ST = 1 + 1 = 2
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
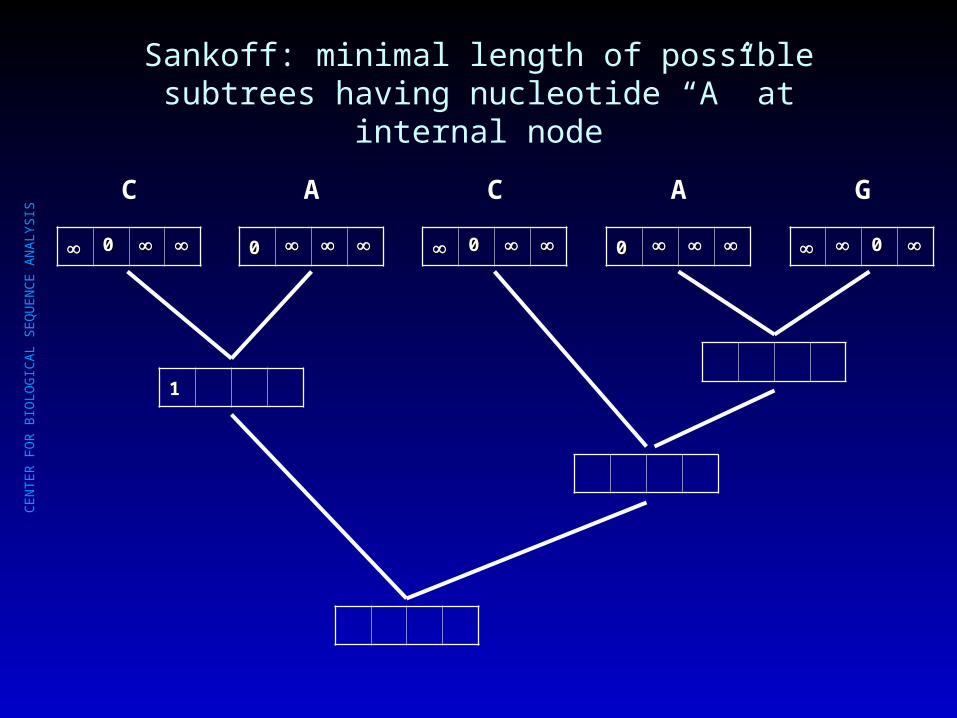
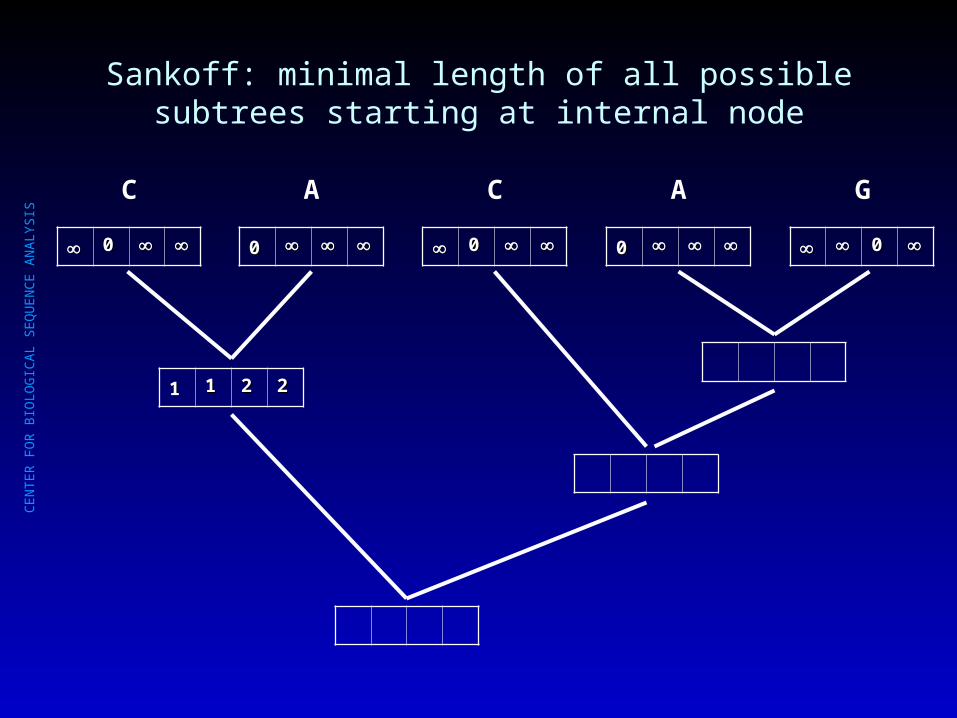
Sankoff: minimal length of all possible subtrees starting at internal node
00
C
00
A
00
C
00
A
00
G
11 11 22 22
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
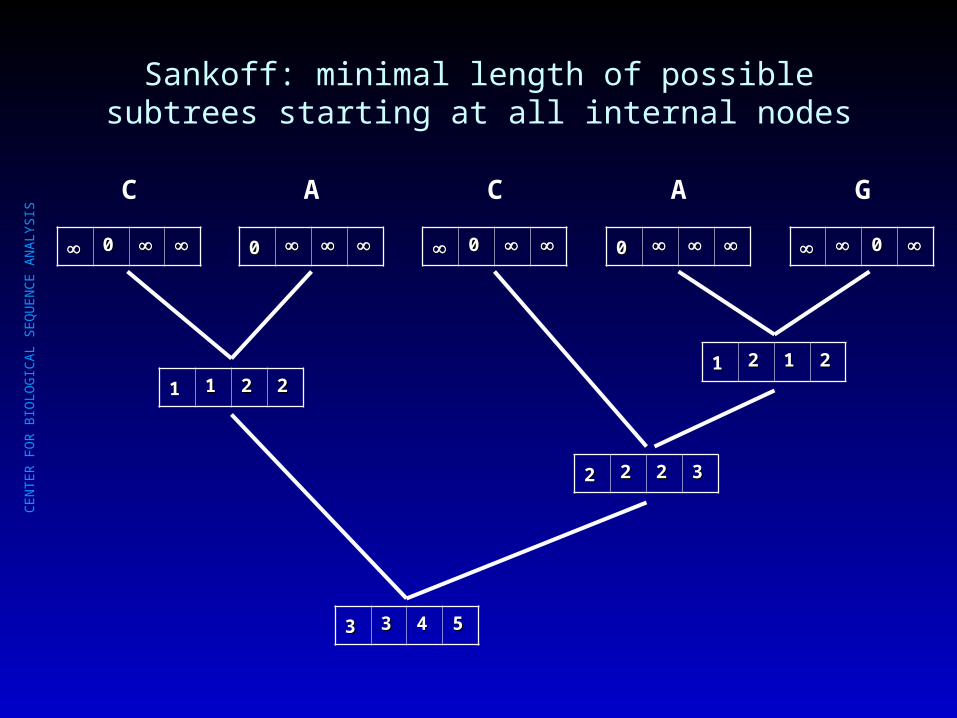
Sankoff: minimal length of possible subtrees starting at all internal nodes
00
C
00
A
00
C
00
A
00
G
11 11 22 2211 22 11 22
22 22 22 33
33 33 44 55
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
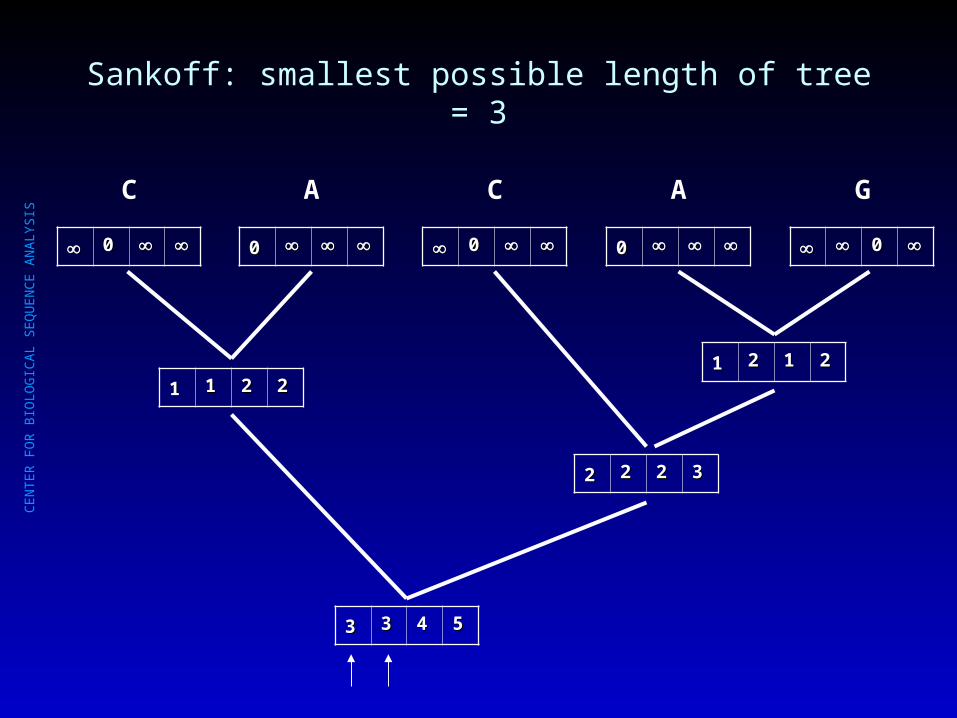
Sankoff: smallest possible length of tree = 3
00
C
00
A
00
C
00
A
00
G
11 11 22 2211 22 11 22
22 22 22 33
33 33 44 55





























