-
7/29/2019 Cell Signalling and Gene Expression Mediated by
RET
1/7
MINISYMPOSIUM
Cell signalling and gene expression mediated by RET
tyrosine kinase
K . K U R O K A W A1 , K . K A W A I1 , M . H A S H I M O T O1 ,
Y . I T O 2 & M . T A K A H A S H I1
From the 1Department of Pathology, and the 2Equipment Center for
Research and Education, Nagoya University Graduate School of
Medicine,
Showa-ku, Nagoya, Japan
Abstract. Kurokawa K, Kawai K, Hashimoto M, ItoY, Takahashi M
(Nagoya University Graduate
School of Medicine, Showa-ku, Nagoya, Japan).
Cell signalling and gene expression mediated by
RET tyrosine kinase (Minisymposium). J Intern Med
2003; 253: 627633.
Germline mutations of the RETproto-oncogene cause
multiple endocrine neoplasia (MEN) 2A or 2B by
different mechanisms. As is the case for other receptor
tyrosine kinases, mutant RET recruits a variety of
signalling molecules via phosphorylated tyrosine
residues present in the kinase domain and carboxy-
terminal tail. As we previously reported, the signalingvia
phosphorylated tyrosine 1062 plays a crucial role
in the transforming activities of both RET-MEN2A
and RET-MEN2B mutant protein. Interestingly, this
single tyrosine residue represents a binding site for
several signalling molecules including SHC, Enigma,
SNT/FRS2, DOK and IRS1 and is responsible for ac-
tivation of the RAS/ERK, PI3-K/AKT, JNK,
p38MAPK and ERK5 signalling pathways. Amongstthese, the
PI3-K/AKT and JNK pathways appeared to
be more strongly activated in the cells expressing
RET-MEN2B than in the cells expressing RET-
MEN2A, suggesting the possibility that these path-
ways may be involved in the disease phenotype. In
addition, RET is alternatively spliced to produce three
isoforms and the splicing site is present just down-
stream of tyrosine 1062. These isoforms play different
roles for the tumour development associated with
MEN 2 or the development of the kidney and the
enteric nervous system. Moreover, using differential
display analysis, we identified several genes whoseexpression is
highly induced by RET-MEN2B mutant
proteins. The differential gene expression by RET-
MEN2A and RET-MEN2B may also be important for
the development of their phenotypes.
Keywords: DOK1, multiple endocrine neoplasia type
2, RET isoforms, SHC, SNT/FRS2, stanniocalcin 1.
Introduction
The RETproto-oncogene encodes a receptor tyrosine
kinase [1, 2] that is essential for the development of
the enteric nervous system and the kidney [3]. The
glial cell line-derived neurotrophic factor (GDNF)
family ligands (GFLs) including GDNF, neurturin,
persephin and artemin exert their physiological
functions via activation of RET tyrosine kinase.
However, GFLs do not bind to the extracellulardomain of RET
directly but require glycosylphos-
phatidylinositol-linked cell surface proteins called
GFRas for complex formation with RET. GFRa family
members (GFRa14) show preferential binding to a
particular GFL and plays specific roles in vivo
through the preferred ligandreceptor complex
formation [4]. Gene ablation studies revealed that
the GDNF/GFRa1/RET complex formation is crucial
Journal of Internal Medicine 2003; 253: 627633
2003 Blackwell Publishing Ltd 627
-
7/29/2019 Cell Signalling and Gene Expression Mediated by
RET
2/7
for the development of the kidney and the enteric
nervous system [5].
RET mutations are responsible for the develop-
ment of several human diseases including papillary
thyroid carcinoma, multiple endocrine neoplasia
(MEN) 2A and 2B, familial medullary thyroid carci-noma (FMTC)
and Hirschsprungs disease (HSCR)
[610]. More than 10 rearranged forms ofREThave
been cloned from sporadic and radiation-associated
papillary thyroid carcinomas [11, 12], whereas
germline point mutations of RET were identified in
MEN2A, MEN2B and FMTC. MEN2A or FMTC
mutations were mostly found in one of six cysteine
residues in the RET extracellular cysteine-rich do-
main [13]. These cysteine mutations induce disul-
phide-linked RET dimerization, leading to its ligand-
independent activation [14, 15]. On the contrary,
the MEN2B mutations were detected at codon 918(methionine to
threonine) in most cases or at codon
883 (alanine to phenylalanine) in fewer than 4%
cases [13], which are present in the catalytic core of
the RET tyrosine kinase domain. These mutations
probably induce conformational change of the kinase
domain, resulting in activation without dimerization
[1517]. However, we do not fully understand the
mechanisms by which the different phenotypes
between MEN2A and MEN2B are caused.
Hirschsprungs disease is a congenital malforma-
tion associated with aganglionosis of the gastroin-
testinal tract. RET mutations are responsible forabout 50% of
familial and 1020% of sporadic cases
of HSCR and a variety of missense, non-sense and
frame shift mutations have been identified along
with its whole coding sequence [1822]. Based on
functional analyses, at least four mechanisms of RET
dysfunction are responsible for the development of
HSCR. First, most mutations identified in the extra-
cellular domain markedly impaired the RET cell
surface expression, probably because of incorrect
folding of the RET protein [2325]. Secondly, a few
mutations in the carboxy-terminal tail impaired the
binding of adaptor proteins such as SHC to RET,resulting in the
defect of RET-mediated RAS/ERK
and PI3-K/AKT signalling [26, 27]. Thirdly, the
mutations in the kinase domain, which were iden-
tified in amino acids conserved amongst members of
the tyrosine kinase family, almost completely abol-
ished the RET tyrosine kinase activity [2831].
Fourthly, some mutations in the kinase domain
severely impaired the activation of PLCc pathway
[31]. Taken together, these findings suggested that
several intracellular signalling pathways via RET
cooperatively function for the normal development
of the enteric nervous system.
Cell signalling mediated by RET
RET is alternatively spliced to produce three isoforms
(designated RET9, RET43 and RET51) that differ in
the carboxy-terminal sequence [32, 33]. There are
16 tyrosines in the intracellular domain of RET9 and
RET43 and 18 tyrosines in that of RET51. Amongst
these, tyrosines 1015 and 1062 (Y1015 and
Y1062) have been shown to be important for cell
signalling mediated by both ligand-dependent and
-independent activation of RET [12, 34]. Y1015
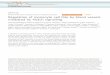
represents a binding site for PLCc [35], and Y1062
represents a binding site for several adaptor proteinssuch as
SHC [3638], SNT/FRS2 [39, 40], IRS1
[41], DOK1 [42], DOK4 and 5 [43], and Enigma
[44] (Fig. 1). As a result, various signalling path-
ways including the RAS/ERK, PI-3K/AKT,
p38MAPK, JNK [45] and ERK5 pathways [46] are
activated mainly via phosphorylated Y1062 in RET.
Consistent with these findings, mutation of Y1062
markedly impaired the transforming activity of all
types of MEN2 mutant proteins [36].
When SHC binds to Y1062 following RET activa-
tion, it further complexes with GRB2 and GAB1/
GAB2 proteins, leading to activation of the PI3-K/AKT signalling
pathway. On the contrary, asso-
ciation of SHC with the GRB2/SOS complex is
responsible for activation of the RAS/ERK pathway
[45, 47] (Fig. 1). Activation of the PI3-K/AKT and
RAS/ERK pathways resulted in the activation of
transcription factors, NFjB and CREB, respectively
[45]. Moreover, binding of SNT/FRS2 to Y1062
leads to the RAS/ERK activation because it has
GRB2 and SHP-2 binding sites in the carboxy-
teminal region [39, 40].
Using a yeast two-hybrid screen, we found that
DOK1 also binds to tyrosine 1062 in RET [42, 48].DOK1 was more
strongly associated with
RET-MEN2B mutant proteins than RET-MEN2A
mutant proteins. As a result, DOK1 was highly
phosphorylated in the cells expressing RET-MEN2B.
DOK1 has six tyrosine residues that represent
binding sites for RAS-GTPase activating protein
(RAS-GAP), leading to suppression of the RAS/ERK
activation. In addition, DOK1 contains a tyrosine
2003 Blackwell Publishing Ltd Journal of Internal Medicine 253:
627633
6 28 K . K UR OK AW A et al.
-
7/29/2019 Cell Signalling and Gene Expression Mediated by
RET
3/7
residue that is a binding site for the NCK adaptor
protein. The association of NCK with DOK1appeared to be
responsible for JNK and c-JUN
activation (Fig. 1). Salvatore et al. [49] reported
that the level of phosphorylation of Y1062 is
increased in PC12 cells expressing RET-MEN2B,
resulting in enhancement of activation of the ERK
and PI3-K/AKT pathways. Taken together with our
findings indicating high levels of activation of JNK
and AKT by RET-MEN2B [42, 48], it is suggested
that the enhanced signalling via Y1062 may be
involved in the MEN2B phenotype.
DOK4 and 5 also bind to Y1062 of RET and
induce ligand-dependent axonal outgrowth ofPC12 cells [43]. DOK4
is expressed broadly in
many tissues, such as brain, heart, lung and
kidney, whereas DOK5 is specifically expressed in
the brain. Because DOK4 and DOK5 associate with
neither RAS-GAP nor NCK, it is interesting to
investigate which signalling pathways are activa-
ted by them.
Regulation of cell signalling via Y1062in RET
Both SHC and SNT/FRS2 bind to pY1062 of RET viatheir
phosphotyrosine-binding (PTB) domains, that
recognize the common consensus motif NKLpY.
Both PTB domains have conformational topology
but do not have sequence homology [50]. So far,
two types of the PTB domains are known. The PTB
domain of SNT/FRS2 has sequence similarity to the
PTB domain of IRS1, so these PTB domains are
called phosphotyrosine-binding domain, insulin
receptor substrate 1-like (PTBI). DOK-family pro-
teins also contain PTBI.The PTB domain of SHC binds to TrkA,
that has
the canonical consensus motif for its binding
(NPXpY, X is any amino acid) in the juxtamembrane
region. The PTBI domain of SNT/FRS2 recognizes
the same consensus motif in TrkA as the PTB
domain of SHC. Consistent with this finding, the
binding mode of SNT/FRS2 PTBI to RET in vitro
resembles that of SHC PTB, depending on phos-
phorylation of Y1062 [39]. Although the PTBI
domain of SNT/FRS2 also binds to fibroblast growth
factor receptor (FGFR) [5154], the consensus
sequence for its binding in FGFR represents a uniquepeptide that
does not contain phosphotyrosine. In
addition, the binding is ligand-independent. The
finding that SNT/FRS2 recognizes different receptors
with different modes suggests the interference of
signalling pathways activated by each receptor [52].
Recently, it was reported that the microdomains of
membrane, lipid rafts, play a role for regulating or
separating cell signalling via SHC and SNT/FRS2
from Y1062 of RET [55, 56]. As GFRas are GPI-
anchored at the carboxy-terminus and SNT/FRS2
has a myristylation sequence at the amino-terminus
[57], these proteins locate on rafts. RET receptortyrosine
kinase locates outside rafts under unstimu-
lated conditions, and appears to be recruited to rafts
by GPI-anchored coreceptor GFRas. As a result, after
stimulation by GDNF, association of RET with SNT/
FRS2 is supposed to lead to sustained ERK activation.
On the contrary, SHC locates outside rafts, and may
be involved in the transient ERK activation as well as
the activation of the PI3-K/AKT pathway [56].
Y1062Y1015
PLC
Signal sequence Cysteine-rich
Transmembrane
Kinase domain
SHC
GAB1/2
GRB2
PI3-K
RET51
RET9RET43
AKT
SOS
SNT/FRS2 DOK1
p38MAPK
ERK5
SHP-2
NCK
JNKRAS
ERK
c-JUN
RAS-GAP
NFB CREB
GRB2++
Fig. 1 Signaling pathways medi-
ated by RET receptor tyrosine
kinase.
2003 Blackwell Publishing Ltd Journal of Internal Medicine 253:
627633
M I N I SY M P O SI U M : C E L L S I G N AL L I N G B Y R E T 6
2 9
-
7/29/2019 Cell Signalling and Gene Expression Mediated by
RET
4/7
The splicing site of three RET isoforms is present
just downstream of Y1062, and their different roles
are recently demonstrated. By NIH 3T3 transfection
assay, we showed that RET51-MEN2A and RET51-
MEN2B mutant proteins have stronger transforming
activity than RET9-MEN2A and RET9-MEN2Bmutant proteins,
respectively [36]. In addition, the
activity of RET43 was very low [26]. These findings
suggested that the sequences downstream of Y1062
are important for binding of adaptor proteins and
that RET51 may contribute more significantly to the
tumour development associated with MEN 2 than
RET9 and RET43. On the contrary, RET9 was
shown to be critical for the development of the
kidney and the enteric nervous system using
targeted mutagenesis mice expressing RET9 or
RET51 only [58], suggesting the possibility that
RET9 and RET51 are involved in the activation of
different signalling pathways in vivo [59].
Computational analysis of SHC PTB domain
binding to each RET isoform
To obtain supportive information about the function
of RET isoforms based on structural biology, we
simulatedthe interaction of the SHCPTB domainwith
the amino acids around Y1062 of each RET isoform
(Fig. 2). The simulations were performed based on the
model of the SHC PTB domain complexed with the
tyrosine-phosphorylated peptide of TrkA [60].
As shown in Fig. 2a, a hydrogen bond is formed
between arginine (R) 67 in the SHC PTB domain
RET51
M1064
2.37NN
H
R207 M1064
2.37 NN
H
R207
RET peptide with tyrosine 1062
SHC PTB domain
(b)
(a)
2.62
R67
pY1062
Y1062F
N
H
RET9
R207R1064
pY1062
2.47
NH
R207R1064
2.47
NH
RET43
A1064R207
A1064R207
Fig. 2 Computational analyses of SHC phosphotyrosine-binding
(PTB) domain binding to RET peptide with tyrosine 1062.
Simulations
of the interaction between SHC PTB domain and RET peptide with
tyrosine 1062 were performed based on the model of the SHC PTB
domain complexed with the tyrosine-phosphorylated peptide of
TrkA [60]. The distances between the functional residues and the
potential
energy of compounds were calculated using the insight II
software package. The compounds were simulated with force field
parameters
based on the consistent valence force field (CVFF). The
group-based cut off, 0.95 nm for the van der Waals and 0.95 nm for
Coulomb
interactions, were introduced. The temperature was set at 298K.
Calculations based on the simulation images were carried out
using
the insight II package. (a) A hydrogen bond between
phosphotyrosine (pY) 1062 in RET9 and arginine (R) 67 in SHC (2.62
A ). When
Y1062 is replaced with phenylalanine (Y1062F, light blue), the
hydrogen bond cannot be formed. (b) The interaction between SHC
PTB domain and three isoforms of RET. Arginine (R) 1064 in RET9
or methionine (M) 1064 in RET51 forms a hydrogen bond with
arginine (R) 207 in the SHC PTB domain (2.47 A and 2.37 A
respectively), but RET 43 does not.
2003 Blackwell Publishing Ltd Journal of Internal Medicine 253:
627633
6 30 K . K UR OK AW A et al.
-
7/29/2019 Cell Signalling and Gene Expression Mediated by
RET
5/7
and phosphotyrosine (pY) 1062 in RET (2.62A).
When Y1062 is replaced by phenylalanine
(Y1062F), the hydrogen bond cannot be formed.
In addition, arginine (R) 1064 in RET9 or methion-
ine (M) 1064 in RET51 forms a hydrogen bond with
arginine (R) 207 in the SHC PTB domain (2.47 A
and 2.37 A respectively), but RET 43 does not (Fig.
2b). These findings are consistent with our previous
data using in vitro binding assay [26, 39]. The
current simulations suggested that the conformation
of the SHC PTB domain binding to RET9 is almost
the same as that to TrkA [60]. However, the actual
conformation of the SHC PTB domain binding to
RET51 might be changed to some degree, because
the distance between methionine (M) 1064 in
RET and arginine (R) 207 in SHC appears to be
too close to form stable interactions between these
molecules.Amongst three RET isoforms, only RET9 has the
consensus sequence for binding of the SHC SH2
domain [38, 61]. This may contribute to the stable
binding of SHC to RET9 in addition to the stable
mode of binding of the SHC PTB domain to RET9.
Gene expression and disease phenotype
We performed a differential display analysis of gene
expression using NIH3T3 cells expressing the
RET-MEN2A or RET-MEN2B mutant proteins [62].
As a result, we identified 130 known genes and 13previously
unidentified sequences whose expression
was affected by each mutant protein. Of 130 known
genes, 29 genes were confirmed to be expressed
differentially by Northern blotting (Table 1). Based
on their expression patterns, they were classified
into four types: (i) 10 genes induced by both RET-
MEN2A and RET-MEN2B mutant proteins (type I),
(ii) six genes induced predominantly by RET-MEN2A
(type II), (iii) five genes induced predominantly by
RET-MEN2B (type III), (iv) eight genes repressed by
RET-MEN2A and RET-MEN2B (type IV). Type I
includes cyclin D1, cofilin, and cathepsin L and Bgenes that are
known to be involved in cell growth,
tumour progression and invasion. In contrast, type
IV includes type I collagen, lysyl oxidase, annexin I,
and tissue inhibitor of matrix metalloproteinase 3
(TIMP3) genes that have been implicated in tumour
suppression. Type II and type III include various
genes with different physiological functions, and it is
unknown whether these gene expressions are really
involved in the tumour development or diseasephenotype.
To see the physiological significance of inducible
genes, we investigated whether these inducible
genes are also induced by GDNF stimulation.
Amongst 21 genes induced by RET-MEN2A and/or
RET-MEN2B, six genes including cyclin D1, cathep-
sin B, cofilin, ring finger protein 11 (RNF11),
integrin-a6, and stanniocalcin 1 (STC1) genes were
also induced in TGW human neuroblastoma cells in
response to GDNF stimulation, although the time
course of the induction was different depending on
the genes [62]. The STC1 gene was highly inducedby both MEN2B
mutant protein and GDNF stimu-
lation. Because STC1 was suggestive of a role in
early skeletal development that is affected in MEN2B
patients, we stained paraffin sections of human
medullary thyroid carcinoma, arising from MEN2A,
MEN2B and sporadic cases. Interestingly, MEN2B
MTC was strongly stained with the STC1 antibody,
whereas MEN2A MTC was weakly stained and
Table 1 Gene expression induced or suppressed by RET-MEN
2A/2B mutant proteins
Type I Type II Type III Type IV
Cyclin D1 EMK2 STC1 Annexin I
RNF11 ITGA6 PheX Annexin IV
Cofilin PKA-RI EIF4G3 COLIA1Foocen TACC3 Neuropsin COLIA2
Cathepsin L PRNPA PLOD2 Lysyl oxidase
Cathepsin B MPT TIMP3
Decorin Pleiotrophin
INFb SDF1a
TB-2 like 1 protein
HSC73
Type I, the genes induced by both RET-MEN2A and MEN2B
mutant proteins.
Type II, the genes induced predominantly by RET-MEN2A mutant
protein.
Type III, the genes induced predominantly by RET-MEN2B
mutant protein.
Type IV, the genes repressed by both RET-MEN2A and MEN2Bmutant
proteins.
RNF11, ring finger protein 11; INFb, interferon b; HSC73,
heat
shock protein 73; EMK2, ELKL motif kinase 2; ITGA6,
integrin-a6;
PKA-RI, protein kinase A regulatory subunit I; TACC3, trans-
forming acidic coiled coil-containing gene family 3; PRNPA,
pri-
on-related protein A; MPT, mitochondrial phosphate
transporter;
STC1, stanniocalcin 1; PheX, phosphate-regulating gene with
homology to endopeptidases on the X chromosome; EIF4G3,
eukaryotic translation initiation factor 4G3; PLOD2,
procollagen-
lysine, 2-oxyglutarate, 5-dioxygenase 2; COLIA1, type I
collagen
a1 chain; COLIA2, type I collagen a2 chain; TIMP3, tissue
inhibitor of metalloproteinases 3; SDF1a, stromal
cell-derived
factor-1a.
2003 Blackwell Publishing Ltd Journal of Internal Medicine 253:
627633
M I N I SY M P O SI U M : C E L L S I G N AL L I N G B Y R E T 6
3 1
-
7/29/2019 Cell Signalling and Gene Expression Mediated by
RET
6/7
sporadic MTC without RET mutation was almost
unstained [62], suggesting a possible role for STC1
in the development of MEN2B phenotype.
Conflict of interest statement
No conflict of interest was declared.
Acknowledgements
We would like to thank members of our laboratory
for helpful scientific advice. We are grateful to
K. Imaizumi and M. Kozuka for their technical
assistance. This work was supported in part by a
grant-in-aid for COE (Center of Excellence) research
from the Ministry of Education, Culture, Science,
Sports and Technology of Japan.
References
1. Takahashi M, Buma Y, Iwamoto T, Inaguma Y, Ikeda H, Hiai
H. Cloning and expression of the ret proto-oncogene enco-
ding a tyrosine kinase with two potential transmembrane
domains. Oncogene 1988; 3: 5718.
2. Takahashi M, Buma Y, Hiai H. Isolation of ret proto-
oncogene cDNA with an amino-terminal signal sequence.
Oncogene 1989; 4: 8056.
3. Schuchardt A, DAgati V, Larsson-Blomberg L, Costantini F,
Pachnis V. Defects in the kidney and enteric nervous system
of mice lacking the tyrosine kinase receptor Ret. Nature
1994; 367: 3803.
4. Airaksinen MS, Titievsky A, Saarma M. GDNF family neu-
rotrophic factor signalling: four masters, one servant? Mol
Cell Neurosci 1999; 13: 3125.
5. Airaksinen MS, Saarma M. The GDNF family: signalling,
biological functions and therapeutic value. Nat Rev Neurosci
2002; 3: 3894.
6. Grieco M, Santoro M, Berlingieri MT et al. PTC is a novel
rearranged form of the ret proto-oncogene and is frequently
detected in vivo in human thyroid papillary carcinomas. Cell
1990; 60: 5563.
7. Mulligan LM, Kwok JBJ, Healey CS et al. Germ-line
mutations
of the RET proto-oncogene in multiple endocrine neoplasia
type 2A. Nature 1993; 363: 45860.
8. Donis-Keller H, Dou SS, Chi D et al. Mutations in the RET
proto-oncogene are associated with MEN 2A and FMTC.
Hum Mol Genet 1993; 2: 856.
9. Hofstra RMW, Landsvater RM, Ceccherini I et al. A
mutation
in the RET proto-oncogene associated with multiple endo-
crine neoplasia type 2B and sporadic medullary thyroid
carcinoma. Nature 1994; 367: 376.
10. Carlson KM, Dou SS, Chi D et al. Single missense mutation
in
the tyrosine kinase catalytic domain of the RET protoonco-
gene is associated with multiple endocrine neoplasia type
2B.
Proc Natl Acad Sci USA 1994; 91: 157983.
11. Jhiang SM. The RET proto-oncogene in human cancers.
Oncogene 2000; 19: 5597.
12. Takahashi M. The GDNF/RET signalling pathway and hu-
man diseases Cytokine Growth Factor Rev 2001; 12: 3673.
13. Eng C. RET proto-oncogene in the development of human
cancer. J Clin Oncol 1999; 17: 38093.
14. Asai N, Iwashita T, Matsuyama M, Takahashi M. Mechanism
of activation of the ret proto-oncogene by multiple
endocrine neoplasia 2A mutations. Mol Cell Biol 1995; 15:
1619.
15. Santoro M, Carlomagno F, Romano A et al. Activation of
RET
as a dominant transforming gene by germline mutations of
MEN2A and MEN2B. Science 1995; 267: 383.
16. Borrello MG, Smith DP, Pasini B et al. RET activation by
germline MEN2A and MEN2B mutations. Oncogene 1995;
11: 24127.
17. Iwashita T, Asai N, Murakami H, Matsuyama M, Takahashi
M. Identification of tyrosine residues that are essential
for
transforming activity of the ret proto-oncogene with MEN2A
or MEN2B mutation. Oncogene 1996; 12: 487.
18. Romeo G, Ronchetto P, Luo Y et al. Point mutations
affecting
the tyrosine kinase domain of the RET proto-oncogene in
Hirschprungs disease. Nature 1994; 367: 378.
19. Edery P, Lyonnet S, Mulligan LM et al. Mutations of the
RET
protooncogene in Hirschsprungs disease. Nature 1994; 367:
3780.
20. Angrist M, Bolk S, Thiel B et al. Mutation analysis of the
RET
receptor tyrosine kinase in Hirschsprung disease. Hum Mol
Genet 1995; 4: 8230.
21. Attie T, Pelet A, Edery P et al. Diversity of RET
proto-onco-
gene mutations in familial and sporadic Hirschsprung
disease. Hum Mol Genet 1995; 4: 1386.
22. Chakravarti A. Endothelin receptor-mediated signalling
in
Hirschsprung disease. Hum Mol Genet 1996; 5: 307.
23. Carlomagno F, De Vita G, Berlingieri MT et al. Molecular
heterogeneity of RET loss of function in Hirschsprungs
disease. EMBO J 1996; 15: 27125.
24. Iwashita T, Murakami H, Asai N, Takahashi M. Mechanism
of Ret dysfunction by Hirschsprung mutations affecting its
extracellular domain. Hum Mol Genet 1996; 5: 15780.
25. Cosma MP, Cardone M, Carlomagno F, Colantuoni V.
Mutations in the extracellular domain cause RET loss of
function by a dominant negative mechanism. Mol Cell Biol
1998; 18: 3329.
26. Ishiguro Y, Iwashita T, Murakami H et al. The role of
amino
acids surrounding tyrosine 1062 in Ret in specific binding
of
the Shc phosphotyrosine-binding domain. Endocrinology
1999; 140: 3998.
27. Geneste O, Bidaud C, De Vita G et al. Two distinct
mutations
of the RET receptor causing Hirschsprungs disease impair
the binding of signalling effecters to a multifunctional
docking site. Hum Mol Genet 1999; 8: 19899.
28. Pasini B, Borrello MG, Greco A et al. Loss of function
effect of
RETmutations causing Hirschsprung disease
Nature Genet
1995; 10: 340.
29. Pelet A, Geneste O, Edery P et al. Various mechanisms
cause
RET-mediated signalling defects in Hirschsprungs disease.
J Clin Invest 1998; 101: 14123.
30. Cosma MP, Panariello L, Quadro L, Dathan NA, Fattoruso
O,
Colantuoni V. A mutation in the RET proto-oncogene in
Hirschsprungs disease affects the tyrosine kinase activity
associated with multiple endocrine neoplasia type 2A and
2B. Biochem J 1996; 314: 397400.
2003 Blackwell Publishing Ltd Journal of Internal Medicine 253:
627633
6 32 K . K UR OK AW A et al.
-
7/29/2019 Cell Signalling and Gene Expression Mediated by
RET
7/7
31. Iwashita T, Kurokawa K, Qiao S et al. Functional analysis
of
RET with Hirschsprung mutations affecting its kinase do-
main. Gastroenterology 2001; 121: 233.
32. Tahira T, Ishizaka Y, Itoh F, Sugimura T, Nagao M. Char-
acterization of ret proto-oncogene mRNAs encoding two
isoforms of the protein product in a human neuroblastoma
cell line. Oncogene 1990; 5: 9102.
33. Myers SM, Eng C, Ponder BAJ, Mulligan LM. Characteriza-
tion of RET proto-oncogene 3-splicing variants and polya-
denylation sites: a novel C-terminus for RET. Oncogene 1995;
11: 20345.
34. Coulpier M, Anders J, Ibanez CF. Coordinated activation
of
autophosphorylation sites in the RET receptor tyrosine kin-
ase importance of tyrosine 1062 for GDNF mediated
neuronal differentiation and survival. J Biol Chem 2002;
277: 1999.
35. Borrello MG, Alberti L, Arighi E et al. The full
oncogenic
activity of Ret/ptc2 depends on tyrosine 539, a docking site
for phospholipase C-c. Mol Cell Biol 1996; 16: 21563.
36. Asai N, Murakami H, Iwashita T, Takahashi M. A mutation
at tyrosine 1062 in MEN2A-Ret and MEN2B-Ret impairs
their transforming activity and association with Shc adaptor
proteins. J Biol Chem 1996; 271: 17649.
37. Arighi E, Alberti L, Torriti F et al. Identification of Shc
docking
site on Ret tyrosine kinase. Oncogene 1997; 14: 7782.
38. Lorenzo MJ, Gish GD, Houghton C et al. RET alternate
spli-
cing influences the interaction of activated RET with the
SH2
and PTB domains of Shc, and the SH2 domain of Grb2.
Oncogene 1997; 14: 7671.
39. Kurokawa K, Iwashita T, Murakami H, Hayashi H, Kawai K,
Takahashi M. Identification of SNT/FRS2 docking site on
RET receptor tyrosine kinase and its role for signal trans-
duction. Oncogene 2001; 20: 19238.
40. Melillo RM, Santoro M, Ong SH et al. Docking protein
FRS2
links the protein tyrosine kinase RET and its oncogenic
forms
with the mitogen-activated protein kinase signalling
cascade.
Mol Cell Biol 2001; 21: 41787.
41. Melillo RM, Carlomagno F, De Vita G et al. The insulin
re-
ceptor substrate (IRS)-1 recruits phosphatidylinositol
3-kin-
ase to Ret: evidence for a competition between Shc and IRS-1
for the binding to Ret. Oncogene 2001; 20: 2018.
42. Murakami H, Yamamura Y, Shimono Y, Kawai K, Kurokawa
K, Takahashi M. Role of Dok1 in cell signalling mediated by
RET tyrosine kinase. J Biol Chem 2002; 277: 327890.
43. Grimm J, Sachs M, Britsch S et al. Novel p62dok family
members, dok-4 and dok-5, are substrates of the c-Ret
receptor tyrosine kinase and mediate neuronal differenti-
ation. J Cell Biol 2001; 154: 3454.
44. Durick K, Gill GN, Taylor SS. Shc and Enigma are both
re-
quired for mitogenic signalling by Ret/ptc2. Mol Cell Biol
1998; 18: 229308.
45. Hayashi H, Ichihara M, Iwashita Tet al.
Characterization ofintracellular signals via tyrosine 1062 in
RET activated by
glial cell line-derived neurotrophic factor. Oncogene 2000;
19: 44675.
46. Hayashi Y, Iwashita T, Murakamai H et al. Activation of
BMK1 via tyrosine 1062 in RET by GDNF and MEN2A
mutation. Biochem Biophys Res Commun 2001; 281: 689.
47. Besset V, Scott RP, Ibanez CF. Signaling complexes and
protein-protein interactions involved in the activation of
the
Ras and phosphatidylinositol 3-kinase pathways by the c-Ret
receptor tyrosine kinase. J Biol Chem 2000; 275: 391566.
48. Murakami H, Iwashita T, Asai N et al. Enhanced phospha-
tidylinositol 3-kinase activity and high phosphorylation
state
of its downstream signalling molecules mediated by Ret with
the MEN 2B mutation. Biochem Biophys Res Commun 1999;
262: 675.
49. Salvatore D, Melillo RM, Monaco C et al. Increased in
vivo
phosphorylation of Ret tyrosine 1062 is a potential patho-
genetic mechanism of multiple endocrine neoplasia type 2B.
Cancer Res 2001; 61: 142631.
50. Forman-Kay JD, Pawson T. Diversity in protein
recognition
by PTB domains. Curr Opin Struct Biol 1999; 9: 695.
51. Xu H, Lee KW, Goldfarb M. Novel recognition motif on fi-
broblast growth factor receptor mediates direct association
and activation of SNT adapter proteins. J Biol Chem 1998;
273: 179890.
52. Ong SH, Guy GR, Hadari YR et al. FRS2 proteins recruit
intracellular signalling pathways by binding to diverse tar-
gets on fibroblast growth factor and nerve growth factor
receptors. Mol Cell Biol 2000; 20: 9789.
53. Dhalluin C, Yan KS, Plotnikova O et al. Structural basis
of
SNT PTB domain interactions with distinct neurotrophic
receptors. Mol Cell 2000; 6: 929.
54. Yan KS, Kuti M, Yan S et al. FRS2 PTB domain
conformation
regulates interactions with divergent neurotrophic
receptors.
J Biol Chem 2002; 277: 170894.
55. Tansey MG, Baloh RH, Milbrandt J, Johnson EM. GFRa-me-
diated localization of RET to lipid rafts is required for
effective
downstream signalling, differentiation, and neuronal survi-
val. Neuron 2000; 25: 6123.
56. Paratcha G, Ledda F, Baars L et al. Released GFRa1
poten-
tiates downstream signalling, neuronal survival, and differ-
entiation via a novel mechanism of recruitment of c-Ret to
lipid rafts. Neuron 2001; 29: 1784.
57. Kouhara H, Hadari YR, Spivak-Kroizman T et al. A lipid-
anchored Grb2-binding protein that links FGF-receptor ac-
tivation to the Ras/MAPK signalling pathway. Cell 1997; 89:
69702.
58. de Graaff E, Srinivas S, Kilkenny C et al. Differential
activities
of the RET tyrosine kinase receptor isoforms during mam-
malian embryogenesis Genes Dev 2001; 15: 24344.
59. Tsui-Pierchala BA, Ahrens RC, Crowder RJ, Milbrandt J,
Johnson EM. The long and short isoforms of Ret function as
independent signalling complexes. J Biol Chem 2002; 277:
346125.
60. Zhou MM, Ravichandran KS, Olejniczak ET et al. Structure
and ligand recognition of the phosphotyrosine binding do-
main of Shc. Nature 1995; 378: 5892.
61. Ohiwa M, Murakami H, Iwashita T et al. Characterization
of
Ret-Shc-Grb2 complex induced by GDNF, MEN 2A, and MEN
2B mutations. Biochem Biophys Res Commun 1997; 237: 74
51.
62. Watanabe T, Ichihara M, Hashimoto Met al.
Characteriza-tion of gene expression induced by RET with MEN2A
or
MEN2B mutation. Am J Pathol 2002; 161: 2456.
Received 12 March 2003; accepted 20 March 2003.
Correspondence: Masahide Takahashi MD, PhD, Department of
Pathology, Nagoya University Graduate School of Medicine,
65 Tsurumai-cho, Showa-ku, Nagoya 466-8550, Japan (fax:
181 52 7442098; e-mail: [email protected]).
2003 Blackwell Publishing Ltd Journal of Internal Medicine 253:
627633
M I N I SY M P O SI U M : C E L L S I G N AL L I N G B Y R E T 6
3 3