Embed Size (px)
Citation preview

CELL CULTURE PROTOCOLS
NAN QINJune, 28, 2012
Division of Clinical NeurochemistryInstitute of Clinical Chemistry and Laboratory Medicine
and the Department of MedicineUniversity of Dresden
Fetscherstr 7401307 Dresden
Germany
1 of 26

Contents
1 PROTOCOLS FOR MPC CELL CULTURE 41.1 Making collagen A flask . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.1.1 Items required . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41.1.2 Procedure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.2 Making up MPC media . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41.2.1 Items required . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41.2.2 Procedure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.3 Feeding MPC cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51.3.1 Procedure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
1.4 Splitting MPC cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51.4.1 Items required . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51.4.2 Procedure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
2 CELL RELEASE ASSAY 62.1 Making up Krebs Buffer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62.2 Making up Krebs Buffer+K . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62.3 Making up Krebs Buffer+K . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62.4 Release . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62.5 HPLC analyst . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
3 TISCHLER LAB PROTOCOL FOR HUMAN PHEOCHROMOCYTOMADISSOCIATION 83.1 General Considerations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83.2 Tumor Dissociation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83.3 Tumor Cell Purification (optional) . . . . . . . . . . . . . . . . . . . . . . . . . . 93.4 Lysis Buffer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93.5 Trypsinization (optional but often desirable) and Long-term Culture . . . . . . . 93.6 Freezing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
4 HUMAN PHEOCHROMOCYTOMA PRIMAY CELL PHENOTYP CON-TROL 11
5 CHROMOSPHERE ISOLATION 125.1 Primary culture . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125.2 Differential Plating . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
6 RNA ISOLATION 136.1 Sample preparation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 136.2 RNA isolation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
7 shRNA TRANSFECTION 147.1 Backgrund . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 147.2 Protocoll . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
8 siRNA Transfection Protocol/Trian 168.1 Transfection of MPC with small interfering RNAs (siRNA) . . . . . . . . . . . . 16
2 of 26

9 Thermo Scientific DharmaFECT Transfection Reagents siRNA TransfectionProtocol 179.1 Cell Plating . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 179.2 Transfection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
10 Western Blotting 1910.1 Selection Guide: Secondary Antibodies . . . . . . . . . . . . . . . . . . . . . . . . 19
10.1.1 Polyclonal anti-Mouse and anti-Rabbit Secondary Antibodies . . . . . . . 1910.1.2 Biotin-Binding Proteins . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
10.2 Nuclear Extraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1910.2.1 Nuclear Extraction G Biosciences . . . . . . . . . . . . . . . . . . . . . . . 19
10.3 Western Blot Protocol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2010.3.1 Stock solution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2010.3.2 Lower Gel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2210.3.3 Upper Gel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
10.4 Protocol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
11 mirvana PARIS Kit 2511.1 Protein Extraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2511.2 RNA Extraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
3 of 26

1 PROTOCOLS FOR MPC CELL CULTURE
1.1 Making collagen A flask
1.1.1 Items required
• Gibco, DPBS # 14190
• Biochrom, Collagen A # L7220
1.1.2 Procedure
• make 4% Collagen A solution in DPBS (fresh)
• put corresponding amount of collagen A solution into incubation flask.
• distribute collagen A solution over bottom of flask
• after 2 hours, remove collagen A solution and wash flask 2 times with DPBS
1.2 Making up MPC media
1.2.1 Items required
• Gibco, Horse Serum # 26050
• Gibco, FBS # 10108165
• Gibco, DPBS # 14190
• Gibco, RPMI 1640 # 52400
• Promo Cell, TNS= Trypsin inhibitor # C-41120
• Promo Cell, Trypsin/EDTA # C-41020
• Antibiotics Aliquot
1.2.2 Procedure
• Clean hood with ethanol and bleach.
• making Madium
– RPMI 1640 contant: 10% Horse Serum, 5% FBS and 0.1% antibiotics
• Remove 75ml of RPMI 1640 from the 500mL bottle (this number represents the totalvolume of constituents to be added to the bottle), and discard into a waste container
• To the RPMI 1640 add the following:
– 50 ml donor horse serum
– 25ml of fetal calf serum
– 0.5mL antibiotics
• Mix the contents by rocking the bottle up and down gently
• Store media in 4◦ C fridge, and monitor for color changes and growths.
4 of 26

1.3 Feeding MPC cells
1.3.1 Procedure
• Clean hood with ethanol and bleach.
• Remove cells from incubator, and monitor media for variables such as color and turbidity
• Remove media from flask and discard
• Add new media to flask and return flask to incubator
• Store media in 4◦ C fridge, and monitor for color changes and growths.
1.4 Splitting MPC cells
1.4.1 Items required
• 70% ethanol & bleach
• Clean 50ml falcon tubes
• Serological pipettes
• Trypsin-EDTA
• DPBS
1.4.2 Procedure
• Clean hood with ethanol and bleach.
• Remove cells from incubator, and monitor media for variables such as color and turbidity
• Remove media from flask and discard
• Wash cell monolayer with 10ml of DPBS (DPBS should be specifically for cell culture useonly). Rotate PBS over monolayer gently (dont want to remove any of the cells).
• Put 4ml of Trypsin-EDTA into flask. Place in incubator for 5 minutes. After this time,the cells should be sufficiently removed from the flask. if not, repeat pipetting by using10 ml pipette.
• Add 4ml TNS
• Replace the contents of flask into the 15ml falcon tube.
• Spin tube for 5 minutes 480g at room temperature.
• Remove tube from centrifuge, and return to hood. Discard supernatant (into waste con-tainer).
• Resuspend cells and split into new flasks. Usually split about 1:4
5 of 26

2 CELL RELEASE ASSAY
2.1 Making up Krebs Buffer
Krebs Buffer:
• 0.126 M NaCl; 7.36 g/L NaCl
• 2.5 mM KCl; 0.186 g/L KCl
• 25 mM NaHCO3; 2.1 g/L NaHCO3
• 1.2 mM NaH2PO4; 0.166 g/L NaH2PO4.H2O
• 1.2 mM MgCl2; 0.244 g/L MgCl2.6H2O
• 25 mM CaCl2; 0.368 g/L CaCl2.2H2O
2.2 Making up Krebs Buffer+K
Krebs Buffer+K:
• 25 mM NaCl; 1.461 g/L NaCl
• 100 mM KCl; 7.455 g/L KCl
• 25 mM NaHCO3; 2.1 g/L NaHCO3
• 1.2 mM NaH2PO4; 0.166 g/L NaH2PO4.H2O
• 1.2 mM MgCl2; 0.244 g/L MgCl2.6H2O
• 25 mM CaCl2; 0.368 g/L CaCl2.2H2O
2.3 Making up Krebs Buffer+K
Krebs Buffer+100µM Nicotine: 1.59 µl Micotine +100 ml Krebs Buffer
2.4 Release
• Cells cultured in 24 well plate
• After 48 hours culture calculation the cell numbers
• Remove media from wells and discard
• Add release buffer 500µL/well
• Return flask to incubator, 10 min at 37◦C.
• Remove release buffer to label tubes
• Add 500µL Perchloride solution into control well
• Remove cell perchloride solution into label tubes
• Release samples are ready for HPLC assay
6 of 26

2.5 HPLC analyst
• Buffer 1: 20 dilution
• 300 µl extraction
• Perchloride Solution 1: 40 dilution
• 90 µl injection
PHEO cell
• Buffer (without dilution) 50 µl extraction
• Perchloride Solution 1: 10 dilution
• 90 µl injection
7 of 26

3 TISCHLER LAB PROTOCOL FOR HUMAN PHEOCHRO-MOCYTOMA DISSOCIATION
3.1 General Considerations
• Individual tumors have distinct characteristics. Try to cover all bases
• Different lots of collagenase can dramatically affect yield. It may be necessary to testseveral lots and stock up on one that works well.
• A modest amount of starting tissue (no more than 2 cm cube) will often give a betteryield than a huge amount.
• Sedimentation at unit gravity is an easy and efficient method for separating dissociatedtumor cells from blood and debris (see #1 under Tumor Dissociation below).
• Plan to freeze dissociated cells right away. Don’t expect to ”grow them up” first becausethey probably won’t grow. Replicate vials of frozen cells can be used to repeat experiments!Similarly, if you plan to inject tumor into mice do it right away. Small minced tissuefragments can be injected sc or ip with a 16 gauge or larger needle.
• To determine whether pheo cells are truly growing, perform double staining for TH andincorporation of BrdU at 2 wks. This is usually the point at which you can stop wastingyour time.
3.2 Tumor Dissociation
• Mince tumor tissue into 2-3mm chunks in a 100 mm dish containing 10 ml calcium- andmagnesium-free Hank’s (CMF HBSS; Gibco cat # 12444-014) in a laminar flow hood. Use2 scalpels, one in each hand, and be sure to slice, not crush the tissue. OCCASIONALLY,this by itself will dislodge a sizable number of tumor cells. Therefore, collect the CMFHBSS (if the chunks they are very bloody rinse in CMF HBSS and collect the rinse topool with the CMF HBSS that was used for mincing), spin ( 250g × 5 min), aspirate supand re-suspend pellet in culture medium with 15% FBS. Fill a 15 ml conical tube withthe suspension, close it and leave it in a vertical position at room temperature for at least1/2 hr. A gray-tan sediment containing the tumor cells will accumulate at the bottom ofthe tube while RBCs and debris remain suspended. Aspirate everything above the cloudof sediment after it looks like the sediment is all down. Examine a drop of the sedimentmicroscopically to decide whether anything from this step is worth keeping.
• Rock the minced tissue pieces in a 15 ml conical tube containing collagenase B solution(1.5 mg/ml, Boehringer Manheim, cat # 1088 823, 0.22 µm filter-sterilized) in HBSS (notCMF) for 45 min at 37◦C. Volume of enzyme soln should be at least 10× volume of tissuepieces. DNAase is often not necessary during collagenase digestion. If a DNA gel beginsto form add DNAse 1 (to final concentration 50 ug/ml, from a 20× frozen stock soln).
• Stand the tube vertically, let the pieces settle out, aspirate enzyme soln. Fill tube gentlywith CMF HBSS, invert, let pieces settle out again and aspirate again. Add 35 ml of CMFHBSS to the tube and this time triturate the tissue pieces in CMF HBSS. Trituration canbe performed first in a 1ml plastic pipette from which the tip has been broken off understerile conditions (snap it off while the pipette is still in its disposable sleeve), then in anintact plastic pipette and then in 9” Pasteur pipettes. Don’t brutalize the cells or tissue.
8 of 26

After a couple of minutes of trituration, all cells that are going to dislodge will have doneso (Chunks that have not completely dissociated can either be discarded or subjected toan additional round of digestion in fresh collagenase). Collect the cell suspension, add afew ml of culture medium with 15% FBS (even a small amount of serum will protect cellsduring centrifugation). Spin, (discard sup), re-suspend pellet in complete medium andsediment the tumor cells as in #1 above. At this point, the cells will be mostly in smallspherical clusters.
3.3 Tumor Cell Purification (optional)
Pheo cells can often be highly enriched by differential attachment or detachment but it is notpredictable whether or not this will work with any particular tumor. Pheo cells dissociated incollagenase or mechanically dislodged during initial mincing often attach very poorly to collagen.However, they attach well after subsequent trypsinization.
• Plate cells in complete medium (RPMI 1640 with L-glutamine, Gibco cat# 11875-093;15% fetal bovine serum, Gibco cat# 10437-028; 100 units/ml penicillin, 100 µg/ml strepto-mycin, Gibco cat# 15140-122 ) in a collagen coated dish (we recommend collagen preparedaccording to Tischler lab collagen protocol- All collagen preps are not equivalent!). Alsotry dish with no collagen. Place dishes in the tissue cultur incubator at 37◦C and examineperiodically. It may become apparent in as little as 1 hour that fibroblast-like cells haveselectively attached. If so, gently pipette off and save the tumor cells. If this rapid effectis not apparent, culture the cells for 2-3 days, then gently spritz the culture medium witha 9” Pasteur pipette to selectively dislodge and collect loosely attached tumor cells.
3.4 Lysis Buffer
add 8.26 g NH4Cl, 1g KHCO3 (or NaHCO3). 0.037g EDTA in 1L water (adjust pH 7,35). afterstanding 5 minutes at RT. The cell suspension was centrifuged with 200 g for 5 minutes at RT.
3.5 Trypsinization (optional but often desirable) and Long-term Culture
Mechanically dislodged cells, cells freshly dissociated in collagenase or cells purified and collectedas above may eventually attach in the absence or presence of collagen. However, trypsinizationserves to break up cell clusters and improve attachment.
• Centrifuge the cells and rinse/spin ×2 in CMF HBSS. Re-suspend pellet in a 15 ml conicaltube in 5 ml trypsin (0.25%, in CMF HBSS without EDTA). Add DNAse 1 (to finalconcentration 50 µg/ml from a 20x frozen stock soln).
• Rock tube for 45 minutes at 37◦C.
• Add 2 ml of complete culture medium (RPMI 1640 with L-glutamine, Gibco cat# 11875-093, 15% fetal bovine serum, Gibco cat# 10437-028, 100 units/ml penicillin, 100 µg/mlstreptomycin, Gibco cat# 15140-122). Centrifuge (500 × g, 5 minutes at room tem-perature). Aspirate sup, re-suspend pelleted cells in 2ml complete medium. Trituratethe cells using a flame constricted (about 50% of original diameter) glass 9 inch pasteurpipette (pre-wetted with complete medium to prevent sticking), dilute as required andplate. Most pheos will attach to collagen after trypsinization. Also, use of McCoy’s 5Amedium instead of RPMI promotes attachment, flattening and neurite outgrowth.
9 of 26

3.6 Freezing
Until recently we have frozen cells in complete culture medium with 15% serum and 10%DMSO. This year our laboratory started using pre-prepared freezing medium (Gibco cat.#11101-011), which so far seems to be giving better results. Freezing should be done in somesort of controlled-rate freezing device and the vials should be kept in liquid N2 for long-termstorage.
10 of 26

4 HUMAN PHEOCHROMOCYTOMA PRIMAY CELL PHE-NOTYP CONTROL
• Day 1: Freeze a small piece fresh tissue for analyzing catechols content.
• Day 2: analyze the primary cell catechols content.
– Take small amount of cell suspension (500 µL).
– Account the cell number from the cell suspension.
– Centrifuge the cell suspension with 200 g for 5 minutes at RT.
– Remove the supernatant. Resuspense the cell pellet with 300 µL perchloride solution.
– Storage the cell pellet perchloride solutions in -80◦C fro HPLC analyze.
• This step will be repeated every 6 days.
11 of 26

5 CHROMOSPHERE ISOLATION
5.1 Primary culture
• Briefly immerse adrenal glands with 70% ethanol, and transfer to PBS.
• Remove the fat tissue on the glands. Perfuse the gland with PBS through vein until mostof blood is flushed out.
• Inject digestion solution (Collagenase 0.3%, 0.01% DNase in PBS) in to gland. Incubatethe tissue under 37◦C for 15 minutes. Repeat this step for 2 more times (45 minutes intatol).
• cut the tissue, remove the medulla. If the tissue is not digested well, roughly cut withscalpel. collect all medulla tissue in PBS. Repeat pipteeing by using 25 ml pipette, and10 ml pipette until the tissue is homogenized.
• Filter the homogenized tissue with sieve that the undissociated tissue will be removed.Further filter it through 100 µm sieve in tunnel and cell strainer.
• Put the filtrate into 50 ml tubes, add PBS until around 45-50 ml. Centrifuge with 200 gfor 8 minutes. Remove the supernatant. Resuspense the pellet with PBS wash again.
• Pool all the pellet together and resuspense in 20-50 ml medium. Filter again with 100 µmcell strainer if aggregate forms.
• Culture the cells in 10% steroid-free FBS in DMEM/F12 (Gibco 31330) and 1% Pen Strep(Gibco 15140).
5.2 Differential Plating
• After overnight culturing, the cells suspense in the medium are removed and transferredto a new flask. Further culture the cells for 1.5 hr. Repeat this step for 2 more times.
• Collect the cells by centrifuging under 200 g for 8 minutes. Count the cell number. Seedthe cells in 7000 cells/ml with 10% steroid-free FBS in DMEM/F12 in low-attachmentplate. Culture the cells for 2 weeks until chromospheres are ready for further experiment.
12 of 26

6 RNA ISOLATION
1ml Tri-reagen is for 20-100 mg tissue, 5-10×106 cell or 10 cm2 cell.
6.1 Sample preparation
• Step 1.
– Tissue: immediately after removal from the animal, the tissue was minced on ice andhomogenized (at RT) wiht 1 ml of tri-reagen.
– Cell: direct add tri-reagen into the incubation’ s well. Repeat pipetting several times.This solution can be stored 1 month at -70◦C.
• Step 2.
– samples with tri-reagen keep in RT for 5 min.
– add 0.2 ml chloroform in 1 ml tri-reagen, the final suspension was shaken vigorouslyfor 15 s and deep RT for 2-15 minutes.
– samples were centrifuged at 12,000 g for 15 minutes at 4◦C.after centrifugation, RNA was present in the aqueous phase whereas DNA and pro-teins were present in the interphase and phenol phase.
∗ red organist phase: protein
∗ interphase: DNA
∗ colorless: RNA
6.2 RNA isolation
• the aqueous phase was transferred to a fresh tube, mixed with 0.5 ml of isopropanol (per1 ml tri-reagen), and then placed at RT for 5-10 minutes.Samples were centrifuged at 12000 g for 10 minutes at at 4◦C.
• the RNA pellet was resuspended in 1 ml 75% ethanol.This solution can be stored 1 week at 4◦C and 1 year at -20◦C.
• RNA pellet vacuum dried for 5-10 min.
• RNA disolve in DEPC-water at 55-60◦C for 10 minutes.
13 of 26

7 shRNA TRANSFECTION
Product:
Santa Cruz
7.1 Backgrund
RNA interference (RNAi) is one of the most exciting discoveries of the past decade in functionalgenomics and proteomics. While first recognized in nematodes as a response to exogenouslyintroduced long double-stranded RNA (dsRNA), it is now clear that RNAi is utilized by mosteukaryotes in vivo for anti-viral defense, transposon activity modulation and gene regulation,and has rapidly become an important research tool for gene silencing. Specifically, RNAi is thepathway by which short interfering RNA (siRNA) or short hairpin RNA (shRNA) are used tosilence the expression of target genes. Compared to siRNA, shRNA offers advantages in silenc-ing longevity and delivery. Upon introduction, the shRNA plasmid DNA enters the cell whereshRNA is transcribed. The shRNA is then cleaved by an RNase III-like enzyme called Dicer intosmall interfering RNA (siRNA), which are short RNA duplexes of 19-21 nucleotides with twonucleotide 3’ overhangs on each strand. The siRNAs are then assembled into endoribonuclease-containing complexes known as RNA-induced silencing complexes (RISCs), unwinding in theprocess. Activated RISCs subsequently bind to complementary transcripts by base pairing in-teractions between the siRNA anti-sense strand and complementary mRNA. The bound mRNAis cleaved and sequence specific degradation of mRNA results in gene silencing. In mammaliancells, introduction of long dsRNA (more than 30 nucleotides) initiates a potent anti-viral re-sponse, exemplified by nonspecific inhibition of protein synthesis and RNA degradation. Themammalian anti-viral response can be bypassed, however, by the introduction of siRNAs orshRNA plasmid DNA.
7.2 Protocoll
• In a six well tissue culture plate, grow cells to a 50-70% confluency in antibiotic-free nor-mal growth medium supplemented with FBS.NOTE: This protocol is recommended for a single well from a 6 well tissue culture plate.Adjust cell and reagent amounts proportionately for wells or dishes of different sizes.NOTE: Healthy and subconfluent cells are required for successful transfection experi-ments. It is recommended to ensure cell viability one day prior to transfection
• Prepare the following solutions:NOTE: The optimal shRNA Plasmid DNA:shRNA Plasmid Transfection Reagent ratioshould be determined experimentally beginning with 1 µg of shRNA Plasmid DNA andbetween 1.0 and 6.0 µl of shRNA Plasmid Transfection Reagent as outlined below. Oncethe optimal shRNA Plasmid DNA:shRNA Plasmid Transfection Reagent ratio has beenidentified for a given cell type, the appropriate amount of shRNA Plasmid DNA/shRNAPlasmid Transfection Reagent complex used per well should be tested to determine whichamount provides the highest level of transfection efficiency. For example, if the opti-mal shRNA Plasmid DNA:shRNA Plasmid Transfection Reagent ratio is 1 µg:1 µl, thenamounts ranging from 0.5 µg/0.5 µl to 2.0 µg/2.0 µl should be tested.
– Solution A: For each transfection, dilute 10 µl of resuspended shRNA Plasmid DNA(i.e. 1 µg shRNA Plasmid DNA) into 90 µl shRNA Plasmid Transfection Medium:sc-108062.
14 of 26

– Solution B: For each transfection, dilute 1 - 6 µl of shRNA Plasmid TransfectionReagent: sc-108061 with enough shRNA Plasmid Transfection Medium: sc-108062to bring final volume to 100 µl. NOTE: Do not add antibiotics to the shRNAPlasmid Transfection Medium: sc-108062.NOTE: Optimal results may be achieved by using siliconized microcentrifuge tubes.
• Add the shRNA Plasmid DNA solution (Solution A) directly to the dilute shRNA PlasmidTransfection Reagent (Solution B) using a pipette. Mix gently by pipetting the solutionup and down and incubate the mixture 15-45 minutes at room temperature.
• Wash the cells twice with 2 ml of shRNA Transfection Medium: sc-108062. Aspirate themedium and proceed immediately to the next step.NOTE: Do not use PBS as the residual phosphate may compete with DNA and bindthe shRNA Plasmid Transfection Reagent, thereby reducing the transfection efficiency.
• For each transfection, add 0.8 ml shRNA Plasmid Transfection Medium to well.
• Add the 200 µl shRNA Plasmid DNA/shRNA Plasmid Transfection Reagent Complex(Solution A + Solution B) dropwise to well, covering the entire layer.
• Gently mix by swirling the plate to ensure that the entire cell layer is immersed in solution.
• Incubate the cells 5-7 hours at 37◦ C in a CO2 incubator or under conditions normallyused to culture the cells. Longer transfection times may be desirable depending on thecell line.
• Following incubation, add 1 ml of normal growth medium containing 2 times the normalserum and antibiotics concentration (2× normal growth medium).
• Incubate the cells for an additional 18-24 hours under conditions normally used to culturethe cells.OPTIONAL: For transient transfection, aspirate media and replace with fresh 1x normalgrowth medium. Assay the cells using the appropriate protocol 24-72 hours after theaddition of fresh medium in the previous step.For selection of stably transfected cells, proceed with puromycin selection as follows:NOTE: The working puromycin concentration for mammalian cell lines ranges from 1-10µg/ml. Prior to using the puromycin antibiotic (sc-108071), titrate the selection agentto determine the optimal concentration for target cell line. Use the lowest concentrationthat kills 100% of non-transfected cells in 3-5 days from the start of puromycin selection.48 hours post-transfection, aspirate the medium and replace with fresh medium containingpuromycin at the appropriate concentration.Approximately every 2-3 days, aspirate and replace with freshly prepared selective media.NOTE: Controls should always be included in shRNA experiments. Control shRNAsare available as 20 µg of plasmid DNA. Each encode a scrambled shRNA sequence thatwill not lead to the specific degradation of any known cellular mRNA. Control shRNAPlasmids include: sc-108060, sc-108065 and sc-108066 (see below).NOTE: For Western blot analysis prepare cell lysate as follows: Wash cells once with PBS.Lyse cells in 300 µl 1× Electrophoresis Sample Buffer (sc-24945) by gently rocking the 6well plate or by pipetting up and down. Sonicate the lysate on ice if necessary. NOTE:For RT-PCR analysis isolate RNA using the method described by P. Chomczynski andN. Sacchi (1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162: 156-159) or a commercially availableRNA isolation kit.
15 of 26

8 siRNA Transfection Protocol/Trian
8.1 Transfection of MPC with small interfering RNAs (siRNA)
For transfections two chemically synthesized duplex siRNAs directed against maus Hif-2 alpha,and two respective control siRNAs (ON-TARGETplus Non-Targeting Pool, ON-TARGETplusCyclophilin B Control Pool). MPC cells were transfected with siRNA according to Invitrogenprotocol. Cells were grown on 100 mm plates until they reached 80% of confluence. Single siR-NAs or combinations of two different siRNA were transfected at a final concentration of 100nMusing Oligofectamine reagent (Invitrogen, Carlsbad, CA) in OptiMEM medium (Invitrogen).For the transfection of one 100 mm plate 32 µl of siRNA (20 µM) duplexes were diluted in 1ml of OptiMEM medium and 48 µl of Oligofectamine reagent in 102 µl of OptiMEM medium,mixed gently and incubated for 5 min at room temperature. Then Oligofectamine solution wasadded to siRNA and mixed, followed by incubation for 15 min at room temperature. Duringthis time normal growth medium was substituted for by OptiMEM medium. Transfection com-plexes were then added slowly to the cells. Transfection medium was incubated with cells for4-6 h and then changed again to normal growth medium. After 24 h, the cells were passagedfor different functional assays, which were performed 72-96h after transfection.
16 of 26

9 Thermo Scientific DharmaFECT Transfection Reagents siRNATransfection Protocol
The following is a general protocol for use of Thermo Scientific DharmaFECT transfectionreagents to deliver siRNA into cultured mammalian cells. The examples given within the pro-tocol are for 96-well plates, and Table 1 provides the transfection reagent volumes for additionalplate types. Table 2 presents recommended DharmaFECT formulations and conditions for themost efficient delivery of siRNA and subsequent silencing (assessed with GAPDH or PPIBsiRNA) in cell lines for which we have performed transfection optimization (96- well format).These results are intended to serve as guidelines for carrying out your own experiments. Success-ful transfection requires careful optimization of conditions. For optimization recommendationssee the Transfection Optimization section on page 2. Below are steps for beginning a transfec-tion experiment with known optimal conditions.Each experiment should include the following samples in triplicate:
• Untreated cells
• Positive control siRNA (targeting an endogenous or reporter gene)
• Negative control siRNA (non-targeting)
• The desired test siRNA
9.1 Cell Plating
Optimal cell densities will vary with growth characteristics that are unique to each cell type andneed to be determined empirically. See Table 2 for cell line specific cell density recommendationsin 96-well format. For larger plate formats you can vary the number of cells plated in proportionto the difference in well surface area.
• Trypsinize and count cells
• Dilute cells in antibiotic-free complete medium to achieve the appropriate plating densityin 100 µL of solution. (Complete medium is medium that the cells are maintained in, andmay contain serum)
• Plate 100 µL of cells into each well of a 96-well plate
• Incubate cells at 37C with 5% CO2 overnight
9.2 Transfection
The following steps will be used for positive control, negative control and test siRNAs. Werecommend using 5-50 nM final siRNA concentrations. The volumes in this protocol are givenfor a single well in a 96-well plate format for 25 nM final siRNA concentration. To transfecttriplicate wells and to account for loss during pipetting, multiply the volumes by 3.5.
• Prepare 5 µM siRNA solution in 1× siRNA buffer or another appropriate RNase-freesolution from your stock solution. (See Basic siRNA Resuspension Protocol for moredetails)
• In separate tubes, dilute the siRNA 1 and the appropriate DharmaFECT transfectionreagent (Tube 2) with serum-free medium.
17 of 26
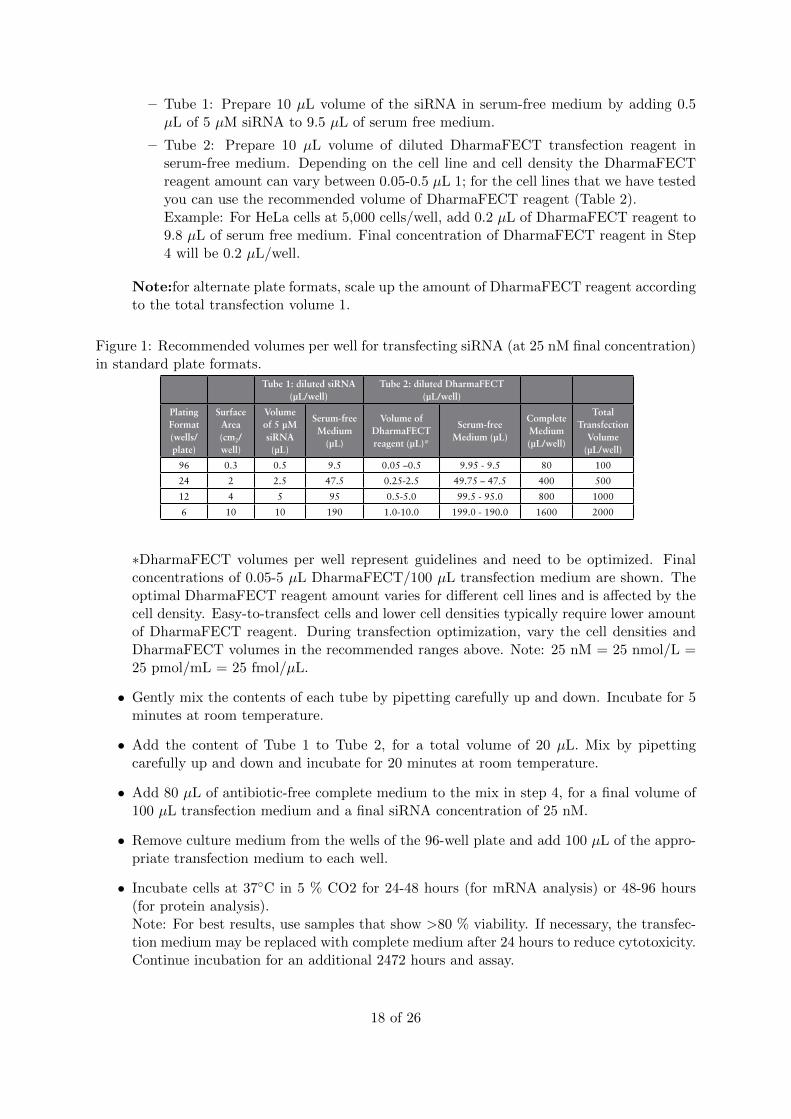
– Tube 1: Prepare 10 µL volume of the siRNA in serum-free medium by adding 0.5µL of 5 µM siRNA to 9.5 µL of serum free medium.
– Tube 2: Prepare 10 µL volume of diluted DharmaFECT transfection reagent inserum-free medium. Depending on the cell line and cell density the DharmaFECTreagent amount can vary between 0.05-0.5 µL 1; for the cell lines that we have testedyou can use the recommended volume of DharmaFECT reagent (Table 2).Example: For HeLa cells at 5,000 cells/well, add 0.2 µL of DharmaFECT reagent to9.8 µL of serum free medium. Final concentration of DharmaFECT reagent in Step4 will be 0.2 µL/well.
Note:for alternate plate formats, scale up the amount of DharmaFECT reagent accordingto the total transfection volume 1.
Figure 1: Recommended volumes per well for transfecting siRNA (at 25 nM final concentration)in standard plate formats.
Tube 1: diluted siRNA (μL/well)
Tube 2: diluted DharmaFECT (μL/well)
Plating Format (wells/plate)
Surface Area (cm2/well)
Volume of 5 μM siRNA (μL)
Serum-free Medium
(μL)
Volume of DharmaFECT reagent (μL)*
Serum-free Medium (μL)
Complete Medium (μL/well)
Total Transfection
Volume (μL/well)
96 0.3 0.5 9.5 0.05 –0.5 9.95 - 9.5 80 100
24 2 2.5 47.5 0.25-2.5 49.75 – 47.5 400 500
12 4 5 95 0.5-5.0 99.5 - 95.0 800 1000
6 10 10 190 1.0-10.0 199.0 - 190.0 1600 2000
Note: for alternate plate formats, scale up the amount of DharmaFECT reagent according to the total transfection volume (Table 1).
3. Gently mix the contents of each tube by pipetting carefully up and down. Incubate for 5 minutes at room temperature.
4. Add the content of Tube 1 to Tube 2, for a total volume of 20 μL. Mix by pipetting carefully up and down and incubate for 20 minutes at room temperature.
5. Add 80 μL of antibiotic-free complete medium to the mix in step 4, for a "nal volume of 100 μL transfection medium and a "nal siRNA concentration of 25 nM.
6. Remove culture medium from the wells of the 96-well plate and add 100 μL of the appropriate transfection medium to each well.
7. Incubate cells at 37°C in 5 % CO2 for 24–48 hours (for mRNA analysis) or 48–96 hours (for protein analysis).Note: For best results, use samples that show >80 % viability. If necessary, the transfection medium may be replaced with complete medium after 24 hours to reduce cytotoxicity. Continue incubation for an additional 24–72 hours and assay.
Transfection optimizationTo obtain the highest transfection ef"ciency with minimal effects on cell viability we recommend the following guidelines in optimizing transfection conditions for each cell line:
The optimization experiment should include at least three cell densities and four DharmaFECT transfection reagent volumes within the range recommended in Table 1. When selecting cell densities to assess, consider the assay and time-point requirements: lower cell densities for long term assays and higher cell numbers for short-term experiments.Use positive and negative control siRNAs at 25 nM "nal concentration as well as untreated cells to "nd conditions that show target mRNA knockdown of > 80 % with the positive control siRNA and > 80 % cell viability. Use these optimal conditions for your subsequent experiments with test siRNAs. Since the siRNA amount for optimal silencing can vary due to intrinsic properties of the target gene, we recommend performing a dose curve transfection with your test siRNA (using a range from 5 to 50 nM) to "nd the optimal siRNA concentration for your test siRNA.
Note: For high throughput siRNA screening in easy-to-transfect cells, we recommend using a wet reverse transfection protocol (See Wet reverse transfection protocol). The DharmaFECT volumes and siRNA amounts for reverse transfection are usually lower than the amounts needed for traditional transfection. Therefore, a transfection optimization should be performed for the protocol that is going to be used for subsequent experiments.
Table 1. Recommended volumes per well for transfecting siRNA (at 25 nM !nal concentration) in standard plate formats.
*DharmaFECT volumes per well represent guidelines and need to be optimized. Final concentrations of 0.05– 5 μL DharmaFECT/100 μL transfection medium are shown. The optimal DharmaFECT reagent amount varies for different cell lines and is affected by the cell density. Easy-to-transfect cells and lower cell densities typically require lower amount of DharmaFECT reagent. During transfection optimization, vary the cell densities and DharmaFECT volumes in the recommended ranges above. Note: 25 nM = 25 nmol/L = 25 pmol/mL = 25 fmol/μL
∗DharmaFECT volumes per well represent guidelines and need to be optimized. Finalconcentrations of 0.05-5 µL DharmaFECT/100 µL transfection medium are shown. Theoptimal DharmaFECT reagent amount varies for different cell lines and is affected by thecell density. Easy-to-transfect cells and lower cell densities typically require lower amountof DharmaFECT reagent. During transfection optimization, vary the cell densities andDharmaFECT volumes in the recommended ranges above. Note: 25 nM = 25 nmol/L =25 pmol/mL = 25 fmol/µL.
• Gently mix the contents of each tube by pipetting carefully up and down. Incubate for 5minutes at room temperature.
• Add the content of Tube 1 to Tube 2, for a total volume of 20 µL. Mix by pipettingcarefully up and down and incubate for 20 minutes at room temperature.
• Add 80 µL of antibiotic-free complete medium to the mix in step 4, for a final volume of100 µL transfection medium and a final siRNA concentration of 25 nM.
• Remove culture medium from the wells of the 96-well plate and add 100 µL of the appro-priate transfection medium to each well.
• Incubate cells at 37◦C in 5 % CO2 for 24-48 hours (for mRNA analysis) or 48-96 hours(for protein analysis).Note: For best results, use samples that show >80 % viability. If necessary, the transfec-tion medium may be replaced with complete medium after 24 hours to reduce cytotoxicity.Continue incubation for an additional 2472 hours and assay.
18 of 26

10 Western Blotting
10.1 Selection Guide: Secondary Antibodies
10.1.1 Polyclonal anti-Mouse and anti-Rabbit Secondary Antibodies
Most primary antibodies are produced in mouse or rabbit host species; therefore, anti-mouseIgG and anti-rabbit IgG are the most popular classes of secondary antibodies. Goat is the hostspecies most easily and frequently used by manufacturers to produce polyclonal anti-mouseand anti-rabbit secondary antibodies. Consequently, goat secondary antibodies against mouseIgG and rabbit IgG are commercially available in the widest variety of forms. Several kindsof anti-mouse and anti-rabbit secondary antibodies from other host species are also available.See the table below for help locating secondary antibodies with specificity for mouse or rabbitantibodies and their conjugation options.
10.1.2 Biotin-Binding Proteins
For applications in which the primary antibody is labeled with a biotin tag, a biotin bindingprotein makes a suitable detection reagent. Both avidin and streptavidin bind very stronglyto biotin and allow a single detection reagent to be used with multiple primary antibodies,regardless of host.
10.2 Nuclear Extraction
10.2.1 Nuclear Extraction G Biosciences
• Add 500µl of ice cold SubCell Buffer-I. Gently vortex to suspend the cells and incubateon ice for 10 minutes.
• Homogenize the cells on ice using tight pestle.
• Add 250µl 3× SubCell Buffer-II and mix by inverting.
• Centrifuge the tube at 700 × g for 10 minutes to pellet the nuclei. Transfer the supernatantto a new tube (cytoplasmic fraction).
• The nuclear pellet can be washed with 500 µl 1 × SubCell Buffer-II (1parts 3 × SubCellBuffer-II + 2 parts SubCell Buffer-I) and centrifuge again as above.
• Add 1M DTT into Nuclear Protein Extraction Buffer-I & Buffer-II to final concentrationof 1mM DTT.
• Suspended the nuclei pellet in 100 µl ice cold Nuclear Protein Extraction Buffer-I.
• Vortex the suspension for 5 seconds.
• Add 100 µl of ice cold Nuclear Protein Extraction Buffer-II and immediately mix thecontent of the tube by inversion.
• Incubate the suspension on ice for 60 minutes, mixing the content every 10 minutes.
• Centrifuge the tube at 22,500 × g for 30 minutes.
• transfer the clear supernatant to a clean tube.
• store the nuclear extract at -70◦C until use.
19 of 26

10.3 Western Blot Protocol
10.3.1 Stock solution
• resolving gel buffer: 1.5 M Tris-HCl (pH 8.8)
Weight Material18.15 g Tris base
Dissolve in deionized water 60 ml adjust pH with 6 N HCl of 8.8 and fill to 100 mL. Storeat 4◦C.
• stacking gel buffer: 0.5 M Tris-HCl, pH: 6.8
Weight Material6 g Tris base
Dissolve in deionized water 60 ml adjust pH with 6 N HCl of 6.8 and fill to 100 mL. Storeat 4◦C.
• 10% (w/v) SDS
Weight Material10 g SDS90 ml water
Dissolve SDS in deionized water and fill to 100 ml.
• 0.5% (w/v) bromophenel blue
Weight Material0.5 g bromophenol blue90 ml water
Dissolve bromophenol blue in deionized water and fill to 100 ml.
• 10× running buffer: 0.25 M tris, 1 M glycin, 1% SDS, pH:8.3
Weight Material30 g Tris Base144 g glycin10 g SDS
Dissolve in deionized water and fill to 1000 ml. Store at 4◦C. Warm the buffer before useto remove possible precipitates. Do not pH.
• 2×Sample Buffer
Weight Material3.55 ml Demonized water1.25 ml 0.5 M Tris-HCl, pH 6.82.5 ml Glycerol2.0 ml 10% SDS0.2 ml 0.5% Bromphenol blue
20 of 26

Add 50 µl of β-Mercaptoethanol to 950 µl of sample buffer before used. Mix the sampleto the sample buffer at least 1:2 proportion and heat to a 95◦C for 4 min.
• Detection Reagent
Name Storage ComponentSolution A 4◦C 200 mL 0.1 M Tris/HCl (pH 8,6) + 50 mg Luminol
(Sigma A4685)Solution B dark, RT 11 mg p-Coumaric acid (Sigma C9008) diluted in
10mL DMSOSolution C 4◦C H2O2 30 %
Mix 1mL Sol.A + 0,3 uL Sol.C + 100 uL Sol.B Incubate membrane for 2 min Exposure.
• Ammoniumpersulfat (APS)
Weight Material500 mg APS5 ml water
Store at -20◦C.
• SDS-PAGE Transfer Buffer
Weight Material25 mM Tris-Base192 mM Glycin20% v/v MethanolpH 8.3
For 1 L of buffer mix 3.03 g of Tris-Base, 14.4 g of glycin and 200 ml of methanol; Bringto 1 L with deionized water. Do not pH.
• 10× TBS
Weight Material24.2 g Tris-Base80 g NaCl
Dissolve in deionized water adjust pH with 6 N HCl and fill to 1 L.
• Wash Buffer
Weight Material10 ml 10×TBS1 ml Tween 20
Fill to 1 L with water.
• Blocking Buffer 5% milk in TBS buffer.
21 of 26
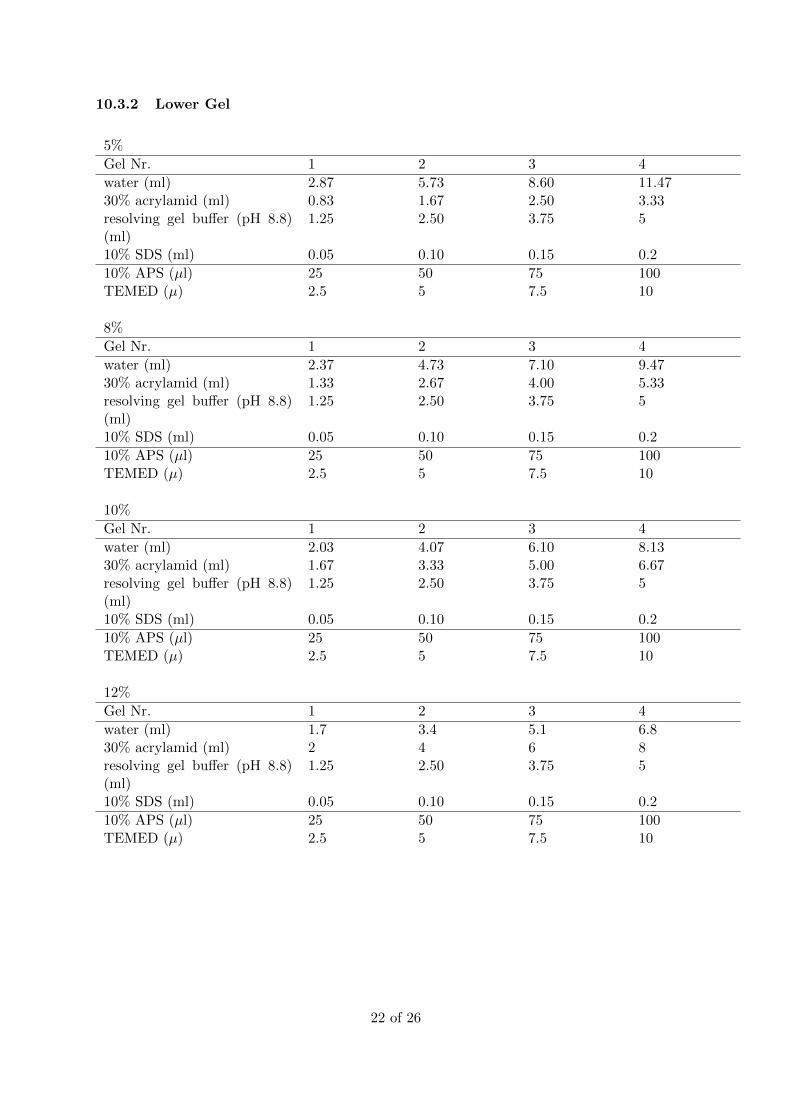
10.3.2 Lower Gel
5%
Gel Nr. 1 2 3 4
water (ml) 2.87 5.73 8.60 11.4730% acrylamid (ml) 0.83 1.67 2.50 3.33resolving gel buffer (pH 8.8)(ml)
1.25 2.50 3.75 5
10% SDS (ml) 0.05 0.10 0.15 0.2
10% APS (µl) 25 50 75 100TEMED (µ) 2.5 5 7.5 10
8%
Gel Nr. 1 2 3 4
water (ml) 2.37 4.73 7.10 9.4730% acrylamid (ml) 1.33 2.67 4.00 5.33resolving gel buffer (pH 8.8)(ml)
1.25 2.50 3.75 5
10% SDS (ml) 0.05 0.10 0.15 0.2
10% APS (µl) 25 50 75 100TEMED (µ) 2.5 5 7.5 10
10%
Gel Nr. 1 2 3 4
water (ml) 2.03 4.07 6.10 8.1330% acrylamid (ml) 1.67 3.33 5.00 6.67resolving gel buffer (pH 8.8)(ml)
1.25 2.50 3.75 5
10% SDS (ml) 0.05 0.10 0.15 0.2
10% APS (µl) 25 50 75 100TEMED (µ) 2.5 5 7.5 10
12%
Gel Nr. 1 2 3 4
water (ml) 1.7 3.4 5.1 6.830% acrylamid (ml) 2 4 6 8resolving gel buffer (pH 8.8)(ml)
1.25 2.50 3.75 5
10% SDS (ml) 0.05 0.10 0.15 0.2
10% APS (µl) 25 50 75 100TEMED (µ) 2.5 5 7.5 10
22 of 26
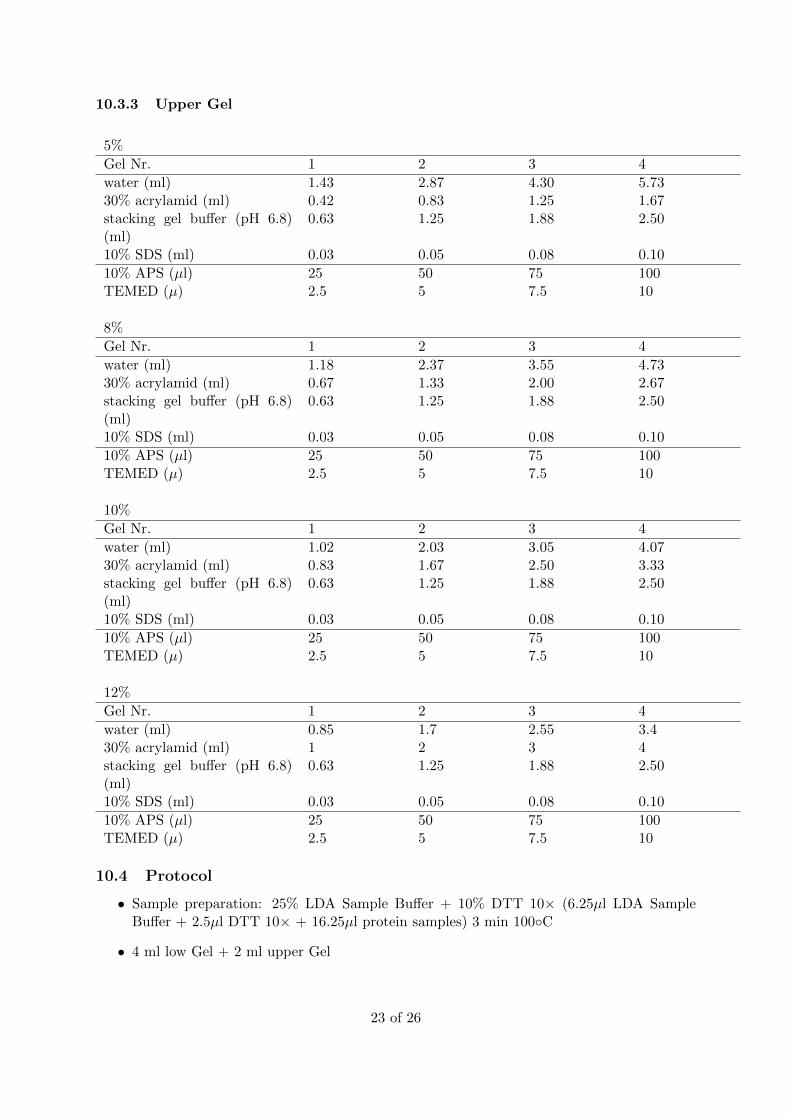
10.3.3 Upper Gel
5%
Gel Nr. 1 2 3 4
water (ml) 1.43 2.87 4.30 5.7330% acrylamid (ml) 0.42 0.83 1.25 1.67stacking gel buffer (pH 6.8)(ml)
0.63 1.25 1.88 2.50
10% SDS (ml) 0.03 0.05 0.08 0.10
10% APS (µl) 25 50 75 100TEMED (µ) 2.5 5 7.5 10
8%
Gel Nr. 1 2 3 4
water (ml) 1.18 2.37 3.55 4.7330% acrylamid (ml) 0.67 1.33 2.00 2.67stacking gel buffer (pH 6.8)(ml)
0.63 1.25 1.88 2.50
10% SDS (ml) 0.03 0.05 0.08 0.10
10% APS (µl) 25 50 75 100TEMED (µ) 2.5 5 7.5 10
10%
Gel Nr. 1 2 3 4
water (ml) 1.02 2.03 3.05 4.0730% acrylamid (ml) 0.83 1.67 2.50 3.33stacking gel buffer (pH 6.8)(ml)
0.63 1.25 1.88 2.50
10% SDS (ml) 0.03 0.05 0.08 0.10
10% APS (µl) 25 50 75 100TEMED (µ) 2.5 5 7.5 10
12%
Gel Nr. 1 2 3 4
water (ml) 0.85 1.7 2.55 3.430% acrylamid (ml) 1 2 3 4stacking gel buffer (pH 6.8)(ml)
0.63 1.25 1.88 2.50
10% SDS (ml) 0.03 0.05 0.08 0.10
10% APS (µl) 25 50 75 100TEMED (µ) 2.5 5 7.5 10
10.4 Protocol
• Sample preparation: 25% LDA Sample Buffer + 10% DTT 10× (6.25µl LDA SampleBuffer + 2.5µl DTT 10× + 16.25µl protein samples) 3 min 100◦C
• 4 ml low Gel + 2 ml upper Gel
23 of 26
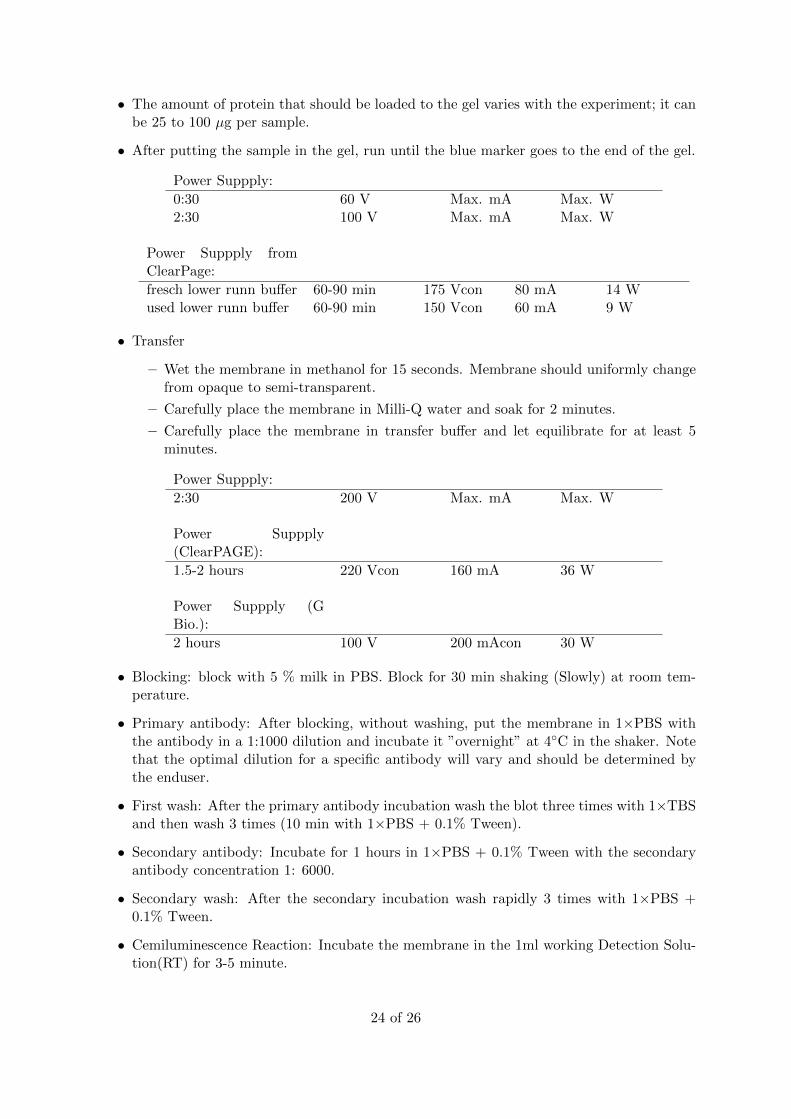
• The amount of protein that should be loaded to the gel varies with the experiment; it canbe 25 to 100 µg per sample.
• After putting the sample in the gel, run until the blue marker goes to the end of the gel.
Power Suppply:
0:30 60 V Max. mA Max. W2:30 100 V Max. mA Max. W
Power Suppply fromClearPage:
fresch lower runn buffer 60-90 min 175 Vcon 80 mA 14 Wused lower runn buffer 60-90 min 150 Vcon 60 mA 9 W
• Transfer
– Wet the membrane in methanol for 15 seconds. Membrane should uniformly changefrom opaque to semi-transparent.
– Carefully place the membrane in Milli-Q water and soak for 2 minutes.
– Carefully place the membrane in transfer buffer and let equilibrate for at least 5minutes.
Power Suppply:
2:30 200 V Max. mA Max. W
Power Suppply(ClearPAGE):
1.5-2 hours 220 Vcon 160 mA 36 W
Power Suppply (GBio.):
2 hours 100 V 200 mAcon 30 W
• Blocking: block with 5 % milk in PBS. Block for 30 min shaking (Slowly) at room tem-perature.
• Primary antibody: After blocking, without washing, put the membrane in 1×PBS withthe antibody in a 1:1000 dilution and incubate it ”overnight” at 4◦C in the shaker. Notethat the optimal dilution for a specific antibody will vary and should be determined bythe enduser.
• First wash: After the primary antibody incubation wash the blot three times with 1×TBSand then wash 3 times (10 min with 1×PBS + 0.1% Tween).
• Secondary antibody: Incubate for 1 hours in 1×PBS + 0.1% Tween with the secondaryantibody concentration 1: 6000.
• Secondary wash: After the secondary incubation wash rapidly 3 times with 1×PBS +0.1% Tween.
• Cemiluminescence Reaction: Incubate the membrane in the 1ml working Detection Solu-tion(RT) for 3-5 minute.
24 of 26

11 mirvana PARIS Kit
11.1 Protein Extraction
• 300µL ice-cold Cell Disruption Buffer for 106 cells
• 100µL ice-cold Cell Disruption Buffer for 10µg Tissue
• Homogenize on ice
• Incubate the buffer on ice for 5 min. 4◦C for 1-2 min at top speed in a microcentrifuge.
11.2 RNA Extraction
• 300µL ice-cold Cell Disruption Buffer for 106 cells
• 100µL ice-cold Cell Disruption Buffer for 10µg Tissue
• Homogenize on ice
• Transfer the lysate that will be used for RNA isolation to a tube containing an equalamount of 2× Denaturing solution at room temp.
• Immediately mix thoroughly.
• Incubate the mixture on ice for 5 min.
• Add a volume of Acid-Phenol:chloroformBe sure to withdraw the bottom phaseequal to the total volume of the sample lysate plus the 2× Denaturing solution.
• Vortex for 30-60 sec to mix.
• Centrifuge for 5 min at maximum speed (10,000× g) at room temp to separate themixture into aqueous and organic phases.
• Remove the aqueous (upper) phase, and transfer it to a fresh tube. Note the volumerecovered.
• Add 1.25 volumes of room temp 100% ethanol to the aqueous phase and mix thoroughly.
• Pipet the lysate/ethanol mixture onto the Filter Cartridge (700µL/ Cartridge).
• Centrifuge for 30 sec.
• Discard the flow-through, and repeat until all of the lysate/ethanol mixture is throughthe filter.
• Apply 700µL miRNA Wash Solution 1 to the Filter Cartridge and cenrifuge for 15 sec.Discard the flow-through.
• Apply 500µL Wash Solution 2/3 and draw it though the Filter Cartridge.
• Repeat with a second 500µL of Wash Solution 2/3.
• After discarding the flow-through, spin the assembly for 1 min.
• Transfer the Filter Cartridge into a fresh Collection Tube. Apply 100µL of 95◦C ElutionSolution or nuclease-free water.
• Centrifuge for 30 sec to recover the RNA.
25 of 26

Name Weight Total CellDisruptionBuffer
Cell Disrup-tion Buffer forRNA
2×Denaturingsolution atroom temp
Acid-Phenol:chloroform(Bottom)
Aqueous Ethanol H2O
mg µL = µL Total µL µL µL 1.25 ×µL
µL
26 of 26





























