Embed Size (px)
Citation preview

Carolyn A. Haller and Elliot L. ChaikofJiantao Xiao, Julianty Angsana, Jing Wen, Sumona V. Smith, Pyong Woo Park, Mandy L. Ford,
Mediated Responses−CellSyndecan-1 Displays a Protective Role in Aortic Aneurysm Formation by Modulating T
Print ISSN: 1079-5642. Online ISSN: 1524-4636 Copyright © 2011 American Heart Association, Inc. All rights reserved.
Greenville Avenue, Dallas, TX 75231is published by the American Heart Association, 7272Arteriosclerosis, Thrombosis, and Vascular Biology
doi: 10.1161/ATVBAHA.111.2421982011;
2012;32:386-396; originally published online December 15,Arterioscler Thromb Vasc Biol.
http://atvb.ahajournals.org/content/32/2/386World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://atvb.ahajournals.org/content/suppl/2011/12/14/ATVBAHA.111.242198.DC1.htmlData Supplement (unedited) at:
http://atvb.ahajournals.org//subscriptions/
at: is onlineArteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document. Question and AnswerPermissions and Rightspage under Services. Further information about this process is available in the
which permission is being requested is located, click Request Permissions in the middle column of the WebCopyright Clearance Center, not the Editorial Office. Once the online version of the published article for
can be obtained via RightsLink, a service of theArteriosclerosis, Thrombosis, and Vascular Biologyin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from

Syndecan-1 Displays a Protective Role in Aortic AneurysmFormation by Modulating T Cell–Mediated Responses
Jiantao Xiao, Julianty Angsana, Jing Wen, Sumona V. Smith, Pyong Woo Park, Mandy L. Ford,Carolyn A. Haller, Elliot L. Chaikof
Objective—Chronic inflammation drives progressive and pathological remodeling inherent to formation of abdominal aorticaneurysm (AAA). Syndecan-1 (Sdc-1) is a cell surface heparan sulfate proteoglycan that displays the capacity to modulateinflammatory processes within the vascular wall. In the current investigation, the role of Sdc-1 in AAA formation wasexamined using 2 models of experimental aneurysm induction, angiotensin II infusion and elastase perfusion.
Methods and Results—Sdc-1 deficiency exacerbated AAA formation in both experimental models and was associated withincreased degradation of elastin, greater protease activity, and enhanced inflammatory cell recruitment into the aorticwall. Bone marrow transplantation studies indicated that deficiency of Sdc-1 in marrow-derived cells significantlycontributed to AAA severity. Immunostaining revealed augmented Sdc-1 expression in a subset of AAA localizedmacrophages. We specifically characterized a higher percentage of CD4� T cells in Sdc-1-deficient AAA, and antibodydepletion studies established the active role of T cells in aneurysmal dilatation. Finally, we confirmed the ability ofSdc-1 macrophage to modulate the inflammatory chemokine environment.
Conclusion—These investigations identify cross-talk between Sdc-1-expressing macrophages and AAA-localized CD4� Tcells, with Sdc-1 providing an important counterbalance to T-cell–driven inflammation in the vascular wall. (ArteriosclerThromb Vasc Biol. 2012;32:386-396.)
Key Words: aneurysms � aortic diseases � glycosominoglycan � leukocytes � macrophages
Abdominal aortic aneurysm (AAA) is a common and life-threatening clinical condition. In the United States, with
approximately 15 000 deaths each year, AAA ranks as the 13thleading cause of death.1 Current treatment strategies for AAAare largely confined to endovascular or open surgical interven-tion or physician surveillance. Despite improvement in mini-mally invasive surgical techniques, the risk of periproceduralmorbidity and mortality and subsequent secondary reinterven-tion remains significant.2 Nonsurgical treatment options wouldbe desirable. Indeed, a number of approaches have been pro-posed to prevent progression of aneurysmal disease during theperiod of aneurysm surveillance, including hemodynamic con-trol and inhibition of inflammation and protease activity.3,4
However, a clinically effective pharmacotherapeutic that limitsor reverses aortic dilatation has yet to be identified. Definingendogenous mediators that serve to dampen the inflammatoryresponse within the setting of AAA may identify new avenuesfor medical therapy.
The syndecans are a family of 4 cell surface proteoglycans(syndecan-1 [Sdc-1], Sdc-2, Sdc-3, and Sdc-4) that display thecapacity to modulate proinflammatory and proteolytic processes
within the vascular wall. The biological function of syndecans isprimarily exerted via pendant glycosaminoglycans, such asheparan sulfate (HS) and chondroitin sulfate, which sequesterand regulate the activity of heparin-binding growth factors,proinflammatory chemokines, and proteases. Intact syndecanectodomains can be released from the cell surface throughproteolytic shedding. This process, which is upregulated withinthe context of inflammation, allows syndecans to exert a biolog-ical effect beyond the confines of the plasma membrane.5
Detailed reviews of syndecan biology can be found elsewhere.6,7
Within the syndecan family, Sdc-1 is emerging as an importantregulator of inflammation. Several studies have documented theprotective role of Sdc-1 in animal models with strong inflam-matory components, such as nephritis,8 toxic shock,9 allergiclung inflammation,10 and myocardial infarction.11 Within thesediverse investigations lies a common thread suggesting that in anenvironment of exaggerated inflammation, Sdc-1 can promote adampened response by interfering with the inflammatory signal-ing cascade.
In adult tissue, Sdc-1 is found predominately expressed onepithelial cells and noncirculating plasma cells. Expression can
Received on: January 17, 2011; final version accepted on: November 28, 2011.From the Department of Surgery (J.X., J.W., S.V.S.) and Emory Transplant Center (M.L.F.), Emory University, Atlanta, GA; Department of
Biomedical Engineering, Georgia Institute of Technology/Emory University, Atlanta, GA (J.A.); Department of Pediatrics, Boston Children’s Hospital(P.W.P.), and Department of Surgery, Beth Israel Deaconess Medical Center (C.A.H., E.L.C.), Harvard Medical School, Boston, MA (P.W.P., C.A.H.,E.L.C.); School of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA (E.L.C.); Wyss Institute of BiologicallyInspired Engineering, Harvard University, Boston, MA (E.L.C.).
Correspondence to Elliot L. Chaikof, MD, PhD, BIDMC, 110 Francis St, Suite 9F, Boston, MA 02215 (e-mail [email protected]) orCarolyn A. Haller, PhD, BIDMC, 110 Francis St, Suite 9F, Boston, MA 02215 (e-mail [email protected]).
© 2011 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol is available at http://atvb.ahajournals.org DOI: 10.1161/ATVBAHA.111.242198
386 at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from

also be induced in additional cell types, such as endothelial cells,smooth muscle cells, fibroblasts, and macrophages. Macrophagespecific Sdc-1 expression is of particular interest, as induction isgoverned through the cAMP/protein kinase A signaling cas-cade.12,13 The recent availability of new cAMP analogs, whichexplicitly target protein kinase A or guanine nucleotide ex-change factor, is generating increased interest in deciphering thespecificity of these downstream effectors.14 Notably, cAMP/protein kinase A specific activation in macrophages has beenreported to be inhibitory toward the production of inflammatorymediators.15 Thus, it appears likely that Sdc-1 is induced onmacrophages in the context of a broad signaling programdesigned to dampen an inflammatory response.
We have previously characterized the spatiotemporal expres-sion for Sdc-1, -2, and -4 in a murine model of angiotensin IIinduced AAA.16 We observed that the expression of Sdc-1 isaugmented during the course of aneurysm formation. In addi-tion, Sdc-1 expression in the aneurysm wall seems restricted toa subset of macrophages. In this investigation, we sought todefine the functional role of Sdc-1 in experimental AAAformation. We report that Sdc-1 has an important protectivefunction in aneurysm formation that appears mediated, at least inpart, through an effect on AAA-localized T cells.
MethodsElastase Perfusion ModelC57BL/6J mice (Jackson Laboratory) and Sdc-1�/� mice (back-crossed 10 times onto a C57BL/6J background, provided by DrPyong Woo Park, Baylor College of Medicine, Houston, TX) werestudied. All experimental procedures were performed in male mice atmaturity (8–12 weeks), according to a protocol approved by EmoryUniversity Institutional Animal Care and Use Committee. Elastaseperfusion was performed as described previously.17 The percentageof increase in abluminal aortic diameter was calculated from thedifference between the preperfusion and final aortic diameter, withAAA arbitrarily defined as the percentage of an increase in aorticdiameter of more than 100%.
Generation of ApoE�/�Sdc-1�/� Mice andAngiotensin II Infusion ModelApolipoprotein E (ApoE)�/�Sdc-1�/� (C57BL/6J background) dou-ble knockout mice were generated by standard crossbreeding exper-iments. Sdc-1�/� breeder males were mated with ApoE�/� females(Jackson Laboratory), and all mice were genotyped by polymerasechain reaction (PCR). Male mice were subsequently maintained onPaigen atherogenic diet (Research Diets) and received a subcutane-ous infusion of angiotensin II (0.75 mg/kg per day) over a 2-weekperiod by mini-osmotic pump (Alza Scientific Products). Systolicblood pressure before and after the implantation of miniosmoticpump was obtained from the mice using a noninvasive tail-cuffsystem (Visitech Systems). The incidence of AAA formation,incidence of fatal aortic rupture, and final aortic diameter at 2 weekswere determined. Total serum cholesterol was measured with Am-plex Red (Molecular Probes).
Light Microscopy and ImmunohistochemistryImmunohistochemistry was performed as described previously.16
The following antibodies were used: Sdc-1 (N-18, Santa CruzBiotechnology), neutrophil (NIMP-R14, Abcam), macrophage(Mac3, BD Biosciences), CD4 (RM4-5, BD), CD8 (53-6.7, BDBiosciences), Foxp3 (FJK-16s, eBioscience). Sections were incu-bated with biotinylated secondary antibodies (Vector Laboratories)followed by alkaline phosphatase streptavidin (Vector Laboratories).Negative controls with isotype IgG were prepared for each specimen.
Spleen sections were used as a positive control tissue for identifica-tion of Foxp3-positive cells. Foxp3-positive cells were counted ineach aortic section by a trained laboratory technician blinded tosample classification. At least 4 sections from each of 3 animals ateach time point in both groups were examined. A mean value forpositively stained cells was determined for each animal, and a meanfor each animal group was then calculated. Acu-Sstain elastic stainkit (Sigma-Aldrich) was used for elastin degradation studies. Doublefluorescent immunostaining was performed as describedpreviously.16
Flow CytometryAortas (pooled from 3–6 individual mice) were excised from belowthe infrarenal arteries to just above the bifurcation after the bloodcontent was flushed. Tissue was finely minced and shaken for 60minutes at 37°C in 1 mL of RPMI 1640 supplemented with 10% fetalcalf serum, 62.5 U/mL collagenase VII (Sigma-Aldrich), and 0.625U/mL Dispase (BD); for Sdc-1 detection, dispase was excluded. Theisolated cells were passed through a 70-�m cell strainer to removedebris and then counted, followed by staining using a standardizedprotocol. The cell antibodies used included anti-Gr1 (RB6-8C5),anti-Mac1� (M1/70), anti-CD3 (145-2C11), anti-CD45 (30-F11),and anti-CD138 (281-2), all from BD. Cell suspensions wereanalyzed by flow cytometry (FACSort, BD) and type-specificnumbers of cells present in each sample were quantified andrecorded.
Gelatin ZymographyAortic tissue extract was prepared in 100 �L of tissue homogenizingbuffer (30 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 10 mmol/LCaCl2, 10 �mol/L E-64, 0.05% Brij35, 0.02% NaN3, and100 mmol/L phenylmethylsulfonyl fluoride). Protein concentrationwas quantified using the BCA protein assay kit (Pierce). Fivemicrograms of aortic tissue extract was run in 10% polyacrylamidecontaining 10% gelatin (Bio-Rad) under nonreducing conditions.The gel was developed (37°C, 3 days) and stained with 0.125%Coomassie Blue. Gelatinolytic activity was quantified by densitom-etry (National Institutes of Health ImageJ software).
Quantitative (Real-Time) ReverseTranscription–PCRMessenger RNA levels within the aortic wall were analyzed withreverse transcription–PCR using 18S rRNA as the internal control.All primers were obtained from Applied Biosystems. Four or 5samples, each containing up to 3 pooled aortas, were obtained fromeach experimental time point. All PCRs were performed in triplicatewith 10 to 25 ng of cDNA using the TaqMan PCR system (AppliedBiosystems). Results were analyzed by comparing RNA level ofsamples with RNA obtained from untreated aortas using the com-parative CT method.
Antibody DepletionMice were made T lymphocytopenic by intraperitoneal injection ofanti-CD3 antibody (50 �g, clone 17A2, Biolegend) at days �1, 4,and 9 after elastase perfusion. Time course of depletion protocol wasverified with Thy-1� (G7, Southern Biotech) staining in the spleen.Splenocytes were harvested and finely minced in RPMI 1640medium supplemented with 5% fetal calf serum, tissue was passedthrough a 100 �m cell strainer, red blood cells were lysed, and totalcell count was recorded. Flow cytometry was used to analyzespleenic T-cell population.
Bone Marrow TransplantationSdc-1�/� and Sdc-1�/� recipients underwent lethal gamma irradia-tion (11 Gy) to eliminate endogenous bone marrow (BM) stem cellsand circulating leukocytes. Using sterile procedures, BM cells wereobtained by flushing femoral and tibial bones from Sdc-1�/� andSdc-1�/� mice. Each irradiated mouse was injected intraorbitallywith 106 BM cells from designated donors. Three groups (8–10mice/group) were studied: (1) Sdc-1�/� BM cells transplanted to
Xiao et al Protective Role of Syndecan-1 in AAA 387
at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from

Sdc-1�/� recipients, (2) Sdc-1�/� BM cells transplanted to Sdc-1�/� recipients, and (3) Sdc-1�/� BM cells transplanted to Sdc-1�/�
recipients. Mice were enrolled in the elastase perfusion model 8weeks after transplantation.
ChemotaxisCD4� T cells were positively selected from splenocytes (MiltenyiBiotec), harvested 8 to 10 days after Listeria monocytogenes (104
CFU/mouse) infection to produce the desired Th1 polarized re-sponse.18,19 Chemotaxis assays were performed using 4�105 cells in5-�m Transwell plates (Corning) with RPMI, 0.1% bovine serumalbumin base media with or without chemokine. In some cases, HS(50 �g/mL) was included in the lower well. The migration responsewas quantified after 4 hours at 37°C in a humidified incubator.
Chemokine ProfileDay 4 thiglycollate elicited peritoneal macrophages were collectedfrom C57BL/6J mice and plated in Dulbecco’s modified Eagle’smedium (10% fetal bovine serum) at 20�106 per 100�20-mm tissueculture dish. Classically activated M1 macrophage were generatedthrough interferon (IFN)-� (100 U/mL)/lipopolysaccharide (10 ng/mL)20 stimulation and Sdc-1 expression was induced with 6-Bnz-cAMP (100 �mol/L).13 Sdc-1 expression was verified with flowcytometry (anti-CD138, 281–2). Serum-free Dulbecco’s modifiedEagle’s medium was added after initial stimulation, and conditionedmedium was collected after 12 hours. Adhered cells were exposed toa 1 mol/L NaCl wash to disrupt any surface electrostatic bindinginteractions. Conditioned media and wash fractions were combined,filtered (0.2 �m), subjected to dialysis and concentration 4-fold(Amicon Ultra 3000 nominal molecular weight limit), and analyzedfor regulated upon activation, normal T expressed and secreted;monocyte chemotactic protein-1; macrophage inflammatory protein-1a; macrophage inflammatory protein-1b; interferon gamma-inducedprotein 10; monokine induced by interferon gamma; macrophage-derived chemokine; and keratinocyte-derived chemokine using theMouse Common Chemokine ELISArray (SA Biosciences). Threeindependent experiments were conducted.
Statistical AnalysisMean and SEM were calculated for each parameter. All data wereanalyzed via 2-tailed Student t test, with the exception of frequencyand mortality contingency tables in the angiotensin II AAA model(Fisher exact test). Values of P�0.05 were considered statisticallysignificant.
ResultsMacrophage-Associated Sdc-1 ExpressionAttenuates Experimental Aneurysm FormationTransient intraaortic perfusion with elastase is a commonexperimental model of AAA with consistent development ofan infrarenal aortic aneurysm 14 days after initial perfusion.17
Immunohistochemistry was used to characterize Sdc-1 ex-pression in concert with the neutrophil and macrophageinfiltrate over the 14-day time course of aneurysm develop-ment. We observed discrete regions of neutrophil infiltrationat 1, 4, and 7 days after elastase infusion; particularly, evidentin both the intima and periadventitia (Supplemental FigureIA–ID, available online at http://atvb.ahajournals.org). The 7-and 14-day inflammatory response was dominated by trans-mural infiltration of macrophages (Supplemental Figure IE–IH). Sdc-1 expression was accentuated during the 14-daytime course, with most Sdc-1-positive cells localized to theadventitia (Supplemental Figure II–IL). Little to no expres-sion of Sdc-1 was detectable in the native aorta (data notshown). Similar patterns of neutrophil and macrophage
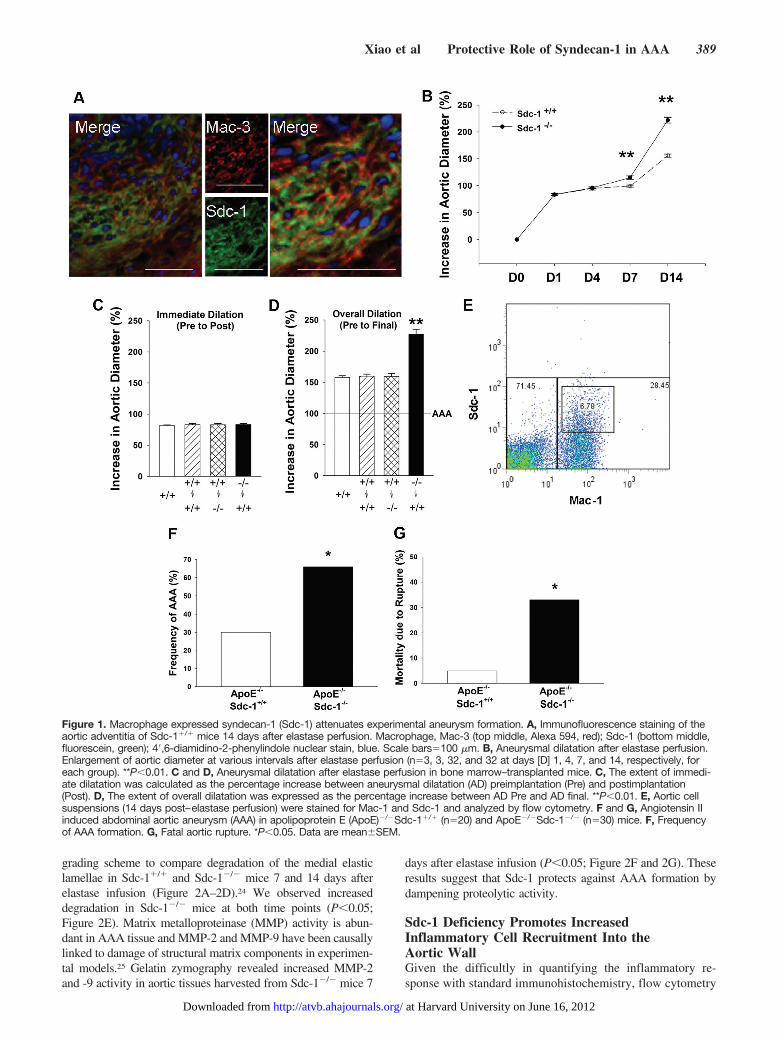
recruitment were observed in Sdc-1-deficient mice (Sup-plemental Figure IM–IT). Immunohistochemical analysisrevealed an association between macrophage and Sdc-1-positive staining, which was confirmed by double immu-nofluorescence staining. As illustrated, Sdc-1 expressioncolocalized with Mac-3-positive cells, consistent with thenotion that Sdc-1 expression is specific to infiltratingmacrophages (Figure 1A).
To study the functional significance of Sdc-1 in thepathogenesis of AAA, we enrolled Sdc-1-deficient mice(Sdc-1�/�, C57BL/6 background) in the elastase perfusionmodel and compared the results with those of wild-typeC57BL/6 mice (Sdc-1�/�). Sdc-1-deficient mice have beenpreviously characterized as healthy, with normal growth,reproduction, tissue morphology, hematologic profile, andserum chemistry parameters.9,21 There was moderate aorticdilatation in both Sdc-1�/� and Sdc-1�/� mice up to 4 daysafter elastase perfusion. Increased dilatation was noted by day7, with AAA (�aortic diameter �100%) in 34% of Sdc-1�/�
and 81% of Sdc-1�/� mice. All mice formed aneurysms byday 14; however, the extent of aortic dilatation was signifi-cantly greater among the Sdc-1�/� group (P�0.01; Figure 1Band Supplemental Table I), suggesting that endogenous Sdc-1expression exerts a protective role in elastase-induced AAA.BM transplantation experiments confirmed that the protectivesource of Sdc-1 expression originates from the circulatingleukocyte population during the development of AAA(P�0.01; Figure 1C and 1D). Finally, to further characterizeSdc-1 expression in AAA tissue, we used flow cytometry(Mac-1/Sdc-1) to examine 14-day aortic tissue digests.Sdc-1� cells were observed as a subset of Mac-1� cells(Figure 1E); significantly, collective observations from dou-ble immunofluorescent staining, BM transplantation, andflow cytometry all suggest that macrophages provide asignificant source of Sdc-1 expression in AAA.
To further test the hypothesis that macrophage Sdc-1expression is protective in AAA, we used a second model ofexperimental aneurysm formation. The murine model ofangiotensin-associated aortic aneurysm is produced by sub-cutaneous administration of angiotensin II into ApoE�/�
mice.22,23 Standard crossbreeding was used to generateApoE�/�Sdc-1�/� double knockout mice, which were en-rolled in this model and responses compared with those ofApoE�/�Sdc-1�/� mice. Animals were euthanized at 3, 7,and 14 days, and aortic tissue was harvested for immunohis-tochemical examination. Sdc-1/macrophage colocalizationwas confirmed by double immunofluorescence staining (Sup-plemental Figure IIA–IIP). Despite equivalent blood pressureresponses and serum cholesterol levels (Supplemental TableII), significantly higher rates of AAA formation (�2-fold)and rupture (�6-fold) were observed in ApoE�/�Sdc-1�/�
mice (P�0.05; Figure 1F and 1G). These data suggest thatthe protective role of macrophage-associated Sdc-1 in AAAis independent of experimental animal model.
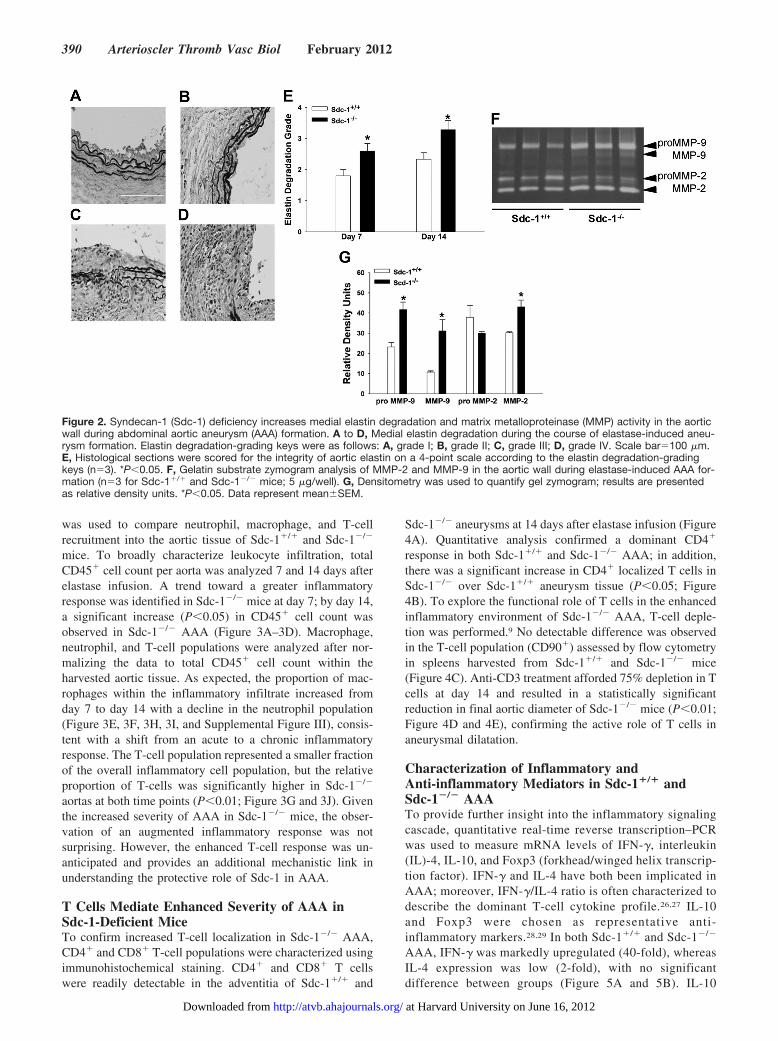
Sdc-1 Deficiency Increases Proteolytic Activity inthe Aortic WallDestruction of medial elastin is a hallmark of AAA pathogene-sis. We stained aortic sections for elastic fibers and used a
388 Arterioscler Thromb Vasc Biol February 2012
at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from
grading scheme to compare degradation of the medial elasticlamellae in Sdc-1�/� and Sdc-1�/� mice 7 and 14 days afterelastase infusion (Figure 2A–2D).24 We observed increaseddegradation in Sdc-1�/� mice at both time points (P�0.05;Figure 2E). Matrix metalloproteinase (MMP) activity is abun-dant in AAA tissue and MMP-2 and MMP-9 have been causallylinked to damage of structural matrix components in experimen-tal models.25 Gelatin zymography revealed increased MMP-2and -9 activity in aortic tissues harvested from Sdc-1�/� mice 7
days after elastase infusion (P�0.05; Figure 2F and 2G). Theseresults suggest that Sdc-1 protects against AAA formation bydampening proteolytic activity.
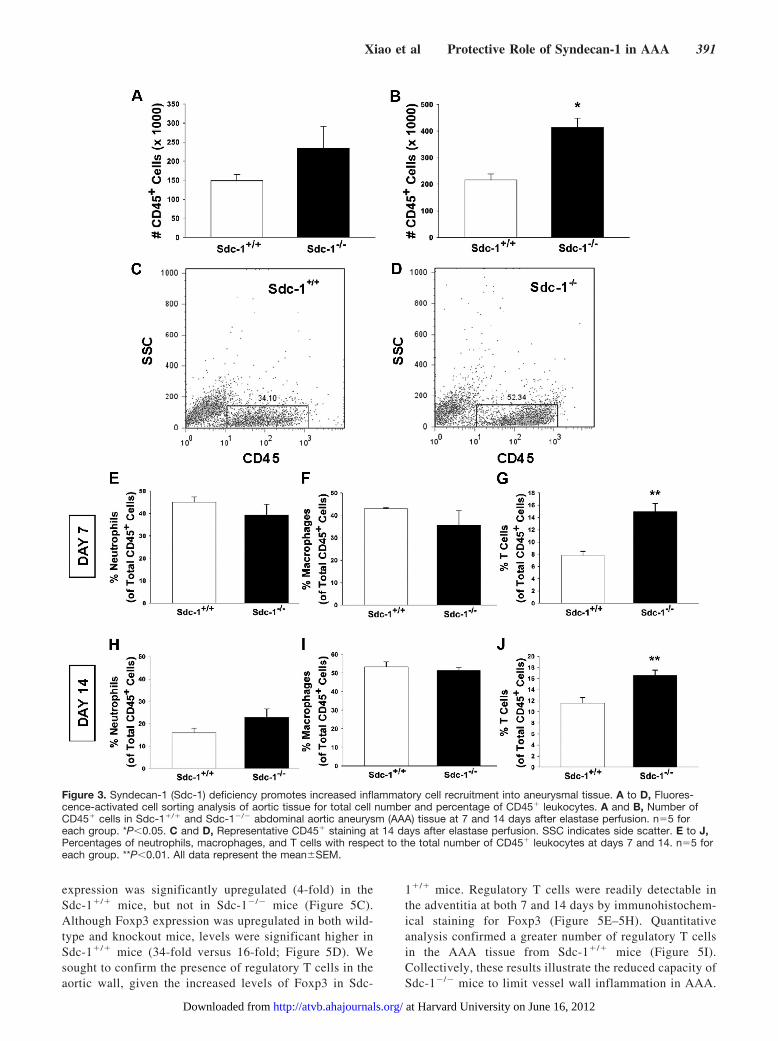
Sdc-1 Deficiency Promotes IncreasedInflammatory Cell Recruitment Into theAortic WallGiven the difficultly in quantifying the inflammatory re-sponse with standard immunohistochemistry, flow cytometry
Figure 1. Macrophage expressed syndecan-1 (Sdc-1) attenuates experimental aneurysm formation. A, Immunofluorescence staining of theaortic adventitia of Sdc-1�/� mice 14 days after elastase perfusion. Macrophage, Mac-3 (top middle, Alexa 594, red); Sdc-1 (bottom middle,fluorescein, green); 4�,6-diamidino-2-phenylindole nuclear stain, blue. Scale bars�100 �m. B, Aneurysmal dilatation after elastase perfusion.Enlargement of aortic diameter at various intervals after elastase perfusion (n�3, 3, 32, and 32 at days [D] 1, 4, 7, and 14, respectively, foreach group). **P�0.01. C and D, Aneurysmal dilatation after elastase perfusion in bone marrow–transplanted mice. C, The extent of immedi-ate dilatation was calculated as the percentage increase between aneurysmal dilatation (AD) preimplantation (Pre) and postimplantation(Post). D, The extent of overall dilatation was expressed as the percentage increase between AD Pre and AD final. **P�0.01. E, Aortic cellsuspensions (14 days post–elastase perfusion) were stained for Mac-1 and Sdc-1 and analyzed by flow cytometry. F and G, Angiotensin IIinduced abdominal aortic aneurysm (AAA) in apolipoprotein E (ApoE)�/�Sdc-1�/� (n�20) and ApoE�/�Sdc-1�/� (n�30) mice. F, Frequencyof AAA formation. G, Fatal aortic rupture. *P�0.05. Data are meanSEM.
Xiao et al Protective Role of Syndecan-1 in AAA 389
at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from
was used to compare neutrophil, macrophage, and T-cellrecruitment into the aortic tissue of Sdc-1�/� and Sdc-1�/�
mice. To broadly characterize leukocyte infiltration, totalCD45� cell count per aorta was analyzed 7 and 14 days afterelastase infusion. A trend toward a greater inflammatoryresponse was identified in Sdc-1�/� mice at day 7; by day 14,a significant increase (P�0.05) in CD45� cell count wasobserved in Sdc-1�/� AAA (Figure 3A–3D). Macrophage,neutrophil, and T-cell populations were analyzed after nor-malizing the data to total CD45� cell count within theharvested aortic tissue. As expected, the proportion of mac-rophages within the inflammatory infiltrate increased fromday 7 to day 14 with a decline in the neutrophil population(Figure 3E, 3F, 3H, 3I, and Supplemental Figure III), consis-tent with a shift from an acute to a chronic inflammatoryresponse. The T-cell population represented a smaller fractionof the overall inflammatory cell population, but the relativeproportion of T-cells was significantly higher in Sdc-1�/�
aortas at both time points (P�0.01; Figure 3G and 3J). Giventhe increased severity of AAA in Sdc-1�/� mice, the obser-vation of an augmented inflammatory response was notsurprising. However, the enhanced T-cell response was un-anticipated and provides an additional mechanistic link inunderstanding the protective role of Sdc-1 in AAA.
T Cells Mediate Enhanced Severity of AAA inSdc-1-Deficient MiceTo confirm increased T-cell localization in Sdc-1�/� AAA,CD4� and CD8� T-cell populations were characterized usingimmunohistochemical staining. CD4� and CD8� T cellswere readily detectable in the adventitia of Sdc-1�/� and
Sdc-1�/� aneurysms at 14 days after elastase infusion (Figure4A). Quantitative analysis confirmed a dominant CD4�
response in both Sdc-1�/� and Sdc-1�/� AAA; in addition,there was a significant increase in CD4� localized T cells inSdc-1�/� over Sdc-1�/� aneurysm tissue (P�0.05; Figure4B). To explore the functional role of T cells in the enhancedinflammatory environment of Sdc-1�/� AAA, T-cell deple-tion was performed.9 No detectable difference was observedin the T-cell population (CD90�) assessed by flow cytometryin spleens harvested from Sdc-1�/� and Sdc-1�/� mice(Figure 4C). Anti-CD3 treatment afforded 75% depletion in Tcells at day 14 and resulted in a statistically significantreduction in final aortic diameter of Sdc-1�/� mice (P�0.01;Figure 4D and 4E), confirming the active role of T cells inaneurysmal dilatation.
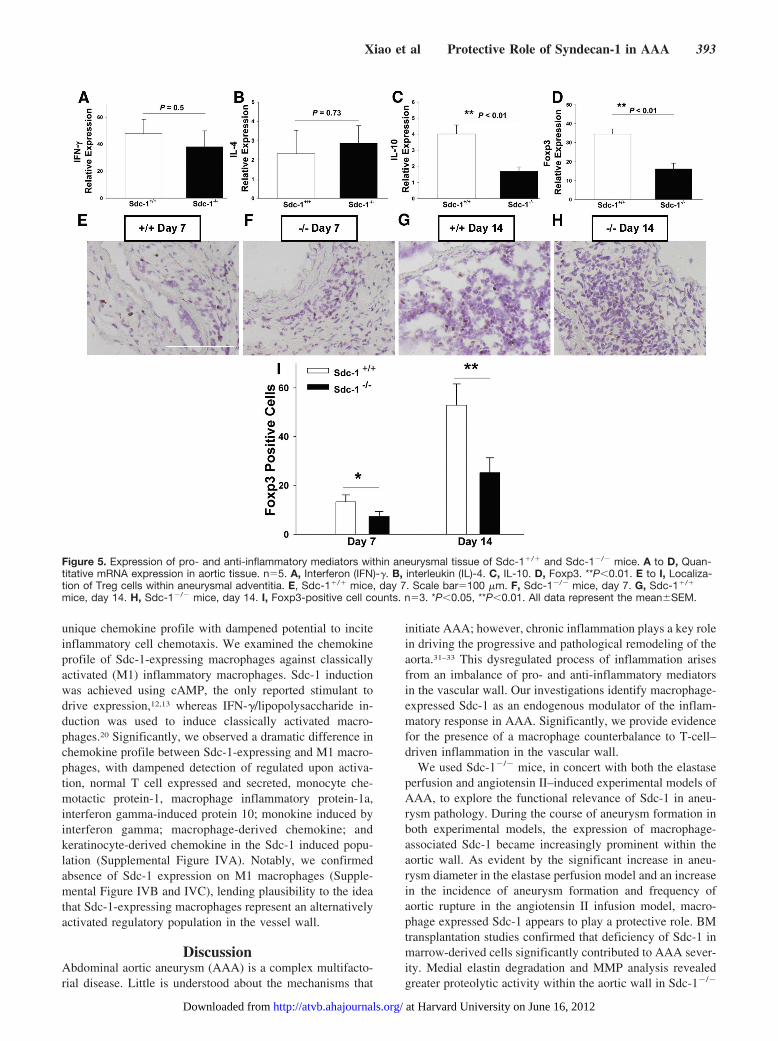
Characterization of Inflammatory andAnti-inflammatory Mediators in Sdc-1�/� andSdc-1�/� AAATo provide further insight into the inflammatory signalingcascade, quantitative real-time reverse transcription–PCRwas used to measure mRNA levels of IFN-�, interleukin(IL)-4, IL-10, and Foxp3 (forkhead/winged helix transcrip-tion factor). IFN-� and IL-4 have both been implicated inAAA; moreover, IFN-�/IL-4 ratio is often characterized todescribe the dominant T-cell cytokine profile.26,27 IL-10and Foxp3 were chosen as representative anti-inflammatory markers.28,29 In both Sdc-1�/� and Sdc-1�/�
AAA, IFN-� was markedly upregulated (40-fold), whereasIL-4 expression was low (2-fold), with no significantdifference between groups (Figure 5A and 5B). IL-10
Figure 2. Syndecan-1 (Sdc-1) deficiency increases medial elastin degradation and matrix metalloproteinase (MMP) activity in the aorticwall during abdominal aortic aneurysm (AAA) formation. A to D, Medial elastin degradation during the course of elastase-induced aneu-rysm formation. Elastin degradation-grading keys were as follows: A, grade I; B, grade II; C, grade III; D, grade IV. Scale bar�100 �m.E, Histological sections were scored for the integrity of aortic elastin on a 4-point scale according to the elastin degradation-gradingkeys (n�3). *P�0.05. F, Gelatin substrate zymogram analysis of MMP-2 and MMP-9 in the aortic wall during elastase-induced AAA for-mation (n�3 for Sdc-1�/� and Sdc-1�/� mice; 5 �g/well). G, Densitometry was used to quantify gel zymogram; results are presentedas relative density units. *P�0.05. Data represent meanSEM.
390 Arterioscler Thromb Vasc Biol February 2012
at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from
expression was significantly upregulated (4-fold) in theSdc-1�/� mice, but not in Sdc-1�/� mice (Figure 5C).Although Foxp3 expression was upregulated in both wild-type and knockout mice, levels were significant higher inSdc-1�/� mice (34-fold versus 16-fold; Figure 5D). Wesought to confirm the presence of regulatory T cells in theaortic wall, given the increased levels of Foxp3 in Sdc-
1�/� mice. Regulatory T cells were readily detectable inthe adventitia at both 7 and 14 days by immunohistochem-ical staining for Foxp3 (Figure 5E–5H). Quantitativeanalysis confirmed a greater number of regulatory T cellsin the AAA tissue from Sdc-1�/� mice (Figure 5I).Collectively, these results illustrate the reduced capacity ofSdc-1�/� mice to limit vessel wall inflammation in AAA.
Figure 3. Syndecan-1 (Sdc-1) deficiency promotes increased inflammatory cell recruitment into aneurysmal tissue. A to D, Fluores-cence-activated cell sorting analysis of aortic tissue for total cell number and percentage of CD45� leukocytes. A and B, Number ofCD45� cells in Sdc-1�/� and Sdc-1�/� abdominal aortic aneurysm (AAA) tissue at 7 and 14 days after elastase perfusion. n�5 foreach group. *P�0.05. C and D, Representative CD45� staining at 14 days after elastase perfusion. SSC indicates side scatter. E to J,Percentages of neutrophils, macrophages, and T cells with respect to the total number of CD45� leukocytes at days 7 and 14. n�5 foreach group. **P�0.01. All data represent the meanSEM.
Xiao et al Protective Role of Syndecan-1 in AAA 391
at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from

Macrophage Sdc-1 Modulates InflammatoryChemokine ProfileBased on our findings that Sdc-1 plays an active role inrestricting the localized T-cell population during aneurysmformation, we elected to investigate the potential for Sdc-1 tomodulate AAA-driven T-cell chemotaxis. Chemokines are smallHS-binding proteins that direct infiltration of leukocytes intoinflamed tissue and are recognized as critical components insustaining chronic inflammation.30 Glycosaminoglycan/chemo-kine binding interactions are well established and thought to bemediated, at least in part, by electrostatic interactions of basicchemokines with negatively charged HS. Such interactionsprovided motivation to test the capacity of HS, as a syndecan-1mimetic, to competitively inhibit T-cell chemotaxis to AAArelevant chemokines. In light of the CD4�/IFN-�-dominantresponse that was characterized during the formation of AAA,we specifically investigated chemotaxis in Th1 polarized CD4�
T cells from Sdc-1�/� mice. Quantitative real-time reversetranscription–PCR was used to identify chemokines of interest(potential to elicit Th1 localization) in elastase-induced aneu-rysm formation. In both Sdc-1�/� and Sdc-1�/� AAA, there
was significant expression of CCL2 (monocyte chemotacticprotein-1), CCL3 (macrophage inflammatory protein-1a), CCL5(regulated upon activation, normal T cell expressed and se-creted), and CXCL9 (MIG); however, no difference in expres-sion was noted between genotypes (data not shown). We testedthe ability of HS to limit Th1 polarized CD4� T-cell chemotaxisto CCL2, CCL3, CCL5, and CXCL9 in a standard Transwellmigration assay. HS effectively limited chemotaxis to CCL2,CCL3, and CXCL9 without influencing basal migration, and wedid not observe an impact on CCL5-driven chemotaxis(P�0.05; Figure 6). Of note, initial studies were performed toensure equivalent chemotactic response in Sdc-1�/� and Sdc-1�/� CD4� T cells, and no inherent differences were observed;in addition, both naïve and activated CD4� T cells fromSdc-1�/� mice were examined for Sdc-1 expression using flowcytometry, and there was no evidence of Sdc-1 expression onCD4� T cells (data not shown). These studies suggest thatmacrophage Sdc-1 may modulate the T-cell inflammatory re-sponse through HS-chemokine binding interactions.
Alternatively, Sdc-1-expressing macrophages may repre-sent a regulatory or M2 macrophage population displaying a
Figure 4. T cells promote abdominal aortic aneurysm (AAA) in syndecan-1 (Sdc-1)-deficient mice. A and B, Characterization of CD4 andCD8 populations in Sdc-1�/� and Sdc-1�/� AAA at 14 days after elastase perfusion. A, Representative immunohistochemical staining inadventitial AAA sections. Scale bars�100 �m. B, CD4 and CD8 cell counts in aortic wall per high-power field. n�3 for each group. *P�0.05,**P�0.01. C to E, Effects of T-cell depletion on AAA formation in Sdc-1�/� mice. C, Number of T cells (Thy-1�) in native spleens and 14 daysafter anti-CD3 antibody treatment. n�3 in both Sdc-1�/� and Sdc-1�/� control group; n�8 in T lymphocytopenic Sdc-1�/� group. **P�0.01.D and E, Aortic diameter measurements were obtained before (Pre) and immediately after (Post) elastase perfusion, with final measurementobtained at 14 days after surgery (Final). D, The extent of immediate dilatation was calculated as the percentage increase between aneurys-mal dilation (AD) preimplantation postimplantation. E, The extent of overall dilatation was expressed as the percentage increase between ADPre and AD Final. **P�0.01. All data represent the meanSEM.
392 Arterioscler Thromb Vasc Biol February 2012
at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from
unique chemokine profile with dampened potential to inciteinflammatory cell chemotaxis. We examined the chemokineprofile of Sdc-1-expressing macrophages against classicallyactivated (M1) inflammatory macrophages. Sdc-1 inductionwas achieved using cAMP, the only reported stimulant todrive expression,12,13 whereas IFN-�/lipopolysaccharide in-duction was used to induce classically activated macro-phages.20 Significantly, we observed a dramatic difference inchemokine profile between Sdc-1-expressing and M1 macro-phages, with dampened detection of regulated upon activa-tion, normal T cell expressed and secreted, monocyte che-motactic protein-1, macrophage inflammatory protein-1a,interferon gamma-induced protein 10; monokine induced byinterferon gamma; macrophage-derived chemokine; andkeratinocyte-derived chemokine in the Sdc-1 induced popu-lation (Supplemental Figure IVA). Notably, we confirmedabsence of Sdc-1 expression on M1 macrophages (Supple-mental Figure IVB and IVC), lending plausibility to the ideathat Sdc-1-expressing macrophages represent an alternativelyactivated regulatory population in the vessel wall.
DiscussionAbdominal aortic aneurysm (AAA) is a complex multifacto-rial disease. Little is understood about the mechanisms that
initiate AAA; however, chronic inflammation plays a key rolein driving the progressive and pathological remodeling of theaorta.31–33 This dysregulated process of inflammation arisesfrom an imbalance of pro- and anti-inflammatory mediatorsin the vascular wall. Our investigations identify macrophage-expressed Sdc-1 as an endogenous modulator of the inflam-matory response in AAA. Significantly, we provide evidencefor the presence of a macrophage counterbalance to T-cell–driven inflammation in the vascular wall.
We used Sdc-1�/� mice, in concert with both the elastaseperfusion and angiotensin II–induced experimental models ofAAA, to explore the functional relevance of Sdc-1 in aneu-rysm pathology. During the course of aneurysm formation inboth experimental models, the expression of macrophage-associated Sdc-1 became increasingly prominent within theaortic wall. As evident by the significant increase in aneu-rysm diameter in the elastase perfusion model and an increasein the incidence of aneurysm formation and frequency ofaortic rupture in the angiotensin II infusion model, macro-phage expressed Sdc-1 appears to play a protective role. BMtransplantation studies confirmed that deficiency of Sdc-1 inmarrow-derived cells significantly contributed to AAA sever-ity. Medial elastin degradation and MMP analysis revealedgreater proteolytic activity within the aortic wall in Sdc-1�/�
Figure 5. Expression of pro- and anti-inflammatory mediators within aneurysmal tissue of Sdc-1�/� and Sdc-1�/� mice. A to D, Quan-titative mRNA expression in aortic tissue. n�5. A, Interferon (IFN)-�. B, interleukin (IL)-4. C, IL-10. D, Foxp3. **P�0.01. E to I, Localiza-tion of Treg cells within aneurysmal adventitia. E, Sdc-1�/� mice, day 7. Scale bar�100 �m. F, Sdc-1�/� mice, day 7. G, Sdc-1�/�
mice, day 14. H, Sdc-1�/� mice, day 14. I, Foxp3-positive cell counts. n�3. *P�0.05, **P�0.01. All data represent the meanSEM.
Xiao et al Protective Role of Syndecan-1 in AAA 393
at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from

mice. Gel zymography alone does not allow us to attribute theincrease in Sdc-1�/� proteolytic activity to a specific cell typewithin the aortic wall. However, several reports have nowdemonstrated that within aortic aneurysm tissue, macro-phages and smooth muscle cells are the primary source ofMMP-9 and MMP-2, respectively.17,25 Flow cytometry ofdigested AAA tissue confirmed increased inflammatory cellrecruitment in Sdc-1�/� samples; accordingly, by absolutenumber, Sdc-1�/� aneurysms contained a greater number ofneutrophils, macrophages, and T cells. We chose to focusfurther efforts on T-cell infiltration after subsequent analysisof individual neutrophil, macrophage, and T-cell populationsas a proportion of total CD45� signal revealed a significantlyhigher percentage of T cells in Sdc-1�/� mice at both 7 and14 days. We characterized a CD4�/IFN-� dominant responsein our elastase-induced model of AAA and used depletionstudies to confirm the role of T cells in aneurysmal dilatation.Furthermore, in reporting dampened expression of IL-10 andFoxp3, we provide evidence that Sdc-1�/� mice display areduced capacity to limit inflammation within the aortic walland, in vitro, we demonstrate the capacity of Sdc-1-expressing macrophages to control the local chemokineenvironment. Thus, we conclude that Sdc-1-expressing mac-rophages serve as a critical component in the vessel wallanti-inflammatory response. Notably, regulatory macro-phages would exert influence through multiple pathwayswithin aneurysm tissue; however, the current report directsfocus toward the specific modulation of the T-cell response.
T lymphocytes have long been recognized as a significantinfiltrate in AAA, yet the specificity of adaptive immunity inaneurysm pathogenesis remains controversial and undefined.33,34
T-cell receptor gene expression has been probed in human tissueto provide evidence for selective, antigen specific activation.Yen et al35 analyzed 5 human explants, reporting polyclonal ornonrestricted diversity, and Platsoucas et al36 examined 10human explants and reported evidence for oligoclonal T-cell
populations, suggesting antigen specific expansion. Many im-munologic triggers have been considered, including microbialinfection, molecular mimicry, and products of vessel wallproteolysis.37 Infiltrating T cells provide a powerful vehicle formediating proinflammatory cytokine release by macrophages.Thus, many studies have sought to characterize the dominantcytokine profile during the course of AAA formation. Xiong etal reported the critical role of CD4� T cells and IFN-� in amouse model of CaCl2-induced aneurysm formation.27 Thepredominant presence of CD4� T cells with high levels of IFN-�or Th1-type transcripts have also been reported in humanAAA.38 In contrast, using a murine model of aortic transplanta-tion, Shimizu et al26 reported that an IL-4 or Th2 dominantresponse was critical to aneurysm formation, and King et al39
reported a protective role for IFN-� in angiotensin-II inducedAAA; both reports suggest that a Th1 response may limitaneurysm formation. In effect, the literature addressing the roleof T lymphocytes in AAA is conflicting and highlights the needfor continued investigation. Moreover, the current paradigmbroadens the realm of T-cell subsets and cautions against simpledefinitions that rely on terminal commitment of Th1 or Th2responses.40,41
HS-chemokine binding interactions are well established.Thus, it is reasonable to hypothesize that Sdc-1 interferes withT-cell infiltration through HS-mediated sequestration of T-cell–specific chemokines. Furthermore, augmented T-cell responseshave been previously reported in Sdc-1�/� mice, and Sdc-1sequestration of T-cell–specific chemokines has been shown toinhibit T-cell migration.9,10 We tested the ability of HS tointerfere with effector CD4� T-cell migration to chemokinesspecifically upregulated in our elastase-induced model of aneu-rysm formation. HS significantly inhibited chemotaxis to CCL2,CCL3, and CXCL9, all potentially important chemokines indriving T-cell migration in AAA formation.42–44 We did notobserve a significant impact on CCL5 migration; however,regulated upon activation, normal T cell expressed and secretedhas been reported to be more discriminatory in glycosaminogly-can binding.45 Adventitial localization and sequential infiltrationof macrophage and T-cell populations is presumed to be criticalin the specific inhibition of the T-cell population by Sdc-1.Macrophage Sdc-1 expression is firmly established in the ad-ventitia, as the T-cell population emerges during aneurysmformation; such spatiotemporal expression is believed to impartsome specificity to HS proteoglycan–chemokine binding inter-actions.6 In addition to chemokine sequestration, it is alsoplausible that Sdc-1-expressing macrophages modulate thechemokine environment in a more direct manner; we chose tocharacterize the chemokine expression profile of Sdc-1 macro-phages against classically activated M1 inflammatory macro-phages. Sdc-1-expressing macrophages display a significantlydampened chemokine profile compared with M1 macrophages,such reduced expression of inflammatory chemokines supportsthe notion that Sdc-1-expressing macrophages function to regu-late the inflammatory response during AAA. Notably, weobserved no evidence of Sdc-1 expression on macrophagesdriven to an M1 phenotype; continued investigations within ourlaboratory are directed at characterizing the specific subtype ofSdc-1-expressing macrophages. Collectively, these findings de-fine a protective role for Sdc-1 expression during the formation
Figure 6. Heparan sulfate (HS)–mediated inhibition of CD4 T-cellchemotaxis to abdominal aortic aneurysm (AAA)–expressedchemokines. Shown is the migration of CD4 cells to CXCL9,CCL2, CCL3, and CCL5 with or without HS (50 �g/mL) in aTranswell migration assay. Chemotactic index was calculatedusing basal migration media (RPMI�0.1% bovine serum albu-min) as a control; there was no difference observed for back-ground movement in basal media with or without HS. n�3.*P�0.05. Data represent the meanSEM.
394 Arterioscler Thromb Vasc Biol February 2012
at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from

of AAA. By modulating the chemokine environment, Sdc-1-expressing macrophages can influence the participation of Tlymphocytes and dampen the magnitude of the inflammatoryresponse. However, an important caveat of our study is that Tlymphocyte participation in AAA may be governed throughmultiple pathways, including trafficking, survival, and prolifer-ation. Although many reports have documented the significanceof chemokine driven lymphocyte trafficking to chronicallyinflamed aortic tissue,43,46–49 alternate hypotheses warrant fur-ther study.
Our in vivo data support the notion that Sdc-1 expressionmay be an important component in a series of programmedevents designed to downregulate the inflammatory responsewithin the aortic wall. Indeed, IL-10 was upregulated only inSdc-1�/� mice, and expression of the regulatory T-cellmarker Foxp3 was much lower in the absence of Sdc-1.Standard immunohistochemistry confirmed the localizationof regulatory T cells to aneurysm tissue, and subsequentquantification revealed a significantly greater presence ofregulatory T cells in Sdc-1�/� AAA. Regulatory T cells canspecifically home to inflamed tissue and effectively suppressboth innate and adaptive immune responses.50 Significantly,regulatory T cells orchestrate suppression of innate immunitythrough release of anti-inflammatory cytokines, such as IL-10and transforming growth factor-�, and they have been impli-cated in dampening the inflammatory potential of neighbor-ing macrophages.51,52 Indeed, regulatory T cells may contrib-ute to elevated IL-10 transcripts observed in Sdc-1�/� AAA.Given reported macrophage/regulatory T-cell cross-talk, it istempting to speculate that polarized macrophage populationsinfluence localization of regulatory T cells to sites of inflam-mation. However, further study is warranted to assess themediators, which govern both trafficking and suppressorfunction of regulatory T cells in aneurysm tissue.
Our study was designed to examine the role of macro-phage-expressed Sdc-1 in AAA. Because syndecans havebeen reported to both augment and inhibit inflammatorysignaling events, predicting the role of Sdc-1 in AAA was notintuitive. Moreover, studying the functional significance ofSdc-1 in macrophages, a cell population fundamentally re-garded as inflammatory in chronic disease, complicatedinterpretation of results. The significance of our findings liesin the seemingly contradictory observation that an endoge-nous anti-inflammatory counterbalance emanates from anAAA localized population of macrophages. However, thepresence of a counterbalance is expected within the currentparadigm of the inflammatory cascade, and macrophages arerecognized as key components in the switch to inflammatoryresolution. Microenvironmentally derived signals give rise tomacrophages that are polarized with specific functional prop-erties, including: classically activated, proinflammatory M1macrophages, and alternatively activated, potentially anti-inflammatory M2 macrophages. Inherent negative feedbackcontrol in macrophage activation and functional plasticitywould be expected to result in an overlapping M1/M2presence in chronic inflammation. In both human and mouseAAA, Sdc-1 expression is augmented in a subset of localizedmacrophages; herein, we present evidence that Sdc-1 isinduced on regulatory macrophages in the context of a
programmed response to dampen inflammation. Indeed, it hasbeen suggested that chemokine modulation is a criticalfunctional attribute in alternatively activated macro-phages.53,54 However, the effectiveness of this negative reg-ulation is most likely limited in a pathological setting. By thevery nature of chronic inflammation, the native processes thatserve to restore homeostasis can be overpowered by theexaggerated degree of the inflammatory insult. Within thisparadox lie new targets for therapeutic intervention. Charac-terizing the molecular mechanisms that regulate the intrinsiccounterbalance is an important step toward the developmentof new approaches to promote the resolution of chronicinflammation in the vascular wall.
Sources of FundingThis work was funded by National Institutes of Health GrantHL060903 (to E.L.C.) and fellowships from the American College ofSurgeons (to S.V.S.) and the American Heart Association (to J.A.).
DisclosuresNone.
References1. Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, Zheng
ZJ, Flegal K, O’Donnell C, Kittner S, Lloyd-Jones D, Goff DC Jr, Hong Y,Adams R, Friday G, Furie K, Gorelick P, Kissela B, Marler J, Meigs J, RogerV, Sidney S, Sorlie P, Steinberger J, Wasserthiel-Smoller S, Wilson M, WolfP; American Heart Association Statistics Committee and Stroke StatisticsCommittee. Heart disease and stroke statistics—2006 update: a report fromthe American Heart Association Statistics Committee and Stroke StatisticsCommittee. Circulation. 2006;113:e85–e151.
2. Brewster DC, Cronenwett JL, Hallett JW Jr, Johnston KW, Krupski WC,Matsumura JS. Guidelines for the treatment of abdominal aortic aneu-rysms: report of a subcommittee of the Joint Council of the AmericanAssociation for Vascular Surgery and Society for Vascular Surgery.J Vasc Surg. 2003;37:1106–1117.
3. Yoshimura K, Aoki H, Ikeda Y, Furutani A, Hamano K, Matsuzaki M.Regression of abdominal aortic aneurysm by inhibition of c-junN-terminal kinase in mice. Ann N Y Acad Sci. 2006;1085:74–81.
4. Prall AK, Longo GM, Mayhan WG, Waltke EA, Fleckten B, ThompsonRW, Baxter BT. Doxycycline in patients with abdominal aortic aneu-rysms and in mice: comparison of serum levels and effect on aneurysmgrowth in mice. J Vasc Surg. 2002;35:923–929.
5. Subramanian SV, Fitzgerald ML, Bernfield M. Regulated shedding ofsyndecan-1 and -4 ectodomains by thrombin and growth factor receptoractivation. J Biol Chem. 1997;272:14713–14720.
6. Bartlett AH, Hayashida K, Park PW. Molecular and cellular mechanismsof syndecans in tissue injury and inflammation. Mol Cells. 2007;24:153–166.
7. Alexopoulou AN, Multhaupt HA, Couchman JR. Syndecans in woundhealing, inflammation and vascular biology. Int J Biochem Cell Biol.2007;39:505–528.
8. Rops AL, Gotte M, Baselmans MH, van den Hoven MJ, Steenbergen EJ,Lensen JF, Wijnhoven TJ, Cevikbas F, van den Heuvel LP, van Kup-pevelt TH, Berden JH, van der Vlag J. Syndecan-1 deficiency aggravatesanti-glomerular basement membrane nephritis. Kidney Int. 2007;72:1204–1215.
9. Hayashida K, Chen Y, Bartlett AH, Park PW. Syndecan-1 is an in vivosuppressor of Gram-positive toxic shock. J Biol Chem. 2008;283:19895–19903.
10. Xu J, Park PW, Kheradmand F, Corry DB. Endogenous attenuation ofallergic lung inflammation by syndecan-1. J Immunol. 2005;174:5758–5765.
11. Vanhoutte D, Schellings MW, Gotte M, Swinnen M, Herias V, Wild MK,Vestweber D, Chorianopoulos E, Cortes V, Rigotti A, Stepp MA, Van deWerf F, Carmeliet P, Pinto YM, Heymans S. Increased expression ofsyndecan-1 protects against cardiac dilatation and dysfunction after myo-cardial infarction. Circulation. 2007;115:475–482.
Xiao et al Protective Role of Syndecan-1 in AAA 395
at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from

12. Yeaman C, Rapraeger AC. Post-transcriptional regulation of syndecan-1expression by cAMP in peritoneal macrophages. J Cell Biol. 1993;122:941–950.
13. Kim C, Wilcox-Adelman S, Sano Y, Tang WJ, Collier RJ, Park JM.Antiinflammatory cAMP signaling and cell migration genes co-opted bythe anthrax bacillus. Proc Natl Acad Sci U S A. 2008;105:6150–6155.
14. Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK,Martinez A, Maenhaut C, Bos JL, Genieser HG, Doskeland SO. cAMPanalog mapping of Epac1 and cAMP kinase: discriminating analogsdemonstrate that Epac and cAMP kinase act synergistically to promotePC-12 cell neurite extension. J Biol Chem. 2003;278:35394–35402.
15. Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cuttingedge: macrophage inhibition by cyclic amp (cAMP): differential roles ofprotein kinase a and exchange protein directly activated by cAMP-1.J Immunol. 2005;174:595–599.
16. Wen J, Wang P, Smith SV, Haller CA, Chaikof EL. Syndecans aredifferentially expressed during the course of aortic aneurysm formation.J Vasc Surg. 2007;46:1014–1025.
17. Pyo R, Lee JK, Shipley JM, Curci JA, Mao D, Ziporin SJ, Ennis TL,Shapiro SD, Senior RM, Thompson RW. Targeted gene disruption ofmatrix metalloproteinase-9 (gelatinase B) suppresses development ofexperimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649.
18. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM.Development of Th1 CD4� T cells through IL-12 produced by listeria-induced macrophages. Science. 1993;260:547–549.
19. Lara-Tejero M, Pamer EG. T cell responses to listeria monocytogenes.Curr Opin Microbiol. 2004;7:45–50.
20. Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical andfunctional characterization of three activated macrophage populations.J Leukoc Biol. 2006;80:1298–1307.
21. Alexander CM, Reichsman F, Hinkes MT, Lincecum J, Becker KA,Cumberledge S, Bernfield M. Syndecan-1 is required for Wnt-1-inducedmammary tumorigenesis in mice. Nat Genet. 2000;25:329–332.
22. Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes ath-erosclerotic lesions and aneurysms in apolipoprotein E-deficient mice.J Clin Invest. 2000;105:1605–1612.
23. Weiss D, Kools JJ, Taylor WR. Angiotensin II-induced hypertensionaccelerates the development of atherosclerosis in apoE-deficient mice.Circulation. 2001;103:448–454.
24. Sun J, Sukhova GK, Yang M, Wolters PJ, MacFarlane LA, Libby P, SunC, Zhang Y, Liu J, Ennis TL, Knispel R, Xiong W, Thompson RW,Baxter BT, Shi GP. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J Clin Invest. 2007;117:3359–3368.
25. Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrixmetalloproteinases 2 and 9 work in concert to produce aortic aneurysms.J Clin Invest. 2002;110:625–632.
26. Shimizu K, Shichiri M, Libby P, Lee RT, Mitchell RN. Th2-predominantinflammation and blockade of IFN-� signaling induce aneurysms inallografted aortas. J Clin Invest. 2004;114:300–308.
27. Xiong W, Zhao Y, Prall A, Greiner TC, Baxter BT. Key roles of CD4�T cells and IFN-� in the development of abdominal aortic aneurysms ina murine model. J Immunol. 2004;172:2607–2612.
28. Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F.CD4�CD25� T(R) cells suppress innate immune pathology throughcytokine-dependent mechanisms. J Exp Med. 2003;197:111–119.
29. Schottelius AJ, Mayo MW, Sartor RB, Baldwin AS Jr. Interleukin-10signaling blocks inhibitor of �B kinase activity and nuclear factor �BDNA binding. J Biol Chem. 1999;274:31868–31874.
30. Charo IF, Ransohoff RM. The many roles of chemokines and chemokinereceptors in inflammation. N Engl J Med. 2006;354:610–621.
31. Pearce WH, Koch AE. Cellular components and features of immuneresponse in abdominal aortic aneurysms. Ann N Y Acad Sci. 1996;800:175–185.
32. Newman KM, Jean-Claude J, Li H, Ramey WG, Tilson MD. Cytokinesthat activate proteolysis are increased in abdominal aortic aneurysms.Circulation. 1994;90:II224–II227.
33. Koch AE, Haines GK, Rizzo RJ, Radosevich JA, Pope RM, RobinsonPG, Pearce WH. Human abdominal aortic aneurysms. Immunophenotypicanalysis suggesting an immune-mediated response. Am J Pathol. 1990;137:1199–1213.
34. Bobryshev YV, Lord RS. Vascular-associated lymphoid tissue (VALT)involvement in aortic aneurysm. Atherosclerosis. 2001;154:15–21.
35. Yen HC, Lee FY, Chau LY. Analysis of the T cell receptor v� repertoirein human aortic aneurysms. Atherosclerosis. 1997;135:29–36.
36. Platsoucas CD, Lu S, Nwaneshiudu I, Solomides C, Agelan A, Ntaoula N,Purev E, Li LP, Kratsios P, Mylonas E, Jung WJ, Evans K, Roberts S, LuY, Layvi R, Lin WL, Zhang X, Gaughan J, Monos DS, Oleszak EL,White JV. Abdominal aortic aneurysm is a specific antigen-driven T celldisease. Ann N Y Acad Sci. 2006;1085:224–235.
37. Kuivaniemi H, Platsoucas CD, Tilson MD III. Aortic aneurysms: animmune disease with a strong genetic component. Circulation. 2008;117:242–252.
38. Galle C, Schandene L, Stordeur P, Peignois Y, Ferreira J, Wautrecht JC,Dereume JP, Goldman M. Predominance of type 1 CD4� T cells inhuman abdominal aortic aneurysm. Clin Exp Immunol. 2005;142:519–527.
39. King VL, Lin AY, Kristo F, Anderson TJ, Ahluwalia N, Hardy GJ,Owens AP III, Howatt DA, Shen D, Tager AM, Luster AD, Daugherty A,Gerszten RE. Interferon-� and the interferon-inducible chemokineCXCL10 protect against aneurysm formation and rupture. Circulation.2009;119:426–435.
40. Bluestone JA, Mackay CR, O’Shea JJ, Stockinger B. The functionalplasticity of T cell subsets. Nat Rev Immunol. 2009;9:811–816.
41. Mucida D, Cheroutre H. The many face-lifts of CD4 T helper cells. AdvImmunol. 2010;107:139–152.
42. Colonnello JS, Hance KA, Shames ML, Wyble CW, Ziporin SJ,Leidenfrost JE, Ennis TL, Upchurch GR Jr, Thompson RW. Transientexposure to elastase induces mouse aortic wall smooth muscle cell pro-duction of MCP-1 and RANTES during development of experimentalaortic aneurysm. J Vasc Surg. 2003;38:138–146.
43. Zhao L, Moos MP, Grabner R, Pedrono F, Fan J, Kaiser B, John N,Schmidt S, Spanbroek R, Lotzer K, Huang L, Cui J, Rader DJ, Evans JF,Habenicht AJ, Funk CD. The 5-lipoxygenase pathway promotes patho-genesis of hyperlipidemia-dependent aortic aneurysm. Nat Med. 2004;10:966–973.
44. MacTaggart JN, Xiong W, Knispel R, Baxter BT. Deletion of CCR2 butnot CCR5 or CXCR3 inhibits aortic aneurysm formation. Surgery. 2007;142:284–288.
45. Kuschert GS, Coulin F, Power CA, Proudfoot AE, Hubbard RE,Hoogewerf AJ, Wells TN. Glycosaminoglycans interact selectivelywith chemokines and modulate receptor binding and cellularresponses. Biochemistry. 1999;38:12959 –12968.
46. Galkina E, Harry BL, Ludwig A, Liehn EA, Sanders JM, Bruce A, WeberC, Ley K. CXCR6 promotes atherosclerosis by supporting T-cell homing,interferon-� production, and macrophage accumulation in the aortic wall.Circulation. 2007;116:1801–1811.
47. Ocana E, Perez-Requena J, Bohorquez JC, Brieva JA, Rodriguez C.Chemokine receptor expression on infiltrating lymphocytes fromabdominal aortic aneurysms: role of CXCR4-CXCL12 in lymphoidrecruitment. Atherosclerosis. 2008;200:264–270.
48. Tang EH, Shvartz E, Shimizu K, Rocha VZ, Zheng C, Fukuda D, Shi GP,Sukhova G, Libby P. Deletion of EP4 on bone marrow-derived cellsenhances inflammation and angiotensin II-induced abdominal aortic an-eurysm formation. Arterioscler Thromb Vasc Biol. 2011;31:261–269.
49. Golledge J, Clancy P, Moran C, Biros E, Rush C, Walker P, Norman P.The novel association of the chemokine CCL22 with abdominal aorticaneurysm. Am J Pathol. 2010;176:2098–2106.
50. Tang Q, Bluestone JA. The Foxp3� regulatory T cell: a jack of all trades,master of regulation. Nat Immunol. 2008;9:239–244.
51. Taams LS, van Amelsfort JM, Tiemessen MM, Jacobs KM, de Jong EC,Akbar AN, Bijlsma JW, Lafeber FP. Modulation of monocyte/macro-phage function by human CD4�CD25� regulatory T cells. HumImmunol. 2005;66:222–230.
52. Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S,Taams LS. CD4�CD25�Foxp3� regulatory T cells induce alternativeactivation of human monocytes/macrophages. Proc Natl Acad Sci U S A.2007;104:19446–19451.
53. D’Amico G, Frascaroli G, Bianchi G, Transidico P, Doni A, Vecchi A,Sozzani S, Allavena P, Mantovani A. Uncoupling of inflammatorychemokine receptors by IL-10: generation of functional decoys. NatImmunol. 2000;1:387–391.
54. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. Thechemokine system in diverse forms of macrophage activation and polar-ization. Trends Immunol. 2004;25:677–686.
396 Arterioscler Thromb Vasc Biol February 2012
at Harvard University on June 16, 2012http://atvb.ahajournals.org/Downloaded from
![Statin Benefits and Risks [Read-Only] - Welcome to URMC · Statin Benefits and Risks ... Arterioscler Thromb Vasc Biol. 2009; ... (but up to 46% in METSIM) • Risk Factors – Age](https://img.pdfslide.us/doc/110x75/5ad579e37f8b9a1a028d2357/statin-benefits-and-risks-read-only-welcome-to-urmc-benefits-and-risks-arterioscler.jpg)


















![Emergent Reversal of Anticoagulation in the ED and ICU ... · PDF fileLovenox (enoxaparin sodum injection) [prescribing information]. 2013 Oct. Thromb Haemost 2015;113:931. ... •](https://img.pdfslide.us/doc/110x75/5a9627c87f8b9ab6188c8799/emergent-reversal-of-anticoagulation-in-the-ed-and-icu-enoxaparin-sodum-injection.jpg)









