Embed Size (px)
Citation preview

A new story about an old gene
Cécile ACQUAVIVA-BOURDAIN
Service des Maladies Héréditaires du Métabolisme
Hospices Civils de LyonFrance

“Assembly of COX in SURF1-p null mutants is blocked at an early step.
Control
In 1999…, Tiranti et al. reported in LSCOX
BMdeC
and in 2013?....
However, detection of residual amounts of fully assembled complex suggests a certain degree of redundancy.” Hum Mol Genet 1999, 13, 2533-2540

Echaniz et al report SURF1 deficiency in CMTCOX
BMdeC
but markedly reduced, amount of fully assembled complex IV.” Neurology 2013, 81, 1-8
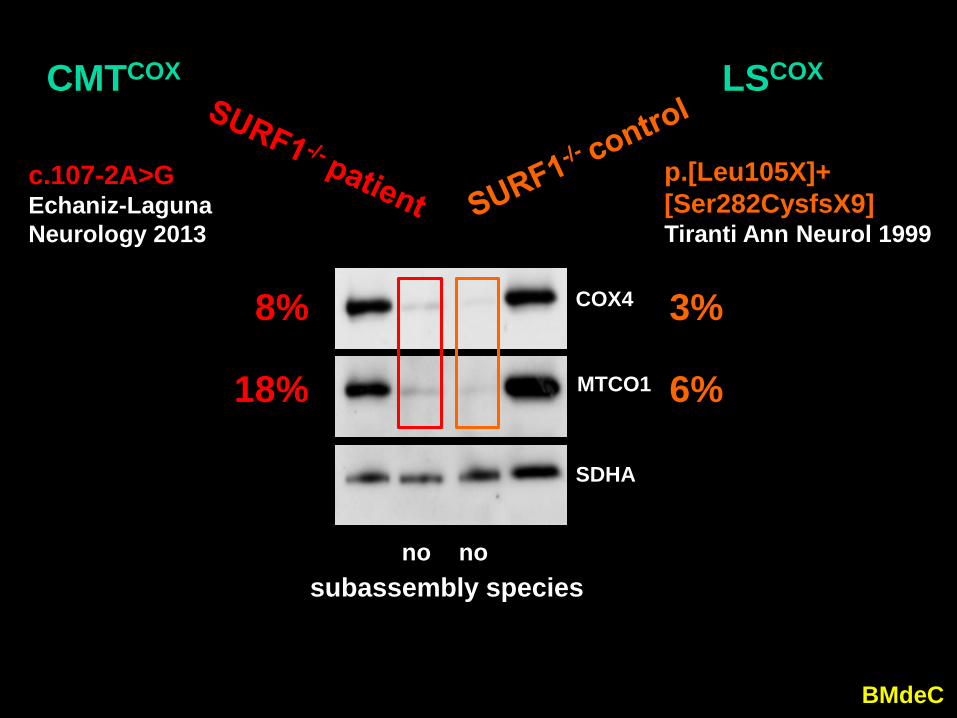
“In the c.107-2A>G samples, the absence of SURF1protein 1 2 3
SURF1
SDHB 1 2 3 4
COX4
MTCO1
SDHA
is associated with a detectable,
BMdeC
nosubassembly species
no
1 2 3 4
COX4
MTCO1
SDHA
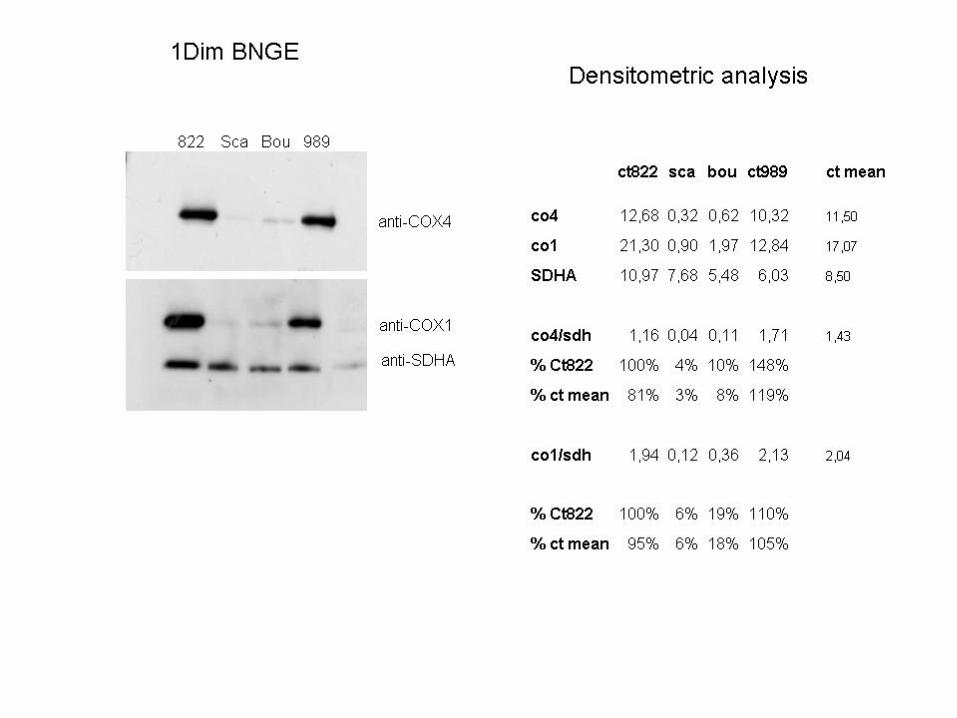
18%
8%
c.107-2A>GEchaniz-LagunaNeurology 2013
CMTCOX
3%
6%
p.[Leu105X]+[Ser282CysfsX9]Tiranti Ann Neurol 1999
LSCOX
BMdeC
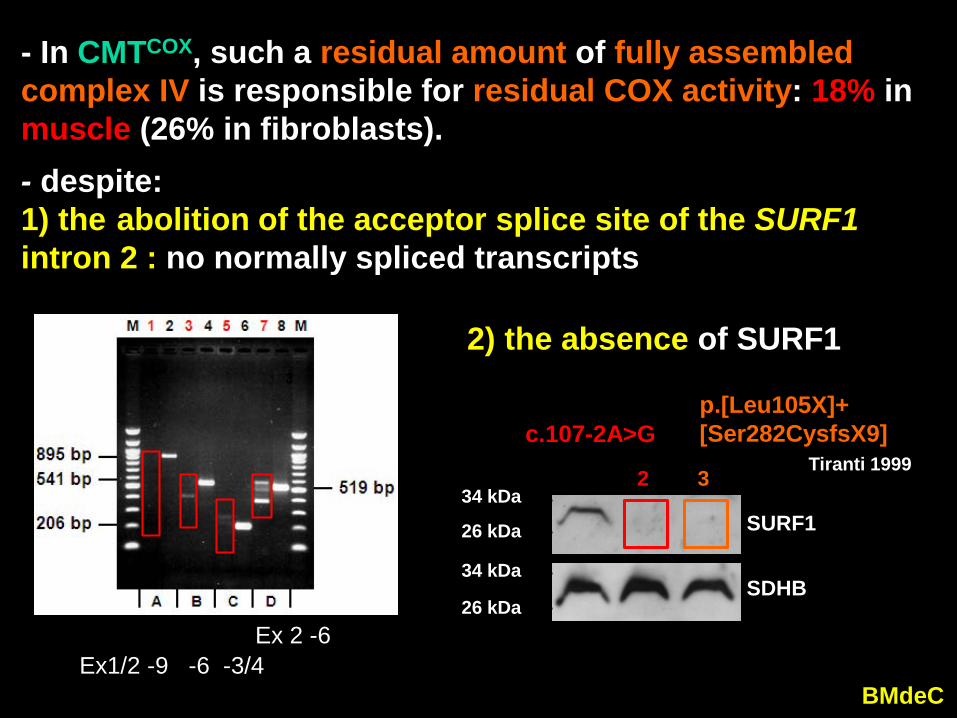
p.[Leu105X]+[Ser282CysfsX9]
Tiranti 1999c.107-2A>G
1 2 3
SURF1
SDHB
34 kDa
34 kDa
26 kDa
26 kDa
- despite: 1) the abolition of the acceptor splice site of the SURF1 intron 2 : no normally spliced transcripts
Ex1/2 -9 -6 -3/4 Ex 2 -6
- In CMTCOX, such a residual amount of fully assembled complex IV is responsible for residual COX activity: 18% inmuscle (26% in fibroblasts).
2) the absence of SURF1
BMdeC
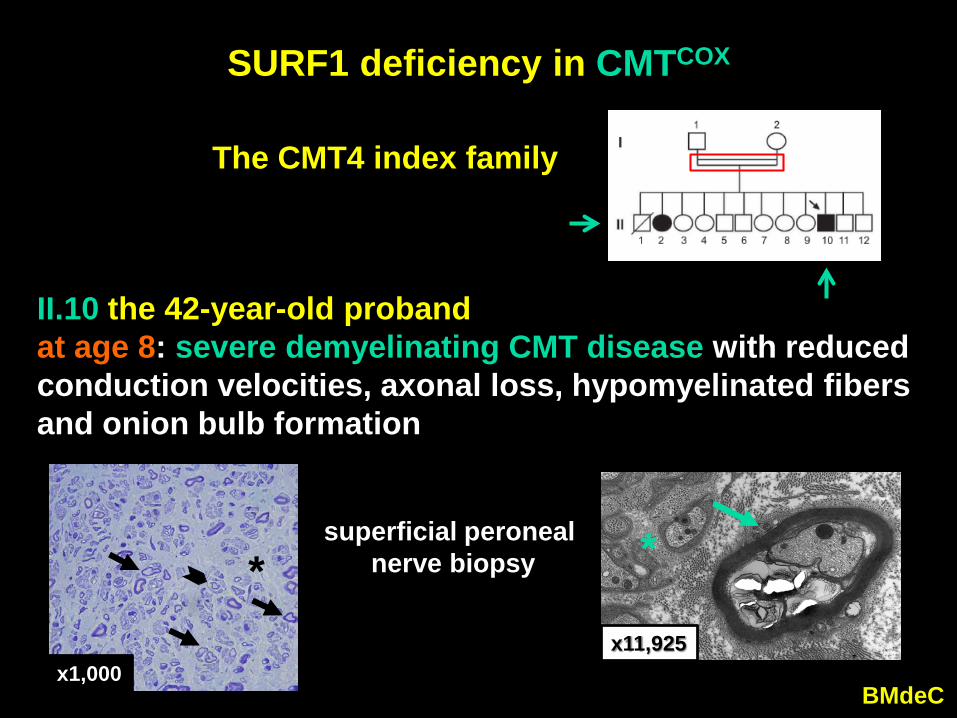
SURF1 deficiency in CMTCOX
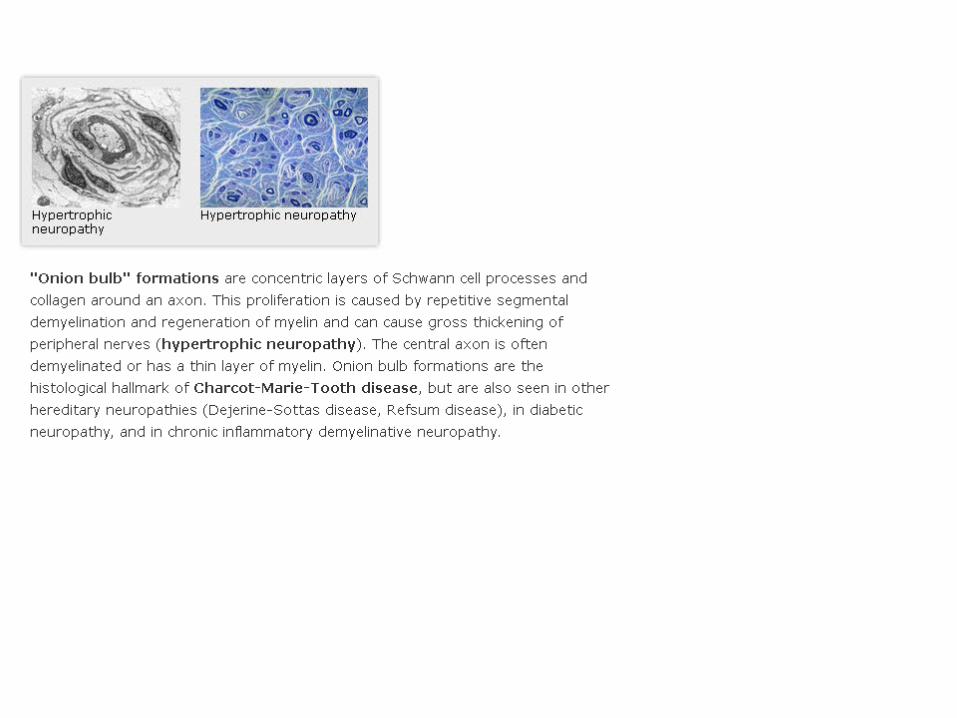
II.10 the 42-year-old probandat age 8: severe demyelinating CMT disease with reduced conduction velocities, axonal loss, hypomyelinated fibers and onion bulb formation
*
x1,000
superficial peronealnerve biopsy
The CMT4 index family
*
x11,925
BMdeC
The CMT4 index family (2)
at age 42: - still able to walk, but no more than 30 m- very slow progression of the CMT disease- nystagmus and mild hearing loss- lactic acidosis (3.3 mmol/l)- no mutation in CMT4-causing genes: PMP22, MPZ, GJB1, GDAP1, PRX, SH3TC2, MTMR2
brain MRIhyperintense lesions in both putamina: typical of LS
MUSCLE and SKIN biopsiesmitochondrial disease suspected
BMdeC
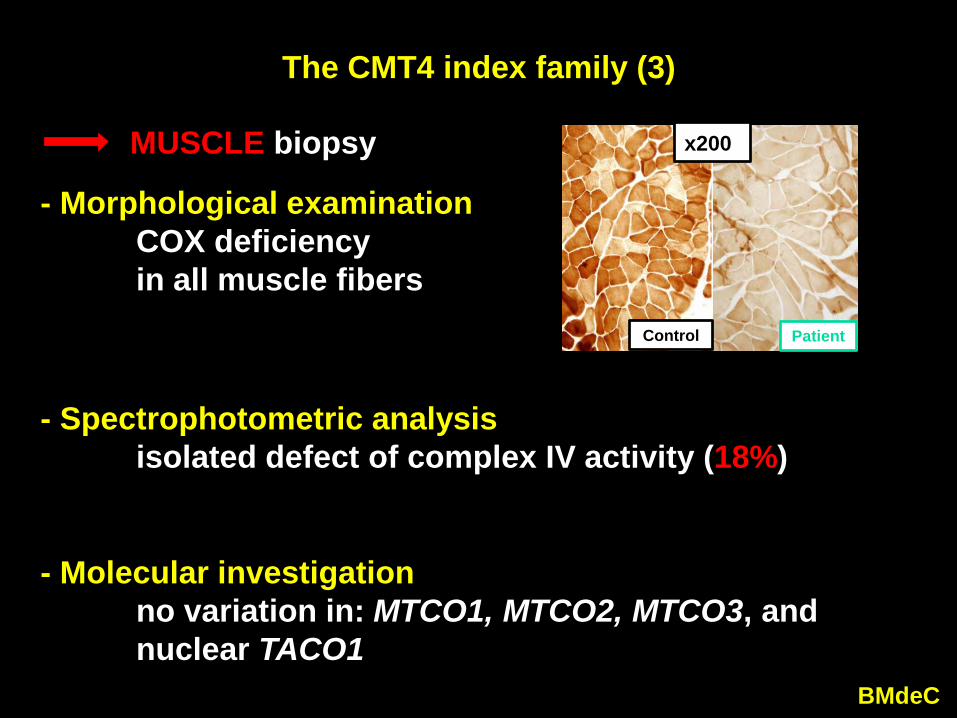
The CMT4 index family (3)
- Morphological examinationCOX deficiency in all muscle fibers
- Spectrophotometric analysisisolated defect of complex IV activity (18%)
- Molecular investigationno variation in: MTCO1, MTCO2, MTCO3, and nuclear TACO1
MUSCLE biopsy
Control Patient
x200)
BMdeC
The CMT4 index family (4)
II.2 the 57-year-old elder sister
before age 10: severe demyelinating polyneuropathy with the same symptoms after age 40: marked cerebellar ataxia, nystagmus, hearing loss, kyphoscoliosis, lactic acidosis
brain MRIunconclusive
MUSCLE biopsywas refused
BMdeC
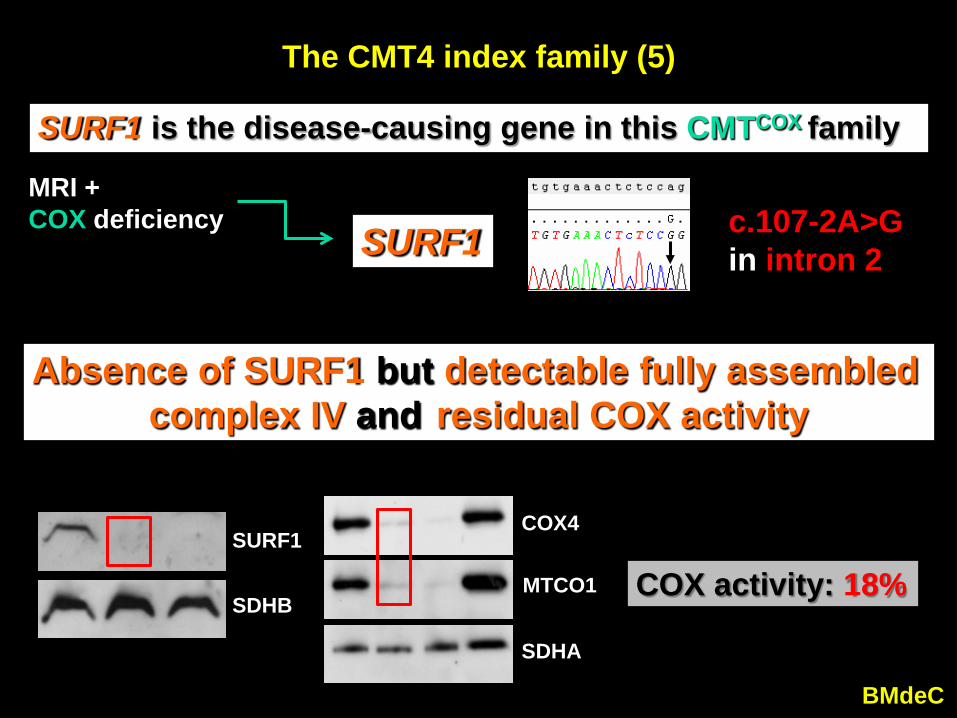
The CMT4 index family (5)
SURF1 is the disease-causing gene in this CMTCOX family
SURF1 c.107-2A>Gin intron 2
MRI + COX deficiency
Absence of SURF1 but detectable fully assembled complex IV and residual COX activity
1 2
SURF1
SDHB
1 2 3
COX4
MTCO1
SDHA
COX activity: 18%

BMdeC
What about SURF1 in other CMT4 patients?
- in 40 unrelated patients with genetically undefined CMT4- another patient with two additional pathogenic variants: p. [Arg192Trp] + [Leu267GlufsX24]at age 3: severe demyelinating CMT disease at age 10: mild cerebellar ataxia + abnormal MRI
• 3 patients with SURF1-associated CMT4 presented with: severe childhood-onset neuropathy,
motor nerve conduction velocities < 25 m/s, and lactic acidosis.
• 2/3 patients had brain MRI abnormalities, and developed cerebellar ataxia years after polyneuropathy.
• 2/41 families (5%) with disease-causing SURF1 variants

- Is the COX defect modulated as reported in yeaststrains with ablated SHY1 (the SURF1 ortholog)(Bestwick 2010)?+ by adaptative changes with interacting partners (other COX assembly factors, cyt c…) + and/or by adaptative mechanisms (mtCu++ level)
- Is the variable severity of the phenotype associatedwith the absence of SURF1 depending on theefficiency of compensatory genetic or epigeneticmechanisms in humans?
SURF1-associated LSCOX or CMTCOX?
BMdeC
- Similar or identical mutations: why LS or CMT?

BMdeC
SURF1 should be systematically screened in patients with: 1) childhood-onset severe demyelinating neuropathy
2) additional features 3) brain MRI abnormalities4) cerebellar ataxia developing years after polyneuropathy.
Echaniz-Laguna A, Ghezzi D, Chassagne M, Mayençon M, Padet S, Melchionda L, Rouvet I,
Lannes B, Bozon D, Latour P, Zeviani M, Mousson de Camaret B
Strasbourg-Lyon Milan
Neurology 2013, 81, 1523-1530

Supplemental slides
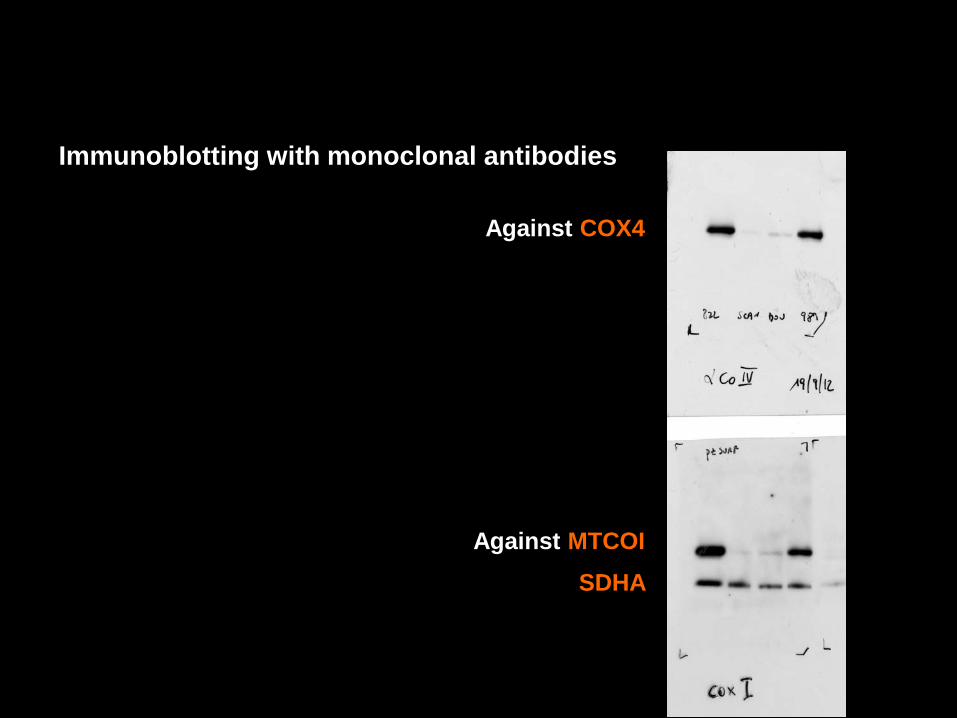
Immunoblotting with monoclonal antibodies
Against MTCOI
Against COX4
SDHA
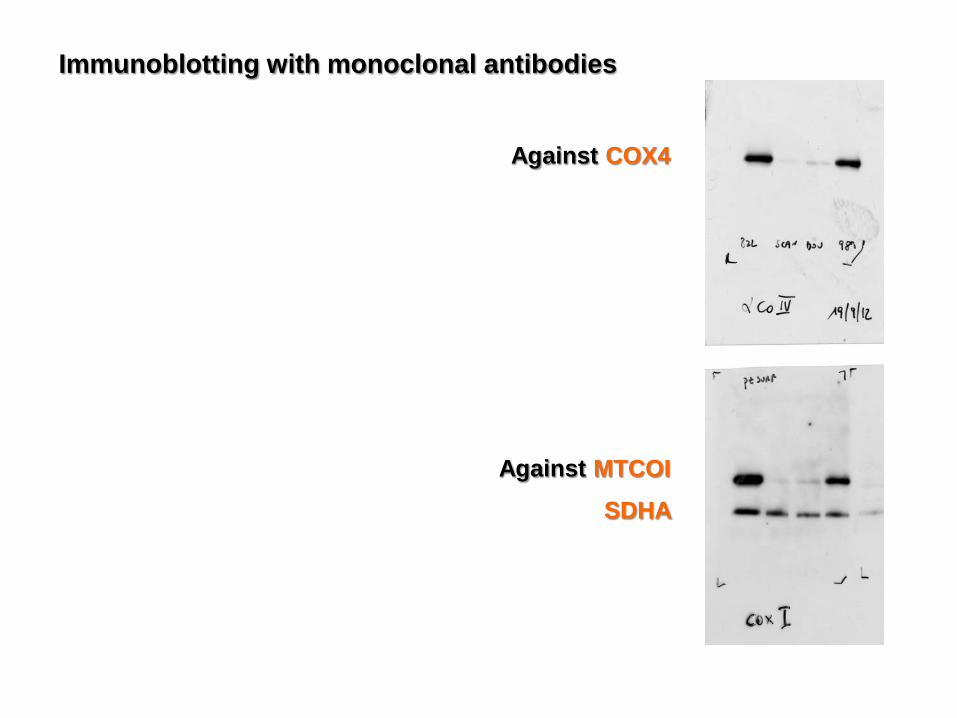
Immunoblotting with monoclonal antibodies
Against MTCOI
Against COX4
SDHA


Classification des CMT
• Formes démyélinisantes: CMT1
► VCM > 38 m/s (nerf médian)
• Formes axonales: CMT2
► VCM < 38 m/s (nerf médian)

Classification des CMT (suite)
• CMT démyélinisant AD: CMT1
• CMT démyélinisant AR: CMT4
• CMT axonal AD: CMT2
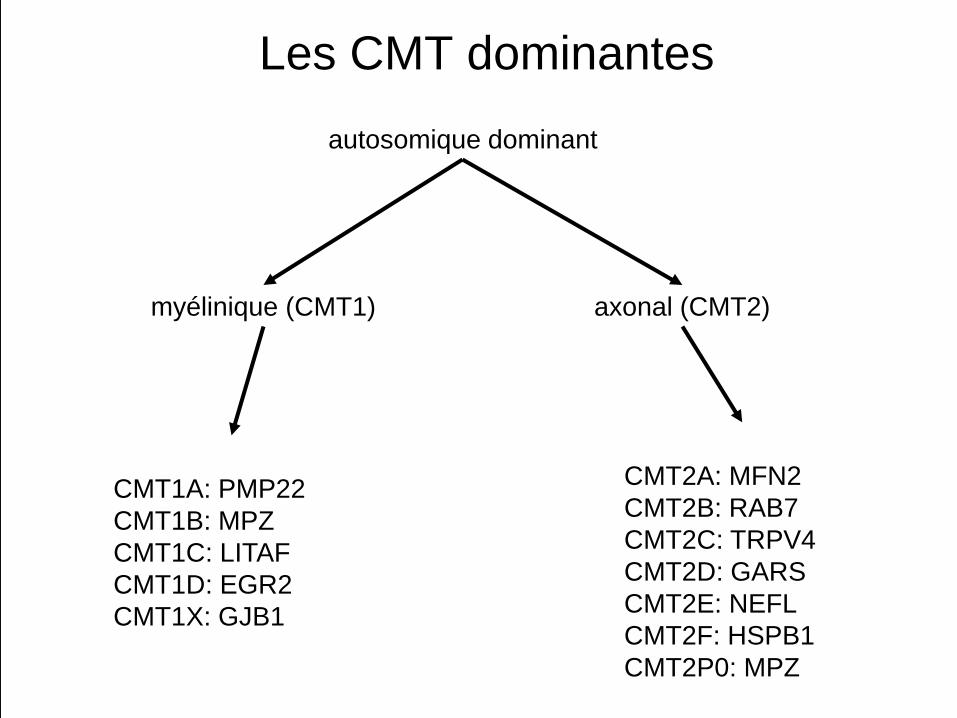
Les CMT dominantes
CMT1A: PMP22CMT1B: MPZCMT1C: LITAFCMT1D: EGR2CMT1X: GJB1
CMT2A: MFN2CMT2B: RAB7CMT2C: TRPV4CMT2D: GARSCMT2E: NEFLCMT2F: HSPB1CMT2P0: MPZ
autosomique dominant
axonal (CMT2)myélinique (CMT1)
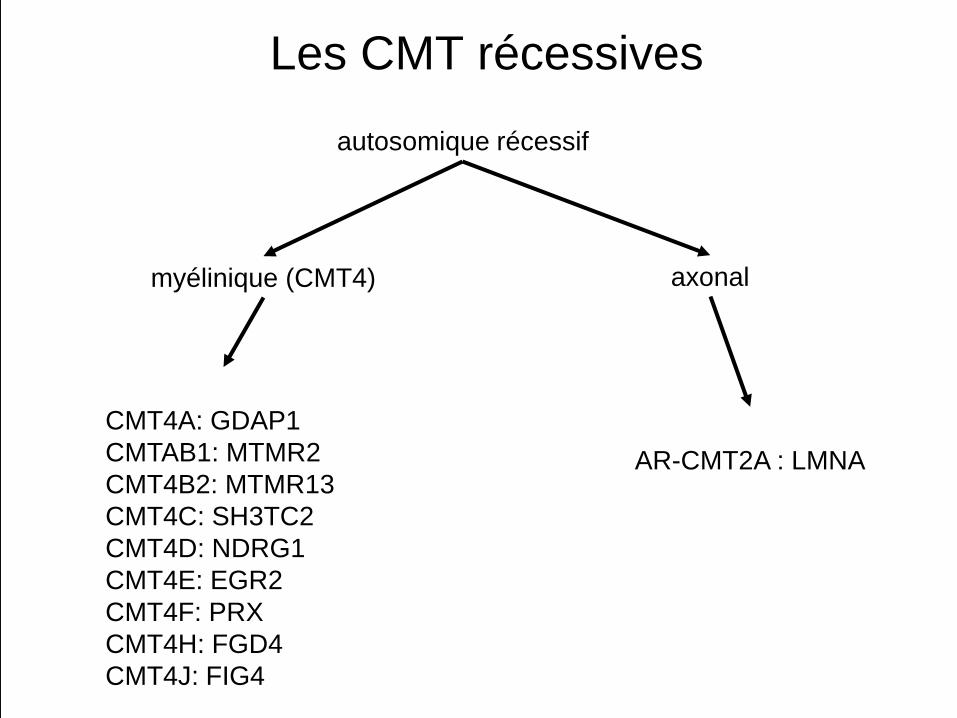
Les CMT récessives
CMT4A: GDAP1CMTAB1: MTMR2CMT4B2: MTMR13CMT4C: SH3TC2CMT4D: NDRG1CMT4E: EGR2CMT4F: PRXCMT4H: FGD4CMT4J: FIG4
AR-CMT2A : LMNA
autosomique récessif
myélinique (CMT4) axonal
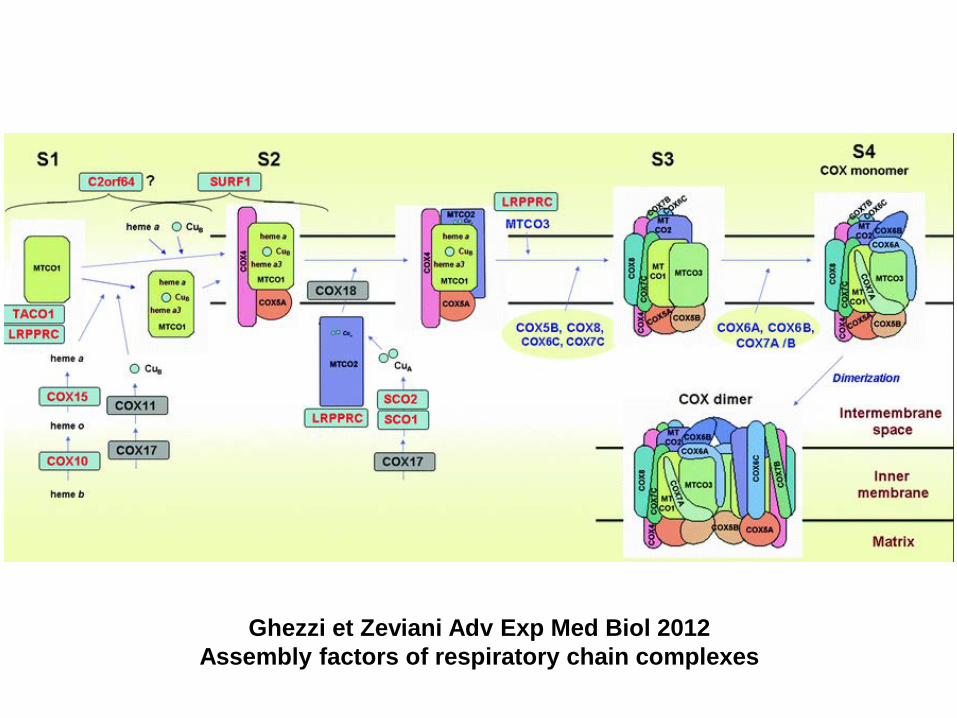
Ghezzi et Zeviani Adv Exp Med Biol 2012 Assembly factors of respiratory chain complexes