Embed Size (px)
Citation preview

DaltonTransactions
PAPER
Cite this: Dalton Trans., 2013, 42, 16056
Received 18th June 2013,Accepted 22nd July 2013
DOI: 10.1039/c3dt51620b
www.rsc.org/dalton
Cationic Group-IV pincer-type complexes forpolymerization and hydroamination catalysis†
Lapo Luconi,a Jerzy Klosin,b Austin J. Smith,b Stéphane Germain,c
Emmanuelle Schulz,c,d Jérôme Hannedouchec,d and Giuliano Giambastiani*a
Neutral ZrIV and Hf IV dimethyl complexes stabilized by unsymmetrical dianionic {N,C,N’} pincer ligands
have been prepared from their corresponding bis-amido complexes upon treatment with AlMe3. Their
structure consists of a central σ-bonded aryl donor group (C) capable of forming robust M–C bonds with
the metal center, enforced by the synergic effect of both the coordination of peripheral donor groups
(N) and the chelating rigid structure of the {N,C,N} ligand framework. Such a combination translates into
systems having a unique balance between stability and reactivity. These ZrIV and Hf IV dimethyl complexes
were converted in situ into cationic species [MIV{N−,C−,N}Me][B(C6F5)4] which are active catalysts for the
room temperature (r.t.) intramolecular hydroamination/cyclization of primary and secondary aminoalk-
enes as well as for the high temperature ethylene–1-octene copolymerizations.
Introduction
Tuning the performance of single site catalysts by designingnovel ligand structures still remains one of the most challen-ging issues for many inorganic and organometallic chemists.Varying the steric and electronic properties of a given ligand’sfamily, while maintaining its donor atom set unchanged, con-stitutes a key tool for controlling and investigating the reacti-vity, stability and catalytic performance at the coordinatedmetal center. Our recent studies on Group-IV organometallicsfor C–C (polymerization and copolymerization)1 and C–N(intramolecular hydroamination)2 bond-forming reactionshave provided original insights into the organometallic reacti-vity and catalysis of pyridylamido early transition metal com-pounds.3 Redrawing their molecular framework (Fig. 1, left vs.right) has resulted in the preparation of unsymmetrical dianio-nic {N,C,N′} pincer-type systems characterized by remarkablethermal stability and catalytic activity.4
The basic structure of the newly synthesized complexes con-sists of a central σ-bonded aryl donor group (C) capable of
forming robust M–C bonds with a given metal center which isenforced by the coordination of peripheral donor groups (N)and the chelating rigid structure of the {N,C,N} framework.Such structural features translate into systems having a uniquebalance between stability and reactivity. While a relatively largenumber of late transition metal complexes stabilized by sym-metrical {N,C,N}-pincer-type ligands have been reported todate,5 much less work has been devoted to the synthesis andcharacterization of their early transition metal counter-parts.5a,6 Indeed, prior to our recent study,4 unsymmetrical{N,C,N′}-systems were rather uncommon and limited to com-plexes based on either palladium7–9 or rhodium.9 Unlike pyri-dylamido systems (Fig. 1, left), pincer-type bis-amido Group-IVcomplexes (Fig. 1, right) have shown only negligible activity inolefin polymerization while being good to excellent candidatesfor the intramolecular hydroamination/cyclization of primary
Fig. 1 Left: Group-IV pyridylamido complexes.1–3 Right: Group-IV {N,C,N’}-pincer complexes;4 (*) this work.
†Electronic supplementary information (ESI) available. CCDC 942401. For ESIand crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt51620b
aInstitute of Chemistry of OrganoMetallic Compounds ICCOM-CNR, Via Madonna
del Piano, 10-50019 Sesto F.no., Florence, Italy.
E-mail: [email protected] R&D, The Dow Chemical Company, 1776 Building, Midland, MI 48674,
USAcEquipe de Catalyse Moleculaire, Université Paris-Sud, ICMMO, UMR 8182, Orsay,
F-91405, FrancedCNRS, Orsay, F-91405, France
16056 | Dalton Trans., 2013, 42, 16056–16065 This journal is © The Royal Society of Chemistry 2013
Publ
ishe
d on
14
Aug
ust 2
013.
Dow
nloa
ded
by U
nive
rsity
of
Vir
gini
a on
07/
11/2
013
05:0
3:57
.
View Article OnlineView Journal | View Issue

aminoalkenes under mild reaction conditions (low reactiontemperature and catalyst loading).4
In this paper we describe the synthesis and characterizationof the dimethyl ZrIV and Hf IV pincer complexes (starting fromthe bis-amido precursors) as well as their activation upon treat-ment with organoboron compounds. The resulting cationicspecies have been investigated with respect to their perform-ance in high-temperature ethylene–1-octene copolymerizationas well as in intramolecular hydroamination of primary andsecondary aminoalkenes. This study also gives useful insightsinto specific organometallic activities of these Group-IV pincercomplexes. In particular, the variety of their activation modes,resulting from the treatment of the dimethyl species withdifferent organoboron compounds as well as the ability of theMIV–CAr (M
IV = ZrIV, Hf IV) bonds to undergo olefin insertion,compared to state-of-the-art Group-IV pyridylamido complexes,will be discussed.1b,10
Results and discussionSynthesis and characterization of [ZrIV(κ3-N,C,N′)Me2] (3) and[HfIV(κ3-N,C,N′)Me2] (4) from the bis-amido derivatives4 (1 and 2)
Conversion of 1 and 2 into their dimethyl analogues is carriedout by following literature procedures already used for pyridyl-amido complexes.11 Treatment of the bis-amido precursors ina cooled toluene solution with 5 equiv. of AlMe3 results in aclean alkylation to give the dimethyl-derivatives 3 and 4 aslight-brown and off-white microcrystals, respectively(Scheme 1). Purification by crystallization from concentratedpentane solutions at −30 °C is used to remove aluminium by-products providing 3 and 4 in excellent isolated yields (86 and93%, respectively).
Complexes 3 and 4 are isolated as air and moisture-sensi-tive microcrystals and are characterized by spectroscopic data(Fig. S1–S4†) and, in the case of 4, X-ray diffraction studies.The 1H and 13C{1H} NMR spectra indicate that both dimethylcomplexes possess Cs symmetry in solution. Both complexesare highly soluble in aromatic hydrocarbons, but only spar-ingly soluble in aliphatic hydrocarbons (n-pentane andn-hexane). While colorless plate-like specimens of 4 are suc-cessfully grown by slow evaporation of a benzene-d6–hexanesolution at r.t., all attempts to isolate crystals of 3 suitable forX-ray analysis failed. An ORTEP representation of the crystalstructure of 4 is given in Fig. 2.
Table 1 lists the main crystal and structural-refinementdata, while selected bond lengths and angles are summarizedin Table 2.
The coordination environment at the metal center consistsof two nitrogen atoms and one carbon atom from the dianio-nic tridentate {N−,C−,N} ligand (κ3-coordination) and twomethyl groups. Similar to parent bis-amido precursors, 4shows a distorted square-pyramidal coordination geometry,with the ligand donor atoms and one methyl group lying equa-torially and the other Me fragment occupying the axial posi-tion.12 The κ3-coordinated fragment is almost planar, with thepyrazolyl moieties slightly tilted away from the phenyl centralcore [θ(C1–C6–N2–N3) = 5.02]. Finally, the Hf(1)–N(1), Hf(1)–N(3) and Hf(1)–C(1) distances fall in a similar range to thosemeasured for related structures.13–15 Complexes 3 and 4 are
Scheme 1 Synthesis of [ZrIV(κ3-N,C,N’)Me2] (3) and [Hf IV(κ3-N,C,N’)Me2] (4).
Fig. 2 Crystal structure of [Hf IV(κ3-N,C,N’)Me2] (4). Thermal ellipsoids aredrawn at the 40% probability level. Hydrogen atoms are omitted for clarity.
Table 1 Crystal data and structure refinement for complex 4
4
CCDC number 942401Empirical formula C24H31N3HfFormula weight 540.01Temperature [K] 100(2)Wavelength [Å] 0.71073Crystal system, space group Orthorhombic, Pbcaa [Å] 10.5097(2)b [Å] 12.3812(2)c [Å] 33.8422(5) Åα [°] 90β [°] 90γ [°] 90V [Å3] 4403.64(13)Z, Dc [g m−3] 8, 1.629Absorption coefficient [mm−1] 4.751F(000) 2144Crystal size [mm] 0.22 × 0.22 × 0.04Θ Range for data collection [°] 1.20–27.50Limiting indices −12 ≤ h ≤ 13
−16 ≤ k ≤ 16−43 ≤ l ≤ 43
Reflections collected/unique 51 747/4968GOF on F2 1.198Data/restraints/parameters 4968/0/259Final R indices [I > 2σ(I)] R1 = 0.0207,
wR2 = 0.0374R indices (all data) R1 = 0.0258
wR2 = 0.0387Largest diff. peak and hole [e Å−3] 0.643 and −1.670
Dalton Transactions Paper
This journal is © The Royal Society of Chemistry 2013 Dalton Trans., 2013, 42, 16056–16065 | 16057
Publ
ishe
d on
14
Aug
ust 2
013.
Dow
nloa
ded
by U
nive
rsity
of
Vir
gini
a on
07/
11/2
013
05:0
3:57
. View Article Online

indefinitely stable when stored under an inert atmosphere, inthe dark, at r.t.
In situ catalyst activation upon treatment with either trityltetrakis(pentafluorophenyl)borate [Ph3C][B(C6F5)4] ortris(pentafluorophenyl)boron B(C6F5)3: synthesis andcharacterization of cationic (3a, 4a) and neutral (3b, 4b)derivatives
The cationic ZrIV and Hf IV complexes 3a and 4a are obtainedupon treatment of 3 and 4 with 1 equiv. of [Ph3C][B(C6F5)4]. Thereactions are performed at r.t. in benzene-d6 and proceedrapidly through the clean abstraction of one methyl group fromthe metal center giving red-orange cationic species 3a and 4a(Scheme 2). Both compounds are barely soluble in apolar hydro-carbons (benzene and toluene) where they rapidly separate outas either dark-red (3a) or orange (4a) semi-solid compounds.
Besides limiting their spectroscopic characterization, thelow solubility of both ion pairs in apolar aromatic solvents issuggestive of weak ion pairing.16 On the other hand, both ionpairs are highly soluble in polar aromatics such as mono- ordihalobenzene (halo = Cl, Br) where they form orange-redsolutions. However, these solvents cause their rapid (within afew minutes) decomposition with the formation of furtheruntreatable brown semi-solid compounds. 3a forms a relativelystable solution in THF-d8 which allows its complete spectro-scopic characterization. As can be inferred from the 1H NMRspectrum of 3a (Fig. S5†), the weak coordinating character ofthe B(C6F5)4
− counterion translates into a Cs-symmetric systemcharacterized by only two non-equivalent Me groups from theiPr fragments and by two well-defined singlets at δ = 0.54 and
5.11 ppm, assigned to Zr–CH3 and ArCH2N, respectively.Finally, the presence of the 1,1,1-triphenylethane side-productis confirmed by a residual singlet at δ = 2.16 ppm. Chemicalconversion of 3 into the cationic system 3a was spectroscopi-cally estimated (1H NMR) to be over 97%.
All attempts to characterize 4a in the same way (from aTHF-d8 solution) were frustrated by its ability to polymerizethe solvent. Although highly soluble in THF, the resultingyellow-orange solution rapidly turns into a gel, thus hamperingthe full complex characterization.
Similar to the procedure outlined above, we have alsostudied the reactivity of 3 and 4 in the presence of 1 equiv. ofthe Lewis acid B(C6F5)3 as an alkyl abstractor. The reaction isperformed at r.t. in benzene-d6 as a solvent and its course ismonitored by 1H NMR spectroscopy. Both complexes reactsmoothly with borane, resulting in the clean and quantitativeformation of new Cs-symmetric species although no resonancecan be assigned to a residual methyl group. In this case, thereaction does not yield the expected cationic complexes[MIV(κ3-N,C,N′)Me][MeB(C6F5)3] (M = ZrIV, Hf IV) but rather pro-duces neutral compounds (3b and 4b, Scheme 2) which arisefrom the C6F5 group transfer.17 Benzene-d6 solutions of 3b and4b proved to be stable over a prolonged period of time at r.t.(and even at higher temperatures) without any apparent altera-tion. Multinuclear NMR spectra (1H, 13C{1H}, 19F{1H} and11B{1H}) of the reaction mixtures showed the progressive disap-pearance (within a few minutes) of the distinctive signalsattributed to the dimethyl precursors and the appearance ofnew peaks from 3b and 4b together with signals ascribed tothe side-products BMe3, Me2B(C6F5), and MeB(C6F5)2 (Fig. S8–S15† and the Experimental section). Such a fast “groupsexchange” is supposed to be responsible for the catalyst de-activation in the hydroamination experiments (vide infra).
Olefin polymerization and copolymerization studies
Following our recent results in the field of polymerization cata-lysis using pyridylamido systems as polymerization catalysts,1
cationic pincers 3a and 4a have been preliminarily evaluatedwith respect to their catalytic performance in room-tempera-ture 1-hexene homo-polymerization.
Unlike cationic pyridylamido catalysts (Fig. 1, left),1b 3a and4a do not show any appreciable catalytic activity for 1-hexenepolymerization under mild reaction conditions. It is note-worthy that, while cyclometallated pyridylamido catalysts typi-cally undergo permanent complex modification via monomer
Table 2 Selected bond distances (Å) and angles (°) for complex 4
4
Hf(1)–N(1) 2.062(2)Hf(1)–N(3) 2.313(2)Hf(1)–C(1) 2.223(3)Hf(1)–C(23) 2.245(3)Hf(1)–C(24) 2.229(3)N(2)–N(3) 1.368(3)N(1)–C(7) 1.477(3)N(1)–Hf(1)–N(3) 142.58(8)N(1)–Hf(1)–C(1) 73.75(9)N(3)–Hf(1)–C(1) 68.84(8)C(1)–C(2)–C(7)–N(1) −13.6(3)C(1)–C(6)–N(2)–N(3) 5.0(3)C(7)–N(1)–C(8)–C(9) −100.3(3)C(7)–N(1)–C(8)–C(13) 78.6(3)
Scheme 2 Catalyst activation upon treatment with [Ph3C][B(C6F5)4] and B(C6F5)3.
Paper Dalton Transactions
16058 | Dalton Trans., 2013, 42, 16056–16065 This journal is © The Royal Society of Chemistry 2013
Publ
ishe
d on
14
Aug
ust 2
013.
Dow
nloa
ded
by U
nive
rsity
of
Vir
gini
a on
07/
11/2
013
05:0
3:57
. View Article Online

insertion into the lateral reactive M–Ar (M = ZrIV, Hf IV) bond,18
3a and 4a do not follow the same reactivity path at their centralM–Ar bond. GC-MS analysis of the hydrolyzed reaction mixturesfrom the stoichiometric reactions of 3a and 4a with 1-hexene atr.t. reveals the presence of a unique peak (m/z = 333; retentiontime: 21.7 min) attributed to the unmodified ligand (Fig. 3 andS16†).19 Looking beyond the simple re-distribution of the donoratom set around the metal center [{C,N,N′} vs. {N,C,N′}, Fig. 1],one may argue that in situ monomer insertion into the M–Arbond (occurring on the pyridylamido systems exclusively) con-stitutes a key element for the generation of kinetically activespecies in low temperature polymerization runs.10
High-temperature ethylene–1-octene copolymerization study
Ethylene–1-octene copolymerization tests have been carriedout at 120 °C in a 2 L batch reactor filled with ethylene and1-octene. Both pre-catalysts 3 and 4 are activated in situ using[HNMe(C18H37)2][B(C6F5)4] (1.2 equiv., relative to pre-catalyst)as an activator. For the sake of completeness of the copolymer-ization study, the Cs-
1b (5) and C1-symmetric20 (6) pyridylamidopolymerization catalysts of state-of-the-art (Fig. 4), having iden-tical denticity but different distributions of the donor atom setaround the metal center [{C,N,N′} vs. {N,C,N′}], have been eval-uated, under identical conditions, for comparison.
Ethylene–1-octene copolymerization data are summarizedin Table 3. Both pincer systems are moderately active in copoly-merization catalysis giving ethylene–1-octene copolymers(linear low-density polyethylenes – LLDPEs) with molecularweights ranging from 143 kDa for 3 to 265 kDa for 4.
In spite of moderate productivities compared to the pyridyl-amido systems 5 and 6 (Table 3, entries 1–3 vs. 4–5), 3 and 4provide copolymers with a relatively high degree of 1-octeneincorporation into the polymer microstructure. Notably, 4gives copolymers with the highest 1-octene incorporationdegree, even better than the best performing pyridylamidocatalyst 6 (up to 12.7 mol%, Table 3, entries 2 vs. 5). Such alevel of incorporation is close to that found in ethylene–1-octene copolymers prepared by Ti-CGC {[(η5-C5Me4)SiMe2-(tBuN)(TiMe2)]}. The latter, which represents a real milestonein the ethylene–α-olefins copolymerization catalysis,21,22
provides ethylene–1-octene copolymers with a co-monomercontent of 13.8 mol% under identical conditions.20
Finally, both pincer complexes form copolymers with higherpolydispersities than those resulting from polyolefins pro-duced by state-of-the-art pyridylamido systems (Table 3, entries1–3 vs. 4–5). Overall, while copolymers produced by pre-catalyst4 show higher co-monomer content together with moderatepolydispersities and monomodal distribution of the polymer
Fig. 3 Left: α-olefin insertion into the lateral M–Ar bond of a state-of-the-art Group-IV amidopyridinate complex18 and GC trace19 (reproduced with permissionfrom Wiley)1b of the reaction solution (catalyst : monomer 1 : 1) after quenching with MeOH. Right: GC trace19 of the reaction solution (catalyst 3a : monomer 1 : 1)after quenching with MeOH.
Fig. 4 State-of-the-art pyridylamido polymerization catalysts.
Dalton Transactions Paper
This journal is © The Royal Society of Chemistry 2013 Dalton Trans., 2013, 42, 16056–16065 | 16059
Publ
ishe
d on
14
Aug
ust 2
013.
Dow
nloa
ded
by U
nive
rsity
of
Vir
gini
a on
07/
11/2
013
05:0
3:57
. View Article Online
molecular weights, the Zr-analogue 3 generates polyolefinswith relatively low co-monomer contents, significantly higherMw/Mn values and polymodal distribution of the polymermolecular weights (Fig. S17 and S18†).
Catalytic activity of cationic pincer complexes in theintramolecular hydroamination of primary and secondaryaminoalkenes23
Our interest in the hydroamination reaction stems from pre-vious studies involving the neutral bis-amido complexes 1 and2 (Scheme 1), which readily and efficiently promote the intra-molecular hydroamination of primary aminoalkenes undermild reaction conditions (i.e., reaction temperature and cata-lyst loading).4,24,25 It was of interest for us to determinewhether the activated 3a,b and 4a,b species could promote thehydroamination reaction as efficiently as neutral bis-amidoparents 1 and 2.4 All catalytic runs (Scheme 3) are performedunder an inert atmosphere in a J-Young NMR tube and thereaction process is monitored by 1H NMR with spectrarecorded at constant reaction times. Under the adopted experi-mental conditions, all hydroamination runs are extremelyselective towards the cyclization product, with no evidence ofany side-product.
Substrate conversions are determined by 1H NMR spec-troscopy using ferrocene as the internal standard (spectraacquired at different reaction times) and are confirmed by gas-chromatography of the final crude reaction mixture (see theExperimental section). All catalytic details and optimized cycli-zation conditions are listed in Table 4.
As Table 4 shows, both cationic systems 3a and 4a are activecatalysts for the intramolecular hydroamination reaction of a
number of model primary and secondary aminoalkenes pro-viding, for selected issues, relatively fast and complete sub-strate conversions already at r.t. (Table 4, entries 2 and 5). Onlyone other cationic Group-IV complex is known to lead to thecomplete cyclization of I under the above-mentioned reactionconditions (i.e., reaction temperature and catalyst load-ing).25c,26 Notably, structurally related {C−,N,N−} cationicGroup-IV pyridylamido complexes, recently reported by someof us,2 typically require more severe reaction conditions andhigh catalyst loadings to promote the cyclization of I, efficien-tly (100 °C, 10 mol% of the catalyst). Comparing different
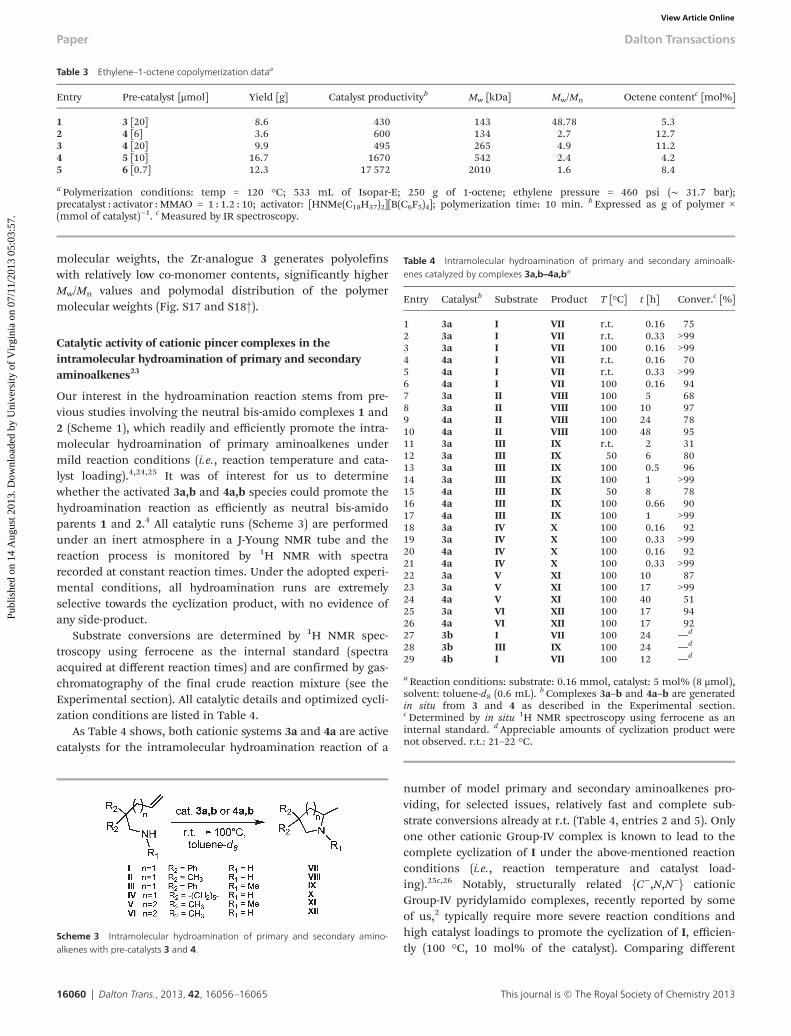
Table 3 Ethylene–1-octene copolymerization dataa
Entry Pre-catalyst [μmol] Yield [g] Catalyst productivityb Mw [kDa] Mw/Mn Octene contentc [mol%]
1 3 [20] 8.6 430 143 48.78 5.32 4 [6] 3.6 600 134 2.7 12.73 4 [20] 9.9 495 265 4.9 11.24 5 [10] 16.7 1670 542 2.4 4.25 6 [0.7] 12.3 17 572 2010 1.6 8.4
a Polymerization conditions: temp = 120 °C; 533 mL of Isopar-E; 250 g of 1-octene; ethylene pressure = 460 psi (∼ 31.7 bar);precatalyst : activator : MMAO = 1 : 1.2 : 10; activator: [HNMe(C18H37)2][B(C6F5)4]; polymerization time: 10 min. b Expressed as g of polymer ×(mmol of catalyst)−1. cMeasured by IR spectroscopy.
Scheme 3 Intramolecular hydroamination of primary and secondary amino-alkenes with pre-catalysts 3 and 4.
Table 4 Intramolecular hydroamination of primary and secondary aminoalk-enes catalyzed by complexes 3a,b–4a,ba
Entry Catalystb Substrate Product T [°C] t [h] Conver.c [%]
1 3a I VII r.t. 0.16 752 3a I VII r.t. 0.33 >993 3a I VII 100 0.16 >994 4a I VII r.t. 0.16 705 4a I VII r.t. 0.33 >996 4a I VII 100 0.16 947 3a II VIII 100 5 688 3a II VIII 100 10 979 4a II VIII 100 24 7810 4a II VIII 100 48 9511 3a III IX r.t. 2 3112 3a III IX 50 6 8013 3a III IX 100 0.5 9614 3a III IX 100 1 >9915 4a III IX 50 8 7816 4a III IX 100 0.66 9017 4a III IX 100 1 >9918 3a IV X 100 0.16 9219 3a IV X 100 0.33 >9920 4a IV X 100 0.16 9221 4a IV X 100 0.33 >9922 3a V XI 100 10 8723 3a V XI 100 17 >9924 4a V XI 100 40 5125 3a VI XII 100 17 9426 4a VI XII 100 17 9227 3b I VII 100 24 —d
28 3b III IX 100 24 —d
29 4b I VII 100 12 —d
a Reaction conditions: substrate: 0.16 mmol, catalyst: 5 mol% (8 μmol),solvent: toluene-d8 (0.6 mL). b Complexes 3a–b and 4a–b are generatedin situ from 3 and 4 as described in the Experimental section.cDetermined by in situ 1H NMR spectroscopy using ferrocene as aninternal standard. d Appreciable amounts of cyclization product werenot observed. r.t.: 21–22 °C.
Paper Dalton Transactions
16060 | Dalton Trans., 2013, 42, 16056–16065 This journal is © The Royal Society of Chemistry 2013
Publ
ishe
d on
14
Aug
ust 2
013.
Dow
nloa
ded
by U
nive
rsity
of
Vir
gini
a on
07/
11/2
013
05:0
3:57
. View Article Online

cyclization precursors under similar reaction conditions(Table 4, entries 2 and 5 vs. 7 and 9, 12 and 15, 22 and 24)makes clear the existence of a significant substrate depen-dence on the cyclization rate. In particular, the diphenyl-sub-stituted precursor I provides the cyclization product VIIquantitatively (entries 1–2) either at r.t. within 20 min or morerapidly upon heating at 100 °C (entry 3). For all other cycliza-tion precursors (II, IV–VI), only more severe reaction con-ditions (from 50 to 100 °C) result in appreciable substrateconversions.
Analysis of the cyclization results shows that the higher thereaction temperature, the higher the substrate conversion andthe shorter the reaction time (entries 12 vs. 13–14, 15 vs.16–17). Furthermore, cyclization of primary aminoalkenes(generally) proceeds more efficiently compared to the cycliza-tion of secondary ones (entries 1 vs. 11, 3 vs. 13, 6 vs. 16 and26 vs. 24). Although this latter trend cannot be considered tobe a real distinctive feature of the catalytic systems used, therelevant reactivity gap observed between I and II, IV–VIsuggests the occurrence of cooperative and positive substrate–complex interactions beyond the simple kinetic Thorpe–Ingoldeffect. DFT calculations are currently ongoing to address boththe existence and the nature of the above mentioned sub-strate–complex interactions and will be reported elsewhere.
Similar to several literature precedents24,25 with Group-IVcatalysts employed in hydroamination reactions, 3a displays(under similar reaction conditions) higher catalytic perform-ances compared to the 4a analogue. Unlike neutral bis-amidoparent precursors, hydroaminations with cationic 3a and 4aproceed efficiently on both primary and secondary aminoalk-enes (entries 1 vs. 3, 3 vs. 13, 6 vs. 17, and 23 vs. 25). All thesepieces of evidence taken together support the hypothesis of8e− amido species as intermediates in the catalytic cycle.25c
Finally, only catalyst activation with [Ph3C][B(C6F5)4] gives cata-lytically (cationic) active forms (3a and 4a) whereas complexesobtained upon catalyst activation with B(C6F5)3 (3b and 4b) donot show any apparent catalytic performance, even after pro-longed reaction times (Table 4, entries 27–29). Such a reactivitytrend is in line with the different activation paths of the di-methyl complexes 3 and 4 as outlined above {[Ph3C][B(C6F5)4]vs. B(C6F5)3}.
Conclusions
The study describes the preparation of cationic unsymmetrical{N,C,N′} Group-IV pincer complexes and demonstrates theirability to promote efficiently intramolecular hydroaminationreactions as well as ethylene–1-octene copolymerizations inhigh temperature solution processes. Unlike neutral bis-amidoprecursors, cationic ZrIV and Hf IV species are active forms inthe hydroamination reaction of primary and secondaryaminoalkenes revealing a remarkable substrate dependence onthe cyclization rate. As a result, only selected substratesundergo quantitative and rapid hydroamination/cyclizationalready at r.t. Aimed at widening the catalysts application
range, cationic ZrIV and Hf IV species have also been investi-gated in ethylene–1-octene copolymerizations. In spite of mod-erate productivities both systems provide copolymers with arelatively high degree of 1-octene incorporation (up to12.7 mol%) similar to that obtained with the best performingstate-of-the-art half-sandwich Ti-CGC. Specifics of the acti-vation modes (generation of cationic species from dimethylprecursors) for these Group-IV pincer complexes are given indetail. Overall the study highlights the ability of robust andthermally stable cationic Group-IV systems to operate efficien-tly within ad hoc catalytic processes under increasing tempera-ture conditions.
ExperimentalGeneral considerations and materials characterization
All air- and/or moisture-sensitive reactions were performedunder an inert atmosphere in flame-dried flasks by using stan-dard Schlenk-type techniques or in a drybox filled with nitro-gen. Benzene and toluene were purified by distillation fromsodium/triglyme benzophenone ketyl and stored over 4 Åmolecular sieves or were obtained by means of an MBraunsolvent purification system. Benzene-d6 and toluene-d8 weredried over sodium/benzophenone ketyl and condensedin vacuo over activated 4 Å molecular sieves prior to use. Aminoalkenes I,27 II,28 III,29 IV,30 V25a and VI28 were prepared accord-ing to literature procedures. All other reagents and solventswere used (unless otherwise stated) as purchased from com-mercial suppliers without further purification. The bis(amido)pincer-complexes [ZrIV(κ3-N,C,N′)(NMe2)2] (1), [Hf IV(κ3-N,C,N′)(NMe2)2] (2) were obtained on a gram scale according to stan-dard procedures described in the literature.4 1D (1H and 13C{1H}) and 2D (COSY H–H, HETCOR H–C) NMR spectra of allorganometallic species were obtained using either a BrukerAvance DRX-400 spectrometer (400.13 and 100.61 MHz for 1Hand 13C, respectively) or a Bruker Avance 300 MHz instrument(300.13 and 75.47 MHz for 1H and 13C, respectively). Chemicalshifts (δ) are reported in ppm relative to trimethylsilane (TMS),referenced to the chemical shifts of residual solvent reson-ances (1H and 13C). The multiplicities of the 13C{1H} NMRspectra were determined on the basis of the 13C{1H} JMODsequence and quoted as: CH3, CH2, CH, and C for primary,secondary, tertiary, and quaternary carbon atoms, respectively.The C, H, N elemental analyses were performed atICCOM-CNR using a Thermo FlashEA 1112 Series CHNS-Oelemental analyzer with an accepted tolerance of ±0.4 unit oncarbon (C), hydrogen (H), and nitrogen (N). Molecular weightdistribution (Mw and Mn) was determined by analysis on acustom Dow-built Robotic-Assisted Dilution High-TemperatureGel Permeation Chromatographer (RAD-GPC). Polymersamples were dissolved for 90 min at 160 °C at a concentrationof 30 mg mL−1 in 1,2,4-trichlorobenzene (TCB) and stabilizedby 300 ppm BHT. They were then diluted to 1 mg mL−1
immediately before a 400 μL aliquot of the sample wasinjected. The GPC utilized two Polymer Labs PLgel 10 μm
Dalton Transactions Paper
This journal is © The Royal Society of Chemistry 2013 Dalton Trans., 2013, 42, 16056–16065 | 16061
Publ
ishe
d on
14
Aug
ust 2
013.
Dow
nloa
ded
by U
nive
rsity
of
Vir
gini
a on
07/
11/2
013
05:0
3:57
. View Article Online

MIXED-B columns (300 × 10 mm) at a flow rate of 2.0 mLmin−1 at 150 °C. Sample detection was performed using a Poly-Char IR4 detector in concentration mode. A conventional cali-bration of narrow polystyrene (PS) standards was utilized, withapparent units adjusted to homo-polyethylene (PE) usingknown Mark–Houwink coefficients for PS and PE in TCB atthis temperature. Thermal characterization was performedusing a TA Instruments Q2000 DSC. 5 mg of each sample wereweighed into a TZero aluminum DSC pan with a lid. A heat :cool : heat profile from −90 °C to 200 °C at 10 °C min−1 wasused, under nitrogen purge. Data analysis was performedusing TA Universal Analysis software. Tg (where appropriate)was calculated by an onset-at-inflection methodology. Tm wasdetermined by calculating Tm(peak) after linear integration ofextrapolation from post-melt plateau. Both Tg and Tm were cal-culated from the second heat cycle.
X-ray data measurement
X-ray intensity data were collected on a Bruker SMART diffract-ometer using MoKα radiation (λ = 0.71073 Å) and an APEXIICCD area detector. Raw data frames were read by the programSAINT31 and integrated using 3D profiling algorithms. Theresulting data were reduced to produce hkl reflections andtheir intensities and estimated standard deviations. The datawere corrected for Lorentz and polarization effects and multi-scan absorption corrections were applied. The structure wassolved (SHELXS-97) and refined (SHELXL-2013) using full-matrix least-squares refinement. The non-H atoms wererefined with anisotropic thermal parameters and H atomswere calculated in idealized positions and refined riding ontheir parent atoms. The refinement was carried out using F2
rather than F values. R1 is calculated to provide a reference tothe conventional R value, but its function is not minimized.Crystals were grown by slow evaporation of a benzene-d6–hexane solvent mixture at r.t. A clear colorless plate-like speci-men of 4 was used for the X-ray crystallographic analysis.
General procedure for the synthesis of complexesZrIV{κ3-N,C,N′}Me2 (3) and HfIV{κ3-N,C,N′}Me2 (4)
A toluene (7 mL) solution of the selected complex MIV{κ3-N,C,N′}(NMe2)2 (MIV = Zr, Hf) (0.5 mmol) was cooled down to−20 °C for 3 h, and a 2 M hexane solution of AlMe3 (2.5 mmol,5 equiv.) was added dropwise. The solution was then allowedto warm at r.t. within 2 h under stirring. Afterwards, the
solvent was removed under reduced pressure and the semi-solid crude material was dried under reduced pressure. Crudecomplexes 3 and 4 were purified by crystallization at −30 °Cfrom concentrated pentane solutions. Brown and off-whitemicrocrystals were collected for 3 and 4, respectively. Suitablecrystals of 4 were grown at r.t. as colorless plate-like specimensfrom a benzene-d6–hexane mixture. Data for 3: (86% isolatedyield); 1H NMR (300 MHz, C6D6, 293 K): δ 0.50 (s, 6H, Me,H23,24), 1.26 (d, 3JHH = 6.8 Hz, 6H, CH(CH3
ACH3B), H19,22), 1.44
(d, 3JHH = 6.8 Hz, 6H, CH(CH3ACH3
B), H18,21), 4.07 (sept, 3JHH =6.8 Hz, 2H, CH(CH3)2, H
17,20), 5.11 (s, 2H, CH2N, H10), 5.65 (m,
1H, CH Ar, H8), 6.47 (m, 1H, CH Ar, H3), 6.82 (m, 1H, CH Ar,H9), 7.03 (m, 2H, CH Ar, H2,4), 7.27–7.30 (4H, CH Ar,H7,13,14,15). 13C{1H} NMR (75 MHz, C6D6, 293 K): δ 23.9(CH(CH3)2, C17,20), 28.0 (CH(CH3
ACH3B), C18,21), 28.1
(CH(CH3ACH3
B), C19,22), 45.2 (CH3, Me, C23,24), 70.0 (C10), 107.3(C3), 107.9 (C8), 122.0 (C2), 124.2 (C13,15), 125.3 (C9), 126.7(C14), 128.9 (C4), 141.1 (C7), 142.1 (C), 143.4 (C), 148.5 (C),156.2 (C), 177.7 (C6). Anal. Calcd (%) for C24H31N3Zr (452.75):C 63.67, H 6.90, N 9.28; found C 63.75, H 6.95, N 9.32. Data for4: (93% isolated yield); 1H NMR (300 MHz, C6D6, 293 K): δ 0.33(s, 6H, Me, H23,24), 1.27 (d, 3JHH = 6.8 Hz, 6H, CH(CH3
ACH3B),
H19,22), 1.46 (d, 3JHH = 6.8 Hz, 6H, CH(CH3ACH3
B, H18,21), 4.08(sept, 3JHH = 6.8 Hz, 2H, CH(CH3)2, H
17,20), 5.24 (s, 2H, CH2N,H10), 5.61 (m, 1H, CH Ar, H8), 6.53 (m, 1H, CH Ar, H2), 6.81(m, 1H, CH Ar, H9), 7.01–7.11 (2H, CH Ar, H3,4), 7.24 (m, 1H,CH Ar, H7), 7.31 (3H, CH Ar, H13,14,15). 13C{1H} NMR (75 MHz,C6D6, 293 K): δ 24.0 (CH(CH3
ACH3B), C18,21), 27.8 (CH(CH3)2,
C17,20), 28.1 (CH(CH3ACH3
B), C19,22), 59.5 (CH3, Me, C23,24), 69.2(C10), 107.4 (C2), 108.0 (C8), 122.5 (C4), 124.2 (C13,15), 125.8 (C9),126.3 (C14), 129.0 (C3), 141.3 (C7), 143.3 (C), 144.0 (C), 148.3 (C),156.2 (C), 187.9 (C6). Anal. Calcd (%) for C24H31HfN3 (540.01): C53.38, H 5.79, N 7.78; found C 53.42, H 5.83, N 7.82.
General procedure for the activation of the dimethylprecursors 3 and 4 with Ph3C
+[B(C6F5)4]−
A solution of Ph3C+[B(C6F5)4]
− (1 equiv.) in C6H6 (1 mL) wasadded in one portion to a dry and degassed solution ofcomplex 3 (0.025 g, 55 μmol) in C6H6 (1 mL) at r.t. The mixturewas stirred at r.t. for 20 min while a dark-red semi-solidmaterial precipitates out. The semi-solid compound was dec-anted from the supernatant and washed with C6H6 (2 ×1.5 mL) and pentane (2 × 1.5 mL). The recovered material wasdried under vacuum to constant weight before being
Paper Dalton Transactions
16062 | Dalton Trans., 2013, 42, 16056–16065 This journal is © The Royal Society of Chemistry 2013
Publ
ishe
d on
14
Aug
ust 2
013.
Dow
nloa
ded
by U
nive
rsity
of
Vir
gini
a on
07/
11/2
013
05:0
3:57
. View Article Online

characterized spectroscopically in THF-d8. Data for 3a: (esti-mated yield >97% – conversion of 3 based on 1H-NMR spec-troscopy). 1H NMR (400 MHz, THF-d8, 293 K, select data): δ0.54 (s, 3H, Me, H23), 1.24 (d, 3JHH = 6.8 Hz, 6H, CH(CH3
ACH3B), H19,22), 1.31 (d, 3JHH = 6.8 Hz, 6H, CH(CH3
ACH3B),
H18,21), 3.65 (sept, 3JHH = 6.8 Hz, 2H, CH(CH3)2, H17,20), 5.11 (s,
2H, CH2N, H10). 13C{1H} NMR (100 MHz, THF-d8, 293 K, select
data): δ 24.1(CH(CH3)2, C17,20), 27.9 (CH(CH3ACH3
B), C18,21),28.6 (CH(CH3
ACH3B), C19,22), 51.7 (CH3, Me, C23,24), 73.4 (C10),
178.7 (C6). 19F{1H} NMR (376.3 MHz, THF-d8, 293 K): δ −131.4(s, o-C6F5), −161.6 (t, p-C6F5, JF–F = 21.3 Hz), −165.5 (m,m-C6F5).
Reaction of 3 and 4 with B(C6F5)3 in C6D6
A solution of B(C6F5)3 (1 equiv.) in benzene-d6 (0.5 mL) wasadded in one portion to a dry and degassed solution of thedimethyl complex (3 or 4) (0.025 g) in benzene-d6 (0.5 mL).The reaction was periodically monitored via 1H-NMR spec-troscopy until complete conversion of the starting materialsinto 3b or 4b (10 min) together with boranes of formula MeB(C6F5)2, Me2B(C6F5) and BMe3. Data for 3b: (estimated yield>98% – conversion of 3 based on multinuclear NMR spec-troscopy). 1H NMR (400 MHz, C6D6, 293 K): δ 0.67 (d, 3JHH =6.8 Hz, 6H, CH(CH3
ACH3B), H19,22), 0.73 (br s, B(CH3)3), 0.96
(br s, B(CH3)2(C6F5)), 1.07 (d, 3JHH = 6.8 Hz, 6H, CH(CH3ACH3
B,H18,21), 1.33 (br s, BCH3(C6F5)2), 3.67 (sept, 3JHH = 6.8 Hz, 2H,CH(CH3)2, H
17,20), 5.28 (s, 2H, CH2N, H10), 5.63 (m, 1H, CH Ar,
H8), 6.37 (m, 1H, CH Ar, H2), 6.73 (m, 1H, CH Ar, H9),6.98–6.93 (2H, CH Ar, H3,4), 7.05–7.03 (2H, CH Ar, H13,15), 7.16(1H, CH Ar, H14), 7.26 (m, 1H, CH Ar, H7). 13C{1H} NMR(100 MHz, C6D6, 293 K): δ 22.4 (CH(CH3)2, C17,20), 27.04(CH(CH3
ACH3B), C18,21), 28.1 (CH(CH3
ACH3B), C19,22), 71.2
(C10), 107.9 (C2), 108.8 (C8), 122.5 (C4), 124.4 (C13,15), 126.3(C9), 128.8 (C14), 130.8 (C3), 135.8 (C), 141.2 (C7), 142.4 (C),148.7 (C), 155.9 (C), 177.7 (C6). 11B{1H} NMR (96.2 MHz, C6D6,293 K): δ 52.5 BCH3(C6F5)2 (61.8%), 62.2 B(CH3)2(C6F5)(30.1%), 67.9 B(CH3)3 (8.1%). 19F{1H} NMR (376.3 MHz, C6D6,293 K): [LZr(C6F5)2] δ −122.3 (brs, 4 F, Fo C6F5), −150.0 (t, 2 F,3J = 19.2 Hz, Fp C6F5), −158.8 (m, 4 F, Fm C6F5); [B(CH3)(C6F5)2]−128.4 (m, 4 F, Fo C6F5), −145.5 (t, 2 F, 3J = 21.0 Hz, Fp C6F5),−159.8 (m, 4, Fm C6F5); [B(CH3)2(C6F5)] −129.4 (m, 2 F, FoC6F5), −149.9 (t, 1 F, 3J = 24.4 Hz, Fp C6F5), −161.0 (m, 2, FmC6F5). Data for 4b: (estimated yield >98% – conversion of4 based on multinuclear NMR spectroscopy). 1H NMR
(400 MHz, C6D6, 293 K): δ 0.68 (d, 3JHH = 6.8 Hz, 6H,CH(CH3
ACH3B), H19,22), 0.73 (br s, B(CH3)3), 0.97 (br s, B
(CH3)2(C6F5)), 1.11 (d, 3JHH = 6.8 Hz, 6H, CH(CH3ACH3
B),H18,21), 1.34 (br s, BCH3(C6F5)2), 3.72 (sept, 3JHH = 6.8 Hz, 2H,CH(CH3)2, H
17,20), 5.48 (s, 2H, CH2N, H10), 5.61 (m, 1H, CH Ar,
H8), 6.46 (m, 1H, CH Ar, H2), 6.75 (m, 1H, CH Ar, H9),7.07–6.97 (5H, CH Ar, H3,4,13,14,15), 7.24 (m, 1H, CH Ar, H7).13C{1H} NMR (100 MHz, C6D6, 293 K): δ 22.7 (CH(CH3)2,C17,20), 27.1 (CH(CH3
ACH3B), C18,21), 27.9 (CH(CH3
ACH3B),
C19,22), 69.0 (C10), 108.1 (C2), 108.9 (C8), 123.2 (C4), 124.3(C13,15), 126.9 (C9), 130.8 (C3), 138.6 (C), 141.4 (C7), 143.2 (C),147.7 (C), 156.3 (C), 192.5 (C6). 11B{1H} NMR (96.2 MHz, C6D6,293 K): δ 52.5 BCH3(C6F5)2 (48%), 62.2 B(CH3)2(C6F5) (48%),67.9 B(CH3)3 (4%). 19F{1H} NMR (376.3 MHz, C6D6, 293 K):[LHf(C6F5)2] δ −122.1 (brs, 4 F, Fo C6F5), −149.5 (t, 2 F, 3J =19.6 Hz, Fp C6F5), −158.8 (m, 4 F, Fm C6F5); [B(CH3)(C6F5)2]−128.4 (m, 4 F, Fo C6F5), −145.4 (t, 2 F, 3J = 21.4 Hz, Fp C6F5),−159.8 (m, 4, Fm C6F5); [B(CH3)2(C6F5)] −129.4 (m, 2 F, FoC6F5), −149.8 (t, 1 F, 3J = 24.4 Hz, Fp C6F5), −161.0 (m, 2,Fm C6F5).
General procedure for intramolecular hydroamination ofaminoalkenes
All catalytic tests were carried out under an inert atmosphereand samples were prepared and handled in an N2-filled drybox. In a general procedure, a solution of 3a or 4a (8 μmol) intoluene-d8 (0.6 mL) at r.t. was activated upon treatment withPh3C
+[B(C6F5)4]− (1 equiv.). The resulting solution was allowed
to stir for a few minutes and then transferred into a vial con-taining the corresponding aminoalkene (0.16 mmol). The reac-tion mixture was then introduced into a J. Young-tap NMRtube and placed in an oil bath heated at 100 °C. The conver-sion of each reaction was monitored spectroscopically (by acomparative integration of selected proton signals on the sub-strate and their corresponding protons on the cyclizationproduct) and gas-chromatographically (by integration of thereagent and the product signals). Selected experiments wereadditionally monitored by 1H NMR spectroscopy using ferro-cene as an internal standard (0.2 mL of a stock 0.17 M ferro-cene solution in toluene-d8). The internal standard wasused to measure the substrate conversion and confirm theappropriate reaction mass balance. For all the scrutinizedissues, the reaction mass balance was confirmed in the limitof the NMR spectrometer experimental error (1H NMR acqui-sition parameters: acquisition time (aq.): 5 s; d1: 8 s; scannumber (ns): 32).
Polymerization procedure
Ethylene–1-octene copolymerization. A stirred 2-liter Parrreactor was used in the polymerizations. All feeds were passedthrough columns of alumina and Q-5™ catalyst (availablefrom Engelhard Chemicals Inc.) prior to introduction into thereactor. Pro-catalyst and cocatalyst (activator) solutions werehandled in the glove box. The reactor was charged with about533 g of mixed alkanes solvent and 250 g of 1-octene comono-mer. The reactor contents were heated to the polymerization
Dalton Transactions Paper
This journal is © The Royal Society of Chemistry 2013 Dalton Trans., 2013, 42, 16056–16065 | 16063
Publ
ishe
d on
14
Aug
ust 2
013.
Dow
nloa
ded
by U
nive
rsity
of
Vir
gini
a on
07/
11/2
013
05:0
3:57
. View Article Online

temperature of 120 °C while 10 μmol of MMAO were added tothe reactor as a scavenger for trace O2 and water. Once at temp-erature, the reactor was saturated with ethylene at 460 psig(3.4 MPa). Catalysts and cocatalysts, as dilute solutions intoluene, were mixed and transferred to a catalyst addition tankand injected into the reactor. The polymerization conditionswere maintained for 10 min with ethylene added on demand.Heat was continuously removed from the reaction vesselthrough an internal cooling coil. The resulting solution wasremoved from the reactor and added to 5 mL of a toluene solu-tion containing approximately 67 mg of a hindered phenolantioxidant (Irganox™ 1010 from Ciba Geigy Corporation) and133 mg of a phosphorus stabilizer (Irgafos™ 168 from CibaGeigy Corporation). Between polymerization runs, a wash cyclewas conducted in which 850 g of mixed alkanes were added tothe reactor and the reactor was heated to 150 °C. The reactorwas then emptied of the heated solvent immediately beforebeginning a new polymerization run. Polymers were recoveredby drying for about 12 h in a temperature-ramped vacuumoven with a final set point of 140 °C.
Acknowledgements
The authors thank the Groupe de Recherche International(GDRI) “Homogeneous Catalysis for Sustainable Development”for support. G. G. also thanks Dr Cristiano Zuccaccia for fruit-ful discussions.
References
1 (a) L. Luconi, G. Giambastiani, A. Rossin, C. Bianchini andA. Lledós, Inorg. Chem., 2010, 49, 6811; (b) L. Luconi,A. Rossin, G. Tuci, I. Tritto, L. Boggioni, J. J. Klosin,C. N. Theriault and G. Giambastiani, Chem.–Eur. J., 2012,18, 671.
2 L. Luconi, A. Rossin, G. Tuci, S. Germain, E. Schulz,J. Hannedouche and G. Giambastiani, ChemCatChem,2013, 5, 1142.
3 For a comprehensive review contribution see:G. Giambastiani, L. Luconi, R. L. Kuhlman andP. D. Hustad, in Olefin Upgrading Catalysis by Nitrogen-based Metal Complexes I, ed. G. Giambastiani andJ. Campora, Springer, London, 2011, pp. 197–282, ISBN978-90-481-3814-2, and references cited therein.
4 L. Luconi, A. Rossin, A. Motta, G. Tuci andG. Giambastiani, Chem.–Eur. J., 2013, 19, 4906.
5 For comprehensive review papers and books on pincer-typecomplexes and their application in catalysis see:(a) M. H. P. Rietveld, D. M. Grove and G. van Koten, NewJ. Chem., 1997, 21, 751; (b) M. Albrecht and G. van Koten,Angew. Chem., Int. Ed., 2001, 40, 3750; (c) M. E. Van DerBoom and D. Milstein, Chem. Rev., 2003, 103, 1759;(d) J. T. Singleton, Tetrahedron, 2003, 59, 1837;(e) D. Morales-Morales, Rev. Soc. Quim. México, 2004, 48,
338; (f ) M. Q. Slagt, D. A. P. Zwieten, A. J. C. M. Moerkerk,R. J. M. K. Gebbink and G. van Koten, Coord. Chem. Rev.,2004, 248, 2275; (g) D. Pugh and A. A. Danopoulos, Coord.Chem. Rev., 2007, 251, 610; (h) D. Morales-Morales andC. M. Jensen, in The Chemistry of Pincer Compounds, Else-vier Science, 2007, p. 450, ISBN: 978-0-444-53138-4;(i) J.-L. Niu, X.-Q. Hao, J.-F. Gong and M.-P. Song, DaltonTrans., 2011, 40, 5135; ( j) N. Selander and K. J. Szabó,Chem. Rev., 2011, 111, 2048.
6 For selected examples of mono- and trianionic {N,C,N}-pincer complexes with early transition metals see:(a) J. G. Donkervoort, J. T. B. H. Jastrzebski, B.-J. Deelman,H. Kooijman, N. Veldman, A. L. Spek and G. van Koten,Organometallics, 1997, 16, 4174; (b) M. Contel, M. Stol,M. A. Casado, G. P. M. van Klink, D. D. Ellis, A. L. Spek andG. van Koten, Organometallics, 2002, 21, 4556; (c) J. Koller,S. Sarkar, K. A. Abboud and A. S. Veige, Organometallics,2007, 26, 5438; (d) S. Sarkar, K. P. McGowan, J. A. Culver,A. R. Carloson, J. Koller, A. J. Peloquin, M. K. Veige,K. A. Abboud and A. S. Veige, Inorg. Chem., 2010, 49, 5143;(e) A. V. Chuchuryukin, R. Huang, M. Lutz, J. C. Chadwick,A. L. Spek and G. van Koten, Organometallics, 2011, 30,2819; (f ) A. V. Chuchuryukin, R. Huang, E. E. van Faassen,G. P. M. van Klink, M. Lutz, J. C. Chadwick, A. L. Spek andG. van Koten, Dalton Trans., 2011, 40, 8887;(g) K. P. McGowan, K. A. Abboud and A. S. Veige, Organo-metallics, 2011, 30, 4949.
7 X.-Q. Hao, Y.-N. Wang, J.-R. Liu, K.-L. Wang, J.-F. Gong andM.-P. Song, J. Organomet. Chem., 2010, 695, 82.
8 For a general review on unsymmetrical {E,C,E′} and {E,C,Z}-pincer PdII complexes, see: I. Moreno, R. SanMartin,B. Inés, M. T. Herrero and E. Domínguez, Curr. Org. Chem.,2009, 13, 878.
9 For a related example of an unsymmetrical {N,C,N′}-pincercomplex incorporating an N-heterocyclic carbene func-tional group, see: N. Schneider, V. César, S. Bellemin-Laponnaz and L. H. Gade, Organometallics, 2005, 24, 4886.
10 R. D. J. Froese, P. D. Hustad, R. L. Kuhlman andT. T. Wenzel, J. Am. Chem. Soc., 2007, 129, 7831.
11 (a) G. M. Diamond, R. F. Jordan and J. L. Petersen, J. Am.Chem. Soc., 1996, 118, 8024; (b) R. T. Boussie,G. M. Diamond, C. Goh, K. A. Hall, A. M. La-Pointe,M. K. Leclerc, V. Murphy, J. A. W. Shoemaker, H. Turner,R. K. Rosen, J. C. Stevens, F. Alfano, V. Busico, R. Cipulloand G. Talarico, Angew. Chem., 2006, 45, 3278.
12 The calculated index of trigonality “τ” for this structure is0.26; see also A. W. Addison, T. N. Rao, J. Reedijk, J. vanRijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984,1349.
13 For related structures containing Hf–amido bonds, see:(a) K. Nienkemper, G. Kehr, S. Kehr, R. Frohlich andG. Erker, J. Organomet. Chem., 2008, 693, 1572; see alsoref. 1b, 2 and 4.
14 For related structures containing Hf–Npyrazolyl bonds, see:(a) A. Otero, J. Fernandez-Baeza, A. Antinolo, J. Tejeda,A. Lara-Sanchez, L. Sanchez-Barba, M. Fernandez-Lopez
Paper Dalton Transactions
16064 | Dalton Trans., 2013, 42, 16056–16065 This journal is © The Royal Society of Chemistry 2013
Publ
ishe
d on
14
Aug
ust 2
013.
Dow
nloa
ded
by U
nive
rsity
of
Vir
gini
a on
07/
11/2
013
05:0
3:57
. View Article Online

and I. Lopez-Solera, Inorg. Chem., 2004, 43, 1350; (b) Seealso ref. 4.
15 For related structures containing Hf–Ar bonds, see:(a) H. Tsurugi, K. Yamamoto and K. Mashima, J. Am. Chem.Soc., 2011, 133, 732; (b) See also ref. 1b, 2, 4 and 6c.
16 C. L. Beswick and T. J. Marks, Organometallics, 1999, 18,2410.
17 (a) R. F. Munhá, M. A. Antunes, L. G. Alves, L. F. Veiros,M. D. Fryzuk and A. M. Martins, Organometallics, 2010, 29,3753; (b) C. Krempner, M. Köckerling, H. Reinke andK. Weichert, Inorg. Chem., 2006, 45, 3203; (c) Z. J. Tonzetichand R. R. Schrock, Polyhedron, 2006, 25, 469;(d) R. M. Gauvin, C. Mazet and J. Kress, J. Organomet.Chem., 2002, 658, 1; (e) S. J. Lancaster and M. Bochmann,Organometallics, 2001, 20, 2093.
18 Such a reactivity translates into a lowering of the symmetryof the active species. For literature precedents see:(a) G. J. Domski, E. B. Lobkovsky and G. W. Coates, Macro-molecules, 2007, 40, 3510; (b) C. Zuccaccia, A. Macchioni,V. Busico, R. Cipullo, G. Talarico, F. Alfano, H. W. Boone,K. A. Frazier, P. D. Hustad, J. C. Stevens, P. C. Vosejpka andK. A. Abboud, J. Am. Chem. Soc., 2008, 130, 10354;(c) C. Zuccaccia, V. Busico, R. Cipullo, G. Talarico,R. D. J. Froese, P. C. Vosejpka, P. D. Hustad andA. Macchioni, Organometallics, 2009, 28, 5445. See alsoref. 1b, 10.
19 GC program: 40 °C for 1 min, 15 °C min−1, 250 °C for20 min.
20 K. A. Frazier, R. D. Froese, Y. He, J. Klosin, C. N. Theriault,P. C. Vosejpka, Z. Zhou and K. A. Abboud, Organometallics,2011, 30, 3318.
21 A. L. McKnight and R. M. Waymouth, Chem. Rev., 1998, 98,2587.
22 (a) T. R. Boussie, G. M. Diamond, C. Goh, K. A. Hall,A. M. LaPointe, M. Leclerc, C. Lund, V. Murphy,J. A. W. Shoemaker, U. Tracht, H. Turner, J. Zhang, T. Uno,R. K. Rosen and J. C. Stevens, J. Am. Chem. Soc., 2003, 125,4306; (b) Dow Chemical Co. invs., Eur. Patent Appl. EP 416815-A2, 1991; J. C. Stevens, F. J. Timmers, D. R. Wilson,G. F. Schmidt, P. N. Nickias, R. K. Rosen, T. R. Boussie,G. M. Diamond, C. Goh, K. A. Hall, A. M. LaPointe,M. Leclerc, C. Lund, V. Murphy, J. A. W. Shoemaker,U. Tracht, H. Turner, J. Zhang, T. Uno, G. W. Knight andS. Lai, Chem. Abstr., 1991, 115, 93163; (c) J. Klosin,W. J. Kruper Jr., P. N. Nickias, G. R. Roof, P. De Waele andK. A. Abboud, Organometallics, 2001, 20, 2663.
23 For recent general reviews on hydroamination catalysis see:(a) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo andM. Tada, Chem. Rev., 2008, 108, 3795; (b) J. Hannedoucheand E. Schulz, Chem.–Eur. J., 2013, 19, 4972.
24 For a selection of neutral Group-IV metal-catalyzed hydro-amination of aminoalkenes see: (a) R. K. Thomson,J. A. Bexrud and L. L. Schafer, Organometallics, 2006, 25,4069; (b) M. C. Wood, D. C. Leitch, C. S. Yeung, J. A. Kozakand L. L. Schafer, Angew. Chem., Int. Ed., 2007, 46, 354;(c) S. Majumder and A. L. Odom, Organometallics, 2008, 27,
1174; (d) A. L. Gott, A. J. Clarke, G. J. Clarkson and P. Scott,Chem. Commun., 2008, 1422; (e) C. Müller, W. Saak andS. Doye, Eur. J. Org. Chem., 2008, 2731; (f ) J. Cho,T. K. Hollis, T. R. Helgert and E. J. Valente, Chem.Commun., 2008, 5001; (g) D. C. Leitch, P. R. Payne,C. R. Dunbar and L. L. Schafer, J. Am. Chem. Soc., 2009,131, 18246; (h) A. L. Reznichenko and K. C. Hultzsch,Organometallics, 2010, 29, 24; (i) J. A. Bexrud andL. L. Schafer, Dalton Trans., 2010, 39, 361; ( j) Y.-C. Hu,C.-F. Liang, J.-H. Tsai, G. P. A. Yap, Y.-T. Chang andT.-G. Ong, Organometallics, 2010, 29, 3357; (k) K. Manna,A. Ellern and A. D. Sadow, Chem. Commun., 2010, 46, 339;(l) K. Manna, S. Xu and A. D. Sadow, Angew. Chem., Int. Ed.,2011, 50, 1865; (m) J. Cho, T. K. Hollis, E. J. Valente andJ. M. Trate, J. Organomet. Chem., 2011, 696, 373; (n) H. Kim,P. H. Lee and T. Livinghouse, Chem. Commun., 2005, 5205;(o) J. A. Bexrud, J. D. Beard, D. C. Leitch and L. L. Schafer,Org. Lett., 2005, 7, 1959; (p) H. Kim, Y. K. Kim, J. H. Shim,M. Kim, M. Han, T. Livinghouse and P. H. Lee, Adv. Synth.Catal., 2006, 348, 2609; (q) D. A. Watson, M. Chiu andR. G. Bergman, Organometallics, 2006, 25, 4731;(r) A. L. Gott, A. J. Clarke, G. J. Clarkson and P. Scott,Organometallics, 2007, 26, 1729; (s) X. Li, S. Haibin andG. Zi, Eur. J. Inorg. Chem., 2008, 1135; (t) G. Zi, Q. Wang,L. Xiang and H. Song, Dalton Trans., 2008, 5930; (u) G. Zi,X. Liu, L. Xiang and H. Song, Organometallics, 2009, 28,1127; (v) G. Zi, F. Zhang, L. Xue, L. Ai and H. Song,J. Organomet. Chem., 2010, 695, 730; (w) G. Zi, F. Zhang,L. Xiang, Y. Chen, W. Fang and H. Song, Dalton Trans.,2010, 39, 4048; (x) T. R. Helgert, T. K. Hollis andE. J. Valente, Organometallics, 2012, 31, 3002; (y) K. Manna,W. C. Everett, G. Schoendorff, A. Ellern, T. L. Windus andA. D. Sadow, J. Am. Chem. Soc., 2013, 135, 7235.
25 For cationic Group-IV metal-catalyzed hydroamination ofaminoalkenes see: (a) P. D. Knight, I. Munslow,P. N. O’Shaughnessy and P. Scott, Chem. Commun., 2004,894; (b) D. V. Gribkov and K. C. Hultzsch, Angew. Chem.,2004, 116, 5659, (Angew. Chem., Int. Ed., 2004, 43, 5542);(c) X. Wang, Z. Chen, X.-L. Sun, Y. Tang and Z. Xie, Org.Lett., 2011, 13, 4758; (d) A. Mukherjee, S. Nembenna,T. K. Sen, S. Pillai Sarish, P. K. Ghorai, H. Ott, D. Stalke,S. K. Mandal and H. W. Roesky, Angew. Chem., 2011, 123,4054, (Angew. Chem., Int. Ed., 2011, 50, 3968).
26 For a selection of seminal papers dealing with hydroamina-tion catalysis by neutral Group-IV complexes under mildreaction conditions see also: ref. 24k, l, y.
27 S. Hong, S. Tian, M. V. Metz and T. J. Marks, J. Am. Chem.Soc., 2003, 125, 14768.
28 J. Y. Kim and T. Livinghouse, Org. Lett., 2005, 7, 1737.29 B.-D. Stubbert and T. J. Marks, J. Am. Chem. Soc., 2007, 129,
4253.30 I. Aillaud, J. Collin, C. Duhayon, R. Guillot, D. Lyubov,
E. Schulz and A. Trifonov, Chem.–Eur. J., 2008, 14, 2189.31 G. M. Sheldrick, SHELXTL 6.1. Crystallographic software
package, Bruker AXS, Inc., Madison, Wisconsin, USA,2008.
Dalton Transactions Paper
This journal is © The Royal Society of Chemistry 2013 Dalton Trans., 2013, 42, 16056–16065 | 16065
Publ
ishe
d on
14
Aug
ust 2
013.
Dow
nloa
ded
by U
nive
rsity
of
Vir
gini
a on
07/
11/2
013
05:0
3:57
. View Article Online





























