Embed Size (px)
Citation preview

CATHEPSIN D DEFICIENCY – MOLECULAR AND
CELLULAR MECHANISMS OF NEURODEGENERATION
Sanna Partanen
Institute of Biomedicine/Biochemistry
Biomedicum Helsinki
University of Helsinki
Finland
The Finnish Graduate School of Neuroscience
ACADEMIC DISSERTATION
To be presented, with the permission of the Medical Faculty of the
University of Helsinki, for public examination in Auditorium XII,
University Main Building, Fabianinkatu 33, 3rd floor,
on November 25th, 2006, at 10 am.
HELSINKI 2006

Cover graphic: reprinted with permission of graphic designer Hanna Selkälä,
YLE TV1 Tahdon asia, 2005.
ISBN 952-92-1264-X (paperback)
ISBN 952-10-3512-9 (PDF)
http://ethesis.helsinki.fi/
Helsinki 2006
Yliopistopaino

SUPERVISED BY
Docent JAANA VESTERINEN
Institute of Biomedicine/Biochemistry
Biomedicum Helsinki
University of Helsinki
Helsinki, Finland
Docent MARC BAUMANN
Protein chemistry/Proteomics unit
Biomedicum Helsinki
University of Helsinki
Helsinki, Finland
REVIEWED BY
Professor JARI KOISTINAHO
A.I. Virtanen Institute for Molecular Sciences
University of Kuopio
Kuopio, Finland
Professor DAN LINDHOLM
Department of Biological and Environmental Sciences
University of Helsinki
Helsinki, Finland
OPPONENT
Professor ELINA IKONEN
Institute of Biomedicine/Anatomy
University of Helsinki
Helsinki, Finland


To Juho, Eevi and Lenni

TABLE OF CONTENTS
ABBREVIATIONS ...........................................................................................1
LIST OF ORIGINAL PUBLICATIONS........................................................5
ABSTRACT.......................................................................................................6
1. INTRODUCTION.................................................................................7
2. REVIEW OF THE LITERATURE ....................................................92.1. LYSOSOMES.........................................................................................9
2.1.1. Lysosomal proteases .................................................................102.1.2. Intracellular transport of soluble lysosomal enzymes...............122.1.3. Lysosomal membrane proteins .................................................142.1.4. Lysosomal storage disorders.....................................................15
2.2. NEURONS AND SYNAPTIC CIRCUITS...........................................16
2.2.1. Neurotransmission ....................................................................182.2.2. Synaptic vesicles .......................................................................182.2.3. Soluble N-ethyl maleimide-sensitive factor attachment protein
receptors (SNAREs)..................................................................202.2.4. Synaptophysin...........................................................................242.2.5. Synaptic organization of the brain ............................................252.2.6. Abnormalities of presynaptic proteins in neuronal diseases.....26
2.3. NEURONAL CEROID-LIPOFUSCINOSES (NCLs)..........................27
2.3.1. Cathepsin D (CTSD) deficiency ...............................................322.3.1.1. CTSD ................................................................................322.3.1.2. CTSD-deficient sheep .......................................................362.3.1.3. CTSD knock-out (-/-) mice ...............................................382.3.1.4. Other CTSD deficiencies ..................................................402.3.1.5. CTSD in Alzheimer’s disease and cancer.........................40
2.3.2. Postulated mechanisms of neurodegeneration in NCLs ...........422.3.2.1. Cell death mechanisms......................................................422.3.2.2. Selectivity of cell death.....................................................47
3. AIMS OF THE STUDY......................................................................51
4. MATERIALS AND METHODS .......................................................53
5. RESULTS AND DISCUSSION .........................................................55

5.1. DEVELOPMENTAL EXPRESSION OF CTSD IN THE RAT BRAIN (I) .............55
5.1.1. mRNA expression.....................................................................555.1.2. Protein expression .....................................................................56
5.2. INTRACELLULAR STABILITY, PROCESSING AND TRANSPORT OF MUTANTD293N MOUSE (m) CTSD IS AFFECTED IN HEK-293 CELLS (II) ............57
5.2.1. Loss of enzyme activity in mutant D293N mCTSD.................575.2.2. Reduced stability of mutant D293N mCTSD ...........................585.2.3. Altered processing of mutant D293N mCTSD.........................595.2.4. Intracellular transport and secretion defect of mutant D293N
mCTSD .....................................................................................60
5.3. CTSD DEFICIENCY IN HUMANS (III).......................................................62
5.3.1. A mutation in the human (h) CTSD gene causing congenitalNCL...........................................................................................62
5.3.2. Truncation of the inactive mutant c.764dupA hCTSD in BHKcells ...........................................................................................63
5.3.3. Lack of the mutant c.764dupA hCTSD in congenital NCLpatients ......................................................................................64
5.4. SYNAPTIC AND THALAMOCORTICAL CHANGES IN CTSD -/- MICE (IV)...65
5.4.1. Regional specificity of thalamocortical changes ......................655.4.2. Loss of neurons and synapses ...................................................685.4.3. Presynaptic protein changes......................................................69
6. CONCLUSIONS AND FUTURE PROSPECTS..............................71
ACKNOWLEDGEMENTS............................................................................73
REFERENCES................................................................................................75


1
ABBREVIATIONS
Asp aspartic acid
ATP adenosine triphosphate
BHK baby hamster kidney
CD-MPR cation-dependent mannose-6-phosphate receptor
CI-MPR cation-independent mannose-6-phosphate receptor
CL curvilinear profile
CNS central nervous system
CONCL congenital ovine neuronal ceroid-lipofuscinosis
CTSB cathepsin B
CTSD cathepsin D
CTSL cathepsin L
DNA deoxy-ribonucleic acid
DRG dorsal root ganglion
E embryonic day
ER endoplasmic reticulum
FP fingerprint profile
GABA gamma-aminobutyric acid
GAD 65/67 glutamic acid decarboxylase 65/67
GFAP glial fibrillary acidic protein
GROD granular osmiophilic deposit
hCTSD human cathepsin D
HEK human embryonic kidney
INCL infantile neuronal ceroid-lipofuscinosis
iNOS inducible nitric oxide synthase
JNCL juvenile neuronal ceroid-lipofuscinosis
kDa kilodalton
LAMP lysosome-associated membrane protein

2
LIMP lysosomal integral membrane protein
LINCL late infantile neuronal ceroid-lipofuscinosis
L-NAME NG-nitro-L-arginine methylester
LSD lysosomal storage disorder
LTP long-term potentiation
M1 primary motor cortex
mCTSD mouse cathepsin D
M6P mannose-6-phosphate
MPR mannose-6-phosphate receptor
mRNA messenger ribonucleic acid
NCL neuronal ceroid-lipofuscinosis
NO nitric oxide
P postnatal day
PCR polymerase chain reaction
PPT1 palmitoyl protein thioesterase-1
RL rectilinear profile
RNA ribonucleic acid
Rt reticular thalamic
RT room temperature
SAP sphingolipid activator protein
S1 primary somatosensory cortex
S1BF primary somatosensory cortex, barrel field
SM Sec1/Munc18
SMT S-methyl-isothiourea
SNAP25 synaptosomal-associated protein of 25 kDa
SNARE soluble N-ethylmaleimide-sensitive factor-attachment
protein receptor
Sub c subunit c
SV synaptic vesicle
Syb synaptobrevin 2

3
Syp synaptophysin
Syt synaptotagmin I
Syx syntaxin 1
TGN trans Golgi network
TPP1 tripeptidyl peptidase-1
TUNEL transferase-mediated dUTP-nick end labelling
VAMP2 vesicle-associated membrane protein 2
VPL ventral posterolateral
VPM ventral posteromedial

4

5
LIST OF ORIGINAL PUBLICATIONS
This thesis is based on the following original publications, which are referred
to in the text by Roman numerals I –IV:
I. Suopanki J, Partanen S, Ezaki J, Baumann M, Kominami E, Tyynelä J
(2000). Developmental changes in the expression of neuronal ceroid
lipofuscinoses-linked proteins. Mol Genet Metab 71(1-2):190-4. Review.**
II. Partanen S, Storch S, Löffler H-G, Hasilik A, Tyynelä J, Braulke T (2003).
A replacement of the active-site aspartic acid residue 293 in mouse cathepsin
D affects its intracellular stability, processing and transport in HEK-293
cells. Biochem J 369:55-62.
III. Siintola E*, Partanen S*, Strömme P, Haapanen A, Haltia M, Maehlen J,
Lehesjoki A-E, Tyynelä J (2006). Cathepsin D deficiency underlies
congenital human neuronal ceroid-lipofuscinosis. Brain 129:1438-45.
IV. Partanen S*, Haapanen A*, Kielar C, Pontikis C, Alexander N, Inkinen T,
Saftig P, Gillingwater TH, Cooper JD, Tyynelä J (2006). Synaptic changes in
the thalamocortical system of cathepsin D deficient mice, a model of
neuronal ceroid-lipofuscinosis [submitted].
* These authors contributed equally to this work.**This publication has also been presented in the thesis of Jaana Suopanki,
PhD.
These articles have been reprinted with the kind permission of their copyright
holders. In addition, some unpublished material is presented.

6
ABSTRACT
Cathepsin D (CTSD) is a lysosomal protease, the deficiency of which is fatal
and associated with neurodegeneration. CTSD knock-out mice, which die at
the age of four weeks, show intestinal necrosis, loss of lymphoid cells and
moderate pathological changes in the brain. An active-site mutation in the
CTSD gene underlies a neurodegenerative disease in newborn sheep,
characterized by brain atrophy without any changes to visceral tissues. The
CTSD deficiences belong to the group of neuronal ceroid-lipofuscinoses
(NCLs), severe neurodegenerative lysosomal storage disorders.
The aim of this thesis was to examine the molecular and cellular mechanisms
behind neurodegeneration in CTSD deficiency. We found the developmental
expression pattern of CTSD to resemble that of synaptophysin and the
increasing expression of CTSD to coincide with the active period of
myelination in the rat brain, suggesting a role for CTSD in early rat brain
development. An active-site mutation underlying the congenital ovine NCL not
only affected enzymatic activity, but also changed the stability, processing and
transport of the mutant protein, possibly contributing to the disease
pathogenesis. We also provide CTSD deficiency as a first molecular
explanation for human congenital NCL, a lysosomal storage disorder,
characterized by neuronal loss and demyelination in the central nervous
system. Finally, we show the first evidence for synaptic abnormalities and
thalamocortical changes in CTSD-deficient mice at the molecular and
ultrastructural levels.
Keywords: cathepsin D, congenital, cortex, lysosomal storage disorder,
lysosome, mutation, neurodegeneration, neuronal ceroid-lipofuscinosis,
overexpression, synapse, thalamus

7
1. INTRODUCTION
The lysosome is the cell’s main digestive compartment into which many kinds
of macromolecules are delivered for degradation. Defects in lysosomal
enzymes induce the accumulation of undegraded molecules in the
endosomal/lysosomal system. Despite the large number and clinical diversity
of lysosomal storage disorders, these diseases share some common features.
They are inherited in an autosomal recessive manner and manifest during early
childhood. Many of them affect the central nervous system (CNS) and have a
progressive neurological phenotype. Neuronal ceroid-lipofuscinoses (NCLs)
are a group of at least ten different neurodegenerative diseases that belong to
the lysosomal storage disorders (Haltia, 2003; III). NCLs are clinically
characterized by psychomotor retardation, epilepsy, blindness and premature
death and pathologically by progressive neuronal loss. The remaining neurons
are filled with an autofluorescent storage material, ceroid-lipofuscin, which
accumulates in lysosome-derived organelles. The proteinaceous ceroid-
lipofuscin is mainly composed of either mitochondrial adenosine triphosphate
(ATP) synthase subunit c (Sub c) or sphingolipid activator proteins (SAPs) and
shows different ultrastructural patterns, depending on the type of NCL disease.
Cathepsin D (CTSD; EC 3.4.23.5) is a lysosomal enzyme belonging to the
pepsin family of aspartic proteases. CTSD possesses two active-site aspartic
acid residues essential for catalytic activity. CTSD is widely distributed in
various mammalian cells (Barrett and Kirsche, 1981), and it participates in, for
example, tissue homeostasis. In addition to its role as a protease, CTSD may
have non-enzymatic functions.
A deficiency in CTSD, which is ultimately fatal, leads to congenital ovine
NCL, the first reported disease arising from a naturally occurring mutation in

8
the CTSD gene (Tyynelä et al., 2000). Newborn CTSD-deficient sheep are
unable to rise or support their head, and they die within a few days. The
phenotype of affected sheep differs considerably from that of a genetically
generated CTSD knock-out (-/-) mouse. CTSD -/- mice are apparently normal
at birth, but they develop intestinal necrosis and a loss of lymphoid cells in the
spleen and thymus at around four weeks of age (Saftig et al., 1995). Affected
sheep, by contrast, show extreme brain atrophy without any changes to visceral
tissues (Tyynelä et al., 2000). In addition, axons in the white matter of CTSD-
deficient sheep are largely devoid of myelin (Tyynelä et al., 2000). In CTSD -/-
mice, the storage material contains mitochondrial ATP synthase Sub c, while in
CTSD-deficient sheep lysosomal SAPs accumulate in the lysosomes.
At present, the mechanisms by which CTSD deficiency causes neuronal death
are unknown. In this thesis, the effects of CTSD deficiency on
neurodegeneration were examined at the molecular and cellular levels. In the
review of the literature, the basic structures and functions of the lysosomes and
neurons, the key elements in neurodegenerative lysosomal diseases, are
introduced. The CTSD enzyme and CTSD deficiencies as members of NCLs
are then discussed. Finally, the potential mechanisms of neuronal death in
NCLs are described. After presenting the results of the studies, conclusions and
future prospects are provided concerning the possible role of CTSD in neuronal
vulnerability.

9
2. REVIEW OF THE LITERATURE
2.1. LYSOSOMES
The term ‘lysosome’, Greek for ‘digestive body’, was first introduced 51 years
ago (de Duve, 1955). Lysosomes are membrane-surrounded cell organelles that
degrade macromolecules entering the lysosomes via autophagocytosis (from
the cytoplasm) or endocytosis (from the extracellular space) or through the
degradative or secretory pathway (Kornfeld and Mellman, 1989). Lysosomes
are identified by their acidic pH, hydrolases with acidic pH optimum and
specific highly glycosylated membrane-associated proteins. The size of
lysosomes varies between < 1 µm (as in neurons) and several microns (as in
macrophages). Lysosomes occur in all mammalian cells except red blood cells.
Different cell types show quantitative differences in the lysosomal
composition, macrophages having a larger fractional volume of lysosomes in
their cytoplasm compared with many other cell types (Steinman et al., 1976).
Recent studies, including proteomic analyses, have revealed that lysosomes
possess at least 50-60 soluble hydrolases (Journet et al., 2002; Sleat et al.,
2005), and a minimum of seven integral membrane proteins (Fig. 1; Eskelinen
et al., 2003). The absence of mannose-6-phosphate receptors (MPRs)
discriminates lysosomes from endosomes and other related vesicular structures.
Lysosomes function in the turnover of cellular proteins, down-regulation of
surface receptors, release of endocytosed nutrients, inactivation of pathogenic
organisms, repair of the plasma membrane and loading of processed antigens
onto major histocompatibility complex (MHC) class II molecules (Eskelinen et
al., 2003).

10
Figure 1. Schematic drawing of the topology of the major lysosomal integral membrane
proteins (modified from Eskelinen et al., 2003, with permission of Elsevier). One neuronal
ceroid-lipofuscinosis-associated integral membrane protein (CLN3) and one soluble lysosomal
enzyme, cathepsin D (CTSD) are included. LIMP= lysosomal integral membrane protein,
LAMP= lysosome-associated membrane protein.
2.1.1. Lysosomal proteases
Lysosomal proteases belong to the family of serine, aspartic or cysteine
proteases. They are ubiquitously expressed, and in a tissue- or cell-specific
manner. Different cell types, the same cell types in different physiological
environments or even different endocytic compartments of the same cell
exhibit different expression patterns of lysosomal enzymes (Brix, 2005). As an
example, one set of lysosomal proteases are upregulated while the others are
V-type H+-ATPase
COOH
COOH
COOH
COOH
COOH
COOH
COOH
NH2
NH2
NH2
NH2
NH2
NH2
NH2
Sialic acid
Cystine
LIMP-2
LAMP-2
LIMP-1
CLN3
LAMP-1
Cystinosin
Sialin
CTSD
ATP
ADP+Pi
H+

11
downregulated in cancer (Roshy et al., 2003). Lysosomal proteases are present
in all vesicles of the endocytic pathway (e.g. endocytic vesicles, early
endosomes, late endosomes, autophagic vacuoles and lysosomes). In certain
cell types, lysosomal proteases are secreted and have important functions at the
cell surface or in the pericellular environment (Andrews, 2000; Brix et al.,
2001; Linke et al., 2002; Roshy et al., 2003). Lysosomal proteolytic enzymes
catalyse the hydrolysis of proteins, usually working as endopeptidases that
cleave peptide bonds within a peptide chain. They participate in bulk protein
degradation within the lysosomes, antigen processing within the early
endosomes, proprotein processing in secretory vesicles and prehormone
processing and degradation of matrix material in the extracellular space.
Recently, lysosomal enzymes have been postulated to initiate apoptotic
processes within the cytosol (Kågedal et al., 2001). To function properly,
lysosomal enzymes have to be synthesized with a functional catalytic site and
must move along a precise intracellular pathway to reach their site of action. At
acidic pH, lysosomal proteases are processed to an active form. If a lysosomal
enzyme undergoes a mutation that prevents correct cellular targeting, it will
either be degraded or secreted from the cell. Lack of precise enzymatic activity
within the lysosome usually leads to a fatal lysosomal storage disorder (LSD),
which causes augmentation of the lysosomal apparatus.
Cathepsins are lysosomal hydrolases that degrade proteins in lysosomes at an
acidic pH [e.g. cathepsin D (CTSD); Fig. 1]. According to their active-site
amino acids, cathepsins are divided into three subgroups [i.e. cysteine (B, C, H,
F, K, L, O, S, V, W), aspartic (D, E) and serine (G) cathepsins; de Duve,
1983]. Cathepsins participate in general protein turnover and can also perform
specific functions in neovascularization (Nakagawa et al., 1998), cell growth
and tissue homeostasis (Saftig et al., 1995; Koike et al., 2000; 2003; Nakanishi
et al., 2001). Interestingly, cathepsins also have a role outside the lysosomes.
When cathepsins are secreted into the extracellular space, they participate in

12
degradation of the extracellular matrix or induction of fibroblast invasive
growth, and when they are released into the cytosol they may execute a
programmed cell death (Koblinski et al., 2000; Fehrenbacher and Jäättelä,
2005; Laurent-Matha et al., 2005). CTSD will be discussed in more detail in
Section 2.3.1.1.
2.1.2. Intracellular transport of soluble lysosomal enzymes
Most lysosomal soluble enzymes are synthesized as N-glycosylated precursors
with a signal recognition sequence that guides them through the membrane of
the endoplasmic reticulum (ER; Lodish, 1999; Fig. 2, step 1). After removal of
the signal peptide, N-linked oligosaccharides undergo extensive processing
before completion of translation (Kornfeld and Kornfeld, 1985).
Monoglycosylated core glycans of a newly synthesized polypeptide bind to the
molecular chaperone calnexin until the protein is properly folded (von Figura
et al., 1998). When entering the Golgi apparatus, the oligosaccharide chains of
lysosomal enzymes are further trimmed and complex sugars and sulphate
groups are added. The mannose-6-phosphate (M6P) recognition marker is
created by N-acetylglucosamine-1-phosphotransferase (phosphotransferase:
Fig. 2, step 2) and by N-acetylglucosamine-1-phosphodieser α-N-
acetylglucosaminidase (uncovering enzyme; Fig. 2, step 3; Lazzarino and
Gabel, 1988). After binding to the mannose-6-phosphate receptors (MPRs),
either to cation-dependent (46 Da; CD-MPR, MPR46) or cation-independent
(300 kDa; CI-MPR, MPR300) MPRs, receptor-ligand complexes exit the trans
Golgi network (TGN) in clathrin-coated vesicles and fuse with membranes of
the early or late endosomal compartment (Kornfeld, 1992; Fig. 2, steps 4 and
5). Low pH dissociates the lysosomal enzymes from the receptors, and the
enzymes are delivered to lysosomes (Fig. 2, step 6). MPRs recycle back to the
TGN to mediate further rounds of transport (Ghosh et al., 2003; Fig. 2, steps 7
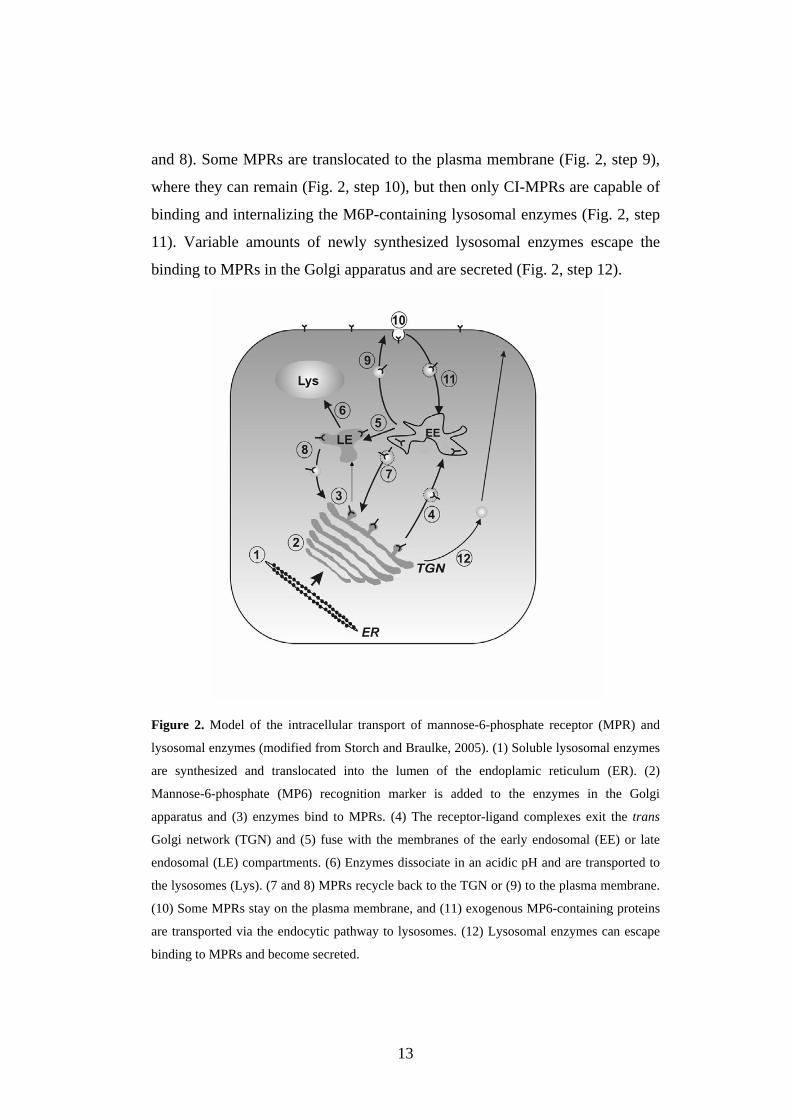
13
and 8). Some MPRs are translocated to the plasma membrane (Fig. 2, step 9),
where they can remain (Fig. 2, step 10), but then only CI-MPRs are capable of
binding and internalizing the M6P-containing lysosomal enzymes (Fig. 2, step
11). Variable amounts of newly synthesized lysosomal enzymes escape the
binding to MPRs in the Golgi apparatus and are secreted (Fig. 2, step 12).
Figure 2. Model of the intracellular transport of mannose-6-phosphate receptor (MPR) and
lysosomal enzymes (modified from Storch and Braulke, 2005). (1) Soluble lysosomal enzymes
are synthesized and translocated into the lumen of the endoplamic reticulum (ER). (2)
Mannose-6-phosphate (MP6) recognition marker is added to the enzymes in the Golgi
apparatus and (3) enzymes bind to MPRs. (4) The receptor-ligand complexes exit the trans
Golgi network (TGN) and (5) fuse with the membranes of the early endosomal (EE) or late
endosomal (LE) compartments. (6) Enzymes dissociate in an acidic pH and are transported to
the lysosomes (Lys). (7 and 8) MPRs recycle back to the TGN or (9) to the plasma membrane.
(10) Some MPRs stay on the plasma membrane, and (11) exogenous MP6-containing proteins
are transported via the endocytic pathway to lysosomes. (12) Lysosomal enzymes can escape
binding to MPRs and become secreted.

14
Several studies have provided evidence of the existence of an alternative,
MPR-independent mechanism of lysosomal targeting (Dittmer et al., 1999).
For example, intracellular trafficking of sphingolipid activator protein
precursor and the GM2-activator protein (the cofactor of β-hexosaminidase A)
to lysosomes is dependent on sortilin (Lefrancois et al., 2005). Sortilin is a
member of the type I Vsp10p superfamily, which comprises a family of
heterogeneous type I transmembrane receptors. In addition, the MPR-
independent pathway of proCTSD to lysosome is stronger in breast cancer cells
than in normal cells or other lysosomal enzymes (Capony et al., 1994).
2.1.3. Lysosomal membrane proteins
Lysosomal membrane participates in acidification of the lysosomal matrix,
mediation of fusion between lysosomes and other organelles, transport of
degraded products to the cytoplasm and sequestration of lysosomal enzymes.
Lysosomal membrane proteins are usually highly glycosylated, and they cover
the inner surface of the lysosomal membranes. Lysosome-associated
membrane proteins LAMP-1 and LAMP-2 as well as lysosomal integral
membrane proteins LIMP-1 and LIMP-2 are the most abundant (over 50% of
the lysosomal membrane mass) lysosomal membrane proteins (Fig. 1). The
heavy glycosylation of both LAMP proteins protects against degradation by
lysosomal hydrolases (Kornfeld and Mellman, 1989; Fukuda, 1991). Knock-
out mouse models of these proteins have revealed that LAMP-1, LAMP-2 and
LIMP-2 are very important for normal cell physiology and are involved in
several pathological conditions. Lysosomal membrane protein transport to the
lysosomes differs from that of the soluble enzymes. After synthesis, LAMPs
and LIMPs are transported from the TGN to endo/lysosomes mainly via an
intracellular pathway (Fukuda, 1991; Höning and Hunziker, 1995). The

15
lysosomal targeting depends on the sorting signal in the cytoplasmic tail
(Peters and von Figura, 1994; Hunziker et al., 1996; Le Borgne et al., 1998).
Less abundant lysosomal membrane proteins take part in acidification of the
vacuolar compartment (V-type H+-ATPase, vacuolar proton pump; Forgac,
1999; Fig. 1) and translocation of amino acids (cystinosin, the cystine
transporter; Fig. 1) and monosaccharides.
2.1.4. Lysosomal storage disorders
In principle, mutations in the genes that encode any of the 50-60 known
soluble lysosomal hydrolases or the seven known integral lysosomal membrane
proteins could cause a lysosomal storage disorder (LSD). At the moment, about
50 different LSDs have been identified in humans, over 40 of which involve
soluble hydrolases. LSDs result from the defective function of a specific
protein, which leads to accumulation of either undegraded substrate(s) or
catabolic products within the lysosome. Most LSDs are inherited in an
autosomal recessive manner, and the disease is progressive and the process
unremitting. LSDs are normally monogenic, but for most LSDs, several
mutations have been found in the same gene in different patients. The
frequency of LSDs is about 1 in 8000 live births (Meikle et al., 1999). LSDs
are classified according to the defective enzyme or protein, rather than on the
basis of the substrate accumulated. LSDs can be grouped as follows: 1)
sphingolipidoses, 2) mucopolysaccharidoses, 3) oligosaccharidoses and
glycoproteinosis, 4) diseases caused by defects in integral membrane proteins
and 5) others (Futerman and van Meer, 2004). Despite the variety of LSDs, the
CNS is involved in most of these. Typical for LSDs is progressive neurological
symptoms such as blindness, mental retardation and motor and sensory
problems (Jeyakumar et al., 2005). For example, mutations in the integral
membrane protein CLN3 and CTSD (Fig. 1) cause NCL, a severe

16
neurodegenerative LSD. CTSD deficiency, the topic of this thesis, will be
discussed in more detailed in Section 2.3.1.
2.2. NEURONS AND SYNAPTIC CIRCUITS
The central nervous system (CNS) is composed of various types of cells,
including neurons and glial cells (astrocytes, oligodendrocytes and microglial
cells). Glial cells are more abundant in the brain than neurons (ratio 3:1). They
do not participate directly in synaptic interactions and electrical signalling, the
main functions of the nervous system, but they have supportive roles in
maintaining the signalling abilities of neurons and they help define synaptic
contacts. The main function of astrocytes is to provide an appropriate chemical
environment for the neurons and to regulate neurotransmission in synapses by
uptaking the released neurotransmitters. Oligodendrocytes form myelin around
axons, which have a role in rapid action potential conduction. Microglial cells
are a kind of scavenger cells that protects the brain against possible pathogens
and intruders and remove cellular material from the site of brain injury or
during normal cell turnover. Microglia and tissue macrophages are of the same
origin and share many properties.
Neurons vary enormously in size and configuration. They consist of a cell
body (soma), several thick dendrites and a long thin axon in the direction of
impulse propagation (Fig. 3). The dendrites together with the cell body provide
the major site for synaptic terminals made by the axonal endings of other nerve
cells. The axon is a unique extension of the neuronal cell body that may travel
a few hundred micrometers or further, depending on the type of neuron and the
species. Neurons contain many of the same organelles as other cells in the
body (Fig. 3). However, the axon lacks the protein synthesis machinery. All the
proteins needed in the axon and synapses must be transported along the axon in
membranous organelles or protein complexes from the cell body where the
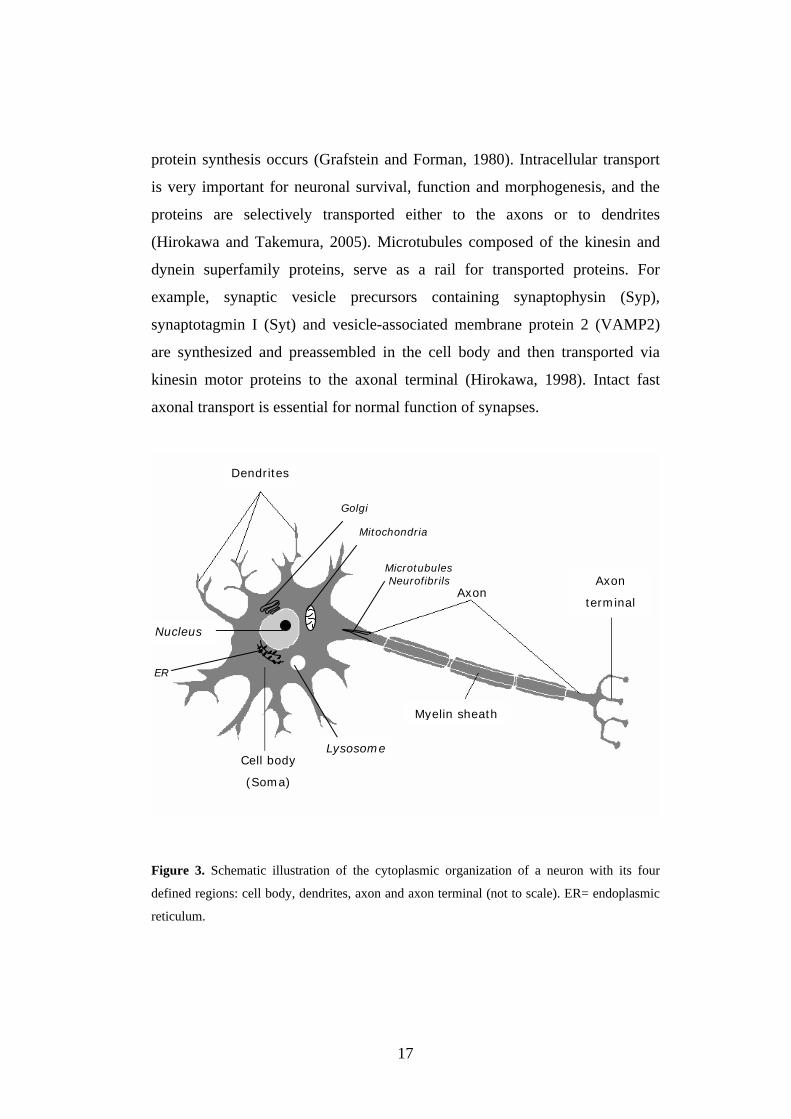
17
protein synthesis occurs (Grafstein and Forman, 1980). Intracellular transport
is very important for neuronal survival, function and morphogenesis, and the
proteins are selectively transported either to the axons or to dendrites
(Hirokawa and Takemura, 2005). Microtubules composed of the kinesin and
dynein superfamily proteins, serve as a rail for transported proteins. For
example, synaptic vesicle precursors containing synaptophysin (Syp),
synaptotagmin I (Syt) and vesicle-associated membrane protein 2 (VAMP2)
are synthesized and preassembled in the cell body and then transported via
kinesin motor proteins to the axonal terminal (Hirokawa, 1998). Intact fast
axonal transport is essential for normal function of synapses.
Figure 3. Schematic illustration of the cytoplasmic organization of a neuron with its four
defined regions: cell body, dendrites, axon and axon terminal (not to scale). ER= endoplasmic
reticulum.
Cell body
(Soma)
Nucleus
ER
Dendrites
Golgi
Mitochondria
AxonAxon
terminal
Microtubules Neurofibrils
Myelin sheath
Lysosome

18
2.2.1. Neurotransmission
Synapses are functional contacts of neighbouring neurons in the CNS.
Synapses are defined as asymmetric junctions composed of a presynaptic
terminal, including neurotransmitter-containing vesicles, a synaptic cleft and a
postsynaptic apparatus with neurotransmitter receptors (Garner et al., 2000).
Synaptic neurotransmission starts when the action potential proceeds to the
presynaptic nerve terminals and induces neurotransmitter release (Katz, 1969).
An action potential triggers Ca2+ influx to the cell, which induces the release of
neurotransmitters from synaptic vesicles (SVs) of nerve terminals into the
synaptic cleft by regulated exocytosis (Südhof, 2004). However, only 10-20%
of action potentials trigger neurotransmitter release (Goda and Südhof, 1997).
After exocytosis, SVs undergo endocytosis, recycling and refilling with
neurotransmitters for a new round of exocytosis (Barker et al., 1972). Recently,
various extracellular and intracellular proteases have been suggested to be
involved in long-lasting regulation of synaptic transmission (Tomimatsu et al.,
2002). One of these is a tissue-type plasminogen activator, an extracellular
serine protease whose expression is induced in the hippocampus during long-
term potentiation (LTP: Qian et al., 1993; Baranes et al., 1998). Neuropsin,
another example of an extracellular matrix serine protease, also has a
regulatory effect on LTP in the hippocampus (Komai et al., 2000). An
intracellular cysteine protease, calpain, seems to play an important role not
only in necrotic and apoptotic cell death but also in axonal and synaptic
plasticity (Chan and Mattson, 1999).
2.2.2. Synaptic vesicles
The function of SVs (~20 nm radius) is to take up and release
neurotransmitters. The SVs that contain neurotransmitters are classified as

19
large dense-core vesicles and small SVs. Because of the small size, SVs
contain few and small proteins. Many of these proteins are present only on a
subset of vesicles or they bind transiently to the vesicles. The proteins in SVs
are involved in neurotransmitter uptake and/or they participate in SV endo- and
exocytosis and recycling. In the classical 'all-or-none' model of exocytosis, a
SV fuses with the presynaptic membrane and releases its content into the
synapse, followed by the recycling of the vesicle membrane (Wightman and
Haynes, 2004). Alternatively, a SV can form a transient fusion pore in the
presynaptic membrane and release only part of its content by 'kiss-and-run'
exocytosis. Small SVs in the nerve terminal undergo a trafficking cycle with
the following steps (Südhof, 2004; Fig. 4): (1) active transport of
neurotransmitters into SVs, (2) SVs’ clustering close to the active zone, (3)
SVs’ docking at the active zone, (4) SVs’ priming and (5) a conversion into a
state of competence for Ca2+-triggered fusion-pore opening. After the fusion-
pore opening, three alternative pathways for endocytosis and recycling of SVs
have been proposed (Richards et al., 2000): (1) “kiss-and-stay”; SVs’
reacidification and refill with neurotransmitters without undocking, thus
remaining in the readily releasable pool, (2) “kiss-and-run”; SVs’ undocking
and recycling locally to refill with transmitters and reacidify and (3) SVs’
endocytosis via clathrin-coated pits and reacidification and refill with
neurotransmitters directly. SVs can also be refilled via an endosomal
intermediate. Large dense-core vesicles primarily undergo ‘all-or-none’
exocytosis, rarely ‘kiss-and-run’ exocytosis. The active transport of all
neurotransmitters into the synaptic vesicles is accomplished by the action of a
vacuolar proton pump that couples ATP hydrolysis with proton translocation,
thus establishing an electrochemical gradient across the vesicle membrane
(Maycox et al., 1988).

20
Figure 4. The synaptic vesicle cycle (modified from Südhof, 2004, with permission of Annual
Reviews). (1) Synaptic vesicles (SVs) are filled with neurotransmitters (NTs) and (2)
clusterized. (3) SVs dock at the active zone and (4) are primed for Ca2+ -triggered (5) fusion-
pore opening. After fusion-pore opening, SVs undergo endocytosis and recycling either via (6)
local reuse, (7) recycling without an endosomal intermediate or (8) clathrin-mediated
endocytosis with (9) recycling through endosomes. Steps in exocytosis are indicated by black
arrows and steps in endocytosis and recycling by grey arrows. Insert: Schematic picture of
SNARE-interacting proteins; synaptosomal-associated protein of 25 kDa (SNAP25), syntaxin
1 (Syx), synaptobrevin 2 (Syb) and synaptotagmin (Syt) in a SV.
2.2.3. Soluble N-ethyl maleimide-sensitive factor attachment protein
receptors (SNAREs)
Synaptic vesicle fusion involves a highly conserved family of proteins termed
SNAREs (soluble N-ethyl maleimide-sensitive factor attachment protein
NT
H+
1
2
3
ATP
4
”Docking
”Priming
NT
Ca2
Ca25
Receptors
8
7
H+
9
Postsynaptic Cell
Presynaptic terminal
”Endocytosis”
6
”Acidification
Active zoneActive zone
SNAP25 Syx
Syb
Syt
”Fusion
Early endosome

21
receptors; Brunger, 2005; Fig. 4; insert). Formation of the SNARE complex,
composed of synaptobrevin (Syb), synaptosomal-associated protein of 25 kDa
(SNAP25) and syntaxin 1 (Syx), is essential for SV exocytosis (Lin and
Scheller, 2000). SNARE proteins can be classified into the t-SNARE (also
called Q-SNARE) proteins: Syx and SNAP25, and the v-SNARE (also called
R-SNARE) protein: Syb (Söllner et al., 1993). t-SNARE and v-SNARE are
located in opposing bilayers, and they interact in a circular array to form
conducting pores in the presence of Ca2+ (Cho et al., 2002). SNAREs are
directly linked to Ca2+-triggered exocytosis, most likely in conjunction with a
Ca2+ sensor, synaptotagmin I (Syt; Davis et al., 1999; Fernandez-Chacon et al.,
2001; Sakaba et al., 2005). The main steps in membrane fusion during
secretion are highly controlled and regulated events (Leabu, 2006). The
selective proteolysis of synaptic SNAREs accounts for the total block of
neurotransmitter release caused by clostridial neurotoxins in vivo (Humeau et
al., 2000; Schiavo et al., 2000).
The SNARE proteins themselves are not sufficient to trigger the membrane
fusion reaction. The action of Sec1/Munc18 (SM)-like proteins seems to be
more fundamental to membrane fusion (Verhage et al., 2000). Most of the SM
proteins interact with the SNARE proteins by binding to Q-SNARE (Toonen
and Verhage, 2003). SM proteins also appear to function as a link between the
SNARE complex and the Rab effectors and other tethering factors (Kauppi et
al., 2004).
Vesicle-associated membrane protein 2 (VAMP2; synaptobrevin; Syb) is an
integral membrane protein of 18 kDa (Trimble et al., 1988, Baumert et al.,
1989) that is palmitoylated at its cysteine residues (Veit et al., 2000). It forms
two mutually exclusive protein complexes, the SNARE complex (Lin and
Scheller, 2000) and a complex with synaptophysin (Syp; Washbourne et al.,
1995). The Syp/Syb complex can be considered a molecular marker for mature

22
synapses (Yelamanchili et al., 2005). Syb can form multimers, but recent
studies have revealed that the homodimerization of Syb is very weak, and thus
Syb homodimers do not have a major role in SNARE complex
oligodimerization in vivo (Bowen et al., 2002).
In Syb knock-out mice, the rates of both spontaneous and Ca2+-triggered SV
fusion were decreased (Schoch et al., 2001), revealing the importance of Syb as
a part of neurotransmission. In Syb-deficient synapses, altered shape and size
of SVs were observed, and stimulus-dependent endocytosis was delayed (Deak
et al., 2004). The proteolysis of Syb by tetanus or botulinum neurotoxin types
B, D, F and G blocks neuroexocytosis (Schiavo et al., 1992, 1993a, 1993b,
1994). Tetanus toxin cleaves Syb also while being bound to Syp or while
existing as a homodimer (Reisinger et al., 2004). Dissociation of the Syp/Syb
complex depends on an elevation of the intracellular Ca2+ concentration after
stimulation with an ionophore (Reisinger et al., 2004). An elevation of the
intracellular Ca2+ concentration does not, however, directly dissociate the
Syp/Syb complex, but requires a cytosolic factor, which is present in cultivated
neurons (Reisinger et al., 2004). Syp/Syb complex formation is dependent on
the presence of the transmembrane domain and C-terminal part of Syb
(Edelmann et al., 1995; Yelamanchili et al., 2005) and is sensitive to reducing
agents and high-salt conditions (Edelmann et al., 1995; Becher et al., 1999a).
The Syp/Syb interaction also strongly depends on the cholesterol content of the
membrane (Mitter et al., 2003). Chronic blockage of glutamate receptors
causes an increase in neurotransmitter release but a decrease in the Syp/Syb
complex (Bacci et al., 2001).
Synaptosomal-associated protein of 25 kDa (SNAP25) is a palmitoylated
membrane protein predominantly localized in the plasma membrane in neural
and neuroendocrine cells where it functions as a part of the SNARE complex
(Hess et al., 1992; Chen and Scheller, 2001). Palmitoylation of SNAP25 is

23
important for initial membrane targeting of the protein (Gonzalo and Linder,
1998). SNAP25 is also found in intracellular membranes (Aikawa et al., 2006).
Intracellular SNAP25 localizes to the recycling endosome (RE) and TGN
compartments and has a role as a t-SNARE at a trafficking step in the
endosomal recycling pathway (Aikawa et al., 2006). Studies of SNAP25
knock-out mice showed that the nervous system of these mice developed
normally in utero even when SNAP25 was missing from the SNARE complex
(Washbourne et al., 2002). Axonal outgrowth, synaptic contact targeting and
action potential-independent, spontaneous transmitter release occurred in
SNAP25 mutant mice, whereas action potential-dependent neurotransmitter
release was totally abolished (Wasbourne et al., 2002). The proteolytic
cleavage of the amino-terminal domain of SNAP25 by calpain suppresses
neurotransmitter release, and calpain inhibitors enhance Ca2+-dependent
glutamate release (Ando et al., 2005).
Syntaxins 1-4 comprise a large family of membrane-associated proteins that
specifically associate with the plasma membrane and regulate trafficking for
exocytosis and/or for insertion of proteins into the plasma membrane (Bennett
et al., 1993; Gaisano et al., 1996; Scales et al., 2000). Syntaxin 1 (Syx) is the
best studied of the syntaxins and the principal isoform associated with synapses
in the brain (Bennett et al., 1992, 1993). Several studies have demonstrated that
Syx interacts with multi-transmembrane proteins such as neurotransmitter
transporters (Deken et al. 2000; Geerlings et al. 2000; Zhu et al. 2005). It can
directly interact with Syb and SNAP25 (Calakos et al., 1994; Pevsner et al.,
1994). This interaction is mediated by a confined domain of Syx that is
adjacent to the transmembrane domain (Calakos et al., 1994). Recently, the
SNAP25/Syx complex has been shown to functionally modulate
neurotransmitter gamma-aminobutyric acid (GABA) reuptake (Fan et al.,
2006).

24
2.2.4. Synaptophysin
Synaptophysin (Syp), with a molecular mass of 38 kDa, is one of the most
abundant SV proteins in brain and neuroendocrine tissue. It has a C-terminal
domain with nine pentapeptide repeats that are phosphorylated by tyrosine
kinases within the nerve terminal (Pang et al., 1988). Some evidence of the
importance of tyrosine phosphorylation in LTP has emerged (Purcell and
Carew, 2003; Kalia et al., 2004). The physiological role of Syp remains
unknown, but the role of Syp in a positive modulation of neuronal exocytosis
has been strongly postulated (Reisinger et al., 2004). Syp serves as a regulator
of the availability of Syb for the SNARE complex (Edelmann et al., 1995;
Becher et al., 1999a). An increase in the amount of the Syp/Syb complex was
observed in kindled rats with enhanced presynaptic activity that served as a
model of human epilepsy (Hinz et al., 2001). Formation of the Syp/Syb
complex increases during neuronal development (Becher et al., 1999a), and it
is absent in neuroendocrine cells and embryonic neurons (Becher et al.,
1999b). Syp knock-out mice do not develop phenotypic changes (Eshkind and
Leube, 1995; McMahon et al., 1996), showing that Syp is not necessarily
required for exocytosis. However, Syp plays a role in regulating activity-
dependent synapse formation (Tarsa and Goda, 2002).
Syp interaction with the GTPase dynamin I, whose activity is essential for SV
endocytosis, regulates clathrin-independent endocytosis of SVs in a Ca2+-
dependent manner (Daly et al., 2000; Marks et al., 2001). Additionally, Syp
functions as a cholesterol-binding protein, and thus, might help to maintain
membrane integrity of SVs during endo- and exocytosis (Thiele et al., 2000).
Syp can form multimers (Thomas et al., 1988; Fykse et al., 1993).

25
2.2.5. Synaptic organization of the brain
Neurons with connecting synapses form circuits that mediate the specific
functional operation of different brain regions. As an example, the circuit that
defines the main thalamocortical loop of the rat somatosensory system is
presented in Fig. 5. The somatosensory information from the peripheral organs
enters the nervous system through the dorsal root ganglion (DRG) cells and is
conveyed to the spinal cord. The thalamus projects to the primary
somatosensory (S1) cortex, which in turn projects to other regions of the
cerebral cortex. Excitatory neurons in the ventral posteromedial (VPM)
thalamic nuclei project mainly to layer IV of the S1 cortex. On their way to the
cortex, the axons of VPM thalamic nuclei also project to the reticular thalamic
(Rt) nuclei. Descending projections in the thalamocortical system feed
information back from the cortex to the thalamus and originate primarily in
layer VI of the S1 cortex. On their way to VPM thalamic nuclei,
corticothalamic projections also send branches to the inhibitory neurons in the
Rt nuclei. Inhibitory neurons in Rt nuclei either project to the VPM nucleus,
activating GABA receptors, or interconnect locally within Rt nuclei.

26
Figure 5. Schematic presentation of the rat thalamocortical system. The somatosensory
information travels from the periphery through the dorsal root ganglia (DRG) and spinal cord
to the thalamus. The ventral posteromedial (VPM) thalamic neurons send their axons to layer
IV in the primary somatosensory (S1) cortex (insert). Descending projections from layer VI of
the S1 cortex (insert) send branches to the reticular thalamic (Rt) nucleus and terminate on
neurons in the VPM nucleus. Arrows indicate the direction of information flow.
2.2.6. Abnormalities of presynaptic proteins in neuronal diseases
Several neurological disorders are characterized by abnormalities in
presynaptic proteins. In the cingulate cortex of schizophrenia patients, elevated
levels of Syx (Honer et al., 1997) and abnormalities in GABA receptors (Benes
et al., 1992) were observed. Qualitative abnormalities in asymmetric synapses
and loss of vesicles with accumulation of abnormal membranous material were
I
II
III
IV
S1
DRG
Spinal cord
Thalamus (VPM)
Rt
V
VI
S1

27
also detected (Miyakawa et al., 1972; Ong and Garey, 1993). Relatively more
SNAP25 and Syx were present in the heterotrimetric SNARE complex of
patients with schizophrenia who committed suicide (Honer et al., 2002). In
traumatic brain injury, increased levels of complexin I (a marker of
axosomatic inhibitory synapses) and complexin II (a marker of axodendritic
and axospinous excitatory synapses) were reported in association with neuronal
loss and the pathophysiology of cerebral damage (Yi et al., 2006). Complexins
are known to bind to SNAREs at the presynaptic terminal and therefore to
regulate neurotransmission (Pabst et al., 2000). In Alzheimer’s disease,
accumulation of SNAP25 immunoreactive material was found in axons,
strongly suggesting an impairment of axonal transport (Dessi et al., 1997).
Furthermore, in juvenile neuroaxonal dystrophy in a Rottweiler and in murine
scrapie, abnormal expression of synaptic proteins was observed (Siso et al.,
2001, 2002). Syp and SNAP25 accumulated in neurons, mainly in the
thalamus, midbrain and pons, and granular deposits of α-synuclein were
present in neurophils of the same areas in murine scrapie (Siso et al., 2002).
2.3. NEURONAL CEROID-LIPOFUSCINOSES (NCLs)
Neuronal ceroid-lipofuscinoses (NCLs) are a group of recessively inherited
lysosomal diseases characterized by progressive neurodegeneration and
premature death. NCLs are among the most common causes of encephalopathy
in children, with an incidence of 1:12500 live births worldwide (Banerjee et al.,
1992). NCL patients exhibit epileptic seizures, progressive mental retardation,
ataxia, myoclonus and visual failure, eventually leading to death (Santavuori et
al. 1998, Haltia, 2003). Based on age of onset, ultrastructural and genetic
findings and clinical characteristics, NCLs have been divided into subtypes.
Ten different NCL disease subtypes currently exist, some with an unknown
genetic background (Table 1). Mutations have been identified in seven distinct

28
genes (Cooper, 2003; III), and the proteins are known as CLN proteins. Three
of these proteins are soluble lysosomal hydrolases (CLN1, CLN2 and CTSD),
and four are thought to be transmembrane proteins (CLN3, CLN5, CLN6 and
CLN8). To date, CLN4, CLN7 and CLN9 genes have not been identified.
Infantile neuronal ceroid-lipofuscinosis (INCL; Santavuori-Haltia disease) is
a lysosomal disorder caused by deficiency in the CLN1 gene encoding
palmitoyl protein thioesterase-1 (PPT1; CLN1; Vesa et al., 1995). PPT1
degrades palmitoylated proteins by deacylating cysteine thioesters, and the
latter accumulate in INCL (Hofmann et al., 2002). Late infantile neuronal
ceroid-lipofuscinosis (LINCL; Jansky-Bielschowsky disease) is a disease
caused by a deficiency in the CLN2 gene encoding tripeptidyl peptidase-1
(TPP1; CLN2; Sleat et al., 1997), a serine protease that cleaves tripeptides
from the amino terminus of small proteins. Juvenile neuronal ceroid-
lipofuscinosis (JNCL; Spielmayer-Vogt, Sjögren disease, Batten disease), the
most common of the NCLs worldwide, is caused by mutations in the CLN3
gene (International Batten Disease Consortium, 1995). CLN3 is an integral
membrane protein of 438 amino acids with 6-11 predicted transmembrane
domains, and it has recently been proposed to be an arginine transporter and to
participate in pH regulation (Pearce, 1999; Kim et al., 2003). CLN5 may exist
as an integral membrane protein or a soluble, glycosylated protein (Savukoski
et al. 1998; Isosomppi et al. 2002). CLN6 is a unique 311-amino acid protein
with 6-7 transmembrane domains (Gao et al., 2002; Wheeler et al., 2002).
CLN8 is a transmembrane protein with a putative function in lipid metabolism
(Ranta et al., 1999; Winter and Ponting, 2002).
Rare cases of congenital human neuronal ceroid-lipofuscinosis have been
reported (Norman and Wood, 1941; Brown et al., 1954; Sandbank, 1968;
Humphreys et al., 1985; Garborg et al., 1987; Barohn et al., 1992; Wisniewski
and Kida, 1992). Congenital human NCL cases were originally reported under
the diagnosis amaurotic familial idiocy. Typically, infants with congenital NCL

29
developed epileptic seizures, decerebrate rigidity and abnormal respiration
patterns within hours after uneventful deliveries. The general autopsy findings
were normal, but the brains were extremely small and firm. Activated
astrocytes and microglia as well as neuronal loss were also detected in the
cerebral cortex of affected children. Some patients showed microcephaly, and
ultrastructural analysis of autopsy material revealed material consistent with
ceroid-lipofuscin, an autofluorescent hydrophobic material. Humphreys et al.
(1985) reported both granular and lamellar ultrastructure of autofluorescent
storage bodies, while Garborg et al. (1987) described only granular osmiophilic
deposits (GRODs).
The ceroid-lipofuscinoses also occur in animals; several forms have been used
extensively as models of analogous diseases in humans. The New Zealand
South Hampshire sheep disease, OCL6, is syntenic with CLN6 (Broom et al.,
1998). The motor neuron degeneration (mnd) mouse is syntenic with CLN8
(Ranta et al., 1999), and the nclf mouse with CLN6 (Bronson et al., 1998). Two
different PPT1 knock-out mouse models of INCL have been produced (Gupta
et al., 2001; Jalanko et al., 2005). Two CLN3 null mutant mice (Mitchison et
al., 1999; Katz et al., 2001) and one Cln3 ex7/8 knock-in mouse (Cotman et al.,
2002) also exist. Moreover, a mouse model of CLN2 has recently been
generated (Sleat et al., 2004). NCL disease has also been found in, for
example, dogs, cats, cattle and horses (Jolly and Walkley, 1997).
A feature common to all NCLs, whether in humans or animals, is the
accumulation ceroid-lipofuscin, mainly in the cytoplasm of neurons, but also to
a certain extent in other cells (Goebel and Wisniewski, 2004). Recently, there
has been discussion about the distinguishing features of lipofuscin and ceroid
(Seehafer and Pearce, 2006). Lipofuscin (“aging pigment”) accumulates in
aging cells, while ceroid (“lipofuscin-like lipopiment”) arises from
pathological condition such as cell stress, disease and malnutrition (Yin, 1996;

30
Terman and Brunk, 1998). Lysosomes that have accumulated lipopigments are
called secondary lysosomes, cytosomes, residual bodies or dense bodies
(Seehafer and Pearce, 2006). Lipopigment storage bodies appear to be an
electron-dense mass surrounded by a typical lysosomal membrane.
Administration of lysosomal enzyme inhibitors, such as leupeptin, to brains of
young rats resulted in an accumulation of lipopigments in their brain,
suggesting that loss of lysosomal enzyme activity can precipitate lipofuscin
development (Ivy et al., 1984). In NCL diseases, the major component of
storage material is protein (Palmer et al., 1986). Depending on the NCL disease
type, the storage material is composed of either SAPs A and D or
mitochondrial ATP synthase Sub c Table 1). The ultrastructural pattern of
lysosomal storage materials in different NCL diseases also varies, being
GRODs, curvilinear profiles (CLs), fingerprint profiles (FPs) and/or rectilinear
profiles (RLs) (Elleder, 1991; Table 1).
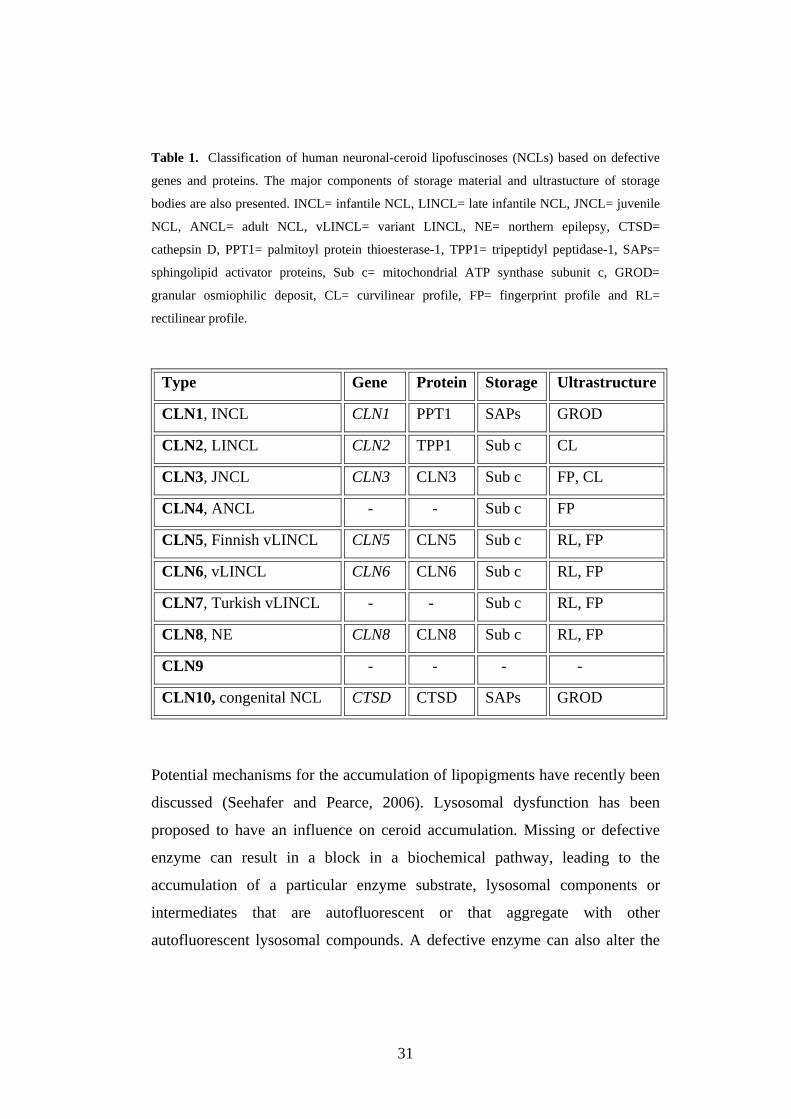
31
Table 1. Classification of human neuronal-ceroid lipofuscinoses (NCLs) based on defective
genes and proteins. The major components of storage material and ultrastucture of storage
bodies are also presented. INCL= infantile NCL, LINCL= late infantile NCL, JNCL= juvenile
NCL, ANCL= adult NCL, vLINCL= variant LINCL, NE= northern epilepsy, CTSD=
cathepsin D, PPT1= palmitoyl protein thioesterase-1, TPP1= tripeptidyl peptidase-1, SAPs=
sphingolipid activator proteins, Sub c= mitochondrial ATP synthase subunit c, GROD=
granular osmiophilic deposit, CL= curvilinear profile, FP= fingerprint profile and RL=
rectilinear profile.
Type Gene Protein Storage Ultrastructure
CLN1, INCL CLN1 PPT1 SAPs GROD
CLN2, LINCL CLN2 TPP1 Sub c CL
CLN3, JNCL CLN3 CLN3 Sub c FP, CL
CLN4, ANCL - - Sub c FP
CLN5, Finnish vLINCL CLN5 CLN5 Sub c RL, FP
CLN6, vLINCL CLN6 CLN6 Sub c RL, FP
CLN7, Turkish vLINCL - - Sub c RL, FP
CLN8, NE CLN8 CLN8 Sub c RL, FP
CLN9 - - - -
CLN10, congenital NCL CTSD CTSD SAPs GROD
Potential mechanisms for the accumulation of lipopigments have recently been
discussed (Seehafer and Pearce, 2006). Lysosomal dysfunction has been
proposed to have an influence on ceroid accumulation. Missing or defective
enzyme can result in a block in a biochemical pathway, leading to the
accumulation of a particular enzyme substrate, lysosomal components or
intermediates that are autofluorescent or that aggregate with other
autofluorescent lysosomal compounds. A defective enzyme can also alter the

32
environment of the lysosomes or the cell itself or disrupt the trafficking
pathway. Another possible mechanism for accumulation of lipopigments is
autophagy, which is known to contribute to protein and organelle turnover.
Autophagy has been shown to be decreased in adult mice with accumulated
lipopigments (Terman, 1995). Because the lipopigments contain lipid oxidation
products, oxidative damage has been postulated to be responsible for the
accumulation of storage material. Both autophagy and oxidative stress as
potential contributors to neuronal death in NCLs will be discussed in detail in
Section 2.3.2.
2.3.1. Cathepsin D (CTSD) deficiency
2.3.1.1. CTSD
Cathepsin D (CTSD; EC 3.4.23.5) is a lysosomal enzyme belonging to the
pepsin family of aspartyl proteases (Tang, 1979). It comprises approximately
11% of the total lysosomal enzymes (Dean and Barrett, 1976) and 90% of the
total brain acid proteinases (Marks and Lajtha, 1965). The expression level of
CTSD varies depending on the tissue, neurons in the CNS possessing abundant
CTSD (Whitaker and Rhodes, 1983; Reid et al., 1986). Like all lysosomal
proteinases, CTSD requires an acidic pH for its activity (Briozzo et al., 1988).
CTSD has been suggested to participate in various biological events such as
cellular protein turnover and degradation of several brain-specific antigens
(Banay-Schwartz et al., 1987). CTSD can activate precursors of biologically
active proteins in prelysosomal compartments of specialized cells (Diment et
al., 1989). The activity and localization of CTSD are changed during aging,
and CTSD is involved in certain pathological conditions, e.g. Alzheimer’s
disease (Matus and Green, 1987; Nakaniski et al., 1994, 1997; Cataldo et al.,
1995). In addition to its role as a protease, CTSD may also have non-enzymatic

33
functions. For example, the inactive proCTSD zymogen functions as a mitogen
in some cancer cell lines (Fusek and Vetvicka, 1994). In the extracellular
space, CTSD may take part in such pathological processes as inflammation
(Barrett, 1977), tumour progression and formation of metastases (Tandon et al.,
1990; Mignatti and Rifkin, 1993). In contrast to other tissue proteases (e.g.
serine proteases and metalloproteinases), no endogenous CTSD tissue
inhibitors have been identified in mammals. However, pepstatin, a specific
inhibitor of aspartic proteases, has often been used for purification of CTSD
and also to study its function in some in vitro systems (Dean, 1975; Conner,
1989).
The mouse Ctsd gene consists of 11 kb of genomic deoxy-ribonucleic acid
(DNA). It is localized in chromosome 4 and contains 9 exons (size 99-823 bp)
and 8 introns (size 94 bp-3.2 kb) (Hetman et al., 1994). The human CTSD gene
is assigned to chromosome 11 (Hasilik et al., 1982). The exon-intron
organization of the CTSD gene is very similar between man and mouse
(Redecker et al., 1991). The main differences are found in promoter
organization and in the length of exons 1 and 9 (Hetman et al., 1994). CTSD,
like other housekeeping genes, is expressed in all tissues (Barrett, 1977), but
the expression level varies between tissues. The transcription factor c-myc, as
well as p53, regulates the transcription of CTSD (Blackwell et al., 1990; Wu et
al., 1998).
The sequence similarity of sheep CTSD is 85% compared with human protein
and 79% compared with mouse protein (Tyynelä et al., 2000; Fig. 6). CTSD
has a bilobed structure, consisting of two evolutionarily related lobes, mainly
made up of β sheets, and a deep active-site cleft. Each of these lobes contains a
key active-site aspartic acid residue (Asp32 and Asp215 in pepsin numbering)
and a single carbohydrate group (Metcalf and Fusek, 1993). Together these

34
aspartates are thought to position and activate a water molecule, which then
hydrolyses the substrate peptide bond. Several studies have revealed that the
mutation of an active-site aspartate in CTSD is sufficient to inactivate the
enzyme without affecting protein processing (Wittlin et al., 1999).
CTSD is synthesized on the rough ER as an inactive prepro-enzyme which is
proteolytically processed to the active, mature form (Hasilik and Neufeld,
1980; Richo and Conner, 1994). An N-terminal signal sequence with length of
20 amino acids mediates the transport of prepro-CTSD across the ER
membrane (Erickson and Blobel, 1979). After removing this presequence, the
propeptide keeps the CTSD inactive during cellular transport (Erickson et al.,
1981). ProCTSD acquires cotranslationally two high-mannose oligosaccharide
chains (Takahashi et al., 1983). Each high-mannose carbohydrate chain
contributes approximately 2 kDa to the mass of the protein. One or both of the
high-mannose chains are phosphorylated in the early Golgi apparatus by
addition of N-acetylglucosamine 1-phosphate (Kornfeld and Mellman, 1989).
Removal of the N-acetylglucosamine in the TGN enables CTSD to bind to
MPRs, which mediate targeting to late endosomes (Kornfeld and Mellman,
1989). This MPR-dependent transport also requires interaction with prosaposin
(Gopalakrishnan et al., 2004). Alternatively, mouse proCTSD is membrane-
associated by a MPR-independent mechanism, for example, macrophages
(Diment et al., 1988).

35
Figure 6. Protein sequence alignment of human, sheep and mouse cathepsin D (CD; Tyynelä
et al., 2000). N-terminal signal sequence and propeptide of prepro-CTSD is cleaved to a single-
chain enzyme, which is further processed into a two-chain form: an N-terminal light chain and
a C-terminal heavy chain. The proteolytic cleavage sites of human CTSD are indicated by
arrows and two catalytically important aspartic acid residues by arrowheads. Identical and
similar amino acids are highlighted in black; similar amino acids are separated from identical
amino acids by bolded white letters.
Endosomal/lysosomal cleavage of the propeptide generates an active single-
chain enzyme of approximately 44 kDa. In humans, but rarely in rodents, the
single-chain enzyme is subsequently cleaved to a two-chain form; to a 30-kDa

36
heavy chain (carboxy-terminal domain) and a 14-kDa lightchain (amino-
terminal domain). The variation in cleavage between humans and rodents
results from species-specific protein sequence differences at the cleavage site
(Yonezawa et al., 1988). Depending on the cell type studied, 2-66% of a newly
synthesized CTSD fails lysosomal sorting and is secreted (Capony et al.,
1994). For instance, breast tumour cells secrete large amounts of proCTSD
(Godbold et al., 1998).
The mechanisms associated with the processing and activation of lysosomal
proteases, including CTSD, remain largely unknown (Ishidoh and Kominami,
2002). proCTSD is capable of acid-dependent autoactivation in vitro, removing
26 residues to yield an active form, designated pseudoCTSD, which is not a
processing intermediate in vivo (Hasilik et al., 1982; Richo and Conner, 1994).
Prosaposin and ceramide promote the autoactivation of proCTSD (Heinrich et
al., 1999; Gopalakrishnan et al., 2004). In vivo, CTSD undergoes several
proteolytic processing steps during biosynthesis (Hasilik and Neufeld, 1980;
Richo and Conner, 1994), mainly by cysteine proteases (Gieselmann et al.,
1985). Two lysosomal cysteine proteases, CTSB and CTSL, have been shown
to be involved in CTSD processing (Felbor et al., 2002; Wille et al., 2004;
Laurent-Matha et al., 2006). One mechanism proposed for the activation of
CTSD is a combination of partial autoactivation and enzyme-assisted
activation to yield a mature enzyme (Conner and Richo, 1992; Larsen et al.,
1993). Recently, however, the mechanism of CTSD maturation has been
demonstrated to be independent of its catalytic activity and autoactivation
(Laurent-Matha et al., 2006).
2.3.1.2. CTSD-deficient sheep
Congenital ovine NCL (CONCL) disease was found in a flock of white
Swedish landrace sheep, on an experimental farm in Northern Sweden (Järplid

37
and Haltia, 1993). The newborn lambs were weak, trembling and unable to
raise and support their bodies. They died within a few days without bottle-
feeding. At autopsy, marked brain atrophy, accompanied by reduced thickness
of the cerebral cortex and a white matter largely devoid of myelin, was
observed (Tyynelä et al., 2000). Hippocampal pyramidal neurons were mostly
degenerated, and the deep layers of the cerebral cortex showed a massive loss
of neurons, reactive astrocytosis and infiltration of macrophages. Particularly
in the deeper layers, axonal enlargements and neurons with abundant storage
material commonly coincided with the most intense neuronal loss. The
thalamus appeared relatively normal, and the cerebellum was less affected,
showing increased thickness of the external granule cell layer. However, some
loss of cerebellar Purkinje cells and internal granular cells was apparent. The
remaining cortical neurons were filled with an autofluorescent storage material
with GRODs (Järplid and Haltia, 1993). The amounts of SAPs A and D were
elevated in CONCL neurons, whereas there was no accumulation of Sub c
(Tyynelä et al., 2000). Activities of many other lysosomal enzymes (e.g. TPP1,
CTSL and CTSB) were increased in the CONCL brain.
A deficiency in the CTSD gene was shown to be the cause of CONCL
(Tyynelä et al., 2000). A single nucleotide missense mutation in the CTSD
gene resulted in the conversion of an active-site aspartic acid residue (Asp 295
according to human CTSD numbering) to asparagine, which led to production
of an inactive but stable protein (Tyynelä et al., 2000). The steady-state level of
CTSD was higher in the CONCL brain than in the control brain (Tyynelä et al.,
2001), suggesting the preservation of a non-enzymatic role of CTSD in
affected sheep. CONCL sheep showed no pathological changes in their
lymphoid organs or gut, thus being in contrast to the findings in CTSD-
deficient mice.

38
2.3.1.3. CTSD knock-out (-/-) mice
CTSD -/- mice were generated by Saftig et al. (1995) by introducing targeting
construct pCDneo4 to disrupt the Ctsd gene. The open reading frame of the
gene in exon 4 was interrupted, and the truncation led to the coding of only the
N-terminal quarter of mature CTSD. Affected mice were apparently normal at
birth. At approximately two weeks of age, CTSD -/- mice showed severe
neurological symptoms, including tremor, epileptic seizures and muscle
rigidity (Koike et al. 2000). CTSD -/- mice subsequently developed atrophic
changes in their lymphoid system and small intestine, dying prematurely at
about 26 days of age (Saftig et al., 1995). The spleen and thymus underwent
massive destruction, with a loss of T and B cells, and the overall architecture of
the ileal mucosa was altered. Apoptotic cell death in the thymus increased in
CTSD -/- mice after the age of two weeks. However, bulk protein degradation
was unchanged, suggesting a compensatory action of cysteine proteinases and
a role for CTSD in limited proteolysis (Saftig et al., 1995). At the terminal
stage, the mean weight of affected mice was only ~60% of that of their wild-
type littermates. Initially, no overt brain pathology was observed in CTSD -/-
mice (Saftig et al., 1995). However, CTSD -/- mice exhibit neuropathologically
many of the characteristics of NCLs, including neuronal deposition of
autofluorescent storage material with granular or fingerprint-type ultrastructure
and accumulation of Sub c (Koike et al., 2000). As in the CONCL brain, the
activities of TPP1 and CTSB were increased in CTSD -/- mouse brains (Koike
et al., 2000).
Recently, several explanations for the pathological changes in CTSD -/- mice
have been postulated. The observation that CTSD -/- mice are blind and the
selective loss of photoreceptor cells in the outer nuclear layer have led to
further investigation of cell death pathways in retinal atrophy of affected
animals (Koike et al., 2000). Results have revealed that CTSD deficiency

39
induces apoptosis of the cells in the outer nuclear layer via caspase 9 and 3
activation, while the loss of the neurons in the inner nuclear layer is mediated
by nitric oxide (NO) from microglial cells (Koike et al., 2003).
Morphologically transformed microglia with large rounded cell bodies
appeared after postnatal day (P) 16 in the cerebral cortex and thalamus of
CTSD -/- mice, and correspondingly, macrophages in the intestine expressed
inducible nitric oxide synthase (iNOS) (Nakanishi et al., 2001). These results
suggest that NO production by microglia and peripheral macrophages
contributes to neuronal apoptosis and intestinal necrosis (Nakanishi et al.,
2001). Interestingly, CTSD -/- mice treated with the competitive NOS inhibitor
NG-nitro-L-arginine methylester (L-NAME) or the iNOS inhibitor S-methyl-
isothiourea (SMT) survived significantly longer than the control littermates and
showed no decrease in their body weight. Moreover, the number of transferase-
mediated dUTP-nick end labelling (TUNEL)-positive cells in the thalamus was
somewhat decreased after treatment. However, neither the L-NAME nor the
SMT treatment prevented the eventual death of affected mice. In addition,
CTSD -/- mice had increased phagocytic activity during the terminal stage. In
CTSD -/- mouse brains, double membrane-bound vacuoles containing part of
the cytoplasm were detected, indicating that these vacuoles are
autophagosomes or autolysosomes (Koike et al., 2000, 2005). This suggests
that autophagy is involved in the abnormal storage accumulation of CNS
neurons in CTSD -/- mice (Koike et al., 2005).
Recently, mechanisms underlying the epileptic seizures of CTSD -/- mice have
been also discussed (Shimizu et al., 2005). In hippocampal slices of CTSD -/-
mice at P20, spontaneous burst discharges were recorded from both the CA1
and CA3 pyramidal cells (Koike et al., 2000). Activated microglia were found
to accumulate in the pyramidal cell layer of the hippocampal CA3 subfield.
Dysfunction of GABAergic interneurons in this region was due to the
accumulation of glutamate decarboxylase (GAD) 67 degradation products in

40
the lysosomes (Shimizu et al., 2005). However, numerical density of
GABAergic interneurons was not markedly changed.
2.3.1.4. Other CTSD deficiencies
American Bulldogs with young-adult-onset NCL exhibited ataxia,
psychomotor retardation and hypermetria, and they died before seven years of
age (Evans et al., 2005). Autofluorescent storage material was present in the
cytoplasm of the neurons throughout the brain and in retinal ganglion cells
(Evans et al., 2005; Awano et al., 2006). The storage bodies with membrane-
bound inclusions had coarsely granular matrices. Brain samples had 36% of
CTSD enzymatic activity left and had an increase (e.g. TPP1) or no change
(e.g. cathepsin H) in 15 other lysosomal enzymes (Awano et al., 2006). These
dogs also had a transition (c.597G>A) in exon 5 that predicts a p.M199I
missense mutation.
A Drosophila NCL model of CTSD deficiency was generated by inactivating
the conserved Drosophila CTSD homologue (Myllykangas et al., 2005).
CTSD-deficient flies exhibited the key features of NCLs. Neurons were filled
with autofluorescent storage inclusions that closely resembled the GRODs
found in human infantile and ovine congenital NCL forms (Myllykangas et al.,
2005). CTSD mutant flies also showed modest age-dependent
neurodegeneration.
2.3.1.5. CTSD in Alzheimer’s disease and cancer
CTSD has also been linked to other diseases such as Alzheimer’s disease and
cancer. In Alzheimer’s disease, alterations in the expression of CTSD occur in
the early phases of disease progression (Cataldo et al., 1995). Genetic variation
in CTSD is associated with an increased risk for Alzheimer’s disease

41
(Papassotiropuolos et al., 2000). CTSD activity is elevated in Alzheimer’s
patients (Schwagerl et al., 1995), and CTSD, like the other cathepsins, is
present in association with amyloid deposits (Bernstein et al., 1989). β-
Amyloid, the main component of cerebral amyloid in Alzheimer’s disease
(Glenner and Wong, 1984; Masters et al., 1985), is formed through proteolysis
by β- and γ-secretases (Haas et al. 1995). CTSD has the specificity required for
both β- and γ-secretase (Ladror et al., 1994; Evin et al., 1995), and it can
degrade β-amyloid protein at a pH between 4 and 6 in soluble human brain
extracts (McDermott and Gibson, 1996). Based on these observations, CTSD
has been suggested to be a potential therapeutic target in Alzheimer’s disease
(Haass and De Strooper, 1999; Vassar et al., 1999).
Several members of the cathepsins, in particular CTSD, B and L, have been
implicated in cancer progression and metastasis (Rochefort and Liaudet-
Coopman, 1999; Koblinski et al., 2000; Joyce et al., 2004; Turk et al., 2004).
In breast cancer, CTSD proenzyme (52 kDa) is abnormally hypersecreted and
this overexpression increases the metastatic potential of the tumour by the
digestion of extracellular matrix (Rochefort, 1992; Ferrandina et al., 1997;
Foekens et al., 1999). High level of CTSD is strongly associated with poor
prognosis in patients with primary breast cancer (Wesley and May, 1996;
Foekens et al. 1999; Scorilas et al. 1999). CTSD mitogenic activity does not
require its catalytic activity (Glondu et al., 2001). The mitogenic response does
not result from MPRs or a putative receptor that is specific to the CTSD
profragment. CTSD has an essential role in the multiple steps of tumour
progression, in stimulating cancer cell proliferation, fibroblast outgrowth and
angiogenesis, as well as in inhibiting tumour apoptosis (Berchem et al., 2002).

42
2.3.2. Postulated mechanisms of neurodegeneration in NCLs
The mechanisms of neuronal death in NCL diseases and in other
neurodegenerative diseases are widely discussed but rather poorly known.
Reasons for the neuronal vulnerability also remain obscure. Excitotoxicity,
dysfunction of mitochondria, oxidative stress and inflammation in NCL disease
progression have been considered to be contributors to neuronal death.
Accumulating data indicate that neurons may die via apoptotic or autophagic
mechanisms. Autophagic and apoptotic cell death processes can occur either
simultaneously or sequentially in degenerating neurons, and the mechanisms of
degeneration may also differ in different compartments of the cell.
2.3.2.1. Cell death mechanisms
Neuronal cell death occurs by at least three different mechanisms: apoptosis,
autophagy and necrosis (Yuan et al. 2003). Apoptosis, “programmed cell
death”, is a major mechanism for active cell death, playing an important role in
the elimination of surplus cells during embryogenesis and maintenance of
tissue cell number. Apoptosis is characterized morphologically by cell
shrinkage and nuclear chromatin condensation, fragmentation of cells into
apoptotic bodies and heterophagocytosis by neighbouring cells. Apoptosis is
defined by DNA laddering.
Apoptosis is regulated by many different intra- and extracellular events. During
apoptosis the cell is degraded through activation of proteases and
endonucleases. Caspases play a central role in the induction and execution of
apoptosis (Cohen, 1997). Lysosomal proteases, such as cathepsins, act as
mediators of apoptosis in several cell systems (Deiss et al., 1996; Ishisaka et
al., 1998; Guicciardi et al., 2000; Kågedal et al., 2001). The function of CTSD

43
in apoptosis is not yet fully understood, but it has been postulated that CTSD
can either prevent or promote apoptosis induced by cytotoxic agents (reviewed
in Liaudet-Coopman et al., 2006). CTSD may mediate apoptosis induced by
oxidative stress (Kågedal et al., 2001). During oxidative stress-induced
apoptosis, mature 34-kDa CTSD is translocated from lysosomal structures into
the cytosol, leading in turn to the mitochondrial release of cytochrome c (Cyt
c) into the cytosol, activation of pro-caspases-9 and -3, in vitro cleavage of Bid
at pH 6.2 or Bax activation independently of Bid cleavage (Roberg et al.,
1999;2002; Liaudet-Coopman et al., 2006; Fig. 6). Cytosol is acidified to pH
6.7-7 in apoptosis (Gottlieb et al., 1996; Matsuyama et al., 2000). However,
CTSD can work at a pH of 6.2, but not above, in vitro (Capony et al., 1987).
Thus, the question about the ability of CTSD to cleave cytosolic substrate(s),
as implicated in the apoptotic cascade, remains still open. Deficiency of CTSD
activity did not alter cell death induction in CONCL fibroblasts (Tardy et al.,
2003). The postulated role of CTSD in apoptosis is presented in Fig. 7.
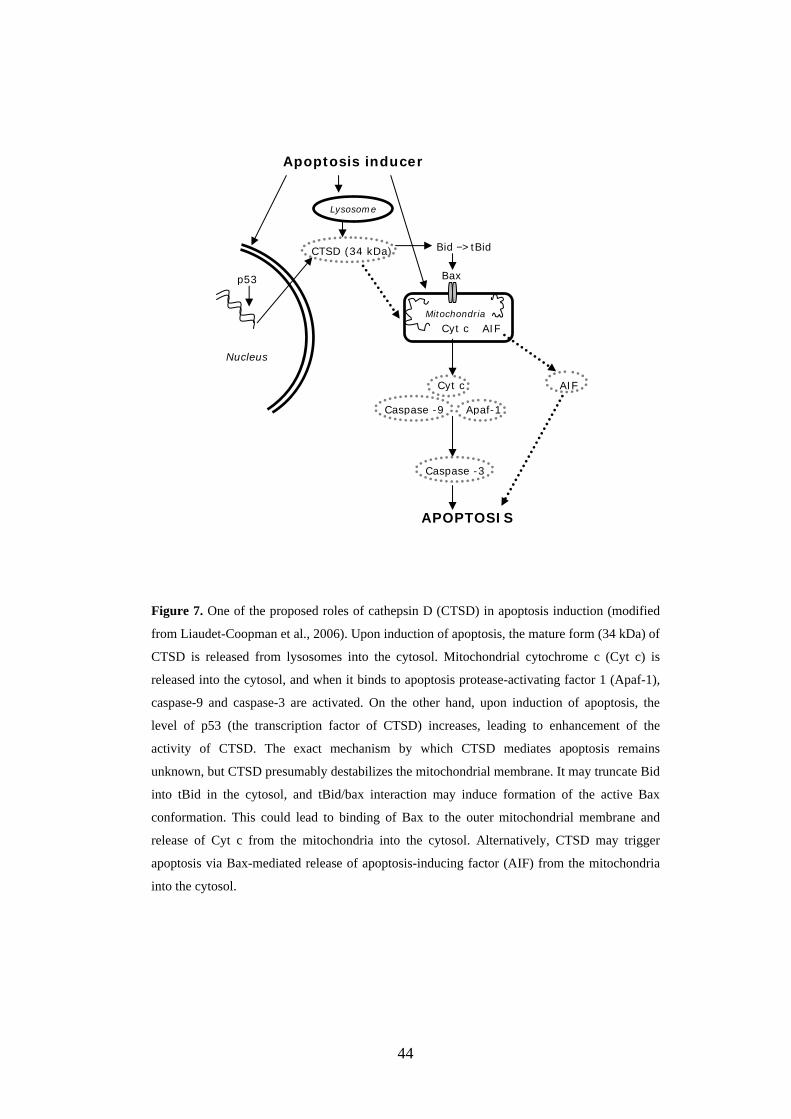
44
Figure 7. One of the proposed roles of cathepsin D (CTSD) in apoptosis induction (modified
from Liaudet-Coopman et al., 2006). Upon induction of apoptosis, the mature form (34 kDa) of
CTSD is released from lysosomes into the cytosol. Mitochondrial cytochrome c (Cyt c) is
released into the cytosol, and when it binds to apoptosis protease-activating factor 1 (Apaf-1),
caspase-9 and caspase-3 are activated. On the other hand, upon induction of apoptosis, the
level of p53 (the transcription factor of CTSD) increases, leading to enhancement of the
activity of CTSD. The exact mechanism by which CTSD mediates apoptosis remains
unknown, but CTSD presumably destabilizes the mitochondrial membrane. It may truncate Bid
into tBid in the cytosol, and tBid/bax interaction may induce formation of the active Bax
conformation. This could lead to binding of Bax to the outer mitochondrial membrane and
release of Cyt c from the mitochondria into the cytosol. Alternatively, CTSD may trigger
apoptosis via Bax-mediated release of apoptosis-inducing factor (AIF) from the mitochondria
into the cytosol.
Apoptosis inducer
Mitochondria
Lysosome
CTSD (34 kDa)
Cyt c
Cyt c
Caspase -9 Apaf-1
Caspase -3
APOPTOSIS
AIF
Bax
Nucleus
p53
AIF
Bid −> tBid

45
Studies on cancer cell lines have shown that overexpressed CTSD prevents
tumour apoptosis in a manner dependent on its catalytic activity (Berchem et
al., 2002). Intriguingly, the same cell lines treated with chemotherapeutic
agents revealed a stimulation of apoptosis independently of CTSD catalytic
activity (Beaujouin et al., 2006). In addition to CTSD -/- studies, experiments
in other NCL mouse models support apoptosis as the main cell death
mechanism in the retina (Cotman et al., 2002; Seigel et al., 2002; Koike et al.,
2003). TUNEL-positive cells were seen in the cerebellum of PPT1-deficient
mice (Gupta et al., 2001). Apoptotic neurons have also been found in the brains
of INCL, LINCL and JNCL patients and in South Hampshire sheep (Lane et
al., 1996; Riikonen et al., 2000), but not in nclf (Bronson et al. 1998) or
CLN3( ex7/8)mice (Cotman et al., 2002). CLN3 protein has been proposed to
have a role in programmed cell death, apparently in the regulation of ceramide
synthesis (Puranam et al., 1999).
Autophagy (“self-eating”), another mechanism leading to cell death, is a
lysosomal degradation pathway for cytoplasmic material. Autophagy has been
discussed as a subtype of apoptosis, although autophagy is believed to
participate in the protection of cells from death (Clarke, 1990). Earlier,
autophagy was described only as a stress condition, an adaptive response of
cells to nutrient deprivation. Autophagy has recently been associated with
much more complex cellular processes such as cellular differentiation, tissue
remodelling, growth control, cell defence and adaptation to adverse
environments (Cuervo, 2004a). Based on the ways that substrates enter the
lysosomal lumen, the following three different forms of autophagy have been
described in mammalian cells (Fig. 8): 1) macroautophagy, or simply
autophagy, 2) microautophagy and 3) chaperone-mediated autophagy (Seglen
and Bohley, 1992; Kim and Klionsky, 2000; Cuervo, 2004b). In
macroautophagy, autophagosomes fuse with lysosomal vesicles, resulting in
degradation of the cytoplasm (Arstila and Trump, 1968). During the maturation

46
process, autophagosomes, also called initial autophagic vacuoles, develop into
late or degradative autophagic vacuoles that contain lysosomal membrane
proteins and enzymes and are acidic (Dunn, 1990). In microautophagy, the
cytoplasmic material is non-selectively uptaken by lysosomes (Ahlberg et al.,
1982; Kopitz et al., 1990) and in chaperone-mediated autophagy, the proteins
with a special signal sequence are transported through the lysosomal
membrane to the lumen of lysosomes (Cuervo and Dice, 1996).
Figure 8. Three forms of autophagy in mammalian cells: macroautophagy, microautophagy
and chaperone-mediated autophagy (modified from Cuervo, 2004a, with permission of
Elsevier). Cytosolic organelles indicated with circles and single proteins with strings.
Peroxisome-specific macroautophagy is called macropexophagy. The arrow with a broken line
indicates the degradation of peroxisomes by microautophagy (micropexophagy), which has
only been described in yeast.
Autophagosome
Peroxisome
Macroautophagy
Microautophagy
Chaperone-mediated autophagy
Lysosome
Membrane receptorCytosolic chaperoneand co-chaperones
Cytosolic protein(substrate)
Lysosomalchaperone

47
Programmed cell death, or autophagic cell death, has been described in
mammary carcinoma cells (Bursch et al., 1996). Changes in the lysosomal
compartment are linked to neuronal death in many neurodegenerative disorders
and transmissible neuronal pathologies (prion diseases) (Nixon et al., 2000;
Larsen and Sulzer, 2002; Liberski et al., 2002). Autophagy plays a role in
neuronal death in CTSD as well as in CTSB and CTSL double-deficient mouse
models of NCLs (Koike et al., 2005). Autophagy is activated and the
development of autophagic vacuoles is also disrupted in Cln3 ex7/8) mice
(Cao et al., 2006). In addition, a disturbance in autophagy may contribute to the
pathogenesis of cancer, liver and immune diseases, pathogen infection,
myopathies and such neurodegenerative disorders as Parkinson’s, Alzheimer’s
and Huntington’s disease (Kegel et al., 2000; Nixon et al., 2000; Stefanis et al.,
2001; Bursch and Ellinger, 2005). Activation of autophagy may facilitate the
removal of intracellular protein aggregates – a hallmark of neurodegenerative
diseases (Jellinger and Stadelmann, 2000; Larsen and Sulzer, 2002). The
increasing number of aggregates, their decreased susceptibility to degradation
by lysosomal enzymes and the maintained activation of autophagy might lead
to cell exhaustion and engagement in programmed cell death type II (Cuervo,
2004a).
2.3.2.2. Selectivity of cell death
Although accumulation of autofluorescent storage material is common for both
neurons and somatic cells of NCL patients (Hofmann and Peltonen, 2001), the
effects of the disease seem to be confined to the CNS. This selectivity may
arise from neurons being post-mitotic, and dysfunctional or dying neurons
having a very limited ability to renew (Mitchison et al., 2004). Another
explanation for selective neuronal vulnerability could be the existence of other
proteins or enzymes that can compensate for the missing gene product outside

48
the CNS (Mitchison et al., 2004). As an example, the missing function of TPP1
can be compensated with cathepsin C in somatic tissue of LINCL (Bernardini
and Warburton, 2002). Alternatively, CLN proteins may play an important role
in synapses; at least PPT1 (Lehtovirta et al., 2001; Suopanki et al., 2002;
Ahtiainen et al., 2003) and CLN3 (Luiro et al., 2001) have been demonstrated
to co-localize with synaptic markers. Although Virmani et al. (2005) did not
observe PPT1 in synapses, they did find a decrease in the amount of synaptic
neurotransmitter vesicles in the neuronal cultures of PPT1 knock-out mice.
Synapse loss or a loss of neurons is also the key contributor to the onset and
progression of several other neurodegenerative diseases (Ironside and
Dearmond, 1994; Sasaki and Maryama, 1994; Selkoe, 2002; Li et al., 2003;
Coleman et al., 2004).
The most sensitive cell types for degeneration in NCLs seem to be the
GABAergic, inhibitory interneurons (Mitchison et al., 2004). GABAergic and
retinal neurons, cells that contain abundant mitochondria, degenerate first in
several NCL diseases, and the loss of GABAergic interneurons has been
detected in several NCL animal models (Cooper et al., 1999; 2002; Lam et al.,
1999; Mitchison et al., 1999; Oswald et al., 2001). In addition, the shape and
number of mitochondria in GABAergic cells of an affected English setter and
OCL6 lambs were altered (March et al., 1995; Walkley et al., 1995). Although
the number of GABAergic interneurons remained the same in CTSD -/- mice
compared with control mice, the amounts of GAD65 and GAD67, the enzymes
needed for GABA synthesis, were decreased in the hippocampus of affected
mice (Shimizu et al., 2005). Moreover, the presence of an auto-antibody to
GAD65 was also reported in CLN3 -/- mice (Chattopadhyay et al., 2002a) and
JNCL patients (Chattopadhyay et al., 2002a; 2002b).
The accumulation of Sub c in OCL6 lambs and other forms of ceroid-
lipofuscinosis first drew attention to a possible association between

49
mitochondria and pathogenesis of NCL diseases. The mitochondrion is the
organelle that is most influenced by oxidative damage, and it is the major
source of free radicals in the cell. A dysfunction in mitochondria leads to
overproduction of free oxygen radicals and oxidative stress. Lysosomes are
involved in recycling or degradation of aged mitochondria via autophagy. The
involvement of mitochondria in NCL disease pathology has been the target of
interest in several studies, which have presented conflicting conclusions.
Mitochondria of OCL6 sheep showed normal oxidative phosphorylation
(Palmer et al., 1992), whereas oxidative phosphorylation was deficient in
muscles of JNCL patients (Majander et al., 1995). Impaired mitochondrial fatty
acid oxidation was found in fibroblasts of LINCL and JNCL patients (Dawson
et al., 1996). Protein glycoxidation was also observed in the brains of LINCL
patients (Hachiya et al., 2006). In addition, a mutation in a PPT1 homologue
affected the number and size of mitochondria and resulted in an abnormality in
mitochondrial morphology in C. elegans (Porter et al., 2005). mRNA and
protein expression of manganese-dependent superoxide dismutase (MnSOD), a
mitochondrial enzyme that helps to reduce the level of reactive oxygen species
(ROS), were significantly increased in CLN6 patients (Heine et al., 2003).
Calpain is a mediator of p53 induction, resulting in mitochondrial membrane
depolarization and caspase-dependent apoptosis (Chan et al., 2002; Sedarous et
al., 2003). Calpain is part of the excitotoxic mechanism linked to
overstimulation of synaptic glutamate receptors.
Chronic excitotoxicity has been postulated to account for selective neuronal
vulnerability in epilepsies and a number of neurodegenerative diseases,
including Alzheimer’s, Parkinson’s, Huntington’s and amyotrophic lateral
sclerosis (Beal, 1995; Hennebury, 1995; Schulz et al., 1995). The reasoning of
the “energy-linked excitotoxicity hypothesis” is that there is a compromise in
the neuron’s ability to produce ATP due to an inherited defect, an
environmental toxin or aging (Jolly and Walkley, 1999). The continued influx

50
of Ca2+, because of the inadequate amount of ATP to maintain the ATPase
pumps, initiates a cascade of events leading to increased free radical formation
and neuronal death (Novelli et al. 1988; Beal, 1995; Hennebury, 1995). Partial
depolarization, the result of activation of glutamate receptor subtypes in a
neuron, relieves the voltage-dependent magnesium ion block in the N-methyl-
D-aspartate (NMDA) channel. There is variable glutamate input to neurons in
the cortex, but it is particularly strong in layer IV of the primate brain, the
major receiving zone of the cortex for excitatory thalamocortical input. This
layer also contains the majority of the cytochrome c oxidase immunoreactivity,
which is supposed to indicate a high metabolic activity (Wong-Riley, 1989).

51
3. AIMS OF THE STUDY
Naturally occurring cathepsin D deficiency in sheep and genetically
manipulated cathepsin D knock-out mice have been described earlier. In this
study specific aims were to:
o Characterize the expression of cathepsin D during rat brain
development.
o Characterize the intracellular stability, processing and transport of
inactive mouse cathepsin D.
o Investigate cathepsin D deficiency as a cause of congenital human
neuronal ceroid-lipofuscinosis.
o Examine the neuropathology and molecular mechanisms of
neurodegeneration in cathepsin D knock-out mice.

52

53
4. MATERIALS AND METHODS
Experimental animals and patient material used in this study
Wistar rats were kept in an animal care facility and treated according to the
ethics guidelines of the European Union. Two independent developmental
series [embryonic day (E)16-P64] of Wistar rat brain tissue and one (P1-P4) of
spleen were collected and analysed. The rat studies were approved by the
Institutional Ethics Committee of the University of Helsinki.
Heterozygous (+/-) CTSD mice were kindly provided by Professor Saftig
(University of Kiel, Germany; Saftig et al., 1995). CTSD knock-out (-/-) mice
were produced by mating. Mice were held in an animal care facility where the
food and water were available ad libitum and the light/dark cycle was 12/12
hours. The brains were dissected at around P24. The study plan was approved
by the Ethics Committee of the University of Helsinki.
Paraffin-embedded brain tissue samples of three human congenital NCL
patients were obtained at routine autopsies. An ethylenedia-minetetraacetic
acid (EDTA) blood sample from one congenital NCL patient was collected for
diagnostic purposes. Clinical data concerning foetal and postnatal development
were retrieved from medical charts. The controls for identified CTSD
mutations consisted of 92 CEPH (Centre d'Etude du Polymorphisme Humain)
individuals. The study plan was approved by an institutional review board of
Helsinki University Central Hospital.
The details of the other experimental methods and materials used are described
in the original publications (I-IV; Table 2). The primer sequences used in these
studies are available from the author on request.
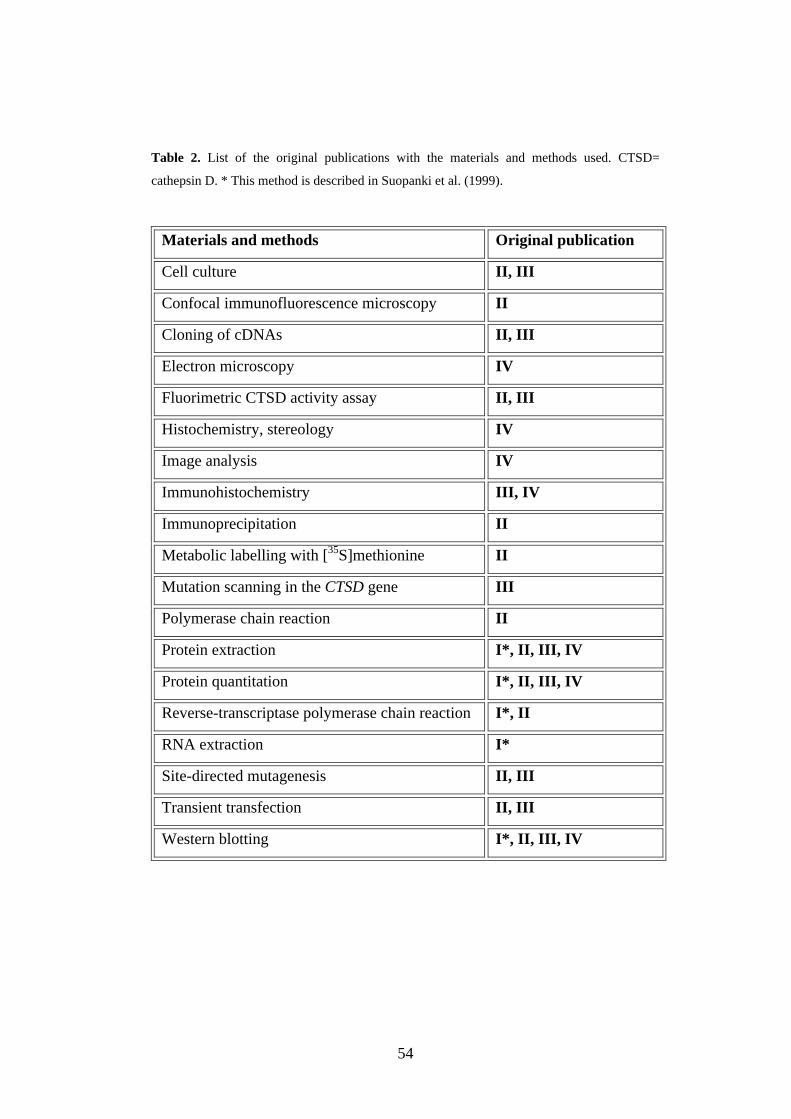
54
Table 2. List of the original publications with the materials and methods used. CTSD=
cathepsin D. * This method is described in Suopanki et al. (1999).
Materials and methods Original publication
Cell culture II, III
Confocal immunofluorescence microscopy II
Cloning of cDNAs II, III
Electron microscopy IV
Fluorimetric CTSD activity assay II, III
Histochemistry, stereology IV
Image analysis IV
Immunohistochemistry III, IV
Immunoprecipitation II
Metabolic labelling with [35S]methionine II
Mutation scanning in the CTSD gene III
Polymerase chain reaction II
Protein extraction I*, II, III, IV
Protein quantitation I*, II, III, IV
Reverse-transcriptase polymerase chain reaction I*, II
RNA extraction I*
Site-directed mutagenesis II, III
Transient transfection II, III
Western blotting I*, II, III, IV

55
5. RESULTS AND DISCUSSION
5.1. Developmental expression of CTSD in the rat brain (I)
Defects in PPT1, TPP1 and CTSD cause NCLs, severe neurodegenerative
disorders, in early childhood. These NCL-associated proteins are all
ubiquitously expressed, showing a typical lysosomal punctuate staining
pattern, in immunohistochemically stained neurons (Ellis and Divor, 1979;
Bernstein et al., 1987; Isosomppi et al., 1999; Suopanki et al., 1999). The aim
of this study was to compare the developmental expression profiles of these
NCL-associated lysosomal enzymes in the rat brain (I). mRNA and protein
levels of PPT1, TPP1 and CTSD were followed to compare expression changes
to different maturation steps of the nervous system.
5.1.1. mRNA expression
CTSD and TPP1 mRNA expressions were relatively constant throughout brain
development (E16-P64; I, Fig. 1A). At early adulthood, CTSD mRNA
expression decreased slightly in the brain, whereas TPP1 mRNA expression
increased (I, Fig. 1A). In the spleen, CTSD and TPP1 mRNA expression
profiles were similar than those in the brain, showing a marginally higher peak
at P15 (I, Fig. 1B). However, only the PPT1 expression profile showed clear
development-dependent changes in both the rat brain and spleen (I, Fig. 1A,
B). mRNA expression of sphingolipid activator protein precursors (proSAPs),
the products of which accumulate in PPT1 deficiency and CTSD deficiency in
sheep and humans, increased throughout the brain development, as previously
reported in mice (I, Fig. 1A; Kreda et al., 1994). By contrast, mRNA level of
proSAP revealed no major changes during the development of the rat spleen (I,
Fig. 1B).

56
CTSD deficiency is found in humans, mice, sheep and dogs. Both rats and
mice have a gestation time of 21 days, while sheep pregnancy lasts for 144-151
days and human pregnancy 266 days. Due to the big variation in gestation
times and life span of these animals, the maturity of the brain at birth differs
considerably between different species. Interestingly, CTSD -/- mice start to
show abnormal behaviour and a pathological phenotype at P14, dying at
approximately P26. CTSD-deficient sheep are very weak and trembling
already at birth (Järplid and Haltia, 1993). They only live for some days and
their brains are very atrophic already at that young age. Myelin biosynthesis in
the rat CNS is greatest between P14 and P30 (Skoff, 1976; Benjamins and
Smith, 1977), whereas myelination in sheep continues several months after
birth. The brains of CONCL sheep and CTSD -/- mice are largely devoid of
myelin. The mRNA expression profile of CTSD in the developing rat brain
suggests that CTSD may have a role in normal development of neurons and
myelination.
5.1.2. Protein expression
At the protein level, the processed form (43 kDa) of the CTSD single chain
was seen already on E16, and it showed a slight increase in expression towards
adulthood (I, Fig. 2). Very low, hardly visible levels of the heavy chain (30
kDa) appeared from P6 onwards. The synaptophysin expression profile
resembled that of the CTSD during rat brain development. Additionally, the
expression of PPT1, TPP1 and CTSD increased in the adult rat brain, while the
expression of growth-associated protein 43 (GAP-43) decreased (I; Fig. 2). The
amounts of the mature forms of PPT1 (37/39 kDa) and TPP1 (46 kDa)
increased towards early adulthood, whereas the amount of TPP1 precursor (67
kDa) decreased after P12 (I; Fig. 2).

57
CTSD-specific activity in the rat brain doubles between postnatal days 1 and
10, with a decline occuring at 6 months (Marks et al., 1975). CTSD undergoes
several proteolytic processing steps during its biosynthesis. In rats, CTSD is
known to exist almost entirely as a single-chain form (43 kDa), and only very
low levels of the 30-kDa heavy chain are produced in lysosomes (Tanaka et al.,
1991). Neurons of rat cortical areas are mainly generated at E16-E21 (Berry et
al., 1964; Berry and Rogers, 1965). Synapse formation in the cerebral cortex in
rats starts at birth and reaches a peak around P26, at the time point when CTSD
-/- mice die. Some lysosomal enzymes underlying different NCLs are known to
localize to synapses (Lehtovirta et al., 2001; Luiro et al., 2001; Suopanki et al.,
2002). In this study, especially the protein expression of PPT1 and CTSD
increased simultaneously with the postnatal period of synaptogenesis,
suggesting a role for these enzymes in neuronal reorganization, myelination
and/or plasticity.
5.2. Intracellular stability, processing and transport of mutant D293N
mouse (m) CTSD is affected in HEK-293 cells (II)
Because of the phenotypic differences between CTSD-deficient sheep and
CTSD -/- mice, the aim of this study was to examine the effect of an active-site
mutation in the transport, processing and stability of the mutant D293N
mCTSD. Western blotting, metabolic labelling and confocal microscopy were
used to investigate the overexpression of mCTSD in HEK-293 cells.
5.2.1. Loss of enzyme activity in mutant D293N mCTSD
First, we developed a novel CTSD activity assay that is based on the cleavage
of 6[(7-amino-4-methylcoumarin-3-acetyl)amino]hexoid acid (AMCA)-

58
conjugated haemoglobin by active CTSD and the measurement of the
fluorescence of the cleavage products. CTSD enzyme activity was previously
measured mainly using radioactively labelled haemoglobin as a substrate
(Hasilik et al., 1982). Acidic pH is required for the activity of CTSD in vitro,
with a pH optimum of 4.5-5.0, using an extracellular matrix as a substrate
(Briozzo et al., 1988). In our novel assay, the optimum activity of CTSD was
obtained at pH 3.9 (II, Fig. 1A), and the activity was almost completely
inhibited by addition of pepstatin A (II, Fig. 1B), as expected from the reports
of Ayoagi et al. (1972). The mutant D293N mCTSD presented an almost
complete lack of enzyme activity (II, Fig. 1B and C), similarly to
enzymatically inactive sheep (Tyynelä et al., 2000) and the mutant D295N
human (h) CTSD (Glondu et al., 2001).
5.2.2. Reduced stability of mutant D293N mCTSD
Interestingly, CTSD in WT-transfected cells revealed threefold the
immunoreactivity of cells transfected with the mutant D293N mCTSD [II, Fig.
1C (inset)]. This result was in contrast to findings in CTSD-deficient sheep,
where the mutant protein seemed to be more stable than the WT protein
(Tyynelä et al., 2000; II, Fig. 2B). The WT and especially the mutant D293N
mCTSD were stabilized by addition of protease inhibitors (pepstatin A and
leupeptin; II, Fig. 2A), suggesting that the proteolytic degradation of the
mutant protein happens partially in lysosomes. However, the experiments on
the mutant D295N hCTSD in rat cancer cells did not reveal instability of the
mutant protein (Glondu et al., 2001). The differences in the stability of the
mutant CTSD proteins in these two studies can be explained by differences in
species (mouse CTSD vs. human CTSD) or in heterologous overexpressing
systems (transiently expressed mouse CTSD in human cells vs. stable
transfected rat cells with human CTSD). Another explanation could lie in the

59
different protease patterns in these different systems: no endogenous CTSD
activity in CONCL cells, low amounts of endogenous CTSD activity in rat
cancer cells (Garcia et al., 1990) and high endogenous CTSD activity in HEK-
293 cells. In this study, mCTSD did not appear in its processed 30-kDa form,
contrary to expectation based on earlier reports of mouse cell experiments
(Pohlmann et al., 1995; Dittmer et al., 1999). This is also most likely the result
of the use of a heterologous overexpressing system.
5.2.3. Altered processing of mutant D293N mCTSD
The mutant D293N mCTSD showed increased electrophoretic mobility
compared with the WT protein [II, Fig. 1C (inset); Fig. 2A) in Western
blottings. A shift of about 1 kDa could also be seen in cell extracts from
affected sheep (II, Fig. 2B). The apparent molecular mass of both the WT and
the mutant D293N mCTSD was reduced by approximately 1.5-2 kDa after
treatment of CTSD [35S]methionine-labelled immunoprecipitates with PNGase
F (II, Fig. 3), revealing that the difference in electrophoretic mobility was not
due to a difference in glycosylation. Since WT and the mutant D293N mCTSD
are equally glycosylated, the molecular mass difference between them is most
probably the result of proteolytic cleavage. The proteolytic processing seems to
be a very early event, as it is detectable after a short 20-min pulse in HEK-293
cells (II, Fig. 4), likely occurring in the ER (II, Fig. 5, lower panels).
Interestingly, in CLN2, a loss of TPP1 activity was accompanied by reduced
expression of the mutant hCLN2 protein (Tsiakas et al., 2004). In addition, a
difference in the electrophoretic mobility was observed in the mutant hCLN2
protein relative to the WT enzyme (Tsiakas et al., 2004). Although the mutated
protein is different in these two neurodegenerative disorders, the sequence of
events leading to aggregation of the mutant protein is apparently the same

60
(Cuervo, 2004a): (1) abnormal conformation (misfolding) of the affected
protein exposes normally hidden hydrophobic residues, (2) the cell responds to
these abnormal proteins by activating the chaperone system (to promote
refolding) and cytosolic proteases (to promote removal) and (3) as levels of the
altered protein increase, the chaperones and proteases become trapped in the
aggregates by themselves (Michalik and Van Broeckhoven, 2003). The active-
site mutation in CTSD can be expected to alter the three-dimensional, compact
structure of the precursor CTSD, allowing proteases to cleave it. Especially the
N-terminal propeptide of CTSD seems to play an important role in CTSD
folding and protection against proteolytic cleavages by interacting with the
active-site residues of CTSD (Dunn, 1997; Khan and James, 1998). Despite
several attempts to sequence the N-terminal and C-terminal segments of
affected sheep CTSD, we could not locate the site of the cleavage (unpublished
data). However, some reports of protease cleavages of CTSD (Erickson and
Blobel, 1983; Wittlin et al., 1999) and also the appearance of the molecular
mass shift in the mature (30-kDa) C-terminal form of the mutant ovine CTSD
(II, Fig. 2B) suggest that the proteolytic processing occurs in the C-terminus of
the mutant CTSD.
5.2.4. Intracellular transport and secretion defect of mutant D293N mCTSD
Clear alterations in the transport of the mutant D293N mCTSD were observed
in the pulse-chase labelling and confocal immunofluorescence microscopy
experiments (II, Figs. 4 and 5). After a 20-min pulse, two protein bands (45
and 43 kDa) were seen in the precipitates of WT cells and also in the mutant
D293N mCTSD extracts (44 and 42 kDa; II, Fig. 4). After a 20-min chase,
both the WT and the mutant D293N mCTSD were seen only in cell extracts,
whereas after a 40-min chase about 70% of the WT mCTSD was present in the
media (II, Fig. 4), but no mutant D293N mCTSD was secreted. After a 100-

61
min chase, about 10% of the mutant D293N mCTSD was secreted. When the
protein synthesis was stopped with cycloheximide and the steady-state
localization of immunofluorescent CTSD was followed after a 6-h chase, the
mutant D293N mCTSD was present mainly in lysosomes, but also to some
extent in other parts of the cells (II, Fig. 5, upper right panel). WT CTSD was,
by contrast, perfectly colocalized with the lysosomal marker LAMP-1 (II, Fig.
5, upper left panel). When the protein synthesis was not prevented, the mutant
D293N mCTSD colocalized with the Golgi-ER marker (protein disulphide
isomerase; PDI), while the WT protein was almost exclusively present in
lysosomes (II, Fig. 5, lower panels).
The time-dependent accumulation of the precursor and intermediate forms of
CTSD has previously been followed in cultured fibroblasts (Gieselmann et al.,
1983; Braulke et al., 1988). A 47-kDa intermediate form of CTSD becomes
detectable after a 60-min chase (10% of total; Gieselmann et al., 1983). At
37°C, newly synthesized CTSD passes the TGN within 30-60 min, is secreted
into prelysosomal organelles within 1-2 h and is processed to a mature form in
lysosomes within 1.5-3 h of synthesis (Braulke et al., 1988). A small fraction
5% of total) of CTSD precursor is normally secreted (Braulke et al., 1988).
The data from pulse-chase labellings and immunofluorescence experiments in
this study showed differences between the mutant D293N and WT mCTSD in
transport rates and folding kinetics. The unfolded, proteolytically cleaved
mutant D293N mCTSD was retarded in the ER.
A logical extension to these studies with the mutant D293N mCTSD was to
create a mouse model with the same active-site mutation. However, the attempt
to create this CTSD-deficient mouse failed. The protein expression could not
be confirmed with Western blotting, even though mRNA expression was
apparent. These mice died at the same age as the CTSD -/- mice, but whether
this was a real phenotype or not remained uncertain (data not published). In

62
addition a Drosophila model for the D293N mutation in CTSD was generated.
Although the protein expression of these flies could be confirmed by Western
blotting (data not shown), the phenotype or the pathology of these flies did not
essentially differ from the CTSD null flies (Myllykangas et al., 2005).
5.3. CTSD deficiency in humans (III)
The clinical and neuropathological pictures of four patients with unclear
disease aetiology were compatible with the diagnosis of congenital NCL
(Humphreys et al., 1985; Garborg et al., 1987). Due to the close resemblance
of neuropathological findings in human and ovine congenital NCLs, the aim of
this study was to screen the CTSD gene for mutations in human patients.
5.3.1. A mutation in the human (h) CTSD gene causing congenital NCL
Nucleotide duplication (c.764dupA) in a homozygous form was found in exon
6 of the CTSD gene in one congenital NCL patient (III, Fig. 1A, B). This was
the only patient from whom we were able to isolate good-quality genomic
DNA. The father of this patient was a heterozygous carrier of the mutation (III,
Fig. 1B). The mutation leads to a premature stop codon (TAC>TAAC) at
position 255 (p.Tyr255X), which predicts a truncation of the protein by 158
amino acids, thus suggesting the mutation to be disease-causing. An additional
nucleotide change, c.845G>A, was found in exon 7 (data not shown). This
mutation was located upstream of the duplication, thus presumably having no
effect on the disease. The close resemblance of the phenotype of this patient
and his siblings and our immunohistochemical findings in all three patients
examined (III, Figs. 3, 4 and 5) strongly suggest that CTSD deficiency is the
cause of congenital NCL in these patients.

63
5.3.2. Truncation of the inactive mutant c.764dupA hCTSD in BHK cells
To examine the effect of the nucleotide duplication (c.764dupA) on the activity
and stability of hCTSD in baby hamster kidney (BHK) cells, the mutant
c.764dupA cDNA of human CTSD was created and cloned into a pcDNA 3.1
expression vector. Transient expression of the mutant c.764dupA hCTSD was
followed by an enzyme activity assay and Western blotting. Activity
measurement showed a relative lack of enzyme activity, resembling the
inactive sheep mutation (c.883G>A) created in the hCTSD cDNA (III, Fig.
2A). When the protein expression was followed by Western blotting, the
formation of truncated mutant c.764dupA hCTSD with a calculated molecular
mass of 27 kDa was found in BHK cell extracts after transient transfection (III,
Fig. 2B). By contrast, WT hCTSD showed the normal processing of CTSD to
the single-chain active form (43 kDa) and the mature form (31 kDa) of the
enzyme (III, Fig. 2B). The mutant c.883G>A hCTSD, generated according to
the reported ovine mutation, was normally processed, but had an increase in
electrophoretic mobility, as previously reported (Tyynelä et al., 2000; II). The
complete lack of CTSD enzyme activity also leads to an early-onset, severe
NCL in mice and sheep (Koike et al., 2000; Tyynelä et al., 2000), while the
missense mutation in the CTSD gene of affected American bulldogs and two
missense mutations in the exons 5 and 9 of the CTSD gene of one human NCL
patient result in a partial inactivation of the enzyme and later-onset CTSD
deficiencies (Awano et al., 2006; Steinfeld et al., 2006). Based on these
findings of different CTSD deficiencies, the activity of CTSD seems to be
crucial for early developmental events and survival.

64
5.3.3. Lack of the mutant c.764dupA hCTSD in congenital NCL patients
Glial fibrillary acidic protein (GFAP)-positive hypertrophic astrocytes were
abundant throughout the brain, particularly in the white matter of a congenital
NCL patient (III, Fig. 3A-C). CD68 staining showed that the microglial
activation was most intense in the deep layers of the cortical grey matter (III,
Fig. 3D-F). Storage material, typical of NCLs, was also found in the congenital
NCL patient’s brain, and it contained the sphingolipid activator protein D (III,
Fig. 4A), which is also the main storage component in ovine congenital NCL
(Tyynelä et al., 2000) and in the infantile form of NCL disease, caused by
mutations in PPT1 (Vesa et al., 1995). Moreover, ultrastructural findings of
congenital NCL patients showed accumulation of GRODs, which is in
agreement with the findings of other CTSD deficiencies (human: Steinfeld et
al., 2006; sheep: Tyynelä et al,. 2000; American bulldogs: Awano et al., 2006;
mice: Koike et al., 2000; Drosophila: Myllykangas et al., 2005).
Immunohistochemical analysis of congenital NCL patients’ brains revealed an
almost complete absence of CTSD immunoreactivity (III, Fig. 5A), in contrast
to the typical lysosomal punctuate CTSD staining in normal neurons (III, Fig.
5B). However, some pale, probably unspecific staining could be seen in
hypertrophic astrocytes (III, Fig. 5B). The appearance of CTSD
immunoreactivity in overexpression studies but not in the
immunohistochemical staining of CTSD can be explained by the difference in
species. When human protein is expressed in hamster cells, the abnormally
shortened mRNA is not degraded by the foreign non-sense-mediated decay
system, as presumably would happen in human cells (Hentze and Kulozik,
1999).

65
The neuropathological picture of all three patients for whom paraffin-
embedded brain tissue was available was very severe and similar. In the
cerebral and cerebellar cortices there was an extreme neuronal loss and
microglial activation and the white matter was almost completely lacking in
myelin and axons, consistent with previous reports in two congenital NCL
patients (Humbreys et al. 1985; Garborg et al. 1987). Interestingly, the myelin
loss in the white matter was also seen in the congenital NCL sheep (Tyynelä et
al., 2000), suggesting a role for active CTSD in proper myelination. Further
evidence for the close relationship between CTSD and myelination comes from
a developmental study where CTSD expression seemed to increase
simultaneously with increased myelination (I).
5.4. Synaptic and thalamocortical changes in CTSD -/- mice (IV)
Involvement of autophagy and nitric oxide (NO) in the neuronal apoptosis of
CTSD -/- mice has been proposed (Nakanishi et al., 2001; Koike et al., 2005),
but the molecular mechanisms behind the neuronal death remain poorly
understood. As we postulated already in Study I, CTSD may play a role in
synaptic reorganization and/or plasticity. The aim of this study was to
characterize the distribution of the neurodegenerative changes in CTSD -/-
mice and the role of CTSD in synapses.
5.4.1. Regional specificity of thalamocortical changes
Visually, CTSD -/- mouse brains appeared relatively normal, despite the slight
reduction in overall brain size compared with control animals (IV; Fig. 1A).
Cavalieri estimates revealed, however, significant decreases in the regional
volumes of, for instance, the cortex, corpus callosum, thalamus and
hippocampus (IV; Fig. 1B). The volume of the cerebellar white matter was also
decreased, while the cerebellar grey matter seemed to be the least affected

66
region (IV; Fig. 1B). Primary motor cortex (M1), primary somatosensory
cortex, barrel field (S1BF) and primary visual cortex (V1) revealed
significantly decreased thicknesses in CTSD -/- mice compared with controls
(IV; Fig. 2B). These changes in S1BF and M1 were concentrated in individual
laminae (IV; Fig. 2A). Laminae I, II, III and V were normal in S1BF of CTSD
-/- mice, in contrast to laminae IV and VI, which were significantly thinner in
CTSD -/- mice than in controls (IV; Fig. 2B). Laminae I, V and VI were
unaffected in M1 of affected mice, while the combined thickness of laminae II
and III was significantly reduced compared with controls (IV; Fig. 2B).
Immunohistochemical staining with an astrocytic marker (GFAP) was highly
increased in CTSD -/- mouse brains, showing the pronounced and widespread
astrocytosis within the CNS of affected mice (IV; Fig. 3A). Particularly intense
GFAP staining was visible in individual thalamic nuclei, including the ventral
posterior (VPM/VPL) thalamic region. In addition, the CA2 subfield of the
hippocampus in CTSD -/- mice was intensively stained with GFAP (IV; Fig.
3B). Immunohistochemical staining with the microglial marker F4/80 showed
a relatively localized staining in CTSD -/- mouse brains. Microglia were the
most abundant in the same thalamic nuclei, e.g. in VPM/VPL, where the
staining for astrocytosis was also the most intense (IV; Fig. 3A). In line profile
analysis, increased intensity of GFAP and F4/80 was localized to laminae IV
and VI of S1BF (IV; Fig. 6B) in CTSD -/- mice. These alterations colocalized
well with a relatively decreased immunoreactivity of the presynaptic protein
synaptophysin (IV; Fig. 6B).
Based on these results, the regional specificity of the neuropathological
changes in the thalamocortical system of CTSD -/- mice is obvious. The
thalamus is evidently a very important target of the pathology in NCL diseases.
Pathological signal intensity changes in individual thalamic nuclei have been
observed in young JNCL patients using in vivo magnetic resonance imaging

67
(Autti et al., 1997). Involvement of selected thalamic nuclei has also been
reported in post-mortem examination of INCL patients (Haltia et al., 1973). In
addition, pronounced thalamic pathology, including glial activation and
neuronal loss, has been described in several mouse models of NCL (Nakanishi
et al., 2001; Bible et al., 2004; Pontikis et al., 2005; Weimer et al., 2006).
However, changes in the thalamocortical system in NCLs have not yet been
reported, although the thalamus and different cortical layers interconnect.
Region-specific astrocytes and microglial responses at the early stages of
pathogenesis in NCL were first described in OCL6 lambs (Oswald et al.,
2005). These changes spread and the accumulation of fluorescence became
more pronounced with increasing age, the degenerative changes reaching the
motor cortex by 12 months. Interestingly, however, microglial activation was
already present shortly after birth in OCL6 sheep, long before degenerative
changes and clinical symptoms became apparent (Oswald et al., 2005). A
similar phenomenon has been seen in a mouse model of CLN3 (Pontikis et al.,
2004). Neuronal atrophy of these affected animals was more pronounced in the
upper layers (II-III), while the amount of fluorescence did not differ
significantly from that within the lower layers (IV-VI) in any cortical areas
examined (Oswald et al., 2005). Progressive glial responses with the layer
specificity have also been noted in a number of other neurodegenerative
disorders (Troost et al., 1993; von Eitzen et al., 1998; Sheng et al., 1998) as
well as in a CLN1 animal model (Bible et al., 2004). This progressive
activation of microglia in interconnected pathways may be indicative of an
ongoing degenerative process in these brain regions. Studies on a mouse model
of CLN1 (Bible et al., 2004) suggest that progressive activation may be
common in NCLs. Astrocytes participate in regulation of glutamate-induced
neurotransmission and excitotoxicity by uptaking glutamate in synapses, and
they may be activated in CTSD -/- mice in an attempted neuroprotective
response, as a result of localized elevation of glutamate concentration (Oliet et

68
al., 2001; Piet et al., 2002). In addition, the ultrastructural finding of putative
astrocytic “pinching” of presynaptic terminals (IV; Fig. 5G) suggests that
astrocytes play an active role in the synapses.
5.4.2. Loss of neurons and synapses
Reactive changes in specific thalamocortical regions suggest that the main
neurodegenerative processes occur in these areas. To examine whether the
number of neurons in these regions was altered, we counted Nissl-stained
somatosensory relay neurons in the VMP/VPL thalamic nuclei and cortical
neurons in laminae IV and VI as well as parvalbumin-positive inhibitory
neurons in the Rt nucleus. The analyses revealed a significant loss of neurons
in the VPM/VPL, but not in the Rt nucleus in CTSD -/- mice compared with
controls (IV; Fig. 4). In S1BF, a marked and significant loss of neurons in
lamina IV was observed (IV; Fig. 4). In lamina VI of S1BF, the number of
neurons appeared slightly, but not significantly decreased (IV; Fig. 4). Despite
no loss of inhibitory neurons in Rt nucleus, the impaired input of feedback
neurons in lamina VI may decrease the modulatory influence of S1BF on the
VPM/VPL.
In more detail, the localized nature of the neuronal loss coincided with the loss
of synapses in the VPM (IV; Fig. 5). A markedly reduced number of synapses
in the VPM of affected mice was observed by electron microscopy, while the
synapse number was the same in the S1BF of CTSD -/- and control mice (IV;
Fig. 5H). Thus, the degenerative changes in the VPM seem to precede those in
the cortex. An electron-dense storage material was present throughout the
VPM in CTSD -/- mice and especially in myelinated axon profiles (IV; Fig.
5C), whereas in S1BF they were rare and much less prominent (IV; Fig. 5D).
Degeneration of axons, with the breakdown of axon membranes, disruption of

69
axonal organelles and unravelling of myelin sheaths was observed in the VPM
of CTSD -/- mice (IV; Fig. 5E), in contrast to the S1BF, which appeared
relatively normal (IV; Fig. 5F).
Pronounced loss of neurons within individual laminae of the somatosensory
cortex in Cln3 ex7/8 mice, another animal model of NCLs, has been noted
(Pontikis et al., 2005). Moreover, gross axonal pathology with disrupted axons
and unravelled myelin sheaths has previously been observed in the corpus
callosum of CTSD -/- mice (Koike et al., 2005). Our ultrastructural evidence
for axonal degeneration further supports the idea of blockage of axonal
transport and accumulation of autophagic vacuoles reported in CTSD -/- mice
(Koike et al., 2005). Under normal physiological conditions, the neurons can
produce NO, which acts as a gaseous neurotransmitter. However, excess
production of NO by activated microglia in CTSD -/- mice (Nakanishi et al.,
2001) may block the axonal transport of synaptic vesicle precursors, thus
contributing to the dysfunction of axonal transport and synapses (Stagi et al.,
2005). The results of this study again emphasize the importance of CTSD in
proper myelination and axonal transport.
5.4.3. Presynaptic protein changes
The first evidence of changes in the presynaptic system of CTSD -/- mice
appeared after staining of parasagittal cryosections with a synaptic marker,
synaptophysin (Syp). Syp staining was increased throughout the cortex and
thalamus of affected mice compared with controls (IV; Fig. 6A). Syp
immunoreactivity displayed an abnormal laminar distribution, forming two
bands of relatively reduced staining intensity in the line profile analysis (IV;
Fig. 6B). As previously discussed, the immunoreactivity alterations of GFAP,
F4/80 and Syp were localized to laminae IV and VI, suggesting a

70
colocalization of glial activation and presynaptic changes. Alterations in the
immunoreactivities of the presynaptic proteins Syp, SNAP25, Syb and Syx
were also observed in the coronal cryosections of CTSD -/- mice (IV; Fig. 7A-
D). Furthermore, the immunoreactive patterns of Syp and Syb were similar in
the threshold analysis (IV; Fig. 7, charts), and thus, some changes in the
distribution and amount of these proteins were expected to be observed in
CTSD -/- mice. Immunostaining of paraffin-embedded sections revealed an
abnormal aggregation of Syp (IV; Fig. 8 B, D) and Syb (IV; Fig. 8 A, C)
stainings, especially in VPM/VPL thalamic nuclei of affected mice. By
contrast, immunoreactive clusters of SNAP25 were observed in the zona
incerta dorsal/ventral (ZID/ZIV) region of the thalamus (IV; Fig. 9D, F) and in
S1BF (IV; Fig. 9C, E) of CTSD -/- mice. Furthermore, Western blotting
showed an increase in the amount of the Syp/Syb complex and a decrease in
the Syp dimer in the cortex and thalamus of CTSD -/- mice compared with
controls (IV; Fig. 10B). Western blotting did not show any radical changes in
the monomeric forms of presynaptic proteins Syp, Syp, SNAP25 or Syx (IV;
Fig. 10A).
It is hardly surprising that Syp and Syb behave similarly because Syp forms a
complex with Syb in mature neurons (Reisinger et al., 2004). However, Syb
cannot simultaneously bind to Syp and participate in the SNARE complex. Syp
serves as a regulator of the availability of Syb for fusion (Edelmann et al.,
1995; Becher et al., 1999a). The increase in the Syp/Syb complex could be
easily associated with excessive neuronal activity and excitotoxicity,
potentially associated with the mechanisms behind the epileptic seizures of
CTSD -/- mice. The level of the Syp/Syb complex has also been reported to be
increased in the kindling model of epilepsy in adult rats (Hinz et al., 2001).

71
6. CONCLUSIONS AND FUTURE PROSPECTS
Based on the main findings of this thesis, we can conclude the following:
o Cathepsin D protein expression increases during rat brain development,
and the results suggest a role for cathepsin D in synaptic reorganization,
plasticity and/or myelination.
o An active-site mutation in the cathepsin D gene affects the activity,
stability, processing, transport and secretion of the mutant protein.
o A duplication mutation, c.794dupA, in the human cathepsin D gene
causes congenital NCL and severe brain atrophy.
o Cathepsin D and lysosomes seem to play an important role in
controlling presynaptic events in the thalamocortical system.
The importance of Cathepsin D (CTSD), as a member of lysosomal proteases,
in the field of NCL research was revealed when the naturally occurring CTSD-
deficient sheep were characterized (Tyynelä et al., 2000) and CTSD knock-out
mice were generated (Saftig et al., 1995). Based on findings in congenital
CTSD-deficient human patients, the activity of CTSD seems to be crucial for
early development and survival. Broad interest in CTSD deficiency as a
possible diagnosis in microcephalic neonates with epileptic seizures has
already offered new patient material, and thus, future studies in this field will
likely focus on investigating new CTSD-deficient human cases.
At the molecular level, the important target of interest in NCL research is the
cell death mechanism and the involvement of synapses in neurodegeneration.
Synapses are the primary pathological target in a wide range of
neurodegenerative diseases, and thus, the synapse needs to be viewed as an
important target for development of novel protective treatments aimed at

72
preventing or slowing the progression of these diseases. Despite their diverse
clinical appearance and underlying genetic causes, synapse degeneration can
be common to many lysosomal storage disorders (reviewed in Jeyakumar et al.,
2005). Two NCL proteins, CLN3 and CLN1, are known to localize in the
synaptic region (Lehtovirta et al., 2001; Luiro et al., 2001; Suopanki et al.,
2002), and TPP1 also colocalizes with presynaptic markers (Partanen et al.,
unpublished data). In addition to our data revealing presynaptic changes in
CTSD -/- mice, evidence of synapse dysfunction in NCLs has recently been
reported in PPT1-deficient neurons in vitro (Virmani et al., 2005).
The specialized morphology of neurons and their synaptic excitability render
the neuron at risk for a variety of insults involving oxidative damage.
Promoters of an important set of genes for synaptic plasticity and vesicular
transport have been shown to be selectively damaged by oxidative stress in
cultured neurons (Lu et al., 2004). This suggests that synaptic alterations
resulting from oxidative stress are potential contributors to neurodegenerative
disease. Data from tissue and experimental models of Alzheimer’s disease
clearly indicate a link between lysosomal disturbance and the protein
accumulation that leads to synaptopathogenesis (Bahr and Bendiske, 2002).
The data presented in this thesis and in studies of other neurodegenerative
diseases strongly suggest that lysosomal defects, which are associated with
mitochondrial dysfunction, impaired axonal transport and increased
intracellular Ca2+ concentration as well as myelination, warrant further
examination. In addition, the role of astrocytes in synapses is an interesting
field of study. In the future, the key mediators of neuronal death and
physiological events in the thalamocortical system will be examined with
proteomic, micro-array and functional imaging methods. The developmental
course of changes in CTSD knock-out mice also requires further
characterization.

73
ACKNOWLEDGEMENTS
This thesis was completed at the Institute of Biomedicine/Biochemistry, University of
Helsinki, during 2000-2006. The present and former directors of the Institute, Professors Olli
Jänne, Ismo Virtanen, Esa Korpi, Tomi Mäkelä and Paavo Kinnunen, are thanked for placing
outstanding research facilities at my disposal. I am indebted to Professor Tomi Mäkelä for
accepting the role of ‘kustos’ at my thesis defense. I also thank Professors Jari Koistinaho and
Dan Lindholm for reviewing my thesis and for insightful comments. Carol Ann Pelli, HonBSc,
I thank for language editing of this thesis. Professors Dan Lindholm and Anna-Elina Lehesjoki
are warmly thanked for being part of my thesis follow-up group. I deeply appreciate Professor
Elina Ikonen for accepting the role of opponent at my thesis defense. The chairman of the
Finnish Graduate School of Neuroscience, Professor Kai Kaila, and the coordinator, Katri
Wegelius, PhD, are thanked for interesting courses and for generous help and support.
My deepest gratitude is due to my supervisor, Docent Jaana Vesterinen. Jaana, I thank you for
trusting me and giving me freedom and responsibility in my project. Thank you for allowing
me to gain valuable experience in teaching medical students, being part of numerous projects
and visiting the labs in Hamburg and London. Even though our strong personalities and
thoughts about how, when and why to run things sometimes conflicted, I greatly appreciate
your efforts in resolving the personal and scientific problems that I faced. I also thank my
second supervisor, Docent Marc Baumann. You have been a father figure to me ever since we
created our own small group. It is not difficult to follow ‘the footprints in the snow’, to
discover where Jaana received her work habits and way of thinking. You are a funny and lively
person with strong opinions.
My present workmates Maria Lindfors, Eeva Kuosmanen, Aleksi Haapanen and Teija Inkinen
as well as my former colleagues Jaana Suopanki, Tuija Järvinen, Katriina Jyrkiäinen, Sirkka
Kanerva and Reeta Korhonen are thanked for sharing weekdays with me. Thank you “Mia” for
your sincerity, your energetic personality and the many laughs and tense moments with the
mice. Eeva, I thank for sharing the wonderfulness of a teacher’s life and for updating me on
celebrity lives. Sorry for all of those pens I “borrowed” and never returned. Aleksi, you amaze
me with your mathematical and medical skills. Thank you for collaboration on the London
project. Our technicians, Teija, Tuija, “Kati” and Sirkka, without your help with everything in
the lab, my results would not look as good as they do. Jaana, I thank for collaboration on my
first publication and for persuading me to carry out my thesis in this group. Reeta, thank you

74
for interesting scientific conversations. Our secretary Kaija Tiilikka is thanked for her cruzial
help with the paper work as well as printer and fax problems.
I owe a debt of gratitude to Professor Thomas Braulke for teaching me new scientific methods
and how to work efficiently and remain persistent during my stay in Hamburg. Thank you also
for taking such good care of me when I was sick, watching the Oscar gala with me to the wee
hours and introducing me to bowling and German culture. Stephan Storch, PhD, is thanked for
acting as a technical supervisor during my year in Hamburg. I am also very grateful for Dr.
Jonathan Cooper for teaching me during my stay in London how to mount specimens perfectly
and how to balance work and family. Professor Andrej Hasilik, I thank for hospitality during
my visit to Marburg and for valuable email communications since then. I am honoured to have
gotten to know Eija Siintola, a Genetic expert and an outstanding scientist. Professor Matti
Haltia, ‘the father of the NCLs’, is thanked for fruitful collaboration.
My parents, Vappu and Veikko, and my younger sister, Milla, are warmly thanked for letting
me make my own choices, for taking care of the kids and for being there whenever needed.
Without you, none of this would have been possible. I am also indebted to my parents-in-law,
Kaarina and Juhani. Thank you for helping with the kids and for scientific opinions and advice
that encouraged me to finish this project. And, together with my brother-in-law, Leo, and my
grandmothers-in-law, Eila and Marjatta, I thank you for taking me into your family and
making me feel significant and loved.
Finally, my love and gratitude are owed to my husband Juho, my daughter Eevi and my son
Lenni. This thesis is dedicated to the three of you. Juho, I am so lucky to have met and married
you. Your continuous optimism motivates me, and you never forget to say those most wanted
words to me: “You are beautiful and clever. I am proud of you and I love you.” My dear
children, Eevi and Lenni, you have given me everything I ever dreamed of. Thank you for
being patient when I have travelled and worked too much. The thousands of power hugs and
kisses and songs and stories that you kindly offer me every day fill my heart with joy.
Helsinki, October 2006
Sanna Partanen

75
REFERENCES
AHLBERG, J., MARZELLA, L. & GLAUMANN, H. 1982, "Uptake and degradation ofproteins by isolated rat liver lysosomes. Suggestion of a microautophagic pathway ofproteolysis.", Lab Invest, vol. 47, pp. 523-532.
AHTIAINEN, L., VAN DIGGELEN, O.P., JALANKO, A. & KOPRA, O. 2003, "Palmitoylprotein thioesterase 1 is targeted to the axons in neurons.", J Comp Neurol, vol. 455, no.3, pp. 368-377.
AIKAWA, Y., XIA, X. & MARTIN, T.F. 2006, "SNAP25, but not syntaxin 1A, recycles viaan ARF6-regulated pathway in neuroendocrine cells", Molecular biology of the cell, vol.17, no. 2, pp. 711-722.
ANDO, K., KUDO, Y. & TAKAHASHI, M. 2005, "Negative regulation of neurotransmitterrelease by calpain: a possible involvement of specific SNAP-25 cleavage", Journal ofneurochemistry, vol. 94, no. 3, pp. 651-658.
ANDREWS, N.W. 2000, "Regulated secretion of conventional lysosomes", Trends in cellbiology, vol. 10, no. 8, pp. 316-321.
AOYAGI, T., MORISHIMA, H., NISHIZAWA, R., KUNIMOTO, S. & TAKEUCHI, T. 1972,"Biological activity of pepstatins, pepstanone A and partial peptides on pepsin, cathepsinD and renin.", J Antibiot (Tokyo), vol. 25, no. 12, pp. 689-694.
ARSTILA, A.U. & TRUMP, B.F. 1968, "Studies on cellular autophagocytosis. The formationof autophagic vacuoles in the liver after glucagon administration.", Am J Pathol, vol. 53,pp. 687-733.
AUTTI, T., RAININKO, R., VANHANEN, S.L. & SANTAVUORI, P. 1997, "Magneticresonance techniques in neuronal ceroid lipofuscinoses and some other lysosomaldiseases affecting the brain", Current opinion in neurology, vol. 10, no. 6, pp. 519-524.
AWANO, T., KATZ, M.L., O'BRIEN, D.P., TAYLOR, J.F., EVANS, J., KHAN, S., SOHAR,I., LOBEL, P. & JOHNSON, G.S. 2006, "A mutation in the cathepsin D gene (CTSD) inAmerican Bulldogs with neuronal ceroid lipofuscinosis", Molecular Genetics &Metabolism, vol. 87, no. 4, pp. 341-348.
BACCI, A., COCO, S., PRAVETTONI, E., SCHENK, U., ARMANO, S., FRASSONI, C.,VERDERIO, C., DE CAMILLI, P. & MATTEOLI, M. 2001, "Chronic blockade ofglutamate receptors enhances presynaptic release and downregulates the interactionbetween synaptophysin-synaptobrevin-vesicle-associated membrane protein 2", Journalof Neuroscience, vol. 21, no. 17, pp. 6588-6596.
BAHR, B.A. & BENDISKE, J. 2002, "The neuropathogenic contributions of lysosomaldysfunction", Journal of neurochemistry, vol. 83, no. 3, pp. 481-489.

76
BANAY-SCHWARTZ, M., DAHL, D., HUI, K.S. & LAJTHA, A. 1987, "The breakdown ofthe individual neurofilament proteins by cathepsin D", Neurochemical research, vol. 12,no. 4, pp. 361-367.
BANERJEE, P., DASGUPTA, A., SIAKOTOS, A.N. & DAWSON, G. 1992, "Evidence forlipase abnormality: high levels of free and triacylglycerol forms of unsaturated fatty acidsin neuronal ceroid-lipofuscinosis tissue", American Journal of Medical Genetics, vol. 42,no. 4, pp. 549-554.
BARANES, D., LEDERFEIN, D., HUANG, Y.Y., CHEN, M., BAILEY, C.H. & KANDEL,E.R. 1998, "Tissue plasminogen activator contributes to the late phase of LTP and tosynaptic growth in the hippocampal mossy fiber pathway", Neuron, vol. 21, no. 4, pp.813-825.
BARKER, L.A., DOWDALL, M.J. & WITTAKER, P.J. 1972, "Choline metabolism in thecerebral cortex of guinea pigs. Stable-bound acetylcholine.", Biochem J, vol. 130, no. 4,pp. 1063-1075.
BAROHN, R.J., DOWD, D.C. & KAGAN-HALLET, K.S. 1992, "Congenital ceroid-lipofuscinosis.[see comment]", Pediatric neurology, vol. 8, no. 1, pp. 54-59.
BARRETT, A.J. 1977, "Human cathepsin D", Advances in Experimental Medicine & Biology,vol. 95, pp. 291-300.
BARRETT, A.J. & KIRSCHE, H. 1981, "Cathepsin B, cathepsin H, cathepsin L.", MethodsEnzymol, vol. 80, pp. 535-561.
BAUMERT, M., MAYCOX, P.R., NAVONE, F., DE CAMILLI, P. & JAHN, R. 1989,"Synaptobrevin: an integral membrane protein of 18,000 daltons present in small synapticvesicles of rat brain", EMBO Journal, vol. 8, no. 2, pp. 379-384.
BEAL, M.F. 1995, "Aging, energy, and oxidative stress in neurodegenerative diseases.", AnnNeurol, vol. 38, no. 3, pp. 357-366.
BEAUJOUIN, M., BAGHDIGUIAN, S., GLONDU-LASSIS, M., BERCHEM, G. &LIAUDET-COOPMAN, E. 2006, "Overexpression of both catalytically active and -inactive cathepsin D by cancer cells enhances apoptosis-dependent chemo-sensitivity",Oncogene, vol. 25, no. 13, pp. 1967-1973.
BECHER, A., DRENCKHAHN, A., PAHNER, I., MARGITTAI, M., JAHN, R. & AHNERT-HILGER, G. 1999a, "The synaptophysin-synaptobrevin complex: a hallmark of synapticvesicle maturation", Journal of Neuroscience, vol. 19, no. 6, pp. 1922-1931.
BECHER, A., DRENCKHAHN, A., PAHNER, I. & AHNERT-HILGER, G. 1999b, "Thesynaptophysin-synaptobrevin complex is developmentally upregulated in cultivatedneurons but is absent in neuroendocrine cells", European journal of cell biology, vol. 78,no. 9, pp. 650-656.

77
BENES, F.M., VINCENT, S.L., ALSTERBERG, G., BIRD, E.D. & SANGIOVANNI, J.P.1992, "Increased GABAA receptor binding in superficial layers of cingulate cortex inschizophrenics.", J Neurosci, vol. 12, no. 3, pp. 924-929.
BENJAMINS, J.A. & SMITH, M.E. 1977, "Metabolism of myelin." in Myelin., ed. P. Morell,Plenum Press, New York, US, pp. 233-270.
BENNETT, M.K., CALAKOS, N. & SCHELLER, R.H. 1992, "Syntaxin: a synaptic proteinimplicated in docking of synaptic vesicles at presynaptic active zones.", Science, vol.257, no. 5067, pp. 255-259.
BENNETT, M.K., GARCIA-ARRARAS, J.E., ELFERINK, L.A., PETERSON, K.,FLEMING, A.M., HAZUKA, C.D. & SCHELLER, R.H. 1993, "The syntaxin family ofvesicular transport receptors", Cell, vol. 74, no. 5, pp. 863-873.
BERCHEM, G., GLONDU, M., GLEIZES, M., BROUILLET, J.P., VIGNON, F., GARCIA,M. & LIAUDET-COOPMAN, E. 2002, "Cathepsin-D affects multiple tumor progressionsteps in vivo: proliferation, angiogenesis and apoptosis", Oncogene, vol. 21, no. 38, pp.5951-5955.
BERNARDINI, F. & WARBURTON, M.J. 2002, "Lysosomal degradation of cholecystokinin-(29-33)-amide in mouse brain is dependent on tripeptidyl peptidase-I: implications for thedegradation and storage of peptides in classical late-infantile neuronal ceroidlipofuscinosis.", Biochem J, vol. 366, no. 2, pp. 521-529.
BERNSTEIN, H.G., KIRSCHKE, H., WIEDERANDERS, B., MULLER, A., RINNE, A. &DORN, A. 1987, "Cathepsin D, B, and H in rat brain as demonstrated byimmunohistochemistry.", Acta Histochem, vol. 82, no. 1, pp. 25-27.
BERNSTEIN, H.G., BRUSZIS, S., SCHMIDT, D., WIEDERANDERS, B. & DORN, A. 1989,"Immunodetection of cathepsin D in neuritic plaques found in brains of patients withdementia of Alzheimer type", Journal fur Hirnforschung, vol. 30, no. 5, pp. 613-618.
BERRY, M., ROGERS, A.W. & EAYRS, J.T. 1964, "Pattern of cell migration during corticalhistogenesis.", Nature, vol. 203, pp. 591-593.
BERRY, M. & ROGERS, A.W. 1965, "The migration of neuroblats in the developing cerebralcortex.", J Anat (London), vol. 99, pp. 691-709.
BIBLE, E., GUPTA, P., HOFMANN, S.L. & COOPER, J.D. 2004, "Regional and cellularneuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model ofinfantile neuronal ceroid lipofuscinosis", Neurobiology of disease, vol. 16, no. 2, pp. 346-359.
BLACKWELL, T.K., KRETZNER, L., BLACKWOOD, E.M., EISENMAN, R.N. &WEINTRAUB, H. 1990, "Sequence-specific DNA binding by the c-Myc protein",Science, vol. 250, no. 4984, pp. 1149-1151.

78
BOWEN, M.E., ENGELMAN, D.M. & BRUNGER, A.T. 2002, "Mutational analysis ofsynaptobrevin transmembrane domain oligomerization", Biochemistry, vol. 41, no. 52,pp. 15861-15866.
BRAULKE, T., HASILIK, A. & VON FIGURA, K. 1988, "Low temperature blocks transportand sorting of cathepsin D in fibroblasts", Biological chemistry Hoppe-Seyler, vol. 369,no. 6, pp. 441-449.
BRIOZZO, P., MORISSET, M., CAPONY, F., ROUGEOT, C. & ROCHEFORT, H. 1988, "Invitro degradation of extracellular matrix with Mr 52,000 cathepsin D secreted by breastcancer cells", Cancer research, vol. 48, no. 13, pp. 3688-3692.
BRIX, K., LINKE, M., TEPEL, C. & HERZOG, V. 2001, "Cysteine proteinases mediateextracellular prohormone processing in the thyroid", Biological chemistry, vol. 382, no.5, pp. 717-725.
BRIX, K. 2005, "Lysosomal ptoteases." in Lysosomes, ed. P. Saftig, 1. edition, LandesBioscience/Eurekah.com, Georgetown,TX, US, pp. 50-59.
BRONSON, R.T., DONAHUE, L.R., JOHNSON, K.R., TANNER, A., LANE, P.W. &FAUST, J.R. 1998, "Neuronal ceroid lipofuscinosis (nclf), a new disorder of the mouselinked to chromosome 9", American Journal of Medical Genetics, vol. 77, no. 4, pp. 289-297.
BROOM, M.F., ZHOU, C., BROOM, J.E., BARWELL, K.J., JOLLY, R.D. & HILL, D.F.1998, "Ovine neuronal ceroid lipofuscinosis: a large animal model syntenic with thehuman neuronal ceroid lipofuscinosis variant CLN6", Journal of medical genetics, vol.35, no. 9, pp. 717-721.
BROWN, N.J., CORNER, B.D. & DOGSON, M.C.H. 1954, "A second case in the samefamily of congenital familial cerebral lipidosis resembling amaurotic family idiocy.",Arch Dis Child, vol. 29, pp. 48-54.
BRUNGER, A.T. 2005, "Structure and function of SNARE and SNARE-interacting proteins",Quarterly reviews of biophysics, vol. 38, no. 1, pp. 1-47.
BURSCH, W., ELLINGER, A., KIENZL, H., TOROK, L., PANDEY, S., SIKORSKA, M.,WALKER, R. & HERMANN, R.S. 1996, "Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) inculture: the role of autophagy", Carcinogenesis, vol. 17, no. 8, pp. 1595-1607.
BURSCH, W. & ELLINGER, A. 2005, "Autophagy--a basic mechanism and a potential rolefor neurodegeneration.", Folia Neuropathol, vol. 43, no. 4, pp. 297-310.
CALAKOS, N., BENNETT, M.K., PETERSON, K.E. & SCHELLER, R.H. 1994, "Protein-protein interactions contributing to the specificity of intracellular vesicular trafficking",Science, vol. 263, no. 5150, pp. 1146-1149.
CAO, Y., ESPINOLA, J.A., FOSSALE, E., MASSEY, A.C., CUERVO, A.M.,MACDONALD, M.E. & COTMAN, S.L. 2006, "Autophagy is disrupted in a knock-in

79
mouse model of juvenile neuronal ceroid lipofuscinosis.", J Biol Chem, vol. 281, no. 29,pp. 20483-20493.
CAPONY, F., MORISSET, M., BARRETT, A.J., CAPONY, J.P., BROQUET, P., VIGNON,F., CHAMBON, M., LOUISOT, P. & ROCHEFORT, H. 1987, "Phosphorylation,glycosylation, and proteolytic activity of the 52-kD estrogen-induced protein secreted byMCF7 cells", J Cell Biol, vol. 104, no. 2, pp. 253-262.
CAPONY, F., BRAULKE, T., ROUGEOT, C., ROUX, S., MONTCOURRIER, P. &ROCHEFORT, H. 1994, "Specific mannose-6-phosphate receptor-independent sorting ofpro-cathepsin D in breast cancer cells", Experimental cell research, vol. 215, no. 1, pp.154-163.
CATALDO, A.M., BARNETT, J.L., BERMAN, S.A., LI, J., QUARLESS, S., BURSZTAJN,S., LIPPA, C. & NIXON, R.A. 1995, "Gene expression and cellular content of cathepsinD in Alzheimer's disease brain: evidence for early up-regulation of the endosomal-lysosomal system", Neuron, vol. 14, no. 3, pp. 671-680.
CHAN, S.L., CULMSEE, C., HAUGHEY, N., KLAPPER, W. & MATTSON, M.P. 2002,"Presenilin-1 mutations sensitize neurons to DNA damage-induced death by amechanism involving perturbed calcium homeostasis and activation of calpains andcaspase-12", Neurobiology of disease, vol. 11, no. 1, pp. 2-19.
CHAN, S.L. & MATTSON, M.P. 1999, "Caspase and calpain substrates: roles in synapticplasticity and cell death", Journal of neuroscience research, vol. 58, no. 1, pp. 167-190.
CHATTOPADHYAY, S., ITO, M., COOPER, J.D., BROOKS, A.I., CURRAN, T.M.,POWERS, J.M. & PEARCE, D.A. 2002a, "An autoantibody inhibitory to glutamic aciddecarboxylase in the neurodegenerative disorder Batten disease", Human moleculargenetics, vol. 11, no. 12, pp. 1421-1431.
CHATTOPADHYAY, S., KRISCENSKI-PERRY, E., WENGER, D.A. & PEARCE, D.A.2002b, "An autoantibody to GAD65 in sera of patients with juvenile neuronal ceroidlipofuscinoses", Neurology, vol. 59, no. 11, pp. 1816-1817.
CHEN, Y.A. & SCHELLER, R.H. 2001, "SNARE-mediated membrane fusion", NatureReviews Molecular Cell Biology, vol. 2, no. 2, pp. 98-106.
CHO, S.J., KELLY, M., ROGNLIEN, K.T., CHO, J., HOERBER, J.K.H. & JENA, B.P. 2002"SNAREs in opposing bilayers interact in a circular array to form conducting pores.",Biophys J, vol. 83, pp. 2522-2527.
CLARKE, P.G. 1990, "Developmental cell death: morphological diversity and multiplemechanisms", Anatomy & Embryology, vol. 181, no. 3, pp. 195-213.
COHEN, G.M. 1997, "Caspases: the executioners of apoptosis", Biochemical Journal, vol.326, no. Pt 1, pp. 1-16.

80
COLEMAN, P., FEDEROFF, H. & KURLAN, R. 2004, "A focus on the synapse forneuroprotection in Alzheimer disease and other dementias.", Neurology, vol. 63, no. 7,pp. 1155-1162.
CONNER, G.E. 1989, "Isolation of procathepsin D from mature cathepsin D by pepstatinaffinity chromatography. Autocatalytic proteolysis of the zymogen form of the enzyme.",Biochem J, vol. 263, no. 2, pp. 601-604.
CONNER, G.E. & RICHO, G. 1992, "Isolation and characterization of a stable activationintermediate of the lysosomal aspartyl protease cathepsin D", Biochemistry, vol. 31, no.4, pp. 1142-1147.
COOPER, J.D., MESSER, A., FENG, A.K., CHUA-COUZENS, J. & MOBLEY, W.C. 1999,"Apparent loss and hypertrophy of interneurons in a mouse model of neuronal ceroidlipofuscinosis: evidence for partial response to insulin-like growth factor-1 treatment",Journal of Neuroscience, vol. 19, no. 7, pp. 2556-2567.
COOPER, J.D., GUPTA, P., BIBLE, E. & HOFMANN, S 2002. "Profound loss of GABAergicinterneurons in the PPT1 knockout mouse model of infantile neuronal ceroidlipofuscinosis.", Neuropathol Appl Neurobiol, vol. 28, pp. 158-159.
COOPER, J.D. 2003, "Progress towards understanding the neurobiology of Batten disease orneuronal ceroid lipofuscinosis", Current opinion in neurology, vol. 16, no. 2, pp. 121-128.
COTMAN, S.L., VRBANAC, V., LEBEL, L.A., LEE, R.L., JOHNSON, K.A., DONAHUE,L.R., TEED, A.M., ANTONELLIS, K., BRONSON, R.T., LERNER, T.J. &MACDONALD, M.E. 2002, "Cln3(Deltaex7/8) knock-in mice with the common JNCLmutation exhibit progressive neurologic disease that begins before birth", Humanmolecular genetics, vol. 11, no. 22, pp. 2709-2721.
CUERVO, A.M. & DICE, J.F. 1996, "A receptor for the selective uptake and degradation ofproteins by lysosomes.", Science, vol. 273, pp. 501-503.
CUERVO, A.M. 2004a, "Autophagy: in sickness and in health", Trends in cell biology, vol.14, no. 2, pp. 70-77.
CUERVO, A.M. 2004b, "Autophagy: many paths to the same end", Molecular & CellularBiochemistry, vol. 263, no. 1-2, pp. 55-72.
DALY, C., SUGIMORI, M., MOREIRA, J.E., ZIFF, E.B. & LLINAS, R. 2000,"Synaptophysin regulates clathrin-independent endocytosis of synaptic vesicles",Proceedings of the National Academy of Sciences of the United States of America, vol.97, no. 11, pp. 6120-6125.
DAVIS, A.F., BAI, J., FASSHAUER, D., WOLOWICK, M.J., LEWIS, J.L. & CHAPMAN,E.R. 1999, "Kinetics of synaptotagmin responses to Ca2+ and assembly with the coreSNARE complex onto membranes.[erratum appears in Neuron 1999 Dec;24(4):1049]",Neuron, vol. 24, no. 2, pp. 363-376.

81
DAWSON, G., KILKUS, J., SIAKOTOS, A.N. & SINGH, I. 1996, "Mitochondrialabnormalities in CLN2 and CLN3 forms of Batten disease", Molecular & ChemicalNeuropathology, vol. 29, no. 2-3, pp. 227-235.
DE DUVE, C. 1983, "Lysosomes revisited", European Journal of Biochemistry, vol. 137, no.3, pp. 391-397.
DE DUVE, C.P.B., GIANETTO, R., WATTIAUX, R. & APPELMANS, F. 1955, "Tissuefractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue.",Biochem J, vol. 60, pp. 604-617.
DEAK, F., SCHOCH, S., LIU, X., SUDHOF, T.C. & KAVALALI, E.T. 2004, "Synaptobrevinis essential for fast synaptic-vesicle endocytosis", Nature cell biology, vol. 6, no. 11, pp.1102-1108.
DEAN, R.T. 1975, "Direct evidence for importance of lysosomes in degradation ofintracellular proteins.", Nature, vol. 257, pp. 414-416.
DEAN, R.T. & BARRETT, A.J. 1976, "Lysosomes", Essays in biochemistry, vol. 12, pp. 1-40.
DEISS, L.P., GALINKA, H., BERISSI, H., COHEN, O. & KIMCHI, A. 1996, "Cathepsin Dprotease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 andTNF-alpha", EMBO Journal, vol. 15, no. 15, pp. 3861-3870.
DEKEN, S.L., BECKMAN, M.L., BOOS, L. & QUICK, M.W. 2000, "Transport rates ofGABA transporters: regulation by the N-terminal domain and syntaxin 1A", Natureneuroscience, vol. 3, no. 10, pp. 998-1003.
DESSI, F., COLLE, M.A., HAUW, J.J. & DUYCKAERTS, C. 1997, "Accumulation ofSNAP-25 immunoreactive material in axons of Alzheimer's disease", Neuroreport, vol. 8,no. 17, pp. 3685-3689.
DIMENT, S., LEECH, M.S. & STAHL, P.D. 1988, "Cathepsin D is membrane-associated inmacrophage endosomes", Journal of Biological Chemistry, vol. 263, no. 14, pp. 6901-6907.
DIMENT, S., MARTIN, K.J. & STAHL, P.D. 1989, "Cleavage of parathyroid hormone inmacrophage endosomes illustrates a novel pathway for intracellular processing ofproteins", Journal of Biological Chemistry, vol. 264, no. 23, pp. 13403-13406.
DITTMER, F., ULBRICH, E.J., HAFNER, A., SCHMAHL, W., MEISTER, T.,POHLMANN, R. & VON FIGURA, K. 1999, "Alternative mechanisms for trafficking oflysosomal enzymes in mannose 6-phosphate receptor-deficient mice are cell type-specific", Journal of cell science, vol. 112, no. Pt 10, pp. 1591-1597.
DUNN, B. 1997, "Splitting image", Natl Struct Biol, vol. 4, pp. 969-972.
DUNN, W.A. Jr, 1990, "Studies on the mechanisms of autophagy: maturation of theautophagic vacuole", Journal of Cell Biology, vol. 110, no. 6, pp. 1935-1945.

82
EDELMANN, L., HANSON, P.I., CHAPMAN, E.R. & JAHN, R. 1995, "Synaptobrevinbinding to synaptophysin: a potential mechanism for controlling the exocytotic fusionmachine", EMBO Journal, vol. 14, no. 2, pp. 224-231.
ELLEDER, M. 1991, "Primary extracellular ceroid type lipopigment. A histochemical andultrastructural study", Histochemical Journal, vol. 23, no. 6, pp. 247-258.
ELLIS, S. & DIVOR, A.R. 1979, "Specificity and lysosomal localization of tripeptidylaminopeptidase.", Fed Proc, vol. 38, pp. 832.
ERICKSON, A.H. & BLOBEL, G. 1983, "Carboxyl-terminal proteolytic processing duringbiosynthesis of the lysosomal enzymes beta-glucuronidase and cathepsin D",Biochemistry, vol. 22, no. 22, pp. 5201-5205.
ERICKSON, A.H. & BLOBEL, G. 1979, "Early events in the biosynthesis of the lysosomalenzyme cathepsin D", Journal of Biological Chemistry, vol. 254, no. 23, pp. 11771-11774.
ERICKSON, A.H., CONNER, G.E. & BLOBEL, G. 1981, "Biosynthesis of a lysosomalenzyme. Partial structure of two transient and functionally distinct NH2-terminalsequences in cathepsin D", Journal of Biological Chemistry, vol. 256, no. 21, pp. 11224-11231.
ESHKIND, L.G. & LEUBE, R.E. 1995, "Mice lacking synaptophysin reproduce and formtypical synaptic vesicles", Cell & Tissue Research, vol. 282, no. 3, pp. 423-433.
ESKELINEN, E.L., TANAKA, Y. & SAFTIG, P. 2003, "At the acidic edge: emergingfunctions for lysosomal membrane proteins", Trends in cell biology, vol. 13, no. 3, pp.137-145.
EVANS, J., KATZ, M.L., LEVESQUE, D., SHELTON, G.D., DE LAHUNTA, A. &O'BRIEN, D. 2005, "A variant form of neuronal ceroid lipofuscinosis in Americanbulldogs", Journal of Veterinary Internal Medicine, vol. 19, no. 1, pp. 44-51.
EVIN, G., CAPPAI, R., LI, Q.X., CULVENOR, J.G., SMALL, D.H., BEYREUTHER, K. &MASTERS, C.L. 1995, "Candidate gamma-secretases in the generation of the carboxylterminus of the Alzheimer's disease beta A4 amyloid: possible involvement of cathepsinD", Biochemistry, vol. 34, no. 43, pp. 14185-14192.
FAN, H.P., FAN, F.J., BAO, L. & PEI, G. 2006, "SNAP-25/syntaxin 1A complex functionallymodulates neurotransmitter GABA reuptake.", J Biol Chem. 2006, vol. 22, no. 281, pp.28174-28184.
FEHRENBACHER, N. & JAATTELA, M. 2005, "Lysosomes as targets for cancer therapy",Cancer research, vol. 65, no. 8, pp. 2993-2995.
FELBOR, U., KESSLER, B., MOTHES, W., GOEBEL, H.H., PLOEGH, H.L., BRONSON,R.T. & OLSEN, B.R. 2002, "Neuronal loss and brain atrophy in mice lacking cathepsinsB and L", Proceedings of the National Academy of Sciences of the United States ofAmerica, vol. 99, no. 12, pp. 7883-7888.

83
FERNANDEZ-CHACON, R., KONIGSTORFER, A., GERBER, S.H., GARCIA, J., MATOS,M.F., STEVENS, C.F., BROSE, N., RIZO, J., ROSENMUND, C. & SUDHOF, T.C.2001, "Synaptotagmin I functions as a calcium regulator of release probability", Nature,vol. 410, no. 6824, pp. 41-49.
FERRANDINA, G., SCAMBIA, G., BARDELLI, F., BENEDETTI PANICI, P., MANCUSO,S. & MESSORI, A. 1997, "Relationship between cathepsin-D content and disease-freesurvival in node-negative breast cancer patients: a meta-analysis.[see comment]", Britishjournal of cancer, vol. 76, no. 5, pp. 661-666.
FOEKENS, J.A., LOOK, M.P., BOLT-DE VRIES, J., MEIJER-VAN GELDER, M.E., VANPUTTEN, W.L. & KLIJN, J.G. 1999, "Cathepsin-D in primary breast cancer: prognosticevaluation involving 2810 patients.[see comment]", British journal of cancer, vol. 79, no.2, pp. 300-307.
FORGAC, M. 1999, "Structure and properties of the vacuolar (H+)-ATPases", Journal ofBiological Chemistry, vol. 274, no. 19, pp. 12951-12954.
FUKUDA, M. 1991, "Lysosomal membrane glycoproteins. Structure, biosynthesis, andintracellular trafficking", Journal of Biological Chemistry, vol. 266, no. 32, pp. 21327-21330.
FUSEK, M. & VETVICKA, V. 1994, "Mitogenic function of human procathepsin D: the roleof the propeptide", Biochemical Journal, vol. 303, no. Pt 3, pp. 775-780.
FUTERMAN, A.H. & VAN MEER, G. 2004, "The cell biology of lysosomal storagedisorders", Nature Reviews Molecular Cell Biology, vol. 5, no. 7, pp. 554-565.
FYKSE, E.M., TAKEI, K., WALCH-SOLIMENA, C., GEPPERT, M., JAHN, R., DECAMILLI, P. & SUDHOF, T.C. 1993, "Relative properties and localizations of synapticvesicle protein isoforms: the case of the synaptophysins", Journal of Neuroscience, vol.13, no. 11, pp. 4997-5007.
GAISANO, H.Y., GHAI, M., MALKUS, P.N., SHEU, L., BOUQUILLON, A., BENNETT,M.K. & TRIMBLE, W.S. 1996 "Distinct cellular locations of the syntaxin family ofproteins in rat pancreatic acinar cells", Mol Biol Cell, vol. 7, no.12, pp. 2019-2027.
GAO, H., BOUSTANY, R.M., ESPINOLA, J.A., COTMAN, S.L., SRINIDHI, L.,ANTONELLIS, K.A., GILLIS, T., QIN, X., LIU, S., DONAHUE, L.R., BRONSON,R.T., FAUST, J.R., STOUT, D., HAINES, J.L., LERNER, T.J. & MACDONALD, M.E.2002, "Mutations in a novel CLN6-encoded transmembrane protein cause variantneuronal ceroid lipofuscinosis in man and mouse", American Journal of Human Genetics,vol. 70, no. 2, pp. 324-335.
GARBORG, I., TORVIK, A., HALS, J., TANGSRUD, S.E. & LINDEMANN, R. 1987,"Congenital neuronal ceroid lipofuscinosis. A case report", Acta Pathologica,Microbiologica, et Immunologica Scandinavica - Section A, Pathology, vol. 95, no. 3, pp.119-125.

84
GARCIA, M., DEROCQ, D., PUJOL, P. & ROCHEFORT, H. 1990, "Overexpression oftransfected cathepsin D in transformed cells increases their malignant phenotype andmetastatic potency", Oncogene, vol. 5, no. 12, pp. 1809-1814.
GARNER, C.C., NASH, J. & HUGANIR, R.L. 2000, "PDZ domains in synapse assembly andsignalling", Trends in cell biology, vol. 10, no. 7, pp. 274-280.
GEERLINGS, A., LOPEZ-CORCUERA, B. & ARAGON, C. 2000, "Characterization of theinteractions between the glycine transporters GLYT1 and GLYT2 and the SNAREprotein syntaxin 1A", FEBS letters, vol. 470, no. 1, pp. 51-54.
GHOSH, P., DAHMS, N.M. & KORNFELD, S. 2003, "Mannose 6-phosphate receptors: newtwists in the tale", Nature Reviews Molecular Cell Biology, vol. 4, no. 3, pp. 202-212.
GIESELMANN, V., POHLMANN, R., HASILIK, A. & VON FIGURA, K. 1983,"Biosynthesis and transport of cathepsin D in cultured human fibroblasts", Journal ofCell Biology, vol. 97, no. 1, pp. 1-5.
GIESELMANN, V., HASILIK, A. & VON FIGURA, K. 1985, "Processing of humancathepsin D in lysosomes in vitro", Journal of Biological Chemistry, vol. 260, no. 5, pp.3215-3220.
GLENNER, G.G. & WONG, C.W. 1984, "Alzheimer's disease: initial report of the purificationand characterization of a novel cerebrovascular amyloid protein", Biochemical &Biophysical Research Communications, vol. 120, no. 3, pp. 885-890.
GLONDU, M., COOPMAN, P., LAURENT-MATHA, V., GARCIA, M., ROCHEFORT, H. &LIAUDET-COOPMAN, E. 2001, "A mutated cathepsin-D devoid of its catalytic activitystimulates the growth of cancer cells", Oncogene, vol. 20, no. 47, pp. 6920-6929.
GODA, Y. & SUDHOF, T.C. 1997, "Calcium regulation of neurotransmitter release: reliablyunreliable?", Current opinion in cell biology, vol. 9, no. 4, pp. 513-518.
GODBOLD, G.D., AHN, K., YEYEODU, S., LEE, L.F., TING, J.P. & ERICKSON, A.H.1998, "Biosynthesis and intracellular targeting of the lysosomal aspartic proteinasecathepsin D", Advances in Experimental Medicine & Biology, vol. 436, pp. 153-162.
GOEBEL, H.H. & WISNIEWSKI, K.E. 2004, "Current state of clinical and morphologicalfeatures in human NCL", Brain Pathology, vol. 14, no. 1, pp. 61-69.
GONZALO, S. & LINDER, M.E. 1998, "SNAP-25 palmitoylation and plasma membranetargeting require a functional secretory pathway", Molecular biology of the cell, vol. 9,no. 3, pp. 585-597.
GOPALAKRISHNAN, M.M., GROSCH, H.W., LOCATELLI-HOOPS, S., WERTH, N.,SMOLENOVA, E., NETTERSHEIM, M., SANDHOFF, K. & HASILIK, A. 2004,"Purified recombinant human prosaposin forms oligomers that bind procathepsin D andaffect its autoactivation", Biochemical Journal, vol. 383, no. Pt. 3, pp. 507-515.

85
GOTTLIEB, R.A., NORDBERG, J., SKOWRONSKI, E. & BABIOR, B.M. 1996, "Apoptosisinduced in Jurkat cells by several agents is preceded by intracellular acidification.", ProcNatl Acad Sci, vol. 93, no. 2, pp. 654-658.
GRAFSTEIN, B. & FORMAN, D.S. 1980, "Intracellular transport in neurons", PhysiologicalReviews, vol. 60, no. 4, pp. 1167-1283.
GUICCIARDI, M.E., DEUSSING, J., MIYOSHI, H., BRONK, S.F., SVINGEN, P.A.,PETERS, C., KAUFMANN, S.H. & GORES, G.J. 2000, "Cathepsin B contributes toTNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release ofcytochrome c.", J Clin Invest, vol. 106, no. 9, pp. 1127-1137.
GUPTA, P., SOYOMBO, A.A., ATASHBAND, A., WISNIEWSKI, K.E., SHELTON, J.M.,RICHARDSON, J.A., HAMMER, R.E. & HOFMANN, S.L. 2001, "Disruption of PPT1or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice", Proceedings of theNational Academy of Sciences of the United States of America, vol. 98, no. 24, pp.13566-13571.
HAAS, C., HUNG, A.Y., CITRON, M., TEPLOW, D.B. & SELKOE, D.J. 1995, "beta-Amyloid, protein processing and Alzheimer's disease", Arzneimittel-Forschung, vol. 45,no. 3A, pp. 398-402.
HAASS, C. & DE STROOPER, B. 1999, "The presenilins in Alzheimer's disease--proteolysisholds the key", Science, vol. 286, no. 5441, pp. 916-919.
HACHIYA, Y., HAYASHI, M., KUMADA, S., UCHIYAMA, A., TSUCHIYA, K. &KURATA, K. 2006, "Mechanisms of neurodegeneration in neuronal ceroid-lipofuscinoses.", Acta Neuropathol (Berl), vol. 111, no. 2, pp. 168-177.
HALTIA, M., RAPOLA, J. & SANTAVUORI, P. 1973, "Infantile type of so-called neuronalceroid-lipofuscinosis. Histological and electron microscopic studies", ActaNeuropathologica, vol. 26, no. 2, pp. 157-170.
HALTIA, M. 2003, "The neuronal ceroid-lipofuscinoses", Journal of Neuropathology &Experimental Neurology, vol. 62, no. 1, pp. 1-13.
HASILIK, A. & NEUFELD, E.F. 1980, "Biosynthesis of lysosomal enzymes in fibroblasts.Synthesis as precursors of higher molecular weight", Journal of Biological Chemistry,vol. 255, no. 10, pp. 4937-4945.
HASILIK, A., VON FIGURA, K., CONZELMANN, E., NEHRKORN, H. & SANDHOFF, K.1982, "Lysosomal enzyme precursors in human fibroblasts. Activation of cathepsin Dprecursor in vitro and activity of beta-hexosaminidase A precursor towards gangliosideGM2.", Eur J Biochem, vol. 125, no. 2, pp. 317-325.
HEINE, C., TYYNELA, J., COOPER, J.D., PALMER, D.N., ELLEDER, M.,KOHLSCHUTTER, A. & BRAULKE, T. 2003, "Enhanced expression of manganese-dependent superoxide dismutase in human and sheep CLN6 tissues.", Biochem J, vol.376, no. 2, pp. 369-376.

86
HEINRICH, M., WICKEL, M., SCHNEIDER-BRACHERT, W., SANDBERG, C., GAHR, J.,SCHWANDNER, R., WEBER, T., SAFTIG, P., PETERS, C., BRUNNER, J.,KRONKE, M. & SCHUTZE, S. 1999, "Cathepsin D targeted by acid sphingomyelinase-derived ceramide.[erratum appears in EMBO J 2000 Jan 17;19(2):315]", EMBO Journal,vol. 18, no. 19, pp. 5252-5263.
HENNEBURY, R.C. 1995, "Excitotoxicity as a consequence of impairment of energymetabolism: The energy-linked excitotoxic hypothesis." in Mitochondria and FreeRadicals in Neurodegenerative Diseases., eds. M.F. Beal, N. Howell & I. Bodis-Wollner,Wiley-Liss, New York, US, pp. 111-143.
HENTZE, M.W. & KULOZIK, A.E. 1999, "A perfect message: RNA surveillance andnonsense-mediated decay", Cell, vol. 96, no. 3, pp. 307-310.
HESS, D.T., SLATER, T.M., WILSON, M.C. & SKENE, J.H. 1992, "The 25 kDasynaptosomal-associated protein SNAP-25 is the major methionine-rich polypeptide inrapid axonal transport and a major substrate for palmitoylation in adult CNS", Journal ofNeuroscience, vol. 12, no. 12, pp. 4634-4641.
HETMAN, M., PERSCHL, A., SAFTIG, P., VON FIGURA, K. & PETERS, C. 1994, "Mousecathepsin D gene: molecular organization, characterization of the promoter, andchromosomal localization", DNA & Cell Biology, vol. 13, no. 4, pp. 419-427.
HINZ, B., BECHER, A., MITTER, D., SCHULZE, K., HEINEMANN, U., DRAGUHN, A. &AHNERT-HILGER, G. 2001, "Activity-dependent changes of the presynapticsynaptophysin-synaptobrevin complex in adult rat brain", European journal of cellbiology, vol. 80, no. 10, pp. 615-619.
HIROKAWA, N. 1998, "Kinesin and dynein superfamily proteins and the mechanism oforganelle transport. [Review] [76 refs]", Science, vol. 279, no. 5350, pp. 519-526.
HIROKAWA, N. & TAKEMURA, R. 2005, "Molecular motors and mechanisms of directionaltransport in neurons", Nature Reviews Neuroscience, vol. 6, no. 3, pp. 201-214.
HOFMANN, S.L. & PELTONEN, L. 2001, "The neuronal ceroid lipofuscinoses." in Themetabolic & molecular basis of inherited disease., eds. C.R. Scriver, A.L. Beaudet, W.S.Sly & D. Valle, 8th edition, McGraw-Hill Inc, New York, US, pp. 3877-3894.
HOFMANN, S.L., ATASHBAND, A., CHO, S.K., DAS, A.K., GUPTA, P. & LU, J.Y. 2002,"Neuronal ceroid lipofuscinoses caused by defects in soluble lysosomal enzymes (CLN1and CLN2)", Current Molecular Medicine, vol. 2, no. 5, pp. 423-437.
HONER, W.G., FALKAI, P., YOUNG, C., WANG, T., XIE, J., BONNER, J., HU, L.,BOULIANNE, G.L., LUO, Z. & TRIMBLE, W.S. 1997, "Cingulate cortex synapticterminal proteins and neural cell adhesion molecule in schizophrenia", Neuroscience, vol.78, no. 1, pp. 99-110.
HONER, W.G., FALKAI, P., BAYER, T.A., XIE, J., HU, L., LI, H.Y., ARANGO, V.,MANN, J.J., DWORK, A.J. & TRIMBLE, W.S. 2002, "Abnormalities of SNAREmechanism proteins in anterior frontal cortex in severe mental illness", Cerebral Cortex,vol. 12, no. 4, pp. 349-356.

87
HONING, S. & HUNZIKER, W. 1995, "Cytoplasmic determinants involved in directlysosomal sorting, endocytosis, and basolateral targeting of rat lgp120 (lamp-I) in MDCKcells", Journal of Cell Biology, vol. 128, no. 3, pp. 321-332.
HUMPHREYS, S., LAKE, B.D. & SCHULZ, C.L. 1985, "Congenital amaurotic idiocy-Apathological, histochemical, biochemical, and ultrastuctural study.", Neuropathol ApplNeurobiol, vol. 11, pp. 475-484.
HUMEAU, Y., DOUSSAU, F., GRANT, N.J. & POULAIN, B. 2000, "How botulinum andtetanus neurotoxins block neurotransmitter release", Biochimie, vol. 82, no. 5, pp. 427-446.
HUNZIKER, W., SIMMEN, T. & HONING, S. 1996, "Trafficking of lysosomal membraneproteins in polarized kidney cells", Nephrologie, vol. 17, no. 7, pp. 347-350.
INTERNATIONAL BATTEN DISEASE CONSORTIUM 1995 "Isolation of a novel geneunderlying Batten disease, CLN3", Cell, vol. 82, pp. 949-957.
IRONSIDE, J.W. & DEARMOND, S. 1994, "Human prion diseases.", Brain Pathol, vol. 4, pp.313-314.
ISHIDOH, K. & KOMINAMI, E. 2002, "Processing and activation of lysosomal proteinases",Biological chemistry, vol. 383, no. 12, pp. 1827-1831.
ISHISAKA, R., UTSUMI, T., YABUKI, M., KANNO, T., FURUNO, T., INOUE, M. &UTSUMI, K. 1998, "Activation of caspase-3-like protease by digitonin-treatedlysosomes", FEBS letters, vol. 435, no. 2-3, pp. 233-236.
ISOSOMPPI, J., HEINONEN, O., HILTUNEN, J.O., GREENE, N.D., VESA, J., UUSITALO,A., MITCHISON, H.M., SAARMA, M., JALANKO, A. & PELTONEN, L. 1999,"Developmental expression of palmitoyl protein thioesterase in normal mice", BrainResearch.Developmental Brain Research, vol. 118, no. 1-2, pp. 1-11.
ISOSOMPPI, J., VESA, J., JALANKO, A. & PELTONEN, L. 2002, "Lysosomal localizationof the neuronal ceroid lipofuscinosis CLN5 protein", Human molecular genetics, vol. 11,no. 8, pp. 885-891.
IVY, G.O., SCHOTTLER, F., WENZEL, J., BAUDRY, M. & LYNCH, G. 1984, "Inhibitorsof lysosomal enzymes: accumulation of lipofuscin-like dense bodies in the brain",Science, vol. 226, no. 4677, pp. 985-987.
JALANKO, A., VESA, J., MANNINEN, T., VON SCHANTZ, C., MINYE, H., FABRITIUS,A.L., SALONEN, T., RAPOLA, J., GENTILE, M., KOPRA, O. & PELTONEN, L.2005, "Mice with Ppt1Deltaex4 mutation replicate the INCL phenotype and show aninflammation-associated loss of interneurons", Neurobiology of disease, vol. 18, no. 1,pp. 226-241.
JARPLID, B. & HALTIA, M. 1993, "An animal model of the infantile type of neuronal ceroid-lipofuscinosis", Journal of inherited metabolic disease, vol. 16, no. 2, pp. 274-277.

88
JELLINGER, K.A. & STADELMANN, C. 2000, "Mechanisms of cell death inneurodegenerative disorders", J Neural Transm Suppl, vol. 59, pp. 95-114.
JEYAKUMAR, M., DWEK, R.A., BUTTERS, T.D. & PLATT, F.M. 2005, "Storage solutions:treating lysosomal disorders of the brain", Nature Reviews Neuroscience, vol. 6, no. 9,pp. 713-725.
JOLLY, R.D. & WALKLEY, S.U. 1997, "Lysosomal storage diseases of animals: an essay incomparative pathology", Vet Pathol, vol. 34, pp. 527-548.
JOLLY, R.D. & WALKLEY, S.U. 1999, "Ovine ceroid lipofuscinosis (OCL6): postulatedmechanism of neurodegeneration", Mol Genet Metab, vol. 66, no. 4, pp. 376-380.
JOURNET, A., CHAPEL, A., KIEFFER, S., ROUX, F. & GARIN, J. 2002, "Proteomicanalysis of human lysosomes: application to monocytic and breast cancer cells",Proteomics, vol. 2, no. 8, pp. 1026-1040.
JOYCE, J.A., BARUCH, A., CHEHADE, K., MEYER-MORSE, N., GIRAUDO, E., TSAI,F.Y., GREENBAUM, D.C., HAGER, J.H., BOGYO, M. & HANAHAN, D. 2004,"Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis duringmultistage tumorigenesis.[see comment]", Cancer Cell, vol. 5, no. 5, pp. 443-453.
KAGEDAL, K., JOHANSSON, U. & OLLINGER, K. 2001, "The lysosomal proteasecathepsin D mediates apoptosis induced by oxidative stress", FASEB Journal, vol. 15, no.9, pp. 1592-1594.
KALIA, L.V., GINGRICH, J.R. & SALTER, M.W. 2004, "Src in synaptic transmission andplasticity", Oncogene, vol. 23, no. 48, pp. 8007-8016.
KATZ, B. 1969, The Release of Neural Transmitter Substances. Liverpool Univ., Liverpool,UK.
KATZ, M.L., SHIBUYA, H. & JOHNSON, G.S. 2001, "Animal models for the ceroidlipofuscinoses", Advances in Genetics, vol. 45, pp. 183-203.
KAUPPI, M., JÄNTTI, J. & OLKKONEN V.M. 2004, The function of Sec1/Munc18 proteins -Solution of the mystery in sight? Springer-Verlag GmbH, Berlin, Germany.
KEGEL, K.B., KIM, M., SAPP, E., MCINTYRE, C., CASTANO, J.G., ARONIN, N. &DIFIGLIA, M. 2000, "Huntingtin expression stimulates endosomal-lysosomal activity,endosome tubulation, and autophagy", Journal of Neuroscience, vol. 20, no. 19, pp.7268-7278.
KHAN, A.R. & JAMES, M.N. 1998, "Molecular mechanisms for the conversion of zymogensto active proteolytic enzymes", Protein Science, vol. 7, no. 4, pp. 815-836.
KIM, J. & KLIONSKY, D.J. 2000, "Autophagy, cytoplasm-to-vacuole targeting pathway, andpexophagy in yeast and mammalian cells", Annual Review of Biochemistry, vol. 69, pp.303-342.

89
KIM, Y., RAMIREZ-MONTEALEGRE, D. & PEARCE, D.A. 2003, "A role in vacuolararginine transport for yeast Btn1p and for human CLN3, the protein defective in Battendisease", Proceedings of the National Academy of Sciences of the United States ofAmerica, vol. 100, no. 26, pp. 15458-15462.
KOBLINSKI, J.E., AHRAM, M. & SLOANE, B.F. 2000, "Unraveling the role of proteases incancer", Clinica Chimica Acta, vol. 291, no. 2, pp. 113-135.
KOIKE, M., NAKANISHI, H., SAFTIG, P., EZAKI, J., ISAHARA, K., OHSAWA, Y.,SCHULZ-SCHAEFFER, W., WATANABE, T., WAGURI, S., KAMETAKA, S.,SHIBATA, M., YAMAMOTO, K., KOMINAMI, E., PETERS, C., VON FIGURA, K. &UCHIYAMA, Y. 2000, "Cathepsin D deficiency induces lysosomal storage with ceroidlipofuscin in mouse CNS neurons", Journal of Neuroscience, vol. 20, no. 18, pp. 6898-6906.
KOIKE, M., SHIBATA, M., OHSAWA, Y., NAKANISHI, H., KOGA, T., KAMETAKA, S.,WAGURI, S., MOMOI, T., KOMINAMI, E., PETERS, C., FIGURA, K., SAFTIG, P. &UCHIYAMA, Y. 2003, "Involvement of two different cell death pathways in retinalatrophy of cathepsin D-deficient mice", Molecular & Cellular Neurosciences, vol. 22, no.2, pp. 146-161.
KOIKE, M., SHIBATA, M., WAGURI, S., YOSHIMURA, K., TANIDA, I., KOMINAMI, E.,GOTOW, T., PETERS, C., VON FIGURA, K., MIZUSHIMA, N., SAFTIG, P. &UCHIYAMA, Y. 2005, "Participation of autophagy in storage of lysosomes in neuronsfrom mouse models of neuronal ceroid-lipofuscinoses (Batten disease).[see comment]",American Journal of Pathology, vol. 167, no. 6, pp. 1713-1728.
KOMAI, S., MATSUYAMA, T., MATSUMOTO, K., KATO, K., KOBAYASHI, M.,IMAMURA, K., YOSHIDA, S., UGAWA, S. & SHIOSAKA, S. 2000, "Neuropsinregulates an early phase of schaffer-collateral long-term potentiation in the murinehippocampus", European Journal of Neuroscience, vol. 12, no. 4, pp. 1479-1486.
KOPITZ, J., KISEN, G.O., GORDON, P.B., BOHLEY, P. & SEGLEN, P.O. 1990,"Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes", Journal ofCell Biology, vol. 111, no. 3, pp. 941-953.
KORNFELD, R. & KORNFELD, S. 1985, "Assembly of asparagine-linked oligosaccharides",Annual Review of Biochemistry, vol. 54, pp. 631-664.
KORNFELD, S. 1992, "Structure and function of the mannose 6-phosphate/insulinlike growthfactor II receptors", Annual Review of Biochemistry, vol. 61, pp. 307-330.
KORNFELD, S. & MELLMAN, I. 1989, "The biogenesis of lysosomes", Annual Review ofCell Biology, vol. 5, pp. 483-525.
KREDA, S.M., FUJITA, N. & SUZUKI, K. 1994, "Expression of sphingolipid activatorprotein gene in brain and systemic organs of developing mice", Developmentalneuroscience, vol. 16, no. 1-2, pp. 90-99.

90
LADROR, U.S., SNYDER, S.W., WANG, G.T., HOLZMAN, T.F. & KRAFFT, G.A. 1994,"Cleavage at the amino and carboxyl termini of Alzheimer's amyloid-beta by cathepsinD", Journal of Biological Chemistry, vol. 269, no. 28, pp. 18422-18428.
LAM, H.D.D., MITCHISON, H.M., GREENE, N.D.E., NUSSBAUM, R.L., MOBLEY, W.C.& COOPER, J.D. 1999, "Pathologic involvement of interneurons in mouse models ofneuronal ceroid lipofuscinosis.", Soc Neurosci Abs, vol. 25, pp. 1593.
LANE, S.C., JOLLY, R.D., SCHMECHEL, D.E., ALROY, J. & BOUSTANY, R.M. 1996,"Apoptosis as the mechanism of neurodegeneration in Batten's disease", Journal ofneurochemistry, vol. 67, no. 2, pp. 677-683.
LARSEN, K.E. & SULZER, D. 2002, "Autophagy in neurons.", Histol Histopathol, vol. 17,pp. 897-908.
LARSEN, L.B., BOISEN, A. & PETERSEN, T.E. 1993, "Procathepsin D cannot autoactivateto cathepsin D at acid pH", FEBS letters, vol. 319, no. 1-2, pp. 54-58.
LAURENT-MATHA, V., MARUANI-HERRMANN, S., PREBOIS, C., BEAUJOUIN, M.,GLONDU, M., NOEL, A., ALVAREZ-GONZALEZ, M.L., BLACHER, S.,COOPMAN, P., BAGHDIGUIAN, S., GILLES, C., LONCAREK, J., FREISS, G.,VIGNON, F. & LIAUDET-COOPMAN, E. 2005, "Catalytically inactive humancathepsin D triggers fibroblast invasive growth", Journal of Cell Biology, vol. 168, no. 3,pp. 489-499.
LAURENT-MATHA, V., DEROCQ, D., PREBOIS, C., KATUNUMA, N. & LIAUDET-COOPMAN, E. 2006, "Processing of human cathepsin D is independent of its catalyticfunction and auto-activation: involvement of cathepsins L and B", Journal ofBiochemistry, vol. 139, no. 3, pp. 363-371.
LAZZARINO, D.A. & GABEL, C.A. 1988, "Biosynthesis of the mannose 6-phosphaterecognition marker in transport-impaired mouse lymphoma cells. Demonstration of atwo-step phosphorylation", Journal of Biological Chemistry, vol. 263, no. 21, pp. 10118-10126.
LE BORGNE, R., ALCONADA, A., BAUER, U. & HOFLACK, B. 1998, "The mammalianAP-3 adaptor-like complex mediates the intracellular transport of lysosomal membraneglycoproteins", Journal of Biological Chemistry, vol. 273, no. 45, pp. 29451-29461.
LEABU, M. 2006, "Membrane fusion in cells: molecular machinery and mechanisms", Journalof Cellular & Molecular Medicine, vol. 10, no. 2, pp. 423-427.
LEFRANCOIS, S., CANUEL, M., ZENG, J. & MORALES, C.R. 2005, "Inactivation ofsortilin (a novel lysosomal sorting receptor) by dominant negative competition and RNAinterference.", Biol Proced Online, vol. 7, pp. 17-25.
LEHTOVIRTA, M., KYTTALA, A., ESKELINEN, E.L., HESS, M., HEINONEN, O. &JALANKO, A. 2001, "Palmitoyl protein thioesterase (PPT) localizes into synaptosomesand synaptic vesicles in neurons: implications for infantile neuronal ceroid lipofuscinosis(INCL)", Human molecular genetics, vol. 10, no. 1, pp. 69-75.

91
LI, J.Y., PLOMANN, M. & BRUNDIN, P. 2003, "Huntington's disease: a synaptopathy?",Trends Mol Med, vol. 9, no. 10, pp. 414-420.
LIAUDET-COOPMAN, E., BEAUJOUIN, M., DEROCQ, D., GARCIA, M., GLONDU-LASSIS, M., LAURENT-MATHA, V., PREBOIS, C., ROCHEFORT, H. & VIGNON,F. 2006, "Cathepsin D: newly discovered functions of a long-standing aspartic protease incancer and apoptosis.", Cancer Lett, vol. 237, no. 2, pp. 167-79.
LIBERSKI, P.P., GAJDUSEK, D.C. & BROWN, P. 2002, "How do neurons degenerate inprion diseases or transmissible spongiform encephalopathies (TSEs): neuronal autophagyrevisited.", Acta Neurobiol Exp, vol. 62, no. 3, pp. 141-147.
LIN, R.C. & SCHELLER, R.H. 2000, "Mechanisms of synaptic vesicle exocytosis", AnnualReview of Cell & Developmental Biology, vol. 16, pp. 19-49.
LINKE, M., HERZOG, V. & BRIX, K. 2002, "Trafficking of lysosomal cathepsin B-greenfluorescent protein to the surface of thyroid epithelial cells involves theendosomal/lysosomal compartment", Journal of cell science, vol. 115, no. Pt 24, pp.4877-4889.
LODISH, H., BERK, A., ZIPURSKY, L.S., MATSUDAIRA, P., BALTIMORE, D. &DARRELL, J.E. 1999, Molecular cell biology. 1st edition, W.H. Freeman & Co, NewYork, US.
LU, T., PAN, Y., KAO, S.Y., LI, C., KOHANE, I., CHAN, J. & YANKNER, B.A. 2004,"Gene regulation and DNA damage in the ageing human brain", Nature, vol. 429, no.6994, pp. 883-891.
LUIRO, K., KOPRA, O., LEHTOVIRTA, M. & JALANKO, A. 2001, "CLN3 protein istargeted to neuronal synapses but excluded from synaptic vesicles: new clues to Battendisease", Human molecular genetics, vol. 10, no. 19, pp. 2123-2131.
MAJANDER, A., PIHKO, H. & SANTAVUORI, P. 1995, "Palmitate oxidation in musclemitochondria of patients with the juvenile form of neuronal ceroid-lipofuscinosis.", Am JMed Genet, vol. 57, no. 2, pp. 298-300.
MARCH, P.A., WURZELMANN, S. & WALKLEY, S.U. 1995, "Morphological alterations inneocortical and cerebellar GABAergic neurons in a canine model of juvenile Battendisease.", Am J Med Genet, vol. 57, no. 2, pp. 204-212.
MARKS, N. & LAJTHA, A. 1965, "Separation of acid and neutral proteinases of brain.",Biochem J, vol. 97, pp. 74-83.
MARKS, N., STERN, F. & LAJTHA, A. 1975, "Changes in proteolytic enzymes and proteinsduring maturation of the brain.", Brain Res, vol. 86, no. 2, pp. 307-322.
MARKS, B., STOWELL, M.H., VALLIS, Y., MILLS, I.G., GIBSON, A., HOPKINS, C.R. &MCMAHON, H.T. 2001, "GTPase activity of dynamin and resulting conformationchange are essential for endocytosis", Nature, vol. 410, no. 6825, pp. 231-235.

92
MASTERS, C.L., SIMMS, G., WEINMAN, N.A., MULTHAUP, G., MCDONALD, B.L. &BEYREUTHER, K. 1985, "Amyloid plaque core protein in Alzheimer disease and Downsyndrome", Proceedings of the National Academy of Sciences of the United States ofAmerica, vol. 82, no. 12, pp. 4245-4249.
MATSUYAMA, S., LLOPIS, J., DEVERAUX, Q.L., TSIEN, R.Y. & REED, J.C. 2000,"Changes in intramitochondrial and cytosolic pH: early events that modulate caspaseactivation during apoptosis.", Nat Cell Biol, vol. 2, no. 6, pp. 318-325.
MATUS, A. & GREEN, G.D. 1987, "Age-related increase in a cathepsin D like protease thatdegrades brain microtubule-associated proteins", Biochemistry, vol. 26, no. 25, pp. 8083-8086.
MAYCOX, P.R., DECKWERTH, T., HELL, J.W. & JAHN, R. 1988, "Glutamate uptake bybrain synaptic vesicles. Energy dependence of transport and functional reconstitution inproteoliposomes", Journal of Biological Chemistry, vol. 263, no. 30, pp. 15423-15428.
MCDERMOTT, J.R. & GIBSON, A.M. 1996, "Degradation of Alzheimer's beta-amyloidprotein by human cathepsin D", Neuroreport, vol. 7, no. 13, pp. 2163-2166.
MCMAHON, H.T., BOLSHAKOV, V.Y., JANZ, R., HAMMER, R.E., SIEGELBAUM, S.A.& SUDHOF, T.C. 1996, "Synaptophysin, a major synaptic vesicle protein, is notessential for neurotransmitter release", Proceedings of the National Academy of Sciencesof the United States of America, vol. 93, no. 10, pp. 4760-4764.
MEIKLE, P.J., HOPWOOD, J.J., CLAGUE, A.E. & CAREY, W.F. 1999, "Prevalence oflysosomal storage disorders", JAMA, vol. 281, no. 3, pp. 249-254.
METCALF, P. & FUSEK, M. 1993, "Two crystal structures for cathepsin D: the lysosomaltargeting signal and active site", EMBO Journal, vol. 12, no. 4, pp. 1293-1302.
MICHALIK, A. & VAN BROECKHOVEN, C. 2003, "Pathogenesis of polyglutaminedisorders: aggregation revisited", Human molecular genetics, vol. 12, no. Spec 2, pp.R173-86.
MIGNATTI, P. & RIFKIN, D.B. 1993, "Biology and biochemistry of proteinases in tumorinvasion", Physiological Reviews, vol. 73, no. 1, pp. 161-195.
MITCHISON, H.M., BERNARD, D.J., GREENE, N.D., COOPER, J.D., JUNAID, M.A.,PULLARKAT, R.K., DE VOS, N., BREUNING, M.H., OWENS, J.W., MOBLEY,W.C., GARDINER, R.M., LAKE, B.D., TASCHNER, P.E. & NUSSBAUM, R.L. 1999,"Targeted disruption of the Cln3 gene provides a mouse model for Batten disease. TheBatten Mouse Model Consortium [corrected]", Neurobiol Dis, vol. 6, no. 5, pp. 321-334.
MITCHISON, H.M. & LIM, M.J. COOPER, J.D. 2004, "Selectivity and types of cell death inthe neuronal ceroid lipofuscinoses.", Brain Pathol, vol. 14, no. 1, pp. 86-96.
MITTER, D., REISINGER, C., HINZ, B., HOLLMANN, S., YELAMANCHILI, S.V.,TREIBER-HELD, S., OHM, T.G., HERRMANN, A. & AHNERT-HILGER, G. 2003,

93
"The synaptophysin/synaptobrevin interaction critically depends on the cholesterolcontent", Journal of neurochemistry, vol. 84, no. 1, pp. 35-42.
MIYAKAWA, T., SUMIYOSHI, S., DESHIMARU, M., SUZUKI, T. & TOMONARI, H.1972, "Electron microscopic study on schizophrenia. Mechanism of pathologicalchanges", Acta Neuropathologica, vol. 20, no. 1, pp. 67-77.
MYLLYKANGAS, L., TYYNELA, J., PAGE-MCCAW, A., RUBIN, G.M., HALTIA, M.J. &FEANY, M.B. 2005, "Cathepsin D-deficient Drosophila recapitulate the key features ofneuronal ceroid lipofuscinoses", Neurobiology of disease, vol. 19, no. 1-2, pp. 194-199.
NAKAGAWA, T., ROTH, W., WONG, P., NELSON, A., FARR, A., DEUSSING, J.,VILLADANGOS, J.A., PLOEGH, H., PETERS, C. & RUDENSKY, A.Y. 1998,"Cathepsin L: critical role in Ii degradation and CD4 T cell selection in the thymus.[seecomment]", Science, vol. 280, no. 5362, pp. 450-453.
NAKANISHI, H., TOMINAGA, K., AMANO, T., HIROTSU, I., INOUE, T. &YAMAMOTO, K. 1994, "Age-related changes in activities and localizations ofcathepsins D, E, B, and L in the rat brain tissues", Experimental neurology, vol. 126, no.1, pp. 119-128.
NAKANISHI, H., AMANO, T., SASTRADIPURA, D.F., YOSHIMINE, Y., TSUKUBA, T.,TANABE, K., HIROTSU, I., OHONO, T. & YAMAMOTO, K. 1997, "Increasedexpression of cathepsins E and D in neurons of the aged rat brain and their colocalizationwith lipofuscin and carboxy-terminal fragments of Alzheimer amyloid precursor protein",Journal of neurochemistry, vol. 68, no. 2, pp. 739-749.
NAKANISHI, H., ZHANG, J., KOIKE, M., NISHIOKU, T., OKAMOTO, Y., KOMINAMI,E., VON FIGURA, K., PETERS, C., YAMAMOTO, K., SAFTIG, P. & UCHIYAMA,Y. 2001, "Involvement of nitric oxide released from microglia-macrophages inpathological changes of cathepsin D-deficient mice", Journal of Neuroscience, vol. 21,no. 19, pp. 7526-7533.
NIXON, R.A., CATALDO, A.M. & MATHEWS, P.M. 2000, "The endosomal-lysosomalsystem of neurons in Alzheimer's disease pathogenesis: a review", Neurochemicalresearch, vol. 25, no. 9-10, pp. 1161-1172.
NORMAN, R.M. & WOOD, N. 1941, "Congenital form of amaurotic family idiocy.", Journalof Neurological Psychiatry, vol. 4, pp. 175-190.
NOVELLI, A., REILLY, J.A., LYSKO, P.G. & HENNEBERRY, R.C. 1988, "Glutamatebecomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energylevels are reduced", Brain research, vol. 451, no. 1-2, pp. 205-212.
OLIET, S.H., PIET, R. & POULAIN, D.A. 2001, "Control of glutamate clearance and synapticefficacy by glial coverage of neurons.[see comment]", Science, vol. 292, no. 5518, pp.923-926.
ONG, W.Y. & GAREY, L.J. 1993, "Ultrastructural features of biopsied temporopolar cortex(area 38) in a case of schizophrenia.", Schizophr Res, vol. 10, no. 1, pp. 15-27.

94
OSWALD, M.J., KAY, G.W. & PALMER, D.N. 2001, "Changes in GABAergic neurondistribution in situ and in neuron cultures in ovine (OCL6) Batten disease", EuropeanJournal of Paediatric Neurology, vol. 5, no. Suppl A, pp. 135-142.
OSWALD, M.J., PALMER, D.N., KAY, G.W., SHEMILT, S.J., REZAIE, P. & COOPER,J.D. 2005, "Glial activation spreads from specific cerebral foci and precedesneurodegeneration in presymptomatic ovine neuronal ceroid lipofuscinosis (CLN6)",Neurobiology of disease, vol. 20, no. 1, pp. 49-63.
PABST, S., HAZZARD, J.W., ANTONIN, W., SUDHOF, T.C., JAHN, R., RIZO, J. &FASSHAUER, D. 2000, "Selective interaction of complexin with the neuronal SNAREcomplex. Determination of the binding regions", Journal of Biological Chemistry, vol.275, no. 26, pp. 19808-19818.
PALMER, D.N., HUSBANDS, D.R., WINTER, P.J., BLUNT, J.W. & JOLLY, R.D. 1986,"Ceroid lipofuscinosis in sheep. I. Bis(monoacylglycero)phosphate, dolichol, ubiquinone,phospholipids, fatty acids, and fluorescence in liver lipopigment lipids", Journal ofBiological Chemistry, vol. 261, no. 4, pp. 1766-1772.
PALMER, D.N., FEARNLEY, I.M., WALKER, J.E., HALL, N.A., LAKE, B.D., WOLFE,L.S., HALTIA, M., MARTINUS, R.D. & JOLLY, R.D. 1992, "Mitochondrial ATPsynthase subunit c storage in the ceroid-lipofuscinoses (Batten disease).", Am J MedGenet, vol. 42, no. 4, pp. 561-567.
PANG, D.T., WANG, J.K., VALTORTA, F., BENFENATI, F. & GREENGARD, P. 1988,"Protein tyrosine phosphorylation in synaptic vesicles", Proceedings of the NationalAcademy of Sciences of the United States of America, vol. 85, no. 3, pp. 762-766.
PAPASSOTIROPOULOS, A., BAGLI, M., KURZ, A., KORNHUBER, J., FORSTL, H.,MAIER, W., PAULS, J., LAUTENSCHLAGER, N. & HEUN, R. 2000, "A geneticvariation of cathepsin D is a major risk factor for Alzheimer's disease.[see comment]",Annals of Neurology, vol. 47, no. 3, pp. 399-403.
PEARCE, D.A., NOSEL, S.A. & SHERMAN, F. 1999, "Studies of pH regulation by Btn1p,the yeast homolog of human Cln3p", Molecular Genetics & Metabolism, vol. 66, no. 4,pp. 320-323.
PETERS, C. & VON FIGURA, K. 1994, "Biogenesis of lysosomal membranes", FEBS letters,vol. 346, no. 1, pp. 108-114.
PEVSNER, J., HSU, S.C., BRAUN, J.E., CALAKOS, N., TING, A.E., BENNETT, M.K. &SCHELLER, R.H. 1994, "Specificity and regulation of a synaptic vesicle dockingcomplex", Neuron, vol. 13, no. 2, pp. 353-361.
PIET, R., POULAIN, D.A. & OLIET, S.H. 2002, "Modulation of synaptic transmission byastrocytes in the rat supraoptic nucleus", Journal of physiology, Paris, vol. 96, no. 3-4,pp. 231-236.
POHLMANN, R., BOEKER, M.W. & VON FIGURA, K. 1995, "The two mannose 6-phosphate receptors transport distinct complements of lysosomal proteins", Journal ofBiological Chemistry, vol. 270, no. 45, pp. 27311-27318.

95
PONTIKIS, C.C., CELLA, C.V., PARIHAR, N., LIM, M.J., CHAKRABARTI, S.,MITCHISON, H.M., MOBLEY, W.C., REZAIE, P., PEARCE, D.A. & COOPER, J.D.2004, "Late onset neurodegeneration in the Cln3-/- mouse model of juvenile neuronalceroid lipofuscinosis is preceded by low level glial activation", Brain research, vol. 1023,no. 2, pp. 231-242.
PONTIKIS, C.C., COTMAN, S.L., MACDONALD, M.E. & COOPER, J.D. 2005,"Thalamocortical neuron loss and localized astrocytosis in the Cln3Deltaex7/8 knock-inmouse model of Batten disease", Neurobiology of disease, vol. 20, no. 3, pp. 823-836.
PORTER, M.Y., TURMAINE, M. & MOLE, S.E. 2005, "Identification and characterization ofCaenorhabditis elegans palmitoyl protein thioesterase1", Journal of neuroscienceresearch, vol. 79, no. 6, pp. 836-848.
PURANAM, K.L., GUO, W.X., QIAN, W.H., NIKBAKHT, K. & BOUSTANY, R.M. 1999,"CLN3 defines a novel antiapoptotic pathway operative in neurodegeneration andmediated by ceramide", Molecular Genetics & Metabolism, vol. 66, no. 4, pp. 294-308.
PURCELL, A.L. & CAREW, T.J. 2003, "Tyrosine kinases, synaptic plasticity and memory:insights from vertebrates and invertebrates.", Trends Neurosci, vol. 26, no. 11, pp. 625-630.
QIAN, Z., GILBERT, M.E., COLICOS, M.A., KANDEL, E.R. & KUHL, D. 1993, "Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling andlong-term potentiation", Nature, vol. 361, no. 6411, pp. 453-457.
RANTA, S., ZHANG, Y., ROSS, B., LONKA, L., TAKKUNEN, E., MESSER, A., SHARP,J., WHEELER, R., KUSUMI, K., MOLE, S., LIU, W., SOARES, M.B., BONALDO,M.F., HIRVASNIEMI, A., DE LA CHAPELLE, A., GILLIAM, T.C. & LEHESJOKI,A.E. 1999, "The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant miceare associated with mutations in CLN8", Nature genetics, vol. 23, no. 2, pp. 233-236.
REDECKER, B., HECKENDORF, B., GROSCH, H.W., MERSMANN, G. & HASILIK, A.1991, "Molecular organization of the human cathepsin D gene", DNA & Cell Biology,vol. 10, no. 6, pp. 423-431.
REID, W.A., VALLER, M.J. & KAY, J. 1986, "Immunolocalization of cathepsin D in normaland neoplastic human tissues", Journal of clinical pathology, vol. 39, no. 12, pp. 1323-1330.
REISINGER, C., YELAMANCHILI, S.V., HINZ, B., MITTER, D., BECHER, A., BIGALKE,H. & AHNERT-HILGER, G. 2004, "The synaptophysin/synaptobrevin complexdissociates independently of neuroexocytosis", Journal of neurochemistry, vol. 90, no. 1,pp. 1-8.
RICHARDS, D.A., GUATIMOSIM, C. & BETZ, W.J. 2000, "Two endocytic recycling routesselectively fill two vesicle pools in frog motor nerve terminals", Neuron, vol. 27, no. 3,pp. 551-559.
RICHO, G.R. & CONNER, G.E. 1994, "Structural requirements of procathepsin D activationand maturation", Journal of Biological Chemistry, vol. 269, no. 20, pp. 14806-14812.

96
RIIKONEN, R., VANHANEN, S.L., TYYNELA, J., SANTAVUORI, P. & TURPEINEN, U.2000, "CSF insulin-like growth factor-1 in infantile neuronal ceroid lipofuscinosis.",Neurology, vol. 54, no. 9, pp. 1828-1832.
ROBERG, K., JOHANSSON, U. & OLLINGER, K. 1999, "Lysosomal release of cathepsin Dprecedes relocation of cytochrome c and loss of mitochondrial transmembrane potentialduring apoptosis induced by oxidative stress", Free radical biology & medicine, vol. 27,no. 11-12, pp. 1228-1237.
ROBERG, K., KAGEDAL, K. & OLLINGER, K. 2002, "Microinjection of cathepsin dinduces caspase-dependent apoptosis in fibroblasts", American Journal of Pathology, vol.161, no. 1, pp. 89-96.
ROCHEFORT, H. 1992, "Cathepsin D in breast cancer: a tissue marker associated withmetastasis", European journal of cancer, vol. 28A, no. 11, pp. 1780-1783.
ROCHEFORT, H. & LIAUDET-COOPMAN, E. 1999, "Cathepsin D in cancer metastasis: aprotease and a ligand", APMIS, vol. 107, no. 1, pp. 86-95.
ROSHY, S., SLOANE, B.F. & MOIN, K. 2003, "Pericellular cathepsin B and malignantprogression", Cancer & Metastasis Reviews, vol. 22, no. 2-3, pp. 271-286.
SAFTIG, P., HETMAN, M., SCHMAHL, W., WEBER, K., HEINE, L., MOSSMANN, H.,KOSTER, A., HESS, B., EVERS, M. & VON FIGURA, K. 1995, "Mice deficient for thelysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosaand profound destruction of lymphoid cells", EMBO Journal, vol. 14, no. 15, pp. 3599-3608.
SAKABA, T., STEIN, A., JAHN, R. & NEHER, E. 2005, "Distinct kinetic changes inneurotransmitter release after SNARE protein cleavage", Science, vol. 309, no. 5733, pp.491-494.
SANDBANK, U. 1968, "Congenital amaurotic idiocy", Pathologia Europaea, vol. 3, no. 2, pp.226-229.
SANTAVUORI, P., VALANNE, L., AUTTI, T., HALTIA, M., PIHKO, H. & SAINIO, K.1998, "Muscle-eye-brain disease: clinical features, visual evoked potentials and brainimaging in 20 patients", European Journal of Paediatric Neurology, vol. 2, no. 1, pp. 41-47.
SASAKI, S. & MARUYAMA, S. 1994, "Synapse loss in anterior horn neurons in amyotrophiclateral sclerosis.", Acta Neuropathol (Berl), vol. 88, no. 3, pp. 222-227.
SAVUKOSKI, M., KLOCKARS, T., HOLMBERG, V., SANTAVUORI, P., LANDER, E.S.& PELTONEN, L. 1998 " CLN5, a novel gene encoding a putative transmembraneprotein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis", NatGenet, vol. 19, no. 3, pp. 286-288.

97
SCALES, S.J., CHEN, Y.A., YOO, B.Y., PATEL, S.M., DOUNG, Y.C. & SCHELLER, R.H.2000, "SNAREs contribute to the specificity of membrane fusion.", Neuron, vol. 26, no.2, pp. 457-464.
SCHIAVO, G., BENFENATI, F., POULAIN, B., ROSSETTO, O., POLVERINO DELAURETO, P., DASGUPTA, B.R. & MONTECUCCO, C. 1992, "Tetanus andbotulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage ofsynaptobrevin.[see comment]", Nature, vol. 359, no. 6398, pp. 832-835.
SCHIAVO, G., SHONE, C.C., ROSSETTO, O., ALEXANDER, F.C. & MONTECUCCO, C.1993a, "Botulinum neurotoxin serotype F is a zinc endopeptidase specific forVAMP/synaptobrevin", Journal of Biological Chemistry, vol. 268, no. 16, pp. 11516-11519.
SCHIAVO, G., ROSSETTO, O., CATSICAS, S., POLVERINO DE LAURETO, P.,DASGUPTA, B.R., BENFENATI, F. & MONTECUCCO, C. 1993b, "Identification ofthe nerve terminal targets of botulinum neurotoxin serotypes A, D, and E", Journal ofBiological Chemistry, vol. 268, no. 32, pp. 23784-23787.
SCHIAVO, G., MALIZIO, C., TRIMBLE, W.S., POLVERINO DE LAURETO, P., MILAN,G., SUGIYAMA, H., JOHNSON, E.A. & MONTECUCCO, C. 1994, "Botulinum Gneurotoxin cleaves VAMP/synaptobrevin at a single Ala-Ala peptide bond", Journal ofBiological Chemistry, vol. 269, no. 32, pp. 20213-20216.
SCHIAVO, G., MATTEOLI, M. & MONTECUCCO, C. 2000, "Neurotoxins affectingneuroexocytosis.", Physiol Rev, vol. 80, no. 2, pp. 717-766.
SCHOCH, S., DEAK, F., KONIGSTORFER, A., MOZHAYEVA, M., SARA, Y., SUDHOF,T.C. & KAVALALI, E.T. 2001, "SNARE function analyzed in synaptobrevin/VAMPknockout mice.[see comment]", Science, vol. 294, no. 5544, pp. 1117-1122.
SCHULZ, J.B., MATHEWS, R.T., JENKINS, B.G., FERRANTE, R.J., SIWEK, D.,HENSHAW, D.R., CIPOLLONI, P.B., MECOCCI, P., KOWALL, N.W., ROSEN, B.R.& BEAL, M.F. 1995, "Blockade of neuronal nitric oxide synthase protects againstexcitotoxicity in vivo.", J Neurosci, vol. 15, pp. 8419-8429.
SCHWAGERL, A.L., MOHAN, P.S., CATALDO, A.M., VONSATTEL, J.P., KOWALL,N.W. & NIXON, R.A. 1995, "Elevated levels of the endosomal-lysosomal proteinasecathepsin D in cerebrospinal fluid in Alzheimer disease", Journal of neurochemistry, vol.64, no. 1, pp. 443-446.
SCORILAS, A., YOTIS, J., PATERAS, C., TRANGAS, T. & TALIERI, M. 1999, "Predictivevalue of c-erbB-2 and cathepsin-D for Greek breast cancer patients using univariate andmultivariate analysis", Clinical Cancer Research, vol. 5, no. 4, pp. 815-821.
SEDAROUS, M., KERAMARIS, E., O'HARE, M., MELLONI, E., SLACK, R.S., ELCE, J.S.,GREER, P.A. & PARK, D.S. 2003, "Calpains mediate p53 activation and neuronal deathevoked by DNA damage", Journal of Biological Chemistry, vol. 278, no. 28, pp. 26031-26038.

98
SEEHAFER, S.S. & PEARCE, D.A. 2006, "You say lipofuscin, we say ceroid: definingautofluorescent storage material", Neurobiology of aging, vol. 27, no. 4, pp. 576-588.
SEGLEN, P.O. & BOHLEY, P. 1992, "Autophagy and other vacuolar protein degradationmechanisms", Experientia, vol. 48, no. 2, pp. 158-172.
SEIGEL, G.M., LOTERY, A., KUMMER, A., BERNARD, D.J., GREENE, N.D.,TURMAINE, M., DERKSEN, T., NUSSBAUM, R.L., DAVIDSON, B., WAGNER, J. &MITCHISON, H.M. 2002, "Retinal pathology and function in a Cln3 knockout mousemodel of juvenile Neuronal Ceroid Lipofuscinosis (batten disease)", Molecular &Cellular Neurosciences, vol. 19, no. 4, pp. 515-527.
SELKOE, D.J. 2002, "Alzheimer's disease is a synaptic failure", Science, vol. 298, no. 5594,pp. 789-791.
SHENG, J.G., GRIFFIN, W.S., ROYSTON, M.C. & MRAK, R.E. 1998, "Distribution ofinterleukin-1-immunoreactive microglia in cerebral cortical layers: implications forneuritic plaque formation in Alzheimer's disease", Neuropathology & AppliedNeurobiology, vol. 24, no. 4, pp. 278-283.
SHIMIZU, T., HAYASHI, Y., YAMASAKI, R., YAMADA, J., ZHANG, J., UKAI, K.,KOIKE, M., MINE, K., VON FIGURA, K., PETERS, C., SAFTIG, P., FUKUDA, T.,UCHIYAMA, Y. & NAKANISHI, H. 2005, "Proteolytic degradation of glutamatedecarboxylase mediates disinhibition of hippocampal CA3 pyramidal cells in cathepsinD-deficient mice", Journal of neurochemistry, vol. 94, no. 3, pp. 680-690.
SISO, S., FERRER, I. & PUMAROLA, M. 2001, "Juvenile neuroaxonal dystrophy in aRottweiler: accumulation of synaptic proteins in dystrophic axons", ActaNeuropathologica, vol. 102, no. 5, pp. 501-504.
SISO, S., PUIG, B., VAREA, R., VIDAL, E., ACIN, C., PRINZ, M., MONTRASIO, F.,BADIOLA, J., AGUZZI, A., PUMAROLA, M. & FERRER, I. 2002, "Abnormalsynaptic protein expression and cell death in murine scrapie", Acta Neuropathologica,vol. 103, no. 6, pp. 615-626.
SKOFF, R.P. 1976, "Myelin deficit in the Jimpy mouse may be due to cellular abnormalities inastroglia", Nature, vol. 264, no. 5586, pp. 560-562.
SLEAT, D.E., DONNELLY, R.J., LACKLAND, H., LIU, C.G., SOHAR, I., PULLARKAT,R.K. & LOBEL, P. 1997, "Association of mutations in a lysosomal protein with classicallate-infantile neuronal ceroid lipofuscinosis", Science, vol. 277, pp. 1802-1805.
SLEAT, D.E., WISEMAN, J.A., EL-BANNA, M., KIM, K.H., MAO, Q., PRICE, S.,MACAULEY, S.L., SIDMAN, R.L., SHEN, M.M., ZHAO, Q., PASSINI, M.A.,DAVIDSON, B.L., STEWART, G.R. & LOBEL, P. 2004, "A mouse model of classicallate-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2gene results in a loss of tripeptidyl-peptidase I activity and progressiveneurodegeneration", Journal of Neuroscience, vol. 24, no. 41, pp. 9117-9126.
SLEAT, D.E., LACKLAND, H., WANG, Y., SOHAR, I., XIAO, G., LI, H. & LOBEL, P.2005, "The human brain mannose 6-phosphate glycoproteome: a complex mixture

99
composed of multiple isoforms of many soluble lysosomal proteins. Erratum in:Proteomics. 2005 May;5(8):2272.", Proteomics, vol. 5, no. 6, pp. 1520-1532.
SOLLNER, T., BENNETT, M.K., WHITEHEART, S.W., SCHELLER, R.H. & ROTHMAN,J.E. 1993, "A protein assembly-disassembly pathway in vitro that may correspond tosequential steps of synaptic vesicle docking, activation, and fusion", Cell, vol. 75, no. 3,pp. 409-418.
STAGI, M., DITTRICH, P.S., FRANK, N., ILIEV, A.I., SCHWILLE, P. & NEUMANN, H.2005, "Breakdown of axonal synaptic vesicle precursor transport by microglial nitricoxide", Journal of Neuroscience, vol. 25, no. 2, pp. 352-362.
STEFANIS, L., LARSEN, K.E., RIDEOUT, H.J., SULZER, D. & GREENE, L.A. 2001,"Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells inducesalterations of the ubiquitin-dependent degradation system, loss of dopamine release, andautophagic cell death", Journal of Neuroscience, vol. 21, no. 24, pp. 9549-9560.
STEINFELD, R., REINHARDT, K., SCHREIBER, K., HILLEBRAND, M., KRAETZNER,R., BRUCK, W., SAFTIG, P. & GARTNER, J. 2006, "Cathepsin D deficiency isassociated with a human neurodegenerative disorder", American Journal of HumanGenetics, vol. 78, no. 6, pp. 988-998.
STEINMAN, R.M., BRODIE, S.E. & COHN, Z.A. 1976, "Membrane flow during pinocytosis.A stereologic analysis.", J Cell Biol, vol. 68, no. 3, pp. 665-687.
STORCH, S. & BRAULKE, T. 2005, "Transport of Lysosomal Enzymes." in Lysosomes, ed.P. Saftig, 1. edition, Landes Bioscience/Eurekah.com, Georgetown,TX, US, pp. 17-26.
SUDHOF, T.C. 2004, "The synaptic vesicle cycle", Annual Review of Neuroscience, vol. 27,pp. 509-547.
SUOPANKI, J., TYYNELA, J., BAUMANN, M. & HALTIA, M. 1999, "Palmitoyl-proteinthioesterase, an enzyme implicated in neurodegeneration, is localized in neurons and isdevelopmentally regulated in rat brain", Neuroscience letters, vol. 265, no. 1, pp. 53-56.
SUOPANKI, J., LINTUNEN, M., LAHTINEN, H., HALTIA, M., PANULA, P., BAUMANN,M. & TYYNELA, J. 2002, "Status epilepticus induces changes in the expression andlocalization of endogenous palmitoyl-protein thioesterase 1", Neurobiology of disease,vol. 10, no. 3, pp. 247-257.
TAKAHASHI, T., SCHMIDT, P.G. & TANG, J. 1983, "Oligosaccharide units of lysosomalcathepsin D from porcine spleen. Amino acid sequence and carbohydrate structure of theglycopeptides", Journal of Biological Chemistry, vol. 258, no. 5, pp. 2819-2830.
TANAKA, F.H., NOGUCHI, Y., KONO, A., HIMENO, M. & KATO, K. 1991, "Isolation andsequencing of a cDNA clone encoding rat liver lysosomal cathepsin D and the structureof three forms of mature enzymes.", Biochem Biophys Res Commun, vol. 179, pp. 190-196.

100
TANDON, A.K., CLARK, G.M., CHAMNESS, G.C., CHIRGWIN, J.M. & MCGUIRE, W.L.1990, "Cathepsin D and prognosis in breast cancer.[see comment]", New EnglandJournal of Medicine, vol. 322, no. 5, pp. 297-302.
TANG, J. 1979, "Evolution in the structure and function of carboxyl proteases", Molecular &Cellular Biochemistry, vol. 26, no. 2, pp. 93-109.
TARDY, C., TYYNELA, J., HASILIK, A., LEVADE, T. & ANDRIEU-ABADIE, N. 2003,"Stress-induced apoptosis is impaired in cells with a lysosomal targeting defect but is notaffected in cells synthesizing a catalytically inactive cathepsin D", Cell Death &Differentiation, vol. 10, no. 9, pp. 1090-1100.
TARSA, L. & GODA, Y. 2002, "Synaptophysin regulates activity-dependent synapseformation in cultured hippocampal neurons", Proceedings of the National Academy ofSciences of the United States of America, vol. 99, no. 2, pp. 1012-1016.
TERMAN, A. 1995, "The effect of age on formation and elimination of autophagic vacuoles inmouse hepatocytes", Gerontology, vol. 41, no. Suppl 2, pp. 319-326.
TERMAN, A. & BRUNK, U.T. 1998, "Lipofuscin: mechanisms of formation and increasewith age", APMIS, vol. 106, no. 2, pp. 265-276.
THIELE, C., HANNAH, M.J., FAHRENHOLZ, F. & HUTTNER, W.B. 2000, "Cholesterolbinds to synaptophysin and is required for biogenesis of synaptic vesicles.[seecomment]", Nature cell biology, vol. 2, no. 1, pp. 42-49.
THOMAS, L., HARTUNG, K., LANGOSCH, D., REHM, H., BAMBERG, E., FRANKE,W.W. & BETZ, H. 1988, "Identification of synaptophysin as a hexameric channel proteinof the synaptic vesicle membrane", Science, vol. 242, no. 4881, pp. 1050-1053.
TOMIMATSU, Y., IDEMOTO, S., MORIGUCHI, S., WATANABE, S. & NAKANISHI, H.2002, "Proteases involved in long-term potentiation", Life Sciences, vol. 72, no. 4-5, pp.355-361.
TOONEN, R.F. & VERHAGE, M. 2003, "Vesicle trafficking: pleasure and pain from SMgenes", Trends in cell biology, vol. 13, no. 4, pp. 177-186.
TRIMBLE, W.S., COWAN, D.M. & SCHELLER, R.H. 1988, "VAMP-1: a synaptic vesicle-associated integral membrane protein", Proceedings of the National Academy of Sciencesof the United States of America, vol. 85, no. 12, pp. 4538-4542.
TROOST, D., CLAESSEN, N., VAN DEN OORD, J.J., SWAAB, D.F. & DE JONG, J.M.1993, "Neuronophagia in the motor cortex in amyotrophic lateral sclerosis",Neuropathology & Applied Neurobiology, vol. 19, no. 5, pp. 390-397.
TSIAKAS, K., STEINFELD, R., STORCH, S., EZAKI, J., LUKACS, Z., KOMINAMI, E.,KOHLSCHUTTER, A., ULLRICH, K. & BRAULKE, T. 2004, "Mutation of theglycosylated asparagine residue 286 in human CLN2 protein results in loss of enzymaticactivity", Glycobiology, vol. 14, no. 4, pp. 15-5C.

101
TURK, V., KOS, J. & TURK, B. 2004, "Cysteine cathepsins (proteases)--on the main stage ofcancer?[comment]", Cancer Cell, vol. 5, no. 5, pp. 409-410.
TYYNELA, J., SOHAR, I., SLEAT, D.E., GIN, R.M., DONNELLY, R.J., BAUMANN, M.,HALTIA, M. & LOBEL, P. 2000, "A mutation in the ovine cathepsin D gene causes acongenital lysosomal storage disease with profound neurodegeneration", EMBO Journal,vol. 19, no. 12, pp. 2786-2792.
TYYNELA, J., SOHAR, I., SLEAT, D.E., GIN, R.M., DONNELLY, R.J., BAUMANN, M.,HALTIA, M. & LOBEL, P. 2001, "Congenital ovine neuronal ceroid lipofuscinosis--acathepsin D deficiency with increased levels of the inactive enzyme", European Journalof Paediatric Neurology, vol. 5, no. Suppl A, pp. 43-45.
VASSAR, R., BENNETT, B.D., BABU-KHAN, S., KAHN, S., MENDIAZ, E.A., DENIS, P.,TEPLOW, D.B., ROSS, S., AMARANTE, P., LOELOFF, R., LUO, Y., FISHER, S.,FULLER, J., EDENSON, S., LILE, J., JAROSINSKI, M.A., BIERE, A.L., CURRAN,E., BURGESS, T., LOUIS, J.C., COLLINS, F., TREANOR, J., ROGERS, G. &CITRON, M. 1999, "Beta-secretase cleavage of Alzheimer's amyloid precursor proteinby the transmembrane aspartic protease BACE.[see comment]", Science, vol. 286, no.5440, pp. 735-741.
VEIT, M., BECHER, A. & AHNERT-HILGER, G. 2000, "Synaptobrevin 2 is palmitoylated insynaptic vesicles prepared from adult, but not from embryonic brain", Molecular &Cellular Neurosciences, vol. 15, no. 4, pp. 408-416.
VERHAGE, M., MAIA, A.S., PLOMP, J.J., BRUSSAARD, A.B., HEEROMA, J.H.,VERMEER, H., TOONEN, R.F., HAMMER, R.E., VAN DEN BERG, T.K., MISSLER,M., GEUZE, H.J. & SUDHOF, T.C. 2000, "Synaptic assembly of the brain in the absenceof neurotransmitter secretion", Science, vol. 287, no. 5454, pp. 864-869.
VESA, J., HELLSTEN, E., VERKRUYSE, L.A., CAMP, L.A., RAPOLA, J., SANTAVUORI,P., HOFMANN, S.L. & PELTONEN, L. 1995, "Mutations in the palmitoyl proteinthioesterase gene causing infantile neuronal ceroid lipofuscinosis", Nature, vol. 376, no.6541, pp. 584-587.
VIRMANI, T., GUPTA, P., LIU, X., KAVALALI, E.T. & HOFMANN, S.L. 2005,"Progressively reduced synaptic vesicle pool size in cultured neurons derived fromneuronal ceroid lipofuscinosis-1 knockout mice", Neurobiology of disease, vol. 20, no. 2,pp. 314-323.
VON EITZEN, U., EGENSPERGER, R., KOSEL, S., GRASBON-FRODL, E.M., IMAI, Y.,BISE, K., KOHSAKA, S., MEHRAEIN, P. & GRAEBER, M.B. 1998, "Microglia andthe development of spongiform change in Creutzfeld-Jacob disease.", Exp Neurol, vol.57, pp. 246-256.
VON FIGURA, K., SCHMIDT, B., SELMER, T. & DIERKS, T. 1998, "A novel proteinmodification generating an aldehyde group in sulfatases: its role in catalysis and disease",Bioessays, vol. 20, no. 6, pp. 505-510.

102
WALKLEY, S.U., MARCH, P.A., SCHROEDER, C.E., WURZELMANN, S. & JOLLY, R.D.1995, "Pathogenesis of brain dysfunction in Batten disease.", Am J Med Genet., vol. 57,no. 2, pp. 196-203.
WASHBOURNE, P., SCHIAVO, G. & MONTECUCCO, C. 1995, "Vesicle-associatedmembrane protein-2 (synaptobrevin-2) forms a complex with synaptophysin",Biochemical Journal, vol. 305, no. Pt 3, pp. 721-724.
WASHBOURNE, P., THOMPSON, P.M., CARTA, M., COSTA, E.T., MATHEWS, J.R.,LOPEZ-BENDITO, G., MOLNAR, Z., BECHER, M.W., VALENZUELA, C.F.,PARTRIDGE, L.D. & WILSON, M.C. 2002, "Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis.[see comment]", Nature neuroscience,vol. 5, no. 1, pp. 19-26.
WEIMER, J.M., CUSTER, A.W., BENEDICT, J.W., ALEXANDER, N.A., KINGSLEY, E.,FEDEROFF, H.J., COOPER, J.D. & PEARCE, D.A. 2006, "Visual deficits in a mousemodel of Batten disease are the result of optic nerve degeneration and loss of dorsallateral geniculate thalamic neurons", Neurobiology of disease, vol. 22, no. 2, pp. 284-293.
WESLEY, B.R. & MAY, F.E.B. 1996, "Cathepsin D and breast cancer.", Eur J Cancer, vol.32, no. A, pp. 15-24.
WHEELER, R.B., SHARP, J.D., SCHULTZ, R.A., JOSLIN, J.M., WILLIAMS, R.E. &MOLE, S.E. 2002, "The gene mutated in variant late-infantile neuronal ceroidlipofuscinosis (CLN6) and in nclf mutant mice encodes a novel predicted transmembraneprotein", American Journal of Human Genetics, vol. 70, no. 2, pp. 537-542.
WHITAKER, J.N. & RHODES, R.H. 1983, "The distribution of cathepsin D in rat tissuesdetermined by immunocytochemistry", American Journal of Anatomy, vol. 166, no. 4,pp. 417-428.
WIGHTMAN, R.M. & HAYNES, C.L. 2004, "Synaptic vesicles really do kiss and run",Nat Neurosci vol. 7, no 4, pp. 321-322.
WILLE, A., GERBER, A., HEIMBURG, A., REISENAUER, A., PETERS, C., SAFTIG, P.,REINHECKEL, T., WELTE, T. & BUHLING, F. 2004, "Cathepsin L is involved incathepsin D processing and regulation of apoptosis in A549 human lung epithelial cells",Biological chemistry, vol. 385, no. 7, pp. 665-670.
WINTER, E. & PONTING, C.P. 2002, "TRAM, LAG1 and CLN8: members of a novel familyof lipid-sensing domains?", Trends in biochemical sciences, vol. 27, no. 8, pp. 381-383.
WISNIEWSKI, K.E. & KIDA, E. 1992, "Congenital ceroid-lipofuscinosis.[comment]",Pediatric neurology, vol. 8, no. 4, pp. 315.
WITTLIN, S., ROSEL, J., HOFMANN, F. & STOVER, D.R. 1999, "Mechanisms and kineticsof procathepsin D activation", European Journal of Biochemistry, vol. 265, no. 1, pp.384-393.

103
WONG-RILEY, M.T. 1989, "Cytochrome oxidase: an endogenous metabolic marker forneuronal activity.", Trends Neurosci, vol. 12, no. 3, pp. 94-101.
WU, G.S., SAFTIG, P., PETERS, C. & EL-DEIRY, W.S. 1998, "Potential role for cathepsin Din p53-dependent tumor suppression and chemosensitivity", Oncogene, vol. 16, no. 17,pp. 2177-2183.
YELAMANCHILI, S.V., REISINGER, C., BECHER, A., SIKORRA, S., BIGALKE, H.,BINZ, T. & AHNERT-HILGER, G. 2005, "The C-terminal transmembrane region ofsynaptobrevin binds synaptophysin from adult synaptic vesicles", European journal ofcell biology, vol. 84, no. 4, pp. 467-475.
YI, J.H., HOOVER, R., MCINTOSH, T.K. & HAZELL, A.S. 2006, "Early, transient increasein complexin I and complexin II in the cerebral cortex following traumatic brain injury isattenuated by N-acetylcysteine", Journal of neurotrauma, vol. 23, no. 1, pp. 86-96.
YIN, D. 1996, "Biochemical basis of lipofuscin, ceroid, and age pigment-like fluorophores",Free radical biology & medicine, vol. 21, no. 6, pp. 871-888.
YONEZAWA, S., TAKAHASHI, T., WANG, X.J., WONG, R.N., HARTSUCK, J.A. &TANG, J. 1988, "Structures at the proteolytic processing region of cathepsin D", Journalof Biological Chemistry, vol. 263, no. 31, pp. 16504-16511.
YUAN, J., LIPINSKI, M. & DEGTEREV, A. 2003, "Diversity in the mechanisms of neuronalcell death", Neuron, vol. 40, no. 2, pp. 401-413.
ZHU, Y., FEI, J. & SCHWARZ, W. 2005, "Expression and transport function of the glutamatetransporter EAAC1 in Xenopus oocytes is regulated by syntaxin 1A", Journal ofneuroscience research, vol. 79, no. 4, pp. 503-508.





























