Embed Size (px)
Citation preview

Catalytic anti-Selective Asymmetric Hydrogenation of a-Amino /3-Keto Esters through Dynamic Kinetic Resolution
Yasumasa Hamada* and Kazuishi Makino
Graduate School of Pharmaceutical Sciences, Chiba University Ya yoi-cho, Inage-ku, Chiba 263-8522, Japan
(Received July 8, 2008; E-mail: [email protected])
Abstract : Asymmetric hydrogenation of a-amino-f3-keto esters using ruthenium (Ru), rhodium (Rh), and iridium (Ir)-chiral phosphine catalysts proceeds anti-selectively through dynamic kinetic resolution to afford
anti-/3-hydroxy-a-amino acids with high enantiomeric purity , which are important intermediates for the synthesis of medicines and natural products. The mechanistic investigation has revealed that the Ru-cat-alyzed asymmetric hydrogenation takes place through the hydrogenation of the double bond in the enol tau-tomer of the substrate and the Rh- and Ir-catalyzed asymmetric hydrogenations proceed through the reduc-tion of the keto tautomer of the substrate.
1. Introduction
Catalytic asymmetric synthesis is still an important and challenging area in modern organic chemistry.' Among syn-thetic methods for obtaining optically pure products, asym-metric synthesis through dynamic kinetic resolution (DKR) is one of the most efficient synthetic methods for obtaining optically pure products.2 As illustrated in Scheme 1, at the
Scheme 1. Asymmetric hydrogenation of racemic ketones through dynamic kinetic resolution.
reaction accompanied by DKR, the chiral center which exists in a substrate can easily racemize under the reaction condi-tions and finally, all the substrates can convert into a diastereomer. By using this method, optically active products with two or more contiguous stereogenic centers can be syn-thesized in theoretically 100% yield from racemic substrates in a stereocontrolled fashion and a single operation. We have been working on the synthesis of biologically active cyclodep-sipeptides from marine origin. In this research, we needed an efficient method for preparation of anti-,3-hydroxy-a-amino acids, which are common structural units widely found as a component of biologically active natural products.4 Typical examples are shown in Figure 1. There are many reports about the synthesis of these amino acids. Those syntheses mainly utilized Sharpless asymmetric epoxidation and di-hydroxylation, enantioselective and diastereoselective aldol reaction, alkylation of the serinal derivatives, Sharpless asym-metric aminohydroxylation, and asymmetric Strecker synthe-sis as the key step. Some of these methods are stereoselective
Figure 1. Naturally-occurring /3-hydroxy-a-amino acids.
but often require careful and tedious handling, and problems with the use of large-scale processes remain to be solved. We envisaged that, when they can be directly synthesized by cat-alytic asymmetric hydrogenation of a-amino-f3-keto esters through DKR, it would become an ideal method for obtain-ing anti-/3-hydroxy-a-amino acids. Therefore, we decided to investigate the development of such a method. In this article, we focus on our developed anti-selective asymmetric hydro-
genation of a-amino-f3-keto esters through DKR, which enables diastereoselective and enantioselective synthesis of anti-f3-hydroxy-a-amino acids.
2, anti-Selective Asymmetric Hydrogenation using
Ru-Axially Chiral Phosphine Catalysts
The asymmetric hydrogenation of racemic ketones
through dynamic kinetic resolution is an efficient method for
obtaining single enantiomers in theoretically 100% yield,
which was originally reported by Noyori and coworkers in
1989.5 In their report, they achieved highly stereoselective
synthesis of syn-f3-hydroxy-a-amino acids from chirally
labile a-acylamino-,8-keto esters. A typical example is shown
in Scheme 2. This asymmetric hydrogenation is highly effi-
cient but is limited to synthesis of only the syn-ƒÀ-hydroxy-
a-amino acids. Therefore, we set out to develop a direct
method for the synthesis of anti-/3-hydroxy-a-amino acids
by the catalytic asymmetric hydrogenation of a-amino-f3-
keto esters through DKR.
We first deliberated the postulated reaction mechanism in
the Noyori's syn-selective asymmetric hydrogenation as
Vol.66 No.11 2008 (21) 1057

Scheme 2. Noyori's syn-selective asymmetric hydrogenation.
Scheme 3. Direct stereodivergent asymmetric hydrogenation.
shown in Scheme 3. The reaction takes place through the six-
membered cyclic transition state 1 by the chelation between
two carbonyl groups of keto and ester functions to provide
the syn-amino acid. We envisioned that, when the substrate
with protection-free amino function was employed, the
hydrogenation should proceed anti-selectively through the
five-membered cyclic transition state 2 by the chelation
between the amino group and the ester carbonyl function,
and lead to an anti-ƒÀ-hydroxy-a-amino acid. Indeed, the
asymmetric hydrogenation of a-amino-113-keto ester
hydrochloride 3 according to the reaction conditions for the
Noyori's syn-selective asymmetric hydrogenation, Ru-(S)-
BINAP in methanol at 50 •Ž for 48 h, produced anti-f3-
hydroxy-a-amino acid ester 4 in almost perfect diastereose-
lectivity and moderate enantioselectivity (Table 1, entry 1)
Table 1. Optimization of anti-selective asymmetric hydrogenation.`'
a) The hydrogenation was carried out by using the catalyst (3.8-4.6 mol%).
b) Yield in two steps. c) Dertermined by 1H NMR analysis.
d) Determined after N-benzoylation by HPLC analysis.
The result clearly shows that the reaction proceeds through
DKR. With this encouraging result in hand, we extensively
surveyed the optimized conditions for the anti-selective asymmetric hydrogenation. Solvent, temperature, and solubi-lity of the substrate were all found to be important factors for the yield and stereoselectivity of the products. Methylene chloride is the solvent of choice for the enantioselectivity, and i-propanol and n-propanol also give satisfactory results
(entries 4, 5, and 7). However, the chemical yield in methy-lene chloride was poor. It seemed due to the poor solubility of the substrate hydrochloride in the solvent. Fortunately, the change of the ester group was found to affect the chemical
yield. Finally, the benzyl ester afforded the satisfactory chem-ical yield and stereoselectivity (entry 10). Table 2 shows the
Table 2. anti-Selective asymmetric hydrogenation.
a) The hydrogenation was carried out by using the catalyst (3.8-4.6 mol%). b) (R)-MeO-BIPHEP was used instead of (S)-BINAP.c) The reaction was carried out at room temperature.
generality of the anti-selective asymmetric hydrogenation using the optimized conditions. The anti-selective asymmetric
hydrogenation was affected by the bulkiness of C4 sub-stituent. The substrates with a secondary alkyl group, such as cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl sub-
stituent, at the a-position of the ketone carbonyl group were the best ones and afforded the anti-products with high
diastereoselectivities and enantioselectivities (entries 1-8).Especially, the cyclohexyl substrate had high reactivity to the
present hydrogenation and the reaction was completed in 6 heven with a substrate/catalyst ratio of 250 under 30 atm of
hydrogen. The thus-obtained (2R,3R)-2-amino-3-cyclo-hexyl-3-hydroxy-propionic acid is a chiral core of the anti-
HIV substance (GW8731401ONO-4128) with a CCR5 antag-onist activity.8 The reaction of the primary alkyl substrate
1058 (22) J. Synth. Org. Chem., Jpn.

was inferior to the secondary one in enantioselectivity. How-ever, the use of (R)-MeO-BIPHEP instead of BINAP , plus lowering the temperature improved its enantioselectivity
(entry 11). The tert-butyl substrate was also inferior in dias-teroselectivity and enantioselectivity under the standard con-ditions, but the hydrogenation in n-propanol turned out to improve the stereoselectivity, leading the product in 89% yield and 79% ee with 96:4 diastereoselectivity (entries 12 and 13). This result suggests that the racemization rate of the tert-butyl substrate in CH2Cl2 is unsatisfactory and the reaction
proceeds through incomplete kinetic resolution. A few months after our report, Genet and coworkers
have reported Ru-axially chiral phosphine catalyzed anti-
selective asymmetric hydrogenation,9 which is essentially the
same as our anti-selective asymmetric hydrogenation.
Incidentally, the required a-amino-/3-keto ester hydro-chlorides are readily made available, as depicted in Scheme 4,
Scheme 4. Synthetic methods for a-amino-keto esters ,
by the following five methods : (a) the acid hydrolysis of a 4-alkoxycarbonyloxazole derived from a carboxylic anhydride and isocyanoacetic acid ester,6' (b) the formation of the oxime from a J3-keto ester and its reduction,9 (c) the base-mediated N-C acyl migration of a N-t-butoxycarbonyl-N-acylglycine ester followed by acidic deprotection,10 (d) the acylation of the benzophenone ketimine derived from a glycine ester in the presence of a strong base followed by acidic hydrolysis,' 1 and (e) the acylation of a N-acylaminomalonic acid half ester and then deprotection.12
We briefly investigated the mechanism of this unique anti-selective asymmetric hydrogenation. In this hydrogena-tion, the substrate a-amino-/3-keto ester presents itself as keto and enol tautomers through tautomerism. A simple
question is which tautomer is hydrogenated. As for the syn-selective asymmetric hydrogenation, Noyori and coworkers have unambiguously elucidated the mechanism using the deu-terium experiment. As in their experiment, we performed the asymmetric hydrogenation of the deuterio substrate.' 3 As depicted in Scheme 5, when the reaction of 5 proceeds through the mechanism of ketone-reduction, the deuterium at the C2 position should remain in the product 6. On the other hand, the hydrogenation of the enol tautomer 7 should
give the deuterium-free amino acid 8 as the major product. The deuterio substrate 10 was first prepared from the cyclo-hexyl substrate 9 by exposure to an excess amount of deuterio-
Scheme 5. Isotope labeling experiment.
Scheme 6. Deuterium experiment.
methanol as shown in Scheme 6. Avoiding the complication
by isotope exchange, the hydrogenation under the standard
conditions was subject to work-up at 50 •Ž for 1 h. The
products ha and lib were obtained in 46% yield and the
HID ratio was 82:18, which clearly supports the hypothesis
that anti-selective asymmetric hydrogenation takes . place
through the hydrogenation of the enol tautomer. As a control
experiment, the syn-selective hydrogenation of the deuterio
a-N-acylamino-fi-keto ester 12 was carried out under condi-
tions of 50 •Ž and 24 h. The HID ratio of the obtained syn-
amino acids 13a and 13b was 34:66, which is the parallel
result to that of Noyori's experiment and supports the mech-
anism of the reduction of the keto tautomer. Although anti-
and syn-selective asymmetric hydrogenations are catalyzed
with the same Ru-axially chiral phosphine complex, the
above results clearly indicate that both the reactions proceed
through substantially different pathways, disclosing a new
aspect of Ru-chiral phosphine catalyzed asymmetric hydro-
genation.
Vol.66 No.11 2008 (23) 1059

3. anti-Selective Asymmetric Hydrogenation using
Iridium-Chiral Phosphine Catalysts
The asymmetric hydrogenation using the Ru-axially chi-ral phosphine described above, proceeds smoothly to afford anti-f3-hydroxy-a-amino acid esters with excellent diastere-omeric and enantiomeric purity. This hydrogenation, howev-er, was limited to the substrates with an alkyl group at the C4
position. In stark contrast, the attempted reaction of the phenyl substrate 14 using Ru-BINAP at 50 t for 48 h in methanol resulted in the formation of the racemic amino acid in a diastereomeric ratio of 93;7 and 31% yield as indicated in Scheme 7. This disappointingly low level of asymmetric
induction prompted us to examine other transition metals for the arlti-selective asymmetric hydrogenation of the a-amino-
,3-keto ester 14. Interestingly, in addition to the known ruthe-nium catalyst, rhodium (Rh) and iridium (Ir) proved to be
potential catalysts for highly anti-selective hydrogenation through DKR. Therefore, we first carried out the optimiza-tion of the Ir-catalyzed anti-selective asymmetric hydrogena-tion, l4
In this hydrogenation, the method for the preparation of the Ir-catalyst was critical. The Ir-catalyst was generated by mixing [Ir(cod)Cl]2 with an axially chiral phosphine in methy-lene chloride at room temperature for 10 min and used after drying without any further purification. Interestingly, the thus-obtained Ir-catalyst was superior to the in situ generat-ed catalyst in the reaction media and the purified Ir-catalyst
prepared according to the procedure of the Crabtree catalyst. Table 3 shows the results of solvent effects and additive effects examined using the most active Ir-catalyst prepared from [Ir(cod)C1]2 with BINAP as described above. Most sol-vents gave an excellent diastereoselectivity with good enan-tioselectivity. Among them, acetic acid showed almost com-
plete diastereoselectivity although the enantioselectivity was low. We employed this impressive diastereoselectivity of acetic acid. In our efforts to improve the enantioselectivity, we were
pleased to find that the presence of sodium acetate affected the enantioselectivity and reaction rate dramatically. Using these conditions, the hydrogenation was completed in the
presence of 3 mol%) of the catalyst for 3 h. The screening of several chiral phosphines revealed that MeO-BIPHEP with electron-donating groups and a large bite angle was the most effective and the enantioselectivity reached 77% ee. Interest-ingly, when phosphines other than the axially chiral phos-
phines (Norphos, DIOP, and PHOX) were employed, the
Table 3. Optimization of Ir-catalyzed asymmetric hydrogenation.
reaction did not take place. In order to further improve the enantioselectivity, additive effects were reexamined. Finally, we found that the addition of an iodide ion source,'s especial-ly sodium iodide, in the preparation of the Ir catalyst led to the maximum enantioselectivity. This method was then applied to various aromatic substrates. The results are shown in Table 4. The anti-selective asymmetric hydrogenation via DKR using 3 mol'%) of the Ir-(S)-MeO-BIPHEP-I catalyst
Table 4. Ir-catalyzed anti-selective asymmetric hydrogenation
1060 (24) J. Synth . Org . Chem ., Jpn.

proceeded with almost complete diastereoselectivities under
100 atm of hydrogen in the presence of sodium acetate (1
equiv) in acetic acid at 27-30 •Ž. Excellent diastereoselectivi-
ties and enantioselectivities were obtained in all aromatic
substrates. The presence of an electron-donating group(s) in
the aromatic nucleus gave positive effects on the enantioselec-
tivity and reaction rate. In contrast, the presence of an elec-
tron-withdrawing group(s), such as halogen, resulted in slow
reaction and somewhat low enantioselectivity. These results
show that the stereoselectivity of the Ir-catalyzed anti-selec-
tive asymmetric hydrogenation is seriously affected by the
electronic environment of the substrates. In order to investi-
gate the potential of this Ir catalyst, the hydrogenation of
some ketonic substrates was attempted. A,B-keto ester, a-N-
acylamino-/3-keto ester, and a-amino acetophenone gave
poor yields and stereoselectivities, respectively. Nonetheless,
the results of the above asymmetric hydrogenation are note-
worthy, not only because the Ir-catalyzed anti-selective
asymmetric hydrogenation gives the complementary results to
that of the Ru-axially chiral phosphine catalyst, but also
because this hydrogenation through dynamic kinetic resolu-
tion is the first example catalyzed by an Ir catalyst. However,
high hydrogen pressure (100 atm) and a tedious degassing
operation by freeze-thaw cycles in the preparation of the cat-
alyst are essential for a smooth reaction and make it difficult
to carry out this hydrogenation in a practical sense.
Therefore, we decided to investigate the development of a second-generation Ir-catalyst for anti-selective asymmetric
hydrogenation and reexamined an additive to the condition using the first-generation catalyst, Ir-(S)-MeO-BIPHEP-I,
without sodium iodide. The results are shown in Table 5.
Table 5. Optimization of the second-generation Ir-catalyst:'
After several experiments on the additive effects, the reaction was found to be accelerated when sodium tetrakis[3,5-bis(tri-fluoromethyl)phenyl]borate (NaBARF)16"7 was added in the
preparation of the catalyst prior to hydrogenation.'. Disap-pointingly, the enantioselectivity was the same level to that of the first-generation Ir-catalyst. Next, we investigated the effects of hydrogen pressure. Surprisingly, we found an unusual relationship between hydrogen pressure and enan-
tioselectivity. Thus, lowering hydrogen pressure enhanced
enantioselectivity (entries 2-6). Under 4.5 atm of hydrogen
the enantioselectivity was improved to 93% ee. Furthermore,
the reaction proceeded even under 1 atm of hydrogen with
similar stereoselectivities in excellent chemical yield. This sec-
ond-generation Ir-catalyst, Ir-(S)-MeO-BIPHEP-BARF
complex, can be readily prepared by mixing [IrCI(cod)]2, (S)-
MeO-BIPHEP, and NaBARF in methylene chloride at 23 •Ž
for 1 h. The catalyst loading can be lowered from 3 mol% to
0.5 mol% without any loss of the yield and diastereo- and
enantioselectivities. A survey of several chiral phosphines
revealed that (S)-MeO-BIPHEP was most efficient in terms
of chemical yield and enantioselectivity. Interestingly, when
the Ir-PHOX catalyst17 developed by the Pfaltz group was
applied to the asymmetric hydrogenation of the a-amino-/3-
keto ester hydrochloride salt 14, the reaction did not take
place. It is noted that this second-generation Ir-catalyst has
remarkable stability and is robust toward moisture and air. In
the preparation of the Ir-catalyst and assembling a reaction
apparatus, the tedious degassing operation by freeze-thaw
cycles and the handling under an inert gas atmosphere are
not necessary. Furthermore, the asymmetric hydrogenation
using the second-generation Ir-catalyst can be carried out
even using a commercially available balloon filled with hydro-
gen gas. For the examination of the scope and limitations, a
series of aromatic substrates with different substituents were
subjected to hydrogenation under the optimized conditions.
The results are shown in Scheme 8. Excellent diastereoselec-
tivities and enantioselectivities were obtained in all aromatic
substrates. Compared to that of the first-generation Ir-cata-
lyst, the halogen-containing substrates afforded somewhat
superior results with respect to enantioselectivity and reactiv-
ity. Hydrogenation was carried out by using the Ir-(S)-MeO-
BIPHEP-BARF catalyst in the presence of sodium acetate (1
equiv) in acetic acid under 4.5 atm of hydrogen at 23 •Ž for
96 h. The introduction of an electron-withdrawing group at
the para or meta position on the phenyl ring resulted in a
slight decrease of the enantionselectivity, but the anti-selec-
tivity was excellent. This cationic Ir complex was also appli-
cable to heteroaromatic substrates containing a sulfur or oxy-
gen atom. In the case of the substrate with an alkyl group,
such as R = isopropyl, at the C4 position, the reaction took
place with side reaction(s) and gave the product in low yield,
but the setereoselectivity was excellent. Surprisingly, hydro-
genation of the hindered substrate with a tent-butyl group
proceeded efficiently to give the anti-/3-hydroxy-a-amino
acid ester with >99:1 diastereoselectivity in quantitative yield
and 91% ee. It is noted that this result is the highest value for
the tort-butyl substrate and is superior to that of the Ru-
BINAP catalyzed anti-selective hydrogenation which we
developed.
A characteristic of the present hydrogenation using thF Ir-(S)-MeO-BIPHEP-BARF catalyst is the dependence of enantioselectivity on the hydrogen pressure. Figure 2 show the relationship between the hydrogen pressure and the enan-tioselectivity. The enantioselectivity decreased with the increase of hydrogen pressure. In order to elucidate the detailed reaction profile and the mechanism of this uniquE asymmetric hydrogenation, we performed kinetic experiment under one atmospheric pressure of hydrogen. The reactior rate of the hydrogenation had each first-order dependence or
Vol.66 No.11 2008 (25) 1061

Scheme 8. anti-Selective asymmetric hydrogenation using the second-generation Ir-catalyst
Figure 2. Plot of H2 pressure vs ee% for Ir-catalyzed asymmetric
hydrogenation via DKR.
the catalyst, hydrogen pressure, and sodium acetate. However,
the concentration of the substrate did not affect the reaction
rate at all. Next, the relationship between hydrogen pressure
and the initial reaction rate was examined. The results are
summarized in Figure 3. Between one and near 15 atmo-
spheric pressures of hydrogen, the reaction rate was linearly
increased together with raising the hydrogen pressure, show-
ing first-order dependence on the pressure. However, the
pressure between 20 and 40 atm did not cause any effect, which suggests that the hydrogen did not participate in the
Figure 3. Plot of H2 pressure vs initial rate for lr-catalyzed
asymmetric hydrogenation via DKR.
rate-determining step of this reaction. Interestingly, when the
pressure was further increased, the reaction rate increased
again in nonlinear fashion from near 40 atmospheric pres-
sures. These results showing the shift of the rate-determining
step suggest that the present hydrogenation using the second-
generation Ir catalyst would proceed through at least two dif-ferent mechanisms.
To gain further insight into the mechanism of this hydro-
genation via DKR, the preliminary isotope labeling experi-
ments were performed as shown in Scheme 9.20 The hydro-
Scheme 9. Isotope experiment using the second-generation I r-catalyst.
genation of deuterio compound 16 in the presence of sodium acetate in deuterioacetic acid under hydrogen (1 atm) for 3 h at 23 t using the second-generation Ir-catalyst, afforded an almost equal mixture of two deuterio 13-hydroxy-a-amino acids 17a and 17b with a ratio of 53:47 in 17% yield. The D!H ratio at the a-position was 100:0, showing no uptake of the gaseous hydrogen at the a-position during the hydro-
genation. However, surprisingly, the product 17b has a deu-terium at the a-position. A question is where the deuterium is derived from. Gotz has reported an interesting phe-nomenon during the reduction of a ketone using the Ir-aminophosphine catalyst.21 In that report, the hydrogen at the amine in the ligand easily exchanges with gaseous hydro-
gen through the Ir catalyst in the equilibration between the amine complex and the amide complex. Although the reac-tion differs substantially from our hydrogenation, this equilib-ration can occur in our case as shown in Scheme 10 and rationalize the generation of the unusual deuterio product 17b. Nonetheless, this result clearly supports that the Ir-cat-alyzed asymmetric hydrogenation of a-amino-/3-keto esters takes place through the reduction of a ketone double bond to
produce J3-hydroxy-a-amino acid esters with anti-stereo-chemistry.
1062 (26) J . Synth . Org . Chem . , Jpn.

Scheme 10. Deuterium exchange mechanism.
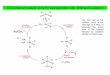
Based on the above knowledge, the catalytic cycles of the
asymmetric hydrogenation using the second-generation Ir-
catalyst under low-pressure conditions can be illustrated as
shown in Scheme 11. The cationic Ir catalyst 18 is formed at
the step of the preparation of the catalyst. Then, hydrogena-
tion of 18 produces the unsaturated Ir catalyst 19 with reduc-
tive elimination of cycooctadiene, which coordinates with the
substrate 14 through the chelation between the oxygen of the
ketone and the nitrogen of the amine function, to yield the
five-membered intermediate 20. Oxidative addition of hydro-
gen to the intermediate generates the dihydride complex 21,
which can easily equilibrate with the amide complex 22. This
equilibration has no effect on the yield and enantioselectivity.
Next, deprotonation from 21 with sodium acetate produces
the amide complex 23. This process would be the rate-deter-
mining step for this hydrogenation from the fact that the
reaction has the first-order dependence on sodium acetate.
Then, the insertion reaction of the carbonyl group in 23, the
reductive elimination of the amide complex , followed by
the ligand exchange reaction together with protonation, fur-nishes the J3-hydroxy-a-amino acid 15 and the regeneration of the real catalyst
4. anti-Selective Asymmetric Hydrogenation using
Rh-Chiral Phosphine Catalysts
In the course of the catalyst screening described above,
we recognized that Rh in combination with chiral phosphines
is also a potential catalyst for this anti-selective asymmetric
hydrogenation. Prior to our investigation, two groups had
examined the Rh-catalyzed DKR approach, which remained
in the stage of preliminary studies due to low conversion
and/or poor enantio- and diastereoselectivities.22 Therefore,
we reexamined optimization for this Rh-catalyzed asymmet-
ric hydrogenation.
We first investigated the effects of several catalyst precur-sors and chiral phosphines using methyl benzoylglycinate hydrochloride (14). Among them, the cationic Rh-catalyst derived from [Rh(nbd)2]BF4 and (R,S)-PPF-P'Bu224 afforded the anti /3-hydroxy-a-amino acid ester in 70% yield with 75% ee and a diastereomeric ratio of 94:6. The bulkier sub-stituent at the benzylic phosphorus of the ligand affected the enantioselectivity. Using the cationic Rh-(R,S)-PPF-P'Bu2 catalyst, we carried out further optimization as shown in Table 6. After some trial and error, we found, surprisingly, that the chemical yield is time-dependent. The reaction time of 30 min was found to be enough for this hydrogenation and
Scheme 11. Catalytic cycle for anti-selective asymmetric hydrogenation using the second-generation Ir-catalyst.
Vol.66 No.11 2008 (27) 1063

Table 6. Rh-catalyzed anti-selective asymmetric hydrogenation
the chemical yield and enantiomeric excess were maximized to 82% yield and 83° ee (entry 3). This result indicates that the hydrogenation is extremely rapid and the product might decompose under the hydrogenation conditions. Pressure of hydrogen had no effect on the enantiomeric excess, but affect-ed the chemical yield (entries 5 and 6). The longer reaction time under moderate hydrogen pressure (5 atm) caused the decomposition of the substrate and had a negative effect on the chemical yield. Using the optimized conditions, we car-ried out asymmetric hydrogenation of several aromatic sub-strates. As shown in Table 7, the reaction time for giving the maximized yield proved to vary for each substrate . Although the yields are moderate, the enantiomeric excess and diastere-oselectivities are satisfactory.
Table 7. anti-Selective asymmetric hydrogenation catalyzed by Rh-(R,S)-PPF-P'Bu7 complex.
In order to elucidate the origin of the extremely high anti-selectivity, we carried out the hydrogenation of the sub-strate with a quaternary carbon at the C2 position, which should reveal whether the reaction proceeds by hydrogenation of the keto form or the enol form. Thus, racemic 2-amino-2-methyl-3-oxo-3-phenylpropionic acid methyl ester (25), a non-enolizable substrate, was subjected to asymmetric hydro-
genation as shown in Scheme 12. The reaction also proceeded under the conditions described above to afford a mixture of two corresponding 18-hydroxy-a-amino acid esters in 17%
yield with a ratio of 73:27 after 1.5 h. The major isomer 26 was found to be 96% ee by the HPLC analysis and confirmed as (2R,3S)-syn by comparisons to the literature value.'-5 Nev-ertheless, this result clearly supports that the Rh-catalyzed asymmetric hydrogenation of the a-amino-,8-keto esters takes place through the reduction of the C=0 double bond to produce J3-hydroxy-a-amino acid esters with anti-stereo-chemistry.
5. Conclusion
The above anti-selective asymmetric hydrogenation of chirally labile a-amino-f3-ketoesters using the Ru-, Ir-, and Rh-chiral phosphine catalysts provides simple and straight-forward access to important anti-18-hydroxy-a-amino acids. These processes are complementary to each other on their scopes and limitations. The Ru-catalyzed asymmetric hydro-
genation of a-amino-f3-ketoesters via DKR is the first example of giving anti-/3-hydroxy-a-amino acids and the Ir-catalyzed asymmetric hydrogenation is the first example of hydrogenation via dynamic kinetic resolution by the Ir cata-lyst. Especially, the second-generation Ir catalyst is robust and can be easily used without care. It is noted that this method does not require special instruments or techniques and can be carried out even by use of the hydrogen-balloon technique. The asymmetric hydrogenation using the Rh-ferrocenylphosphine is not only the first practically successful example of Rh-catalyzed dynamic kinetic resolution, but also demonstrates new potential of Rh-catalyzed asymmetric syn-thesis. The products, anti-,3-hydroxy-a-amino acids, are use-ful as building blocks for the synthesis of various pharmaceu-tical and natural products.
Acknowledgements
We would like to thank all of our coworkers whose names
appear in the references for their dedication, intellectual con-
tribution, and hard work. Our work was partially supported
by Grants-in-Aids from the Ministry of Education, Culture,
Sports, Sciences and Technology, Japan.
References
1) For reviews on asymmetric hydrogenation, see: (a) Ohkuma, T.;
1064 (28) J. Synth . Org . Chem ., Jpn.

Kitamura, M.; Noyori, R. In Ccualytic Asymmetric Synthesis. 2nd ed. (Ed.: I. Ojima), Wiley-VCH, 2000; p 1. (b) Blaser. H.-U.; Malan, C.; Pugin, B.: Spindler, F.; Steiner, H, Studer, M. Adv: Synth. Catal. 2003, 345, 103.
2) For reviews on dynamic kinetic resolution, see: (a) Noyori, R.; Tokunaga. M.; Kitamura, M. Bull. Chem. Soc. Jpn. 1995, 68, 36. (b) Ward, R. S. Tetvhedrat: Asymmetry 1995, 6, 1475. (c) Pel-lissier, H. Tetrabedron 2003, 59, 8291. (d) Vedejs, E.; Jure, M. Angew. Chem. Int. Ed. 2005, 44, 3974. (e) Pellissicr, H. Tetrahe-dron 2008, 64, 1563. (f) Okano, K.; Yasuda, M. H. Speciality Chemicals Maga:ine 2007, 27, 48. (g) Fogassy, E. Organic &
Bianolecular Chemistry 2006, 4, 3011. (h) Baeckvall, J.-E. Cur-rent Opinion in Chemical Biology 2007, 11, 226.
3) For recent examples, see: (a) Hara, S.; Makino, K.; Hamada. Y. Peptide Science 2005, 2006, 39. (b) Hara, S.; Makino, K.:
Hamada, Y. Tetrahedron Lett. 2006, 47, 1081. (c) Makino, K.: Jiang, H.: Suzuki, T.; Hamada, Y. Tetrahedron: Asrnnnetrr 2006, 1
7, 1644. (d) Yoshitomi, Y.; Makino, K.; Hamada, Y. Org. Lett. 2007, 9, 2457. (e) Hara, S.; Nakata, E.: Makino, K.; Hamada.
Y. Peptide Science 2007, 2008, 27. (f) Hamada, Y.; Shioiri, T. Chem. Rev. 2005, 105, 4441.
4) For a review on synthesis of ƒÀ-hydroxy-ƒ¿-amino acids, see:
Makino, K.; Hamada, Y. J. Synth. Org. Chem., Jpn 2005, 63.
1198.
5) Noyori, R.; Ikeda, T.; Ohkuma, T.; Widhalm, M.; Kitamura. M.; Takaya, Akutagawa, S.; Sayo, N.; Saito. T.; Taketomi. T.; Kumobayashi, H. J Am. Chem. Soc. 1989, 111, 9134.
6) For the synthesis of ƒÀ-hydroxy-ƒ¿-amino acids using dynamic
kinetic resolution, see: (a) Ref. 5. (b) Genet, J.-P.; Mallart, S.;
Juge, S. French Patent 8911159, 1989. (c) Mashima, K.; Matsu-
mura, Y.; Kusano, K.; Kumobayashi, H.; Sayo, N.: Hori, Y.;
Ishizaki, T.; Akutagawa, S.; Takaya, H. J. Chem. Soc., Chem.
Commun. 1991, 609. (d) Genet, J.-P.; Pinel, C.: Mallart, S.;
Juge, S.; Thorimbert, S.; Laffitte, J. A. Tetrahedron: Asymmetry
1991, 2, 555. (e) Kitamura, M.: Tokunaga, M.; Noyori, R. J
Am. Cheat Soc. 1993, 115, 144. (f) Mashima, K.; Kusano, K.;
Sato,N.; Matsumura, Y.; Nozaki, K.: Kumobayashi, H.; Sayo,
N.; Hori, Y.: Ishizaki, T.; Akutagawa, S.; Takaya, H. J. Org.
Chem. 1994, 59, 3064. (g) Genet, J.-P.: de Andrade. M. C. C.;
Ratovelomanana-Vidal, V. Tetrahedron Lett. 1995, 36, 2063. (h)
Coulon, E.; de Andrade, M. C. C.; Ratovelomanana-Vidal, V.;
Genet, J.-P. Tetrahedron Lett. 1998, 39, 6467. (i) Makino, K.;
Okamoto, N.; Hara, 0.; Hamada, Y. Tetrahedron: As rnnnetry,
2001, 12, 1757. (j) Mohar, B.; Valleix, A.; Desmurs, J.-R.; Fele-
mez, M.; Wagner, A.; Mioskowski, C. Cheer. Canine. 2001, 2572.
7) (a) Makino, K.; Goto, T.; Hiroki, Y.: Hamada, Y. Angeu. Chem. Int. Ed. 2004, 43, 882. (b) Hamada, Y.; Makino, K. World
Patent W02005/005371 Al, 2005.8) (a) Maeda, K.; Nakata, H.; Ogata, H.; Koh, Y.; Miyakawa, T.;
Mitsuya, H. Current Opinion in Pharmacology, 2004, 4, 447. (b) Maeda, K.: Nakata, H.; Koh, Y.; Miyakawa, T.; Ogata, H.; Takaoka, Y.; Shibayama, S.; Sagawa, K.; Fukushima, D.; Moravek, J.; Koyanagi, Y.; Mitsuya, H. J Vial. 2004, 78, 8654.
9) Modant, C.; Dunkelmann. P.; Ratovelomanana-Vidal, V.; Genet, J.-P. Eur. J Org. Chem. 2004, 3017.
10) Hara, O.: Ito, M.; Hamada. Y. Tetrahedron Lett. 1998. 39. 5537.11) Singh, J.; Gordon, T. D.; Earley, W G.; Morgan, B. A. Tetrahe-
dron Left. 1993, 34, 211.12) Krysan, D. J. Tetrahedron Lett. 1996, 37, 3303.13) Makino, K.; Goto, T.; Maeda, T.; Hamada, Y. in preparation.14) Makino, K.; Hiroki, Y.; Hamada. Y. J Am. Chem Soc. 2005,
127, 5784.15) For additive effects of I-, see: (a) Spindler, F.; Pugin, B.; Blaser..
H.-U. Angeu. Client. hit. Eel. 1990, 29, 558. (b) Morimoto, T.: Nakajima, N.; Achiwa, K. Chem. Phar, n. Bull. 1994, 42, 1951. (c) Morimoto, T.; Nakajima, N.; Achiwa, K. Synlett, 1995, 748. (d) Xiao, D.; Zhang, X. Angel. Client. Int. Ed. 2001, 40, 3425. (e) Wang, W.-B.; Lu, S.-M.: Yang, P.-Y.; Han, X.-W.; Zhou.. Y.-G. J Am. Chem. Soc. 2003, 1255, 10536.
16) Nishida, H.; N. Takada, N.: Yoshiniura, M.: Sonoda, T.: Kobayashi, H. Bull. Cheat. Soc Jprt. 1984, 57, 2600.
17) (a) Lightfoot, A.; Schnider, P.; Pfaltz, A. Angeu . Chem. Int. Ed. 1998, 37, 2897. (b) Pfaltz, A.; Blankenstein, J.; Hilgraf, R.: Hormann, E.; McIntyre, S.; Menges, F.: Sclionleber, M.; Smidt.
S. P.: Wustenberg, B.: Zimmermann, N. Adr. Srntlr. Catal. 2003, 345. 33.
18) Makino, K.; Iwasaki, M.; Hamada, Y. Org Lett. 2006, 8, 4573.19) For hydrogen pressure effects on enantioselectivity using a Ir
complex, see: Perry, M. C.; Cui, X.; Powell, M. T.; Hon, D,-R.: Reibenspies, J. H.: Burgess, K. J. Am. Chem. Soc. 2003, 125, 113.
20) Hamada, Y.; Maeda, T.; Iwasaki, M.; Makino, K. in prepara-tion.
21) Dahlenburg, L.; GOtz, R. Euc J Inorg, Chem. 2004, 888.22) (a) Ishizumi, K.; Terashima, T.; Kojima, A. Jpn Kokai Tokkro
Koho, Jpn, 1990. H02-172956. (b) Genet, J. P.: Pinel, C.; Mal-lart, S.; Juge, S.; Thorimbert, S..; Laffitte, J. A. Tetrahedron: Asrrnnietrr 1991, 2, 555.
23) Makino, K.; Fujii, T.; Hamada, Y. Tetrahedron: Asymmetry 2006, 17, 481.
24) (a) Togni, A.; Breutel, C.; Schnyder, A.; Spindler, F.; Landcrt. H.; Tijiani, A. J Am. Chem. Soc. 1994, 116, 4062. (b) Togni, A. Angetc. Chem. Int. Ed. 1996, 35, 1475.
25) (a) Schollkopf, U.; Groth, U.; Hitt-twig, W. Liehigs, Ann. Chem.
1981, 2407. (b) Avenoza, A.: Cativiela, C.; Corzana, F.; Pere-rina, J. M. Zurbano, M. M. Tetrahedron: Asrnunetry 2000, 11,
2195.
PROFILE
Yasumasa Hamada is professor of Phar-maceutical Chemistry at Chiba Univer-sity. He was born in Hokkaido in 1949 and received his BS degree (1973) for Toyama University and MS degree (1975) from University of' Tokyo. He joined Nagoya City University as an Assistant professor in 1977. After obtaining his Ph.D degree in 1982 and one year postdoctoral work with Prof. E. J. Corey at Harvard University in 1985, he was promoted to Associate Professor at Nagoya City University in 1988. In 1995, he moved to Chiba Uni-versity to take up his present position. He received the PSJ (Pharmaceutical Society of Japan) Award for Young Sci-entists in 1990. His research interests include the development of new meth-ods and reagents for use in organic syn-thesis and total synthesis of biologically active natural products.