Embed Size (px)
Citation preview

Theoretical and Experimental Chemistry, Vol. 40, No. 3, 2004
CATALYSIS BY HYDROGEN CARBONATE AND SILICATE ANIONS OF
THE OXIDATION OF DIETHYL SULFIDE WITH
HYDROGEN PEROXIDE IN AQUEOUS AND
AQUEOUS–ALCOHOLIC MEDIA
UDC 541.127-128:546.215:546:264:547.379.1V. L. Lobachev, V. A. Savelova, and T. M. Prokop’eva
The kinetics of the oxidation of diethyl sulfide (Et2S) with hydrogen peroxide, catalyzed by hydrogen carbonate
and silicate anions, agree with the assumption that peroxymonocarbonate (HCO4
− ) and peroxymonosilicate
( HSiO4
− ) anions are formed as intermediates. The rate of reaction of Et2S with HCO4
− is about 100 times as
fast as with H2O2. Transfer to aqueous–alcoholic solutions leads to an increase in the solubility of Et2S while
retaining the catalytic effects.
Key words: diethyl sulfide, oxidation, kinetics, catalysis, mechanism.
The creation of reagents for the rapid and irreversible destruction of pesticides, chemically active components ofpoisonous substances, derivatives of sulfides, requires a search for new systems which work via oxidative mechanisms.Derivatives of hypochlorous acid which are widely used for the utilization of sulfides, the chloramines are themselvesextremely toxic and aggressive substances [1]. At the present time in so-called “green” technological processes hydrogenperoxide has replaced highly toxic chlorine compounds. However hydrogen peroxide is less effective as an oxidizing agent inthe destruction of sulfides than derivatives of hypochlorous acid. One of the promising methods of activating hydrogenperoxide is to convert it into peroxy acids. Hydrogen carbonates [2], silicates [3], nitrites [4], molybdates [5, 6], phthalates [7],and other compounds may be used as activators for H2O2. The formation of peroxy acids, carried out under mildly basicconditions, are accompanied by the formation highly nucleophilic hydroperoxy anions. Hence such systems, which are mildreagents with a wide range of reactivity, should not only effectively oxidize sulfides but should also destroy toxic esters andacid chlorides of phosphoric acid by a nucleophilic mechanism. From this point of view the H2O2–activator systems are uniquebecause they can be used for the decomposition not only of sulfides but also ecotoxicant substrates belonging to differentclasses of organic compounds.
The objective of the present work was to study the reactivity of hydrogen peroxide in water and aqueous–alcoholicmedia with variable composition, to study the effect of activators, hydrogen carbonate and silicate, on the rate of oxidation of
diethyl sulfide (Et2S), and also to choose the optimal conditions for the decomposition of Et2S as a model for 2,2′-dichloroethylsulfide (mustard gas, yperite).
The use of aqueous–alcoholic mixture not only allows the rate and selectivity of the oxidation of Et2S to be regulated,but it also considerably increases its solubility. This is particularly important because toxic sulfides are frequently practicallyinsoluble in water.
0040-5760/04/4003-0161 ©2004 Plenum Publishing Corporation 161
___________________________________________________________________________________________________L. M. Litvinenko Institute of Physical Organic and Coal Chemistry, National Academy of Sciences of Ukraine, 70 R.
Luxemburg Vul., 83114 Donets’k, Ukraine. E-mail: [email protected]. Translated from Teoreticheskaya iÉksperimental’naya Khimiya, Vol. 40, No.3, pp. 157-161, May-June, 2004. Original article submitted March 3, 2004.

EXPERIMENTAL
Starting materials and reagents. Diethyl sulfide (Et2S) was synthesized by a known method [8]. To prepare theworking solutions alcohols purified according to [9], 35% hydrogen peroxide solution, ammonium hydrogen carbonate(chemically pure) and doubly distilled water were used. Sodium silicate (Na2SiO3·9H2O) was prepared by a known method[10].
Kinetics. The reaction of diethyl sulfide with hydrogen peroxide in aqueous and aqueous–alcoholic media was studied
by the kinetic distribution method [11] by loss of the substrate from the gas phase with [H2O2] >> [Et2S] at 25 °C:
− =1
2
2
[ ]
[ ][ ]
Et S
Et SOx
g
gd
dk
τ λ = +k[ ]/( )Ox 1 αλ
where kλ is the measured rate of loss of the substrate from the gas phase with the ratio λ = Vg/Vl of the volume of gas and
solution in the reactor; k is the liquid phase second order rate constant; α = [Et2S]g / [Et2S]l, the equilibrium coefficient for thedistribution of the substrate between the gas and solution. To measure the concentration of the substrate in the gas phase GLC
was used with toluene, which is inert under these conditions, as internal standard. The value of α, which is necessary for the
calculation of the liquid phase rate constant, was found from the dependence of 1/kλ on λ using Eq. (1).The working solutions were kept for 30 min before introduction of the substrate in order to establish the equilibrium:
(2)HCO3− + H2O2 ↔ HCO4
− + H2O
for studying the oxidation of Et2S in the H2O2/NH4HCO3 system.Results were worked up using the Origin 5.0 program with minimization of the sums of squares of the difference
between the experimental and calculated results.
RESULTS AND DISCUSSION
Reactions in aqueous solutions. In aqueous solutions (phosphate buffer, µ = 0.2) in the range of concentrationstudied, [H2O2] = 0.01-0.1 mol/L, the consumption of Et2S was first order in substrate (Eq. (1) was followed up to a conversionof substrate > 80%) and first order in peroxide. The dependence of the second order rate constant (kH O2 2
) on pH is shown inFig. 1. Since for the acid dissociation for hydrogen peroxide:
H2O2 ↔Ka
HO2− + H+
pKa = 11.5 [12], both H2O2 and HO2− , which are strong two electron oxidants (E 0 = 1.77 and 0.87 V respectively [12]), may be
considered to be potentially active in the oxidation of Et2S under the studied conditions:
(4)Et2S + H2O2 → Et2S=O + H2O,
(5)Et2S + HO2− → Et2S=O + HO–.
If only H2O2 is active, then the dependence of the observed rate constant on acidity is described by the equation
(6)kH O2 2= k4[H
+]/([H+] + Ka).
162
(1)
(3)
If loss of diethyl sulfide occurs via routes (4) and (5), the expression for kH O2 2should have the form
(7)kH O2 2= (k4[H
+] + k5Ka)/([H+] + Ka).
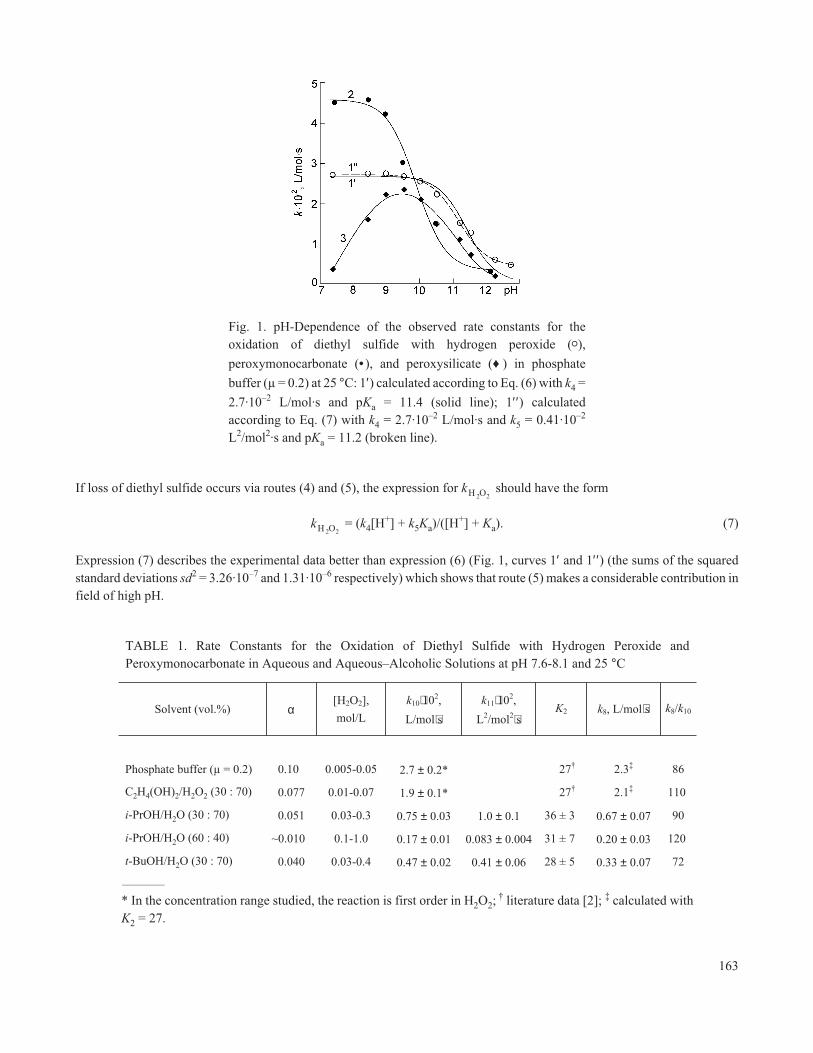
Expression (7) describes the experimental data better than expression (6) (Fig. 1, curves 1′ and 1′′ ) (the sums of the squaredstandard deviations sd2 = 3.26·10–7 and 1.31·10–6 respectively) which shows that route (5) makes a considerable contribution infield of high pH.
163
TABLE 1. Rate Constants for the Oxidation of Diethyl Sulfide with Hydrogen Peroxide andPeroxymonocarbonate in Aqueous and Aqueous–Alcoholic Solutions at pH 7.6-8.1 and 25 °C
Solvent (vol.%) α[H2O2],
mol/L
k10⋅102,
L/mol⋅sk11⋅102,
L2/mol2⋅sK2 k8, L/mol⋅s k8/k10
Phosphate buffer (µ = 0.2) 0.10 0.005-0.05 2.7 ± 0.2* 27† 2.3‡ 86
Ñ2Í4(ÎÍ)2/Í2Î2 (30 : 70) 0.077 0.01-0.07 1.9 ± 0.1* 27† 2.1‡ 110
i-PrOH/H2O (30 : 70) 0.051 0.03-0.3 0.75 ± 0.03 1.0 ± 0.1 36 ± 3 0.67 ± 0.07 90
i-PrOH/H2O (60 : 40) ~0.010 0.1-1.0 0.17 ± 0.01 0.083 ± 0.004 31 ± 7 0.20 ± 0.03 120
t-BuOH/H2O (30 : 70) 0.040 0.03-0.4 0.47 ± 0.02 0.41 ± 0.06 28 ± 5 0.33 ± 0.07 72
________
* In the concentration range studied, the reaction is first order in H2O2;† literature data [2]; ‡ calculated with
K2 = 27.
Fig. 1. pH-Dependence of the observed rate constants for theoxidation of diethyl sulfide with hydrogen peroxide (o),
peroxymonocarbonate (• ), and peroxysilicate (♦ ) in phosphate
buffer (µ = 0.2) at 25 °C: 1′) calculated according to Eq. (6) with k4 =
2.7·10–2 L/mol·s and pKa = 11.4 (solid line); 1′′ ) calculatedaccording to Eq. (7) with k4 = 2.7·10–2 L/mol·s and k5 = 0.41·10–2
L2/mol2·s and pKa = 11.2 (broken line).

Addition of HCO3− ([HCO3
− ] < [H2O2]) leads to an increase in the rate of oxidation of diethyl sulfide. In the range pH
7-9 the catalytic effect of hydrogen carbonate kCO = (k – kH O2 2)/ [HCO3
− ] is independent of the acidity of the media, but it drops
sharply at pH >9 (Fig. 1, curve 2).
According to [2], the catalytic activity of hydrogen carbonate is connected to the formation of peroxymonocarbonate
HCO4− by Eq. (2). Peroxymonocarbonate is a strong two electron oxidizing agent (E 0 = 1.8 V [2]) and oxidizes sulfides to
sulfoxides:
Taking (2) into account, the expression for rate constant for the catalytic route has the form
(9)kCO = k8[H2O2]/([H2O] + K2[H2O2]).
In the concentration ranges studied ([H2O2] = 0.005-0.05 mol/L and [NH4HCO3] = 0.001-0.01 mol/L) the catalyticreaction is first order in both peroxide and hydrogen carbonate, but taking into account the data from [2] that the value for theequilibrium constant does not depend on the composition of the solvent and is the same (K2 = 27) for aqueous andaqueous–alcoholic media, the value of k8 = 2.3 L/mol·s can be calculated (Table 1). According to [2] the explanation of the
decrease in the rate of the catalytic pathway at pH > 9 is the deprotonation of the anion HCO4− , the value of the pKa for which is
close to the corresponding to the value for the anion HSO5− (9.4).
Silicate may be another potential activator of hydrogen peroxide. A number of reactions of H2O2 which are catalyzed
by addition of Na2SiO3 are known, for example the decomposition of benzyl in t-BuOH/H2O systems at pH > 7 and 45 °C [3].The catalytic effect and the characteristics of the kinetics of this reaction were explained by the suggestion of equilibriumformation of peroxysilicate from H2O2 and Na2SiO3.
We have found that addition of sodium silicate (0.01 mol/L) at pH 7.5-11.5 leads to an increase in the rate of oxidationof Et2S, but the observed catalytic effect kSi = k(k – kH O2 2
)/[Na2SiO3] in this case is considerably lower than hydrogencarbonate (kSi < kCO, Fig. 1, curve 3). An extreme relation for the dependence of kSi on the acidity was observed for the
mechanism catalyzed by silicate with a maximum at pH ≈ 9.5 (Fig. 1, curve 3). Taking into account literature data [3] and byanalogy with reactions catalyzed by hydrogen carbonate, it may be assumed that the catalytic effect of Na2SiO3 is connected in
this case with the intermediate formation of the peroxymonosilicate anion HOOSiO2− anion which is the immediate active
particle in the catalytic pathway for the oxidation of Et2S.Reactions in aqueous–alcoholic media. Transfer into aqueous–alcoholic media led to an increase in the solubility of
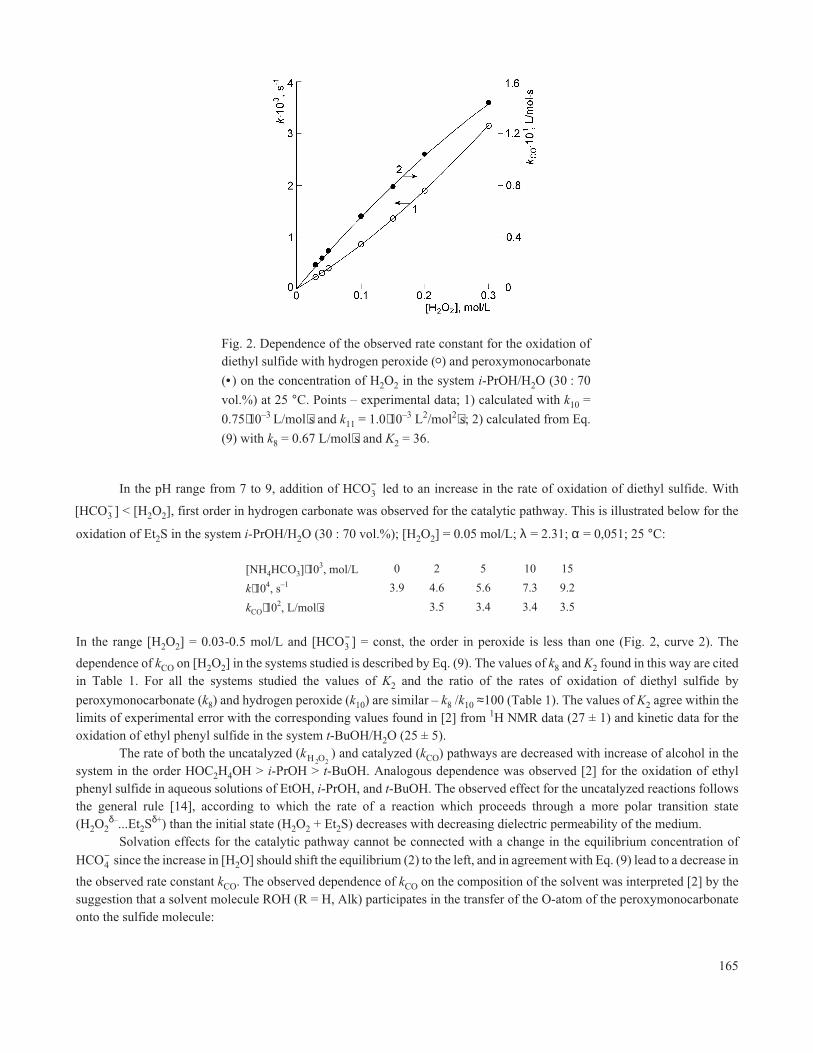
diethyl sulfide (the values of 1/α, Table 1). In the absence of hydrogen carbonate the reaction of diethyl sulfide with hydrogenperoxide is first order in substrate but the observed order in H2O2 is greater than one (Fig. 2, curve 1). This agrees with literaturedata [2] on the oxidation of ethyl phenyl sulfide and 2-chloroethyl phenyl sulfide in aqueous–alcoholic media and indicates thepresence of parallel reactions which are first and second order in H2O2
(10)Et2S + H2O2 → Et2S=O + H2O,
(11)Et2S + 2H2O2 → Et2S=O + H2O2 + H2O.
According to [2] the second order in H2O2 observed in the oxidation of sulfides in aqueous–alcoholic and aprotic mediais connected with the acid-catalytic (electrophilic) effect of the second molecule of hydrogen peroxide, the acidity of which(pKa = 11.5 [12]) is considerably greater than that of H2O (pKa = 15.7) and ROH (pKa > 15, [2, 13]). The values of k10 and k11
found from the dependence of the observed rate constant (k) on [H2O2] are cited in Table 1.
164
O COH
O–
O+ S
Et
EtS
Et
EtO + HCO3.– (8)
In the pH range from 7 to 9, addition of HCO3− led to an increase in the rate of oxidation of diethyl sulfide. With
[HCO3− ] < [H2O2], first order in hydrogen carbonate was observed for the catalytic pathway. This is illustrated below for the
oxidation of Et2S in the system i-PrOH/H2O (30 : 70 vol.%); [H2O2] = 0.05 mol/L; λ = 2.31; α = 0,051; 25 °C:
[NH4HCO3]⋅103, mol/L 0 2 5 10 15
k⋅104, s–1 3.9 4.6 5.6 7.3 9.2
kCO⋅102, L/mol⋅s 3.5 3.4 3.4 3.5
In the range [H2O2] = 0.03-0.5 mol/L and [HCO3− ] = const, the order in peroxide is less than one (Fig. 2, curve 2). The
dependence of kCO on [H2O2] in the systems studied is described by Eq. (9). The values of k8 and K2 found in this way are citedin Table 1. For all the systems studied the values of K2 and the ratio of the rates of oxidation of diethyl sulfide by
peroxymonocarbonate (k8) and hydrogen peroxide (k10) are similar – k8 /k10 ≈100 (Table 1). The values of K2 agree within thelimits of experimental error with the corresponding values found in [2] from 1H NMR data (27 ± 1) and kinetic data for theoxidation of ethyl phenyl sulfide in the system t-BuOH/H2O (25 ± 5).
The rate of both the uncatalyzed (kH O2 2) and catalyzed (kCO) pathways are decreased with increase of alcohol in the
system in the order HOC2H4OH > i-PrOH > t-BuOH. Analogous dependence was observed [2] for the oxidation of ethylphenyl sulfide in aqueous solutions of EtOH, i-PrOH, and t-BuOH. The observed effect for the uncatalyzed reactions followsthe general rule [14], according to which the rate of a reaction which proceeds through a more polar transition state(H2O2
δ–...Et2Sδ+) than the initial state (H2O2 + Et2S) decreases with decreasing dielectric permeability of the medium.
Solvation effects for the catalytic pathway cannot be connected with a change in the equilibrium concentration of
HCO4− since the increase in [H2O] should shift the equilibrium (2) to the left, and in agreement with Eq. (9) lead to a decrease in
the observed rate constant kCO. The observed dependence of kCO on the composition of the solvent was interpreted [2] by thesuggestion that a solvent molecule ROH (R = H, Alk) participates in the transfer of the O-atom of the peroxymonocarbonateonto the sulfide molecule:
165
Fig. 2. Dependence of the observed rate constant for the oxidation ofdiethyl sulfide with hydrogen peroxide (o) and peroxymonocarbonate
(• ) on the concentration of H2O2 in the system i-PrOH/H2O (30 : 70
vol.%) at 25 °C. Points – experimental data; 1) calculated with k10 =
0.75⋅10–3 L/mol⋅s and k11 = 1.0⋅10–3 L2/mol2⋅s; 2) calculated from Eq.
(9) with k8 = 0.67 L/mol⋅s and K2 = 36.

The role of the solvent during the oxidation process is that water or an alcohol serves as a proton donor which facilitates theseparation of the hydrogen carbonate anion from the peroxymonocarbonate. The important role of the proton transfer from thesolvent is confirmed by the presence of a solvation kinetic isotope effect (kH /kD = 1.5) for the oxidation of ethyl phenyl sulfidein EtOH/H2O solutions [2].
So the hydrogen carbonate anion is an effective catalyst for the oxidation of organic sulfides – the second order rate
constant for HCO4− (k8) is approximately 100 times as great as for H2O2 (k10). Transfer from aqueous to aqueous–alcoholic
solutions makes possible an increase in the solubility of sulfides which are frequently practically insoluble in water. At thesame time a noticeable decrease in the rate of oxidation of Et2S is observed. In this connection the ethylene glycol/water systemis of particular interest, for which the decrease in rate in comparison with aqueous solutions is negligible, and the catalyticeffect of hydrogen carbonate is sufficiently great.
This research was carried out with partial support of grant UC2-2489-DO-03 from the Civil Research and
Development Fund of the USA (CRDF). The authors thank Professor C. A. Bunton (University of California, Santa Barbara,
USA) for helpful discussion of the results.
REFERENCES
1. V.-C. Yang, J. A. Baker, and J. R. Ward, Chem. Rev., 92, 1729-1743 (1992).2. D. E. Richardson, H. Yao, K. M. Frank, and D. A. Bennett, J. Am. Chem. Soc., 122, No. 8, 1729-1739 (2000).3. A. Blaschette, Z. Anorg. Allgem. Chem., 384, No. 2, 177-183 (1971).4. S. Vayssié and H. Elias, Angew. Chem., 37, No. 15, 2088-2090 (1998).5. M. Chiarini, N. D. Gillitt, and C. A. Bunton, Langmuir, 18, 3836-3842 (2002).6. C. A. Bunton and N. D. Gillitt, J. Phys. Org. Chem., 15, No. 1, 29-36 (2002).7. B. Segues, E. Peres, J. Rico-Lattes, et al., Bull. Soc. Chim. France,133, 925-937 (1996).8. Weygand-Kil’getag, Experimental Methods in Organic Chemistry [Russian translation], Khimiya, Moscow (1969).9. A. Weissberger, E. Proskauer, J. Riddick, and E. Tups, Organic Solvents. Physical Properties and Methods of
Purification [Russian translation], Izd. Inostr. Lit., Moscow (1958).10. Yu. V. Karyakin and I. I. Angelov, Pure Chemical Substances [in Russian], Khimiya, Moscow (1974).11. E. S. Rudakov, Reactions of Alkanes with Oxidants, Metal Complexes, and Radicals in Solutions [in Russian], Nauk.
Dumka, Kiev (1985).12. F. A. Cotton and G. Wilkinson, Modern Inorganic Chemistry [Russian translation], Mir, Moscow (1969), Vol. 2.13. C. K. Saures, W. P. Jencks, and S. Groh, J. Am. Chem. Soc., 97, No. 16, 5546-5553 (1975).14. J. Gordon, Organic Chemistry of Electrolyte Solutions [Russian translation], Mir, Moscow (1979).
166
HCO4 + ROH + Et2S– O CO
O–
OH
H OR
SEt2
Et2S O + HCO3 + ROH.–(12)



















![Hydrogen Sulfide in Aqueous Solvents - SRDATA at NIST · PDF fileHydrogen Sulfide in Aqueous Solvents 3 COHPONENTS: 1. Hydrogen sulfide; H2S; [7783-06-4] 2. Water; H20; [7732-18-5]](https://img.pdfslide.us/doc/110x75/5ab843c37f8b9a684c8c99d0/hydrogen-sulfide-in-aqueous-solvents-srdata-at-nist-sulfide-in-aqueous-solvents.jpg)









