Embed Size (px)
Citation preview

nature chemical biology | VOL 9 | JUNE 2013 | www.nature.com/naturechemicalbiology 351
news & views
BCL-XL is an anti-apoptotic member of the B-cell lymphoma 2 (BCL-2) protein family that governs
cellular commitment to apoptosis at the mitochondria and prevents unwanted cell death1. Pathologic overexpression of the anti-apoptotic BCL-2 family proteins subverts the natural apoptotic response and contributes to tumor initiation and progression as well as to resistance to chemotherapy. Consequently, anti-apoptotic BCL-2 proteins have become attractive targets for anticancer therapy2,3. The development of specific inhibitors for this attractive target class has been challenging. Lessene et al.4 report the discovery of a new and selective inhibitor of BCL-XL.
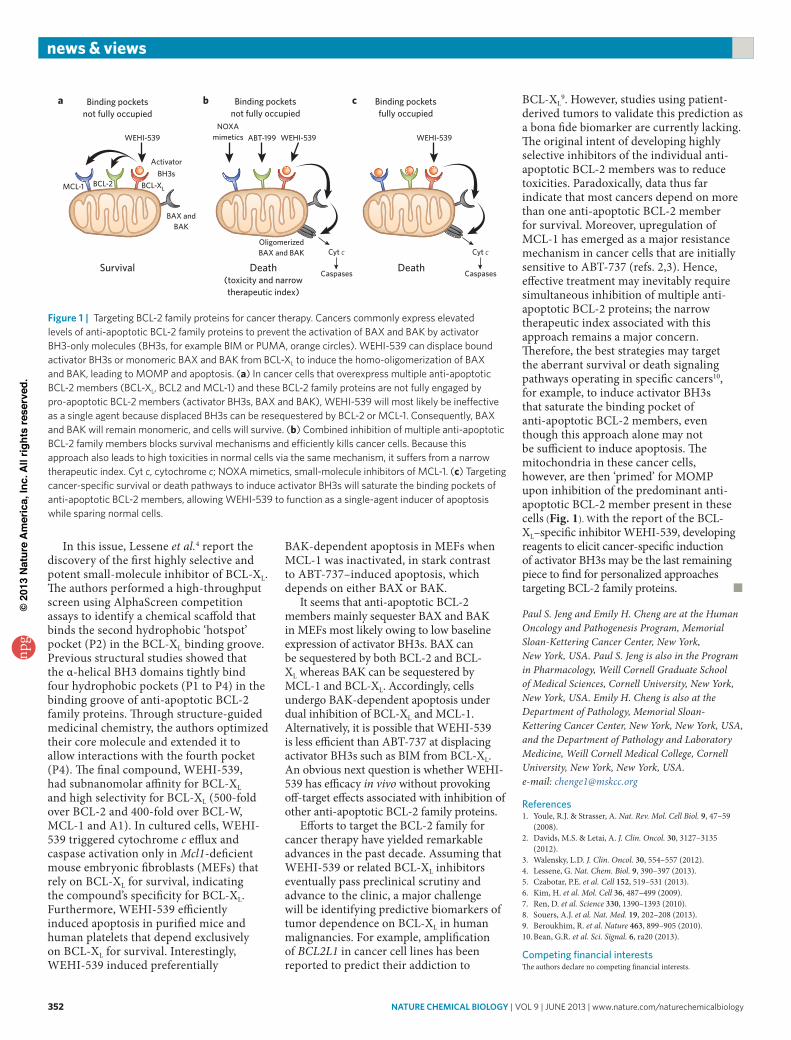
Multiple apoptotic signals culminate in permeabilizing the mitochondrial outer membrane (MOM), resulting in the release of apoptogenic factors such as cytochrome c and SMAC to activate caspases1. BAX and BAK are essential effectors responsible for mitochondrial outer membrane permeabilization (MOMP), whereas BCL-2, BCL-XL and MCL-1 preserve mitochondrial integrity (Fig. 1). The third BCL-2 subfamily, BH3-only proteins (BH3s), promote apoptosis by either
activating BAX and BAK (activator BH3s) or inactivating BCL-2, BCL-XL and MCL-1 (inactivator BH3s)1,5. In healthy cells, BAX and BAK are generally monomeric and inactive either through autoinhibition (BAX) or because they are held in check by associated proteins such as anti-apoptotic BCL-2 proteins. In response to apoptotic signals, activator BH3s, including truncated BID (a BH3-interacting domain death agonist), BIM and PUMA, bind BAX and BAK to induce structural reorganization and oligomerization of BAX and BAK at the MOM for MOMP5–7. Anti-apoptotic BCL-2 family proteins prevent apoptosis by sequestering either activator BH3s or monomeric BAX and BAK, thus preventing the oligomerization and activation of BAX and BAK1. In both scenarios, the protein-protein interactions are mediated by the hydrophobic groove of anti-apoptotic BCL-2 members and by the BH3 domain of proapoptotic BCL-2 members. The ability of anti-apoptotic BCL-2 proteins to sequester proapoptotic BCL-2 members can be further modulated by ‘inactivator’ BH3s through high-affinity competitive binding1.
Cancer cells often overexpress the anti-apoptotic BCL-2 family proteins through
mutations such as chromosomal translocation involving BCL2 or amplification of BCL2L1 (BCL-XL) and MCL1 (refs. 2,3). Given that the hydrophobic binding groove of anti-apoptotic BCL-2 family proteins is critical for their prosurvival function, targeted inhibition of this groove can induce apoptosis in cancer cells by liberating the trapped activator BH3s or multidomain BAX and BAK. ABT-737, the first successful inhibitor of anti-apoptotic BCL-2 family proteins, and its oral bioavailable analog ABT-263 (Navitoclax) function like BAD mimetics that bind and inhibit BCL-2, BCL-XL and BCL-W but not MCL-1 or A1. Activity of ABT-263 was observed in the clinic for lymphoid malignancies, especially in BCL-2–addicted chronic lymphocytic leukemia2,3. The BCL-2–specific inhibitor ABT-199 was developed because inhibition of BCL-XL by ABT-263 caused dose-dependent thrombocytopenia due to the dependence of platelets on BCL-XL for survival8. By analogy, the development of selective BCL-XL inhibitors should be useful for treating tumors that are addicted to BCL-XL while avoiding toxicities associated with BCL-2 inhibition.
CANCER THERAPEUTICS
Pulling the plug on BCL-XLBCL-XL and other anti-apoptotic BCL-2 family proteins serve as guardians of cellular survival. A new selective small-molecule inhibitor of BCL-XL is developed to promote apoptosis in cancers that are addicted to BCL-XL.
Paul S Jeng & Emily H Cheng
adsorption of the CBM occurs in an ordered way. The authors then interpret lower W as indicating a preference for recognition of amorphous regions of the substrate and higher W as indicating a preference for crystalline regions of the substrate (Fig. 1b). This evaluation is a critical advance of their work because previous classical single-molecule techniques have not been able to identify the preferred binding regions of CBMs. Fox et al.1 further used the W values to design fusion cellulases with different CBMs, and they compared the degrees of synergism for all combinations of the fusion proteins. Interestingly, there is an apparent optimum value of DW (the difference between the W values of two CBMs) to obtain maximum synergy, indicating that higher synergy is obtained when enzymes work on tasks that are separate but not too dissimilar.
Recent real-time observation of morphological changes during enzymatic decomposition of plant cell walls8 and single-molecule analysis of cellulases9,10 have been helpful in uncovering the molecular mechanisms of individual enzymes. However, the mechanisms of synergistic hydrolysis by cocktails of cellulases have been a fairly untrodden field for cellulase researchers. Now, the work of Fox et al.1 provides a clue of the synergy by providing a direct visualization of this process and offering guidance as to how to prepare effective enzyme cocktails. Thus, the earlier approaches might be characterized as ‘evaluating individual performance’, whereas the new work gives us a clue as to ‘how to improve teamwork’ and takes a big step toward developing effective biorefineries. ■
Kiyohiko Igarashi is at the Department of Biomaterial Sciences, Graduate School of Agricultural and Life Sciences, The University of Tokyo, Tokyo, Japan. e-mail: [email protected]
Published online 7 April 2013; doi:10.1038/nchembio.1237
References1. Fox, J.M. et al. Nat. Chem. Biol. 9, 356–361 (2013).2. Himmel, M.E. et al. Science 315, 804–807 (2007). 3. Vaaje-Kolstad, G. et al. Science 330, 219–222 (2010). 4. Reese, E.T. et al. J. Bacteriol. 59, 485–497 (1950).5. Teeri, T.T. et al. Biochem. Soc. Trans. 26, 173–178 (1998).6. Cantarel, B.L. et al. Nucleic Acids Res. 37, D233–D238
(2009). 7. Betzig, E. et al. Science 313, 1642–1645 (2006). 8. Ding, S.-Y. et al. Science 338, 1055–1060 (2012). 9. Igarashi, K. et al. J. Biol. Chem. 284, 36186–36190 (2009). 10. Igarashi, K. et al. Science 333, 1279–1282 (2011).
Competing financial interestsThe author declares no competing financial interests.
npg
© 2
013
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.
352 nature chemical biology | VOL 9 | JUNE 2013 | www.nature.com/naturechemicalbiology
news & views
In this issue, Lessene et al.4 report the discovery of the first highly selective and potent small-molecule inhibitor of BCL-XL. The authors performed a high-throughput screen using AlphaScreen competition assays to identify a chemical scaffold that binds the second hydrophobic ‘hotspot’ pocket (P2) in the BCL-XL binding groove. Previous structural studies showed that the α-helical BH3 domains tightly bind four hydrophobic pockets (P1 to P4) in the binding groove of anti-apoptotic BCL-2 family proteins. Through structure-guided medicinal chemistry, the authors optimized their core molecule and extended it to allow interactions with the fourth pocket (P4). The final compound, WEHI-539, had subnanomolar affinity for BCL-XL and high selectivity for BCL-XL (500-fold over BCL-2 and 400-fold over BCL-W, MCL-1 and A1). In cultured cells, WEHI-539 triggered cytochrome c efflux and caspase activation only in Mcl1-deficient mouse embryonic fibroblasts (MEFs) that rely on BCL-XL for survival, indicating the compound’s specificity for BCL-XL. Furthermore, WEHI-539 efficiently induced apoptosis in purified mice and human platelets that depend exclusively on BCL-XL for survival. Interestingly, WEHI-539 induced preferentially
BAK-dependent apoptosis in MEFs when MCL-1 was inactivated, in stark contrast to ABT-737–induced apoptosis, which depends on either BAX or BAK.
It seems that anti-apoptotic BCL-2 members mainly sequester BAX and BAK in MEFs most likely owing to low baseline expression of activator BH3s. BAX can be sequestered by both BCL-2 and BCL-XL whereas BAK can be sequestered by MCL-1 and BCL-XL. Accordingly, cells undergo BAK-dependent apoptosis under dual inhibition of BCL-XL and MCL-1. Alternatively, it is possible that WEHI-539 is less efficient than ABT-737 at displacing activator BH3s such as BIM from BCL-XL. An obvious next question is whether WEHI-539 has efficacy in vivo without provoking off-target effects associated with inhibition of other anti-apoptotic BCL-2 family proteins.
Efforts to target the BCL-2 family for cancer therapy have yielded remarkable advances in the past decade. Assuming that WEHI-539 or related BCL-XL inhibitors eventually pass preclinical scrutiny and advance to the clinic, a major challenge will be identifying predictive biomarkers of tumor dependence on BCL-XL in human malignancies. For example, amplification of BCL2L1 in cancer cell lines has been reported to predict their addiction to
BCL-XL9. However, studies using patient-
derived tumors to validate this prediction as a bona fide biomarker are currently lacking. The original intent of developing highly selective inhibitors of the individual anti-apoptotic BCL-2 members was to reduce toxicities. Paradoxically, data thus far indicate that most cancers depend on more than one anti-apoptotic BCL-2 member for survival. Moreover, upregulation of MCL-1 has emerged as a major resistance mechanism in cancer cells that are initially sensitive to ABT-737 (refs. 2,3). Hence, effective treatment may inevitably require simultaneous inhibition of multiple anti-apoptotic BCL-2 proteins; the narrow therapeutic index associated with this approach remains a major concern. Therefore, the best strategies may target the aberrant survival or death signaling pathways operating in specific cancers10, for example, to induce activator BH3s that saturate the binding pocket of anti-apoptotic BCL-2 members, even though this approach alone may not be sufficient to induce apoptosis. The mitochondria in these cancer cells, however, are then ‘primed’ for MOMP upon inhibition of the predominant anti-apoptotic BCL-2 member present in these cells (Fig. 1). With the report of the BCL-XL–specific inhibitor WEHI-539, developing reagents to elicit cancer-specific induction of activator BH3s may be the last remaining piece to find for personalized approaches targeting BCL-2 family proteins. ■
Paul S. Jeng and Emily H. Cheng are at the Human Oncology and Pathogenesis Program, Memorial Sloan-Kettering Cancer Center, New York, New York, USA. Paul S. Jeng is also in the Program in Pharmacology, Weill Cornell Graduate School of Medical Sciences, Cornell University, New York, New York, USA. Emily H. Cheng is also at the Department of Pathology, Memorial Sloan-Kettering Cancer Center, New York, New York, USA, and the Department of Pathology and Laboratory Medicine, Weill Cornell Medical College, Cornell University, New York, New York, USA. e-mail: [email protected]
References1. Youle, R.J. & Strasser, A. Nat. Rev. Mol. Cell Biol. 9, 47–59
(2008).2. Davids, M.S. & Letai, A. J. Clin. Oncol. 30, 3127–3135
(2012).3. Walensky, L.D. J. Clin. Oncol. 30, 554–557 (2012).4. Lessene, G. Nat. Chem. Biol. 9, 390–397 (2013).5. Czabotar, P.E. et al. Cell 152, 519–531 (2013).6. Kim, H. et al. Mol. Cell 36, 487–499 (2009).7. Ren, D. et al. Science 330, 1390–1393 (2010).8. Souers, A.J. et al. Nat. Med. 19, 202–208 (2013).9. Beroukhim, R. et al. Nature 463, 899–905 (2010).10. Bean, G.R. et al. Sci. Signal. 6, ra20 (2013).
Competing financial interestsThe authors declare no competing financial interests.
Figure 1 | Targeting BCL-2 family proteins for cancer therapy. Cancers commonly express elevated levels of anti-apoptotic BCL-2 family proteins to prevent the activation of BAX and BAK by activator BH3-only molecules (BH3s, for example BIM or PUMA, orange circles). WEHI-539 can displace bound activator BH3s or monomeric BAX and BAK from BCL-XL to induce the homo-oligomerization of BAX and BAK, leading to MOMP and apoptosis. (a) In cancer cells that overexpress multiple anti-apoptotic BCL-2 members (BCL-XL, BCL2 and MCL-1) and these BCL-2 family proteins are not fully engaged by pro-apoptotic BCL-2 members (activator BH3s, BAX and BAK), WEHI-539 will most likely be ineffective as a single agent because displaced BH3s can be resequestered by BCL-2 or MCL-1. Consequently, BAX and BAK will remain monomeric, and cells will survive. (b) Combined inhibition of multiple anti-apoptotic BCL-2 family members blocks survival mechanisms and efficiently kills cancer cells. Because this approach also leads to high toxicities in normal cells via the same mechanism, it suffers from a narrow therapeutic index. Cyt c, cytochrome c; NOXA mimetics, small-molecule inhibitors of MCL-1. (c) Targeting cancer-specific survival or death pathways to induce activator BH3s will saturate the binding pockets of anti-apoptotic BCL-2 members, allowing WEHI-539 to function as a single-agent inducer of apoptosis while sparing normal cells.
Binding pocketsnot fully occupied
Survival Death(toxicity and narrowtherapeutic index)
Death
Binding pocketsnot fully occupied
Binding pocketsfully occupied
a b c
WEHI-539
ActivatorBH3s
BAX andBAK
MCL-1 BCL-2 BCL-XL
WEHI-539ABT-199NOXA
mimetics WEHI-539
OligomerizedBAX and BAK Cyt c
Caspases
Cyt c
Caspases
npg
© 2
013
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.





























