Embed Size (px)
Citation preview

Advanced Drug Delivery Reviews 65 (2013) 80–88
Contents lists available at SciVerse ScienceDirect
Advanced Drug Delivery Reviews
j ourna l homepage: www.e lsev ie r .com/ locate /addr
Cancer nanomedicines: So many papers and so few drugs!☆,☆☆
Vincent J. Venditto, Francis C. Szoka Jr. ⁎Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, CA 94143-0912, USADepartment of Pharmaceutical Chemistry, School of Pharmacy, University of California, San Francisco, CA 94143-0912, USA
☆ This review is part of the Advanced Drug DeliveryAnniversary issue — Advanced Drug Delivery: Perspecti☆☆ This work was supported by NIH R01 GM06185was funded by an NIH Ruth L. Kirschstein Natio1F32AI095062-01A1.
⁎ Corresponding author at: Department of BioengineeUniversity of California, San Francisco, CA 94143-0912,fax: +1 415 476 0688.
E-mail address: [email protected] (F.C. Szoka).
0169-409X/$ – see front matter © 2012 Elsevier B.V. Alhttp://dx.doi.org/10.1016/j.addr.2012.09.038
a b s t r a c t
a r t i c l e i n f oArticle history:Accepted 20 September 2012Available online 1 October 2012
Keywords:CamptothecinLiposomesMonoclonal antibodiesPlatinum therapyPolymers
This review identifies a timeline to nanomedicine anticancer drug approval using the business model ofinventors, innovators and imitators. By evaluating the publication record of nanomedicine cancer therapeuticswe identified a trend of very few publications prior to FDA approval. We first enumerated the publicationsrelated to cancer involving polymers, liposomes or monoclonal antibodies and determined the number ofcitations per publication as well as the number of published clinical trials among the publications. Combiningthese data with the development of specific nanomedicines, we are able to identify an invention phaseconsisting of seminal papers in basic science necessary for the development of a specific nanomedicine.The innovation phase includes the first report, the development and the clinical trials involving thatnanomedicine. Finally, the imitation phase begins after approval when others ride the wave of success byusing the same formulation for new drugs or using the same drug to validate other nanomedicines. Wethen focused our analysis on nanomedicines containing camptothecin derivatives, which are not yetapproved including two polymers considered innovations and one liposomal formulation in the imitationphase. The conclusion that may be drawn from the analysis of the camptothecins is that approved drugsreformulated in polymeric and liposomal cancer nanomedicines have a more difficult time navigatingthrough the approval process than the parent molecule. This is probably due to the fact that for most current-ly approved drugs, reformulating them in a nanocarrier provides a small increase in performance that largepharmaceutical companies do not consider being worth the time, effort and expense of development. Italso appears that drug carriers have a more difficult path through the clinic than monoclonal antibodies.The added complexity of nanocarriers also deters their use to deliver new molecular entities. Thus, thenew drug candidates that might be most improved by drug delivery in nanocarriers are not formulated inthis fashion.
© 2012 Elsevier B.V. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 802. Small molecule drugs vs. nanomedicines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 813. By the numbers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 814. The timeline of drug approval . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 825. Nanomedicines in the pipeline . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 836. Lessons learned moving forward . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
Reviews theme issue on “25thves and Prospects.”1, R21 AI093135. VJ Vendittonal Research Service Award
ring and Therapeutic Sciences,USA. Tel.: +1 415 476 3895;
l rights reserved.
1. Introduction
Richard Feynman proposed the concept of the nanosurgeons andnanodrug delivery devices in his talk “There's Plenty of Room at theBottom”, where he urged researchers to develop nanodevices capableof interacting with the body at the cellular level [1]. Today, over fiftyyears after Feynman's talk, nanotechnology has become a significant

81V.J. Venditto, F.C. Szoka Jr. / Advanced Drug Delivery Reviews 65 (2013) 80–88
focus of government spending leading to numerous publicationsand patents, but few cancer nanomedicines. Many of the cancernanomedicines currently being investigated utilize small molecule cy-totoxic agents discovered during the 1960s including the platinates,camptothecins and adriamycins, which suffer from poor solubility,poor pharmacokinetics and various adverse side effects. Indeed, of thetraditional low molecular weight drugs, only 5% that reach the clinicaltrial stage ever make it to the market [2]. Nanomedicines addressthese issues by improving the solubility and tailoring the release ratesto improve the pharmacokinetics of the drugwith the promise of reduc-ing the adverse side effects [3]. However, from the vast compositionalspace of nanoparticles, liposomes, polymers and proteins developed totreat cancer, success is limited to just a few nanomedicines [4]. Eachof these classes of nanotherapeutics has had varying success at eachstage of the development process. Recent reviews on nanomedicinehave identified the list of nanocarriers, which have made their wayfrom the bench to FDA approved cancer therapies [4,5].
Taking a bibliographic approach [6] we have evaluated a few ofthe first nanotherapeutics approved by the FDA for cancer therapyand identified the seminal papers that led to the development ofthese therapies. From these publications we generated a time linefor each of the therapeutics to identify if there were similaritiesamong various approaches that might be associated with ultimateFDA approval. These therapeutics might be classified using the busi-ness model of inventors, innovators and imitators (Economist.com/blogs/schumpeter (http://www.economist.com/blogs/schumpeter)).Inventors are those who create new technologies necessary for drugdevelopment. Innovators are those who use these invented technolo-gies to develop a new drug. Once the first drug of the class has beenapproved, many imitators try to reproduce these results with otherdrugs or other drug delivery systems leading to a dramatic increasein publications. As pointed out elsewhere, imitation can be a goodbusiness strategy and by extension a good research strategy if theold is remixed with the new (Economist.com/blogs/schumpeter(http://www.economist.com/blogs/schumpeter)). However, in thedrug delivery field even imitation might not lead to a better formula-tion of a new drug.
Identifying a timeline from invention to innovation andfinally imita-tion is our goal. By looking at the publication statistics for the field wehope to identify trends that resulted in FDA approved nanomedicinesfor the beneficial treatment of cancer patients.
2. Small molecule drugs vs. nanomedicines
The platinates, camptothecins and adriamycins along with manyother cytotoxic agents suffer from poor water solubility, poor pharma-cokinetics and numerous adverse side effects. These small moleculetherapeutics achieve approval when the therapeutic benefits outweighthe adverse side effects. For example, camptothecin failed to achieveFDA approval due to the unpredictable side effects, however, chemicalmodification with a charged carboxylase-cleavable carbamate pro-duced irinotecan (Camptosar©) [7]. This prodrug-forming chemicalmodification improved the water solubility and enabled more efficientdosing strategies with fewer adverse side effects as compared tocamptothecin. The improved solubility and dosing facilitated accelerat-ed FDA approval of irinotecan in 1996 and full approval in 1998.Although the approval process identified irinotecan as a beneficial ther-apeutic for patients, adverse reactions persist, such as serious diarrhea.
Nanocarriers, such as polymers and liposomes, also aim to im-prove the solubility and efficacy while decreasing the adverse side ef-fects. Ideally, these aims are achieved by administering the activeagent systemically in a protected fashion with tailored release at thesite of interest. Improved delivery also comes in the form of the en-hanced permeation and retention (EPR) effect [8], which enables im-proved circulation of the nanocarrier with accumulation through thecharacteristic leaky vasculature at the site of the tumor. This strategy
allows for a lower effective dose to be administered to the patient anda higher dose eventually arriving at the tumor. Potential nanocarrierscapable of formulating nearly every small molecule therapeutic haveinfrequently achieved approval suggesting that the development pro-cess for small molecule drugs and nanocarriers is vastly different.
While small molecule and nanocarrier therapeutics are evaluatedfor efficacy and side effects, nanocarriers are also evaluated for eachof the components within the formulation. Other concerns withnanocarriers that must be investigated prior to FDA approval includein vivo aggregation, recognition by the reticuloendothelial system lead-ing to immune activation and controlled release of the therapeutic. Thisis further complicated when the therapeutic is covalently attached tothe drug carrier as in the case of many polymers. The issues affectingdrug delivery devices are underscored by the lack of nanocarriers ap-proved by the FDA. The short list of polymers approved for cancer ther-apy in the U.S. includes Gliadel©, a poly(anhydride) implantable waferapproved for glioblastoma [9], and a number of PEGylated proteins [10]with Zinostatin stimalmer© (SMANCS) approved in Japan [11]. We donot consider the first two of these therapeutics as nanomedicinesbecause Gliadel© is not a nanocarrier and PEGylated proteins arenanosized prior to PEGylation. The absence of polymer nanomedicinesreceiving FDA approval is surprising due to the vast compositionalspace of polymer architectures and the burgeoning research within thefield [12]. On the other hand, the list of liposomal drugs approved bythe FDA for cancer therapy,with the active component and the companyin parentheses, includeDoxil© (Doxorubicin; Johnson& Johnson; recent-ly reviewed by Barenholz [13]), DaunoXome© (Daunorubicin; Gilead),Mepact© (Muramyl tripeptide; IDM Pharma SAS) and Depocyt©
(Cytarabine; Pacira Pharmaceuticals) with Myocet© (Doxorubicin;Enzon) approved in Europe and Canada [14]. Depocyt© is technicallynot a nanomedicine because the multi-lamellar vesicles are between 3and 30 μm in size.
Monoclonal antibodies are nanomedicines that bridge the gap be-tween small molecules and polymer or liposomal drug delivery vehi-cles. Some antibodies function as active agents, while others act asnanocarriers delivering drug conjugates to the tumor. Antibodies maythen be considered ideal nanomedicines because they take advantageof the size and solubility of polymer and liposomal drug carriers andthe unimolecular characteristics of small molecule therapeutics. This isevident by the success that monoclonal antibody therapy has had in re-cent years [15]. Currently, there are eleven antibodies approved fortherapy listed here with the target and company in parentheses includ-ing Rituxan© (CD20; Genentech), Herceptin© (HER2; Genentech/Roche), Campath© (CD52; Genzyme), Zevalin© (CD20; labeled withY-90; Biogen-Idec), Bexxar© (CD20; labeled with I-131; Coriza/GlaxoSmithKline), Erbitux© (EGFR; Imclone/Lilly), Avastin© (VEGF;Genentech/Roche), Vectibix© (EGFR; Amgen), Arzerra© (CD20;Genmab), Yervoy© (CTLA-4; Bristol-Myers Squibb), Adcetris© (CD30;Seattle Genetics) and Mylotarg© (CD33; labeled with calicheamicin;Wyeth/Pfizer) pulled from the market in 2010.
3. By the numbers
To first compare each of the fields we evaluated the publication re-cord of cancer therapeutics over time. The publication data reportedhere reflects the citations available through SciFinder Scholar as ofApril 2012. Manuscripts published in 2012 were not included. Thetotal number of publications annually, excluding reviews, wasobtained for cancer as a whole, polymeric cancer therapy or liposomalcancer therapy. Clinical trials were then enumerated by limiting eachof these searches accordingly using SciFinder Scholar delimiters(listed in Table 1). Publications of small molecule drugs weresearched in the same manner and limited by polymeric or liposomaltherapy to identify the subset of publications dealing with each ofthese areas. Average number of citations per publication was used

Table 1Comparison of publications, citations and percentage of clinical trials in cancer forpolymer, liposome and monoclonal antibody therapeutics.a
Cancerb Platinatesb Camptothecinsc
All Publications 2,850,000 19,000 10,000Clinical trials 86,000 (3%) 2100 (11%) 1500 (15%)
Polymers Publications 9000 500 400Clinical trials 95 (1%) 4 (1%) 5 (1%)Citations perpublicationb
11.67 13.84 11.70c
Liposomes Publications 12,000 600 300Clinical trials 500 (4%) 90 (15%) 14 (5%)Citations perpublicationb
9.83 8.80 5.79c
Monoclonalantibodies
Publications 35,000 – –
Clinical trials 2800 (8%) – –
Citations perpublicationb
7.36 – –
a Search terms — All Cancer: cancer NOT reviews; All Cancer Platinates: cancer ANDplatinum therapy NOT reviews; All Cancer Camptothecins: cancer AND camptothecinNOT review; Cancer Polymers: cancer AND polymer NOT review; Cancer PolymerPlatinates: cancer AND polymer AND platinum therapy NOT review; Cancer PolymerCamptothecins: cancer AND polymer AND camptothecin NOT review; Cancer Liposomes:cancer AND liposome NOT review; Cancer Liposomal Platinates: cancer AND liposomeAND platinum therapy; Cancer Liposomal Camptothecins: cancer AND liposome ANDcamptothecin; Cancer Monoclonal Antibodies: cancer AND monoclonal antibody therapyNOT review; Clinical trials: identified using the above search strings and the clinical trialselection in SciFinder Scholar.
b Citations per publication reported between the years 1985 and 2005.c Camptothecin citations per publication reported between the years 1995 and 2005.
82 V.J. Venditto, F.C. Szoka Jr. / Advanced Drug Delivery Reviews 65 (2013) 80–88
to evaluate the publications in each area by dividing the number ofcitations by the total number of publications.
In all, there have been nearly three million cancer publicationsdating back to the late 1800s. Notably, there were only 10,000 publi-cations in total prior to 1945, but over 140,000 in 2011 alone. The dra-matic increase in the number of publications after World War IIcoincided with the National Cancer Institute Act of 1944 and the dis-covery that chemical warfare agents were leading to lymphoid andmyeloid suppression and thus could be exploited for use in cancertherapy [16]. Of the three million total publications since 1945,86,000 involve clinical trials, which amount to about 3% of all publica-tions. Certainly many more clinical trials have been performed thathave not been published or were not identified by SciFinder Scholaras clinical trials, but we have decided to use this dataset to providea representative but relative estimate of clinical trials to enable a gen-eral comparison between the modalities.
We evaluated the relative impact or influence of liposomes, poly-mers and monoclonal antibodies, by calculating the average numberof citations per publication using data from the years between 1985and 2005 (Table 1). We selected this time window because prior to1985 there are fewer than 50 publications in each field providing,too few data points to accurately infer any information. Whereas, inthe years after 2005, over 40% of papers do not have a citation com-pared to 30% between 1985 and 2005. During this period, the cita-tions per publication for polymers and liposomes were 11.67±0.60and 9.83±0.66, respectively. Although a trend exists, this differenceis not statistically significant. Citations per publication for monoclonalantibodies, on the other hand, are significantly higher than both poly-mers (pb0.0001) and liposomes (pb0.05) with 7.36±0.53 citationsper publication as determined by a Kruskal–Wallis one-way ANOVA.Interestingly, this trend is inversely related to the number of FDA ap-proved drugs on the market.
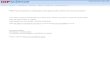
Taking a closer look at each of the fields, synthetic polymeric can-cer therapeutics date back to the early 1960s with fewer than 9000publications and only 95 published clinical trials, which accounts foronly 1% of publications in this field (Fig. 1A). Liposomes have a totalof over 12,000 publications dating back to the late 1960s with over
500 clinical trials or more than 4% of publications (Fig. 1B). Monoclo-nal antibodies on the other hand have over 35,000 papers and 2800clinical trials or 8% published since 1976. Interestingly, it took only10 years (1986) for the total number of antibody publications to ex-ceed 1000, whereas liposomes took 19 years (1991) and polymerstook 36 years (1995). Enumerating clinical trials and the time thateach field take to publish 1000 papers is simple and appears totrack directly with the number of drugs approved by the FDA. It isalso clear that there are very few drug delivery related publicationsin a field prior to drug approval.
4. The timeline of drug approval
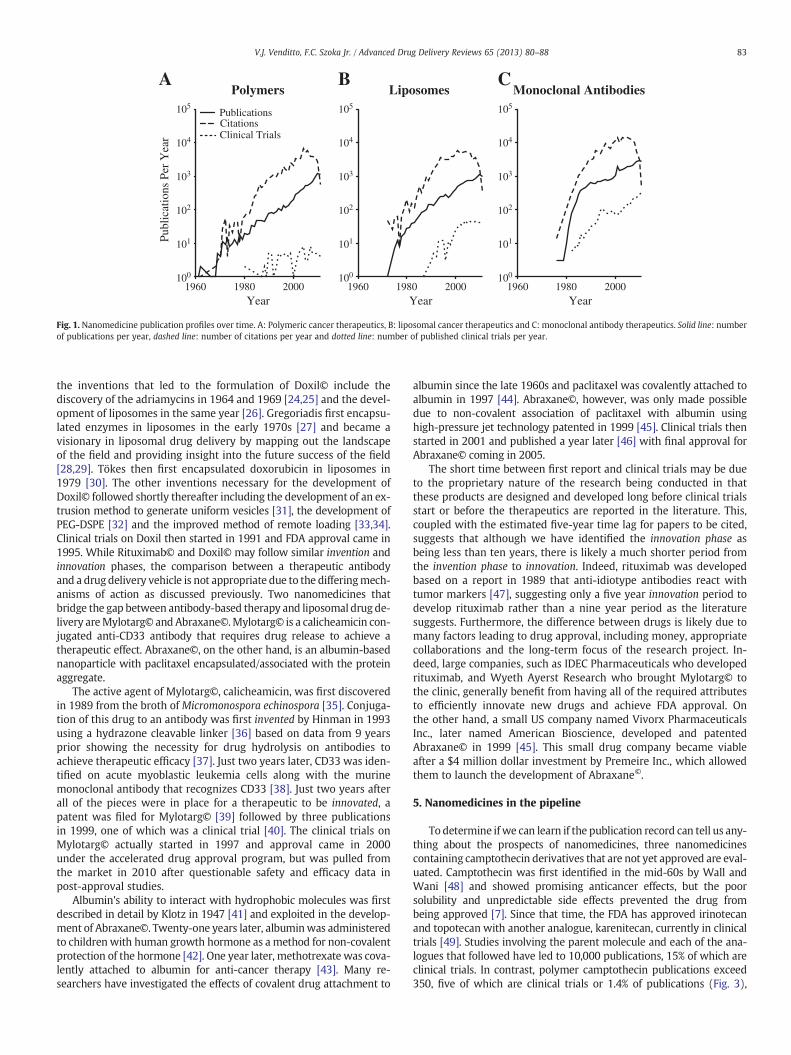
To more closely identify the publication history of the approvednanomedicines, we analyze the publications that came before andafter drug approval. From these publications we are able to identifythe inventors, innovators and imitators within a field. The inventorscreate the technology necessary for drug development including celltargeting markers, protein engineering technology, drug discovery,etc. The innovators remix the invented technology into a drug formu-lation and navigate the approval process. The imitators exploit thetechnology of the newly approved drug to ride the wave of successthat preceded them. We identify the seminal publications duringthe invention phase critical for the development of each specific ther-apeutic. The innovation phase begins after the final inventionwas pub-lished and helps to determine the time that it took for thoseinventions to become a published therapeutic candidate. We alsoidentify the number of years that each nanomedicine takes to enterclinical trials and the number of years from when clinical trialsbegin until drug approval. Imitation comes thereafter as demonstrat-ed by the dramatic increase in publications, which include new po-tential therapeutics as well as basic research to identify mechanismsof action or new indications for the same drug. The invention, innova-tion and imitation phases are overlaid on the publication plot in Fig. 2for polymers, liposomes and monoclonal antibodies.
In 1997, rituximab became the first monoclonal antibody ap-proved for cancer therapy. Four publications involving rituximabwere produced in 1994, one of which was a clinical trial, followedby twelve publications in 1997. Since 1997 there have been 11,500publications involving rituximab. This is in comparison to the wholefield of anticancer monoclonal antibody research, which achieved7400 publications prior to the approval of rituximab and 27,000 pub-lications since then. Certainly, many basic science publications otherthan the first twelve rituximab publications led to the developmentand discovery of rituximab, but it is clear that once this antibodywas identified, there was a very focused route to deliver the antibodyinto the clinic.
The invention phase for rituximab is highlighted by the identifica-tion of murine monoclonal antibodies that target CD20 on B-cells[17,18] followed by the technique of generating humanized anti-bodies [19,20]. Finally, the identification of CD20 expression on Bcell lymphomas [21] led Reff et al. to exploit these developmentsand generate a humanized anti-CD20 monoclonal antibody fortreating B cell lymphoma [22]. This handful of seminal publicationsprior to the initial patent application for rituximab in 1993 [23] canbe accurately attributed to the generation and approval of rituximabin 1997. Furthermore, based on these publication dates, the innova-tion phase lasted about nine years where the multiple inventionswere combined to generate the final monoclonal antibody that wasapproved by the FDA for the treatment of cancer. Since then, manymore antibody therapies have been approved, as the imitation phasehas proven quite fruitful in this example.
To draw a comparison to liposomes the innovation phase startedshortly after the final invention, due to the focus of the team to geta liposomal therapeutic into the clinic. The number of publications in-volving liposomal doxorubicin prior to 1995 was quite limited and
Polymers
PublicationsCitationsClinical Trials
A B CLiposomes Monoclonal Antibodies
100
101
102
103
104
105
100
101
102
103
104
105
100
101
102
103
104
105
Publ
icat
ions
Per
Yea
r
1960 1980 2000Year
1960 1980 2000Year
1960 1980 2000Year
Fig. 1. Nanomedicine publication profiles over time. A: Polymeric cancer therapeutics, B: liposomal cancer therapeutics and C: monoclonal antibody therapeutics. Solid line: numberof publications per year, dashed line: number of citations per year and dotted line: number of published clinical trials per year.
83V.J. Venditto, F.C. Szoka Jr. / Advanced Drug Delivery Reviews 65 (2013) 80–88
the inventions that led to the formulation of Doxil© include thediscovery of the adriamycins in 1964 and 1969 [24,25] and the devel-opment of liposomes in the same year [26]. Gregoriadis first encapsu-lated enzymes in liposomes in the early 1970s [27] and became avisionary in liposomal drug delivery by mapping out the landscapeof the field and providing insight into the future success of the field[28,29]. Tökes then first encapsulated doxorubicin in liposomes in1979 [30]. The other inventions necessary for the development ofDoxil© followed shortly thereafter including the development of an ex-trusion method to generate uniform vesicles [31], the development ofPEG-DSPE [32] and the improved method of remote loading [33,34].Clinical trials on Doxil then started in 1991 and FDA approval came in1995. While Rituximab© and Doxil© may follow similar invention andinnovation phases, the comparison between a therapeutic antibodyand a drug delivery vehicle is not appropriate due to the differingmech-anisms of action as discussed previously. Two nanomedicines thatbridge the gap between antibody-based therapy and liposomal drug de-livery areMylotarg© and Abraxane©.Mylotarg© is a calicheamicin con-jugated anti-CD33 antibody that requires drug release to achieve atherapeutic effect. Abraxane©, on the other hand, is an albumin-basednanoparticle with paclitaxel encapsulated/associated with the proteinaggregate.
The active agent of Mylotarg©, calicheamicin, was first discoveredin 1989 from the broth of Micromonospora echinospora [35]. Conjuga-tion of this drug to an antibody was first invented by Hinman in 1993using a hydrazone cleavable linker [36] based on data from 9 yearsprior showing the necessity for drug hydrolysis on antibodies toachieve therapeutic efficacy [37]. Just two years later, CD33 was iden-tified on acute myoblastic leukemia cells along with the murinemonoclonal antibody that recognizes CD33 [38]. Just two years afterall of the pieces were in place for a therapeutic to be innovated, apatent was filed for Mylotarg© [39] followed by three publicationsin 1999, one of which was a clinical trial [40]. The clinical trials onMylotarg© actually started in 1997 and approval came in 2000under the accelerated drug approval program, but was pulled fromthe market in 2010 after questionable safety and efficacy data inpost-approval studies.
Albumin's ability to interact with hydrophobic molecules was firstdescribed in detail by Klotz in 1947 [41] and exploited in the develop-ment of Abraxane©. Twenty-one years later, albuminwas administeredto children with human growth hormone as a method for non-covalentprotection of the hormone [42]. One year later, methotrexate was cova-lently attached to albumin for anti-cancer therapy [43]. Many re-searchers have investigated the effects of covalent drug attachment to
albumin since the late 1960s and paclitaxel was covalently attached toalbumin in 1997 [44]. Abraxane©, however, was only made possibledue to non-covalent association of paclitaxel with albumin usinghigh-pressure jet technology patented in 1999 [45]. Clinical trials thenstarted in 2001 and published a year later [46] with final approval forAbraxane© coming in 2005.
The short time between first report and clinical trials may be dueto the proprietary nature of the research being conducted in thatthese products are designed and developed long before clinical trialsstart or before the therapeutics are reported in the literature. This,coupled with the estimated five-year time lag for papers to be cited,suggests that although we have identified the innovation phase asbeing less than ten years, there is likely a much shorter period fromthe invention phase to innovation. Indeed, rituximab was developedbased on a report in 1989 that anti-idiotype antibodies react withtumor markers [47], suggesting only a five year innovation period todevelop rituximab rather than a nine year period as the literaturesuggests. Furthermore, the difference between drugs is likely due tomany factors leading to drug approval, including money, appropriatecollaborations and the long-term focus of the research project. In-deed, large companies, such as IDEC Pharmaceuticals who developedrituximab, and Wyeth Ayerst Research who brought Mylotarg© tothe clinic, generally benefit from having all of the required attributesto efficiently innovate new drugs and achieve FDA approval. Onthe other hand, a small US company named Vivorx PharmaceuticalsInc., later named American Bioscience, developed and patentedAbraxane© in 1999 [45]. This small drug company became viableafter a $4 million dollar investment by Premeire Inc., which allowedthem to launch the development of Abraxane©.
5. Nanomedicines in the pipeline
To determine if we can learn if the publication record can tell us any-thing about the prospects of nanomedicines, three nanomedicinescontaining camptothecin derivatives that are not yet approved are eval-uated. Camptothecin was first identified in the mid-60s by Wall andWani [48] and showed promising anticancer effects, but the poorsolubility and unpredictable side effects prevented the drug frombeing approved [7]. Since that time, the FDA has approved irinotecanand topotecan with another analogue, karenitecan, currently in clinicaltrials [49]. Studies involving the parent molecule and each of the ana-logues that followed have led to 10,000 publications, 15% of which areclinical trials. In contrast, polymer camptothecin publications exceed350, five of which are clinical trials or 1.4% of publications (Fig. 3),
InventionInnov.
Imitation
Liposomal Drug Encapsulation
Liposomes InventedAdriamycin Discovered
Adriamycin Liposomes
Extrusion Method Developed
DOXIL FDA Approved
Optimized Remote LoadingPEG-liposome Patented
DOXIL Clinical Trial
InIIventittonInIInov.
ImIIitiiatttitton
Liposomal Drug Encapsulation
Liposomes InventedAdriamycin Discovered
Adriamycin Liposomes
Extrusion Method Developed
DOXIL FDA Approved
Optimized Remote LoadingPEG-liposome Patented
DOXIL Clinical Trial
InnovationInvention
InIInovatittonInIIventitton
19601980
20000
1000
2000Publications Per Year
30000
1000
2000
Publications Per Year
30000
1000
2000
Publications Per Year
3000
Camptothecin Discovered
Cyclodextrin EnzymaticallySynthesized
Cyclodextrin-camptothecin Polymer Synthesized
Cyclodextrin Polymers Synthesized
Clinical Trials Started
InnovationIm
itationInvention
Anti-CD20 Murine Antibodies
Rituximab IdentifiedRituximab Clinical Trial
Rituximab FDA Approved
CD20 Expression on Lymphoma
Humanized AntibodyTechnology
InIInovatittonImII
itiiatttittonInIIventitton
Anti-CD20 Murine Antibodies
Rituximab Identififf edRituximab Clinical Trial
Rituximab FDA Approved
CD20 Expression on Lymphoma
Humanized AntibodyTechnology
Monoclonal A
ntibodies
Liposom
es
Polym
ersABC
Year
19601980
2000Y
ear
19601980
2000Y
ear
Fig.2.Invention,innovationand
imitation
timeline
overlappedon
theplot
ofpublications
peryear
with
seminalpublications
highlighted.
84V.J.V
enditto,F.C.SzokaJr./
Advanced
Drug
Delivery
Reviews65
(2013)80
–88
85V.J. Venditto, F.C. Szoka Jr. / Advanced Drug Delivery Reviews 65 (2013) 80–88
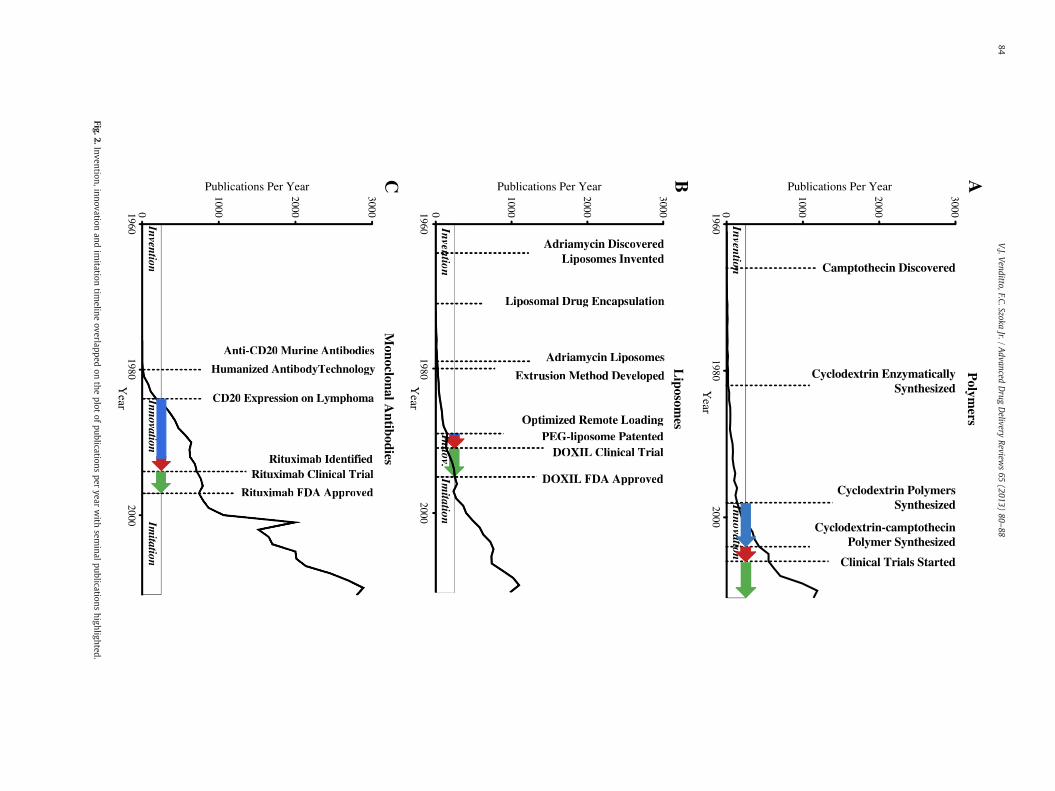
while liposomal formulations containing a camptothecin derivativehave received just over 300 papers with 14 clinical trials or 4.7%(Fig. 3). We also identified the publication statistics of platinates,which follow similar trends as summarized in Table 1 and representedgraphically in Fig. 3, but not discussed here.
There are many factors leading to the imbalance of clinical trialswhen comparing polymers to liposomes even though they have asimilar number of publications. One reason is that polymer re-searchers focus on the appropriate drug attachment to optimize thepharmacokinetics, while many liposomal formulations may be readilygenerated without the requirement to devise new chemical linkageswhat are in essence new molecular entities. Thus liposomes allowfor a wider range of release rates of the parent compound withoutgenerating new molecular entities. This enables efficacy studies tobe directly correlated to the liposome properties and formulationcharacteristics.
Evaluation of the publication record of two not yet approved poly-meric drugs was then performed to compare their trajectories tothe approved drugs. These polymer nanocarriers are two of themore promising nanomedicines currently in clinical trials and areboth based on the delivery of camptothecin. One nanocarrier (CRLX101) is a cyclodextrin polymer containing camptothecin currently inPhase II clinical trials with Cerulean Pharma Inc. [50] and the other(EZN-2208) is a branched PEG construct by Enzon also in Phase IIclinical trials. Due to the use of the same parent drug, the inventionphase with these two nanomedicines overlap.
The inventions that lead to CRLX 101 include the patent for linearcyclodextrin copolymers in 1998 [51] which was published as a cat-ionic transfection reagent in 2003 [52]. The polymer was thenredesigned to prevent cytotoxicity while including a carboxylic acidfor camptothecin attachment [53] and optimized for efficacy in micea year later [54]. Phase I clinical trials with CRLX 101 started in2006 and proceeded into phase II trials in 2008, which are still ongo-ing and to be completed in April 2013 [55]. The number of publica-tions leading to phase two trials is limited to just six publications,one of which is a clinical trial. While CRLX 101 is being investigatedby Cerulean Pharma Inc. the original polymer was developed in aca-demia in collaboration with Insert Therapeutics Inc., now Calando.Calando then licensed the rights of CRLX 101 to Cerulean Pharma
Polymer Publications
Polymer Clinical Trials
A
1990 2000 2010100
101
102
103Camptothecins
Year
Cum
ulat
ive
Publ
icat
ions
Fig. 3. Polymer and liposomal cumulative publication profiles with small molecule drugs. Aderivative and B: publication profile of polymer and liposomal therapeutics containing platipublications and dotted lines: clinical trials.
Inc., but it is clear that the focus of Davis et al. and the industrial col-laborations have pushed cyclodextrin polymers into the clinic rapidlyfor camptothecin delivery as well as siRNA delivery.
Enzon's PEG-camptothecin construct follows a more circuitousroute due to poor phase II efficacy results received in 2005. PEG-camptothecinwasfirst reported in 1998 by Greenwald [56] as amethodto improve solubility of the drug and as a method to improve lactonestability. This version of pegylated camptothecin consisted of a linear40 kDa poly(ethylene glycol) with two camptothecin moieties at eachend. Five more publications followed the original report in 1998,which led to a Phase II clinical trial in 2003, however, poor efficacydata led Enzon to pull the compound from clinical trials. Afterreevaluating the observed issues from the first construct theyreformulated the compound with a branched 40 kDa poly(ethyleneglycol) containing a camptothecin derivative at each of four ends [57].The camptothecin derivative employed is the metabolite of irinotecanand with four drug moieties improves the drug loading in this system.This construct entered the clinic in 2007 and is currently in phase II clin-ical trials with four publications thus far.
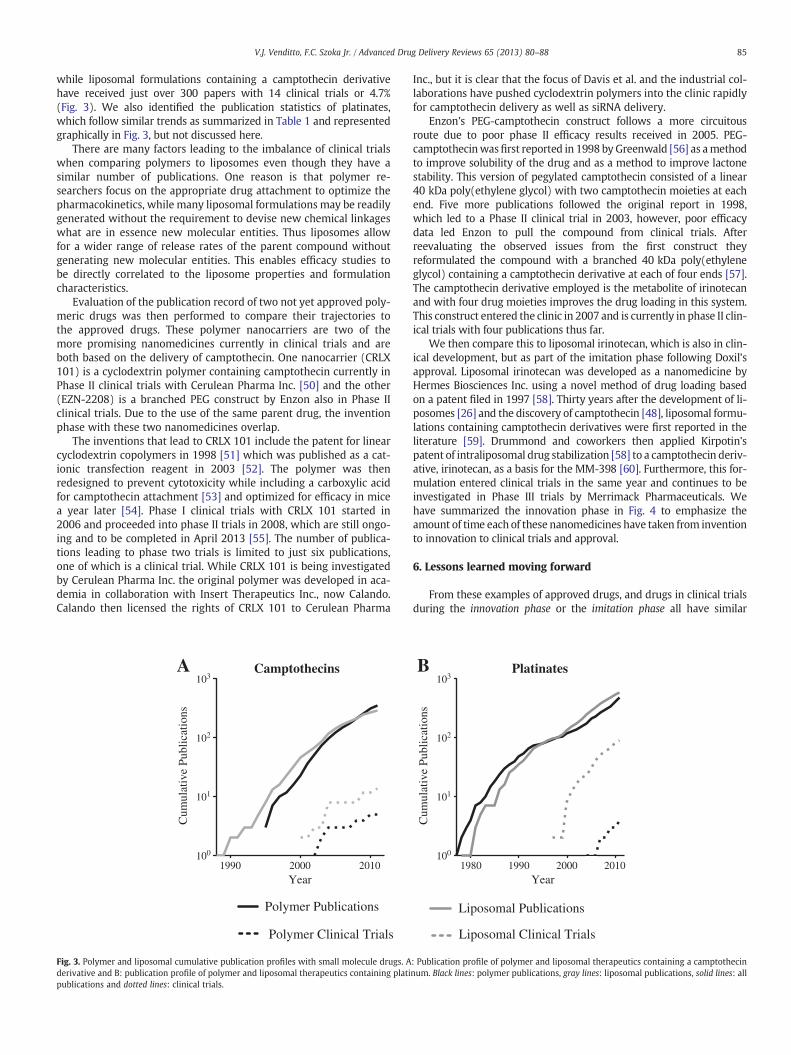
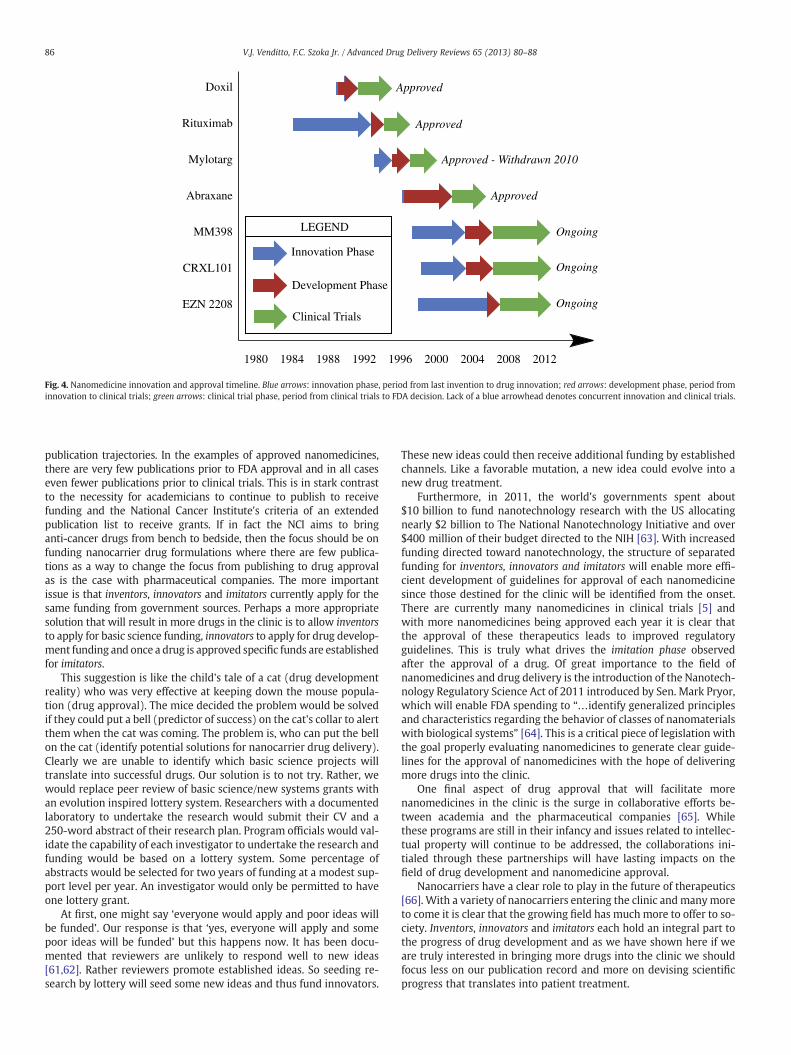
We then compare this to liposomal irinotecan, which is also in clin-ical development, but as part of the imitation phase following Doxil'sapproval. Liposomal irinotecan was developed as a nanomedicine byHermes Biosciences Inc. using a novel method of drug loading basedon a patent filed in 1997 [58]. Thirty years after the development of li-posomes [26] and the discovery of camptothecin [48], liposomal formu-lations containing camptothecin derivatives were first reported in theliterature [59]. Drummond and coworkers then applied Kirpotin'spatent of intraliposomal drug stabilization [58] to a camptothecin deriv-ative, irinotecan, as a basis for the MM-398 [60]. Furthermore, this for-mulation entered clinical trials in the same year and continues to beinvestigated in Phase III trials by Merrimack Pharmaceuticals. Wehave summarized the innovation phase in Fig. 4 to emphasize theamount of time each of these nanomedicines have taken from inventionto innovation to clinical trials and approval.
6. Lessons learned moving forward
From these examples of approved drugs, and drugs in clinical trialsduring the innovation phase or the imitation phase all have similar
1980 1990 2000 2010
Platinates
Year
Cum
ulat
ive
Publ
icat
ions
Liposomal Publications
Liposomal Clinical Trials
B
100
101
102
103
: Publication profile of polymer and liposomal therapeutics containing a camptothecinnum. Black lines: polymer publications, gray lines: liposomal publications, solid lines: all
Doxil
Rituximab
Mylotarg
Abraxane
CRXL101
1980 1984 1988 1992 1996 2000 2004 2008 2012
MM398 LEGEND
Innovation Phase
Development Phase
Clinical Trials
Approved
Approved
Approved - Withdrawn 2010
Approved
Ongoing
Ongoing
EZN 2208 Ongoing
Fig. 4. Nanomedicine innovation and approval timeline. Blue arrows: innovation phase, period from last invention to drug innovation; red arrows: development phase, period frominnovation to clinical trials; green arrows: clinical trial phase, period from clinical trials to FDA decision. Lack of a blue arrowhead denotes concurrent innovation and clinical trials.
86 V.J. Venditto, F.C. Szoka Jr. / Advanced Drug Delivery Reviews 65 (2013) 80–88
publication trajectories. In the examples of approved nanomedicines,there are very few publications prior to FDA approval and in all caseseven fewer publications prior to clinical trials. This is in stark contrastto the necessity for academicians to continue to publish to receivefunding and the National Cancer Institute's criteria of an extendedpublication list to receive grants. If in fact the NCI aims to bringanti-cancer drugs from bench to bedside, then the focus should be onfunding nanocarrier drug formulations where there are few publica-tions as a way to change the focus from publishing to drug approvalas is the case with pharmaceutical companies. The more importantissue is that inventors, innovators and imitators currently apply for thesame funding from government sources. Perhaps a more appropriatesolution that will result in more drugs in the clinic is to allow inventorsto apply for basic science funding, innovators to apply for drug develop-ment funding and once a drug is approved specific funds are establishedfor imitators.
This suggestion is like the child's tale of a cat (drug developmentreality) who was very effective at keeping down the mouse popula-tion (drug approval). The mice decided the problem would be solvedif they could put a bell (predictor of success) on the cat's collar to alertthem when the cat was coming. The problem is, who can put the bellon the cat (identify potential solutions for nanocarrier drug delivery).Clearly we are unable to identify which basic science projects willtranslate into successful drugs. Our solution is to not try. Rather, wewould replace peer review of basic science/new systems grants withan evolution inspired lottery system. Researchers with a documentedlaboratory to undertake the research would submit their CV and a250-word abstract of their research plan. Program officials would val-idate the capability of each investigator to undertake the research andfunding would be based on a lottery system. Some percentage ofabstracts would be selected for two years of funding at a modest sup-port level per year. An investigator would only be permitted to haveone lottery grant.
At first, one might say ‘everyone would apply and poor ideas willbe funded’. Our response is that ‘yes, everyone will apply and somepoor ideas will be funded’ but this happens now. It has been docu-mented that reviewers are unlikely to respond well to new ideas[61,62]. Rather reviewers promote established ideas. So seeding re-search by lottery will seed some new ideas and thus fund innovators.
These new ideas could then receive additional funding by establishedchannels. Like a favorable mutation, a new idea could evolve into anew drug treatment.
Furthermore, in 2011, the world's governments spent about$10 billion to fund nanotechnology research with the US allocatingnearly $2 billion to The National Nanotechnology Initiative and over$400 million of their budget directed to the NIH [63]. With increasedfunding directed toward nanotechnology, the structure of separatedfunding for inventors, innovators and imitators will enable more effi-cient development of guidelines for approval of each nanomedicinesince those destined for the clinic will be identified from the onset.There are currently many nanomedicines in clinical trials [5] andwith more nanomedicines being approved each year it is clear thatthe approval of these therapeutics leads to improved regulatoryguidelines. This is truly what drives the imitation phase observedafter the approval of a drug. Of great importance to the field ofnanomedicines and drug delivery is the introduction of the Nanotech-nology Regulatory Science Act of 2011 introduced by Sen. Mark Pryor,which will enable FDA spending to “…identify generalized principlesand characteristics regarding the behavior of classes of nanomaterialswith biological systems” [64]. This is a critical piece of legislation withthe goal properly evaluating nanomedicines to generate clear guide-lines for the approval of nanomedicines with the hope of deliveringmore drugs into the clinic.
One final aspect of drug approval that will facilitate morenanomedicines in the clinic is the surge in collaborative efforts be-tween academia and the pharmaceutical companies [65]. Whilethese programs are still in their infancy and issues related to intellec-tual property will continue to be addressed, the collaborations ini-tialed through these partnerships will have lasting impacts on thefield of drug development and nanomedicine approval.
Nanocarriers have a clear role to play in the future of therapeutics[66]. With a variety of nanocarriers entering the clinic andmanymoreto come it is clear that the growing field has much more to offer to so-ciety. Inventors, innovators and imitators each hold an integral part tothe progress of drug development and as we have shown here if weare truly interested in bringing more drugs into the clinic we shouldfocus less on our publication record and more on devising scientificprogress that translates into patient treatment.

87V.J. Venditto, F.C. Szoka Jr. / Advanced Drug Delivery Reviews 65 (2013) 80–88
Acknowledgments
We thank Dr. Colin L. Walsh (UCSF) and Matthew R. Tiffany(UCSF) for helpful discussion and suggestions regarding the contentof the paper.
References
[1] R.P. Feynman, There's plenty of room at the bottom, Eng. Sci. (CalTech) 23 (1960)22–36.
[2] D. Sarker, P. Workman, Pharmacodynamic biomarkers for molecular cancer ther-apeutics, Adv. Cancer Res. 96 (2007) 213–268.
[3] E. Blanco, A. Hsiao, A.P. Mann, M.G. Landry, F. Meric-Bernstam, M. Ferrari,Nanomedicine in cancer therapy: innovative trends and prospects, Cancer Sci.102 (2011) 1247–1252.
[4] F. Alexis, E.M. Pridgen, R. Langer, O.C. Farokhzad, Nanoparticle technologies forcancer therapy, in: M. Schafer-Korting (Ed.), Drug Delivery, Handbook of Experi-mental Pharmacology, 197, Springer-Verlag, Berlin, 2010, pp. 55–86.
[5] R. Duncan, R. Gaspar, Nanomedicine(s) under the microscope, Mol. Pharmaceutics8 (2011) 2101–2141.
[6] F.D. Ledley, L.M. McNamee, Patterns of technological innovation in biotechnology,Nature Biotechnol. in press.
[7] V.J. Venditto, E.E. Simanek, Cancer therapies utilizing the camptothecins: a reviewof the in vivo literature, Mol. Pharmaceutics 7 (2010) 307–349.
[8] Y. Matsumura, H. Maeda, A new concept for macromolecular therapeutics in can-cer chemotherapy: mechanism of tumoritropic accumulation of proteins and theantitumor agent Smancs, Cancer Res. 72 (1986) 6387–6392.
[9] H. Brem, H.C. Lawson, The development of new brain tumor therapy utilizing thelocal and sustained delivery of chemotherapeutic agents form biodegradablepolymers, Cancer 86 (1999) 197–199.
[10] M. Morpurgo, G. Pasut, F.M. Veronese, PEGylated proteins as cancer therapeutics,in: V.P. Torchilin (Ed.), Delivery of Protein and Peptide Drugs in Cancer, WorldScientific Publishing Co., Singapore, 2006, pp. 85–110.
[11] H. Maeda, SMANCS and polymer-conjugated macromolecular drugs: advantagesin cancer chemotherapy, Adv. Drug Deliv. Rev. 46 (2001) 169–185.
[12] M.E. Fox, F.C. Szoka, J.M. Fréchet, Soluble polymer carriers for the treatment of cancer:the importance of molecular architecture, Acc. Chem. Res. 42 (2009) 1141–1151.
[13] Y. Barenholz, Doxil - the first FDA-approved nano-drug: lessons learned, J. Con-trol. Release 160 (2012) 117–134.
[14] P.D. Marcato, N. Duran, New aspects of nanopharmaceutical delivery systems,J. Nanosci. Nanotechnol. 8 (2008) 1–14.
[15] L.M. Weiner, J.C. Murray, C.W. Shuptrine, Antibody-based immunotherapy ofcancer, Cell 148 (2012) 1081–1084.
[16] L.S. Goodman, M.M. Wintrobe, Nitrogen mustard therapy, J. Am. Med. Assoc. 132(1946) 126–132.
[17] P. Stashenko, L.M. Nadler, R. Hardy, S.F. Schlosssman, Characterization of a humanB lymphocyte-specific antigen, J. Immunol. 125 (1980) 1678–1685.
[18] L.M. Nadler, J. Ritz, R. Hardy, J.M. Pesando, S.F. Schlossman, P. Stashenko, A uniquecell surface antigen identifying lymphoid malignancies of B cell origin, J. Clin.Invest. 67 (1981) 134–140.
[19] S.L. Morrison, M.J. Johnson, S.A. Herzenberg, V.T. Oi, Chimeric human antibodymolecules: mouse antigen-binding domains with human constant region do-mains, Proc. Natl. Acad. Sci. U. S. A. 81 (1984) 6851–6855.
[20] G.L. Boulianne, N. Hozumi, M.J. Shulman, Production of functional chimaericmouse/human antibody, Nature 312 (1984) 643–646.
[21] K. Horibe, L.M. Nadler, Human B cell associated antigens defined by monoclonalantibodies, in: E.L. Heise (Ed.), Lymphocyte Surface Antigens, American Societyfor Histocompatability and Immunogenetics, New York, 1984, pp. 309–323.
[22] M.E. Reff, K. Carner, K.S. Chambers, P.C. Chinn, J.E. Leonard, R. Raab, R.A. Newman,N. Hanna, D.R. Anderson, Depletion of B cells in vivo by a chimeric mouse humanmonoclonal antibody to CD20, Blood 83 (1994) 435–445.
[23] D.R. Anderson, N. Hanna, J.E. Leonard, R.A. Newman, M.E. Reff, W.H. Rastetter,Therapeutic application of chimeric and radiolabeled antibodies to human B lym-phocyte restricted differentiation antigen for treatment of B cell lymphoma, in,United States Patent 5,736,137 (1998).
[24] F. Arcamore, G. Franceschi, P. Orezzi, G. Cassinelli, W. Barbieri, R. Mondelli, Dau-nomycin. I. The structure of daunomycinone, J. Am. Chem. Soc. 86 (1964) 15–19.
[25] F. Arcamore, G. Cassinelli, G. Fantini, A. Grein, P. Orezzi, C. Pol, C. Spalla, Adriamycin,14-hydroxydaimomycin, a new antitumor antibiotic from S. peucetius var. caesius,Biotechnol. Bioeng. 11 (1969) 1101–1110.
[26] A.D. Bangham, R.W. Horne, Negative staining of phospholipids and their structur-al modification by surface-active agents as observed in the electron microscope,J. Mol. Biol. 8 (1964) 660–668.
[27] G. Gregoriadis, P.D. Leathwood, B.E. Ryman, Enzyme entrapment in liposomes,FEBS Lett. 14 (1971) 95–99.
[28] G. Gregoriadis, The carrier potential of liposomes in biology and medicine, NewEngl. J. Med. 295 (1976) 704–710.
[29] G. Gregoriadis, The carrier potential of liposomes in biology and medicine, NewEngl. J. Med. 295 (1976) 765–770.
[30] E.A. Forssen, Z.A. Tokes, In vitro and in vivo studies with adriamycin liposomes,Biochim. Biophys. Res. Commun. 91 (1979) 1295–1301.
[31] F. Szoka, F. Olson, T. Heath, W. Vail, E. Mayhew, D. Papahadjopoulos, Preparationof unilamellar liposomes of intermediate size (0.1–0.2 mumol) by a combination
of reverse phase evaporation and extrusion through polycarbonate membranes,Biochim. Biophys. Acta 3 (1980) 559–571.
[32] M.C. Woodle, M.C., F.J. Martin, A. Yau-Yang, C.T. Redmann, Liposomes with en-hanced circulation time, in, United States Patent 5,013,556 (1991).
[33] Y. Barenholz, Loading and controlled release of amphiphatic molecules to andfrom liposomes, in, United States Patent 5,192,549 (1993).
[34] G. Haran, R. Cohen, L.K. Bar, Y. Barenholz, Transmembrane ammonium sulfategradients in liposomes produce efficient and stable entrapment of amphiphaticweak base, Biochim. Biophys. Acta 1151 (1993) 201–215.
[35] M.D. Lee, J.K. Manning, D.R. Williams, N.A. Kuck, R.T. Testa, D.B. Borders,Calicheamicins, a novel family of antitumor antibiotics. III. Isolation, purificationand characterization of calicheamicins beta 1 Br, gamma 1 Br, alpha 2I, alpha 3I,beta 1I and delta 1I, J. Antibiot. 42 (1989) 1070–1087.
[36] L.M. Hinman, P.R. Hamann, R. Wallace, A.T. Menendez, F.E. Durr, J. Upeslacis,Preparation and characterization of monoclonal antibody conjugates of thecalicheamicins: a novel and potent family of antitumor antibiotics, Cancer Res.53 (1993) 3336–3342.
[37] A. Trouet, M. Masquelier, R. Baurain, D. Deprez-De Campeneere, A covalent link-age between daunorubicin and proteins that is stable in serum and reversible bylysosomal hydrolases, as required for a lysosomotropic drug-carrier conjugate: invitro and in vivo studies, Proc. Natl. Acad. Sci. U. S. A. 79 (1982) 626–629.
[38] J.D. Griffin, D. Linch, K. Sabbath, P. Larcom, S.F. Schlossman, A monoclonal anti-body reactive with normal and leukemic human myeloid progenitor cells, Leuk.Res. 8 (1984) 521–534.
[39] P.R. Hamann, L. Hinman, I. Hollander, R. Holycomb, W. Hallett, H.-R. Tsou, M.J.Weiss, Enediyne derivatives useful for the synthesis of conjugates ofmethyltrithio antitumor agents, in United States Patent 5,739,116 (1998).
[40] E.L. Sievers, F.R. Appelbaum, R.T. Spielberger, S.J. Forman, D. Flowers, F.O. Smith,K. Shannon-Dorcy, M.S. Berger, I.D. Bernstein, Selective ablation of acute myeloidleukemia using antibody-targeted chemotherapy: a phase I study of an anti-CD33calicheamicin immuoconjugate, Blood 93 (1999) 3678–3684.
[41] I.M. Klotz, The effects of salts and proteins on the spectra of some dyes and indi-cators, Chem. Rev. 41 (1947) 373–399.
[42] P.J. Collipp, R.D. Snyder, Short asthmatic children and human growth hormone:evaluation of albumin-bound growth hormone, Clin. Pediatr. 7 (1968) 659–664.
[43] G. Magnenat, R. Schindler, H. Isliker, Transport of cytostatic agents by the plasmaproteins. III. In vitro antitumor action of cytostatic-azoprotein conjugates, Eur. J.Cancer 5 (1969) 33–40.
[44] F. Dosio, P. Brusa, P. Crosasso, S. Arpicco, L. Cattel, Preparation, characterizationand properties in vitro and in vivo of paclitaxel–albumin conjugate, J. Control.Res. 47 (1997) 293–304.
[45] N.P. Desai, N.P., C. Tao, A. Yang, L. Louie, T. Zheng, Z. Yao, P. Soon-Shiong, S. Magdassi,Protein stabilized pharmacologically active agents, methods for the preparationthereof and methods for the use thereof, in, United States Patent 5,916,596, (1999).
[46] N.K. Ibrahim, N. Desai, S. Legha, P. Soon-Shiong, R.L. Theriault, E. Rivera, B.Esmaeli, S.E. Ring, A. Bedikian, G.N. Hortobagyi, J.A. Ellerhorst, Phase I and phar-macokinetic study of ABI-007, a cremophor-free, protein-stabilized, nanoparticleformulation of paclitaxel, Clin. Cancer Res. 8 (2002) 1038–1044.
[47] R.A. Miller, S. Hart, M. Samoszuk, C. Coulter, S. Brown, D. Czerwinski, J.Kelkenberg, I. Roystin, R. Levy, Shared idiotypes expressed by human B-cell lym-phomas, New Engl. J. Med. 321 (1989) 851–857.
[48] M.E. Wall, M.C. Wani, C.E. Cooke, K.H. Palmer, A.T. McPhail, G.A. Sim, The isolationand structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitorfrom Camptotheca acuminata, J. Am. Chem. Soc. 88 (1966) 3888–3890.
[49] A. Daud, N. Valkov, B. Centeno, J. Derderian, P. Sullivan, P. Munster, P. Urbas, R.C.DeConti, E. Berghorn, Z. Liu, F.H. Hausheer, D. Sullivan, Phase II trial of karenitecinin patients with malignant melanoma: clinical and translational study, Clin. CancerRes. 11 (2005) 3009–3016.
[50] A.V. Yurkovetskiy, R.J. Fram, XMT-1001, a novel polymeric camptothecinpro-drug in clinical development for patients with advanced cancer, Adv. DrugDelivery Rev. 61 (2009) 1193–1202.
[51] M.E. Davis, H. Gonzalez, S. Hwang, Linear cyclodextrin copolymers, in, UnitedStates Patent 6,509,323 (2003).
[52] H. Gonzalez, S.J. Hwang, M.E. Davis, New class of polymers for the delivery ofmacromolecular therapeutics, Bioconjug. Chem. 10 (1999) 1068–1074.
[53] J. Cheng, K.T. Khin, G.S. Jensen, A. Liu, M.E. Davis, Synthesis of linearbeta-cyclodextrin-based polymers and their camptothecin conjugates, Bioconj.Chem. 14 (2003) 1007–1017.
[54] J. Cheng, K.T. Khin, M.E. Davis, Antitumor activity of beta-cyclodextrin polymer–camptothecin conjugates, Mol. Pharmaceutics 1 (2004) 183–193.
[55] M.E. Davis, Design and development of IT-101, a cyclodextrin-containing polymerconjugate of camptothecin, Adv. Drug Deliv. Rev. 61 (2009) 1189–1192.
[56] R.B. Greenwald, A. Pendri, C.D. Conover, C. Lee, Y.H. Choe, C. Gilbert, A. Martinez, J.Xia, D. Wu, M. Hsue, Camptothecin-20-PEG ester transport forms: the effect ofspacer groups on antitumor activity, Bioorg. Med. Chem. 6 (1998) 551–562.
[57] P. Sapra, P. Kraft, M. Mehlig, J. Maltby, H. Zhao, L.M. Greenberger, I.D. Horak, Markedtherapeutic efficacy of a novel polyethylene glycol-SN38 conjugate, EZN-2208, in xe-nograft models of B-cell non-Hodgkin's lymphoma, Haematologica 94 (2009)1456–1459.
[58] D. Kirpotin, Compound-loaded liposomes and methods for their preparation, in,United States Patent, 6,110,491 (2000).
[59] Y. Sadzuka, S. Hirotsu, S. Hirota, Effect of liposomalization on the antitumor activ-ity, side-effects and tissue distribution of CPT-11, Cancer Lett. 127 (1998) 99–106.
[60] D.C. Drummond, C.O. Noble, Z. Guo, K. Hong, J.W. Park, D.B. Kirpotin, Develop-ment of a highly active nanoliposomal irinotecan using a novel intraliposomalstabilization strategy, Cancer Res. 66 (2006) 3271–3277.

88 V.J. Venditto, F.C. Szoka Jr. / Advanced Drug Delivery Reviews 65 (2013) 80–88
[61] M. Eisenhart, The paradox of peer review: admitting too much or allowing toolittle? Res. Sci. Educ. 32 (2002) 241–255.
[62] S. Thurner, R. Hanel, Peer-review in a world with rational scientists: toward se-lection of the average, Eur. Phys. J. 84 (2011) 707–711.
[63] National Science and Technology Council, Committee on Technology, in: TheNanotechnology Initiative — Supplement to the President's 2012 Budget, 2011.
[64] Nanotechnology Regulatory Science Act of 2011, S. 1662, 112th Cong. (2011).[65] M. Ratner, Pfizer reaches out to academia— again, Nat. Biotechnol. 29 (2011) 3–4.[66] National Institutes of Health, NIH launches collaborative program with industry
and researchers to spur therapeutic development, NIH News, May 3, 2012.