Embed Size (px)
Citation preview

Dissertatio
n
Building blocks for multinuclear
near-ir luminescent lanthanide complexes
Caroline Bischof
Ruhr-Universität Bochum
Faculty of Chemistry and Biochemistry


Building blocks for multinuclearnear-IR luminescent lanthanide complexes
by
Caroline Bischof
A dissertation submitted in partial satisfaction
of the requirements for the degree of a
Doctor of Philosophy (Dr. rer. nat.) in Chemistry
at the
Faculty of Chemistry and Biochemistry
Ruhr-Universität Bochum
March 2013


Committee in charge:
Dr. Michael Seitz
Prof. Dr. Martin Feigel
Thesis defense date:
April 12th 2013

The present work was performed in between October 2009 until March 2013
at the Chair of Inorganic Chemistry I at the Ruhr-University Bochum
(Germany) under the supervision of Dr. Michael Seitz.
I hereby declare, on oath, that I have written the present dissertation by my own
and have not used other than the acknowledged resources and aids.

Acknowledgement
Although only my name appears on the title page, there have been many people who
contributed to this work and to the continuous learning process I went through dur-
ing the past 3.5 years. I am very grateful for their help and I would like to thank
particularly the following people:
Dr. Michael Seitz for the supervision of this work, the given scientific freedom in pur-
suing my ideas and valuable lessons on organic chemistry. I would further like to
thank him for his great support and the chance to participate in several international
conferences.
Prof. Feigel for kindly acting as second referee of my doctoral thesis.
Prof. Nils Metzler-Nolte for his continuous support over many years.
My former Bachelor student Felix Stog and my former Master students Julia Scholten
and Simon Trosien, for working with me on the deuterated cryptates.
Martin Gartmann, Gregor Barchan and Hans Jochen Hauswald for measuring several
NMR spectra for me and, more importantly, for helping out when the spectromet-
ers and me had a different opinions of what to do with my sample.
Bauke Albada, Nicola Alzakhem, Christine Doffek, Sven Hennig and Hendrik Pfeiffer for
proof-reading this thesis.
Dr. Klaus Merz and Vera Vasylyeva for their help with X-ray crystals determinations.
I would also like to thank Herr Klaus for letting me supervise students in the Ad-
vanced Inorganic Chemical Practical, the introduction to numerous good reads and
lots of cheerful laughter.

Gundula Talbot for her lending me her ear, for helpful advices and for smiling.
My climbing friends for experimenting with me in the areas of gravity and vertical
limits and producing visible results.
My bio-connection: Sabrina Beck, Rebecca Gentek, Jan Gleichhagen, Sven Hennig, Alex-
ander Neuhaus, Steffen van der Wal and Bauke Albada for their input on biochemical
matters and for sharing their different perspectives with me. I particularly thank Jan
for providing litres of different buffers. Also, thank you Sabrina and Becci for being
“my girls”!
My colleagues from the ACI, especially Jan Dittrich, Tuba Güden, Nina Hüsken, Qian
Kun, Mingyan Ma, Julia Norman, Hendrik Pfeiffer, Lukasz Raszeja and Thomas Sowik for
making the past years a colourful and memorable time, and also for sharing lots of
delicious food. I particularly thank Hendrik and Thomas for their mental support.
My partners in crime: David Bulfield, David Köster, Malay Patra, Anna Sosniak, Felix
Stog and Jessica Wahsner for being such wonderful labmates and supportive friends.
Whenever I faced a problem or just wanted to share the latest climbing story, they
were at my side listening, discussing, helping out, cheering up and laughing.
My faithful companion Nicola Alzakhem, who is an outstandingly talented chemist and
has been a patient teacher for me. With him, I celebrated clean NMR spectra, waltzed
through the lab, discussed organic chemistry from A to Z, and went for numerous
“walkabouts” to lectures, glass containers and countries. I consider myself very lucky
to have him in my life.
Ein besonderer Dank gilt meiner Familie, vor allem meinen Brüdern und meinen El-
tern. Sie haben mich stets ermuntert frohen Mutes meinen Weg zu gehen und dafür
Sorge getragen, dass ich im Gedankennebel meiner Promotion nicht die wesentlichen
Dinge im Leben aus den Augen verliere.
Last but not least I am grateful for the financial support from the International Max
Planck Research School in Chemical Biology and from the Ruhr University Research
School.

for Jürgen and Claus Bischof,
who were once joking about “Acetan Hydrid”


“ The only true voyage of discovery would be not to visit new landscapes
but to possess other eyes, to behold the universe through the eyes of another...”Marcel Proust, La Prisonnière

Contents
1 Introduction 1
1.1 Imaging in biological systems . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.1.1 Imaging in medicine and biology . . . . . . . . . . . . . . . . . . 1
1.1.2 Optical imaging . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2 Fluorescent probes for optical imaging . . . . . . . . . . . . . . . . . . . 4
1.2.1 Luminescence, fluorescence and phosphorescence . . . . . . . . . 4
1.2.2 Properties of fluorescent probes . . . . . . . . . . . . . . . . . . . 5
1.2.3 Classification of fluorophores . . . . . . . . . . . . . . . . . . . . . 7
1.3 Near-IR optical imaging with lanthanides . . . . . . . . . . . . . . . . . . 10
1.3.1 Upconversion processes . . . . . . . . . . . . . . . . . . . . . . . . 11
1.3.2 Activators and sensitisers . . . . . . . . . . . . . . . . . . . . . . . 11
1.3.3 Upconversion nanophosphors . . . . . . . . . . . . . . . . . . . . 13
1.3.4 Upconversion in molecular lanthanide complexes . . . . . . . . . 13
2 Concept of the project 16
3 Development of binuclear cryptates 18
3.1 Deuterated cryptates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
3.1.1 Retrosynthetic analysis . . . . . . . . . . . . . . . . . . . . . . . . 21
3.1.2 Synthesis of building blocks and cryptates . . . . . . . . . . . . . 22
3.1.3 Photophysical properties . . . . . . . . . . . . . . . . . . . . . . . 27
3.2 Functionalised cryptates . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
3.2.1 Retrosynthetic analysis of functionalised cryptates . . . . . . . . 34
3.2.2 Synthesis of carboxy functionalised cryptates . . . . . . . . . . . 35
3.2.3 Synthesis of amino functionalised cryptates . . . . . . . . . . . . 39
3.3 Binuclear cryptates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
3.3.1 Strategy for the coupling of cryptates . . . . . . . . . . . . . . . . 45
3.3.2 Functionalised cryptates for coupling reactions . . . . . . . . . . 45
3.3.3 First approaches towards the coupling of cryptates . . . . . . . . 47
3.3.4 Preparation of the first binuclear TBP cryptate . . . . . . . . . . . 48

4 Chemical unclicking of amide bonds 51
4.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
4.2 Concept . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
4.3 Preliminary work . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
4.4 Retrosynthetic analysis of Unclick serine . . . . . . . . . . . . . . . . . . 56
4.5 Investigation of different Unclick systems . . . . . . . . . . . . . . . . . . 57
4.5.1 Fmoc-Unclick serine . . . . . . . . . . . . . . . . . . . . . . . . . . 57
4.5.2 Acetyl-Unclick-serine . . . . . . . . . . . . . . . . . . . . . . . . . 60
4.5.3 Acetyl-Unclick-serine via Boc-cycloserine . . . . . . . . . . . . . . 62
4.5.4 Fmoc-Gly-Unclick-serine . . . . . . . . . . . . . . . . . . . . . . . 66
5 Summary 70
6 Experimental Section 74
6.1 General . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
6.2 Luminescence measurements . . . . . . . . . . . . . . . . . . . . . . . . . 75
6.3 Simple bipyridine building blocks . . . . . . . . . . . . . . . . . . . . . . 76
6.4 Bipyridine ester building blocks . . . . . . . . . . . . . . . . . . . . . . . 87
6.5 Nitro and amino bipyridine building blocks . . . . . . . . . . . . . . . . 93
6.6 Simple macrocycles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
6.7 Nitro and amino functionalised macrocycles . . . . . . . . . . . . . . . . 99
6.8 Simple deuterated cryptates . . . . . . . . . . . . . . . . . . . . . . . . . . 101
6.9 Carboxylic acid ester functionalised crypates . . . . . . . . . . . . . . . . 104
6.10 Amino functionalised cryptates . . . . . . . . . . . . . . . . . . . . . . . . 106
6.11 Derivatives of functionalised cryptates . . . . . . . . . . . . . . . . . . . . 108
6.12 Lanthanide complexes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
6.13 Fmoc-protected serines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
6.14 Acetyl-protected serines . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
6.15 Boc-protected serines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
6.16 Other serines and related molecules . . . . . . . . . . . . . . . . . . . . . 125
7 Bibliography 129
Appendix i
A Data for crystal structures . . . . . . . . . . . . . . . . . . . . . . . . . . . i
B Publication list . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xiii

List of abbreviations
AA any amino acid
Boc tert-butyloxycarbonyl
CT computed tomography
CFP cyan fluorescent protein
DELFIA dissociation enhanced lanthanide fluorescence immunoassay
DIPEA N,N’-diisopropylethylenediamine
DMF dimethylformamide
DMSO dimethylsulfoxide
DOTA 1,4,7,10-tetraazacyclododecane-
1,4,7,10-tetraacetic acid
EDC 1-ethyl-3-(3-dimethyllaminopropyl)carbodiimide
EI electron impact mass spectrometry
ESI electro-spray ionisation mass spectrometry
ETU energy transfer upconversion
FAB fast atom bombardment mass spectrometry
FRET fluorescence resonance energy transfer
Fmoc 9-fluorenylmethyloxycarbonyl
GFP green fluorescent protein
HATU 1-[bis(dimethylamino)methylene]-1H-1,2,3-
triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
MRI magnetic resonance imaging
PET positron emission tomography
PBS phosphate-buffered saline
PDB protein data bank
QD quantum dots
RFP red fluorescent protein
rt room temperature
SPECT single-photon emission computed tomography
SPPS solid phase peptide synthesis
TBP tris(bipyridine)

TFA trifluoroacetic acid
THF tetrahydrofurane
TLC thin layer chromatography
TMS trimethylsilyl
TRF time-resolved detection
UC upconversion
YFP yellow fluorescent protein

1 Introduction
1.1 Imaging in biological systems
1.1.1 Imaging in medicine and biology
Bioimaging is a collective term for a wide range of visualisation techniques in medi-
cine and modern biology. These techniques are used for the non-invasive visualisa-
tion of morphological and physiological details of living systems. Hence, bioimaging
methods are powerful tools for the diagnosis of diseases and also for the understand-
ing of biological processes. Bioimaging comprises
• anatomical imaging:
the creation of images of anatomic structures (organs or tissues),
• functional imaging:
the studies of organ functions during physiological stimulation,
• molecular imaging:
the in vivo probing of biological processes on a molecular level.
Anatomical and functional imaging together form the field of classical diagnostic med-
ical imaging. The commonly applied methods are based on ultrasound, X-ray, radi-
onuclides or nuclear magnetic resonance. The scope of information gathered by em-
ploying these techniques can even be extended by the use of contrast agents. Import-
ant advantages and potential risks of these methods are summarised in Table 1.1. [1–4]
The desire to investigate biological processes in detail led to the development of
imaging techniques, that allow to probe details on a molecular level. Molecular ima-
ging plays a vital role in drug discovery, in fundamental biological research and for the
characterisation of disease-related molecular events. Applications range from simple
cell localisation to the visualisation of complex events. For example, protein-protein
interactions can be monitored with positron emission tomography (PET), and receptor
assays or enzymatic pathways can be probed with radiotracers. Optical imaging
provides an alternative to these methods and is often used for the visualisation of
structures and pathways in biological systems.
1

1.1 Imaging in biological systems
Table 1.1: Benefits and limitations of methods in diagnostic medical imaging [4,5]
Imaging method Benefits Limitations
Ultrasound low patient risk; restricted to superficial structuresquick examination poor spatial resolution (0.1-1mm) [6]
X-ray and X-ray versatile diagnosis of bones exposition to ionising radiation hascomputed tomography (CT) or organs to be kept low
Radionuclide imaging high sensitivity; incorporation exposition to radiation released by(PET, SPECT) or attachment to substances for radionuclides has to be kept low;
specific accumulation possible; poor spatial and temporal resolutionallows whole body scanning
Magnetic resonance imaging does not require cannot be used for patients with(MRI) ionising radiation metal-containing implants due to
large magnetic field strengths
1.1.2 Optical imaging
Optical technologies rely on the use of light and provide essential tools for non-
invasive, high-resolution imaging . [7,8] The detected light can either originate from
the target structure itself or from reporter molecules, so-called optical probes. In the
first case, the observed reflection of light or change of polarisation is translated into an
image. However, the commonly low intrinsic contrast of the investigated structures
often limits the amount of gained information. A much better signal-to-background
ratio and, thus, a higher degree of information can be achieved by the utilisation of
optical probes. The most frequently used reporter molecules are fluorescence probes,
which are molecules that emit light from an electronically excited state upon relaxa-
tion to a ground state. Advanced fluorescent probes for medical diagnostics possess
three functional parts: [3]
• a signalling component,
• a carrier with suitable pharmacokinetics,
• and a targeting moiety for the delivery to a specific structure.
Such probes are incorporated into the system of interest and are excited by an external
excitation source. Usually, the excitation and emission wavelengths of these probes
are in the visible region of the electromagnetic spectrum. The performance of optical
probes is often optimised by maximising or differentiating the target signal e.g. by
the use of activatable imaging probes.
According to the underlying chemical mechanisms, activatable imaging probes are di-
vided into three classes. [3] First, there are small molecules with an intrinsic activation
2

1 Introduction
mechanism (Figure 1.1a). They are highly specific and react with an analyte or un-
dergo an intramolecular alteration. This results in a modification in the absorption
or emission characteristics (e.g. intensity, wavelength). The second class comprises
large molecules, whose luminescence is quenched due to deaggregation (Figure 1.1b).
This process might be a result of changed environmental conditions (e.g. pH) or the
interaction with an enzyme. Such macromolecules can be selectively accumulated
in target structures, which results in high resolutions. However, due to their size,
they are slowly metabolised and slowly excreted from the body. The last class are
assemblies of macromolecular targeting moieties and small molecule signalling moi-
eties (Figure 1.1c). They combine the advantages of the two former classes for they
provide high resolutions after selective activation. Typical activatable imaging probes
are short peptides or nucleic acids strands. [9–11]
++
a)
b)
c)
Figure 1.1: Activatable optical imaging probes. Blue and red structures are intact and altered optical
probes, respectively. Grey spheres stand for non-luminescent molecules and white struc-
tures for analytes. a) Small molecules with an intrinsic activation mechanism. b) Large
molecules, whose luminescence is quenched due to deaggregation. c) Molecular assemblies
of macromolecules with small signalling moieties.
Optical imaging with suitable probes provides several advantages, e.g. high sens-
itivity, low toxicity, low cost, the possibility to target designated structures and the
option of selective signal activation or quenching. [3,12] Hence, this technique repres-
ents an interesting alternative to the use of radioisotopes, which are often employed
for the detection and quantification of molecules in a biological environment. [13–17]
Applications of fluorescence probes offer a broad selection of emission penetration
depths and spatial resolutions, ranging from micrometers to centimetres. Thus, ver-
satile structures from small viruses to eukaryotic cells and large tissues can be mon-
itored. Important optical in vivo imaging techniques are listed in Table 1.2.
3

1.2 Fluorescent probes for optical imaging
Table 1.2: Optical imaging techniques [8]
Technique Contrasta Depth Common Clinical
wavelength potential
Microscopic resolutionEpi A, Fl 20 µm visible experimentalConfocal Fl 500 µm visible experimentalTwo-photon Fl 800 µm visible yes
Mesoscopic resolutionOptical projection tomography A, Fl 15 µmb visible noOptical coherence tomography S 2 mm vis. + nIR yesLaser speckle imaging S 1 mm vis. + nIR yes
Macroscopic resolution intrinsicHyperspectral imaging A, S, Fl <5 mm visible yesEndoscopy A, S, Fl <5 mm visible yesPolarization imaging A, S <1.5 cm vis. + nIR yesFl. reflectance imaging (FRI) A, Fl <7 mm nIR yesDiffuse optical tomography (DOT) A, Fl <20cm nIR yes
Macroscopic resolution molecularFl. resonance imaging (FRI) A, Fl <7 mm nIR yesFl. molecular tomography (FMT) Fl <20 cm nIR yesBioluminescence imaging (BLI) E <3 cm 500-600 nm no
a A=absorption, E=emission, S=scattering, Fl=fluorescence. b In cleared specimen.
The development of clinical optical imaging techniques means the extension of ex-
isting assays and the attempt to translate in vitro or 2D applications into in vivo or
optical tomography (3D) techniques. The underlying methods are already limited
due to photobleaching of the probes as well as strong absorption, light scattering and
autofluorescence in biological specimens. The novel techniques also have to overcome
weak tissue penetration and master the challenge to obtain high-resolution images of
cells located within deep tissues. [12] Hence, optical imaging is still regarded a highly
experimental technique. [7] One option to solve the mentioned problems and to expand
applications from through-skin-visualisation of superficial tissues (e.g., breast, [18,19]
lymph nodes [20,21]) is the use of endoscopy [20,21] or surgery [22,23].
1.2 Fluorescent probes for optical imaging
1.2.1 Luminescence, fluorescence and phosphorescence
Luminescence means the emission of light by a substance, when an electron returns
from an electronically excited state to the ground state. Depending on the spin before
and after this transitions, two categories are distinguished (Table 1.3).
4

1 Introduction
During fluorescence, transitions are spin-allowed (∆S=0) and, consequently, the emis-
sion rates are fast. In contrast, in the case of phosphorescence, the spin changes during
the transition from the triplet excited state (T1) to the ground state (S0). This process
is spin-forbidden (∆S>0) and, hence, emission rates are slow. Since the time-scale
of this process is significantly longer than the autofluorescence in biological systems,
time-resolved detection (TRD) procedures can be employed. During these experi-
ments, the background fluorescence is allow to decay to negligible levels before the
phosphorescence signal is measured. [24,25] A general problem of phosphorescence is
that the triplet state might be subject to photochemical reactions, which destroy the
luminophore (photobleaching).
Table 1.3: Comparison of different types of luminescence [24]
Fluorescence Phosphorescence
Spin selection rule∆S=0 ∆S>0
(allowed) (forbidden)
Transition path S0 → S1 → S1 S0 → S1ISC→ T1 → S0
Emission ratesfast slow
107 − 109 s−1 1− 103 s−1
Lifetimesshort long
0.1 µs − 1 ns 1 s − 1 ms
1.2.2 Properties of fluorescent probes
Most commonly, fluorescence probes are excited with photons from an external light
source and relax back to the ground state under the emission of usually less energetic
photons. [7,8] For this processes to be efficient, certain factors have to be taken into
account.
The wavelengths of absorption and emission are the first properties to be considered.
High energy radiation (blue, green), which in general can cause tissue damage, has
a short tissue penetration depth and is therefore only suitable for the imaging of
superficial structures. [26] Lower energy excitation (yellow, red) provides an increased
penetration depth, but also goes along with strong autofluorescence, since biological
specimens mainly absorb in this spectral region (Figure 1.2). [27] An advantageous
combination of deep tissue penetration and low autofluorescence can be achieve by
the use of near-IR radiation. However, it was also reported that its use can cause tissue
heating. [28] Obviously, the use of every sort of radiation possesses advantages and
5

1.2 Fluorescent probes for optical imaging
drawbacks and, therefore, wavelengths always have to be adjusted to the respective
specimen. Still, in every case the absorption and emission spectra should be separated
by large Stoke’s shifts to allow for an efficient optical separation of excitation and
fluorescence. [3]
Figure 1.2: Wavelength-dependent absorption coefficient of normally oxygenated tissue (saturation of
70%) with 50% water, 15% lipids and 50 mM hemoglobin concentration of 50 mM. [8]
The intensities of absorption and emission are also of importance. The absorption
intensity is a measure for the excitation probability. When it increases, less light is
required for the excitation of the probe, which simultaneously reduces the odds of
tissue damage. The fluorescence intensity, which is determined by the product of
quantum yield and extinction coefficient, reflects the brightness of a probe. With
higher fluorescence intensity, deeper penetration depths and higher signal-to-noise
ratios can be achieved. However, the latter is often only attained using larger emitters
(e.g. green fluorescent protein, quantum dots). [3] In addition to these photophysical
requirements, one also has to consider certain chemical properties of the probes.
Regarding the stability, the main problems are photobleaching and metabolisation.
Photobleaching can be reduced by decreasing the light intensity (e.g. by reducing
the number of scans), and simultaneous utilisation of highly sensitive video cameras
or photographic films. Another approach is the design photostable probes or repeat
probe injection. In vivo stability is much more difficult to achieve, because fluorescence
probes are often decomposed in the presences of lysosomes and enzymes and lose
their fluorescence properties. Still, for some compounds, degradation can be desired,
so they can be metabolised and excreted. [3,29]
6

1 Introduction
One last factor that should be considered is that fluorescent probes can induce
an alteration of pharmacokinetics. When several fluorophores or large molecules (e.g.
nanoparticles or quantum dots) are attached to the target structure, its bioavailabilty,
distribution and clearance can be significantly altered. As a consequence, size and
form have to be considered carefully, keeping in mind the target structure. [3]
1.2.3 Classification of fluorophores
Fluorescence probes can be divided into three major classes: genetically encoded
fluorophores, inorganic materials and small molecule fluorophores. [3]
The class of genetically encoded fluorophores [3,30,31] comprises fluorescent molecules
that naturally occur in eukaryotic cells. The most prominent examples is the green
fluorescent protein (GFP) (Figure 1.3a). The fluorophore of this protein, an imidazoline
moiety (λex 488 nm, λem 509 nm), is derived from sequential serine-tyrosine-glycine
residues, [32–34] and is embedded into a beta-barrel. Mutations or circular permuta-
tions of these or other amino acids result in new spectral variants e.g. blue fluores-
cence (Cyan Fluorescent Protein, CFP). [32,35] Other representatives of this group are
the endogenous yellow (YFP) and red fluorescent proteins (RFP), artificial endogen-
ous proteins with “unnatural wavelengths” (e.g. in the near-IR), and certain fusion
proteins. All these molecules are relatively large (30-50 kDa), which limits their tar-
geted delivery and potentially affects normal protein functions. Fluorescent proteins
have usually broad excitation and emission spectra as well as low quantum yields.
Still, they can be obtained without cumbersome synthetic procedures and possess ex-
cellent photostability. These important advantages mostly explain their routine use
in functional and molecular imaging e.g. for site-specific labelling of proteins or for
monitoring gene expression. However, translation of these applications for clinical
use is highly impracticable, since the DNA code for the respective protein needs to be
transferred into the host cells.
The most commonly used inorganic materials for bioimaging are quantum dots (QD)
(Figure 1.3b). [3,12,30,31] These are semiconductor nanocrystals, which have sizes ran-
ging from 2 to 50 nm, typically consist of CdSe or CdTe cores and a ZnS coating. [36]
Their emission wavelength can be tuned by changing the particle size, where an in-
crease in size results in a red-shift of emission. QD are commercially available in
various sizes with emissions across the visible spectrum. They possess superb photo-
physical properties, e.g. high photostability, high fluorescence quantum yields (>80%
in organic solvents), broad excitation spectra and narrow emission bandwidths. All
these factors contribute to excpetionally high signal-to-noise ratios. In addition, their
7

1.2 Fluorescent probes for optical imaging
functionalisation is also feasible e.g. to enhance water solubility or to attach bio-
molecules to them. [37] Therefore, QD have become regularly used fluorophores for
microscopy [38] and for the monitoring of analytes in living cells. [39] There are two
drawbacks which limit the use of quantum dots. On the one hand, they contain
heavy metals (Se, Cd), which are potentially cytotoxic. On the other hand, their size
often exceeds the renal excreation limit (~6 nm). Instead, they are mostly excreted
through liver and bile without metabolism which implies a prolonged residence time
in the blood. If they are attachted to targetting moieties, the clearance from the body
is even more delayed. In addition to quantum dots, carbon nanotubes and gold nan-
oparticles are also promising inorganic materials for in vivo imaging. However, they
also raise concerns regarding biodegradability and toxicity, which still impedes their
clinical use. [40] Other important inorganic materials are lanthanide-based luminescent
nanoparticles, which will be discussed in section 1.3.3.
10 Å
CdSe coreZnS shell
β-barrel
imidazoline
10 Å
a) b) c)
OHN NH
O
O
Cl
ca. 20-500 Å
Figure 1.3: Typical representatives for different classes of fluorophores. (a) The green fluorescent
protein [41] (PDB Code 1GFL, genetically encoded fluorophore), (b) a simple quantum dot [42]
(inorganic materials) and (c) Rhodamine 6G (small-molecule fluorophore).
The class of small-molecule fluorophores [3,31,43] comprises a large number of organic and
inorganic compounds with low molecular weights (300-2000 g/mol). The majority
of them are commercially available organic dyes with core structures of cyanines [44],
boradiazaindacenes [45], coumarins [46], and xanthenes [47,48]. Well-known representat-
ives of organic fluorophores are fluoresceins and rhodamines (Figure 1.3c), which are
both derived from a xanthene motif. Small molecule fluorophores can have emis-
sion wavelengths from blue to near-infrared, and thus cover the greater part of the
electromagnetic spectrum. Depending on the molecule, the emission wavelength or
intensity can also be influenced by pH, solvent or the presence of metal ions. This
sensitive emission as well as their small size and monodispersity make them versatile
8

1 Introduction
tools for biomolecular labelling, cellular staining or the indication of environmental
factors. However, limitations may arise from their susceptibility to photobleaching,
lacking stability under physiological conditions, strong background fluorescence in
the visible spectrum and broad emission bands. [49]
Prominent examples for inorganic small-molecule fluorophores are transition metal
complexes such as [Ru(bpy)3]Cl2, and lanthanide complexes with macrocyclic lig-
ands, such as cryptates [50–53] (Figure 1.4a) or 1,4,7,10-tetraazacyclododecane-1,4,7,10-
tetraacetic acid (DOTA) [54,55] (Figure 1.4b). Lanthanides exhibit elementspecific lu-
minescence, which covers the whole spectral range from UV to near-IR. However,
they possess low absorption coefficients (<10 M−1 cm−1) due to Laporte-forbidden
transitions within the 4f orbitals. [56] As a result, excited states are not populated read-
ily, leading to weak emission intensities and limiting the detection level. Hence, the
surrounding of the lanthanide has to be equipped with a strongly absorbing chromo-
phore. This can be the ligand itself or a moiety attached to it. [57]
N
N
N
NN
N
N
N
Ln
3+ -
N N
NN
O
O
O
O
O
OO
O Ln
a) b)
Figure 1.4: Lanthanide-based small molecule fluorophores. Structures of lanthanide complexes with
a trisbipyridine (TBP) cryptand (a) and a DOTA ligand (b).
Irradiation of this "antenna" at a suitable wavelength leads to the excitation of a
singlet excited state (S0), followed by intersystem crossing to a triplet excited state (T1)
(Figure 1.5). Subsequent energy transfer to matching lanthanide emissive states res-
ults in efficient sensitation of luminescence (Figure 1.5c). [58] The observed lanthanide
emission bands are narrow (full width half maximum <12 nm), [59] since the electronic
states are not subject to coupling with vibrational modes. Moreover, the f-electrons are
shielded by outlying 5s and 5p electrons, so that ligand field effects marginally influ-
ence the spectroscopic properties. Consequently, lanthanides are mostly insensitive to
changes in their coordination sphere. Concentration dependent self-absorption does
also not occur to a great extend, since absorption and emission bands are separated
by large Stoke’s shifts (~200 nm). [24,25] Overall, their unique photophysical properties
make molecular lanthanide complexes excellent candidates for optical probes, which
can even be used for time-resolved measurements.
9

1.3 Near-IR optical imaging with lanthanides
Ligand Ln3+
S1
T1
S0
4f*
emis
sion
abso
rptio
nEne
rgy
Figure 1.5: Jablonski diagram that illustrates the antenna effect. The dashed-dotted, dashed, dotted,
and full arrows represent photon excitation, energy transfer, multiphonon relaxation, and
emission processes, respectively.
Thus, they form a low-cost alternative to inorganic materials. Typical applications
of luminescent lanthanide complexes are DELFIA (dissociation enhanced lanthanide
fluorescence immunoassay) and time-resolved FRET (fluorescence resonance energy
transfer) assays. [60–62] The biggest challenge in employing lanthanide complexes is to
find suitable antenna ligands and protect the lanthanide efficiently from quenching
molecules in its proximity.
1.3 Near-IR optical imaging with lanthanides
Optical imaging techniques that rely on lanthanide luminescence often employ probes
that emit in the visible region e.g. Eu3+ or Tb3+ complexes. [15–17,63,64] However, there is
a growing interest in the combination of the excellent photophysical properties of the
lanthanides with the advantages of near-IR radiation. [65,66] As mentioned previously,
the usefulness or near-IR light in bioimaging arises from its deep penetration depth
and the minor autofluorescence in this spectral window (650-900 nm). Furthermore,
the light scattering decreases with increasing wavelengths, which can also contribute
to a higher sensitivity of the probe.
There are two different options to combine the merits of lanthanide luminescence and
near-IR radiation. On the one hand, near-IR emission e.g. from Yb3+, Nd3+ and Er3+
complexes can be sensitised, using biologically compatible, visible light. [67–70] On the
other hand, lanthanides can convert near-IR long-wavelength excitation into shorter-
wavelength emission through upconversion processes. In contrast to the former case,
such systems (usually inorganic materials) are preferably used for bioimaging, since
their emission is less prone to non-radiative deactivation and, simultaneously, they
provide the chance to excite and detect in the near-IR region. [49,71]
10

1 Introduction
1.3.1 Upconversion processes
During simple linear optical processes (Figure 1.6 left), an activator in its ground state
absorbs a photon, which leads to the population of an excited state. Radiative de-
activation then results in the emission of light. It is also possible, that the activator
interacts with a nearby sensitiser to get promoted to an excited state, while the sens-
itiser relaxes to its ground state.
In a non-linear optical process (Figure 1.6 right), the activator is first promoted to
a metastable excited level by absorption of a pump photon. Non-radiative energy
transfer to this system by a nearby sensitiser leads to the promotion of the activator
to a higher excited state. [72–75] The sequential absorption of two or more photons of
lower energy (e.g. near-IR) thus produces emission of higher energy (e.g. visible
light). This process is referred to as upconversion (UC) luminescence. It is considered
a non-linear process, since the luminescence intensity is proportional to the square of
the laser power (I ∝ P2).
G
E1
G
E1
G
E1
E2
Ene
rgy
Upconversion luminescenceLuminescence
AS AS
Figure 1.6: Energy schemes illustrating the difference between linear and non-linear optical pro-
cesses. A and S stand for activator and sensitiser, respecivley. Ground and excited states
are marked with G and E1 or E2, respectively. In simple systems, activators are excited dir-
ectly (left) or by energy transfer from a sensitiser (middle). Upconversion can be achieved
by energy transfer upconversion (ETU, right). The dashed-dotted, dashed, dotted, and full
arrows represent photon excitation, energy transfer, multiphonon relaxation, and emission
processes, respectively.
1.3.2 Activators and sensitisers
In general, all lanthanides with multiple long-lived metastable excited levels can act
as activators. However, the energetic distances between ground state, metastable in-
termediate level and excited level have to be approximately the same for an efficient
energy transfer (Figure 1.7). For example, in an Er3+ ion the energy gaps between
11

1.3 Near-IR optical imaging with lanthanides
4 I15/2 and 4 I11/2 (10 350 cm−1) and between the excited states 4 I11/2 and 4F7/2 (10 370
cm−1) are very similar. Hence, 4F7/2 can be populated readily by non-linear energy
transfer from a sensitiser. Subsequent non-radiative deactivation leads to the popu-
lation of lower-lying metastable levels (e.g. 2H11/2, 4S3/2, 4F9/2), which relax to the
ground state under UC emission. In addition to Er3+, the lanthanides Tm3+ and Ho3+
are also frequently used activators. [49]
The sensitiser, which can be either a lanthanide, a transition metal or an organic
chromophore has to be located in the proximity of the activator. The most often used
sensitiser is Yb3+, because it has just one excited level and a higher absorption cross
section than most other lanthanides. Yb3+ absorbs low-energy photons at around
980 nm due to the 2F7/2 → 2F5/2 transition, which is resonant with f-f-transitions of
Er3+, Tm3+ and Ho3+.
Particularly interesting combinations for bioimaging are Yb/Er (λexc 980 nm, λem
655 nm) and Yb/Tm (λexc 980 nm, λem 800 nm), which have both, excitation as well
as emission wavelengths, in the near-IR. [49,59,76]
4F7/2
2H11/24S3/2
4F9/2
4I9/2
4I11/2
4I13/2
4I15/22F7/2
2F5/2
0
5
10
15
20
E/(
10
3cm
-1)
Yb3+ Er3+
980
nm
655
nm
540
nm
520
nm
Figure 1.7: Energy transfer mechanisms for UC processes involving an Yb3+ sensitiser and an Er3+
activator using 980 nm excitation. The dashed-dotted, dashed, dotted, and full arrows
represent photon excitation, energy transfer, multiphonon relaxation, and near-IR emission
processes, respectively.
Regardless of the specific lanthanide combination, it is important that the sensitiser
content is much lower than the activator content to avoid an energy loss during cross-
relaxation. In addition, non-radiative deactivation by other molecules (e.g. solvents)
has to be avoided. In practice, this is achieved by the controlled doping of inorganic
matrices with lanthanides. [49,55]
12

1 Introduction
1.3.3 Upconversion nanophosphors
Upconversion nanophosphors are lanthanide-doped crystalline materials, [38] that can
have different shapes (e.g., nanospheres, nanorods, nanocubes or nanoplates) and
sizes (sub-nm to sub-µm range). Under continuous wave excitation at 980 nm, they
generate violet to near-IR emission. Their UC efficiency and emission colour can be
adjusted by the choice of host lattice (material, structure) and dopant ions (concentra-
tion, combination). [59,76,77]
The host lattice has to meet several requirements, such as consistent defined shape
and nanoscale size, low cytotoxicity, water stability and low lattice phonon ener-
gies to prevent non-radiative deactivation processes. Commonly used host matrices
are transparent crystalline materials based on fluorides, oxides, chlorides, bromides,
oxysulfides, phosphates or vanadates. These materials also contain alkali metals (e.g.
Na+) or rare earth elements (e.g. Y3+, La3+, Gd3+) as counter ions. Among the re-
ported lattices, hexagonal 100 nm [NaYF4:Yb,Er] nanocrystals proved to be the most
efficient matrix with absolute UC luminescence efficiencies of up to 0.3%. The dopants
are embedded into the host lattice in relatively low concentrations (usually <2 mol%)
to create sufficient distances between neighbouring ions. [49,60,76]
The most important benefits of nanophosphors are their photostable luminescence
and minimised background autofluorescence. Additionally, they can be excited with
an inexpensive continuous wave near-IR diode laser and in comparison with quan-
tum dots, they possess low toxicity. Drawbacks, which limit their standardised use are
the lack of generalised protocols for synthesis and modification, and also the low UC
luminescence efficiency due to vibrational deactivation and surface defects. [59]
Nevertheless, upconversion nanophosphors are already in use in several applications
as alternatives to quantum dots or organic fluorophores. They are used in vitro for
immunochromatographic assays, bioaffinity assays, and homogeneous bioanalytical
assays based on FRET. [49,76,77] Since their emission has a deeper penetration depth
than quantum dots, upconversion nanophosphors were successfully applied in vivo
for small-animal imaging e.g. to visualise tumours, the lymphatic and vascular system
or to track cells. [59]
1.3.4 Upconversion in molecular lanthanide complexes
There are only few precedences for systems, which combine the advantages of small
molecule fluorophores with the benefits of upconverted near-IR emission. In com-
parison with UC nanophosphors, the reported compounds also contain a lanthanide
13

1.3 Near-IR optical imaging with lanthanides
activator, but the sensitisers are either organic chromophores or transition metals.
When organic chromophores are employed as sensitisers, multiphoton absorption of
the organic moiety causes the population of its singlet excited state. By definition, this
intersystem crossing and energy transfer to matching lanthanide emissive states (e.g.
of Eu, Tb, Nd, Er, Tm) then results in sensitised emission (Figure 1.8a). This process
displays a special variant of the antenna effect, for which low energy radiation can
be employed. It was observed for homonuclear lanthanide complexes and also in
lanthanide coordination polymers. [78–82]
In the recently reported heteronuclear complex developed by Piguet et al., a trans-
ition metal served as the sensitiser. Its development was the result of a sophisticated
molecular design, which took into account three requirements for efficient upconver-
sion processes. First, the activator has to be surrounded by at least two equidistant
sensitisers. Second, the activator has to be protected from high-frequency oscillators
to reduce energy loss due to non-radiative relaxation of 4f excited levels.
sensitiser activator
Si S1 T1
S0
sensitiser sensitiser
a) b)
4f*
emis
sion
activator
NN
N N
N
NN NN
NNCr
Er
Cr
540 nm
750 nm 750 nm
Ligand Ln3+
Figure 1.8: Upconversion luminescence in molecular systems. (a) Jablonski diagram that illustrates
molecular upconversion complexes with organic sensitisers. The dashed-dotted, dashed,
dotted, and full arrows represent photon excitation, energy transfer, multiphonon relax-
ation, and emission processes, respectively. (b) Heteronuclear upconversion complex de-
veloped by Piguet et al., in which Cr3+ served as the sensitiser. Two of the three ligands are
omitted for clarity. [83]
Third, the excited state lifetimes of the sensitiser have to be long enough for the in-
tramolecular energy transfer to occur. In the reported complex, a central Er3+ activator
is embedded between two peripherical chromium(III) sensitisers (Figure 1.8b). The
latter possess a sufficiently long excited state lifetimes to enable the energy transfer to
the activator. Hence, when the transition metal is irradiated with near-IR light, the in-
termetallic non-linear energy transfer results in lanthanide-centered near-IR emission.
14
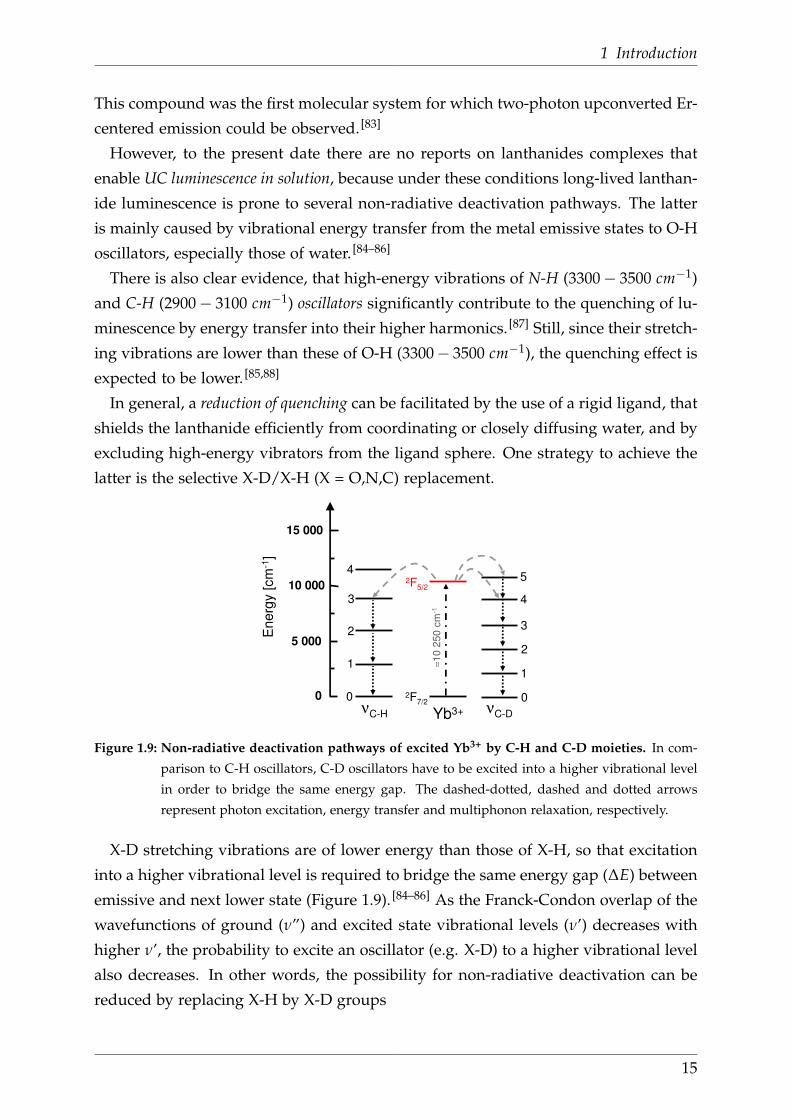
1 Introduction
This compound was the first molecular system for which two-photon upconverted Er-
centered emission could be observed. [83]
However, to the present date there are no reports on lanthanides complexes that
enable UC luminescence in solution, because under these conditions long-lived lanthan-
ide luminescence is prone to several non-radiative deactivation pathways. The latter
is mainly caused by vibrational energy transfer from the metal emissive states to O-H
oscillators, especially those of water. [84–86]
There is also clear evidence, that high-energy vibrations of N-H (3300− 3500 cm−1)
and C-H (2900− 3100 cm−1) oscillators significantly contribute to the quenching of lu-
minescence by energy transfer into their higher harmonics. [87] Still, since their stretch-
ing vibrations are lower than these of O-H (3300− 3500 cm−1), the quenching effect is
expected to be lower. [85,88]
In general, a reduction of quenching can be facilitated by the use of a rigid ligand, that
shields the lanthanide efficiently from coordinating or closely diffusing water, and by
excluding high-energy vibrators from the ligand sphere. One strategy to achieve the
latter is the selective X-D/X-H (X = O,N,C) replacement.
Ene
rgy
[cm
-1]
≈10
250
cm
-1
Yb3+
2F7/2
2F5/2
0
5 000
10 000
15 000
4
3
2
1
0νC-H
4
3
2
1
0
5
νC-D
Figure 1.9: Non-radiative deactivation pathways of excited Yb3+ by C-H and C-D moieties. In com-
parison to C-H oscillators, C-D oscillators have to be excited into a higher vibrational level
in order to bridge the same energy gap. The dashed-dotted, dashed and dotted arrows
represent photon excitation, energy transfer and multiphonon relaxation, respectively.
X-D stretching vibrations are of lower energy than those of X-H, so that excitation
into a higher vibrational level is required to bridge the same energy gap (∆E) between
emissive and next lower state (Figure 1.9). [84–86] As the Franck-Condon overlap of the
wavefunctions of ground (ν”) and excited state vibrational levels (ν’) decreases with
higher ν’, the probability to excite an oscillator (e.g. X-D) to a higher vibrational level
also decreases. In other words, the possibility for non-radiative deactivation can be
reduced by replacing X-H by X-D groups
15

2 Concept of the project
The long-term goal of this project is the development of a monodispers, molecular
upconversion system in form of a binuclear lanthanide complex (Figure 2.1). The
covalent linkage of the two complexes brings the activator and the sensitiser of an
upconversion system in a close and defined distance from each other, independent
from the concentration of the binuclear compound in solution. The envisaged system
should be excited with near-IR radiation, followed by emission of light of shorter
wavelengths.
655 nm980 nm
Yb Er
D
D D
D
Figure 2.1: Model for a molecular upconversion complex.
The molecular upconversion complex has to meet two important requirements. On
the one hand, non-radiative deactivation by X-H oscillators in the proximity of the
lanthanide has to be minimised. On the other hand, sufficient stability of the lanthan-
ide complexes in a biological environment has to be ensured. Both challenges can be
addressed by employing tris(bipyridine) (TBP) cryptands (Figure 2.2).
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
Figure 2.2: Molecular structure of a dinuclear TBP cryptand
16

2 Concept of the project
These ligands reportedly form kinetically stable complexes with different lanthan-
ides and sensitise their emission. [50,89,90] Furthermore, TBP cryptands efficiently pro-
tect the coordinated metal from interactions with surrounding solvent molecules,
which potentially cause luminescence quenching. [52] These ligands are also devoid
of N-H and O-H oscillators and convenient protocols for bipyridine perdeuteration
can be employed for the replacement of C-H groups. [91]
As part of the development of such molecular upconversion systems, the achievement
of three specific goals was aimed in this work.
• First, the evaluation of the influence of C-H oscillators on near-IR emissive
lanthanide TBP cryptates. This implies the synthesis and photophysical invest-
igation of different deuterated lanthanide TBP cryptates.
• Second, the introduction of functional groups to TBP cryptates.
• Third, the selective coupling of suitably functionalised TBP cryptates.
17

3 Development of binuclear cryptates
The envisaged binuclear system should be accessible from functionalised, deuterated
tris(bipyridine) cryptates. As mentioned before, these cryptates possess a high kinetic
stability and efficiently sensitise lanthanide luminescence. [50,89] These properties can
both be further improved by the introduction of N-oxides (Figure 3.1). [92,93] While
initial studies primarily employed the simple compound, later experiments were con-
ducted with the rigid, oxidised analogue. It is worth mentioning, that the introduction
of an axially chiral bipyridine-N,N’-dioxide moiety to the helical structure of such a
cryptate results in the formation of two different enantiomers. Still, within this work
only the racemic mixtures thereof were synthesised.
N
N
N
N
N
N
N
NOO
N
N
N
N
N
N
N
N
Figure 3.1: Structures of tris(bipyridine) (TBP) cryptates. Left: (bpy.bpy.bpy). Right: (bpy.bpy.bpy)O2 .
The strategy for the development of binuclear cryptates comprised the success-
ive preparation and characterisation of functionalised and deuterated cryptates (Fig-
ure 3.2). At a later stage, coupling experiments to the binuclear compound had to be
conducted.
a) b) c)
D
D
D
D
Figure 3.2: Milestones for the development of binuclear cryptates The blue and grey structures stand
for TBP cryptands, the yellow spheres stand for a coordinated metal. a) Deuterated cryptate.
b) Cryptate with a functional group. c) Deuterated cryptate with a functional group.
18

3 Development of binuclear cryptates
First, a deuterated cryptand (Figure 3.2a) had to be prepared. The apparent ap-
proach was to apply literature known protocols for perdeuteration, [91] and to find
novel procedures for the partial deuteration of bipyridines. These deuterated build-
ing blocks were assembled to the desired cryptands, whose effect on the luminescence
performance of different near-IR emissive lanthanides was evaluated.
Second, suitable functional groups that allow for selective coupling reactions were
introduced to the cryptand (Figure 3.2b). Since reportedly carboxyl functionalised
cryptates can be successfully coupled to other molecules (e.g. antibodies, DNA, pro-
teins), [94] a carboxylic acid ester was regarded as most promising functional group
for this work. Amino groups can also enable coupling to amino acids or to carboxyl
functionalised cryptates via an amide bond and were therefore chosen as alternative
functional group. While the synthesis of carboxylic acid ester derivatives is already
known in the literature, [95,96] novel synthetic pathways for the preparation of amino
functionalised cryptands had to be developed.
For the preparation of the rigid, functionalised cryptates, again the chirality had
be taken into account. There are two possibilities to incorporate an N,N’-dioxide and
a functional group into one cryptate (Figure 3.3). In the first case, one bipyridine
features both structural elements, so that the axially chiral bipyridine represents the
only stereogenic element within the cryptate. Consequently, racemic mixtures are
obtained. In the second case, the N,N’-dioxide and the functional group are attached
to different bipyridines. Hence, all three bipyridine moieties are inequivalent and
one of them is unsymmetrical, which creates an unusual form of chirality. The latter
adds to the axial chirality of the N,N’-dioxide, so that mixtures of diastereomers are
expected which were, however, not aimed to be separated within this work.
N
N
N
N
N
N
N
N
X
OO
N
N
N
N
N
N
N
NOO
X
Figure 3.3: Possible structures of functionalised, rigid TBP cryptates. When N,N’-dioxide and a func-
tional group X are locate at the same bipyridine, racemic mixtures are obtained (left). When
they are located at different bipyridines different diastereomers result (right).
Third, a functionalised, deuterated cryptand had to be synthesised (Figure 3.2c).
Also, the consistency of the photophysical properties of the corresponding lanthanide
complexes, compared to the unfunctionalised counterpart, had to be verified.
19

Last, protocols for the selective coupling of two functionalised cryptands had to be
developed (Figure 3.4). In case of the cryptates that carry an N,N-dioxide moiety, the
coupling to a binuclear compound is anticipated to further increases the stereochem-
ical complexity.
+
Figure 3.4: Selective coupling of two cryptates. The grey structures stand for TBP cryptands, the
yellow spheres represent any coordinated metal.
20

3 Development of binuclear cryptates
3.1 Deuterated cryptates
3.1.1 Retrosynthetic analysis
Deuterated TBP cryptands can be derived from deuterated building blocks, which can
be assembled in two different ways. On the one hand, it is possible to react a bipyrid-
ine moiety with an azamacrocycle, where the latter is obtained from two identical
bipyridine building blocks (Scheme 3.1 top). On the other hand, three bipyridines
with aminomethyl and bromomethyl groups can be connected directly to the target
compound in one step (Scheme 3.1 bottom).
N
NH2N
H2N
N
NBr
Br
2 +
N
N
N
N
N
N
N
N
2N
N
N
N
N
N
H
H
N
NBr
Br
+N
NBr
Br
OR
[Dx] [Dy] [Dy] [Dy]
[Dx] [Dy]
[Dz]
Scheme 3.1: Retrosynthetic analysis of a deuterated TBP cryptand. Dx, Dy and Dz stand for any
deuteration level
Both strategies have to be employed to combine different deuterated and undeuter-
ated bipyridines and, thus, preparing a series of isotopologic cryptates. As candid-
ates for deuterated building blocks, perdeuterated bipyridines and those deuterated
in benzylic positions were considered as the most promising (Figure 3.5).
N
NZ
Z
D D
D D
N
N
DD
DD
D
D
Z
Z
D D
D D
N
NZ
Z
OR
[Dx/y]
Figure 3.5: Required deuterated bipyridine building blocks. Dx,y stands for the deuteration level (D4
or D10) and Z for Br or NH2.
21

3.1 Deuterated cryptates
3.1.2 Synthesis of building blocks and cryptates
Undeuterated bipyridines
The simple, undeuterated building blocks were prepared from commercially available
2-amino-6-picoline (1) (Scheme 3.2), starting with a Sandmeyer reaction. In accord-
ance to the literature procedure, 1 was diazotised in the presence of NaNO2 and a
mineral acid (HBr) to generate a labile diazonium salt. The diazonium salt directly
underwent a SNAr reaction with Br− to give 2 in 58% yield after distillation. [97]
N
N
N
N
N
N
Br
Br
N
Z
a
b c d
Z=NH2
Z=Br
OO
N
N
NH
HN
N
N
e
1
2
3 4 5 6
Scheme 3.2: Synthesis of undeuterated building blocks. (a) i. HBr, Br2, rt→-20 °C; ii. NaNO2, NaOH,
-20 °C→rt (58%). (b) i. dry toluene, Raney-Ni, reflux; ii. H2O, 40 °C (17%). (c) mCPBA,
CHCl3, 0 °C→rt (80%). (d) i. TFA anhydride, dry CH2Cl2, reflux; ii. dry DMF/THF, LiBr,
rt (48%). (e) i. EtOH, tosylamide monosodium salt, reflux; ii. conc. H2SO4, 110 °C (57%).
In the next step, 2 was subjected to a Nickel catalysed reductive coupling to give
3. This reaction proceeded in two steps (Scheme 3.3). First, The two-fold oxidative
addition of 2-bromo-6-picoline (2) to Ni0 and coupling of the pyridine rings gave a
NiI I complex, which was hydrolysed to release of 6,6’-dimethyl-2,2’-bipyridine (3). [98]
N Br N N N NNi
Br Br
Raney-Ni H2O
Ni(H2O)2Br2
2 3
Scheme 3.3: Nickel-catalysed reductive coupling of 2-bromo-6-picoline (2) to 6,6’-dimethyl-2,2’-
bipyridine (3).
3 was then treated with meta-chloroperbenzoic acid (mCPBA) to form the corres-
ponding N,N’-dioxide 4. Traces of the reactant and the mono-oxidised side product
were removed by column chromatography to afford the pure product in an excellent
yield of 80%. [99]
22

3 Development of binuclear cryptates
The obtained N,N’-dioxide 4 was treated with trifluoroacetic acid (TFA) anhydride
to generate a trifluoroacetate species via a Boekelheide rearrangement. The resulting
reactive intermediate had to be handled under the strict exclusion of moisture to
avoid the hydrolysis to the corresponding, non-reactive alcohols. Following addition
of LiBr at room temperature yielded 5 in 48% yield after column chromatography. [100]
This nucleophilic substitution requires the strict exclusion of water and the use of an
aprotic solvent like DMF to solvate the lithium ions and generate “naked”, highly
nucleophilic bromide ions.
Finally, two equivalents of 5 were assembled to an azamacrocycle in a two-step
procedure. [101] For this purpose, an ethanolic solution of the bipyridine building block
(5) was first heated at reflux in the presence of tosylamide monosodium salt. [101] After
isolation of the tosylated macrocycle, the amine was then deprotected under acidic
conditions (H2SO4) to give the desired macrocycle 6 in 57% yield.
Partially deuterated bipyridines
The partially deuterated bipyridine units were derived from previously prepared 6,6’-
dimethyl-2,2’-bipyridine (3) (Scheme 3.4). At the beginning, 3 was converted into the
corresponding dicarboxylic acid 7 via a CrO3-mediated oxidation in the presence of
H2SO4. [102]
N
N
N
N
Z
Z
a
b Z=OH
Z=OEt
N
N
Z
Z
c
d Z=OH
Z=Br
O
O
DD
DD
eZ=NH2 3HBr H2O
3 7
8
9
10
11
Scheme 3.4: Synthesis of building blocks deuterated in benzylic position. (a) conc. H2SO4, 65 °C,
CrO3, 70 °C. (b) EtOH, conc. H2SO4, reflux (73% over two steps). (c) NaBD4, CD3OD,
rt (65%, >99%D). (d) PBr3, dry DMF, rt (92%, 98%D). (e) i. CHCl3, urotropine, re-
flux; ii. H2O/EtOH/HBr), 75 °C→rt (61%, 97%D).
Subsequent treatment with EtOH under acidic conditions gave the corresponding
ethyl ester 8 [103] in 73% over two steps. In the following key step, the ester was
reduced with NaBD4 in CD3OD to generate a bipyridine with selectively deuterated
23

3.1 Deuterated cryptates
benzylic positions. The pure product (9) was obtained in 65% yield after column
chromatography. The overall deuteration level of >99% was confirmed using ESI
mass spectrometry.
Nucleophilic substitution of the hydroxyl groups by bromine was achieved by treat-
ment of 9 with PBr3 in DMF and gave 10 in an excellent yield of 92% (98%[D]). Fi-
nally, a Delépine reaction was used for the synthesis of 11. [104] This reaction pro-
ceeded via an SN2 reaction of 10 with urotropine, followed by acidic hydrolysis
(HBr/EtOH/H2O) of the urotropine salt and gave the desired primary amine (61%
yield, 97% [D]).
Remarkably, the deuteration levels of the cryptate building blocks 10 and 11 are just
a little lower than for 9, although no deuterated solvents or reagents were used after
the reaction with NaBD4.
Perdeuterated bipyrdines
All perdeuterated compounds were prepared employing procedures developed by
J. Wahsner (Scheme 3.5). [105] Their synthesis started with the base-catalysed deu-
terium exchange of previously synthesised N,N’-dioxide 4. In analogy to the pro-
cedure for 2,2’-bipyridine, this reaction was performed in an autoclave with D2O as
deuterium source and NaOD as base. [91]
c Z=OAc
Z=OHd
Z=Br
N
N
N
N
Z
Z
a bN
N
e
[D10][D12]
OO
OO
Z=NH2 3HBr H2O
4 12 13
14
15
16
Scheme 3.5: Synthesis of benzylic deuterated building blocks. (a) NaOD (40 wt% in D2O), D2O,
autoclave, 150 °C (78%,>99%D). (b) Ac2O, reflux (57%, 98%D). (c) K2CO3, dry EtOH, rt
(57%, 98%D). (d) PBr3, dry DMF (8 ml), rt (53%, 99%D). (e) i. CHCl3, urotropine, re-
flux; ii. H2O/EtOH/HBr), 75 °C→rt (73%, 98%D).
After a total reaction time of four days, the crude product was purified with column
chromatography to give the desired perdeuterated compound 12 in 78% yield. The
excellent overall-deuteration level of over 99% was confirmed by ESI mass spectro-
24

3 Development of binuclear cryptates
metry (Figure 3.6). Despite no deuterated reagents or solvents were used after the
H/D exchange, deuteration levels did not decrease significantly during the following
four synthetic steps.
1 5 3 1 7 0 1 8 7 2 0 4 2 2 1 2 3 8 2 5 5 2 7 2 2 8 9
2 2 8 2 2 9 2 3 0 2 3 1
O ON N
D 1 2
E x a c t m a s s : 2 2 8 . 1 7
m / z
2 2 8 . 9 8[ M + H ] +
Figure 3.6: ESI+ mass spectrum of 12 after deuteration in an autoclave with D2O/NaOD, which con-
firms an overall deuteration level >99%.
In analogy to the procedures for the undeuterated compound, [99] 12 was reacted
in a standard Boekelheide rearrangement with acetic anhydride to give 13, followed
by saponification with K2CO3 in EtOH provided the alcohol 14 in 56% yield after
column chromatography. [105] Nucleophilic substitution (SN2) of the hydroxyl groups
by treatment with PBr3 gave the brominated analogue 15 in 53% yield (99% [D]) after
purification. [105] Last, 15 was subjected to a Delépine reaction, which provided the
bis(methylamine) 16 in 73% yield with an overall deuteration level of 98%. [105] Slow
cooling of the reaction mixture also afforded crystals, which were suitable for a X-ray
single crystal structure determination (Figure 3.7).
Figure 3.7: X-ray crystal structure of [D10]-6,6’-bis(aminomethyl)-2,2’-bipyridine trihydrobromide hy-
drate (16, monoclinic, P21/n). Ellipsoids are drawn at 50% probability level.
25

3.1 Deuterated cryptates
Synthesis of isotopologic cryptates
With the necessary building blocks in hand, a series of isotopologic sodium cryptates
was prepared (Scheme 3.6). As depicted, the [D4]-cryptate 17 was obtained by the
reaction of bipyridine moiety 10 with macrocyle 6.
N
N
N
N
N
N
N
NNa Br
+N
N
NH
HN
N
N
+N
N
Br
Br[Dx]
H2N
H2N
N
N
[Dy]
x=4
x=10
x=4
x=10
2
[Dy][Dx]
[Dx]
x=4, y=0
x=y=4
x=y=10
x=4 x=0
a
a
N
N
Br
Br[D4]
N
N
N
N
N
N
N
NNa Br
[D4]
10
10
10 6
15
18
11
16
17
19
20
21
Scheme 3.6: Synthesis of the isotopologic cryptates. Top: [D4]-(bpy.bpy.bpy). Bottom: [D8]-, [D12]-,
and [D30]-(bpy.bpy.bpy). (a) Na2CO3, CH3CN, reflux (40-58%,>97%[D]).
On the contrary, the [D8]-, [D12]-, and [D30]-analogues 19, 20, and 21 were obtained
from 6,6’-bis(bromomethyl)- and 6,6’-bis(aminomethyl) bipyridines as illustrated. In
all cases, suspensions of the respective building blocks with Na2CO3 in CH3CN were
heated at reflux and, after purification by column chromatography, products were
isolated in yields between 40% and 58% with deuteration levels >97%. In comparison,
previously reported deuterated DOTA ligands did not exceed a deuteration degree of
93%. [88] One exemplary ESI mass spectrum for 21 is shown in Figure 3.8.
Finally, the sodium cryptates were converted into the corresponding lanthanide
complexes by refluxing them in CH3CN in the presence of YbCl3·6H2O or NdCl3·6H2O,
respectively. The obtained complexes were characterised by analytical HPLC as well
as positive ESI mass spectrometry.
26

3 Development of binuclear cryptates
2 0 0 4 0 0 6 0 0 8 0 0 1 0 0 0
6 2 4 6 2 6 6 2 8 6 3 0
N
N
NN
N
N
N
N N a
[ D 3 0 ]E x a c t m a s s : 6 2 7 . 4 4
m / z
6 2 7 . 2 5[ M ] +
6 2 7 . 2
6 2 6 . 3
6 2 4 . 3
6 2 8 . 2
6 2 9 . 3
6 2 5 . 3
Figure 3.8: ESI+ mass spectrum of 21, which confirms an overall deuteration level of 98%
In this way, two sets of isotopologic [Dx]-TBP lanthanide cryptates (Ln = Yb, Nd)
were obtained. For the completion of the series (Figure 3.9), the [D0]-, [D10]- and
[D20]-TBP cryptates were also prepared within the Seitz group. [105]
3+
Ln = Nd, Yb
[Dx] = [D0] [D4], [D8], [D12] [D10], [D20], [D30]
N
N
N
N
N
N
N
NLn
[Dx]
Figure 3.9: Series of isotopologic lanthanide cryptates. The [D4]-, [D8]-, and [D12]-TBP cryptates
possess either one, two or three benzylically deuterated bipyridines. [D10]-, [D20]-, and
[D30]-TBP cryptates contain either one, two or three perdeuterated bipyridines.
3.1.3 Photophysical properties
For the investigation of the luminescence behaviour of the selectively deuterated
cryptates, [D6]-DMSO for [Dx]-Nd and D2O for [Dx]-Yb were chosen as solvents in or-
der to minimize quenching effects in solution. Thus, the most intensive luminescence
signals with monoexponential decay profiles could be measured. First, emission spec-
tra of the lanthanide cryptates were recorded, which showed the expected bands in
the near-IR region (Figure 3.10).
27
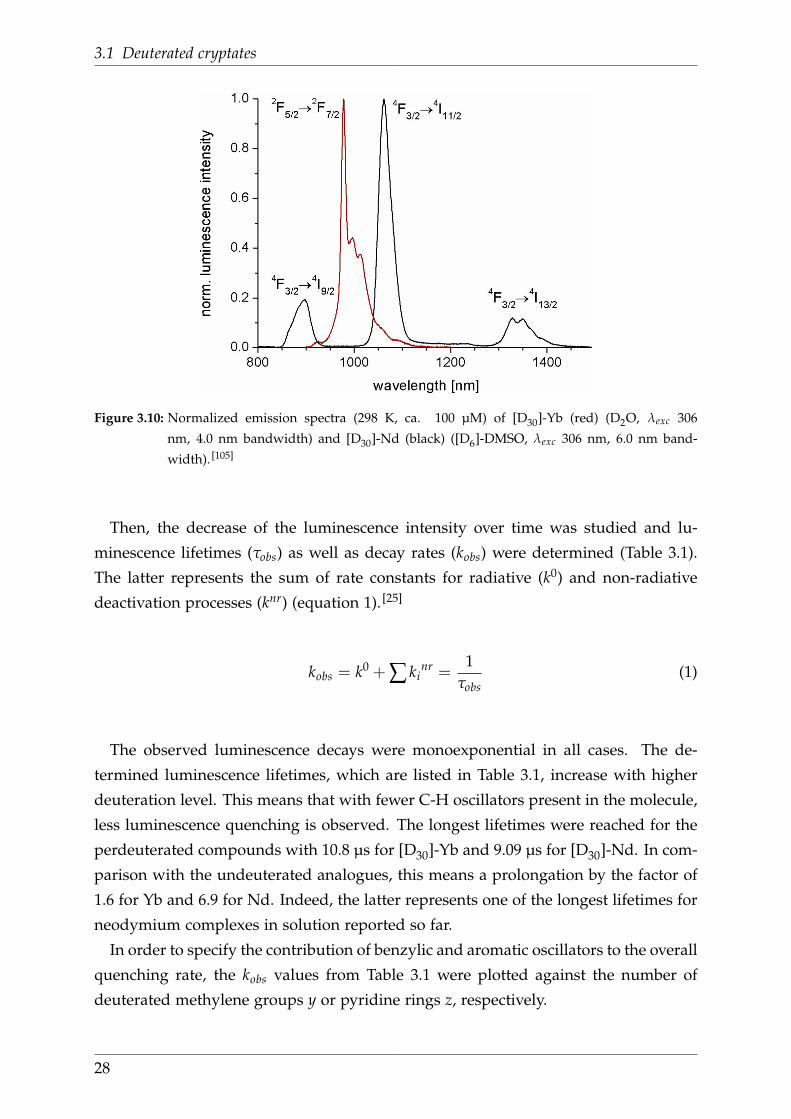
3.1 Deuterated cryptates
Figure 3.10: Normalized emission spectra (298 K, ca. 100 µM) of [D30]-Yb (red) (D2O, λexc 306
nm, 4.0 nm bandwidth) and [D30]-Nd (black) ([D6]-DMSO, λexc 306 nm, 6.0 nm band-
width). [105]
Then, the decrease of the luminescence intensity over time was studied and lu-
minescence lifetimes (τobs) as well as decay rates (kobs) were determined (Table 3.1).
The latter represents the sum of rate constants for radiative (k0) and non-radiative
deactivation processes (knr) (equation 1). [25]
kobs = k0 + ∑ kinr =
1τobs
(1)
The observed luminescence decays were monoexponential in all cases. The de-
termined luminescence lifetimes, which are listed in Table 3.1, increase with higher
deuteration level. This means that with fewer C-H oscillators present in the molecule,
less luminescence quenching is observed. The longest lifetimes were reached for the
perdeuterated compounds with 10.8 µs for [D30]-Yb and 9.09 µs for [D30]-Nd. In com-
parison with the undeuterated analogues, this means a prolongation by the factor of
1.6 for Yb and 6.9 for Nd. Indeed, the latter represents one of the longest lifetimes for
neodymium complexes in solution reported so far.
In order to specify the contribution of benzylic and aromatic oscillators to the overall
quenching rate, the kobs values from Table 3.1 were plotted against the number of
deuterated methylene groups y or pyridine rings z, respectively.
28

3 Development of binuclear cryptates
Table 3.1: Luminescence lifetimes τobs and excited state deactivation rates kobs of
Yb (D2O, λexc 335 nm, λem 980 nm) and Nd complexes ([D6]-DMSO,
λexc 306 nm, λem 1064 nm)
y=no. z=no. of deuter- τobs kobs calcd. kobsComplex of CD2 ated pyridine rings µsa [102 ms−1] [102 ms−1]b
[D12]-Yb 6 0 9.6 1.0 1.0[D8]-Yb 4 0 8.6 1.2 1.2[D4]-Yb 2 0 7.5 1.3 1.3[D0]-Yb 0 0 6.7 1.5 1.5[D10]-Yb 2 2 7.7 1.3 1.3[D20]-Yb 4 4 9.2 1.1 1.1[D30]-Yb 6 6 11.0c 0.93c -
[D12]-Nd 6 0 5.4 1.9 2.0[D8]-Nd 4 0 2.5 4.0 3.9[D4]-Nd 2 0 1.7 5.8 5.7[D0]-Nd 0 0 1.3 7.6 7.6[D10]-Nd 2 2 1.8 5.5 5.6[D20]-Nd 4 4 2.8 3.5 3.5[D30]-Nd 6 6 9.1c 1.1c -a Estimated error: ±15% for τobs > 3 µs and ±20% for τobs < 3 µsb Calculated using the parameters obtained from the global fitting procedurec value not used for the global fit
The 2D-projections reveal for both series of lanthanide complexes a linear correla-
tion as depicted exemplarily in Figure 5.2. In contrast to the commonly employed,
error-prone two-point calibrations (perdeuterated vs. undeuterated), this linearity is
established using multiple data points. The plotted values were modeled globally
with a three-dimensional planar fit function (equation 2).
kobs = k0 − y∆kbenzyl − z∆kpy (2)
k0 kobs of [D0]-Ln∆kbenzyl , ∆kpy quenching rate difference for the two/three C-H
oscillators of one benzylic methylene group or one pyridine ring
It should be noted, that the [D30]-Ln complexes were not included in the fitting
procedures, because at the time the obtained data (e.g, triplet level of [D30]-Gd or the
emission band shape of [D30]-Yb), suggested a different photophysical behaviour of
the perdeuterated cryptates.
29

3.1 Deuterated cryptates
Figure 3.11: Deactivation rates kobs of Nd complexes for the two series [D0]-[D4]-[D8]-[D12] (∆) and
[D0]-[D10]-[D20] (∇) with the corresponding 2D-projections (solid lines) of the 3D-global
fit planes. Yellow spheres stand for Nd, grey and blue structures represent undeuterated
and deuterated bipyridine moities, respectively. [105]
The rates (∆k) of the different oscillator groups (benzylic C-H, aromatic C-H) ob-
tained by the fitting procedures are shown in Table 3.2. Independent of the lanthanide,
benzylic C-H oscillators have a stronger impact on luminescence quenching than aro-
matic C-H moieties. Moreover, the deactivation rates are always greater for Nd com-
pared to Yb because of the smaller energy gap between emissive state and highest
receiving state (∆ENd ~5400 cm−1 vs. ∆EYb ~10 250 cm−1).
Table 3.2: Quenching rate differences ∆k for benzylic
and aromatic C-(H/D) oscillator groups in
[Dx]-Yb and [Dx]-Nda
k0 ∆kbenzyl ∆kpy kobsComplex [102ms-1]a [ms-1]a [ms-1]a R2
[Dx]-Yb 1.5 7.7 2.2 0.997[Dx]-Nd 7.6 94.1 9.4 0.997
a Estimated error: ±25%
30

3 Development of binuclear cryptates
In comparison to literature data, the determined deactivation rate ∆kbenzyl for Yb
luminescence of 7.7 ms−1 lies within the range of previously reported values (3-13
ms−1) for methylene groups close to Yb (~3.5-4.5 Å). [88,106]. All other values are
unprecedented.
To further deconvolute the contributions of individual C-H oscillators, two factors
were considered: the Franck-Condon overlaps with lanthanide excited states, and the
distance of an oscillator from the metal centre (rLn−H). The overlap factors were re-
garded to be similar within a particular group of C-H oscillators (either benzylic or
aromatic). This assumption simplifies the calculations, because then the quenching
rate of an oscillator only depends on its distance from the metal centre. Typical val-
ues for the distances rLn−H were obtained from the Cambridge Structural Database7
(CSD, version 5.31, February 2010) of structurally similar compounds as indicated in
Figure 3.12. The results of this search as well as overall averages of the respective
distances are listed in Table 3.3 to Table 3.6.
A B
N N
H3H4
H5
LnN
Heq
N
Hax
Ln**
*
*
H4
H5
H3
*
*
*
*
*
(Ln = Nd, Yb)
Figure 3.12: Structural motifs used for the Cambridge Structural Database search to obtain typical
distances for rLn−H . Positions allowed for substitutions are marked with an asterisc.
Table 3.3: Average distances rNd−H in
crystal structures with motif A
CSD Code Nd-H3 Nd-H4 Nd-H3
[Å] [Å] [Å]
XISFOV 5.573 6.346 5.525XIFMAA 5.542 6.338 5.479XAQLIL 5.539 6.270 5.471XAKWAH 5.440 6.291 5.578QIRDOK 5.571 6.332 5.486NAZHOM 5.546 6.310 5.483LEWNUX 5.580 6.339 5.531IDUQAA 5.555 6.312 5.487GUHJAU 5.492 6.270 5.466DOJHIU 5.577 6.337 5.512DICNUZ 5.560 6.330 5.501AKIPIT 5.542 6.307 5.491
Overall avg. 5.543 6.315 5.501
Table 3.4: Average distances rYb−H in
crystal structures with motif A
CSD Code Yb-H3 Yb-H4 Yb-H3
[Å] [Å] [Å]
XEWVUQ 5.390 6.172 5.396XEPLIN 5.360 6.155 5.386XEFBAM 5.440 6.227 5.442XAWSAQ 5.398 6.240 5.373RENXIR 5.300 6.095 5.344PAFBII 5.402 6.180 5.394LEWNIL 5.399 6.175 5.432LEGDAD 5.377 6.144 5.345JAPQOG 5.369 6.140 5.357DEFTEO 5.416 6.190 5.390
Overall avg. 5.385 6.172 5.386
31

3.1 Deuterated cryptates
Table 3.5: Average distances rNd−H
in crystal structures with
motif B
CSD Code Nd-Heq Nd-Hax
[Å] [Å]
AYEJAP 3.569 4.372BEJTIU 3.622 4.443HIZHIH 3.617 4.374LARGOB 3.512 4.347
Overall avg. 3.580 4.384
Table 3.6: Average distances rYb−H
in crystal structures with
motif B
CSD Code Yb-Heq Yb-Hax
[Å] [Å]
QAXYAP 3.356 4.202REPCAR 3.572 4.275YIPJIR 3.639 4.277YIPJOX 3.616 4.184
Overall avg. 3.546 4.235
For the theoretical deconvolution, one has to bear in mind that vibrational energy
transfer from the lanthanide ion to an oscillator is inversely proportional to the sixth
power of the distance. This can be derived from the general energy transfer formula
of Förster. [107,108]
∆ki ∝ (rLn−H)−6 (3)
Additionally, it was assumed that individual contributions to the overall quenching
rate are additive (equation 4).
∆ktot = ∑i
∆ki (4)
With these considerations and the determined average distances, the quenching
rates of individual C-H oscillators (∆ki) were calculated according to equation 5.
∆ki =∆ktot · (rLn−H)
−6
∑i(rLn−H)−6 (5)
The used parameters and results are summarised in Table 3.7 for Nd and Table 3.8
for Yb. The values for ∆ki are estimates for quenching rate differences, meaning
the rate by which single C-H groups quench lminescence stronger than single C-D
groups. Figure 3.13 also gives a graphical representation of the calculated estimates.
32

3 Development of binuclear cryptates
Table 3.7: Nd deconvolution of individual C-H deactivation rates ∆ki
based on typical crystal structure values for rLn−H .
benzylic H aromatic HHeq Hax H3 H4 H5
rNd−H [Å] 3.580a 4.384a 5.543b 6.315b 5.501b
(rNd−H)−6 10−5[Å]−6 47.5 14.1 3.45 1.58 3.61
∆ktot [ms−1] ��94.1c�� ����9.4c����∆ki [ms−1] 72.6 21.5 3.8 1.7 3.9a, b, c values taken from Table 3.5, Table 3.3,Table 3.2, respectively
Table 3.8: Yb deconvolution of individual C-H deactivation rates ∆ki
based on typical crystal structure values for rLn−H .
benzylic H aromatic HHeq Hax H3 H4 H5
rYb−H [Å] 3.567a 4.235a 5.393b 6.183b 5.389b
(rYb−H)−6 10−5[Å]−6 50.3 17.3 4.10 1.81 4.10
∆ktot [ms−1] ��7.7c�� ����2.2c����∆ki [ms−1] 5.7 2.0 0.9 0.4 0.9a, b, c values taken from Table 3.6, Table 3.4,Table 3.2, respectively
For both lanthanides, similar trends are observed. Among the aromatic oscillators,
the C-H groups in 3 and 5 position quench about twice as much as those in 4 posi-
tion. Among the benzylic oscillators, deactivation rates of closer lying equatorial C-H
groups are about the factor three higher compared to further away axial C-H groups.
Figure 3.13: Estimates for quenching rate differences in (∆ki in ms−1) for individual C-(H/D) oscillat-
ors in the Nd (upper value) and Yb complexes (lower value). [105]
33

3.2 Functionalised cryptates
3.2 Functionalised cryptates
3.2.1 Retrosynthetic analysis of functionalised cryptates
Monofunctionalised cryptates are usually obtained from a bipyridine and a macro-
cycle. Since either of the two reactants can carry the functional group, two possible
combinations result (Scheme 3.7). The first was used for the synthesis of the majority
of the functionlised cryptates, while the second was only applied for a cryptate that
possesses a functional group and an N,N’-dioxide moiety at different bipyridine arms.
N
N
N
N
N
N
N
N
N
N
N
N
N
N
H
H
N
NBr
Br
+
OR
Z
N
N
N
N
N
N
H
H
N
NBr
Br
+
Z
Z
Scheme 3.7: Retrosynthetic analysis of a TBP cryptand with a functional group X.
The functionalised macrocycle, which is required for the second outlined syn-
thesis, can be prepared from a simple and a functionalised bipyridine as depicted
in Scheme 3.8.
N
N
N
N
N
N
H
H
N
NNH2
NH2Z
N
NBr
Br
+
Z
Scheme 3.8: Retrosynthetic analysis of an azamacrocycle with a functional group X.
34

3 Development of binuclear cryptates
3.2.2 Synthesis of carboxy functionalised cryptates
Undeuterated building blocks and cryptates
The synthesis of carboxy functionalised cryptates started with the preparation of func-
tionalised bipyridines (Scheme 3.9). 2-(Tributylstannyl)-6-methylpyridine (22) was de-
rived from previously prepared 2. [109] In this conversion, a halogen-metal replacement
using n-BuLi was followed by a metal exchange with Bu3SnCl. Instead of purification
by column chromatography as reported in the literature, a fractional vacuum distilla-
tion was performed. Although this procedure is tedious, it reduces the contact with
toxic tin compounds and provided 22 in 60% yield and excellent purity (Figure 3.14).
Following Stille coupling with an isonicotinic acid methyl ester triflate 23 provided
bipyridine 24 in 59% after two-fold purification by column chromatography. [109] Com-
pound 24 was treated with mCPBA to provide the corresponding N,N’-dioxide 25 in
71% yield after column chromatography. The N,N’-dioxide 25 was subjected to a
Boekelheide reaction with TFA, followed by treatment with anhydrous LiBr. The res-
ulting methyl ester functionalised bipyridine building block 26 was obtained in 53%
yield. Compound 27 was prepared from 26 by treatment with peroxytrifluoroacetic
acid. This strong oxidising agent was generated in situ from urea hydrogen peroxide,
which represents a solid source of anhydrous H2O2, and TFA anhydride. [110,111]
N
OTf
MeOOCN
NN
Z
a
c
Z=Br
Z=SnBu3
MeOOC
b+
N
N
Br
Br
MeOOC
N
N
MeOOC
OO
Br
Br
N
N
MeOOC
OO
e
d
2
22
23 24 25
2627
Scheme 3.9: Synthesis of bipyridine building blocks that carry a methyl ester. (a) i. n-BuLi, dry THF,
−78 °C; ii. Bu3SnCl, −78 °C→rt (60%). (b) PdCl2(PPh3)2, PPh3, dry xylene, reflux (59%).
(c) mCPBA, CH2Cl2, 0 °C→rt (71%). (d) i. TFA anhydride, dry CH2Cl2, reflux; ii. dry
DMF/THF, LiBr, rt (53%). (e) urea·H2O2, CH2Cl2, TFA anhydride, 0 °C→rt (31%).
35

3.2 Functionalised cryptates
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5f1 (ppm)
1
2
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5f1 (ppm)
36.4
66.
9836
.56
17.9
7
3.28
1.05
0.98
1.00
9.00
6.15
6.64
5.82
2.90
0.94
0.88
0.92
0.50.70.91.11.31.51.71.9f1 (ppm)
Figure 3.14: 1H NMR spectra (200 MHz, CDCl3) of 22 (below) and the major by-product (above) of the
halogen-metal exchange reaction.
Following the literature procedures, the reactions (Scheme 3.10) of the bipyridines
28 and 29 with macrocycle 6 finally provided the cryptates 28 [95] and 29 [112] in 36%
and 39% yield after column chromatography.
N
N
N
N
N
N
N
NNa
N
N
COOMe
OO
Br
Br
N
N
N
N
N
N
N
NNa O
O
Br
Br
COOMe
COOMe
+N
N
NH
HN
N
N
aN
N
Br
Br
COOMe
N
N
NH
HN
N
N
+ b
6 26 28
6 27 29
Scheme 3.10: Synthesis of cryptates with a carboxylic acid methyl ester. (a) Na2CO3, CH3CN, reflux
(36%). (b) Na2CO3, CH3CN, reflux (39%).
36

3 Development of binuclear cryptates
Deuterated building blocks and cryptate
In the next stage of development, it was investigated if the deuteration protocols can
also be applied to the carboxy functionalised cryptates. Cryptate 28 was chosen as
the model system for this endeavour. The synthesis of its perdeuterated analogue
30 was envisaged by the coupling of two perdeuterated precursors, with one being
the previously synthesised [D20]-macrocycle 31 and the other being a perdeuterated
functionalised bipydine 32.
aN
N
MeOOC
OO
[D11]
N
N
MeOOC
OO
b N
N
MeOOC
d
[D9]
OAc
OAc
N
N
MeOOC
c
[D9]
Br
Br
N
N
MeOOC
OO
[D9]
Br
Br
25 33 34 32
32
Scheme 3.11: Synthesis of a deuterated bipyridine building block that carries a methyl ester. (a)
i. NaOD (40 wt% in D2O), D2O, autoclave, 150 °C. ii. conc. H2SO4, MeOH, reflux
(62%, 98%D). (b) Ac2O, reflux (67%, 98%D). (c) HBr (47%) in HOAc, reflux. (d) i. TFA
anhydride, dry CH2Cl2, reflux; ii. dry DMF/THF, LiBr, rt (45%, >99%).
For the synthesis of a perdeuterated, functionalised bipyridine (Scheme 3.11), the
same strategy as for the simple bipyridines (see section 3.1.2) was applied. [105] Perdeu-
teration was performed in an autoclave with D2O as deuterium source and NaOD as
base. This reaction was repeated twice to ensure a sufficiently high deuteration level
of the crude product. Due to the basic conditions employed in this step, the methyl
ester was saponified and had to be restored using a Fisher esterificaton. Thus, the
desired product 33 was obtained in 62% yield with a deuteration level of 98%D. Sub-
sequent Boekelheide rearrangment with acetic anhydride gave 34 in 67% yield (98%D)
after two-fold column chromatography. However, the nucleophilic substitution of the
acetoxy group using HBr/HOAc as bromine source did not work well. [95]. In repeated
attempts this reaction only yielded small amounts of 32 beside the mono-brominated
analogue and the corresponding dialcohol, which is the hydrolysis product of 34.
37

3.2 Functionalised cryptates
Hence, the alternative reaction of 33 with TFA anhydride, followed by a treatment
with LiBr was tried. This approach was successful and 32 could be isolated in 45%
yield with a deuteration level >99%, as determined from its ESI mass spectrum.
The prepared carboxy-functionalised, perdeuterated building block 32 was reacted
with the previously synthesised perdeuterated macrocyle 31 using standard condi-
tions for the synthesis of TBP cryptates. As a result, the sodium complex was obtained
in 26% (98%D) yield after column chromatography.
+N
N
NH
HN
N
N
aN
N
Br
Br
N
N
N
N
N
N
N
NM
COOMeCOOMe
bM=Yb3+, n=3
[D9][D20] [D29]
M=Na+, n=1
nXX=Cl or Br
31 32 35
36
Scheme 3.12: Synthesis of deuterated sodium and ytterbium cryptates with a methyl ester function-
ality. (a) Na2CO3, CH3CN, reflux (26%, 98%D). (b) YbCl3·6H2O, CH3CN, reflux.
Subsequent reaction with YbCl3·6H2O afforded the corresponding ytterbium crypt-
ate 36, which was purified by precipitation from its methanolic solution. For this
compound, the ESI mass spectrum only showed the signal for the reactant and not
for the triple charged product. However, the recorded steady state emission spectrum
(Figure 3.15) confirmed the expected near-IR emission of ytterbium. Consequently, the
synthesis of the first perdeuterated functionalised ytterbium cryptate was successful.
8 5 0 9 0 0 9 5 0 1 0 0 0 1 0 5 0 1 1 0 0 1 1 5 0
0 . 2
0 . 4
0 . 6
0 . 8
1 . 0 2 F 5 / 2 2 F 7 / 2
relati
ve em
ission
inten
sity
w a v e l e n g t h i n n m
9 7 5 . 5 n m
9 9 8 . 5 n m1 0 1 0 . 5 n m
Figure 3.15: Steady state emission spectrum of 36. The spectrum was recorded at room temperature
with an excitation wavelength of 312 nm in D2O.
38

3 Development of binuclear cryptates
3.2.3 Synthesis of amino functionalised cryptates
Simple building blocks and cryptate
For the preparation of a amino functionalised TBP cryptate, a macrocycle and a
bipyridine with an amino group were required.
N
N
Z
N
N
N
N
N
N
a b
O2N
N
N
c
Z
d Z=NH2
Z=NHAc
Z=NH2, NHAc
e
OO
O O
3 37 38 39
40
Scheme 3.13: Synthetic approach towards an amino functionalised bipyridine building block. (a)
mCPBA, CHCl3, 0 °C→rt (81%). (b) i. H2SO4/HNO3, 100 °C; ii. aq. NaOH, rt (61%).
(c) NaBH4, Pd/C, MeOH, 0 °C→rt (68%). (d) AcCl, py, rt (37%). (e) mCPBA, CH2Cl2,
0 °C→rt.
The multistep synthesis of the latter (Scheme 3.13) started with the reaction of 6,6’-
dimethyl-2,2’-bipyridine (3) with one equivalent of mCPBA to give mono-N-oxide 37
in 81% yield after column chromatography. [99] The activated pyridine ring was select-
ively nitrated by treatment with a mixture of nitric and sulfuric acid at elevated tem-
peratures (100 °C). [113] Aqueous workup of the reaction mixture led to precipitation
of the nitro functionalised bipyridine (38), which was isolated in 61% yield. The nitro
group was subsequently reduced to the corresponding amine with Pd/NaBH4. [114]
The clean product 39 (Figure 3.16 bottom) was obtained in 68% yield after aqueous
workup and was used without further purification.
Compound 39 was susbsequently treated with two equivalents of mCPBA. How-
ever, TLC and 1H NMR spectrum of the isolated residue indicated that the presumed
N,N’-dioxide was not formed. Instead the applied conditions only caused reoxidition
of the amino to a nitro group. Indeed, there is already literature precedence for the
closely related hydrogen peroxide induced oxidation of amino groups attached to a
pyridine. [115]
Accordingly, a modification of the synthetic strategy was necessary and an addi-
tional step was introduced to protect the amino function with an acetyl group. While
the acetylation with acetic anhydride failed, [116] the alternative reaction of 39 with
acetyl chloride in dry pyridine was more successful, [117] and gave 40 in 37% yield.
However, despite purification by column chromatography, the 1H NMR of the isol-
ated material confirmed the presence of acetyl containing impurities (Figure 3.16 top).
39

3.2 Functionalised cryptates
1.82.02.22.42.62.83.03.23.43.63.84.04.24.46.26.46.66.87.07.27.47.67.88.08.28.48.68.8f1 (ppm)
1
2
3.0
1.0
1.0
0.9
0.8
1.0
1.0
2.9
3.3
1.7
1.0
1.0
1.0
1.0
1.0
5.9
CDCl3
CDCl3
EtOH
Figure 3.16: 1H NMR spectra of 39 (400 MHz, blue trace) and 40 (250 MHz, red trace) in CDCl3.
Nevertheless, oxidation of 40 was attempted by treatment with mCPBA, but inde-
pendent of increasing amounts of oxidising agent or prolonged reaction times, 1H
NMR and ESI mass spectra only proved the formation of the mono-N-oxide.
Consequently, the strategy was changed again and the conversion of the nitro to
an amino group was shifted from an early stage of the synthesis pathway to the
end. This means, that the novel primary synthetic target was the formation of a nitro
functionalised cryptate, which should be reduced to the amino functionalised cryptate
in the last step. Hence, a nitro functionalised bipyridine building block was required
for the cryptate synthesis (Scheme 3.14).
N
N
O2N
bN
N
Br
Br
O2N
N
N
O
O2N
a OO
38 41 42
Scheme 3.14: Synthesis of a nitro functionalised bipyridine building block. (a) mCPBA, CHCl3,
0 °C→rt (82%). (b) i. TFA anhydride, dry CH2Cl2, reflux; ii. dry DMF/THF, LiBr, rt
(56%).
40
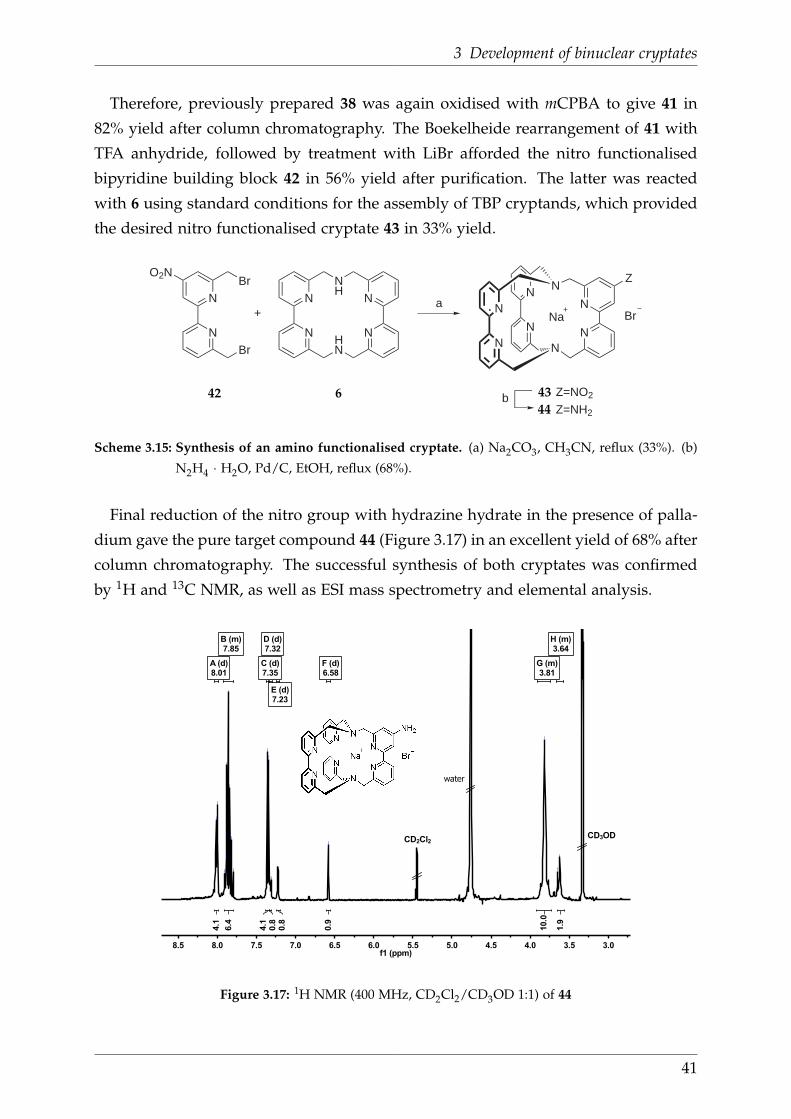
3 Development of binuclear cryptates
Therefore, previously prepared 38 was again oxidised with mCPBA to give 41 in
82% yield after column chromatography. The Boekelheide rearrangement of 41 with
TFA anhydride, followed by treatment with LiBr afforded the nitro functionalised
bipyridine building block 42 in 56% yield after purification. The latter was reacted
with 6 using standard conditions for the assembly of TBP cryptands, which provided
the desired nitro functionalised cryptate 43 in 33% yield.
+N
NBr
N
N
NH
HN
N
N
a
N
N
N
N
N
N
N
NNa Br
O2NBr Z
b Z=NO2
Z=NH2
42 6 43
44
Scheme 3.15: Synthesis of an amino functionalised cryptate. (a) Na2CO3, CH3CN, reflux (33%). (b)
N2H4 · H2O, Pd/C, EtOH, reflux (68%).
Final reduction of the nitro group with hydrazine hydrate in the presence of palla-
dium gave the pure target compound 44 (Figure 3.17) in an excellent yield of 68% after
column chromatography. The successful synthesis of both cryptates was confirmed
by 1H and 13C NMR, as well as ESI mass spectrometry and elemental analysis.
3.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
1.9
10.0
0.9
0.8
0.8
4.1
6.4
4.1
A (d)8.01
B (m)7.85
C (d)7.35
D (d)7.32
E (d)7.23
F (d)6.58
G (m)3.81
H (m)3.64
3.62
3.65
3.80
3.80
3.82
3.83
6.58
6.58
7.22
7.23
7.31
7.33
7.34
7.36
7.80
7.82
7.84
7.84
7.84
7.86
7.86
7.88
7.88
7.89
7.90
8.00
8.02
CD2Cl2
water
CD3OD
Figure 3.17: 1H NMR (400 MHz, CD2Cl2/CD3OD 1:1) of 44
41

3.2 Functionalised cryptates
A cryptate with a rigid N,N’-dioxide moiety
For the development of an analogous rigid, amino functionalised cryptate, one has
to keep in mind that the N,N’-dioxides and the amino group cannot be present at
the same bipyridine (see section 3.2.3). During the synthesis, oxidising or reducing
agents affect the pyridine and amino nitrogens similarly and, on that account, these
structural features have to be introduced with different building blocks (Figure 3.18).
N
N
N
N
N
N
N
NNa Br
N
N
N
N
N
N
N
NNa Br
H2N
D30
OO
OO
45 46
Figure 3.18: Rigid TBP cryptates. Left: deuterated parent compound developed by Doffek et al. [93]
Right: envisaged functionalised analogue.
Since it was difficult to prepare an N,N’-dioxidised macrocycle, the second retrosyn-
thetic pathway (see Scheme 3.7) was applied. Accordingly, a functionalised building
block first has to be coupled to a simple bipyridine to form a macrocycle. The latter
can subsequently react with an N,N’-dioxide moiety to build the desired cryptate.
While the nitro functionalised bipyridine 42 was already at hand, its counterpart for
the macrocycle synthesis was prepared from previously synthesised 5 (Scheme 3.16
left).
N
N
N
N
a
bZ=NHTs
Z
Z
Br
Br
N
N
c
Br
Br
OO
Z=NH2 3HBr H2O 518
47
48
Scheme 3.16: Synthesis of additional required bipyridine building blocks for a functionalised rigid
cryptate. (a) i. CHCl3, urotropine, reflux; ii. H2O/EtOH/HBr, 75 °C→rt (50%). (b) aq.
NaOH, TosCl, Et2O, 0 °C→rt (49%). (c) urea · H2O2, CH2Cl2, TFA anhydride, 0 °C→rt
(84%).
A Delépine reaction was used for the conversion to amine 18, followed by pro-
tection of the primary amine with tosyl chloride to give 47 in 49% yield. [112] The
42

3 Development of binuclear cryptates
bipyridine-N,N’-dioxide building block was prepared from the same precursor by
treatment with peroxytrifluoroacetic acid (Scheme 3.16 right). [118] In comparison to
the literature known oxidation of 5 with mCPBA, [92] this reaction is less tedious and
gave 48 in relatively high yield (84%) after column chromatography.
After their successful preparation, the three building blocks were assembled step-
wise, starting with the reaction of 47 and 42 under slightly basic conditions. [112]
Thereby, the tosyl protecting groups prevented multiple substitution at the amines
of 47, so that the macrocycle and not the cryptate was obtained. Acid-mediated de-
protection of the amines gave the poorly soluble nitrated macrocycle 49 in 52% yield.
N
N
NH
HN
N
N
a
NO2
+N
NNHTs
NHTsN
NBr
Br
N
N
NH
HN
N
N
NH2
NO2
N
N
N
N
N
N
N
NNa Br
H2N
c
b
OO
47 42 49
50
48
46
Scheme 3.17: Synthesis of a rigid, amino functionalised TBP cryptand. (a) i. DMF, K2CO3, 50 °C→rt;
ii. H2SO4, 110 °C (52%). (b) N2H4 · H2O, Pd/C, EtOH, reflux (85%) (c) Na2CO3, CH3CN,
reflux (8%).
In analogy to the procedure for amino cryptate 44, compound 49 was reduced with
N2H4 · H2O to the corresponding amino macrocycle. The isolated crude product 50
was used without further purification for the final step, during which it was coupled
to the third building block (48). As a result, the desired cryptate 46 was formed, which
was isolated in 8% yield after purification. As for the other amino functionalised
cryptate, the identity of the target compound was confirmed by 1H and 13C NMR,
ESI mass spectrometry as well as elemental analysis. It is worth mentioning, that the1H spectrum of 46 (Figure 3.19) shows numerous signals and is far more complicated
than that of 44. This can be explained by the existence of diastereomers.
43

3.2 Functionalised cryptates
As mentioned previously (see Figure 3.3), the cryptate is devoid of symmetry ele-
ments, because all three bipyridine moieties are inequivalent and one of them is un-
symmetrical. Hence, 46 possesses a special form of chirality. The latter adds to the
axial chirality of bpyridine-N,N’-dioxide and, as a consequence, different diastereo-
mers result. Because of their great similarity it was not possible to unambiguously
distinguish these species spectroscopically.
3.03.23.43.63.84.04.24.44.64.85.05.25.45.65.86.06.26.46.66.87.07.27.47.67.88.0f1 (ppm)
2.0
2.9
4.3
2.0
0.7
0.8
0.9
3.3
1.7
10.0
A (d)4.45
B (m)7.74
C (m)7.50
D (m)7.39
E (m)7.01
F (s)6.62
G (m)4.30
H (m)3.85
I (m)3.45
J (m)3.14
3.28
3.30
3.33
3.37
3.39
3.46
3.49
3.51
3.52
3.75
3.80
3.82
3.84
3.87
3.88
3.93
4.28
4.30
4.32
4.43
4.48
6.62
6.98
6.99
7.02
7.03
7.32
7.35
7.36
7.38
7.38
7.38
7.39
7.41
7.42
7.45
7.48
7.49
7.49
7.51
7.52
7.52
7.59
7.60
7.61
7.62
7.63
7.64
7.64
7.66
7.67
7.67
7.70
7.70
7.73
7.74
7.76
7.76
7.77
7.79
7.80
7.81
7.82
7.82
7.83
7.83
7.83
7.84
7.85
7.86
7.88
CD2Cl2
CD3ODwater
Figure 3.19: 1H NMR (250 MHz, CD2Cl2/CD3OD 1:1) of 46
44

3 Development of binuclear cryptates
3.3 Binuclear cryptates
3.3.1 Strategy for the coupling of cryptates
There are different possibilities to connect two cryptates (Figure 3.20). The shortest
linkage is an amide bond, for which two cryptates with different functional groups
are required. However, from a synthetic point of view, this approach is rather cum-
bersome, since both coupling partners have to be synthesised in sufficient amounts.
Furthermore, an amide bond formation with an aromatic amino group is often prob-
lematic due to its low nucleophilicity.
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
NNa
O
NH
O
NHNH
OO
NHNH
O
NHNH
O
Na
Figure 3.20: Molecular architecture of binuclear TBP cryptates. The dotted line stands for the possible
linkages, which are shown below.
In comparison, the connection via one or two ethylendiamine bridges only involves
aliphatic amino groups and one functionalised cryptate. Consequently, this type of
linkage was favoured for the first coupling reactions of two TBP cryptates. It is also
worth noting, that only undeuterated, less precious cryptates were used for the de-
velopment of these reactions.
3.3.2 Functionalised cryptates for coupling reactions
The synthesis of cryptates suitable for coupling reactions started with the preparation
of the two required coupling partners from the methyl ester functionalised cryptate
28 (Scheme 3.18). On the one hand, the methyl ester 28 was saponified with NaOH
to give the corresponding carboxyl functionalised cryptate 51 (80% yield). [119] On
45
3.3 Binuclear cryptates
the other hand, the reaction of 28 with ethylenediamine under anhydrous conditions
resulted in the formation of amine 52.
N
N
N
N
N
N
N
NNa
N
N
N
N
N
N
N
NNa
O
NH
NH2
O
OMe
a
Br
N
N
N
N
N
N
N
NNa
O
Br
O
b
2851 52
Scheme 3.18: Synthesis of the coupling partners from a common precursor. (a) aq. NaOH, MeOH,
40 °C (80%). (b) ethylendiamine, dry MeOH, 0 °C→rt (87%).
The crude product of this reaction was purified with reversed-phase column chro-
matography using silanised silica (RP-2) as solid phase and CH2Cl2/MeOH (15:1 v/v)
as eluent. For thin layer chromatography, the employed RP-18 plates were developed
first with iodine to confirm the presence of the cryptate and, subsequently, with nin-
hydrin to detect the primary amine. This purification could be performed on a 50 mg
scale and enabled the isolation of the pure 52 in 87% yield.
3.03.23.43.63.84.04.26.87.07.27.47.67.88.08.28.48.68.89.0f1 (ppm)
1
2
3
7.3
1.7
2.7
4.9
10.9
1.2
11.7
5.2
1.1
5.4
4.3
1.3
1.1
2.2
2.2
11.7
4.9
6.0
4.2
1.0
1.0
CDCl3
MeOH CD3OD
CD3OD
Figure 3.21: 1H NMR spectra of 28 (200 MHz, CDCl3), 51 (250 MHz, CD2Cl2/CD3OD 1:1) and 52 (250
MHz, CD2Cl2/CD3OD 1:1).
46

3 Development of binuclear cryptates
The direct comparison of the 1H NMR spectra of 28, 51 and 52 (Scheme 3.18) re-
veals, that both derivatisation reactions go along with the loss of the methyl ester
moiety (peak at 4.00 ppm). While no new signal are observed in the spectrum of the
carboxylic acid 51, the 1H NMR spectrum of amine 52 displays two novel triplets,
which could be assigned to the two methylene groups of the ethylenediamine.
3.3.3 First approaches towards the coupling of cryptates
In the first attempt to form a binuclear TBP cryptate, it was tried to couple two methyl
ester functionalised cryptates (28) with one equivalent of ethylenediamine. Still, this
reaction only gave the monocoupled product 52. Therefore, the selective coupling of
the latter with a carboxyl functionalised cryptate 51 was tried.
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
NNa
O
NH
HN
O
Na
Br
Br
53
Figure 3.22: Binuclear TBP cryptate, which was observed in the ESI mass spectrum.
The bottle-neck for all amide bond formations is the choice of an appropriate ac-
tivating agent, which enhances the electrophilicity of the carboxylate group. On that
account, several commonly used compounds were tested for this coupling reaction.
When employing oxalyl chloride, DCC/HOBt or EDCI/HOBt , no product formation
was observed. In contrast, after activation of acid 51 with HATU in the presence of
DIPEA or NEt3, ESI mass spectra indicated the the formation of the binuclear com-
pound 53. Nevertheless, it was not possible to gain sufficient amounts of this species
for a complete characterisation. It was concluded that major obstacles for this syn-
thetic pathway are the inefficient activation of the carboxylic acid, and the loss of
material during the purification procedure, which might be related to stability issues.
47

3.3 Binuclear cryptates
3.3.4 Preparation of the first binuclear TBP cryptate
To overcome the described problems, the synthetic strategy for the preparation of the
binuclear cryptates was modified regarding two important points. First, instead of
coupling to a carboxyl functionalised cryptate, it was aimed to connect to ethylene-
diamine linked cryptates via a carbamide. This way, one circumvents the difficult
activation of the carboxylic acid of a cryptate and, in addition, the synthetic effort
is reduced, since only one sort of functionalised cryptate needs to be prepared. As
second alteration, the cryptates were equipped with an N,N’-dioxide moiety which is
known to increase their stability. [93] The resulting novel target structure is in shown
in Figure 3.23.
N
N
N
N
N
N
N
NNa
Br
O
NH
HN
N
N
N
N
N
N
N
NNa
Br
O
NH
HN
OOO
OO
54
Figure 3.23: Revised target structure of a binuclear TBP cryptate.
The ethylenediamine linked cryptate 55 was prepared by treatment of 29 with neat
ethylenediamine. Purification with reversed-phase column chromatography using the
above discussed conditions afforded the pure compound in an excellent yield of 76%.
a
N
N
N
N
N
N
N
NNa O
OBr
N
N
N
N
N
N
N
NNa O
OBr
O
O
O
NH
NH2
29 55
Scheme 3.19: Preparation of a rigid, ethylenediamine linked cryptate. (a) ethylendiamine, neat,
0 °C→rt (76%).
For the coupling to the binuclear compound (Scheme 3.20), six equivalents of 55
were treated with one equivalent of the urea synthon triphosgene, which was chosen
as a safer and more practical alternative to phosgene. The reaction was performed un-
der anhydrous conditions in the presence of DIPEA and yielded the target compound
besides an isocyanate by-product. The latter was removed with two-fold column
48

3 Development of binuclear cryptates
chromatography, so that target molecule 54 could be isolated in 50% yield. Successful
separation was confirmed by 1H NMR spectroscopy, whereas the spectra could be
assigned to the respective species with the aid of their ESI mass spectra (Figure 3.24).
O
O
O
Cl
ClCl
Cl
ClCl+ a6
N
N
N
N
N
N
N
NNa O
OBr
O
NH
NH2
55
54
Scheme 3.20: Coupling of two ethylenediamine linked cryptates to a binculear TBP cryptate. (a)
DIPEA, dry CH2Cl2, 0 °C→rt (50%).
The 1H NMR specta of reactant 55 and product 54 are shown in Figure 3.25. As
expected, the spectra do not differ in terms of integral ratios, since no new protons
were introduced during the reaction. However, the shape and the complexity of the
whole spectrum is altered significantly after the coupling. The product shows a larger
number of overlapping peaks and it is also noticable, that the signals in the aromatic
region are more condensed. In summary, the synthesis and purification of the first
binuclear TBP cryptate was successful.
6 0 0 7 0 0 8 0 0 9 0 0 1 0 0 0 1 1 0 0 1 2 0 0 1 3 0 0 1 4 0 0 1 5 0 0 1 6 0 0
E x a c t M a s s o f 5 4 : 1 6 1 4 . 3 9
1 4 9 1 . 8 9 [ M + H - 2 N a - B r ] +
7 4 1 . 0 1 i s o c y a n a t e b y p r o d u c t
7 2 8 . 5 5 [ M - 2 B r ] 2 +
m / zFigure 3.24: ESI+ MS spectrum of 54
49

3.3 Binuclear cryptates
3.03.23.43.63.84.04.24.46.87.07.27.47.67.88.08.28.48.6f1 (ppm)
1.9
4.5
3.9
4.1
2.1
4.0
1.1
1.0
9.2
0.9
0.9
19.0
8.1
0.8
4.2
0.8
1.9
6.9
4.2
17.0
1.9
0.8
CD3OD
CD3OD
Figure 3.25: 1H NMR spectra (250 MHz, CD2Cl2/CD3OD 1:1) of 55 (top) and 54 (bottom).
50

4 Chemical unclicking of amide bonds
4.1 Introduction
The first part of this work focussed on the development of binuclear cryptates to
prepare a promising molecular environment for an upconversion system. This part
deals with the design of a synthetic concept, that allows to selectively disconnect
binuclear lanthanide cryptates and thus switch off potential upconversion emission.
As it was outlined in the previous section, cryptates can be directly connected via
an amide bond or a short linker. Another approach would be the integration of two
cryptates into a tripeptide. In this case, the cleavage of a peptide bond would also
result in a separation of two cryptates as depicted in Figure 4.1.
Ln1
AA1 AA2 AA3
Ln2 Ln1
AA1 AA2 AA3
Ln2
+
Figure 4.1: Envisaged selective cleavage of a tripeptide with two cryptates. Blue structures stand for
cryptands, yellow spheres for lanthanides, and grey spheres for amino acids.
With the potential application as optical probe in biological systems in mind, two
restrictions were made towards the synthetic concept. First, the trigger of the cleav-
age had to be bioorthogonal, meaning that it is not associated with and chemically
inert within a biological system. [120] Second, the reaction itself should be a straight-
forward, simple chemical transformation, which proceeds under mild conditions (e.g.
temperatures between 25 °C and 40 °C, aqueous solutions, neutral pH). With these
requirements fulfilled, this idea of selective bond cleavage (“Unclick chemistry”) rep-
resents the counterpart of the well-established “Click chemistry”. [121]
51

4.2 Concept
4.2 Concept
Specific peptide bonds are routinely cleaved with proteolytic enzymes. [122] As an al-
ternative, non-enzymatic, chemical methods were developed, which take advantage
of the nature of certain amino acid residues. [123] In contrast to proteases, these mo-
lecules represent the smallest possible entity to cleave an amide bond and, therefore,
are expected to marginally disturb other molecules within a biological environment.
In order to keep Unclick chemistry as simple as possible, an artificial amino acid, that
possesses a specially protected side chain, was chosen as the key molecule.
R1
R1
+
Figure 4.2: The principle of Unclick chemistry.
The unprotected side chain is thought to contain a nucleophilic moiety, which at-
tacks the amide bond in an intramolecular cyclisation reaction. As a result, the amide
is cleaved and the cyclic form of the key molecule, as well as an amino acid or pep-
tide with a free N-terminus is obtained. The designated side-chain protecting group
masks the nucleophilic moiety, until it is removed by applying a bioorthogonal chem-
ical trigger (Figure 4.2, key and lock). Therewith, it provides a precise spatial and
temporal control over the reaction.
The molecular design of the Unclick amino acid was inspired by L-asparaginyl-
residues. Peptides, which possess the latter moiety, undergo an intramolecular re-
action at basic pH (7.4-13.8), during which the side chain amide nitrogen nucleo-
philically attacks the amide carbon. As a result, the peptide bond is cleaved and a
C-terminal succinimidyl-residue as well as a free N-terminus are formed. [124]
52

4 Chemical unclicking of amide bonds
HN
PG NH
O
O
NH2
R
ONH
OHN
H2N R+5-exo-trig PG
Figure 4.3: Core structure of the Unclick amino acid and envisaged peptide bond cleavage. PG =
protecting group.
The envisaged nucleophilic moiety of the target molecule was also thought to pos-
sess an amino group, which is however deprotonated under physiological conditions
(pH 7.4). Organic hydroxyl amines display pKa values beyond 7.4 (e.g. N-methyl hy-
droxylamine 5.96, N,O-dimethyl hydroxylamine 4.75), [125] and, hence, an aminoserine
was chosen as the lead structure for the Unclick amino acid. The hydroxylamine ni-
trogen of this molecule is expected to react intramolecularly with the amide bond
to form an O-alkyl hydroxamic acid (Figure 4.3). This 5-exo-trig reaction is kinetically
and entropically favoured and, moreover, it is driven by the protonation of the formed
amine in a physiological environment.
HN
PG NH
O
O
NH
R
HN
PG NH
O
O
N O
O
N3
HN
PG NH
O
O
N O
O
NH2
R R
PPh3
Staudinger reaction
Figure 4.4: Structure of “Unclick serine” and deprotection of the Azoc side chain protecting group.
PG = protecting group.
The hydroxylamine was thought to be protected with a bioorthogonal azide-based
carbamate group (Azoc), [126] which is reportedly stable under solid phase peptide
synthesis (SPPS) conditions and orthogonal to Fmoc and Boc. Furthermore, it is se-
lectivley and rapidly deprotected by treatment with bioorthogonal PPh3. For the
introduction of Azoc to the aminoserine, the N- and C-terminus also have to be pro-
tected and the hydroxylamine nitrogen has to be monoalkylated in order to prevent
side reactions.
53

4.3 Preliminary work
4.3 Preliminary work
Before the preparation of Unclick serine was initiated, it was elucidated, if basic re-
quirements for the selective cleavage of cryptate carrying peptides are fulfilled. On
the one hand, it had to be verified that cryptates can be efficiently conjugated to amino
acids. On the other hand, it had to be validated that a 5-exo-trig reaction is general
possible within such a system.
Preparation of an amino acid cryptate conjugate
The conjugation of cryptates to amino acids was already pursued by N. Alzakhem,
who coupled L-lysine to a rigid, carboxyl functionalised TBP cryptand. [112] During
this work, he was faced problems with the activation of the carboxylic acid attached
to the cryptate, which considerably limited the yield of the coupling reaction.
On the account of his findings, a different approach was chosen, which involved
the side chain coupling of L-glutamic acid to the ethylenediamine linked, rigid TBP
cryptate 55. As depicted in Scheme 4.1, the employed L-glutamic acid beared an N-
terminal Fmoc protecting group, which is usually required for the standard coupling
of amino acids using solid phase peptide synthesis (SPPS). Moreover, this moiety as
well as the C-terminal tert-butyl ester are orthogonal to the Azoc group of Unclick
serine.
N
N
N
N
N
N
N
NNa
Br
O
NH
HN O
FmocHNO
OtBu
N
N
N
N
N
N
N
NNa
Br
O
NH
NH2
O
FmocHNO
OtBu
HO
+a O
OOO
55 56
Scheme 4.1: Coupling of a rigid, functionalised TBP cryptate to L-Glu. (a) HATU, DIPEA, DMF,
0 °C→rt (52%).
The reaction of 55 with Fmoc-Glu-OtBu was facilitated by HATU in the presence of
DIPEA and resulted in formation of the amino acid conjugate 56, which was isolated
in 52% yield after two-fold column chromatography. Identity and excellent purity of
the product could be confirmed by ESI mass spectrometry, and by 1H NMR spetro-
scopy (Figure 4.5). Both spectra only displayed the expected signals for pure 56.
54

4 Chemical unclicking of amide bonds
1.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
9.5
2.4
2.1
10.5
4.3
6.4
1.0
1.1
8.3
1.3
15.2
1.0
1.0
CH2Cl2
CHCl3
N-H N-H
enCH2
benz.CH2
GluCH2
GluCH2
Fmoc& Glu & cryp
tBu
N-Harom.CH2
arom.CH2
Fmoc
Figure 4.5: 1H NMR (250 MHz, CDCl3) of 56.
5-exo-trig test reaction
To evaluate the chances for a 5-exo-trig reaction to occur in Unclick serine, the struc-
turally related, commercially available 1-phenyl-3-pyrazolidinone (57) was chosen as
a suitable model system for test reactions. In comparison to Unclick serine, this com-
pound possesses a hydrazine instead of a hydroxylamine moiety, and no N-terminus.
At first, the starting material 57 was reacted with HCl/MeOH to form methyl 3-(1-
phenylhydrazinyl)propanoate hydrochloride (58). Then, the latter was treated with
aqueous NaHCO3 to remove the hydrochloride and, therewith, enhance the nucleo-
philicity of the terminal nitrogen.
NNH
O
Ph
O
O
NPh
NH2 HCl
a
b
57 58
Scheme 4.2: Reversible ring opening of phenylpyrazolidinone. (a) HCl/MeOH, 0°C→rt. (b)
NaHCO3, CHCl3, D2O.
55

4.4 Retrosynthetic analysis of Unclick serine
The behaviour of the free hydrazine in CDCl3, [D6]-DMSO or D2O was followed
with 1H NMR spectroscopy. While no spectral changes were observed in organic
solution, the spectrum of the aqueous mixture after two hours at room temperature
proved the recovery of 57. This result encouraged the working hypothesis regarding
the 5-exo-trig reaction.
4.4 Retrosynthetic analysis of Unclick serine
An N-terminal protected Unclick serine (59, Scheme 4.3) can be derived from an Azoc-
free, N-terminal protected aminoserine (60). This precursor is accessible by ring open-
ing of 61. The required N-methylated, protected cyclic amino acid may arise from the
selective protection and methylation of D-cycloserine (62).
ONH
OH2N
ON
OHN
PG
HN
PG OMe
O
O
HN
HN
PG OH
O
O
N O
O
N3
methylationprotectionAzoc
protectionringopening
59 60 61 62
Scheme 4.3: Retrosynthetic analysis of Unclick serine.
56

4 Chemical unclicking of amide bonds
4.5 Investigation of different Unclick systems
4.5.1 Fmoc-Unclick serine
Fmoc-protected Unclick serine (63) was prepared from commercially available D-
cycloserine in six steps, during which the Fmoc and the Azoc protecting group were
successively introduced to the molecule.
ONH
OHN
Z
ON
OHN
Fmoc
HN
Fmoc OMe
O
O
HN
HN
Fmoc OMe
O
O
N O
O
Z
HCl
b c d
a Z=H
Z=Fmoce Z=Cl
Z=N3
62
6465 66
67
63
Scheme 4.4: Synthesis of Fmoc-Unclick-serine. (a) 1. BSA, dry CH2Cl2, rt; 2. pyridine, Fmoc-OSu, dry
CH2Cl2, 0 °C→rt (84%). (b) MeI, K2CO3, acetone, reflux (70%). (c) HCl/MeOH, 0 °C→rt
(97%). (d) chloromethyl chloroformate, NEt3, dry CH2Cl2, -20 °C (15%). (e) NaN3, DMF,
rt (44%).
Fmoc protection of D-cycloserine (62) was performed according to the literature
procedure of Gordeev et al. [127] Accordingly, the reactant was treated with N-(9-
fluorenyl-methoxycarbonyloxy) succinimide (Fmoc-OSu) in the presence of pyridine,
which functioned as a proton acceptor, and N,O-bis(trimethylsilyl) acetamide (BSA)
to mask the lactam nitrogen. Due to the poor solubility of the obtained crude product,
workup and purification were cumbersome and Fmoc-cycloserine (64) could only be
isolated in low yield (15%). However, a minor modification of the procedure signific-
antly improved the step. When the reaction was not carried out at room temperature
with solid BSA, but at 0 °C with BSA dissolved in CH2Cl2, the product could be
obtained in high yield (84%) and purity, after a short and simple work up.
In the next step, the lactame nitrogen of 62 was alkylated with MeI in the presence
of K2CO3. By carefully adjusting the reaction time, it was possible to facilitate methyl-
ation and, simultaneously, prevent Fmoc deprotection, so that 65 could be isolated in
70% yield after column chromatography. The regioselectivity of this reaction was in-
directly confirmed by the analytic data of the follow up product. The methanolysis
of the lactam with HCl/MeOH gave the acyclic aminoserine hydrochloride 66 in 97%
yield after coevaporation with MeOH.
57

4.5 Investigation of different Unclick systems
The introduction of the Azoc protection group was realised using the two-step liter-
ature procedure of Pothukanuri et al. [126] First, 66 was reacted with chloromethyl chlo-
roformate at -20 °C to give 67 in 15% after column chromatography. In the second
step, 67 was treated with NaN3 to substitute the chloro moiety. In order to reveal
the best solvent for this exchange, test runs were performed in deuterated CH3CN,
DMSO as well as DMF, and monitored with 1H NMR spectroscopy. It was found that
formation of product 63 proceeded very slowly in deuterated CH3CN and DMSO. In
contrast, when DMF was used, the conversion was completed after two hours, how-
ever, significant amounts of a side product were observed after three hours. Hence,
the nucleophilic substitution was performed on larger scale in DMF with a reaction
time of two hours to obtain 63 in 44% yield.
ON
OHN
Fmoc
HN
Fmoc OMe
O
O
HN
HN
Fmoc OMe
O
O
N O
O
N3
a
HN
Fmoc OMe
O
O
HN HCl
b
63
68 65
66
Scheme 4.5: Attempted Unclick reaction. (a) PPh3, [D8]-THF/H2O (9:1). (b) aq. NaHCO3, CHCl2(83%).
To investigate the proposed Unclick mechanism, a solution of Fmoc-Unclick serine
(63) in [D8]-THF/H2O (9:1) was treated with PPh3 (Scheme 4.5 (a)), and the reac-
tion was followed with 1H NMR spectroscopy. It was expected, that the induced
Staudinger reaction triggers Azoc deprotection, and that the resulting free hydroxyl
amine 68 cyclises to 65. Indeed, the spectra indicated a rapid loss of Azoc, but also
the retention of the methyl ester. Thereupon, it was explored, if the cyclisation is pH
dependent by repeating the reaction in mixtures of [D8]-THF/PBS (9:1) at pH 5.2, 7.0
and 8.8. However, the resulting 1H NMR spectra strongly resembled the former one.
58

4 Chemical unclicking of amide bonds
In order to prove the assumption, that the common product of the reactions was 68,
compound 66 treated with aqueous NaHCO3 in CH2Cl2 to remove the hydrochloride
(Scheme 4.5 (b)). The 1H NMR spectra before and after base treatment as well as the
desired cyclic structure are displayed in Figure 4.6.
3.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
1
2
3
2.6
3.2
1.7
1.3
2.1
0.9
0.7
4.4
2.1
2.2
Fmoc
Fmoc
Fmoc
0.9
1.1
1.8
1.1
0.8
0.6
4.0
2.0
2.0
2.75
3.33
1.45
2.40
1.31
1.60
0.70
4.29
2.37
2.32
CH2Cl2 OMe N-Me
N-Me
N-Me
+ NaHCO3
NH
NH
NH
CH2Cl2
CHCl3
CHCl3
CHCl3
OMe
Figure 4.6: 1H NMR of 65 (top, 250 MHz, CDCl3), 66 (middle, 200 MHz, CDCl3) and 68 (bottom, 250
MHz, CDCl3).
The spectrum, which is obtained after the reaction with NaHCO3, is in principle
identical with the spectra obtained from the test reactions. It displays the signal for
the methyl ester and, in comparison to its precursor with the hydrochloride (66), an
upfield shifted N-methyl signal. It was concluded, that Fmoc-Unclick serine (68) fails
to undergo the 5-exo-trig reaction, which might be due to the steric hinderance of the
bulky Fmoc group or the electronic structure of the carbamate moiety.
59

4.5 Investigation of different Unclick systems
4.5.2 Acetyl-Unclick-serine
The comparably small acetyl group was chosen as alternative to the Fmoc protecting
group. In comparison to the carbamate, the amide causes a strong electron withdraw-
ing effect, which is not diminished by the donation of a lone pair from an alkoxy
substituent. As a result, the C-terminus of the acetyl-protected amino acid is more
electrophilic, which was presumed to facilitate the ring closure.
ONH
OHN
Z
ON
OHN
Ac
HN
Ac OMe
O
O
HNHCl
b c
a Z=H
Z=Ac
62
6970 71
Scheme 4.6: Synthesis of Acetyl-aminoserine hydrochloride. (a) Ac2O, MeOH, rt (58%). (b) MeI,
K2CO3, acetone, reflux (27%). (c) HCl/MeOH, 0 °C→rt.
The synthesis of Acetyl-Unclick serine started with the N-terminal protection of
D-cycloserine (62) (Scheme 4.6). Therefore, 62 was treated with Ac2O in refluxing
methanol. [128] Purification of the crude product by recrystallisation in EtOH afforded
69 in the form of crystals, which were suitable for X-ray crystal structure analysis
(Figure 4.7).
Figure 4.7: X-ray crystal strcture of D-acetylcycloserine (69, orthorhombic, P212121). Ellipsoids are
drawn at 50% probability level.
Subsequently, compound 69 was treated with MeI to selectively alkylate the ring
nitrogen. Due to the poor solubility of the reactant and, despite some effort to op-
timise the reaction and the following work up procedure, the methylated product 70
could only be isolated in low yields (max. 27%). Nevertheless, it was possible to
obtain crystals suitable for X-ray crystal structure determination by layering an eth-
60
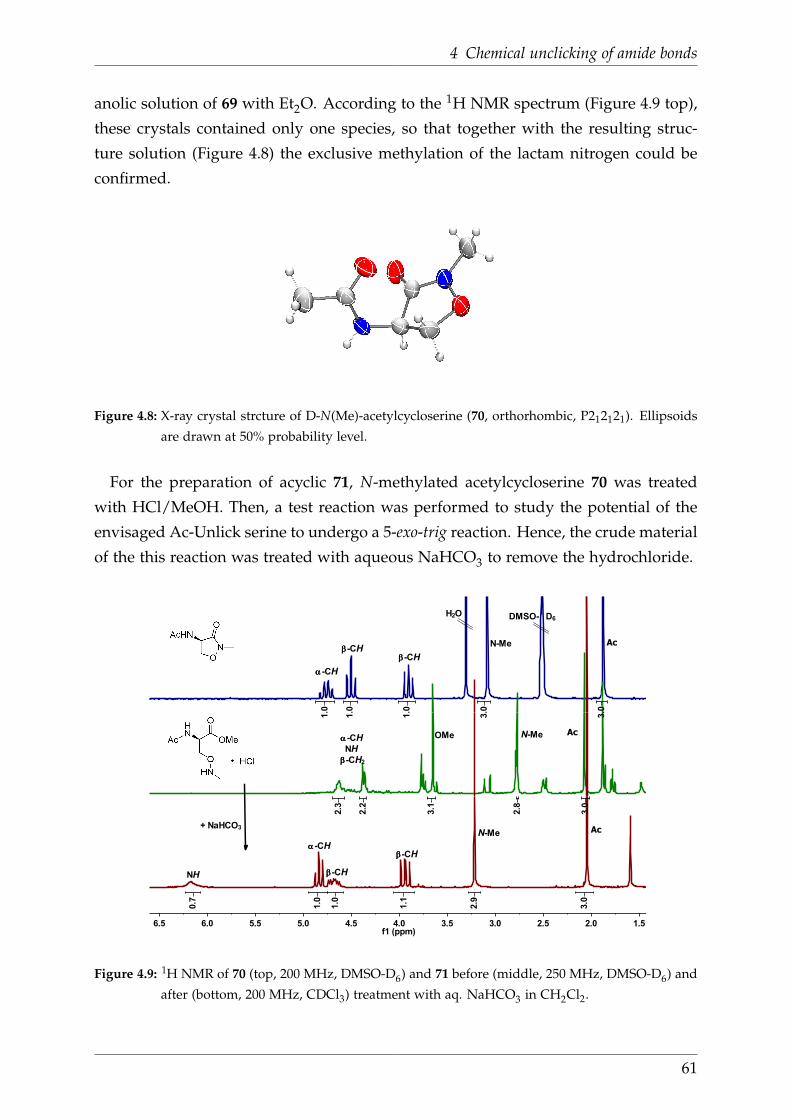
4 Chemical unclicking of amide bonds
anolic solution of 69 with Et2O. According to the 1H NMR spectrum (Figure 4.9 top),
these crystals contained only one species, so that together with the resulting struc-
ture solution (Figure 4.8) the exclusive methylation of the lactam nitrogen could be
confirmed.
Figure 4.8: X-ray crystal strcture of D-N(Me)-acetylcycloserine (70, orthorhombic, P212121). Ellipsoids
are drawn at 50% probability level.
For the preparation of acyclic 71, N-methylated acetylcycloserine 70 was treated
with HCl/MeOH. Then, a test reaction was performed to study the potential of the
envisaged Ac-Unlick serine to undergo a 5-exo-trig reaction. Hence, the crude material
of the this reaction was treated with aqueous NaHCO3 to remove the hydrochloride.
1.52.02.53.03.54.04.55.05.56.06.5f1 (ppm)
1
2
3
3.0
2.9
1.1
1.0
1.0
0.7
NH
aa -CHbb -CH
bb -CH
N-Me Ac
3.0
2.8
3.1
2.2
2.3
aa -CH NHbb -CH2
N-Me
AcOMe
aa -CH
bb -CHbb -CH
Ac
3.0
3.0
1.0
1.0
1.0
H2O DMSO- D6
+ NaHCO3
N-Me
Figure 4.9: 1H NMR of 70 (top, 200 MHz, DMSO-D6) and 71 before (middle, 250 MHz, DMSO-D6) and
after (bottom, 200 MHz, CDCl3) treatment with aq. NaHCO3 in CH2Cl2.
61

4.5 Investigation of different Unclick systems
The comparison of the 1H NMR spectra (Figure 4.9) of 71 before and after base
treatment is difficult, since different solvents were used. Still, it reveals that the latter
reaction causes a loss of the methyl ester signal. Also, the sharp signals of the product,
which resemble those of the 1H NMR spectrum of 70, suggest the recovery of the cyclic
structure.
Since these results were very promising, it was further intended to prepare a larger
amount of 71. However, multiple repetition of the methanolysis of 70 applying the
initial as well as different reaction conditions did not yield the pure product as ex-
pected, but mixtures with unpredictable amounts of an unidentified by-product. Due
to the poor solubility of this mixtures in organic solvents, neither purification nor the
subsequent reaction with chloromethyl chloroformate were successful.
4.5.3 Acetyl-Unclick-serine via Boc-cycloserine
Because of the synthetic unavailability of acetyl-protected Unclick serine 72, the mo-
lecular structure of the target molecule and the synthetic approach were reconsidered.
The resulting novel synthesis sequence does however not allow N-methylation of
the hydroxylamine, which also implies a slightly different target structure (73) (Fig-
ure 4.10).
HN
Ac OMe
O
O
HN O
O
N3
HN
Ac OMe
O
O
N O
O
N3
72 73
Figure 4.10: Structures of the previous (left) and the new synthetic target (right).
Therefore, it was planned to successively protect D-cycloserine with Boc and also
with chloromethyl carbamate (chloroc) prior to the ring opening (Figure 4.10). Taking
this new synthetic pathway (Scheme 4.7), D-cycloserine was first reacted with Boc2O
to give the N-terminal protected amino acid (74) in 54% yield after column chromato-
graphy. [129,130] The subsequent reaction with chloromethyl chloroformate afforded the
diprotected D-cycloserine (75) in 56% after purification. Treatment of the latter with
a HCl/ MeOH not only caused ring opening of the reactant, but also the removal of
Boc to quantitatively yield 76. Thus, the new reaction sequence provides the chance
to modify the N-terminal protecting group at a late stage of the synthetic pathway.
62
4 Chemical unclicking of amide bonds
ONH
OH2N
ONH
OHN
BocO
N
OHN
Boc
H2NOMe
O
O
HN O
O
Cl
O
O
Cl
HN
Ac OMe
O
O
HN O
O
Z
e
a
Z=Cl
Z=N3
b c d
62 74 75 76 77
73
Scheme 4.7: Synthesis of Ac-Unclick serine without the N-methyl group. (a) Boc2O, NEt3, THF/H2O,
0 °C→rt (54%) (b) chloromethyl chloroformate, NEt3, dry CH2Cl2, -20 °C (56%). (c)
HCl/MeOH, 0 °C→rt (quantitative). (d) Ac2O/DIPEA/DMF, rt (47%). (e) NaN3, DMF, rt.
Unexpectedly, the acetylation of 76 appeared to be very difficult and several differ-
ent reaction conditions were tested to optimise this reaction (Table 4.1).
Table 4.1: Overview over tested reaction conditions for the acetylation of 76
reagent base solvent temperature time remark
Ac2O - MeOH rt 2 h incomplete acetylationloss of chloroc during purification
AcCl NEt3 CHCl3 0°C→rt 2 h no acetylationloss of chloroc
AcCl NEt3 CH2Cl2 0°C→rt 1 h three acetyl signals in 1H NMRno separation possible
Ac2O DIPEA DMF rt 15 min simultaneous additionmoderate yield
Ac2O DIPEA DMF rt 15 min successive additionvery low yield
Ac2O NEt3 CH2Cl2 rt 3 h several acetyl signals in 1H NMRpartial loss of chloroc
Ac2O NEt3 CH2Cl2 -50°C, 3 h, then three acetyl signals in 1H NMRthen rt overnight partial loss of chloroc
When Ac2O was used, the resulting 1H NMR spectrum of the crude product indic-
ated incomplete acetylation. Purification with column chromatography to remove the
reactant and traces of Ac2O was not successful, but only led to the loss of chloroc.
Alternatively, the use of AcCl in the presence of a non-nucleophilic base was tested,
employing conditions reported by Griesbeck et al. for the synthesis of N-acetylamino
acid methyl esters. [131] Accordingly, 76 was treated with two equivalents of NEt3 to
remove the hydrochloride and to deprotonate the N-terminal nitrogen. Then, AcCl
was added at low temperature to prevent multiple acetylation of the same nitrogen.
63

4.5 Investigation of different Unclick systems
However, this reaction completely failed to produce 77. In contrast, when CHCl3was used as reaction medium, the isolated crude material showed several acetyl and
methylene groups in the 1H NMR spectrum. Still, further purification with column
chromatography was not successful.
Since the reactions with AcCl were rather discouraging, attention was turned back
to the use of Ac2O, while keeping the idea of employing a non-nucleophilic base. A
simple method that combines both is the utilisation of a capping agent, which is a
mixture of Ac2O and DIPEA in DMF that is normally used in solid phase peptide
synthesis to cap unreacted amino acids. Indeed, treatment of 76 with this mixture,
followed by repeated column chromatography of the crude product provided 77 in
47% yield. In contrast, the successive addition of DIPEA and Ac2O provided the
acetylated product in only 21% yield after column chromatography.
The reaction could not be further improved by replacement of DIPEA with NEt3 and
simultaneous extension of the reaction time. Addition of Ac2O at low temperature
did also not aid product formation. Instead, a partial loss of the chloroc group and
multiple acetylation was observed.
1.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.0f1 (ppm)
1
2
2.8
3.0
2.1
1.3
1.6
3.0
3.0
1.8
1.0
1.7
0.8
0.8
CDCl3
NH
-CH2-Cl
aa -CH
bb -CH2
OMe Ac
NH
-CH2-N3
aa -CH
bb -CH2
OMe AcCDCl3
Figure 4.11: 1H NMR of 77 (top, 250 MHz, CD3OD) and of 73 (bottom, 200 MHz, CDCl3).
64

4 Chemical unclicking of amide bonds
After optimisation of the acetylation, 77 was finally converted into Ac-Unclick ser-
ine (73) by treatment with NaN3. As the 1H NMR spectra show (Figure 4.11), the
substitution of the chloro moiety by an azide group causes an upfield shift of the
methylene protons, while the remaining signals show the same chemical shifts and
integrals as before. However, the result of this test reaction could not be reproduced
when this reaction was repeated on larger scale. The analytical data of the obtained
products always indicated a loss or decomposition of the chloroc moiety.
65

4.5 Investigation of different Unclick systems
4.5.4 Fmoc-Gly-Unclick-serine
For the repeated reconsideration of the Unclick concept, all previous findings were
taken into account. To this point, the following was known:
• Fmoc-protected (cyclo)serine derivatives provide good solubility in organic media, can be purifiedwith reasonable effort and are obtained in satisfying yields.
• Fmoc(N-Me) Unclick serine does not undergo the attempted ring closure after Azoc removal,presumably due to the presence of a bulky carbamate.
• Ac-protected (cyclo)serine derivatives provide very limited solubility in organic media, are usuallypurified by crystallisation and are isolated in low yields.
• Although it was shown, that ring closure in Ac(N-Me) aminoserine occurs readily, its base labilityalso prevents the introduction of Azoc.
• Boc-protected cycloserine derivatives are comparable to Fmoc analogues in solubility, purificationeffort and obtained yield.
• Boc-cycloserine-Chloroc is deprotected during the acidic ring opening. Subsequent acetylation ischallenging and the Cl/N3 replacement is not reproducible. Especially the lack of N-methylationseems to favour side reactions.
From the gathered knowledge, the requirements for an improved Unclick system
were derived. First, an Fmoc or Boc protecting group is necessary to provide sufficient
solution in organic media. This is equally important for the reactions as well as puri-
fication procedures and has, in general, a positive influence on the yield. Second, the
N-terminus has to be equipped with an amide group which causes a stronger electron
withdrawing effect than a carbamate and, consequently, enhances the electrophilicity
of the C-terminus. Third, the hydroxyl carbamate nitrogen has to be methylated to
prevent side reactions. A lead structure, which fulfils all criteria, is shown in Fig-
ure 4.12. It can be obtained by coupling of D-cycloserine to L-glycine, followed by
methylation, ring opening, and Azoc introduction.
NH
O
OR
O
HN
O
OH
O
N O
O
N3
Figure 4.12: The newly developed Unclick system possesses three important features, which are expec-
ted to contribute to a high solubility in organic media (carbamate), a readily occuring ring
closure (N-terminal amide) and a reduced chance of side reactions (N-methylation).
66

4 Chemical unclicking of amide bonds
The synthesis of this dipeptide was attempted via the trimethylsilyl/acyl chloride
method, which was adapted from a procedure of De et al. [132] This rather uncommon
method for peptide coupling in solution was chosen, since silylation of both nitrogens
of 62 significantly increases the solubility of the starting material in organic media. [133]
In addition, when treated with a carboxylic acid chloride, the very good leaving group
trimethylsilylchloride is formed as a byproduct during peptide coupling. [134]
The peptide coupling was prepared by the reaction of Fmoc-Gly-OH (78) with
oxalyl chloride to form acyl chloride 79. At the same time, D-cycloserine (62) was
silylated with BSA to 80 in a solvent mixture of N-methyl-2-pyrrolidone (NMP) and
CH2Cl2. The isolated acid chloride and an excess of pyridine were added to the latter
reaction mixture at low temperature to form dipeptide (81), which was isolated in
42% yield after column chromatography.
+Cl
OCl
+O
Si
NSi
NH
Fmoc
O
OH
NH
Fmoc
O
HN
ONH
O
O
NH
Fmoc
O
Cl
ONH
H2NO
ON
HN
O
TMSTMS
a
b
c78 79
62 80
81
Scheme 4.8: Synthesis of Fmoc-glycine-cycloserine via the trimethylsilyl/acyl chloride method. (a)
neat, 0 °C. (b) BSA, NMP, dry CH2Cl2, rt. (c) pyridine, 0 °C→rt (42%).
The subsequent reactions (Scheme 4.9) were performed in analogy to the synthesis
pathway of Fmoc-Unclick serine. At first, the ring nitrogen was alkylated with MeI in
acetone under slightly basic conditions to yield the compound 82 in 67% yield after
column chromatography.
Following treatment with HCl/MeOH resulted in the formation of two products,
which were separated by column chromatography and identified as 83 (50% yield)
and Fmoc-Gly-OMe (16% yield). This observation implies a partial decomposition
of the dipeptide, which is accompanied by acidic esterification of Fmoc-glycine-OH.
Indeed, it is well-known, that the applied conditions can be used for the C-terminal
esterification unprotected amino acid, [135,136] but are also strong enough to cause the
partial cleave of peptide bonds. [137,138]
67

4.5 Investigation of different Unclick systems
NH
Fmoc
O
HN
ON
O
NH
Fmoc
O
HN
ONH
O
NH
Fmoc
O
HN
O
O
NH
ONH
Fmoc
O
HN
O
O
N
O
O
O
Z
a
Z=Cl
Z=N3
b
d
c
81 82
8384
85
Scheme 4.9: Synthetic pathway for the synthesis of Fmoc-Gly-Unclick serine. (a) MeI, K2CO3, acetone,
reflux (67%). (b) 1.25M HCl/MeOH, 0°C → RT (50%). (c) chloromethyl chloroformate,
NEt3, CH2Cl2, -20°C (71%). (d) NaN3, DMF, rt (72%).
Retrospectively, this acidic amide hydrolysis might also explain the previous ob-
servation of several products after treatment of 70 with HCl/MeOH. It is also worth
mentioning that aminoserine 83 is highly unpolar, which allowed its purification with
column chromatography and, therewith, suggests the absence of a hydrogen chloride
is formed. This hypothesis was strengthened by a test reaction during which 83 was
dissolved in CH2Cl2 and treated with an excess of aqueous NaHCO3. Even after four
days, no changes in the 1H NMR spectrum occurred, which would go along with the
removal of the hydrogen chloride (observed for Fmoc-protected aminoserine) or ring
closure (observed for Ac-protected aminoserine).
Regardless, Azoc introduction was attempted as usual by a two-step procedure.
First, 83 was reacted with chloromethyl chloroformate in CH2Cl2 at -20°C to give 84,
which was obtained in 71% yield after column chromatography. Subsequent reaction
with NaN3 provided 85, which was isolated in 72% yield after purification.
Finally, with the target compound at hand, it was studied, if a 5-exo-trig reaction can
be induced. Therefore, Fmoc-Gly-Unclick serine was treated with PPh3 in aqueous
THF and the reaction was monitored with 1H NMR spectroscopy. Unfortunately, the
resulting 1H NMR spectrum revealed (Figure 4.13), that the product of this reaction
was the unprotected aminoserine 83.
68
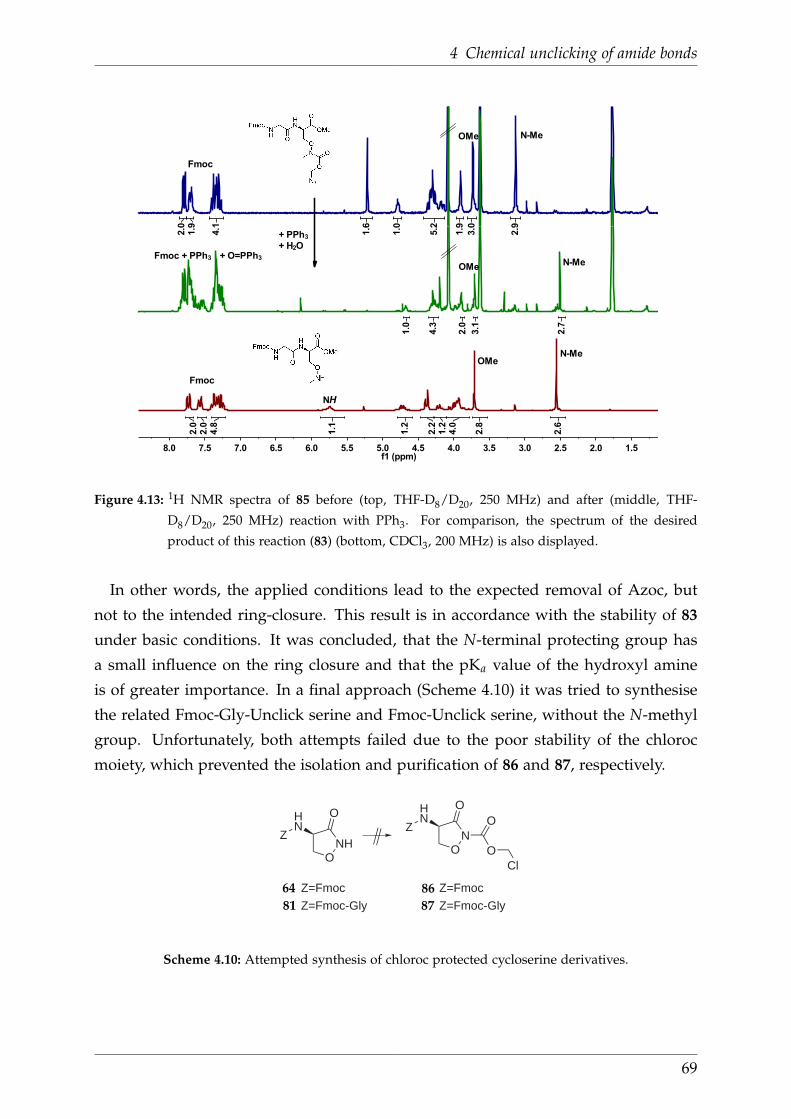
4 Chemical unclicking of amide bonds
1.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
1
2
3
2.9
3.0
1.9
5.2
1.0
1.6
4.1
1.9
2.0
2.7
3.1
2.0
4.3
1.0
2.6
2.8
4.0
1.2
2.2
1.2
1.1
4.8
2.0
2.0
+ PPh3+ H2O
Fmoc
Fmoc
Fmoc + PPh3 + O=PPh3
NH
OMe
OMe
OMe N-Me
N-Me
N-Me
Figure 4.13: 1H NMR spectra of 85 before (top, THF-D8/D20, 250 MHz) and after (middle, THF-
D8/D20, 250 MHz) reaction with PPh3. For comparison, the spectrum of the desired
product of this reaction (83) (bottom, CDCl3, 200 MHz) is also displayed.
In other words, the applied conditions lead to the expected removal of Azoc, but
not to the intended ring-closure. This result is in accordance with the stability of 83
under basic conditions. It was concluded, that the N-terminal protecting group has
a small influence on the ring closure and that the pKa value of the hydroxyl amine
is of greater importance. In a final approach (Scheme 4.10) it was tried to synthesise
the related Fmoc-Gly-Unclick serine and Fmoc-Unclick serine, without the N-methyl
group. Unfortunately, both attempts failed due to the poor stability of the chloroc
moiety, which prevented the isolation and purification of 86 and 87, respectively.
ONH
OHN
ZO
N
OHN
Z
O
O
Cl
Z=Fmoc
Z=Fmoc-Gly
Z=Fmoc
Z=Fmoc-Gly
64
81
86
87
Scheme 4.10: Attempted synthesis of chloroc protected cycloserine derivatives.
69

5 Summary
The first part of this work dealt with the development of a binuclear ligand for a
lanthanide-based, near-IR emissive upconversion complex. The structure of this sys-
tem is based on tris(bipyridine) (TBP) cryptands. These ligands form stable, lumin-
escent lanthanide complexes, are devoid of N-H and O-H oscillators and protect the
coordinated metal efficiently from other oscillators e.g. in the solvent. Within this
work, it was aimed to evaluate, if exclusion of C-H groups from the TBP ligand re-
duces the quenching of lanthanide-centered near-IR luminescence. In addition, it was
intended to introduce functional groups to TBP cryptates and to selectively couple
them to a binuclear ligand.
To address the first matter, efficient protocols for the synthesis of selectively deuter-
ated TBP cryptates were developed. In comparison to the undeuterated analogue, the
preparation of the isotopologues required just one additional synthetic step during
which the H/D exchange was performed using a readily available, cheap deuterium
sources (D2O or NaBD4). Thus, it was possible to prepare a series of isotopologic
cryptates (Figure 5.1) with excellent overall deuteration levels (>97%).
N
N
N
N
N
N
N
N
DD
DD
DD D D
D D DD
DD
DDDD
D D
Na Br
N
N
N
N
N
N
N
N
D D DD
DD
DDDD
Na Br
N
N
N
N
N
N
N
N
D D
DDD D
DDDD
D D
Na Br
Figure 5.1: Examples of synthesised isotopologic cryptates
Photophysical studies of the corresponding Nd3+ and Yb3+ complexes revealed,
that a decreasing number of C-H groups goes along with prolonged luminescence
lifetimes and, hence, with reduced quenching (Figure 5.2). Accordingly, the smallest
quenching rates were observed for the perdeuterated complexes. Compared to the
undeuterated counterparts, the determined luminescence lifetimes were increased by
70

5 Summary
the factor 1.6 for Yb3+ and 6.9 for Nd3+, the latter being among the longest lifetimes
for Nd3+ complexes in solution reported so far. Moreover, the deactivation rate contri-
butions of individual C-H groups were accurately quantified. This study represented
the first systematic investigation of C-H induced luminescence quenching in near-IR
emissive lanthanide cryptates.
Figure 5.2: Decrease of luminescence quenching with increasing degree of deuteration
For the second aim, the selective coupling of functionalised TBP cryptates, carboxy-
lic acid esters and amino groups were regarded the most attractive functional groups.
While the carboxy functionalised cryptates were prepared employing reported pro-
cedures, two novel synthetic pathways were developed, which enabled the prepara-
tion of previously unknown amino functionalised cryptates. Furthermore, the above
mentioned deuteration protocols could be successfully adapted for the synthesis of
the first deuterated carboxy functionalised cryptate (98%D) (Figure 5.3).
N
N
N
N
N
N
N
N
DD
DD
DD D D
DDDD D
DD
DDDD
DD
DD
D
D
DD
D D
Na
COOMe
Br
Figure 5.3: Synthesised perdeuterated functionalised cryptate
71

The synthesis of a binuclear cryptate was initially attempted by the successive coup-
ling of two carboxy functionalised cryptates to one ethylenediamine linker. However,
this approach only provided minimal yields, so that an alternative pathway was pur-
sued. Therefore, two carboxy functionalised cryptates were linked to one ethylene-
diamine each and then coupled via a carbamide. Using this procedure, it was possible
for the first time to successfully prepare, isolate and characterise a dinuclear TBP
crpytate (Figure 5.4).
N
N
N
N
N
N
N
NNa
Br
O
NH
HN
N
N
N
N
N
N
N
NNa
Br
O
NH
HN
OOO
OO
Figure 5.4: Synthesised binuclear cryptate
The second part of this work dealt with the development of a molecular system, that
allows the straightforward selective cleavage of peptide bonds by applying a bioortho-
gonal, chemical trigger. This simple chemical transformation, which proceeds under
mild conditions, represents the counterpart of the well-established “Click chemistry”
and was therefore termed “Unclick chemistry”.
The key molecule of this system was a D-aminoserine with a bioorthogonal azide
carbamate (Azoc) protecting group in its side chain. Within this work, it was aimed
to synthesise such an “Unclick”-serine and to prove the PPh3-mediated Azoc depro-
tection, as well as 5-exo-trig reactions of this molecule. Hence, several D-aminoserines
with different N-terminal protecting groups (Fmoc, acetyl, Boc) or residues (Fmoc-
Glu) were prepared. While it was not possible to prove Azoc deprotection and 5-exo-
trig reaction with the same molecule, still two important goals were achieved.
HN
Fmoc OMe
O
O
HN
HN
Fmoc OMe
O
O
N O
O
N3
PPh3
H2O
Figure 5.5: Selective Azoc-deprotection of Fmoc-protected Unclick serine
72

5 Summary
Using the Fmoc protected D-aminoserine it was shown, that Azoc can be easily
introduced to this amino acid and that deprotection with PPh3 readily occurs under
physiological conditions (Figure 5.5). In addition, it was found that the 5-exo-trig
reaction indeed proceeds within acetyl protected D-aminoserine (Figure 5.6).
HN
Ac O
O
O
NHO
NH
OHN5-exo-trig Ac
Figure 5.6: Ringclosure of an acetyl-protected aminoserine
73

6 Experimental Section
6.1 General
Chemicals were purchased from commercial suppliers and used as received unless
stated otherwise. Deuterated solvents/reagents had deuterium contents > 99.8% with
the exception of NaBD4 (> 98%D). CH3CN for the synthesis of the cryptates was
HPLC grade and was stored under nitrogen. DMF for the synthesis of cryptate amino
acid derivatives was SPPS grade. Dry DMF for other reactions was used as pur-
chased. THF was dried using an MBraun Solvent Purification System. Other solvents
were dried by standard procedures (EtOH (Mg)), CH2Cl2 (CaH2), MeOH (Mg), ethyl-
enediamine (KOH), pyridine (KOH)). Air-sensitive reactions were carried out under
a dry, dioxygen-free atmosphere of N2 using Schlenk technique. Column chroma-
tography was performed with silica gel 60 (Merck, 0.063-0.200 mm) or silica gel 60
silanised (Merck, 0.063-0.200 mm). Analytical thin layer chromatography (TLC) was
done on silica gel 60 F254 plates (Merck, coated on aluminium sheets) or silica gel C18
plates (Machery-Nagel, pre-coated on aluminium sheets, Alugram RP-18 W/UV254)
Product spots were visualized by UV light at 254 nm, and subsequently developed
using I2 vapour or ninhydrin solution as appropriate. Overall deuteration levels were
established by deconvolution of the corresponding mass spectra. ESI mass spectro-
metry was done using Bruker Daltonics Esquire6000. NMR spectra were measured
at room temperature on either a Bruker DPX-200, Bruker DPX-250, or a DRX-400. 1H
NMR spectra were recorded at 200 MHz, 250 MHz, or 400 MHz and 13C NMR at
50.3 MHz, 62.9 MHz, or 100.6 MHz, respectively. The chemical shifts δ are repor-
ted in parts per million (ppm) downfield of tetramethylsilane (TMS), using residual
protonated solvent as internal standard. For NMR spectra recorded in a mixture of
CD2Cl2/CD3OD (1:1), the pure solvent mixture was referenced to tetramethylsilane
and the CD3OD peak (1H NMR: 3.33 ppm, 13C NMR: 49.07 ppm) used as secondary
reference within the sample. Coupling constants J are quoted to the nearest 0.1 Hz.
The abbreviations used in the description of resonances are: s (singlet), d (doublet), t
(triplet), q, (quartet), br (broad), m (multiplet).
74

6 Experimental Section
IR spectra were aquired on a Bruker Tensor 27 FT-IR spectrometer equipped with a
diamond ATR setup. Analytical reversed-phase HPLC was performed on Lichrospher
RP-18e (Merck, 125 × 4mm-5µm, flow rate: 1 ml min−1, UV detection: 300 nm).
X-ray crystal structure determinations were performed by placing the respective
crytal on a glass fiber using perfluoropolyether oil. The measurement was carried
out on an Oxford Xcalibur 2 diffractometer using monochromated Mo Kα radiation.
Frames corresponding to an arbitrary hemisphere of data were collected using ω
scans. The structure was solved within the Wingx [139] package using direct meth-
ods (SIR92 [140]) and expanded using Fourier techniques (SHELXL-97 [141]). Hydrogen
atoms were included but not refined.
6.2 Luminescence measurements
Corrected steady state emission spectra were acquired on a PTI Quantamaster QM4
spectrofluorimeter using 1.0 cm quartz cuvettes with ca. 100 µM solutions at 298 K.
D2O and [D6]-DMSO (NMR grade, 99.9%D) were purchased from commercial sup-
pliers and used as received. The excitation light source was a 75 W continuous xenon
short arc lamp. Emission spectra were collected at 90° to the excitation beam using a
PTI R928 photomultiplier tube (operated at -1000 V) as the detector. Spectral selection
was achieved by single grating monochromators (excitation:1200 grooves/mm, blazed
at 300 nm; visible emission: 1200 grooves/mm, blazed at 400 nm; near-IR emission:
600 grooves/mm, blazed at 1200 nm).
Luminescence lifetimes were measured with the same spectrofluorimeter equipped
with an additional Q4 phosphorescence module. The light source for these measure-
ments was a xenon flash lamp (Hamamatsu L4633: 10 Hz repetition rate, pulse width
ca. 1.5 µs FWHM). Emission was measured at 90° using a PTI P1.7R detector mod-
ule (Hamamatsu PMT R5509-72 with a Hamamatsu C9525 power supply operated
at -1500 V and a Hamamatsu liquid N2 cooling unit C9940 set to -80 °C). Lifetime
data analysis (deconvolution, statistical parameters, etc.) was performed using the
software package FeliX32 from PTI. The instrument response function (IRF) for the
spectral deconvolution procedure was determined using a dilute aqueous dispersion
of colloidal silica (Ludox® AM-30). The given values are averages of three independ-
ent measurements. The global fitting of the lifetime data was performed with Origin
Pro 8G using the “nonlinear surface fit” function constrained to a first order polyno-
mial in y and z: kobs(y,z) = k0 - y ∆kbenzyl - z ∆kpy.
75

6.3 Simple bipyridine building blocks
6.3 Simple bipyridine building blocks
Undeuterated compounds
2-Bromo-6-picoline (2)
N Br2
2 was prepared according to the literature procedure of Maheswari Palanisamy et al. [97]
In a 3-necked round bottom flask equipped with a dropping funnel and mechanical
stirrer, 2-aminopicoline (1) (50.0 g, 0.462 mol, 1.0 equiv) was added in small portions
to gently stirred HBr (47%, 300 ml) at room temperature. The obtained clear yellow
solution was cooled to -20 °C and cooled Br2 (207.0 g, 66.3 ml, 1.295 mol, 2.8 equiv),
was added dropwise over a period of 40 min while the temperature was maintained.
During addition, the solution turned from orange to red and finally a bright yellow
suspension was obtained. After stirring for 90 min at -20 °C, aqueous NaNO2 (86.0 g
in 125 ml water, 1.247 mol, 2.7 equiv) was added dropwise via a dropping funnel (gas
formation!). The brown suspension was allowed to warm to 15 °C and then stirred
for 45 min. Then, it was cooled to -20 °C again and aqueous NaOH (335 g in 500 ml
water) was added dropwise while the temperature was kept between −10 °C and
−20 °C. Subsequently, the mixture was allowed to warm to room temperature and
then stirred for another hour. After extraction with CHCl3, the organic layer was
dried over MgSO4 and the solvent removed in vacuo to leave a brown oil behind. The
crude product was destilled in vacuo (7.1 mbar, 70-72 °C, bath 120 °C) to yield 2 as
colourless liquid.
Yield: 45.7 g (0.266 mol, 58%). 1H NMR (250 MHz, CDCl3): δ 7.42 (t, 3J = 7.7 Hz, 1H),
7.29 (d, 3J = 7.8 Hz, 1H), 7.10 (d, 3J = 7.5 Hz, 2.53 (s, 3H) ppm.
6,6’-Dimethyl-2,2’-bipyridine (3)
N N3
3 was prepared according to the literature procedure of Rode et al. [98] Under nitrogen
and with vigorous stirring, a solution of 2-bromo-6-picoline (2) (21.9 g, 127.1 mmol,
76

6 Experimental Section
2.0 equiv) in dry toluene (100 ml) was added dropwise to Raney-Nickel (3.7 g, 63.6
mmol, 1.0 equiv) over the course of 10 min. The resulting reaction mixture was heated
at reflux for 18 h (bath: 125 °C) to obtain a dark violet suspension, which was filtered
over a Schlenk frit. The violet solid was washed with dry toluene (2 × 40 ml) and then
dried in vacuo. The blue filtrate was treated with HCl/H2O to neutralise remaining
Ni prior to disposal. To hydrolyse the obtained nickel complex, the violet solid was
added to 40 °C warm water (250 ml) in small portions and stirred for 2 h, whereas
the colour turned from violet to green. The suspension was filtered until the filter
was plugged and the aqueous layer was extracted with CHCl3 (2 × 200 ml). The dark
sticky residue was washed with CHCl3 (5 × 100 ml) and filtered again. The combined
organic extracts were dried over MgSO4 and the solvent removed in vacuo to leave
the brown crude product behind (3.58 g). Recrystallisation from boiling petrolether
yielded 3 as colourless crystals.
Yield: 2.0 g (11.1 mmol, 17%). 1H NMR (250 MHz, CDCl3): δ 8.19 (t, 3J = 7.8 Hz, 2H),
7.68 (d, 3J = 7.7 Hz, 2H), 7.15 (d, 3J = 7.6 Hz, 2.63 (s, 6H) ppm.
6,6’-Dimethyl-2,2’-bipyridine-N,N’-dioxide (4)
N NO O
4
4 was prepared according to the literature procedure of Newkome et al. [99] With cool-
ing in an ice bath, a solution of m-chloroperoxybenzoic acid (18.0 g of 77 wt%, 13.9 g
pure, 80.3 mmol, 2.5 equiv) in CHCl3 (450 ml) was added dropwise to a solution of 3
(6.0 g, 32.6 mmol, 1.0 equiv) in CHCl3 (450 ml) over the course of 3 h. The reaction
mixture was allowed to slowly warm to room temperature and stirred overnight.
Then, the organic phase was extracted with saturated NaHCO3 (10 ml), aqueous
Na2S2O3 (10 ml), dried over MgSO4 and evaporated to dryness. The crude product
was purified by column chromatography (SiO2, CH2Cl2/MeOH 24:1→ 9:1) to give 4
as an off-white solid.
Yield: 5.7 g (26.2 mmol, 80%). Rf = 0.38 (SiO2, CH2Cl2/MeOH 9:1, detection UV).
MS (ESI+): m/z (%) 217.04 (7, [M+H]+), 238.98 (100, [M+Na]+), 254.92 (6, [M+K]+).1H NMR (200 MHz, CHCl3): δ 7.39–7.31 (m, 4H), 7.27–7.16 (m, 2H), 2.58 (s, 6H) ppm.13C NMR (50.3 MHz, CHCl3): δ 149.7, 143.6, 126.8, 125.4, 124.5, 17.9 ppm.IR (FT-ATR):
ν̃ 3074 (w, C-Harom), 3046 (w, C-Harom), 2953 (w, CH3), 2910 (w, CH3), 1701 (w), 1608 (w,
77

6.3 Simple bipyridine building blocks
C=Carom), 1560 (w, C=Carom), 1475 (m), 1443 (m), 1360 (m), 1263 (s, N-oxide), 1246 (s),
848 (s), 743 (s) cm-1.
6,6’-Bis(bromomethyl)-2,2’-bipyridine (5)
NN
BrBr5
5 was prepared in analogy to a procedure of Psychogios et al. [100] Under nitrogen,
TFA anhydride (55 ml) was added quickly via syringe to a yellow solution of 4 (2.7 g,
12.6 mmol, 1.0 equiv) in dry CH2Cl2 (55 ml). The orange reaction mixture was heated
at reflux for 1.5 h before the solvents were removed in vacuo. The orange residue
was dissolved in a mixture of dry DMF/THF (2:1, 60 ml) and the obtained solution
was added to solid LiBr (11.9 g, 137.4 mmol, 11.0 equiv), which was dried before-
hand in vacuo at 180 °C for 5 h. The reaction mixture was stirred at room temperat-
ure overnight before the solvents were removed in vacuo at 60 °C. The bright orange
residue was dissolved in CH2Cl2 (200 ml) and washed with water (3 × 200 ml). The
combined organic layers were dried over MgSO4 and evaporated to dryness. The
crude product was dry-loaded onto silica and purified by column chromatography
(SiO2, CH2Cl2/MeOH 100:1) to give 5 as a bright yellow solid.
Yield: 2.1 g (6.0 mmol, 48%). Rf = 0.68 (SiO2, CH2Cl2/MeOH 100:1, detection UV).
MS (ESI+): m/z (%) 362.85 (60, [M+Na]+), 338.81 (26, [M+H+CF3CO]+). 1H NMR
(250 MHz, CDCl3): δ 8.39 (dd, 3J = 8.0 Hz, 4J = 1.1 Hz, 1H), 7.82 (t, 3J = 7.8 Hz, 1H),
7.47 (dd, 3J = 7.7 Hz, 4J = 1.1 Hz, 1H), 4.63 (s, 2H) ppm. 13C NMR (50.3 MHz, CDCl3):
δ 156.4, 155.7, 138.1, 123.7, 120.7, 34.2 ppm. IR (FT-ATR): ν̃ 3039 (w, C-Harom), 2974 (w,
CH2), 1579 (m, C=Carom), 1567 (m, C=Carom), 1438 (m), 1260 (s), 1080 (m, C-Br), 993 (m),
806 (s), 749 (s), 632 (s, C-Br) cm-1.
6,6’-Bis(bromomethyl)-2,2’-bipyridine-N,N’-dioxide (48)
NNOO
BrBr48
78

6 Experimental Section
48 was prepared by a modified procedure of Caron et al. [118] Under nitrogen and with
cooling in an ice bath, urea-hydrogen peroxide (415.3 mg, 4.414 mmol, 3 equiv) was
added as a solid to a solution of 5 (502.0 mg, 1.468 mmol, 1 equiv) in dry CH2Cl2(30 ml) followed by dropwise addition of TFA anhydride (1.511 g, 1 ml, 7.194 mmol,
4.9 equiv) over the course of 15 min. The reaction mixture was slowly warmed to
room temperature and stirred for 2 d. The obtained clear yellow solution was diluted
with CH2Cl2 (50 ml) before sat. Na2S2O3 (15 ml) was added. After stirring for 1 h,
the phases were separated and the aqueous layer extracted with CH2Cl2 (3 × 50 ml).
The combined organic extracts were dried over MgSO4 and the solvent removed in
vacuo. The yellow crude product was dry-loaded onto silica and purified by column
chromatography (SiO2, CH2Cl2/MeOH 50:1→ 24:1) to give 48 as a white solid.
Yield: 462.8 mg (1.237 mmol, 84%). Rf = 0.17 (SiO2, CH2Cl2/MeOH 24:1, detec-
tion UV). MS (ESI+): m/z (%) 372.96 (5, [M+H]+), 394.86 (49, [M+Na]+). 1H NMR
(200 MHz, CDCl3): δ 7.68–7.64 (m, 1H), 7.64–7.61 (m, 1H), 7.61–7.58 (m, 1H), 7.57–7.54
(m, 1H), 7.35 (s, 1H), 7.31 (s, 1H), 4.73 (s, 4H) ppm. IR (FT-ATR): ν̃ 3067 (w, C-Harom),
3048 (w, C-Harom), 3023 (w, C-Harom), 2980 (w, CH2, 1477 (w), 1410 (m), 1384 (s), 1250
(s, N-oxide), 1205 (s), 887 (m), 839 (s), 790 (s), 646 (m, C-Br) cm-1.
6,6’-Bis(aminomethyl)-2,2’-bipyridine trihydrobromide hydrate (18)
NN
H2N
H2O
3 HBr
NH218
18 was prepared according to the literature procedure. [104] 5 (1.5 g, 4.4 mmol, 1 equiv)
in CHCl3 (60 ml) was heated at reflux until dissolution. Then, a solution of hexa-
methylenetetramine (1.3 g, 9.4 mmol, 2.15 equiv) in CHCl3 (40 ml) was added drop-
wise. After refluxing for 3 h, the mixture was cooled to room temperature and stored
at 8 °C overnight. The precipitate was collected on a Buchner funnel, washed with
a small amount of CHCl3 and dried in vacuo. The white solid was suspended in
H2O/EtOH/47% HBr (16 ml, 62 ml, 10 ml) and stirred at 75 °C until dissolution.
Upon cooling in an ice bath, 18 crystallised as slightly yellow needles, which were
filtered off, washed with a small amount of cold EtOH and dried in vacuo.
Yield: 1.1 g (2.2 mmol, 50%). MS (ESI+): m/z (%) 215.01 (100, [M+H2O-3HBr]+).1H NMR (200 MHz, D2O): δ 8.35 (d, 3J = 8.0 Hz, 2H), 8.04 (dd, 3J = 8.3 Hz, 3J = 7.3
Hz, 2H), 7.53 (d, 3J = 7.8 Hz, 2H), 4.43 (s, 4H) ppm. 13C NMR (50.3 MHz, D2O): δ
79

6.3 Simple bipyridine building blocks
152.3, 151.2, 141.0, 124.3, 122.3, 42.4 ppm. IR (FT-ATR): ν̃ 3306 (w, NH+3 ), 3030 (m,
CH3), 2928 (m, CH3), 1612 (m, C=Carom), 1592 (m, C=Carom), 1561 (m, NH+3 ), 1523 (m,
C=Carom), 1508 (m, NH+3 ), 1420 (m), 1265 (m), 1183 (m), 995 (m), 884 (s), 795 (s) cm-1.
6,6’-Bis((N-tosyl)aminomethyl)-2,2’-bipyridine (47)
NN
TsHNNHTs47
47 was prepared according to the literature procedure. [142] 18 (950 mg, 2.0 mmol,
1 equiv) was added to a solution of NaOH (672 mg, 16.8 mmol, 8.5 equiv) in H2O
(6.5 ml). With vigorous stirring, a solution of tosyl chloride (827 mg, 4.3 mmol,
2.2 equiv) in Et2O (3 ml) was added at 0 °C before the yellow suspension was al-
lowed to warm to room temperature. After stirring overnight, the precipitate was
collected on a Buchner funnel and dried in vacuo at 60◦C for 1 d to give 47 as a white
solid.
Yield: 512.4 mg (0.98 mmol, 49%). MS (ESI+): m/z (%) 523.09 (78, [M+H]+), 545.04
(100, [M+Na]+) . 1H NMR (200 MHz, CDCl3): δ 8.17 (d, 3J = 7.8 Hz, 2H), 7.80–7.69
(m, 6H), 7.24–7.13 (m, 6H), 5.81 (t, 3J = 4.8 Hz, 2H), 4.34 (d, 3J = 5.2 Hz, 4H), 2.35 (s,
6H) ppm. 13C NMR (50.3 MHz, CDCl3): δ 154.8, 154.3, 143.6, 137.9, 136.8, 129.8, 127.3,
122.2, 120.0, 47.5, 21.6 ppm. IR (FT-ATR): ν̃ 3261 (w, C-Harom), 1575 (w, C=Carom), 1437
(w), 1337 (m, R-SO2-N), 1160 (s, R-SO2-N), 1074 (m), 898 (w), 812 (m), 798 (m), 667 (s)
cm-1.
2,2’-bipyridine-6,6’dicarboxylic acid (7)
NN
HOOHOO
7
7 was prepared in analogy to the procedure of Alyapyshev et al. [102] 3 (12.1 g, 65.7 mmol,
1.0 equiv) was added in portions to conc. H2SO4 (140 ml) and the mixture was heated
to 65 °C (bath temperature). CrO3 (29.5 g, 295.0 mmol, 4.5 equiv) was added at a
rate that the internal temperature did not rise above 70 °C. After complete addition,
the dark solution was heated to 70 °C (bath temperature) for additional 60 min. The
80

6 Experimental Section
mixture was poured onto crushed ice (ca. 350 g), the precipitate was collected on a
filter, washed with cold water, and dried in vacuo at 100 °C for several hours. The
crude product 7 was obtained as a light-yellow solid. The isolated material was used
for the following reaction without further purification.
Yield: 14.5 g. 1H NMR (200 MHz, DMSO−D6): δ 8.76 (dd, 3J = 7.6 Hz, 4J = 1.1 Hz, 2
H), 8.21 (t, 3J = 7.6 Hz, 2 H), 8.15 (dd, 3J = 7.6 Hz, 3J = 1.1 Hz, 2 H) ppm.
2,2’-bipyridine-6,6’dicarboxylic acid diethyl ester (8)
NN
EtOOEtOO
8
8 was prepared in analogy to the reported methyl ester of Blake et al. [103] 7 was
suspended in dry EtOH (300 mL), conc. H2SO4 (30 mL) was added cautiously, and
the mixture was refluxed for 20 h. At 0 °C, a saturated solution of Na2CO3 was added
dropwise until pH 7. The aqueous phase was extracted with CHCl3 (3 × 200 mL), the
combined organic phases were dried over MgSO4, and concentrated. The product 8
was obtained as a colorless solid.
Yield: 14.4 g (47.8 mmol, 73% over two steps). 1H NMR (200 MHz, CDCl3): δ 8.77 (d,3J = 7.8 Hz, 2 H), 8.15 (d, 3J = 7.8 Hz, 2 H), 7.98 (t, 3J = 7.8 Hz, 2H), 4.50 (q, 3J = 7.1
Hz, 4 H), 1.47 (t, 3J = 7.1 Hz, 6 H) ppm.
Deuterated compounds
[D4]-6,6’-(Bis(hydroxymethyl)-2,2’-bipyridine (9)
N N
HOOH
DD
DD
9
9 was prepared according to the literature procedure for the undeuterated compound
of Newkome et al. [99] Solid NaBD4 (357 mg, 8.52 mmol, 4.0 equiv) was added in
81

6.3 Simple bipyridine building blocks
portions to a suspension of 8 (0.64 g, 2.13 mmol, 1.0 equiv) in CD3OD (5 ml). The
resulting slightly pink solution was stirred at room temperature for 20 h before D2O
(10 ml) was added and the pH adjusted to 6 through addition of 2 M DCl (in D2O).
Then, the mixture was extracted with CHCl3 (4× 25 ml), the combined organic phases
dried over MgSO4 and the solvent removed in vacuo. The remaining residue was dry-
loaded onto silica and subjected to column chromatography (SiO2, CH2Cl2/MeOH
9:1, detection UV) to yield 9 as a colorless solid.
Yield: 303 mg (1.38 mmol, 65%, overall deuteration level: 99%). Rf = 0.08 (SiO2,
CH2Cl2/MeOH 9:1, detection UV). MS (ESI+): m/z (%) 221.0 (26, [M+H]+), 242.9 (100,
[M+Na]+). 1H NMR (250 MHz, CD3OD): δ 8.22 (d, 3J = 7.8 Hz, 2H), 7.88 (t, 3J = 7.8
Hz, 2H), 7.51 (d, 3J = 7.7 Hz, 2H) ppm. 13C NMR (62.9 MHz, CD3OD): δ 161.9, 156.6,
138.9, 121.8, 120.9, 65.3 (quint, J = 22.0 Hz) ppm.
[D4]-6,6’-Bis(bromomethyl)-2,2’-bipyridine (10)
N N
BrBr
DD
DD
10
Under nitrogen and with cooling in an ice bath, PBr3 (0.60 ml, 1.72 g, 6.36 mmol,
5.0 equiv) was added dropwise via syringe to a solution of 9 (280 mg, 1.27 mmol,
1.0 equiv) in dry DMF (8 ml). The yellow-brown suspension was stirred at room
temperature for 20 h before the volatiles were removed under reduced pressure, and
H2O (30 ml) was added cautiously with ice cooling. The pH was adjusted to 6 with
sat. NaHCO3 and the aqueous phase was extracted with CHCl3 (4 × 50 ml). The
combined organic layers were dried over MgSO4 and the solvent removed in vacuo.
The light-yellow solid residue was dry-loaded onto silica and subjected to column
chromatography (SiO2, CH2Cl2/MeOH 100:1, detection UV) to yield 10 as a colorless
solid.
Yield: 404 mg (1.17 mmol, 92%, overall deuteration level: 98%). Rf = 0.60 (SiO2,
CH2Cl2/MeOH 100:1, detection UV) . MS (ESI+): m/z (%) 304.1 (83), 346.8 (100,
[M+H]+). 1H NMR (250 MHz, CD2Cl2): δ 8.39 (d, 3J = 7.9 Hz, 2H), 7.83 (t, 3J =
7.8 Hz, 2H), 7.47 (d, 3J = 7.7 Hz, 2H) ppm. 13C NMR (62.9 MHz, CDCl3): δ 156.3,
155.5, 138.0, 123.6, 120.3, 34.0 (quint, J = 23.2 Hz) ppm.
82

6 Experimental Section
[D4]-6,6’-Bis(aminomethyl)-2,2’-bipyridine trihydrobromide hydrate (11)
NN
H2N
H2O
3 HBr
NH2
DD
DD
11
11 was prepared according to the literature procedure for the undeuterated compound
of Wang et al. [104] 10 (82.6 mg, 239 µmol, 1.0 equiv) was suspended in CHCl3 (3.1 mL)
and heated at reflux until dissolution. Then, a solution of hexamethylenetetramine
(72.4 mg, 516 µmol, 2.16 equiv) in CHCl3 (2 mL) was added dropwise. After refluxing
for 3 h, the reaction mixture was cooled to room temperature and stored at room
temperature overnight. The precipitate formed was collected on a Buchner funnel,
washed with a small amount of CHCl3 and dried in vacuo. Subsequently, the white
solid was suspended in H2O/EtOH/47% HBr) (0.7 ml, 2.9 ml, 0.50 ml) and stirred at
75 °C until dissolution. After storing the mixture at room temperature for 1 h and at
0 °C for 30 min, the precipitate was collected, washed with EtOH and dried in vacuo.
11 was obtained as a light-yellow solid.
Yield: 69.5 mg (145 µmol, 61%, overall deuteration level: 97%). MS (ESI+): m/z (%)
219.0 (48, [M+H]+), 282.1 (44), 311.0 (100). 1H NMR (250 MHz, D2O): δ 8.29 (d, 3J =
8.0 Hz, 2H), 8.00 (t, 3J = 8.0 Hz, 2H), 7.49 (d, 3J = 8.0 Hz, 2H) ppm.
[D12]-6,6’-Dimethyl-2,2’-bipyridine-N,N’-dioxide (12)
N NCD3D3C
D
DDDD
D
O O12
12 was prepared in analogy to the literature procedure of Cook et al., [91] using the
conditions of J. Wahsner. [105] A solution of NaOD (40 wt% in D2O, 3 ml, 44.4 mmol,
6.5 equiv) was added to a suspension of 4 (1.5 g, 6.8 mmol) in D2O (30 ml). The res-
ulting reaction mixture was heated in a stainless steel autoclave for 48 h (bath 150 °C)
to give a brown suspension, which was extracted with CH2Cl2 (3 × 180 ml). The
organic phase was dried over MgSO4, the solvent removed in vacuo and the obtained
yellow crude product was reacted again under the same conditions. After extraction
of the second reaction mixture with CH2Cl2 (3 × 180 ml), the white crude product
83

6.3 Simple bipyridine building blocks
was purified by column chromatography (SiO2, CH2Cl2/MeOH 15:1) to afford 12 as
white solid.
Yield: 1.2 g (5.3 mmol, 78%, overall deuteration level: >99%). Rf = 0.21 (SiO2,
CH2Cl2/MeOH 9:1, detection UV). MS (ESI+): m/z (%) 228.97 (100, [M+H]+).
[D10]-6,6’-(Bis(acetoxymethyl)-2,2’-bipyridine (13)
N ND
DDDD
D
AcOOAc
DD
DD
13
13 was prepared according to the literature procedure for the undeuterated compound
of Newkome et al., [99] using the conditions of J. Wahsner. [105] Under nitrogen, 12 (1.2
g, 5.3 mmol) was dissolved in Ac2O (30 ml) and heated to 120 °C for 16 h. Then,
the solvent was removed in vacuo and the yellow crude product purified by column
chromatography (SiO2, CH2Cl2/MeOH 75:1 → 50:1) to obtain 13 as slightly yellow
powder.
Yield: 0.9 g (3.0 mmol, 57%, overall deuteration level: 98%). Rf = 0.46 (SiO2, CH2Cl2/MeOH 24:1, detection UV). MS (ESI+): m/z (%) 310.99 (100, [M+H]+), 332.96 (19,
[M+Na]+).
[D10]-6,6’-(Bis(hydroxymethyl)-2,2’-bipyridine (14)
N ND
DDDD
D
HOOH
DD
DD
14
14 was prepared according to the literature procedure for the undeuterated compound
of Newkome et al., [99] using the conditions of J. Wahsner. [105] Under nitrogen, anhyd-
rous K2CO3 (1.4 g, 138.2 mmol, 3.5 equiv) was added to a slightly beige suspension
of 13 (0.9 g, 3.0 mmol, 1.0 equiv) in dry EtOH (20 ml). The yellow reaction mixture
was stirred at room temperature for 1 h, then filtered through a paper filter and the
solid was washed with CH2Cl2/MeOH (9:1, 100 ml). After removal of the solvent,
the light-yellow crude product was dry-loaded onto silica and purified by column
chromatography (SiO2, CH2Cl2/MeOH 9:1) to afford 14 as colourless solid.
84

6 Experimental Section
Yield: 0.4 g (1.7 mmol, 57%, overall deuteration level: 98%). Rf = 0.46 (SiO2, CH2Cl2/MeOH 9:1, detection UV). MS (ESI+): m/z (%) 226.98 (5, [M+H]+), 248.91 (100,
[M+Na]+).
[D10]-6,6’-Bis(bromomethyl)-2,2’-bipyridine (15)
N ND
DDDD
D
BrBr
DD
DD
15
15 was prepared according to the literature procedure for the undeuterated compound
of Rodriguez-Ubis et al., [89] using the conditions of J. Wahsner. [105] Under nitrogen,
PBr3 (2.3 g, 8.5 mmol, 5.0 equiv) was added carefully via syringe to a yellow solution
of 14 (0.4 g, 1.7 mmol, 1.0 equiv) in DMF (15 ml). The red reaction mixture was stirred
at room temperature for 1 d, while it turned first dark brown and then olive green.
After removal of the solvent in vacuo, H2O (50 ml) was added cautiously with cooling
in an ice bath and the pH was adjusted to 6 with aq. NaHCO3. The aqueous phase
was extracted with CHCl3 (4 × 100 ml), the organic layer dried over MgSO4 and the
solvent removed in vacuo to afford a slightly yellow crude product. Purification by
column chromatography (SiO2, CH2Cl2/MeOH 9:1) yielded the 15 as a white solid.
Yield: 0.3 g (0.9 mmol, 53%, overall deuteration level: 99%). Rf = 0.81 (SiO2, CH2Cl2/MeOH 9:1, detection UV). MS (ESI+): m/z (%) 352.94 (67, [M+H]+), 374.86 (100,
[M+Na]+).
[D10]-6,6’-Bis(aminomethyl)-2,2’-bipyridine trihydrobromide trihydrate (16)
NN
H2N
H2O
3 HBr
NH2
DD
D
D D
D
DD
DD
16
16 was prepared according to the literature procedure for the undeuterated compound
of Wang et al., [104] using the conditions of J. Wahsner. [105] 15 (100 mg, 0.284 mmol,
1.0 equiv) was suspended in CHCl3 (4 ml) and heated at reflux until dissolution.
Then, a solution of hexamethylenetetramine (85.6 mg, 0.611 mmol, 2.25 equiv) in
85

6.3 Simple bipyridine building blocks
CHCl3 (2.5 ml) was added dropwise. After refluxing for 6 h, the reaction mixture was
cooled to room temperature and stored at 8 °C overnight. The precipitate formed was
collected on a Buchner funnel, washed with a small amount of CHCl3 and dried in
vacuo. Subsequently, the white solid was suspended in H2O/EtOH/47% HBr (1.0 ml,
4.1 ml, 0.68 ml) and stirred at 75 °C until dissolution. Upon cooling in an ice bath, 16
crystallised as slightly yellow needles, which were filtered off, washed with a small
amount of cold EtOH and dried in vacuo.
Yield: 100.1 mg (0.206 mmol, 73%, overall deuteration level: 98%). MS (ESI+): m/z
(%) 225.11 (100, [M+H]+).
86

6 Experimental Section
6.4 Bipyridine ester building blocks
Undeuterated compounds
2-(Tributylstannyl)-6-methylpyridine (22)
NSnBu3
22
22 was prepared according to the literature procedure of Mathieu et al. [109] Under
nitrogen and at -78 °C, n-butyllithium (10.3 g, 100 ml, 160 mmol, 1.1 equiv) was ad-
ded dropwise to a slightly yellow solution of 2-bromo-picoline (2) (25.1 g, 146 mmol,
1.0 equiv) in dry THF (350 ml). The obtained red solution was stirred for another
90 min at -78 °C. Then, freshly distilled tributyltinchloride (61.5 g, 52 ml, 175 mmol,
1.2 equiv) was added dropwise and the brown reaction mixture was allowed to warm
to room temperature and stirred overnight. After addition of water (100 ml), the
phases were separated and the aqueous layer extracted with Et2O (3 × 150 ml). The
combined organic phases were dried over MgSO4 and the solvent removed in vacuo.
The reddish brown oil was subjected to vacuum destillation (p ~8·10-2 mbar) to separ-
ate the slightly yellow byproducts (bp: 70 °C and 90 °C) from 22, which was isolated
as colourless oil (bp: 120 °C).
Yield: 33.1 g (87 mmol, 60%). 1H NMR (200 MHz, CDCl3): δ 7.36 (t, 3J = 7.4 Hz, 1H),
7.19 (d, 3J = 7.4 Hz, 1H), 6.96 (dd, 3J = 7.7 Hz, 4J = 0.9 Hz, 1H), 2.56 (s, 3H), 1.72–1.46
(m, 6H), 1.44–1.25 (m, 6H), 1.23–0.97 (m, 6H), 0.90 (t, 3J = 7.2 Hz, 9H) ppm.
6,6’-Dimethyl-2,2’-bipyridine-4-carboxylic acid methyl ester (24)
N
OMe
N
O
24
24 was prepared according to the literature procedure of Mathieu et al. [109] Under ni-
trogen, PdCl2(PPh3)2 (1.2 g, 1.7 mmol, 0.05 equiv), PPh3 (0.9 g, 3.3 mmol, 0.1 equiv),
2-methyl-6-(toluene-4-sulfonyloxy)isonicotinic acid methyl ester (10.0 g, 33.3 mmol,
1.0 equiv), and 22 (12.8 g, 33.4 mmol, 1.0 equiv) were suspended in dry xylene
(250 ml). The yellow reaction mixture was degassed by four freeze-pump-thaws cycles
87

6.4 Bipyridine ester building blocks
and then heated at reflux for 39 h. The obtained dark brown mixture was allowed to
warm to room temperature and poured into saturated EDTA solution (250 ml). The
aqueous layer was extracted with CH2Cl2 (3 × 500 ml), the combined organic extracts
dried over MgSO4 and the solvent removed in vacuo. The resulting deep red oil was
subjected to column chromatography (SiO2, CH2Cl2/MeOH 75:1 → 50:1) to give the
crude product as dark orange solid. Second purification by column chromatography
(SiO2, CH2Cl2/MeOH 100:1) afforded 24 as a slightly yellow solid.
Yield: 4.8 g (19.6 mmol, 59%). Rf = 0.38 (SiO2, CH2Cl2/MeOH 24:1, detection UV) 1H
NMR (200 MHz, CDCl3): δ 7.37 (d, 4J = 0.7 Hz, 1H), 8.20 (d, 3J = 7.8 Hz, 1H), 7.75–7.62
(m, 2H), 7.17 (d, 3J = 7.3 Hz, 1H), 3.97 (d, 4J = 1.3 Hz, 3H), 2.68 (s, 3H), 2.64 (s, 3H)
ppm.
6,6’-Dimethyl-2,2’-bipyridine-4-carboxylic acid methyl ester-N,N’-dioxide (25)
NN
OOMe
O O25
25 was prepared in analogy to the procedure for compound 4. With cooling in an ice
bath, a solution of m-chloroperoxybenzoic acid (1.8 g of 77 wt%, 1.4 pure, 8.1 mmol,
2.0 equiv) in CH2Cl2 (80 ml) was added dropwise to a solution of 24 (1.0 g, 4.1 mmol,
1.0 equiv) in CH2Cl2 (80 ml) over the course of 1.5 h. The pale yellow solution was
allowed to slowly warm to room temperature and stirred for 48 h. After extraction
of the reaction mixture with saturated NaHCO3 (20 ml) and aqueous Na2S2O3 (2 g in
75 ml, 3 × 20 ml), the organic layer was dried over MgSO4 and the solvent removed
in vacuo. The crude product was dry-loaded onto silica and purified by column chro-
matography (SiO2, CH2Cl2/MeOH 15:1) to afford 25 as a colourless solid.
Yield: 0.8 g (2.9 mmol, 71%). Rf = 0.38 (SiO2, CH2Cl2/MeOH 9:1, detection UV).
MS (ESI+): m/z (%) 274.97 (6, [M+H]+), 296.89 (100, [M+Na]+), 312.83 (5, [M+K]+).1H NMR (200 MHz, CDCl3): δ 7.96 (d, 4J = 0.8 Hz, 2H), 7.44–7.27 (m, 3H), 3.91 (s,
3H), 2.59 (s, 3H), 2.58 (s, 3H) ppm. 13C NMR (50 MHz, CDCl3): δ 164.36, 150.2, 150.1,
143.8, 143.3, 127.3, 126.9, 125.9, 125.7, 125.1, 124.9, 52.8, 18.0 ppm. IR (FT-ATR): ν̃ 3073
(w, C-Harom), 2956 (w, CH3), 2917 (w, CH3), 1720 (s, C=O), 1629 (w, C=Carom), 1554 (w,
C=Carom), 1425 (m), 1327 (m), 1258 (s, N-oxide), 1236 (s), 936 (m), 899 (m), 848 (m), 764
(s) cm-1.
88

6 Experimental Section
6,6’-bis(bromomethyl)-2,2’-bipyridine-4-carboxylic acid methyl ester (26)
N N
Br
OOMe
Br26
26 was prepared in analogy to the procedure of Psychogios et al. [100] Under nitro-
gen, TFA anhydride (3 ml) was added quickly via syringe to a yellow solution of 25
(145.4 mg, 0.530 mmol, 1.0 equiv) in dry CH2Cl2 (3 ml). The yellow reaction mixture
was heated at reflux for 1.5 h before the volatiles were removed in vacuo. The obtained
orange residue was dissolved in a mixture of dry DMF/THF (1:1, 6 ml) and, upon ice
cooling, added to solid LiBr (460.3 mg, 5.300 mmol, 10.0 equiv), which was dried
beforehand in vacuo at 180 °C for 3h. After stirring the reaction mixture at room tem-
perature overnight, the solvents were removed in vacuo. The bright orange residue
was dissolved in CH2Cl2 (5 ml), washed with water (3 × 5 ml) and the combined
organic layers were dried over MgSO4 before evaporation to dryness. The isolated
crude product was dry-loaded onto silica and purified by column chromatography
(SiO2, CH2Cl2/MeOH 100:1) to give the product 26 as a yellow solid.
Yield: 112.8 mg (0.282 mmol, 53%). Rf = 0.64 (SiO2, CH2Cl2/MeOH 100:1, detec-
tion UV). MS (ESI+): m/z (%) 258.99 (42, [reactant-O+H]+), 336.88 (46, [M-Br-O+H]+),
400.74 (100, [M+H]+), 422.69 (100, [M+K]+). 1H NMR (200 MHz, CDCl3): δ 8.92 (d, 4J
= 1.4 Hz, 1H), 8.42 (dd, 3J = 7.9 Hz, 4J = 1.0 Hz, 1H), 8.04 (d, 4J = 1.4 Hz, 1H), 7.90
(t, 3J = 7.8 Hz, 1H), 7.56 (dd, 3J = 7.7 Hz, 4J = 0.9 Hz, 1H), 4.73 (s, 2H), 4.69 (s, 2H),
4.02 (s, 4H) ppm. 13C NMR (50 MHz, CDCl3): δ 165.4, 157.6, 156.6, 156.4, 154.4, 139.9,
138.5, 124.4, 123.1, 121.0, 120.2, 53.0, 33.6, 33.4 ppm. IR (FT-ATR): ν̃ 3077 (w, C-Harom),
2950 (w, CH3), 2848 (w, CH3), 1726 (s, C=O), 1584 (w, C=Carom), 1566 (m, C=Carom),
1425 (m), 1405 (m), 1227 (s), 1202 (s), 991 (m), 912 (w), 826 (m), 771 (s), 652 (s) cm-1.
6,6’-bis(bromomethyl)-2,2’-bipyridine-4-carboxylic acid methyl ester-N,N-dioxide (27)
N N
Br
OOMe
BrO O
27
89

6.4 Bipyridine ester building blocks
27 was prepared in analogy to the procedure of Sieser et al. [143] Under nitrogen and
with cooling in an ice bath, urea/H2O2 adduct (2.1 g, 22.5 mmol, 3 equiv) was added
in portions to a suspension of 26 (3.0 g, 7.5 mmol, 1.0 equiv). Then, TFA anhydride
was added dropwise over the course of 15 min, the mixture was allowed to warm to
room temperature and stirred for 2 d. After dilution with CH2Cl2 (150 ml), a solution
of saturated Na2S2O3 was added and the mixture stirred for 1 h. The phases are
separated, the aqueous layer washed with CH2Cl2 (3 × 200 ml), the combined phases
dried over MgSO4 and evaporated to dryness. The isolated crude product was then
dry-loaded onto silica and purified by column chromatography (SiO2, CH2Cl2/MeOH
50:1) to give 27 as a colourless solid.
Yield: 1.0 g (2.3 mmol, 31%). Rf = (SiO2, CH2Cl2/MeOH 50:1, detection UV+I2 va-
pour). 1H NMR (200 MHz, CDCl3): δ 8.23 (d, 4J = 2.5 Hz, 1H), 8.14 (d, 4J = 2.4 Hz,
1H), 7.67 (dd, 3J = 7.5 Hz, 4J = 1.9 Hz, 1H), 7.59–7.50 (m, 1H), 7.34 (t, J = 7.9 Hz, 1H),
4.73 (s, 2H), 4.70 (s, 2H), 3.96 (s, 3H) ppm.
Deuterated compounds
[D11]-6,6’-Dimethyl-2,2’-bipyridine-4-carboxylic acid methyl ester-N,N’-dioxide (33)
N NCD3D3C
D
DDD
D
OOMe
O O33
Deuteration was performed in analogy to the procedure for compound 12 and esterifi-
cation was performed in analogy to the literature procedure for the undeuterated
dicarboxylic acid. [144] A solution of NaOD (40 wt% in D2O, 1.5 ml, 22.2 mmol, 7.5
equiv) was added to a suspension of 25 (0.8 g, 2.9 mmol, 1.0 equiv) in D2O (15 ml).
The resulting reaction mixture was heated in a stainless steel autoclave for 68 h (bath
150 °C) to give a brown suspension, which was neutralised with DCl (7.6 M in D2O)
and rotavapped to dryness to leave a beige solid behind. Reaction (48 h) and workup
were repeated to yield the carboxylic acid derivative as a beige solid. The latter was
suspended in MeOH (15 ml) and conc. H2SO4 (2 ml) and heated at reflux for 24 h.
After cooling to room temperature, the brownish yellow residue was dissolved in wa-
ter (20 ml) and neutralised cautiously with 10% NaOH. After extraction with CH2Cl2(3 × 100 ml), the organic layer was dried over MgSO4 and the crude product dry-
loaded onto silica. Purification by column chromatography afforded 33 as colourless
90

6 Experimental Section
solid.
Yield: 0.5 g (1.8 mmol, 62%, overall deuteration level: 98%). MS (ESI+): m/z (%) 286.25
(36, [M+H]+), 308.11 (100, [M+Na]+).
[D9]-6,6’-(Bis(acetoxymethyl)-2,2’-bipyridine-4-carboxylic acid methyl ester (34)
N ND
DDD
D
AcOOAc
DD
OOMe
DD
34
34 was prepared in analogy to the procedure for compound 13. Under nitrogen, 33
(0.5 g, 1.8 mmol) was dissolved in Ac2O (25 ml) and heated at reflux for 16 h. Then,
the solvent was removed in vacuo and the yellow crude product was purified twice
by column chromatography (SiO2, CH2Cl2/MeOH 50:1 and CH2Cl2/MeOH 100:1) to
obtain 34 as slightly yellow solid.
Yield: 0.4 g (1.2 mmol, 67%, overall deuteration level: 98%). Rf = 0.79 (SiO2, CH2Cl2/MeOH 9:1, detection UV). MS (ESI+): m/z (%) 390.04 (100, [M+Na]+).
[D9]-6,6’-bis(bromomethyl)-2,2’-bipyridine-4-carboxylic acid methyl ester (32)
N ND
DDD
D
BrBr
DD
DD
OOMe
32
32 was prepared analogous to compound 5. Under nitrogen, TFA anhydride (10 ml)
was added quickly via syringe to a yellow solution of 33 (1.0 g, 3.5 mmol, 1.0 equiv)
in dry CH2Cl2 (10 ml). The orange reaction mixture was heated at reflux for 1.5 h
before the volatiles were removed in vacuo. The obtained residue was dissolved in
a mixture of dry DMF/THF (1:1, 10 ml) and then added upon ice cooling to solid
LiBr (3.0 g, 35.0 mmol, 10.0 equiv), which was dried beforehand in vacuo at 180 °C for
1 h. After stirring the reaction mixture at room temperature overnight, the solvents
were removed in vacuo. The bright orange residue was dissolved in CH2Cl2 (20 ml),
washed with water (5 × 20 ml) and the combined organic layers were dried over
91

6.4 Bipyridine ester building blocks
MgSO4 before evaporation to dryness. The isolated crude product was dry-loaded
onto silica and purified by column chromatography (SiO2, CH2Cl2/MeOH 100:1) to
give 32 as a yellow solid.
Yield: 644 mg (1.57 mmol, 45%, overall deuteration level: >99%). MS (ESI+) m/z (%)
409.78 [M+H]+, 431.74 [M+Na]+. 1H NMR (400 MHz, CDCl3): δ 4.00 (s, 3H) ppm.13C NMR (101 MHz, CDCl3): δ 165.4, 157.3, 156.8, 156.5, 154.6, 139.6, 137.6 (t, J = 24.7
Hz), 123.7 (t, J = 24.2 Hz), 122.5 (t, J = 25.8 Hz), 120.3 (t, J = 25.8 Hz), 119.7 (t, J = 25.6
Hz), 52.9, 34.0–32.6 (m) ppm.
92

6 Experimental Section
6.5 Nitro and amino bipyridine building blocks
6,6’-Dimethyl-2,2’-bipyridine-N-oxide (37)
NNO
37
37 was prepared according to the literature procedure of Newkome et al. [99] With
cooling in an ice bath, a solution of m-chloroperoxybenzoic acid (1.3 g of 77 wt%, 1.0
g pure, 5.6 mmol, 1.0 equiv) in CHCl3 (200 ml) was added dropwise to a solution of 3
(1.0 g, 5.4 mmol, 1.0 equiv) in CHCl3 (200 ml) over the course of 3 h. The reaction mix-
ture was allowed to slowly warm to room temperature and stirred overnight. Then,
the reaction mixture was washed with saturated NaHCO3 (20 ml), dried over MgSO4
and evaporated to dryness. The yellow crude product was subjected to column chro-
matography (SiO2, CH2Cl2/MeOH 100:1 → 9:1) to separate 37 as a white solid from
the reactant (Rf = 0.50) and the N,N’-dioxide byproduct (Rf = 0.24).
Yield: 0.88 g (4.39 mmol, 81%). Rf = 0.42 (SiO2, CH2Cl2/MeOH 9:1, detection UV).1H NMR (200 MHz, CDCl3): δ 8.57 (d, 3J = 7.9 Hz, 1H), 8.02–7.94 (m, 1H), 7.69 (t, 3J =
7.8 Hz, 1H), 7.31–7.27 (m, 1H), 7.25–7.15 m (m, 2H), 2.62 (s, 3H), 2.56 (s, 3H) ppm.
4-Nitro-6,6’-dimethyl-2,2’-bipyridine-N-oxide (38)
NN
NO2
O O38
38 was prepared according to the literature procedure of Mukkala et al. [113] A solution
of 37 (849 mg, 4.24 mmol) in H2SO4/HNO3 (4:3, 14 ml) was heated to 100 °C for 4 h.
After cooling to room temperature, the reaction mixture was poured into crushed ice
(~ 40 g) and the pH was adjusted to 6 with aqueous NaOH. The resulting precipitate
was collected on a Buchner funnel, washed with cold water and dried in vacuo to give
38 as white solid.
Yield: 629 mg (2.57 mmol, 61%). MS (ESI+): m/z (%) 246.14 (10, [M+H]+), 267.96 (100,
[M+Na]+). 1H NMR (200 MHz, CDCl3): δ 8.96 (d, 3J = 3.3 Hz, 1H), 8.59 (d, 3J = 8.0
Hz, 1H), 8.10 (d, 3J = 3.3 Hz, 1H), 7.74 (t, 3J = 7.8 Hz, 1H), 7.32–7.27 (m, 1H), 2.65 (s,
3H), 2.63 (s, 3H) ppm. 13C NMR (50 MHz, CHCl3): δ 158.9, 152.2, 151.8, 148.6, 147.8,
93

6.5 Nitro and amino bipyridine building blocks
136.8, 124.9, 122.3, 120.2, 118.8, 24.7, 18.9 ppm. IR (FT-ATR): ν̃ 3043 (w, C-Harom), 1606
(w, C=Carom), 1524 (s, N=O), 1497 (w, C=Carom), 1337 (s, N=O), 1282 (s, N-oxide), 1101
(m), 848 (m), 781 (s), 738 (s) cm-1.
4-Nitro-6,6’-dimethyl-2,2’-bipyridine-N,N-dioxide (41)
NN
NO2
O O41
41 was prepared according to the procedure developed by N. Alzakhem. [112] With
cooling in an ice bath, a solution of m-chloroperoxybenzoic acid (4.4 g of 77 wt%,
3.4 g pure, 19.6 mmol, 1.5 equiv) in CHCl3 (100 ml) was added dropwise to a solution
of 38 (3.2 g, 13.1 mmol, 1.0 equiv) in CHCl3 (100 ml) over the course of 1 h. The
reaction mixture was allowed to warm to room temperature and stirred overnight.
Then, the reaction mixture was washed with saturated NaHCO3 (10 ml) and saturated
Na2S2O3 (10 ml). The combined aqueous layers were extracted with CHCl3 (3 ×20 ml). The combined organic phases were dried over MgSO4 and the solvent was
removed in vacuo. The crude product was purified twice by column chromatography
(SiO2, CH2Cl2/MeOH 24:1 → 9:1 and CH2Cl2/MeOH 24:1 → 9:1) to give pure 41 as
a yellow solid.
Yield: 2.8 g (10.8 mmol, 82%). Rf = 0.36 (SiO2, CH2Cl2/MeOH 9:1, detection UV).
MS (ESI+): m/z (%) 261.96 (2, [M+H]+), 283.87 (100, [M+Na]+), 299.80 (2, [M+K]+).1H NMR (200 MHz, CDCl3): δ 8.27–8.17 (m, 2H), 7.49–7.27 (m, 3H), 2.62 (s, 3H), 2.59
(s, 3H) ppm. 13C NMR (50 MHz, CDCl3): δ 151.3, 150.3, 144.6, 142.1, 140.6, 127.8,
125.6, 124.9, 120.6, 120.2, 18.4, 18.0 ppm. IR (FT-ATR): ν̃ 3043 (w, C-Harom), 1606 (w,
C=Carom), 1524 (s, N=O), 1497 (w, C=Carom), 1337 (s, N=O), 1280 (s, N-oxide), 1101 (m),
849 (m), 781 (s), 746 (s) cm-1.
4-Nitro-6,6’-bis(bromomethyl)-2,2’-bipyridine (42)
NN
BrBr
NO2
42
94

6 Experimental Section
42 was prepared in analogy to the procedure for compound 5. Under nitrogen, TFA
anhydride (17 ml) was added quickly via syringe to a yellow solution of 41 (1.0 g,
3.9 mmol, 1.0 equiv) in dry CH2Cl2 (17 ml). The orange reaction mixture was heated
at reflux for 2 h before the solvents were removed in vacuo. The orange residue was
dissolved in a mixture of dry DMF/THF (1:1, 26 ml) and the obtained solution was ad-
ded to solid LiBr (3.3 g, 38.2 mmol, 10.0 equiv), which was dried beforehand in vacuo
at 180 °C for 4 h. The reaction mixture was stirred at room temperature overnight be-
fore the solvents were removed in vacuo at 60 °C. The yellow residue was suspended
in CH2Cl2 (70 ml) and washed with water (3 × 70 ml). The combined organic layers
were dried over MgSO4 and evaporated to dryness. The crude product was dry-
loaded onto silica and purified by column chromatography (SiO2, CH2Cl2/MeOH
100:1) to give 42 as a bright yellow solid.
Yield: 840 mg (2.17 mmol, 56%). Rf = 0.48 (SiO2, CH2Cl2/MeOH 9:1, detection UV).
MS (ESI+): m/z (%) 360.17 (100), 374.11 (15), 392.09 (6), 408.05 (5, [M+Na]+). 1H NMR
(200 MHz, CDCl3): δ 9.10 (d, 4J = 2.0 Hz, 1H), 8.43 (dd, 3J = 7.9 Hz, 4J = 1.0 Hz, 1H),
8.17 (d, 4J = 2.0 Hz, 1H), 7.89 (t, 3J = 7.8 Hz, 1H), 7.57 (dd, 3J = 7.8 Hz, 4J = 1.0 Hz,
1H), 4.71 (s, 2H), 4.66 (s, 2H) ppm. 13C NMR (50 MHz, CDCl3): δ 159.3, 158.7, 157.0,
156.0, 153.2, 138.4, 125.0, 120.9, 116.0, 113.4, 33.6, 32.6 ppm. IR (FT-ATR): ν̃ 3085 (w,
C-Harom), 1607 (w, C=Carom), 1571 (m, C=Carom), 1530 (s, N=O), 1407 (m), 1353 (s, N=O),
1263 (m), 1217 (m), 1077 (m), 819 (m), 741 (s), 639 (s, C-Br) cm-1.
4-Amino-6,6’-dimethyl-2,2’-bipyridine (39)
NN
NH2
39
39 was prepared in analogy to the literature procedure for 6-amino-2,2’-bipyridine. [114]
Under nitrogen and with cooling in an ice bath, Pd/C (120 mg) was added to a solu-
tion of 38 (517 mg, 2.11 mmol, 1.0 equiv) in dry MeOH (12 ml). Sodium borohydride
(250 mg, 6.09 mmol, 3.0 equiv) was added as a solid in small portions, which resul-
ted in gas evolution and complete dissolution of the starting material. After stirring
the reaction mixture at room temperature overnight, the catalyst was removed by fil-
tration and the solvent evaporated to dryness. The remaining yellow residue was
dissolved in water (30 ml) and extracted with EtOAc (5 × 50 ml). The combined or-
ganic layers were dried over MgSO4 and the solvent was removed in vacuo to obtain
95

6.5 Nitro and amino bipyridine building blocks
39 as a sticky yellow solid.
Yield: 286 mg (1.44 mmol, 68%). 1H NMR (250 MHz, CDCl3): δ 8.10 (d, 3J = 7.8 Hz,
1H), 7.62 (t, 3J = 7.7 Hz, 1H), 7.45 (d, 3J = 2.1 Hz, 1H), 7.10 (d, 3J = 7.6 Hz, 1H), 6.36
(d, 3J = 2.1 Hz, 1H), 4.22 (s, 2H), 2.59 (s, 3H), 2.45 (s, 3H) ppm.
4-Acetylamino-6,6’-dimethyl-2,2’-bipyridine (40)
NN
NHAc
40
40 was prepared analogy to the literature procedure for 6-(acetyl)amino-2,2’-bipyridine
of Hapke et al. [117] Under nitrogen, Me2bpyNH2 (39) (43.6 mg, 0.219 mmol, 1.0 equiv)
was dissolved in dry pyridine (5 ml), which was freshly distilled over KOH before-
hand. Acetyl chloride (41.2 mg, 36 µl, 0.526 mmol, 2.4 equiv) was added dropwise
and the yellow solution was stirred at room temperature overnight. The reaction was
quenched by cautious addition of water (5 ml) and the mixture was extracted with
CH2Cl2 (2 × 20 ml). The combined organic phases were dried over MgSO4 and the
solvent was removed in vacuo. The crude product was dry-loaded onto silica and
purified by column chromatography (SiO2, CH2Cl2/MeOH 30:1) to give 40 as yellow
sticky solid.
Yield: 19.4 mg (0.080 mmol, 37%). Rf = 0.46 (SiO2, CH2Cl2/MeOH 9:1, detection UV).1H NMR (400 MHz, CDCl3): δ 8.15 (d, 3J = 7.8 Hz, 1H), 7.99 (d, 4J = 1.9 Hz, 1H), 7.96
(br s, 1H), 7.80 (s, 1H), 7.67 (t, 3J = 7.7 Hz, 1H), 7.14 (d, 3J = 7.6 Hz, 1H), 2.58 (s, 3H),
2.57 (s, 3H), 2.11 (s, 3H) ppm. 13C NMR (50 MHz, CDCl3): δ 169.2, 159.8, 157.9, 156.9,
155.7, 154.1, 146.2, 137.3, 123.5, 118.5, 113.0, 108.3, 25.0, 24.8, 24.7 ppm.
96

6 Experimental Section
6.6 Simple macrocycles
Undeuterated compounds
(bpy.bpy) (6)
NNH
N
NHN
N
6
6 was prepared according to the literature procedure of Bottino et al. [101] Under ni-
trogen, a solution of 5 (11.5 g, 33.6 mmol, 1.0 equiv) in EtOH (1.5 l) was added to a
suspension of tosylamide monosodium salt [101] (13.0 g, 67.2 mmol, 2.0 equiv). While
the reaction mixture was heated at reflux for 36 h (bath 98 °C), the tosylated aza mac-
rocycle precipitated. After cooling the mixture to 0 °C for 1 h, the white solid was
collected on a Buchner funnel and dried in vacuo (crude product: 5.8 g). The latter
was dissolved in H2SO4 (80 ml) and heated to 110 °C for 3 h. After cooling to room
temperature and additional cooling with an ice bath, water (10 ml) was added cau-
tiously. The clear brown solution was treated with 50% NaOH solution until the pH
was above 13 and a beige clowdy suspension has formed, which was extracted with
DCM (3 × 300 ml). The combined organic layers were dried over MgSO4 and the
solvent removed in vacuo to yield 6 as a white solid.
Yield: 3.8 g (9.5 mmol, 57%). 1H NMR (200 MHz, CDCl3): δ 7.70 (d, 3J = 7.7 Hz, 4H),
7.39 (t, 3J = 7.7 Hz, 4H), 6.91 (d, 3J = 7.5 Hz, 4H), 4.07 (s, 8H) ppm. 13C NMR (50 MHz,
CDCl3): δ 159.2, 156.1, 136.5, 122.4, 119.3, 56.3 ppm. MS (ESI+): m/z (%) 395.15 (53,
[M+H]+), 417.07 (100, [M+Na]+).
97

6.6 Simple macrocycles
Deuterated compounds
[D20]-(bpy.bpy) (31)
N
NHN
N
N
NH
DD
DD
DD
D D
D D
D D
D D
DD
DD
DD
31
31 was prepared in analogy to the literature procedure for the undeuterated com-
pound of Bottino et al., [101] using the conditions of J. Wahsner. [119] Under nitrogen, a
solution of 15 (90 mg, 0.26 mmol, 1.0 equiv) in EtOH (5 ml) was added to a suspension
of tosylamide monosodium salt [101] (99 mg, 0.51 mmol, 2.0 equiv). While the reaction
mixture was heated at reflux for 24 h (bath 98 °C), the tosylated aza macrocycle pre-
cipitated. The white solid was collected on a Buchner funnel and dried in vacuo (crude
product: 51 mg). The latter was dissolved in D2SO4 (98 wt% in D2O, 99.5%D, 5 ml)
and heated to 110 °C for 2 h. After cooling to room temperature and additional cool-
ing with an ice bath, water (1 ml) was added cautiously and the mixture neutralised
with an aqueous solution of NaOH (excess). Then, the solution was extracted with
CH2Cl2 (3 × 30 ml), the organic layer dried over MgSO4 and the solvent removed in
vacuo to yield 31 as a white solid.
Yield: 43 mg (0.10 mmol, 77%, overall deuteration level: 98%). MS (ESI+): m/z (%)
415.14 (100, [M+H]+), 437.10 (71, [M+Na]+).
98

6 Experimental Section
6.7 Nitro and amino functionalised macrocycles
(bpy.bpy)NO2 (49)
NNH
N
NHN
N
NO2
49
49 was prepared according to the procedure of N. Alzakhem. [112] Under nitrogen,
42 (118 mg, 0.31 mmol, 1.0 equiv) was dissolved in DMF (8 ml). Upon addition of
K2CO3 (635 mg, 4.59 mmol, 15.0 equiv) the yellow solution turned dark orange. After
addition of 47 (160 mg, 0.31 mmol, 1.0 equiv) as a solid, the cloudy pink mixture
was stirred at 50 °C overnight and for another 2 d at room temperature, whereas a
beige solid precipitated from a brown solution. Then, water (20 ml) was added, the
precipitate was collected on a Buchner funnel and shortly dried on the filter. Sub-
sequently, the crude product was treated with H2SO4 (4 ml) and the brown solution
heated at 110 °C. After 3 h, the reaction mixture was poured onto crushed ice (10 g)
and, upon cooling, treated with 50% NaOH until the pH was above 13. The obtained
beige clowdy suspension was extracted with CHCl3 (3 × 100 ml). The combined or-
ganic layers were dried over MgSO4 and the solvent removed in vacuo to yield 49 as
an off-white solid.
Yield: 70 mg (0.16 mmol, 52%). 1H NMR (200 MHz, CDCl3): δ 7.79–7.65 (m, 3H),
7.50–7.28 (m, 4H), 7.26–7.19 (m, 1H), 6.96–6.86 (m, 2H), 6.76 (d, 3J = 7.8 Hz, 1H), 4.18
(s, 2H), 4.10 (s, 4H), 4.06 (s, 2H) ppm. MS (ESI+): m/z (%) 395.17 (7, [M-NO2+H]+),
417.14 (22, [M-NO2+Na]+, 440.11 (68, [M+H]+), 462.04 (100, [M+Na]+).
99

6.7 Nitro and amino functionalised macrocycles
(bpy.bpy)NH2 (50)
NNH
N
NHN
N
NH2
50
Under nitrogen, Pd (10%)/C (20 mg) was added to a solution of 49 (49.7 mg, 0.113 mmol,
1.0 equiv) in dry EtOH (50 ml). Then, hydrazine hydrate (55 µl, 57 mg, 1.14 mmol,
10.0 equiv) in EtOH (10 ml) was added and the reaction mixture heated at reflux
overnight. After cooling to room temperature, the dark suspension was filtered
through a paper filter and the solvent removed in vacuo. The isolated crude product
was reacted again using the same conditions, amounts of reagents and solvent. After
cooling to room temperature, the reaction mixture was filtered again through a paper
filter and after solvent removal, 50 was isolated a yellowish brown solid.
Yield: 39.5 mg (0.096 mmol, 85%). 1H NMR (250 MHz, CD3OD): δ 7.92–7.67 (m, 6H),
7.35–7.18 (m, 3H), 7.09–7.03 (m, 1H), 6.52–6.47 (m, 1H), 4.10–3.99 (m, 6H), 3.91–3.84
(m, 2H) ppm. MS (ESI+): m/z (%) 410.04 (100, [M+H]+), 431.96 (40, [M+Na]+).
100

6 Experimental Section
6.8 Simple deuterated cryptates
General procedure for the synthesis of the sodium cryptates: Under nitrogen, the
building blocks and Na2CO3 (10-14 equiv) were suspended in HPLC grade CH3CN.
The mixture was heated at reflux for 23-45 h, filtered cold, and the solvent removed
in vacuo. The residue was subjected to column chromatography to yield the desired
cryptate as a colorless solid.
[D4]-[Na(bpy.bpy.bpy)]Br (17)
N
N
N
N
N
N
N
N
DD
DD
Na Br
17
6 (45.0 mg, 114 µmol, 1.0 equiv), 10 (41.0 mg, 114 µmol, 1.0 equiv), Na2CO3 (121 mg,
1.14 mmol, 10 equiv), CH3CN (80 ml). Column chromatography (SiO2, CH2Cl2/
MeOH 15:1→ 9:1).
Yield: 31 mg (45 µmol 40%, overall deuteration level: 99%). Rf = 0.19 (SiO2, CH2Cl2/
MeOH 9:1, detection UV+I2 vapour). MS (ESI+): m/z (%) 601.1 (10, [M+H]+). 1H
NMR (250 MHz, CDCl3): δ 7.99–7.70 (m, 12H), 7.44–7.21 (m, 6H), 3.84 (s, 8H) ppm.13C NMR (62.9 MHz, CDCl3): δ 158.6, 155.3, 138.2, 124.1, 120.4, 58.8 (t, J = 20.5 Hz)
ppm.
[D8]-[Na(bpy.bpy.bpy)]Br (19)
N
N
N
N
N
N
N
N
D D
DD
DD
D D
Na Br
19
18 (52.0 mg, 109 µmol, 1.0 equiv), 10 (75.8 mg, 218 µmol, 2.0 equiv), CH3CN (250 ml),
Na2CO3 (162 mg, 1.53 mmol, 14 equiv). Column chromatography (SiO2, CH2Cl2/
101

6.8 Simple deuterated cryptates
MeOH 9:1).
Yield: 38 mg (55 µmol, 51%, overall deuteration level: > 99%). MS (ESI+): m/z (%) =
607.1 (100, [M+H]+). 1H NMR (250 MHz, CDCl3): δ 8.04–7.68 (m, 12H), 7.52–7.15 (m,
6H), 3.84 (s, 4H) ppm. 13C NMR (62.9 MHz, CDCl3): δ 158.5, 155.3, 138.2, 124.1, 120.4,
58.8 (t, J = 20.5 Hz)ppm.
[D12]-[Na(bpy.bpy.bpy)]Br (20)
N
N
N
N
N
N
N
N
D D
DDDD
DDDD
D D
Na Br
20
11 (62.0 mg, 129 µmol, 1.0 equiv), 10 (89.6 mg, 259 µmol, 2.0 equiv), CH3CN (120 ml),
Na2CO3 (191 mg, 1.81 mmol, 14 equiv). Column chromatography (SiO2, CH2Cl2/
MeOH 15:1→ 9:1).
Yield: 52 mg (75 µmol, 58%, overall deuteration level: > 99%). MS (ESI+): m/z (%) =
609.2 (100, [M]+). 1H NMR (250 MHz, CDCl3): δ 7.95–7.72 (m, 12 H), 7.39–7.25 (m, 6
H) ppm. 13C NMR (62.9 MHz, CDCl3): δ 158.5, 155.3, 138.2, 124.1, 120.4, 58.8 (t, J =
20.5 Hz) ppm.
[D30]-[Na(bpy.bpy.bpy)]Br (21)
N
N
N
N
N
N
N
N
DD
DD
DD D D
DDDD D
D
DD
DDDD
DD
DD
D
D
DD
D D
Na Br
21
21 was prepared according to the procedure of J. Wahsner. [105] 15 (108 mg, 0.29 mol,
2.0 equiv), 16 (70 mg, 0.14 mmol, 1.0 equiv), Na2CO3 (215 mg, 2.03 mmol, 14 equiv).
Column chromatography (SiO2, CH2Cl2/MeOH 24:1→ 15:1).
102

6 Experimental Section
Yield: 54 mg (0.08 mmol, 57%, overall deuteration level: 98%). Rf = 0.79 (SiO2,
CH2Cl2/MeOH 15:1, detection UV+I2 vapour). MS (ESI+): m/z (%) 627.25 (100, [M-
Br]+).
103

6.9 Carboxylic acid ester functionalised crypates
6.9 Carboxylic acid ester functionalised crypates
Undeuterated compounds
[Na(bpy.bpy.bpyCOOMe)]Br (28)
N
N
N
N
N
N
N
NNa Br
COOMe
28
26 (66.0 mg, 165.0 µmol, 1.0 equiv), 6 (64.3 mg, 163.0 µmol, 1.0 equiv), NaCO3
(173.1 mg, 1.633 mmol, 10.0 equiv), CH3CN (200 ml). Column chromatography (SiO2,
CH2Cl2/ MeOH 15:1→ 9:1).
Yield: 42.7 mg (58.0 µmol, 36%). 1H NMR (200 MHz, CDCl3): δ 8.39 (s, 1H), 8.02–7.73
(m, 11H), 7.51–7.30 (m, 5H), 4.00 (s, 3H), 3.95 (s, 2H), 3.89 (s, 7H) ppm.
[Na(bpy.bpy.bpyCOOMeO2
)]Br (29)
N
N
N
N
N
N
N
NNa Br
COOMe
OO
29
27 (1.0 g, 2.3 mmol, 1.0 equiv), 6 (0.9 g, 2.3 mmol, 1.0 equiv), NaCO3 (2.5 g, 23.1 mmol,
10.0 equiv), CH3CN (3 l). The reactants and solvent were divided into four identical
batches. Column chromatography (SiO2, CH2Cl2/MeOH 15:1→ 9:1).
Yield: 688 mg (0.90 mmol, 39%). Rf = 0.45 (SiO2, CH2Cl2/MeOH 9:1, detection UV+I2
vapour). MS (ESI+): m/z (%) 687.17 (5, [M-Br]+). 1H NMR (250 MHz, CD2Cl2/CD3OD):
δ 8.32 (d, 4J = 2.5 Hz, 1H), 8.25 (d, 4J = 2.5 Hz, 1H), 7.95–7.87 (m, 8H), 7.85–7.76 (m,
2H), 7.66–7.56 (m, 1H), 7.51–7.41 (m, 4H), 4.35 (dd, 2J = 11.8 Hz, 4J = 4.8 Hz, 2H),
3.98 (s, 3H), 3.95–3.84 (m, 3H), 3.65–3.54 (m, 3H), 3.52–3.48 (m, 2H), 3.47–3.43 (m, 2H),
3.40 (s, 1H) ppm. 13C NMR (50.3 MHz, CDCl3): δ 163.6, 158.8, 158.4 157.8, 157.6,
157.1, 157.0, 156.4, 156.2, 148.6, 148.5, 145.5, 144.0, 138.32, 138.30, 130.2, 128.6, 126.9,
126.8, 125.4, 125.3, 124.74, 124.68, 122.2, 122.1, 121.3, 121.2, 60.9, 60.70, 60.66, 60.62,
104

6 Experimental Section
54.6, 54.3, 53.1 ppm. 3 signals are missing, probably since signals are overlapping due to
the large number of non-equivalent carbons in this molecule. Anal. Calcd. (Found) for
C38H32BrN8NaO4 · 2 H2O (Mr = 803.64): C, 56.79 (56.61); H, 4.52 (4.28); N, 13.94
(13.73).
Deuterated compounds
[D29]-[Na(bpy.bpy.bpyCOOMe)]Br (35)
N
N
N
N
N
N
N
N
DD
DD
DD D D
DDDD D
COOMe
DD
DDDD
DD
DD
D
D
DD
D D
Na Br
35
32 (25.3 mg, 62 µmol, 1.0 equiv), 31 (25.7 mg, 62 µmol, 1.0 equiv), Na2CO3 (66.1 mg,
624 µmol, 10.0 equiv), CH3CN (50 ml). Column chromatography (SiO2, CH2Cl2/
MeOH 24:1→ 15:1).
Yield: 11.9 mg (16 µmol, 26%, overall deuteration level: 98%). Rf = 0.25 (SiO2,
CH2Cl2/MeOH 15:1, detection UV+I2 vapour). MS (ESI+): m/z (%) 684.19 (100, [M-
Br]+). 13C NMR (101.6 MHz, CD2Cl2/CD3OD): δ 166.2, 160.9, 159.7, 159.4, 159.3,
157.4, 157.4, 156.2, 156.2, 155.4, 140.3, 138.9, 138.7, 138.5, 138.2, 124.6, 124.3, 124.1,
121.0, 120.8, 120.5, 59.8, 59.6, 59.6, 59.5, 59.4, 59.2, 52.4 ppm.
105

6.10 Amino functionalised cryptates
6.10 Amino functionalised cryptates
[Na(bpy.bpy.bpyNO2)]Br (43)
N
N
N
N
N
N
N
NNa Br
NO2
43
6 (200 mg, 0.51 mmol, 1.0 equiv), 42 (196 mg, 0.51 mmol, 1.0 equiv), Na2CO3 (536 mg,
5.10 mmol, 10.0 equiv), CH3CN (300 ml). Column chromatography (SiO2, CH2Cl2/
MeOH/NH3 9:1:0.05).
Yield: 129 mg (0.17 mmol, 33%). Rf = 0.47 (SiO2, CH2Cl2/MeOH/NH3 9:1:0.05, de-
tection UV+I2 vapour). MS (ESI+): m/z (%) 642.23 (100, [M-Br]+). 1H NMR (200 MHz,
CD2Cl2/CD3OD): 8.61 (d, 4J = 1.9 Hz, 1H), 8.14–7.78 (m, 11H), 7.41 (dd, 3J = 7.6 Hz, 4J
= 1.0 Hz, 1H), 7.37–7.28 (m, 4H), 4.37 (s, 2H), 4.00 (s, 2H), 3.92–3.79 (m, 8H) ppm. 13H
NMR (50.3 MHz, CD2Cl2/CD3OD): 163.0, 160.0, 159.3, 159.1, 159.0, 156.3, 156.0, 156.0,
154.3, 139.3, 139.0, 138.9, 125.7, 124.7, 124.6, 121.7, 121.1, 121.0, 116.7, 113.6, 60.1, 60.0,
59.9 ppm. Signals are missing, probably since signals are overlapping due to the large number
of non-equivalent carbons in this molecule. Anal. Calcd. (Found) for C36H29BrN9NaO2 ·1 H2O (Mr = 740.58): C, 58.38 (58.50); H, 4.22 (4.28); N, 17.02 (16.80).
[Na(bpy.bpy.bpyNH2)]Br (44)
N
N
N
N
N
N
N
NNa Br
NH2
44
Under nitrogen and with cooling in an ice bath, Pd (10%)/C (20 mg) was added to
a solution of 43 (51.1 mg, 71 µmol, 1.0 equiv) in dry EtOH (50 ml). Then, hydrazine
hydrate (172 µl, 177.4 mg, 3.543 mmol, 50 equiv) was added and the reaction mixture
heated at reflux overnight. After cooling to room temperature, the dark suspension
was filtered through a paper filter and the solvent removed in vacuo. The isolated
crude product was reacted again using the same conditions, amounts of reagents and
106

6 Experimental Section
solvent. After cooling to room temperature, the reaction mixture was filtered again
through a paper filter and, subsequently, through a short pad of celite. After solvent
removal, 44 was isolated as a yellow solid.
Yield: 31.8 mg (43 µmol, 61%). MS (ESI+): m/z (%) 612.09 (100, [M-Br]+). 1H NMR
(400 MHz, CD2Cl2/CD3OD): δ 8.01 (d, 3J = 7.9 Hz, 4H), 7.92–7.79 (m, 6H), 7.35 (d, 3J =
7.7 Hz, 4H), 7.32 (d, 3J = 7.4 Hz, 1H), 7.23 (d, 4J = 2.2 Hz, 1H), 6.58 (d, 4J = 2.0 Hz, 1H),
3.90–3.74 (m, 10H), 3.67–3.58 (m, 2H) ppm. 13C NMR (101.6 MHz, CD2Cl2/CD3OD):
δ 160.2, 160.1, 160.1, 160.0, 159.5, 159.4, 156.7, 156.7, 156.6, 139.2, 139.2, 124.9, 121.4,
121.4, 120.9, 120.8, 109.9, 109.9, 106.8, 106.8, 60.8, 60.7, 60.6, 60.6, 60.6, 60.5 ppm. Anal.
Calcd. (Found) for C36H31BrN9Na · 2.5 H2O (Mr = 737.63): C, 58.62 (58.75); H, 4.92
(4.80); N, 17.09 (16.90).
[Na(bpy.bpyNH2 .bpyO2)]Br (46)
N
N
N
N
N
N
N
NNa Br
H2N
OO
46
6 (105.9 mg, 0.259 mmol, 1.0 equiv), 48 (151.4 mg, 0.405 mmol, 1.5 equiv), Na2CO3
(412.1 mg, 3.888 mmol, 15.0 equiv), CH3CN (400 ml). Column chromatography (SiO2,
CH2Cl2/MeOH/NH3 9:1:0.05).
Yield: 17.6 mg (22 µmol, 8%). Rf = 0.12 (SiO2, CH2Cl2/MeOH/NH3 9:1:0.05, detec-
tion UV+I2 vapour). MS (ESI+): m/z (%) 644.08 (100, [M-Br]+). 1H NMR (250 MHz,
CD2Cl2/CD3OD): δ 7.90–7.57 (m, 10H), 7.54–7.47 (m, 2H), 7.47–7.30 (m, 3H), 7.04–6.97
(m, 1H), 6.62 (s, 1H), 4.45 (d, 2J = 11.8 Hz, 1H), 4.33–4.26 (m, 2H), 4.08–3.59 (m, 4H),
3.54–3.33 (m, 3H), 3.25–3.12 (m, 2H) ppm. 13C NMR (101.6 MHz, CD2Cl2/CD3OD):
δ 159.6, 159.0, 158.8, 158.2, 158.1, 157.5, 149.6, 149.5, 149.4, 146.0, 145.9, 145.8, 139.0,
138.7, 138.5, 130.3, 127.9, 127.8, 127.8, 127.1, 125.1, 124.7, 122.8, 122.8, 122.1, 121.6,
110.4, 109.9, 107.9, 107.2, 62.0, 61.9, 61.8, 61.7, 61.6, 61.5 ppm. Anal. Calcd. (Found)
for C36H31BrN9NaO2 · 3.5 H2O (Mr = 787.64): C, 54.90 (54.50); H, 4.86 (4.40); N, 16.00
(16.45).
107

6.11 Derivatives of functionalised cryptates
6.11 Derivatives of functionalised cryptates
Carboxylic acid functionalised cryptates
General procedure for the synthesis of carboxylic acid functionalised cryptates: A
solution of NaOH in H2O was added to a solution of the carboxylic acid ester func-
tionalised cryptate (1 equiv) in MeOH. While stirring the reaction mixture for 3 h
at 40 °C, the colourless solution turned yellow. After the solvent was removed in
vacuo, the crude product was dry-loaded onto silica and purified by column chroma-
tography (SiO2, CH2Cl2/MeOH 2:1). The desired cryptate was isolated as a colorless
solid.
[Na(bpy.bpy.bpyCOO)]Br (51)
N
N
N
N
N
N
N
NNa
COO
51
51 was prepared according to the literature procedure of J. Wahsner. [119] 28 (36.3 mg,
49 µmol, 1.0 equiv), MeOH (6 ml), NaOH (10.7 mg, 267 µmol, 5.5 equiv), H2O (2 ml).
Yield: 25.2 mg (39 µmol, 80%). Rf = 0.82 (SiO2, CH2Cl2/MeOH 2:1, detection UV+I2
vapour). 1H NMR (250 MHz, CD2Cl2/CD3OD): δ 8.45 (s, 1H), 8.04 (d, 3J = 7.7 Hz,
1H), 7.99–7.93 (m, 4H), 7.89–7.80 (m, 5H), 7.79–7.77 (m, 1H), 7.37–7.27 (m, 5H), 3.83 (s,
12H) ppm.
Ethylenediamine coupled cryptates
[Na(bpy.bpy.bpyC(O)en)]Br (52)
N
N
N
N
N
N
N
NNa
Br
O
NH
NH2
52
108

6 Experimental Section
Under nitrogen and with cooling in an ice bath, 28 (51.4 mg, 70 µmol, 1.0 equiv)
was suspended in dry MeOH (6 ml) and subsequently added to freshly dried ethyl-
enediamine (768 µl, 691.2 mg, 11.501 mmol, 150 equiv). The reaction mixture was
allowed to warm to room temperature and stirred overnight. After removal of the
volatiles, the yellow crude product was dry-loaded onto silanised silica and subjected
to column chromatography (silanised SiO2, CH2Cl2/MeOH 15:1) to give pure 52 as
an off-white solid.
Yield: 46.8 mg (61 µmol, 87%). Rf = 0.31 (SiO2, CH2Cl2/MeOH 24:1, detection
UV+ninhydrin). MS (ESI+): m/z (%) 683.19 (100, [M-Br]+). 1H NMR (250 MHz,
CD2Cl2/CD3OD): δ 8.57 (s, 1H), 8.29 (d, 3J = 8.1 Hz, 1H), 8.00–7.93 (m, 4H), 7.91–7.79
(m, 6H), 7.38–7.27 (m, 5H), 3.97–3.78 (m, 12H), 3.73 (t, 3J = 5.7 Hz, 2H), 3.22 (t, 3J = 5.7
Hz, 2H) ppm.
[Na(bpy.bpy.bpyC(O)enO2
)]Br (55)
N
N
N
N
N
N
N
NNa
Br
O
NH
NH2
OO
55
Under nitrogen and with cooling in an ice bath, 29 (250.7 mg, 0.327 mmol, 1.0 equiv)
was added as a solid to freshly dried ethylenediamine (3.5 ml, 3.15 g, 52.41 mmol,
150 equiv). The slightly red suspension was allowed to warm to room temperature
and stirred for 2 d before the volatiles were removed in vacuo. The crude product was
dry-loaded onto silanised silica and subjected to column chromatography (silanised
SiO2, CH2Cl2/MeOH 24:1) to give pure 55 as an off-white solid.
Yield: 197.0 mg (0.248 mmol, 76%). Rf = 0.48 (SiO2, CH2Cl2/MeOH 4:1, detection
UV+ninhydrin). MS (ESI+): m/z (%) 357.99 (34, [M-Br+H]2+), 715.10 (100, [M-Br]+).1H NMR (250 MHz, CD2Cl2/CD3OD): δ 8.48 (d, 4J = 2.3 Hz, 1H), 8.35 (d, 4J = 2.6
Hz, 1H), 7.96–7.82 (m, 9H), 7.79 (dd, 3J = 7.8 Hz, 4J = 2.0 Hz, 1H), 7.62–7.53 (m, 1H),
7.51–7.42 (m, 4H), 4.43–4.30 (m, 2H), 4.02–3.85 (m, 4H), 3.83–3.60 (m, 4H), 3.59–3.39
(m, 4H), 3.30–3.20 (m, 2H) ppm. 13C NMR (62.9 MHz, CD2Cl2/CD3OD): δ 165.4,
159.4, 159.3, 158.5, 158.5, 157.7, 157.6, 157.1, 149.0, 145.5, 145.4, 139.0, 139.0, 138.7,
130.3, 130.1, 128.4, 128.2, 127.0, 126.3, 125.1, 122.7, 122.0, 121.9, 61.6, 61.5, 61.5, 61.4,
55.3, 55.1, 40.2, 38.2 ppm. 7 signals are missing, probably since signals are overlapping
109

6.11 Derivatives of functionalised cryptates
due to the large number of non-equivalent carbons in this molecule. Anal. Calcd. (Found)
for C39H36BrN10NaO3 · 4 H2O (Mr = 867.72): C, 53.98 (53.85); H, 5.11 (4.92); N, 16.14
(16.00).
Other
([Na(bpy.bpy.bpyC(O)enO2
)]Br)2C=O (54)
N
N
N
N
N
N
N
NNa
BrOO
O
NH
HN
N
N
N
N
N
N
N
NNa
BrOO
O
NH
HN
O
54
Under nitrogen, 55 (49.7 mg, 63 µmol, 6.0 equiv) was suspended in dry CH2Cl2(15 ml). With cooling in an ice bath, DIPEA (10.86 µl, 8.1 mg, 63 µmol, 6.0 equiv)
was added to give a nearly clear solution. Then, a solution of triphosgene (3.2 mg,
10 µmol, 1.0 equiv) in dry CH2Cl2 was added dropwise at 0 °C before the reaction
mixture was allowed to warm to room temperature and stirred for 5 d. The volatiles
were removed in vacuo and the bright green product was purified twice by column
chromatography (SiO2, CH2Cl2/MeOH/NH3 9:1:0.05 and CH2Cl2/MeOH 15:1→ 6:1)
to give the desired dimer 54 as a colourless solid.
Yield: 8.3 mg (5 µmol, 50%). Rf = 0.30 (SiO2, CH2Cl2/MeOH 6:1, detection UV+I2
vapour). MS (ESI+): m/z (%) 728.55 (100, [M-2Br]2+), 741.01 (8, isocyanate byproduct),
1491.89 (6, [M+H-2Na-Br]+). 1H NMR (250 MHz, CD2Cl2/CD3OD): δ 8.12 (d, 4J =
2.6 Hz, 1H), 8.09–8.04 (m, 2H), 8.02–7.82 (m, 17H), 7.81–7.60 (m, 4H), 7.56–7.41 (m,
7H), 7.41–7.34 (m, 2H), 7.17 (t, 3J = 7.8 Hz, 1H), 4.38–4.12 (m, 4H), 4.11–3.98 (m, 1H),
3.97–3.71 (m, 8H), 3.68–3.36 (m, 19H) ppm.
110

6 Experimental Section
[Na(bpy.bpy.bpyC(O)enO2
)-Glu]Br (56)
N
N
N
N
N
N
N
NNa Br
O
NH
HN O
FmocHNO
OtBu
OO
56
56 (11.1 mg, 26 µmol, 1.0 equiv) was dissolved in SPPS grade DMF (1 ml). With
cooling in an ice bath, the free acid was activated with HATU (11.9 mg, 31 µmol,
1.2 equiv) and DIPEA (11.2 µl, 8.4 mg, 65 µmol, 2.5 equiv). After stirring for 10 min,
55 (20.2 mg, 25 µmol, 1.0 equiv) in SPPS grade DMF (1 ml) was added and the reaction
mixture was allowed to slowly warm to room temperature. After stirring overnight,
the volatiles were removed in vacuo and the yellow crude product dry-loaded onto
silica. Twofold purification by column chromatography (SiO2, CH2Cl2/MeOH/NH3
9:1:0.05 and CH2Cl2/MeOH 24:1→ 9:1) affored 56 as a colourless solid.
Yield: 15.2 mg (13 µmol, 52%). Rf = 0.17 (SiO2, CH2Cl2/MeOH 24:1, detection UV+I2
vapour). MS (ESI+): m/z (%) 1122.64 (100, [M-Br]+). 1H NMR (250 MHz, CDCl3): δ
8.25 (s, 1H), 8.13 (s, 1H), 7.88–7.59 (m, 15H), 7.52 (br s, 1H), 7.44–7.31 (m, 8H), 6.76 (br
s, 1H), 6.02 (br s, 1H), 4.42–4.16 (m, 6H), 4.02–3.88 (m, 4H), 3.76–3.13 (m, 10H), 2.39 (s,
2H), 1.65 (s, 2H), 1.46 (s, 9H) ppm.
111

6.12 Lanthanide complexes
6.12 Lanthanide complexes
[Dx]-[Ln(bpy.bpy.bpy)]
N
N
N
N
N
N
N
N
DD
DD
DD D D
DDDD D
D
DD
DDDD
DD
DD
D
D
DD
D D
Ln Br
N
N
N
N
N
N
N
N
R1 R1
R2R2
R3 R3
R3R3R2R2
R1 R1
Ln Br
R1=D; R2,R3=H
R1,R2=D; R3=H
R1,R2,R3=D Ln = Nd, Yb
88
89
90
91
General procedure for the synthesis of [Dx]-Ln: The sodium cryptate (1.0 equiv) and
LnCl3·6H2O (1.05 equiv, 99.9%) were heated at reflux in dry CH3CN (ca. 0.25 ml per
µmol sodium cryptate) for 20 h. The solvent was removed in vacuo, the residue dis-
solved in a minimum amount of MeOH and the solution was layered with Et2O until
the mixture became slightly turbid. After storing the suspension at 4 °C overnight,
the colourless solid was collected on a membrane filter (nylon, 0.45 µm), and washed
with Et2O to yield the lanthanide complex as a colourless to faintly yellow powder
after drying under reduced pressure (yield: 50-70%). The purity of the compounds
was checked by analytical HPLC. In addition, the complexes were characterized by
positive ESI mass spectrometry.
HPLC: The only relevant impurity occasionally observed in the lanthanide complexes
was the corresponding sodium cryptate (starting material for the complexation reac-
tion). Analytical reversed-phase HPLC was performed in each case to rule out this
contaminant. All isotopologic complexes showed the same HPLC properties. The
complexes [Dx]-Yb are decomposing under the HPLC conditions on the timescale of
the run but [Dx]-Na is NOT present initially.
ESI+ Mass Spectrometry: Characteristic peaks for the lanthanide complexes: The iso-
topic patterns found are rather complicated due to the natural isotopic distributions
of both lanthanoids (6 isotopes for Yb and 7 for Nd) and the different deuteration
levels of the cryptands. The following table only lists the most intense peaks for the
corresponding manifolds.
112

6 Experimental Section
Table 6.1: HPLC program for Nd cryptates
(A: H2O (1% TFA, v/v), B: CH3CN
(HPLC gradient grade))
min %A %B
0 85 15
5 85 15
19 45 55
25 45 55
40 85 15
50 85 15
Table 6.2: HPLC program for Yb cryptates
(A: H2O (0.1% HCOOH, v/v), B:
CH3CN (HPLC gradient grade))
min %A %B
0 50 50
5 50 50
25 75 25
35 75 25
50 50 50
60 50 50
Table 6.3: Major ESI+ MS peaks of Nd cryptates
[Dx]-Nd [M+Cl-]2+ [M+2Cl-]+
[D4]-Nd 378.5 791.8
[D8]-Nd 380.4 795.9
[D12]-Nd 382.5 799.9
[D30]-Nd 390.5 819.9
Table 6.4: Major ESI+ MS peaks of Yb cryptates
[Dx]-Yb [M+e-]2+ [M+e-+2H2O]+
[D4]-Yb 378.5 393.4
[D8]-Yb 377.0 395.4
[D12]-Yb 379.0 397.5
[D30]-Yb 387.5 404.5
[D30]-[Tm(bpy.bpy.bpy)]3+ 3X-
N
N
N
N
N
N
N
N
DD
DD
DD D D
DDDD D
D
DD
DDDD
DD
DD
D
D
DD
D D
Tm 3X3
92
21 (11.6 mg, 16 µmol, 1.0 equiv), Tm(NO3)3·5H2O (7.8 mg, 17 µmol, 1.05 equiv),
CH3CN (6 ml). Yield: 5.2 mg.
113

6.12 Lanthanide complexes
[D29]-[Yb(bpy.bpy.bpyCOOMe]3+ 3X-
N
N
N
N
N
N
N
N
DD
DD
DD D D
DDDD D
COOMe
DD
DDDD
DD
DD
D
D
DD
D D
Yb 3X3
36
35 (6.7 mg, 8.76 µmol, 1.0 equiv), YbCl3·6H2O (3.73 mg, 9.63 µmol, 1.0 equiv), CH3CN
(5 ml). Yield: 7.4 mg.
114

6 Experimental Section
6.13 Fmoc-protected serines
Fmoc-D-cycloserine (64)
ONH
OHN
Fmoc
64
Fmoc-D-cylcoserine was prepared by a modified procedure of Gordeev et al.. [127] Un-
der nitrogen, N,O-bis(trimethylsilyl)acetamide (45 ml, 37.4 g, 0.184 mol, 2.5 equiv)
was added in one portion to a white suspension of D-cycloserine (62) (7.5 g, 0.073
mol, 1.0 equiv) in dry CH2Cl2 (70 ml). The white reaction mixture was stirred for
45 min at room temperature whereas the colour changed to slightly yellow upon
dissolution. Then, pyridine (120 ml) was added with a dropping funnel over the
course of 15 min and the mixture was stirred for another 15 min. Under nitrogen,
N-(9-fluorenylmethoxycarbonyloxy)succinimide (25.0 g, 0.074 mol, 1.0 equiv) in dry
CH2Cl2 (120 ml) was added via a dropping funnel at 0 °C over the course of 1 h.
The clear yellow reaction mixture was allowed to warm slowly to room temperature
overnight. The solvents were removed in vacuo. The yellow solid was dissolved in
EtOAc (350 ml) and stirred for 60 min. Undissolved particles were filtered off and the
bright yellow filtrate was extracted with HCl (pH 2, 3 × 50 ml) and brine (3 × 50 ml).
The combined organic extracts were dried over MgSO4 and the solvent removed in
vacuo. The slightly beige solid was suspended in EtOAc/hexanes (1:9, 200 ml), stirred
overnight and filtered to give 64 as white powder. If necessary, impurities of acetic
acid can be removed by recrystallisation from water.
Yield: 19.8 g (0.061 mol, 84%). MS (FAB+): m/z 179.1 (100, [dibenzofulvene-H]+), 325.2
(18, [M+H]+), 347.2 (11, [M+Na]+). 1H NMR (200 MHz, DMSO-D6): δ 11.43 (s, 1H),
7.98–7.81 (m, 2H), 7.71 (d, 3J = 7.1 Hz, 2H), 7.50–7.26 (m, 4H), 4.67–4.43 (m, 2H), 4.34
(d, 3J = 6.6 Hz, 2H), 4.25 (d, 3J = 6.3 Hz, 1H), 3.95 (t, 3J = 7.9 Hz, 1H) ppm. 13C NMR
(50.3 MHz, DMSO-D6): δ 170.4, 156.0, 143.8, 140.8, 127.7, 127.1, 125.2, 120.1, 71.7, 65.9,
52.8, 46.6 ppm. IR (FT-ATR): ν̃ 3312 (w, N-H), 3050 (w, C-Harom), 2963 (w, CH3), 1692
(s, C=O), 1540 (m, C=Carom), 1448 (w), 1386 (w), 1272 (m), 1181 (w), 1105 (w), 1092 (w),
1018 (w), 931 (w), 759 (m), 738 (s) cm-1.
115

6.13 Fmoc-protected serines
Fmoc-D-cycloserine-N(Me)·5H2O (65)
ON
OHN
Fmoc
65
Methyl iodide (1.57 ml, 3.6 g, 25.2 mmol, 1.2 equiv) and K2CO3 (3.3 g, 23.7 mmol,
1.1 equiv) were added to a solution of 64 (6.8 g, 21.0 mmol, 1.0 equiv) in acetone
(200 ml). The reaction mixture was heated at reflux for 45 min and then filtered hot.
After removal of the solvent in vacuo, the crude product was dry-loaded onto silica
and subjected to column chromatography (SiO2, CH2Cl2/MeOH 100:1 → 25:1). The
second major fraction contained the product 65, which was isolated as slightly yellow
solid.
Yield: 5.2 g (14.7 mmol, 70%). Rf = 0.50 (SiO2, CH2Cl2/MeOH 25:1). MS (EI+): m/z
165.0 (35, [fluorenyl-H]+), 178.0 (100, [dibenzofulvene-H]+), 196.0 (6, [fluorenylme-
thyl+H]+), 338.0 (8, [M]+.). MS (FAB+): m/z 179.1 (100, [dibenzofulvene-H]+), 339.1
(55, [M+H]+), 361.1 (30, [M+Na]+). 1H (200 MHz, CDCl3): δ 7.76 (d, 3J = 7.3 Hz, 2H),
7.58 (d, 3J = 7.1 Hz, 2H), 7.35 (ddd, 2J = 13.5 Hz, 2J = 10.6 Hz, 3J = 6.5 Hz, 4H), 5.53
(s, br, 1H), 4.80–4.55 (m, 2H), 4.49–4.37 (m, 2H), 4.28–4.15 (m, 1H), 4.09–3.94 (m, 1H),
3.20 (s, 3H) ppm. 13C NMR (50.3 MHz, CDCl3): δ 165.7, 156.4, 143.8, 141.4, 127.8,
127.2, 125.1, 120.1, 72.9, 67.4, 53.5, 47.1, 31.9 ppm. IR (FT-ATR): ν̃ 3266 (w, C-Harom),
3056 (w, C-Harom), 1709 (s, C=O), 1683 (s, C=O), 1543 (m, C=Carom), 1449 (w), 1271 (m),
1171 (m), 1103 (w), 1020 (m), 1001 (m), 965 (m, N-O), 918 (m), 756 (m), 738 (w), cm-1.
Anal. Calcd. (Found) for C19H18N2O4·0.5 MeOH (Mr = 354.38): C, 66.09 (65.85), H,
5.69 (5.16), N, 7.90 (7.70).
Fmoc-D-serine-O-aminomethyl-OMe·HCl (66)
HN
Fmoc OMe
O
O
HN
HCl
66
With cooling in an ice bath, 65 (4.6 g, 12.9 mmol, 1.0 equiv) was suspended in ice cold
HCl/MeOH (1.25 M, 150 ml). The reaction mixture was allowed to warm to room
temperature and stirred overnight before the solvents were removed in vacuo. The
116

6 Experimental Section
obtained residue was coevaporated with MeOH (2 × 50 ml) to give 66 as a colourless
solid.
Yield: 5.1 g (12.6 mmol, 97%). MS (EI+): m/z 165.0 (60, [fluorenyl-H]+), 178.0 (100,
[dibenzofulvene-H]+), 196.0 (12, [fluorenylmethanol+H]+), 354.9 (59, [M-CH2]+., 368.9
(59, [M-H]+.). 1H (200 MHz, CD3OD): δ 6.24 (d, 3J = 6.9 Hz, 2H), 6.10 (d, 3J = 6.9 Hz,
2H), 5.91–5.70 (m, 4H), 3.06 (d, 3J = 4.9 Hz, 1H), 2.86 (dd, 3J = 6.2 Hz, 3J = 3.3 Hz, 4H),
2.67 (t, 3J = 6.5 Hz, 1H), 2.21 (s, 3H), 1.75 (q, 3H) ppm. 13C NMR (50 MHz, CDCl3 ):
δ 169.2, 156.4, 143.8, 141.4, 127.9, 127.2, 125.3, 120.1, 100.1, 67.7, 53.4, 47.6, 35.6 ppm.
IR (FT-ATR): ν̃ 3259 (w, C-Harom), 2952 (w, CH3), 2368 (w), 1718 (br m, C=O), 1523 (w,
C=Carom), 1477 (w), 1448 (m), 1406 (w), 1213 (m), 1034 (m, N-O), 759 (m), 739 (s), 646
(m) cm-1. Anal. Calcd. (Found) for C20H22N2O5·HCl (Mr = 406.86): C, 59.04 (58.85),
H, 5.70 (5.86), N, 6.89 (6.61).
Fmoc-D-serine-O-aminomethyl-OMe (68)
HN
Fmoc OMe
O
O
HN68
A solution of 66 (1.0 g, 2.5 mmol, 1.0 equiv) in CH2Cl2 (100 ml) and washed with sat.
NaHCO3 (3 × 300 ml). The organic layer was dried over MgSO4, the solvent removed
in vacuo and the remaining residue dry-loaded onto silica. Purification by column
chromatography (SiO2, CH2Cl2/MeOH 75:1) afforded 68 as slightly yellow oil.
Yield: 0.77 g (2.07 mmol, 83%). Rf = 0.2 (SiO2, CH2Cl2/MeOH 50:1). 1H (250 MHz,
CDCl3): δ 7.77 (d, 3J = 7.3 Hz, 2H), 7.66–7.59 (m, 2H), 7.47–7.28 (m, 4H), 4.62–4.49 (m,
1H), 4.49–4.32 (m, 2H), 4.26 (t, 3J = 7.2 Hz, 1H), 4.14–3.93 (m, 2H). 4.63 (t, 3J = 5.0 Hz,
1H), 4.42 (dd, 3J = 6.2 Hz, 3J = 3.3 Hz, 4H), 4.24 (t, 3J = 6.5 Hz, 1H), 3.78 (s, 3H), 2.69
(s, 3H) ppm.
117

6.13 Fmoc-protected serines
Fmoc-D-serine-O-aminomethyl-N-chloromethylcarbonyl-OMe (67 )
HN
Fmoc OMe
O
O
N O
O
Cl67
Under nitrogen, 66 (5.0 g, 12.3 mmol, 1.0 equiv) was suspended in dry CH2Cl2(100 ml) and cooled to -30 °C. Dry triethylamine (5.9 ml, 4.3 g, 41.8 mmol, 3.4 equiv)
and chloromethyl chloroformiate (1.4 ml, 2.0 g, 15.5 mmol, 1.3 equiv) were added
slowly. The stirred mixture was allowed to warm to -20 °C and kept at this temper-
ature for 45 min. The reaction was quenched by cautious addition of water (5 ml).
The mixture was acidified with aq. HCl (1 M, pH 3-4), and extracted with EtOAc
(3 × 180 ml). The combined organic extracts were washed with NaHCO3 (3 × 40 ml)
and brine (3 × 40 ml), and dried over MgSO4. The solvent was removed in vacuo,
the crude product dry-loaded onto silica gel and purified by column chromatography
(SiO2, gradient: hexanes/EtOAc 5:1→ 2:1) to afford 67 as colourless oil.
Yield: 0.9 g (2.1 mmol, 17%). Rf = 0.65 (SiO2, hexanes/EtOAc 1:1). MS (ESI+) m/z
371.07 (7, [reactant+H]+), 451.07 (2, [M+Na-Cl]+), 463.05 (2, [M+H]+), 485.02 (100,
[M+Na]+). 1H NMR (200 MHz, CDCl3) δ 7.77 (d,3J = 7.4 Hz, 2H), 7.69–7.59 (m, 2H),
7.48–7.27 (m, 4H), 5.81–5.72 (m, 2H), 4.65–4.08 (m, 6H), 3.79 (s, 3H), 3.17 (s, 3H) ppm.13C NMR (50.3 MHz, CDCl3) δ 170.1, 156.1, 154.8, 144.0, 143.8, 141.4, 127.8, 127.2,
125.2, 120.1, 74.0, 71.0, 67.3, 53.4, 52.9, 47.2, 36.4 ppm. IR (FT-ATR): ν̃ 3337 (w, N-H),
3050 (w, C-Harom), 2952 (w, CH3), 1720 (br s, C=O), 1516 (m, C=Carom), 1447 (m), 1372
(w), 1327 (m), 1294 (m), 1208 (s), 1141 (s), 1053 (s, N-O), 878 (w), 758 (s), 740 (s, C-Cl),
705 (m), 621 (m) cm-1. Anal. Calcd. (Found) for C22H23ClN2O7 (Mr = 462.88): C, 57.09
(57.53), H, 5.01 (5.32), N, 6.05 (6.28).
118

6 Experimental Section
Fmoc-D-serine-O-aminomethyl-N(Azoc)-OMe (63)
HN
Fmoc OMe
O
O
N O
O
N363
Sodium azide (169.1 mg, 2.60 mmol, 1.5 equiv) was added to a solution of 67 (796.4 mg,
1.72 mmol, 1.0 equiv) in DMF (6 ml) and the mixture was stirred for 3 h at room tem-
perature. The reaction was quenched by cautious addition of water (5 ml) to give first
a clear and then a cloudy mixture. After removal of the solvents in vacuo, the residue
was dissolved in EtOAc (100 ml), washed with water (2× 30 ml) and brine (2× 30 ml).
The organic layer was dried over MgSO4 and the solvent removed in vacuo. The crude
product was dry-loaded onto silica and purified by column chromatography (SiO2,
CH2Cl2/EtOAc 5:1→ 2:1) to give 63 as a yellow oil.
Yield: 352.3 mg (0.75 mmol, 44%). Rf = 0.31 (SiO2, CH2Cl2/EtOAc 9:1). MS (ESI+) m/z
462.00 (7, [67+H]+), (491.98 (100, [M+Na]+). 1H NMR (200 MHz, CDCl3) δ 7.77 (d, 3J
= 7.3 Hz), 7.63 (m, 3J = 6.0 Hz, 2H), 7.46 - 7.28 (m, 4H), 6.11 (s, 1H), 5.17 (s, 1H), 4.62
- 4.06 (m, 6H), 3.79 (s, 3H), 3.17 (s, 3H) ppm. 13C NMR (62.5 MHz, CDCl3) δ 170.2,
156.5, 156.2, 144.00, 143.8, 141.4, 127.8, 127.2, 125.2, 120.2, 73.9, 67.3, 53.4, 52.9, 47.2,
36.4 ppm. IR (FT-ATR): ν̃ 3335 (w, N-H), 3051 (w, C-Harom), 2952 (w, CH3), 2360 (w),
2156 (w), 2109 (m, azideasym), 1715 (br s, C=O), 1609 (w), 1517 (m, C=Carom), 1449 (m),
1338 (m), 1294 (m), 1208 (s, azidesym), 1143 (s), 1055 (s, N-O), 1032 (s), 920 (m), 759 (s),
739 (s) cm-1. Anal. Calcd. (Found) for C22H23ClN5O7 (Mr = 469.45): C, 56.29 (55.83),
H, 4.94 (5.11), N, 14.92 (14.65).
119

6.14 Acetyl-protected serines
6.14 Acetyl-protected serines
Ac-D-cycloserine (69)
ONH
OHN
Ac
69
Acetyl-cycloserine was prepared by a procedure of Howard et al.. [128] D-Cycloserine
(5.0 g, 49.0 mmol, 1.0 equiv) was suspended in MeOH (250 ml) and Ac2O (5.5 g, 5.1 ml,
53.9 mmol, 1.1 equiv) was added dropwise over the course of 10 min. The colourless
reaction mixture was stirred at room temperature for 2 h until total dissolution of
the reactant. After removal of the solvent in vacuo, the white crude product was
recrystallised from boiling EtOH (~75 ml). Upon slow cooling to room temperature,
69 was obtained as white crystals, which were suitable for X-ray crystal structure
analysis.
Yield: 4.1 g (28.2 mmol, 58%). MS (FAB+): m/z 145.1 (52, [M+H]+), 167.0 (15,
[M+Na]+). 1H NMR (200 MHz, DMSO-D6): δ 11.47 (s, 1H), 8.40 (d, 3J = 7.5 Hz,
1H), 4.81 - 4.65 (m, 1H), 4.49 (t, 3J = 8.4 Hz, 1H), 3.89 (dd, 3J = 9.6 Hz, 3J = 8.4 Hz, 1H),
1.86 (s, 3H) ppm. 13C NMR (50 MHz, DMSO-D6): δ 170.5, 169.6, 72.5, 51.1, 22.3 ppm.
IR (FT-ATR): ν̃ 3298 (m, N-H), 2960 (m, CH3), 2808 (w, C-H), 2722 (m, CH2), 1700 (s,
C=O), 1637 (s, C=O), 1556, 1368 (s, CH3bend), 1306, 1035, 777 (m, CH2bend) cm-1. Anal.
Calcd. (Found) for C5H8N2O3 (Mr = 144.13): C, 41.67 (41.85), H, 5.59 (5.75), N, 19.44
(19.46).
Ac-D-cycloserine-N(Me) (70)
ON
OHN
Ac
70
Under nitrogen, methyliodide (2.5 g, 17.3 mmol, 1.2 equiv) and K2CO3 (2.2 g, 16.1
mmol, 1.1 equiv) were added to a solution of 69 (2.0 g, 13.9 mmol, 1.0 equiv) in acetone
(300 ml). The reaction mixture was heated at reflux for 45 min (bath temperature
75 °C) and then filtered hot. After cooling to room temperature, the solvent was
removed in vacuo to leave a white sticky solid behind. The crude product was dry-
loaded onto silica and subjected to column chromatography (SiO2, CH2Cl2/MeOH
120

6 Experimental Section
25:1 → 10:1) to remove unreacted 69 (0.39 g, Rf = 0.1 (SiO2, CH2Cl2/MeOH 15:1) and
obtain pure 70 as a white solid.
Yield: 0.6 g (3.8 mmol, 27%). Rf = 0.36 (SiO2, CH2Cl2/MeOH 15:1). MS (EI+): m/z
158.1 (13, [M+.]). 1H NMR (200 MHz, DMSO-D6): δ 8.44 (d, 3J = 7.5 Hz, 1H), 4.75 (dd,3J = 16.9 Hz, 3J = 8.6 Hz, 1H), 4.49 (t, 3J = 8.4 Hz, 1H), 3.89 (dd, 3J = 9.6 Hz, 3J = 8.4
Hz, 1H), 3.07 (s, 3H), 1.86 (s, 3H) ppm. 13C NMR (100.6 MHz, DMSO-D6): δ 169.5,
169.4, 70.3, 51.1, 31.9, 22.3 ppm. IR (FT-ATR): ν̃ 3280 (m, N-H), 2932 (w, CH3), 1704 (s,
C=O), 1632 (s, C=O), 1533 (s), 1362 (w), 1300 (s, CH3bend), 1094 (m), 705 (m, CH2bend)
cm-1. Anal. Calcd. (Found) for C6H10N2O3 (Mr = 158.16): C, 45.57 (45.76), H, 6.37
(6.72), N, 17.71 (17.80).
Ac-D-serine-O-aminomethyl-OMe·HCl (71)
HN
Ac OMe
O
O
HN
HCl
71
A solution of 70 (68 mg, 0.43 µmol) in ice cold HCl/MeOH (1.25 M, 70 ml) was allowed
to warm to room temperature and stirred overnight. After solvent removal in vacuo,
71 was obtained as a sticky yellow solid, which was used without further purification.
Ac-D-serine-O-amino-N-chloromethylcarbonyl-OMe (77)
HN
OMe
O
O
HN O
O
Cl
Ac
77
A mixture of 93 (100 mg, 0.38 mmol, 1.0 equiv) and capping agent (6% DIPEA & 5%
Ac2O in DMF, 1420 µl, 1.2 equiv & 2.0 equiv) was shaken for 15 min and, subsequently,
the solvent was removed in vacuo to leave a sticky orange residue behind. This crude
product was dry-loaded onto silica and purified by column chromatography twice
(SiO2, CH2Cl2/MeOH 9:1; SiO2, CH2Cl2/MeOH 24:1) to give 77 as a yellow oil.
121

6.14 Acetyl-protected serines
Yield: 48.3 mg (0.180 mmol, 47%). Rf = 0.50 (SiO2, CH2Cl2/MeOH 9:1, detection UV).
MS (ESI+): m/z 291.03 (100, [M+Na]+). 1H NMR (200 MHz, CDCl3): δ 8.45 (br s, 1H),
6.64 (br s, 1H), 5.83–5.70 (m, 2H), 5.00–4.82 (m, 1H), 4.26–4.01 (m, 2H), 3.79 (s, 3H),
2.09 (s, 2H) ppm.
122

6 Experimental Section
6.15 Boc-protected serines
Boc-D-cycloserine
ONH
OHN
Boc
74
74 was prepared by the literature procedure of Thorsteinsson et al. [130] and purified
according to the literature procedure of Wolfe et al. [129] With cooling in an ice bath,
triethylamine (863 mg, 1189 µl, 8.53 mmol, 1.0 equiv) was added dropwise to a solu-
tion of D-cycloserine (871 mg, 8.53 mmol, 1.0 equiv) in THF/H2O (1:1, v/v, 20 ml) and
stirred at 0 °C for 1 h. Then, a solution of di-tert-butyldicarbonate (1.86 g, 8.53 mmol,
1.0 equiv) in THF (8 ml) was added dropwise to the ice cold solution over the course
of 2 h. The reaction mixture was slowly warmed to room temperature and stirred
overnight before the solvent was removed in vacuo. The carefully dried, off-white,
sticky residue was dry-loaded onto silica and purified by a short column chromato-
graphy (SiO2, EtOAc/hexanes 2:1) to afford 74 as a fluffy white solid.
Yield: 925.5 mg (4.577 mmol, 54%). Rf = 0.38 (SiO2, EtOAc/hexanes 2:1, detection
UV). MS (ESI+): m/z 225.04 (100, [M+Na]+). 1H NMR (200 MHz, CDCl3): δ 5.11 (br
s, 1H, NH), 4.79 (t, 3J = 8.2 Hz, 1H), 4.67–4.51 (m, 1H), 4.09 (dd, 3J = 10.6 Hz, 3J = 8.2
Hz, 1H), 1.46 (s, 9H, BocCH3) ppm. 13C NMR (50.3 MHz, CDCl3): δ 171.1, 155.8, 81.0,
75.1, 52.9, 28.4 ppm. IR (FT-ATR): ν̃ 3314 (m, C(O)N-H), 2982 (w, CH3), 1716 (s, Boc
C=O), 1680 (s, C=O), 1539 (m), 1366 (m, C-(CH)3), 1252 (m), 1164 (s, C(O)-O) cm-1.
Boc-D-serine-N-chloromethylcarbonyl-OMe
ON
OHN
Boc
O
O
Cl75
75 was prepared by employing the conditions for the introduction of an (chlorometh-
oxy)carbamate protection group reported by Pothukanuri et al. [126] Under nitrogen,
a solution of 74 (82.3 mg, 0.407 mmol, 1.0 equiv) in dry CH2Cl2 (8 ml) was cooled
to -30 °C before dropwise addition of triethylamine (124.4 mg, 172 µl, 1.221 mmol,
3.0 equiv) and chloromethyl chloroformate (60.4 mg, 42 µl, 0.468 mmol, 1.15 equiv).
123

6.15 Boc-protected serines
The colourless solution was warmed to -20 °C and stirred at this temperature for 1
h . The reaction was quenched by cautious addition of H2O (5 ml) and acidified to
pH 3-4 with HCl (pH 2). After separation of the phases, the aqueous layer was ex-
tracted with EtOAc (3 × 20 ml), the combined organic phases dried over MgSO4 and
the solvents removed in vacuo. The crude product was dry-loaded onto silica and
purified by column chromatography (SiO2, hexane/EtOAc 2:1) to yield 75 as a sticky
white solid.
Yield: 67.7 mg (0.230 mmol, 56%). Rf = 0.25 (SiO2, hexane/EtOAc 2:1, detection UV).
MS (ESI+): m/z 317.12 (60, [M+Na]+), 349.03 (100, [M+Na+MeOH]+). 1H NMR (200
MHz, CDCl3): δ 5.88 (s, 2H, -CH2-), 5.06 (br s, 1H, NH), 4.84 (t, 3J = 8.0 Hz, 1H), 4.77–
4.60 (m, 1H), 4.22 (dd, 3J = 10.9 Hz, 3J = 7.9 Hz, 1H), 1.46 (s, 9H, BocCH3). 13C NMR
(50 MHz, CDCl3): δ 165.7, 145.6, 81.6, 73.1, 71.1, 53.5, 28.3 ppm. IR (FT-ATR): ν̃ 3425
(m, C(O)N-H), 3368 (m, C(O)N-H), 2918 (m, CH2), 2850 (w, CH3), 1787 (w, chlorozoc
C=O), 1760 (s, Boc C=O), 1698 (s, C=O), 1585 (s), 1441 (m), 1367 (m, C-(CH)3), 1283 (s),
1240 (s), 1157 (s, C(O)-O), 1096 (s), 962 (s), 873 (s) cm-1.
D-serine-O-amino-N-chloromethylcarbonyl-OMe
H2NOMe
O
O
HN O
O
Cl76
75 (18.6 mg, 0.062 mmol, 1.0 equiv) was dissolved in ice-cold HCl/MeOH (1.25 M,
5 ml), slowly warmed to room temperature and stirred overnight. The solvents were
removed in vacuo and the residue coevaporated with MeOH (2 × 50 ml) to obtain 76
as a colourless solid.
Yield: quantitative. MS (ESI+): m/z 227.05 (100, [M-Cl]+). 1H NMR (200 MHz, CDCl3):
δ 5.85 (s, 2H), 4.45–4.38 (m, 1H), 4.34–4.29 (m, 2H), 3.87 (s, 3H) ppm. 13C NMR
(50.3 MHz, CD3OD): δ 168.2, 157.3, 74.5, 71.9, 54.1, 53.1 ppm. IR (FT-ATR): ν̃ 2957 (m,
CH2), 1742 (s, C=O), 1570 (w), 1442 (m, CH2), 1364 (m, CH3), 1302 (s, C-Cl), 1236 (s),
1105 (s, C(=O)-O), 972 (m) cm-1.
124

6 Experimental Section
6.16 Other serines and related molecules
Phenylpyrazolidinone hydrochloride (58)
O
O
NPh
NH2
58
A yellow solution of 1-phenyl-3-pyrazolidinone (1.0 g, 6.1 mmol, 1.0 equiv) in ice
cold HCl/MeOH (1.25 M, 150 ml) was allowed to warm slowly to room temperature
overnight. After removal of the solvents in vacuo, the remaining sticky yellow solid
was dissolved in chloroform (50 ml) and extracted with sat. NaHCO3 (3 × 50 ml).
The organic phase was dried over MgSO4 and the solvent removed in vacuo to give 58
as yellow oil.
Yield: 0.9 g (4.7 mmol, 77%). 1H NMR (250 MHz, CD3OD): δ 7.51–7.42 (m, 2H), 7.28
(dd, 3J = 7.8 Hz, 3J = 6.0 Hz, 3H), 3.76 (t, 3J = 6.6 Hz, 2H), 3.68 (s, 3H), 2.63 (t, 3J = 6.6
Hz, 2H) ppm.
Fmoc-glycine-D-cycloserine (81)
ONH
HN
O
ONH
Fmoc
81
The procedure was adapted from a similar reaction reported by De et al. [132] Under
nitrogen and with cooling in an ice bath, oxalyl chloride (8.3 ml, 12.4 g, 100 equiv) was
added dropwise to Fmoc-Gly-OH (291 mg, 0.98 mmol, 1.0 equiv). The bright yellow
mixture was stirred for 2 h at room temperature until the whole starting material
dissolved and gas evolution ceased. Then, the volatiles were removed in vacuo to give
the acyl chloride as pale yellow solid. Also under nitrogen, D-cycloserine (100 mg,
0.98 mmol, 1.0 equiv) was suspended in dry CH2Cl2 (15 ml), before NMP (2.8 ml) and
N,O-bis(trimethylsilyl) acetamide (0.6 ml, 498 mg, 2.45 mmol, 2.5 equiv) were added
dropwise. After stirring at room temperature for 1 h, a clear colourless solution was
obtained, which was treated with pyridine (1.6 ml). Then, a solution of the acid
chloride in CH2Cl2 (6 ml) was added dropwise over the course of 15 min which
resulted in a colour change from yellow over orange to deep red. After stirring at
125

6.16 Other serines and related molecules
room temperature for 1.5 d, the solvents were removed in vacuo with gentle heating
(~50 °C). The dark orange residue was dissolved in EtOAc to give a yellow solution,
which was washed with aq. HCl (pH 2, 3 × 15 ml). The organic layer was dried over
MgSO4 and rotavapped to dryness to leave a yellow residue. This crude product was
dryloaded onto silica and purified by column chromatography (SiO2, CH2Cl2/MeOH,
50:1→ 15:1) to give 81 as a white foamy solid.
Yield: 157.7 mg (0.413 mmol, 42%). Rf = 0.25 (SiO2, CH2Cl2/MeOH 24:1, detection
UV). 1H NMR (250 MHz, DMSO-D6): δ 8.45 (d, 3J = 7.6 Hz, 1H), 7.89 (d, 3J = 7.2 Hz,
2H), 7.72 (d, 3J = 7.5 Hz, 2H), 7.54 (t, 3J = 6.1 Hz, 1H), 7.47–7.28 (m, 4H), 4.83–4.61 (m,
1H), 4.50 (t, 3J = 8.4 Hz, 1H), 4.33–4.18 (m, 3H), 3.92 (dd, 3J = 9.8 Hz, 3J = 8.4 Hz, 1H),
3.66 (d, 3J = 6.1 Hz, 2H) ppm.
Fmoc-glycine-D-cycloserine-N(Me) (82)
ON
HN
O
ONH
Fmoc
82
Methyl iodide (0.26 ml, 0.6 g, 4.2 mmol, 1.3 equiv) was added dropwise to a slightly
yellow suspension of 81 (1.2 g, 3.1 mmol, 1.0 equiv) and K2CO3 (0.5 g, 3.9 mmol,
1.2 equiv) in acetone (35 ml). The reaction mixture was heated at reflux for 1 h,
filtered hot and finally the solvent was removed in vacuo. The obtained brown foamy
residue was dry-loaded onto silica and purified by column chromatography (SiO2,
CH2Cl2/MeOH 50:1→ 30:1) to afford 82 as a white foamy residue.
Yield: 0.8 g (2.1 mmol, 68%). Rf = 0.60 (SiO2, CH2Cl2/MeOH 9:1, detection UV).1H NMR (200 MHz, CDCl3): δ 7.76 (d, 3J = 6.9 Hz, 2H), 7.58 (d, 3J = 7.2 Hz, 2H),
7.45–7.35 (m, 2H), 7.32 (dd, 3J = 7.3 Hz, 4J = 1.4 Hz, 2H), 6.71 (br s, 1H), 5.45 (br s,
1H), 4.80–4.74 (m, 2H), 4.48–4.39 (m, 2H), 4.22 (t, J = 6.8 Hz, 1H), 4.03–3.84 (m, 3H),
3.21 (s, 3H) ppm.
Fmoc-gylcine-D-serine-O-aminomethyl-OMe·HCl (83)
NH
Fmoc
O
HN
O
O
NH
O
HCl
83
126

6 Experimental Section
With cooling in an ice bath, 1.25 M HCl/MeOH (20 ml) was added dropwise to 82
(430 mg, 1.09 mmol). The clear colourless reaction mixture was allowed to slowly
warm to room temperature. After stirring overnight, the solvent was removed in
vacuo with gentle heating to 40 °C to leave a colourless sticky residue behind. The
crude product was dry-loaded onto silica and subjected to column chromatography
(SiO2, CH2Cl2/MeOH 50:1→ 9:1) to separate 83 from the byproduct Fmoc-Gly-OMe,
both being sticky, colourless solids.
Yield (83): 232 mg (0.54 mmol, 50%). Rf = 0.61 (SiO2, CH2Cl2/MeOH 50:1, detection
UV+I2 vapour). 1H NMR (200 MHz, CDCl3): δ 7.75 (d, 3J = 7.0 Hz, 2H), 7.58 (d, 3J =
7.2 Hz, 2H), 7.44–7.27 (m, 4H), 5.76 (br s, 1H), 4.73 (ddd, 3J = 7.6 Hz, 3J = 4.1 Hz, 3J =
3.2 Hz, 1H), 4.40 (d, J = 6.4 Hz, 2H), 4.28–4.15 (m, 1H), 4.11–3.83 (m, 4H), 3.72 (s, 3H),
2.57 (s, 3H) ppm.
Yield (Fmoc-Gly-OMe): 55 mg (0.176 mmol, 16%). Rf = 0.57 (SiO2, CH2Cl2/MeOH 9:1,
detection UV+I2 vapour). 1H NMR (200 MHz, CDCl3): δ 7.77 (d, 3J = 6.8 Hz, 2H), 7.60
(d, 3J = 7.2 Hz, 2H), 7.46–7.36 (m, 2H), 7.36–7.27 (m, 2H), 5.30 (s, 1H), 4.42 (d, 3J = 6.5
Hz, 2H), 4.32–4.17 (m, 1H), 4.00 (d, 3J = 5.6 Hz, 2H), 3.77 (s, 3H) ppm.
Fmoc-glycine-D-serine-O-aminomethyl-N-chloromethylcarbonyl-OMe (84)
NH
Fmoc
O
HN
O
O
N
O
O
O
Cl84
Under nitrogen, 83 (66.2 mg, 0.155 mmol, 1.0 equiv) was dissolved in dry CH2Cl2(1.5 ml) and the solution was cooled to -30 °C. Then, dry NEt3 (62 µl, 44.8 mg,
0.0443 mmol, 2.8 equiv) and chloromethyl chloroformate (15 µl, 22.2 mg, 0.172 mmol,
1.1 equiv) were added dropwise, before the reaction was warmed to -20 °C and stirred
at this temperature for 1 h until the reaction was completed (followed with TLC). The
reaction was quenched by addtion of water (1 ml) and acidified to pH 3-4 with aq.
HCl (pH 2). The two phases were separated and the aqueous layer extracted with
EtOAc (3 × 10 ml). The combined organic phases were dried over MgSO4 and rota-
vapped to dryness. The crude product was then dryloaded onto silica and purified
by column chromatography (SiO2, CH2Cl2/MeOH 50:1 → 24:1) to yield pure 84 as a
colourless, smelly sticky solid.
127

6.16 Other serines and related molecules
Yield: 57.1 mg (0.110 mmol, 71%). Rf = 0.43 (SiO2, CH2Cl2/MeOH 9:1, detection UV).1H NMR (200 MHz, CDCl3): δ 7.75 (d, J = 6.9 Hz, 2H), 7.60 (d, J = 7.2 Hz, 2H), 7.44–
7.28 (m, 4H), 5.70 (s, 2H), 5.62 (m, 1H), 4.80 (dt, J = 8.0 Hz, J = 3.2 Hz, 1H), 4.53–4.34
(m, 3H), 4.30–4.17 (m, 1H), 4.13–3.93 (m, 3H), 3.75 (s, 3H), 3.13 (s, 3H) ppm.
Fmoc-glycine-D-aminoserine-N(Me)-Azoc methyl ester (85)
NH
Fmoc
O
HN
O
O
N
O
O
O
N385
Sodium azide (8.80 mg, 0.135 mmol, 1.5 equiv) was added as a solid to a solution
of 84 (46.4 mg, 0.089 mmol, 1.0 equiv) in DMF (1 ml). The reaction mixture was
stirred for 3 h at room temperature whereupon the colour changed from colourless
to slightly pink and the solution gets turbid due to the precipitation of NaCl. After
solvent removal the crude product was directly subjected to column chromatography
(SiO2, CH2Cl2/MeOH 50:1). The product (85) was eluted as 4th fraction and isolated
as colourless sticky solid.
Yield: 33.5 mg (0.064 mmol, 72%). Rf = 0.64 (SiO2, CH2Cl2/MeOH 9:1, detection
UV+I2 vapour). 1H NMR (250 MHz, CDCl3): δ 7.76 (d, 3J = 7.2 Hz, 2H), 7.60 (d, 3J =
7.6 Hz, 2H), 7.45–7.27 (m, 4H), 5.54 (br s, 1H), 5.12 (s, 2H), 4.79 (dd, 3J = 7.8 Hz, 3J =
3.6 Hz, 1H), 4.50 (dd, 2J = 10.6 Hz, 3J = 2.8 Hz, 1H), 4.45–4.39 (m, 2H), 4.58–4.34 (m,
3H), 4.24 (t, J = 7.0 Hz, 1H), 4.11–3.95 (m, 3H), 3.76 (s, 3H), 3.14 (s, 3H) ppm.
128

7 Bibliography
[1] V. Ntziachristos, A. Leroy-Willig, B. Tavitian, Textbook of in vivo Imaging in Ver-
tebrates 1st ed., Wiley-Interscience, Chichester, 2007, xvi–xvii.
[2] R. Weissleder, U. Mahmood, Radiology 2001, 219, 316–333.
[3] H. Kobayashi, M. Ogawa, R. Alford, P. L. Choykeeter, Y. Urano, Chem. Rev. 2010,
110, 2620–2640.
[4] P. Armstrong, M. Wastie, A. Rockall, Diagnostic Imaging 6th ed., Wiley-
Interscience, Chichester, 2009, 1–16.
[5] C. A. Boswell, M. W. Brechbiel, Nucl. Med. Biol. 2007, 34, 757–778.
[6] J. T. Bushberg, J. A. Seibert, E. M. Leidholdt, J. M. Boone, The Essential Physics of
Medical Imaging 2nd ed., (Eds.: J.-R. John, A. Snyder, T. DeGeorge), Lippincott
Williams & Wilkins, Philadelphia, 2002, 13–16.
[7] C. H. Contag, in Molecular Imaging: Principles and Practice (Ed.: R. Weissleder,
B. D. Ross, A. Rehemtulla, S. S. Gambhir), PMPH-USA, Shelton, 1st ed., 2010,
118–138.
[8] R. Weissleder, V. Ntziachristos, Nat. Med. 2003, 9, 123–128.
[9] A. Oser, W. K. Roth, G. Valet, Nucleic Acids Res. 1988, 16, 1181–1196.
[10] J. Coates, P. G. Sammes, G. Yahioglu, R. M. West, A. J. Garman, J. Chem. Soc.,
Chem. Commun. 1994, 2311–2312.
[11] P. Gottlieb, E. Hazum, E. Tzehoval, M. Feldman, S. Segal, M. Fridkin, Biochem.
Biophys. Res. Commun. 1984, 119, 203–211.
[12] N. Loménie, D. Racoceanu, A. Gouaillard, Advances in Bio-Imaging: from Physics
to Signal Understanding Issues (Ed.: J. Kacprzyk), Springer, Berlin and Heidel-
berg, 2012, 37–54.
[13] E. Soini, I. Hemmila, P. Dahlen, Ann. Biol. Clin. 1990, 48, 567–571.
[14] R. A. Evangelista, A. Pollak, B. Allore, E. F. Templeton, R. C. Morton, E. P.
Diamandis, Clin. Biochem. 1988, 21, 173–178.
[15] E. Lopez, C. Chypre, B. Alpha, G. Mathis, Clin. Chem. 1993, 39, 196–201.
129

7 Bibliography
[16] G. Mathis, Clin. Chem. 1995, 41, 1391–1397.
[17] E. F. Dickson, A. Pollak, E. P. Diamandis, Photochem. Photobiol. 1995, 27, 3–19.
[18] V. Ntziachristos, A. G. Yodh, M. Schnall, B. Chance, Proc. Natl. Acad. Sci. U. S.
A. 2000, 97, 2767–2772.
[19] A. Corlu, R. Choe, T. Durduran, M. A. Rosen, M. Schweiger, S. R. Arridge, M. D.
Schnall, A. G. Yodh, Opt. Express 2007, 15, 6696–6716.
[20] H. Kobayashi, Y. Hama, Y. Koyama, T. Barrett, C. A. S. Regino, Y. Urano, P. L.
Choyke, Nano Lett. 2007, 7, 1711–1716.
[21] Y. Hama, Y. Koyama, Y. Urano, P. L. Choyke, H. Kobayashi, J. Invest. Dermatol.
2007, 127, 2351–2356.
[22] Y. Hama, Y. Urano, Y. Koyama, M. Kamiya, M. Bernardo, R. S. Paik, M. C.
Krishna, P. L. Choyke, H. Kobayashi, Neoplasia 2006, 8, 607–612.
[23] R. A. Sheth, R. Upadhyay, L. Stangenberg, R. Sheth, R. Weissleder, U. Mahmood,
Gynecol. Oncol. 2009, 112, 616–622.
[24] J.-C. G. Bünzli, Chem. Lett. 2009, 38, 104–109.
[25] D. Parker, J. A. G. Williams, J. Chem. Soc., Dalton Trans. 1996, 3613–3628.
[26] Y. M. W. Janssen, B. V. Houten, P. J. A. Borm, B. T. Mossman, Lab. Invest. 1993,
69, 261–274.
[27] T. Schaeffter, Prog. Drug. Res. 2005, 62, 15–81.
[28] K. Smith, The Science of Photobiology 1st ed., Plenum Press, New York, 1977.
[29] W. Mason, Fluorescent and Luminescent Probes for Biological Activity: A Practical
Guide to Technology for Quantitative Real-Time Analysis 2nd ed., Biological Tech-
niques Series, Academic Press, London, 1999, 17–39.
[30] A. Diaspro, Optical Fluorescence Microscopy: From the Spectral to the Nano Dimen-
sion 1st ed., Springer, Berlin and Heidelberg, 2011, 111–130.
[31] A. P. Neal, C. Schultz, in Protein Targeting with Small Molecules: Chemical Biology
Techniques and Applications (Ed.: H. Osada), Wiley-Interscience, Hoboken, 1st
ed., 2009, 91–113.
[32] N. C. Shaner, P. A. Steinbach, R. Y. Tsien, Nat. Methods 2005, 2, 905–909.
[33] K. Brejc, T. K. Sixma, P. A. Kitts, S. R. Kain, R. Y. Tsien, M. Ormo, S. J. Remington,
Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 2306–2311.
[34] L. Zhang, H. N. Patel, J. W. Lappe, R. M. Wachter, J. Am. Chem. Soc. 2006, 128,
4766–4772.
[35] D. M. Chudakov, S. Lukyanov, K. A. Lukyanov, Trends Biotechnol. 2005, 23, 605–
130

7 Bibliography
613.
[36] B. P. Aryal, D. E. Benson, J. Am. Chem. Soc. 2006, 128, 15986–15987.
[37] D. B. Cordes, S. Gamsey, B. Singaram, Angew. Chem., Int. Ed. 2006, 45, 3829–
3832.
[38] X. Wang, M. J. Ruedas-Rama, E. A. H. Hall, Anal. Lett. 2007, 40, 1497–1520.
[39] I. L. Medintz, H. T. Uyeda, E. R. Goldman, H. Mattoussi, Nat. Mater. 2005, 4,
435–446.
[40] J.-H. Park, L. Gu, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia, M. J. Sailor, Nat.
Mater. 2009, 8, 331–336.
[41] F. Yang, L. G. Moss, G. N. Phillips, Nat. Biotechnol. 1996, 14, 1246–1251.
[42] Picture source: http://www.photonics.com/images/spectra/features/2007/May/Quantum
Dots_Fig5_CoreShell.jpg (last access 21st Jan 2013).
[43] L. D. Lavis, R. T. Raines, ACS Chem. Biol. 2008, 3, 142–155.
[44] Z. Zhu, Dyes Pigm. 1995, 27, 77–111.
[45] A. Loudet, K. Burgess, Chem. Rev. 2007, 107, 4891–4932.
[46] H. E. Katerinopoulos, Curr. Pharm. Des. 2004, 10, 3835–3852.
[47] C. C. Woodroofe, M. H. Lim, W. Bu, S. J. Lippard, Tetrahedron 2005, 61, 3097–
3105.
[48] W.-C. Sun, K. R. Gee, D. H. Klaubert, R. P. Haugland, J. Org. Chem. 1997, 62,
6469–6475.
[49] F. Wang, X. Liu, Chem. Soc. Rev. 2009, 38, 976–989.
[50] B. Alpha, J. M. Lehn, G. Mathis, Angew. Chem. 1987, 99, 259–261.
[51] B. Alpha, R. Ballardini, V. Balzani, J. M. Lehn, S. Perathoner, N. Sabbatini, Pho-
tochem. Photobiol. 1990, 52, 299–306.
[52] J. M. Lehn, Acc. Chem. Res. 1978, 11, 49–57.
[53] B. Alpha, V. Balzani, J. M. Lehn, S. Perathoner, N. Sabbatini, Angew. Chemie 1987,
99, 1310–1311.
[54] E. Brucher, Z. Baranyai, G. Tircso, RSC Drug Discovery Ser. 2012, 15, 208–260.
[55] J.-C. Bünzli, C. Piguet, Chem. Soc. Rev. 2005, 34, 1048–1077.
[56] S. Comby, J.-C. G. Bünzli 2007, Vol. 37, pp. 217–470.
[57] V. Balzani, F. Scandola, in Supramolecular Photochemistry, Ellis Horwood,
Chichester, Ch. 12, 1991.
[58] S. Weissmann, J. Chem. Phys. 1942, 10, 214–221.
131

7 Bibliography
[59] J. Zhou, Z. Liub, F. Li, Chem. Soc. Rev. 2012, 41, 1323–1349.
[60] M. H. V. Werts, in Lanthanide Luminescence - Photophysical, analytical and biological
aspects (Ed.: O. Wolfbeiss, P. Hänninen, H. Härmä), Springer, Berlin-Heidelberg,
Springer Series on Fluorescence, 2011, 133–160.
[61] K. Matsumoto, T. Nojima, H. Sano, K. Majima, Macromol. Symp. 2002, 186, 117–
121.
[62] H. Bazin, M. Preaudat, E. Trinquet, G. Mathis, Spectrochim. Acta, Part A 2001,
57A, 2197–2211.
[63] E. Soini, H. Kojola, Clin. Chem. 1983, 29, 65–68.
[64] I. Hemmila, S. Dakubu, V. M. Mukkala, H. Siitari, T. Lovgren, Anal. Biochem.
1984, 137, 335–343.
[65] J.-C. Bünzli, S. V. Eliseeva, J. Rare Earths 2010, 28, 824–842.
[66] S. Faulkner, S. J. A. Pope, B. P. Burton-Pye, Appl. Spectrosc. Rev. 2005, 40, 1–31.
[67] M. D. Ward, Coord. Chem. Rev. 2007, 251, 1663–1677.
[68] M. H. V. Werts, Sci. Prog. 2005, 88, 101–131.
[69] V. Bulach, F. Sguerra, M. W. Hosseini, Coord. Chem. Rev. 2012, 256, 1468–1478.
[70] A. D’Aleo, F. Pointillart, L. Ouahab, C. Andraud, O. Maury, Coord. Chem. Rev.
2012, 256, 1604–1620.
[71] F. Wang, D. Banerjee, Y. Liu, X. Chen, X. Liu, Analyst 2010, 135, 1839–1854.
[72] F. Auzel, C. R. Acad. Sci., Ser. A B 1966, 263B, 819–821.
[73] F. Auzel, C. R. Seances Acad. Sci., Ser. B 1966, 262B, 1016–1019.
[74] V. Ovsyankin, P. Feofilov, Zh. Eksp. Teor. Fiz. 1966, 4, 471–474.
[75] F. Auzel, Chem. Rev. 2004, 104, 139–173.
[76] D. E. Achatz, R. Ali, O. S. Wolfbeis, in Luminescence Applied in Sensor Science (Ed.:
L. Prodi, M. Montalti, N. Zaccheroni), Springer, Topics in Current Chemistry,
2011, 29–50.
[77] T. Soukka, T. Rantanen, K. Kuningas, Ann. N. Y. Acad. Sci. 2008, 1130, 188–200.
[78] G.-L. Law, in Tomorrow’s chemistry today - Concepts in nanoscience, organic materials
and environmental chemistry (Ed.: B. Pignataro), Wiley-VCH, Weinheim, 2nd ed.,
2008, 161–184.
[79] K.-L. Wong, W.-M. Kwok, W.-T. Wong, D. L. Phillips, K.-W. Cheah, Angew.
Chem., Int. Ed. 2004, 43, 4659–4662.
[80] K.-L. Wong, G.-L. Law, W.-M. Kwok, W.-T. Wong, D. L. Phillips, Angew. Chem.,
132

7 Bibliography
Int. Ed. 2005, 44, 3436–3439.
[81] X. Xiao, J. P. Haushalter, G. W. Faris, Opt. Lett. 2005, 30, 1674–1676.
[82] N. Tancrez, C. Feuvrie, I. Ledoux, J. Zyss, L. Toupet, H. L. Bozec, O. Maury, J.
Am. Chem. Soc. 2005, 127, 13474–13475.
[83] L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K. R. Kittilstved, A. Aebischer,
J.-C. G. Bünzli, A. Hauser, C. Piguet, Angew. Chem. Int. Ed. 2011, 50, 4108–4112.
[84] J. L. Kropp, M. W. Windsor, J. Chem. Phys. 1963, 39, 2769–2770.
[85] J. L. Kropp, M. W. Windsor, J. Chem. Phys. 1965, 42, 1599–1609.
[86] J. L. Kropp, M. W. Windsor, J. Chem. Phys. 1966, 45, 761–762.
[87] R. S. Dickins, D. Parker, A. S. de Sousa, J. A. G. Williams, Chem. Commun. 1996,
697–698.
[88] A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S.
de Sousa, J. A. G. Williams, M. Woods, J. Chem. Soc., Perkin Trans. 2 1999, 493–
504.
[89] J. C. Rodriguez-Ubis, B. Alpha, D. Plancherel, J. M. Lehn, Helv. Chim. Acta 1984,
67, 2264–2269.
[90] S. Faulkner, A. Beeby, M. C. Carrie, A. Dadabhoy, A. M. Kenwright, P. G.
Sammes, Inorg. Chem. Commun. 2001, 4, 187–190.
[91] M. J. Cook, A. P. Lewis, G. S. G. McAuliffe, V. Skarda, A. J. Thomson, J. L.
Glasper, D. J. Robbins, J. Chem. Soc., Perkin Trans. 2 1984, 1293–1301.
[92] J. M. Lehn, C. O. Roth, Helv. Chim. Acta 1991, 74, 572–578.
[93] C. Doffek, N. Alzakhem, M. Molon, M. Seitz, Inorg. Chem. 2012, 51, 4539–4545.
[94] J. M. Lehn, G. Mathis, B. Alpha, R. Deschenaux, E. Jolu, Heterocycle complexes
with rare earth metals as fluorescent labels and their preparation (EP 321353 A1), 1989.
[95] F. Havas, N. Leygue, M. Danel, B. Mestre, C. Galaup, C. Picard, Tetrahedron 2009,
65, 7673–7686.
[96] I. Bkouche-Waksman, J. Guilhem, C. Pascard, B. Alpha, R. Deschenaux, J. M.
Lehn, Helv. Chim. Acta 1991, 74, 1163–70.
[97] U. Maheswari Palanisamy, K. Lappalainen, M. Sfregola, S. Barends, P. Gamez,
U. Turpeinen, I. Mutikainen, P. van Wezel Gilles, J. Reedijk, Dalton Trans. 2007,
3676–3683.
[98] T. Rode, E. Breitmaier, Synthesis 1987, 574–575.
[99] G. R. Newkome, W. E. Puckett, G. E. Kiefer, V. D. Gupta, Y. Xia, M. Coreil, M. A.
Hackney, J. Org. Chem. 1982, 47, 4116–4120.
133

7 Bibliography
[100] N. Psychogios, J.-B. Regnouf-de Vains, H. M. Stoeckli-Evans, Eur. J. Inorg. Chem.
2004, 2514–2523.
[101] F. Bottino, M. Di Grazia, P. Finocchiaro, F. R. Fronczek, A. Mamo, S. Pappalardo,
J. Org. Chem. 1988, 53, 3521–3529.
[102] M. Y. Alyapyshev, V. A. Babain, N. E. Borisova, R. N. Kiseleva, D. V. Safronov,
M. D. Reshetova, Mendeleev Commun. 2008, 18, 336–337.
[103] A. J. Blake, N. R. Champness, P. V. Mason, C. Wilson, Acta Crystallogr., Sect. C:
Cryst. Struct. Commun. 2007, C63, m280–m282.
[104] Z. Wang, J. Reibenspies, R. J. Motekaitis, A. E. Martell, J. Chem. Soc., Dalton
Trans. 1995, 1511–1518.
[105] C. Bischof, J. Wahsner, J. Scholten, S. Trosien, M. Seitz, J. Am. Chem. Soc. 2010,
132, 14334–14335.
[106] M. P. O. Wolbers, F. C. J. M. Van Veggel, B. H. M. Snellink-Ruel, J. W. Hofstraat,
F. A. J. Guerts, D. N. Reinhoudt, J. Chem. Soc., Perkin Trans. 2 1998, 2141–2150.
[107] V. L. Ermolaev, E. B. Sveshnikova, Chem. Phys. Lett. 1973, 23, 349–354.
[108] T. Forster, Ann. Physik [6 Folge] 1948, 2, 55–75.
[109] J. Mathieu, A. Marsura, Synth. Commun. 2003, 33, 409–414.
[110] J. Agrawal, R. Hodgson, Organic Chemistry of Explosives Wiley-Interscience,
Chichester, 2007, 17–18.
[111] C. J. Moody, J. L. O’Connell, Chem. Commun. 2000, 1311–1312.
[112] N. Alzakhem, Stable Lanthanide Cryptates as Building Blocks for Solid Phase Peptide
Synthesis - Synthesis, Functionalization and Photophysical Properties, Dissertation,
Ruhr-Universität Bochum, 2013.
[113] V. M. Mukkala, J. J. Kankare, Helv. Chim. Acta 1992, 75, 1578–1592.
[114] Z. Zhou, G. H. Sarova, S. Zhang, Z. Ou, F. T. Tat, K. M. Kadish, L. Echegoyen,
D. M. Guldi, D. I. Schuster, S. R. Wilson, Chem.–Eur. J. 2006, 12, 4241–4248.
[115] M. Karramkam, F. Hinnen, F. Vaufrey, F. Dolle, J. Labelled Compd. Radiopharm.
2003, 46, 979–992.
[116] E. Delfourne, F. Darro, N. Bontemps-Subielos, C. Decaestecker, J. Bastide,
A. Frydman, R. Kiss, J. Med. Chem. 2001, 44, 3275–3282.
[117] M. Hapke, H. Staats, I. Wallmann, A. Luetzen, Synthesis 2007, 2711–2719.
[118] S. Caron, N. M. Do, J. E. Sieser, Tetrahedron Lett. 2000, 41, 2299–2302.
[119] J. Wahsner, Isotopencodierte Lanthanoid-Kryptate für die quantitative Proteomik, Mas-
ter thesis, Ruhr-Universität Bochum, 2011.
134

7 Bibliography
[120] P. F. Van Swieten, M. A. Leeuwenburgh, B. M. Kessler, H. S. Overkleeft, Org.
Biomol. Chem. 2005, 3, 20–27.
[121] J. M. Baskin, C. R. Bertozzi, QSAR Comb. Sci. 2007, 26, 1211–1219.
[122] R. Garrett, C. Grisham, Biochemistry 4th ed., Brooks/Cole Cengage Learning,
Boston, 2007, 103–105.
[123] R. Lundblad, Chemical Reagents for Protein Modification 3rd ed., CRC Press, Boca
Raton, 2010, 269–279.
[124] R. Reid, Peptide and Protein Drug Analysis Drugs and the Pharmaceutical Sciences
Series, Marcel Dekker, New York, 1999, 260–262.
[125] T. Bissot, R. Parry, D. Campbell, J. Am. Chem. Soc. 1957, 79, 796–800.
[126] S. Pothukanuri, N. Winssinger, Org. Lett. 2007, 9, 2223–2225.
[127] M. F. Gordeev, G. W. Luehr, H. C. Hui, E. M. Gordon, D. V. Patel, Tetrahedron
1998, 54, 15879–15890.
[128] J. C. Howard, J. Org. Chem. 1981, 46, 1720–1723.
[129] S. Wolfe, M.-C. Wilson, M.-H. Cheng, G. V. Shustov, C. I. Akuche, Can. J. Chem.
2003, 81, 937–960.
[130] T. Thorsteinsson, M. Masson, T. Jarvinen, T. Nevalainen, T. Loftsson, Chem.
Pharm. Bull. 2002, 50, 554–557.
[131] A. G. Griesbeck, S. Bondock, J. Lex, J. Org. Chem. 2003, 68, 9899–9906.
[132] P. De, G. Koumba Yoya, P. Constant, F. Bedos-Belval, H. Duran, N. Saffon,
M. Daffe, M. Baltas, J. Med. Chem. 2011, 54, 1449–1461.
[133] U. Sreenivasan, R. K. Mishra, R. L. Johnson, J. Med. Chem. 1993, 36, 256–263.
[134] W. Fuller, V. Yalamoori, in Methods of organic chemistry (Houben-Weyl), Vol. E 22a
(Ed.: M. Goodman, A. Felix, L. Moroder, C. Toniolo), Georg Thieme Verlag,
Stuttgart, 4. ed., 2004.
[135] A. Prox, F. Weygand, Peptides, Proc. Eur. Peptide Symp. 8th, Noordwijk, Neth. 1967,
158–172.
[136] T. Wieland, G. Lueben, H. Ottenheym, J. Faesel, J. X. De Vries, A. Prox,
J. Schmid, Angew. Chem., Int. Ed. Engl. 1968, 7, 204–208.
[137] R. R. Burgess, in Methods in Enzymology, Vol. 463 (Guide to protein purification)
(Ed.: M. P. Burgess, Richard R. Deutscher), Elsevier/Academic Press, Oxford,
2nd ed., 2009.
[138] S. Rastogi, Biochemistry 3rd ed., Tata McGraw-Hill, Delhi, 2010.
[139] L. J. Farrugia, J. Appl. Crystallogr. 1999, 32, 837–838.
135

7 Bibliography
[140] A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, J. Appl. Crystallogr.
1993, 26, 343–50.
[141] G. M. Sheldrick, in Institut für Anorganische Chemie der Universität Göttingen.
[142] H. Duerr, K. Zengerle, H. P. Trierweiler, Z. Naturforsch., B: Chem. Sci. 1988, 43,
361–367.
[143] S. Caron, N. M. Do, J. E. Sieser, Tetrahedron Lett. 2000, 41, 2299–2302.
[144] L. Della Ciana, W. J. Dressick, A. Von Zelewsky, J. Heterocycl. Chem. 1990, 27,
163–165.
136

Appendix
A Data for crystal structures
Compound 16
Identification code 6,6’-bis(aminomethyl)-2,2’-bipyridinetrihydrobromide trihydrate
Empirical formula C12H19Br3N4OFormula weight 475.04Temperature 173(2)Wavelength 0.71073 Å
Crystal system, space group monoclinic P2(1)/nUnit cell dimensions a = 11.377(9) Å alpha = 90 deg.
b = 6.818(5) Å beta = 104.657(15) deg.c = 22.214(19) Å gamma = 90 deg.
Volume 1667(2) Å-3
Z, Calculated density 4 1.893 Mg/m3
Absorption coefficient 7.261 mm−1
F(000) 928Crystal size 0.200 x 0.200 x 0.200 mm
Theta range for data collection 2.97 to 25.00 deg.Limiting indices -13<=h<=13, -8<=k<=8, -26<=l<=26Reflections collected / unique 13687 / 2924 [R(int) = 0.1790]
Completeness to theta = 25.00 99.6%Absorption correction NoneRefinement method Full-matrix least-squares on F2
Data / restraints / parameters 2924 / 0 / 217Goodness-of-fit on F2 1.002Final R indices [I>2sigma(I)] R1 = 0.0403, wR2 = 0.1024
R indices (all data) R1 = 0.0442, wR2 = 0.1055Largest diff. peak and hole 1.121 and -0.718 e.Å-3
i

A Data for crystal structures
Table 7.1: Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (A2 x 103) for
16. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
x y z U(eq)
Br(1) 6177(1) 7095(1) 2433(1) 25(1)Br(2) 2231(1) 403(1) 1796(1) 25(1)Br(3) -4832(1) 18223(1) -885(1) 29(1)N(3) -54(3) 7251(4) 1081(2) 16(1)O(1) 2227(3) 5190(5) 1702(2) 25(1)C(3) 2404(4) 7053(5) 82(2) 19(1)N(4) 1558(3) 7163(4) 399(2) 17(1)N(1) -799(4) 7262(7) 2607(2) 25(1)N(2) 3803(3) 7225(5) 1126(2) 26(1)C(8) -494(3) 7519(5) 459(2) 17(1)
C(11) 42(4) 7534(5) -562(2) 23(1)C(12) 2143(4) 7157(5) -562(2) 25(1)C(13) -2453(4) 8036(6) 647(2) 23(1)C(14) -1729(4) 7879(5) 232(2) 20(1)C(15) -1955(4) 7753(6) 1284(2) 22(1)C(16) -47(4) 6976(6) 2161(2) 25(1)C(17) -734(3) 7357(5) 1500(2) 17(1)C(19) 386(4) 7398(5) 77(2) 16(1)C(20) 3707(4) 6820(6) 466(2) 27(1)C(26) 933(4) 7390(6) -893(2) 26(1)
Table 7.2: Bond lengths [Å] for 16
Bond lengths [Å]
N(3)-C(17) 1.354(5)N(3)-C(8) 1.357(5)C(3)-N(4) 1.330(5)C(3)-C(12) 1.389(6)C(3)-C(20) 1.519(6)N(4)-C(19) 1.354(5)N(1)-C(16) 1.478(6)N(2)-C(20) 1.469(6)C(8)-C(14) 1.389(6)C(8)-C(19) 1.470(6)C(11)-C(19) 1.377(5)C(11)-C(26) 1.398(7)C(12)-C(26) 1.395(6)C(13)-C(14) 1.387(6)C(13)-C(15) 1.398(6)C(15)-C(17) 1.377(6)C(16)-C(17) 1.502(5)
Symmetry transformations used to generate equivalent atoms
ii

Appendix
Table 7.3: Bond lengths [A] and angles [deg] for 16
Angle [deg]
C(17)-N(3)-C(8) 124.4(4)N(4)-C(3)-C(12) 123.2(4)N(4)-C(3)-C(20) 116.3(4)C(12)-C(3)-C(20) 120.5(4)C(3)-N(4)-C(19) 118.4(4)N(3)-C(8)-C(14) 118.1(4)N(3)-C(8)-C(19) 116.8(3)C(14)-C(8)-C(19) 125.0(3)C(19)-C(11)-C(26) 118.8(4)C(3)-C(12)-C(26) 118.3(4)C(14)-C(13)-C(15) 120.3(4)C(13)-C(14)-C(8) 119.2(4)C(17)-C(15)-C(13) 119.6(4)N(1)-C(16)-C(17) 112.8(4)N(3)-C(17)-C(15) 118.2(4)N(3)-C(17)-C(16) 114.6(4)C(15)-C(17)-C(16) 127.2(4)N(4)-C(19)-C(11) 122.6(4)N(4)-C(19)-C(8) 115.1(3)C(11)-C(19)-C(8) 122.3(4)N(2)-C(20)-C(3) 110.9(4)
C(12)-C(26)-C(11) 118.8(4)
iii

A Data for crystal structures
Table 7.4: Anisotropic displacement parameters (A2 x 103) for 16. The anisotropic displacement factor
exponent takes the form: -2 π2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ]
U11 U22 U33 U23 U13 U12
Br(1) 24(1) 30(1) 24(1) 2(1) 9(1) 1(1)Br(2) 29(1) 22(1) 22(1) -1(1) 3(1) -2(1)Br(3) 31(1) 35(1) 21(1) 3(1) 6(1) -6(1)N(3) 15(2) 16(2) 18(2) 0(1) 3(1) -1(1)O(1) 36(2) 20(2) 20(2) 3(1) 9(1) 2(1)C(3) 23(2) 15(2) 23(2) -2(1) 12(2) 0(1)N(4) 18(2) 15(2) 19(2) 2(1) 8(1) 1(1)N(1) 26(2) 34(2) 16(2) 0(2) 7(2) -3(2)N(2) 21(2) 26(2) 28(2) 7(2) 4(2) 1(2)C(8) 22(2) 11(2) 18(2) -3(1) 5(2) -5(1)
C(11) 29(2) 19(2) 17(2) 1(2) 2(2) -2(2)C(12) 37(3) 17(2) 25(2) -2(2) 16(2) -3(2)C(13) 15(2) 25(2) 25(2) -2(2) 0(2) 0(2)C(14) 20(2) 20(2) 18(2) 0(2) -1(2) -1(1)C(15) 22(2) 26(2) 21(2) -3(2) 10(2) -1(2)C(16) 20(2) 38(2) 19(2) 2(2) 7(2) 3(2)C(17) 20(2) 14(2) 19(2) 0(1) 9(2) -1(1)C(19) 22(2) 12(2) 16(2) -1(1) 6(2) -2(1)C(20) 21(2) 31(2) 32(2) 3(2) 11(2) 4(2)C(26) 38(3) 21(2) 18(2) -3(2) 8(2) -4(2)
iv

Appendix
Compound 69
Identification code acetylcycloserine
Empirical formula C5H8N2O3Formula weight 144.13Temperature 173(2) KWavelength 0.71075 Å
Crystal system, space group orthorhombic P212121Unit cell dimensions a = 4.985(11) Å alpha = 90 deg.
b = 9.67(2) Å beta = 90 deg.c = 13.62(3) Å gamma = 90 deg.
Volume 657(2) Å-3
Z, Calculated density 4 1.458 Mg/m3
Absorption coefficient 0.121 mm−1F(000) 304Crystal size 0.36 x 0.12 x 0.12 mm
Theta range for data collection 2.58 to 24.98 deg.Limiting indices -3<=h<=5, -11<=k<=11, -16<=l<=9Reflections collected / unique 2375 / 1137 [R(int) = 0.0292]
Completeness to theta = 24.98 99.4 %Max. and min. transmission 0.9855 and 0.9572Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 1137 / 0 / 95Goodness-of-fit on F2 1.081Final R indices [I>2sigma(I)] R1 = 0.0388, wR2 = 0.0920
R indices (all data) R1 = 0.0440, wR2 = 0.0954Absolute structure parameter 0(2)Largest diff. peak and hole 0.190 and -0.167 e.Å-3
v

A Data for crystal structures
Table 7.5: Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (A2 x 103) for
xxx. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
x y z U(eq)
O(1) 622(3) 9497(2) 1577(1) 33(1)O(2) 4515(4) 7204(2) 3257(1) 35(1)N(3) 2461(4) 6568(2) 2700(1) 27(1)N(4) 4435(4) 8813(2) 860(2) 25(1)O(5) 1302(4) 6237(2) 1113(1) 38(1)C(6) 2698(4) 6770(2) 1749(2) 23(1)C(7) 2247(4) 9600(2) 894(2) 24(1)C(8) 5005(4) 7781(2) 1599(2) 24(1)C(9) 1840(6) 10611(3) 77(2) 34(1)
C(10) 5470(5) 8303(2) 2633(2) 30(1)
Table 7.6: Bond lengths [Å] for acetylcycloserine
Bond lengths [Å]
O(1)-C(7) 1.237(3)O(2)-N(3) 1.415(3)O(2)-C(10) 1.441(3)N(3)-C(6) 1.316(4)N(3)-H(3) 0.8800N(4)-C(7) 1.330(4)N(4)-C(8) 1.446(3)N(4)-H(1) 0.86(3)O(5)-C(6) 1.225(3)C(6)-C(8) 1.523(4)C(7)-C(9) 1.496(4)
C(8)-C(10) 1.515(5)C(8)-H(8) 1.0000
C(9)-H(9A) 0.9800C(9)-H(9B) 0.9800C(9)-H(9C) 0.9800
C(10)-H(10A) 0.9900C(10)-H(10B) 0.9900
Symmetry transformations used to generate equivalent atoms
vi

Appendix
Table 7.7: Bond lengths [A] and angles [deg] for acetylcycloserine
Angle [deg]
N(3)-O(2)-C(10) 104.2(2)C(6)-N(3)-O(2) 113.4(2)C(6)-N(3)-H(3) 123.3O(2)-N(3)-H(3) 123.3C(7)-N(4)-C(8) 122.1(2)C(7)-N(4)-H(1) 117.9(18)C(8)-N(4)-H(1) 118.9(18)O(5)-C(6)-N(3) 125.6(2)O(5)-C(6)-C(8) 127.1(2)N(3)-C(6)-C(8) 107.22(19)O(1)-C(7)-N(4) 121.2(2)O(1)-C(7)-C(9) 121.5(2)N(4)-C(7)-C(9) 117.3(2)
N(4)-C(8)-C(10) 116.6(2)N(4)-C(8)-C(6) 112.9(2)C(10)-C(8)-C(6) 101.8(2)N(4)-C(8)-H(8) 108.4C(10)-C(8)-H(8) 108.4C(6)-C(8)-H(8) 108.4
C(7)-C(9)-H(9A) 109.5C(7)-C(9)-H(9B) 109.5
H(9A)-C(9)-H(9B) 109.5C(7)-C(9)-H(9C) 109.5
H(9A)-C(9)-H(9C) 109.5H(9B)-C(9)-H(9C) 109.5O(2)-C(10)-C(8) 104.6(2)
O(2)-C(10)-H(10A) 110.8C(8)-C(10)-H(10A) 110.8O(2)-C(10)-H(10B) 110.8C(8)-C(10)-H(10B) 110.8
H(10A)-C(10)-H(10B) 108.9
vii

A Data for crystal structures
Table 7.8: Anisotropic displacement parameters (A2 x 103) for acetylcycloserine. The anisotropic dis-
placement factor exponent takes the form: -2 π2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ]
U11 U22 U33 U23 U13 U12
O(1) 33(1) 32(1) 36(1) 4(1) 11(1) 7(1)O(2) 48(1) 35(1) 22(1) 6(1) -8(1) -16(1)N(3) 30(1) 28(1) 24(1) 3(1) -2(1) -10(1)N(4) 28(1) 28(1) 19(1) 6(1) 7(1) 4(1)O(5) 47(1) 38(1) 27(1) -1(1) -11(1) -8(1)C(6) 27(1) 19(1) 23(1) 0(1) -2(1) 5(1)C(7) 25(1) 21(1) 26(1) -1(1) 1(1) 0(1)C(8) 23(1) 25(1) 24(1) 4(1) 2(1) 4(1)C(9) 44(2) 31(1) 28(1) 5(1) -5(1) 5(1)C(10) 34(1) 31(1) 25(1) 5(1) -3(1) -8(1)
viii

Appendix
Compound 70
Identification code N-methyl-acetylcycloserine
Empirical formula C6 H10 N2 O3Formula weight 158.16Temperature 293(2) KWavelength 0.71075 Å
Crystal system, space group orthorhombic P212121Unit cell dimensions a = 5.2295(9) Å alpha = 90 deg.
b = 8.3281(15) Å beta = 90 deg.c = 17.934(3) Å gamma = 90 deg.
Volume 781(2) Å-3
Z, Calculated density 4 1.345 Mg/m3
Absorption coefficient 0.109 mm−1F(000) 336Crystal size 0.49 x 0.06 x 0.04 mm
Theta range for data collection 2.70 to 24.98 deg.Limiting indices -6<=h<=6, -9<=k<=9, -21<=l<=21Reflections collected / unique 6687 / 1373 [R(int) = 0.0578]
Completeness to theta = 24.98 99.6 %Max. and min. transmission 0.9962 and 0.9483Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 1373 / 0 / 140Goodness-of-fit on F2 1.075Final R indices [I>2sigma(I)] R1 = 0.0463, wR2 = 0.0969
R indices (all data) R1 = 0.0592, wR2 = 0.1042Absolute structure parameter 1(2)Largest diff. peak and hole 0.133 and -0.152 e.Å-3
ix

A Data for crystal structures
Table 7.9: Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (A2 x 103) for
xxx. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
x y z U(eq)
C(1) 461(8) 4094(5) 5584(2) 64(1)C(2) 993(5) 4683(3) 4815(1) 46(1)C(3) 3652(5) 6369(3) 4026(1) 44(1)C(4) 1435(5) 7134(3) 3613(1) 43(1)C(5) 35(7) 7082(5) 2279(2) 60(1)C(6) 4642(5) 5167(4) 3456(2) 53(1)N(1) 2949(4) 5730(3) 4738(1) 47(1)N(2) 1589(4) 6684(3) 2905(1) 49(1)O(1) -243(4) 4244(2) 4272(1) 61(1)O(2) -209(4) 8028(2) 3865(1) 63(1)O(3) 3749(4) 5724(3) 2746(1) 57(1)
Table 7.10: Bond lengths [Å] for N-methyl-acetylcycloserine
Bond lengths [Å]
C(1)-C(2) 1.490(4)C(1)-H(2) 0.99(4)C(1)-H(3) 0.92(5)C(1)-H(1) 1.00(5)C(2)-O(1) 1.224(3)C(2)-N(1) 1.351(3)C(3)-N(1) 1.432(3)C(3)-C(4) 1.516(4)C(3)-C(6) 1.521(4)C(3)-H(5) 0.90(3)C(4)-O(2) 1.223(3)C(4)-N(2) 1.326(3)C(5)-N(2) 1.425(3)C(5)-H(8) 1.00(4)C(5)-H(6) 0.98(4)C(5)-H(7) 0.96(4)C(6)-O(3) 1.434(3)C(6)-H(9) 1.01(3)
C(6)-H(10) 0.95(3)N(1)-H(4) 0.84(3)N(2)-O(3) 1.413(3)
Symmetry transformations used to generate equivalent atoms
x

Appendix
Table 7.11: Bond lengths [A] and angles [deg] for acetylcycloserine
Angle [deg]
C(2)-C(1)-H(2) 116(2)C(2)-C(1)-H(3) 115(3)H(2)-C(1)-H(3) 109(3)C(2)-C(1)-H(1) 113(2)H(2)-C(1)-H(1) 107(3)H(3)-C(1)-H(1) 94(3)O(1)-C(2)-N(1) 120.7(2)O(1)-C(2)-C(1) 122.6(3)N(1)-C(2)-C(1) 116.6(2)N(1)-C(3)-C(4) 113.3(2)N(1)-C(3)-C(6) 116.2(2)C(4)-C(3)-C(6) 102.0(2)N(1)-C(3)-H(5) 107.7(16)C(4)-C(3)-H(5) 108.7(15)C(6)-C(3)-H(5) 108.6(16)O(2)-C(4)-N(2) 124.6(2)O(2)-C(4)-C(3) 127.8(2)N(2)-C(4)-C(3) 107.6(2)N(2)-C(5)-H(8) 108(2)N(2)-C(5)-H(6) 108(2)H(8)-C(5)-H(6) 111(3)N(2)-C(5)-H(7) 110(2)H(8)-C(5)-H(7) 106(3)H(6)-C(5)-H(7) 114(3)O(3)-C(6)-C(3) 105.8(2)O(3)-C(6)-H(9) 110.8(17)C(3)-C(6)-H(9) 108.0(17)
O(3)-C(6)-H(10) 105(2)C(3)-C(6)-H(10) 111(2)H(9)-C(6)-H(10) 116(3)C(2)-N(1)-C(3) 121.7(2)C(2)-N(1)-H(4) 117(2)C(3)-N(1)-H(4) 122(2)C(4)-N(2)-O(3) 113.73(19)C(4)-N(2)-C(5) 130.8(2)O(3)-N(2)-C(5) 115.3(2)N(2)-O(3)-C(6) 105.27(18)
xi
A Data for crystal structures
Table 7.12: Anisotropic displacement parameters (A2 x 103) for acetylcycloserine. The anisotropic dis-
placement factor exponent takes the form: -2 π2 [ h2 a*2 U11 + ... + 2 h k a* b* U12 ]
U11 U22 U33 U23 U13 U12
C(1) 80(2) 58(2) 53(2) 14(2) -3(2) -8(2)C(2) 47(1) 46(1) 45(1) 4(1) -10(1) 0(1)C(3) 40(1) 51(2) 42(1) 0(1) -6(1) -3(1)C(4) 47(1) 42(1) 40(1) 3(1) 4(1) 1(1)C(5) 60(2) 82(2) 39(2) 13(2) -4(1) 13(2)C(6) 40(1) 67(2) 52(2) -3(1) -6(1) 9(1)N(1) 49(1) 58(1) 34(1) 1(1) -13(1) -10(1)N(2) 47(1) 64(2) 37(1) 0(1) 2(1) 20(1)O(1) 61(1) 67(1) 54(1) 1(1) -19(1) -16(1)O(2) 73(1) 67(1) 49(1) 1(1) 10(1) 27(1)O(3) 52(1) 78(1) 42(1) -7(1) 3(1) 24(1)
xii

Appendix
B Publication list
Publications with peer review process
A1. Understanding the quenching effects of aromatic C-H- and C-D-oscillators in near-IR
lanthanoid luminescence
C. Doffek, N. Alzakhem, C. Bischof, J. Wahsner, T. Gueden, J. Lügger, C. Platas-
Iglesias, M. Seitz*, J. Am. Chem. Soc. 2012, 134, 16413-16423.
A2. Anomalous reversal of aromatic C-H and C-D quenching efficiencies in luminescent
praseodymium cryptates
J. Scholten, G.A. Rosser, J. Wahsner, N. Alzakhem, C. Bischof, F. Stog, A. Beeby,
M. Seitz*, J. Am. Chem. Soc. 2012, 134, 13915-13917.
A3. The dependence of the photophysical properties on the number of 2,2’-bipyridine units in
a series of luminescent Europium and Terbium cryptates
N. Alzakhem, C. Bischof, M. Seitz*, Inorg. Chem. 2012, 51, 9343-9349
A4. Quantification of C-H quenching in near-IR luminescent ytterbium and neodymium
cryptates
C. Bischof, J. Wahsner, J. Scholten, S. Trosien, M. Seitz* , J. Am. Chem. Soc. 2010,
132, 14334-143353.
A5. A [4+2] mixed ligand approach to ruthenium DNA metallointercalators [Ru(tpa)(N-
N)](PF6)2 using a tris(2-pyridylmethyl)amine (tpa) capping ligand
S. Seeberg (nee Kraft), C. Bischof, A. Loos, S. Braun, N. Jafarova, U. Schatz-
schneider*, J. Inorg. Biochem. 2009, 103, 1126-1134.
A6. [N,N’-Bis(salicylidene)-1,2-phenylenediamine]metal complexes with cell death promoting
properties
A. Hille, I. Ott, A. Kitanovic, I. Kitanovic, H. Alborzina, E. Lederer, S. Wölfl,
N. Metzler-Nolte, S. Schäfer, W.S. Sheldrick, C. Bischof, U. Schatzschneider, R.
Gust*, J. Biol. Inorg. Chem. 2009, 14, 711-725.
The following above mentioned publications have evolved from my doctoral disserta-
tion: A1, A2, A3 and A4.
xiii

B Publication list
Publications without peer review process
Conference talks
B1. C. Bischof: Selective deuteration of Lehn cryptands and its influence on near-IR lumin-
escent lanthanides. Utrecht University and IMPRS Joint Symposium on Chemical
Biology in Utrecht, Netherlands. May 2011.
B2. C. Bischof: Quantification of C-H quenching in near-IR luminescent lanthanide crypt-
ates. 7th Coordination Chemistry Meeting in Stuttgart, Germany. March 2011.
B3. C. Bischof: Synthesis of ruthenium CO releasing molecules and their behaviour un-
der physiological conditions. 12th JCF Spring Symposium in Goettingen, Germany.
March 2010.
B4. C. Bischof: Synthesis and characterisation of ruthenium complexes with photolabile CO
and NO ligands. 4th workshop of DFG research group 630 (“Biological function of
organometallic compounds”) in Bergisch Gladbach, Germany. October 2007.
Conference posters
C1. C. Bischof: UnClicking biorelevant bonds with a chemical trigger. Challenges in Or-
ganic Chemistry and Chemical Biology (ISACS7) in Edinburgh, Scotland. June
2012.
C2. C. Bischof: Quantification of C-H quenching in near-IR luminescent lanthanide crypt-
ates. 242nd ACS National Meeting in Denver/CO, USA. September 2011.
C3. C. Bischof: Selective Deuteration of Lanthanoid Bipyridine Cryptates. 39th Interna-
tional Conference on Coordination Chemistry in Adelaide, Australia. July 2010.
The following above mentioned publications have evolved from my doctoral disserta-
tion: B1, B2, C1, C2 and C3
xiv





























