Embed Size (px)
Citation preview

Cancer Letters 332 (2013) 83–93
Contents lists available at SciVerse ScienceDirect
Cancer Letters
journal homepage: www.elsevier .com/locate /canlet
Brucine, an indole alkaloid from Strychnos nux-vomica attenuatesVEGF-induced angiogenesis via inhibiting VEGFR2 signaling pathwayin vitro and in vivo
0304-3835/$ - see front matter � 2013 Elsevier Ireland Ltd. All rights reserved.http://dx.doi.org/10.1016/j.canlet.2013.01.012
⇑ Corresponding author. Tel.: +91 011 29554327; fax: +91 011 29554503.E-mail address: [email protected] (S. Saraswati).
Sarita Saraswati a,⇑, S.S. Agrawal b
a Genome Research Laboratory, Delhi Institute of Pharmaceutical Sciences and Research, Pushp Vihar Sec-3, MB Road, New Delhi 110 017, Indiab Amity University, Sector-125, Noida – 201 303 (UP), India
a r t i c l e i n f o a b s t r a c t
Article history:Received 26 November 2012Received in revised form 8 January 2013Accepted 9 January 2013
Keywords:Vascular endothelial growth factorreceptor-2SignalingAngiogenesisMatrigel
In this study, we investigated the mechanism of brucine in tumor angiogenesis. We found that brucineinhibits VEGF-induced cell proliferation, chemotactic motility, and the formation of capillary-likestructures in HUVECs in a dose-dependent manner. Brucine suppresses VEGF- induced p-VEGFR2 kinaseactivity and inhibits neovascularization in vivo. Brucine inhibits the downstream protein kinases ofVEGFR2, including Src, FAK, ERK, AKT and mTOR. And further downregulates levels of VEGF, NO, IL-6,IL-8, TNF-a and IFN-c in HUVECs. Taken together, our study suggests that brucine potently suppressesangiogenesis by targeting VEGFR2 activation and may be a viable drug candidate in anti-angiogenesisand anti-cancer therapies.
� 2013 Elsevier Ireland Ltd. All rights reserved.
1. Introduction VEGF is viewed as an attractive therapeutic target for the devel-
Angiogenesis, the formation of new vessels from pre-existingvasculature, is an important mechanism used by tumors to pro-mote growth and metastasis [1] and is tightly regulated by an intri-cate balance between stimulators and inhibitors [2]. Out of thenumerous growth factors and cytokines that have been shownangiogenic effects, vascular endothelial growth factor (VEGF), aglycoprotein that has mitogenic activity on vascular endothelialcells, is one of the most critical and specific angiogenic factors reg-ulating normal physiological and pathological neovascularizationsuch as tumor angiogenesis [3]. VEGF exerts its biological actionsby binding to its two receptor tyrosine kinases expressed on endo-thelial cells, namely, VEGFR1 (Flt-1), required mainly for mitogenicand chemotactic responses and VEGFR2 (KDR/Flk-1), which con-tributes to endothelial cell morphogenesis [4]. VEGFRs activationlead to the activation of diverse intracellular signaling molecules,including Src family kinase [5], focal adhesion kinase (FAK) [6,7]extracellular signal-regulated kinases (ERKs), phosphoinositide 3-kinase/AKT kinase, and mammalian target of rapamycin (mTOR)/ribosomal protein S6 kinase (p70S6K) [8] that promote the prolif-eration, migration, differentiation, and survival of endothelial cellsin the pre-existing vasculature. Thus, VEGFR2 and those intracellu-lar signaling molecules appear to be critical targets for the suppres-sion of tumor angiogenesis [9].
opment of novel anticancer agents [3], and numbers of clinical tri-als focused on providing the effect of anti-angiogenic therapies areongoing worldwide in parallel. The National Institutes of Health(NIH) website provides a basic summary of anti-angiogenic drugsthat were or are still currently under clinical investigations(http://www.cancer.gov/clinicaltrials/developments/antiangio-ta-ble). These include monoclonal antibodies targeting VEGF ligandsor VEGFRs [10], soluble receptors that sequester ligands [11] andsmall molecule inhibitors that inhibit kinase activity [12]. Threedrugs developed for their anti-angiogenic actions, bevacizumab(Avastin�), sunitinib malate (Sutent�, SU11248) and sorafenib(Nexavar�, BAY 43-9006), have been approved by the United StatesFood and Drug Administration for treatment of patients with spe-cific types of cancer – all three inhibit VEGF signaling by blockingVEGF ligand or VEGFR [13]. However, serious side effects, such ashypertension, bleeding and gastrointestinal perforation, have beenassociated with currently available anti-VEGF agents, limiting theirchronic use [13]. Hence, there is an urgent need to find a moleculethat can be more specific and less toxic for the treatment of cancer,particularly agents that exhibit activity against drug-resistantstrains, completely sterilize the infection, or shorten the durationof drug therapy and thus promote drug compliance.
Brucine (Fig. 1A), a natural plant alkaloid, isolated from seeds ofStrychnos nux-vomica L. (Loganiaceae), has been shown to possess avariety of biological activities [14]. Preliminary studies suggestcytotoxic [14,15], anti-proliferative [16], antitumor [14] and
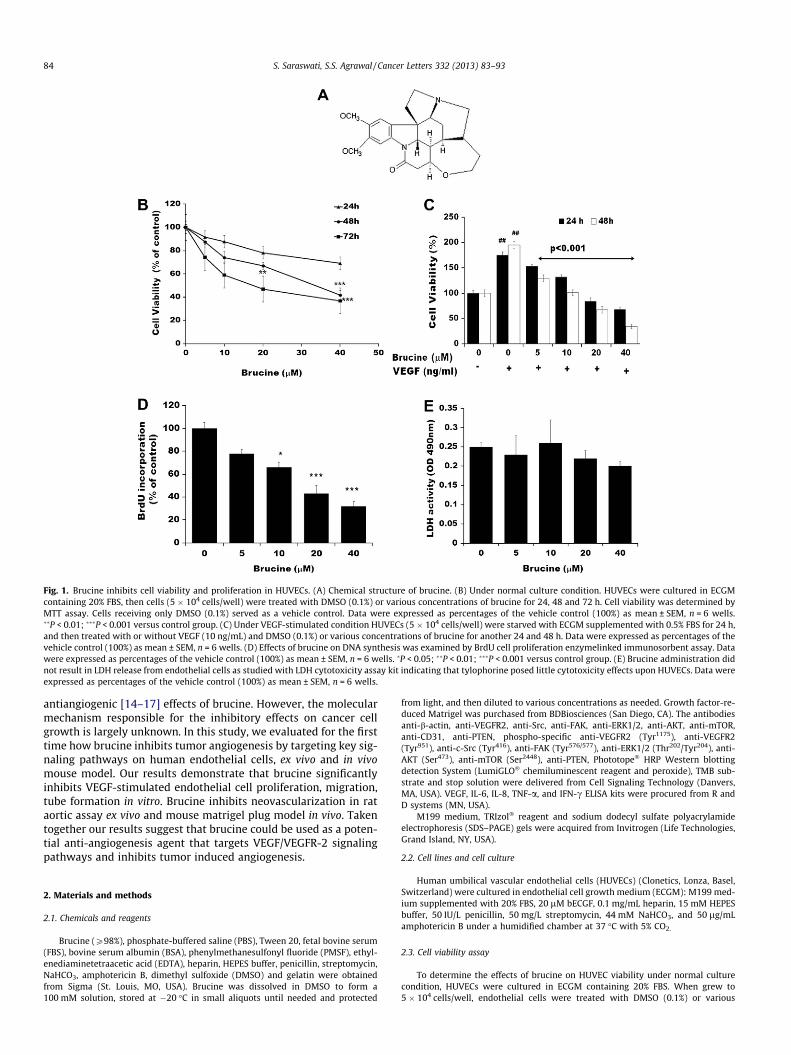
Fig. 1. Brucine inhibits cell viability and proliferation in HUVECs. (A) Chemical structure of brucine. (B) Under normal culture condition. HUVECs were cultured in ECGMcontaining 20% FBS, then cells (5 � 104 cells/well) were treated with DMSO (0.1%) or various concentrations of brucine for 24, 48 and 72 h. Cell viability was determined byMTT assay. Cells receiving only DMSO (0.1%) served as a vehicle control. Data were expressed as percentages of the vehicle control (100%) as mean ± SEM, n = 6 wells.��P < 0.01; ���P < 0.001 versus control group. (C) Under VEGF-stimulated condition HUVECs (5 � 104 cells/well) were starved with ECGM supplemented with 0.5% FBS for 24 h,and then treated with or without VEGF (10 ng/mL) and DMSO (0.1%) or various concentrations of brucine for another 24 and 48 h. Data were expressed as percentages of thevehicle control (100%) as mean ± SEM, n = 6 wells. (D) Effects of brucine on DNA synthesis was examined by BrdU cell proliferation enzymelinked immunosorbent assay. Datawere expressed as percentages of the vehicle control (100%) as mean ± SEM, n = 6 wells. �P < 0.05; ��P < 0.01; ���P < 0.001 versus control group. (E) Brucine administration didnot result in LDH release from endothelial cells as studied with LDH cytotoxicity assay kit indicating that tylophorine posed little cytotoxicity effects upon HUVECs. Data wereexpressed as percentages of the vehicle control (100%) as mean ± SEM, n = 6 wells.
84 S. Saraswati, S.S. Agrawal / Cancer Letters 332 (2013) 83–93
antiangiogenic [14–17] effects of brucine. However, the molecularmechanism responsible for the inhibitory effects on cancer cellgrowth is largely unknown. In this study, we evaluated for the firsttime how brucine inhibits tumor angiogenesis by targeting key sig-naling pathways on human endothelial cells, ex vivo and in vivomouse model. Our results demonstrate that brucine significantlyinhibits VEGF-stimulated endothelial cell proliferation, migration,tube formation in vitro. Brucine inhibits neovascularization in rataortic assay ex vivo and mouse matrigel plug model in vivo. Takentogether our results suggest that brucine could be used as a poten-tial anti-angiogenesis agent that targets VEGF/VEGFR-2 signalingpathways and inhibits tumor induced angiogenesis.
2. Materials and methods
2.1. Chemicals and reagents
Brucine (P98%), phosphate-buffered saline (PBS), Tween 20, fetal bovine serum(FBS), bovine serum albumin (BSA), phenylmethanesulfonyl fluoride (PMSF), ethyl-enediaminetetraacetic acid (EDTA), heparin, HEPES buffer, penicillin, streptomycin,NaHCO3, amphotericin B, dimethyl sulfoxide (DMSO) and gelatin were obtainedfrom Sigma (St. Louis, MO, USA). Brucine was dissolved in DMSO to form a100 mM solution, stored at �20 �C in small aliquots until needed and protected
from light, and then diluted to various concentrations as needed. Growth factor-re-duced Matrigel was purchased from BDBiosciences (San Diego, CA). The antibodiesanti-b-actin, anti-VEGFR2, anti-Src, anti-FAK, anti-ERK1/2, anti-AKT, anti-mTOR,anti-CD31, anti-PTEN, phospho-specific anti-VEGFR2 (Tyr1175), anti-VEGFR2(Tyr951), anti-c-Src (Tyr416), anti-FAK (Tyr576/577), anti-ERK1/2 (Thr202/Tyr204), anti-AKT (Ser473), anti-mTOR (Ser2448), anti-PTEN, Phototope� HRP Western blottingdetection System (LumiGLO� chemiluminescent reagent and peroxide), TMB sub-strate and stop solution were delivered from Cell Signaling Technology (Danvers,MA, USA). VEGF, IL-6, IL-8, TNF-a, and IFN-c ELISA kits were procured from R andD systems (MN, USA).
M199 medium, TRIzol� reagent and sodium dodecyl sulfate polyacrylamideelectrophoresis (SDS–PAGE) gels were acquired from Invitrogen (Life Technologies,Grand Island, NY, USA).
2.2. Cell lines and cell culture
Human umbilical vascular endothelial cells (HUVECs) (Clonetics, Lonza, Basel,Switzerland) were cultured in endothelial cell growth medium (ECGM): M199 med-ium supplemented with 20% FBS, 20 lM bECGF, 0.1 mg/mL heparin, 15 mM HEPESbuffer, 50 IU/L penicillin, 50 mg/L streptomycin, 44 mM NaHCO3, and 50 lg/mLamphotericin B under a humidified chamber at 37 �C with 5% CO2.
2.3. Cell viability assay
To determine the effects of brucine on HUVEC viability under normal culturecondition, HUVECs were cultured in ECGM containing 20% FBS. When grew to5 � 104 cells/well, endothelial cells were treated with DMSO (0.1%) or various

S. Saraswati, S.S. Agrawal / Cancer Letters 332 (2013) 83–93 85
concentrations of brucine (5, 10, 20, 40 lM) for 24, 48 and 72 h. Cell viability wasdetermined by MTT assay as described previously (14). Three independent experi-ments were performed from six replicates of each experiments.
Next, we determine the effects of brucine on HUVEC viability under VEGF-in-duced condition. HUVECs (5 � 104 cells/well) were starved with ECGM containing0.5% FBS for 24 h. After the pre-incubation, cells were treated with or without VEGF(10 ng/mL) and DMSO (0.1%) or various concentrations of brucine (5, 10, 20, 40 lM)and incubated for another 24 and 48 h. Cell viability was also quantified by MTT as-say. Cells receiving only DMSO (0.1%) served as a vehicle control. Inhibition percent-age was expressed as percentage of the vehicle control (100%). The results were themeans calculated from six replicates of each experiment. Three independent exper-iments were performed.
2.4. BrdU incorporation assay
DNA synthesis was determined by bromodeoxyuridine(BrdU) labeling assayusing Cell Proliferation ELISA, BrdU (colorimetric) kit. In brief, 5 � 104 HUVECsper well (100 lL) in ECGM + 20% FBS) were seeded in a gelatin coated for overnightattachment. Then the cultivated medium was replaced with serum-free mediumsupplemented with 10 ng/mL VEGF as well as different concentrations of brucine(5, 10, 20, 40 lM) in a final volume of 100 lL/well. After 24 h, cells were labeledwith BrdU (2 h, 37 �C, 4 h), incubated with Fix Denat solution (30 min, 20 �C), andreincubated with Anti-BrdU POD (90 min, 20 �C). The absorbance was read at450 nm in a microplate reader (Biorad, USA). The assay was repeated three timesindependently.
2.5. Lactate dehydrogenase (LDH) toxicity assay
The LDH release assay was performed using a cytotoxicity detection kit plus(LDH) (Roche Diagnostics) according to the manufacturer’s instructions. In brief,HUVECs were seeded in 96-well plate at a density of 5 � 104 cells per well. Afterincubation with various concentrations of brucine (5, 10, 20, 40 lM) for 24 h, cellsupernatants were collected and analyzed. The absorbance of formed formazanwas read at 490 nm on a microplate reader (Biorad, USA). The LDH content of eachsample was calculated according to the following formula: Cytotoxicity(%) = [(experimental value � low control)/(high control � low control)] � 100. Theassay was repeated three times independently.
2.6. Endothelial cell migration assay: wound healing
HUVECs were allowed to grow to full confluence in 6-well plates pre-coatedwith 0.1% gelatin and then starved with ECGM containing 0.5% FBS for 6 h to inac-tivate cell proliferation. After that, cells were wounded with pipette tips andwashed with PBS. ECGM supplemented with 0.5% FBS was added into the wells withor without VEGF (10 ng/mL) and DMSO (0.1%) or various concentration of brucine(5, 10, 20, 40 lM). Images of cells were taken using an inverted microscope (EclipseTS100, Nikon, Japan) at 100 �magnification after 10 h of incubation in an humidi-fied atmosphere with 5% CO2 at 37 �C. The migrated cells were observed from threerandomly selected fields and quantified by manual counting. Cells receiving onlyDMSO (0.1%) served as a vehicle control. Inhibition percentage was expressed aspercentage of the vehicle control (100%). The assay was repeated three timesindependently.
2.7. Endothelial cell invasion assay
Cell invasion assay was performed using Transwell chambers with 6.5 mmdiameter polycarbonate membrane (8-lm-sized pores). Upper surfaces ofTranswell inserts were coated with matrigel. The bottom chamber of the apparatuscontained 600 lL of endothelial cell medium supplemented with 10 ng/mL VEGF orbrucine at various concentrations (5, 10, 20, 40 lM). The HUVECs (100 lL) wereadded to the upper chamber (2 � 104 cells/well) and incubated in endothelial cellmedium. After 24 h incubation at 37 �C, non-invasive cells on the upper membranesurfaces were removed by wiping with cotton swabs. Cell invasion was quantifiedby counting cells on the lower surface using phase contrast microscope (EclipseTS100, Nikon, Japan) at 100 �magnification. The results were the means calculatedfrom three replicates of each experiment. The assay was repeated three timesindependently.
2.8. Endothelial cell capillary-like tube formation assay
Matrigel™ basement membrane matrix (growth factor reduced) (BD Biosci-ences, San Jose, CA, USA) was thawed at 4 �C, pipetted into pre-chilled 24-wellplates (100 lL matrigel/well), and incubated at 37 �C for 45 min. HUVECs werefirstly incubated in ECGM supplemented with 0.5% FBS for 10 h and then treatedwith DMSO (0.1%) or various concentrations of brucine (5, 10, 20, 40 lM), for30 min before seeding. Cells were collected and placed onto the layer of matrigel(5 � 104 cells/well) in 1 mL of ECGM supplemented with 0.5% FBS, followed bythe addition of VEGF (10 ng/mL). After 6 h of incubation with 5% CO2 at 37 �C, thenetwork-like structures of endothelial cells were examined under an inverted
microscope (Eclipse TS100, Nikon, Japan) at 100 �magnification. The tube-likestructures were defined as endothelial cord formations that were connected at bothends [18]. Branching points in three random fields per well was quantified by man-ual counting. Cells receiving only DMSO (0.1%) served as a vehicle control. Inhibi-tion percentage was expressed as percentage of the vehicle control (100%). Theassay was repeated three times independently.
2.9. Total RNA extraction, reverse transcription and quantitative real-time PCR
The effects of brucine on VEGFR2 were determined by quantitative real-time PCR(qRT-PCR). HUVECs (5 � 104 cells/well) were seeded in 24-well plates, and starvedwith ECGM containing 0.5% FBS for 24 h. After the pre-incubation, cells were treatedwith or without VEGF (50 ng/mL) and DMSO (0.1%) or various concentrations of bru-cine (5, 10, 20, 40 lM) for another 24 h. Then cells were harvested in TRIzol� reagent,and their RNA was extracted, reconstituted in DEPC-treated water, and checked forintegrity by agarose-gel electrophoresis. RNA samples were quantified at OD260/
280, and RNA was introduced to reverse transcribe to single-stranded cDNA usingPrimeScript™ RT reagent kit (TaKaRa, Otsu, Shiga, Japan), followed by qTR-PCR usingthe SYBR� Premix Ex Taq™ (TaKaRa, Otsu, Shiga, Japan) in Mastercycler� ep gradientrealplex2 Real-Time PCR System (Eppendorf, Hamburg, Germany).
The reverse-transcribed RNA was primed with oligonucleotides specific forVEGFR2 (Flk-1) (forward: 50-GACTGTGGCGAAGTGTTTTTGA-30 and reverse: 50-GTGCAGGGGAGGGTTGGCGTAG-30), and b-actin (forward: 50-GTGCGGGACATCAAG-GAGAA-30 and reverse: 50-AGGAAGGAGGGCTGGAAGAG-30) (Applied Biosystems,Carlsbad, CA, USA).
The PCR program was set as below: 94 �C, 3 min, (94 �C, 30 s; 55 �C, 30 s; 72 �C;1 min; read plate) � 30 cycles; 72 �C for 5 min. A standard curve for each gene wasgenerated to monitor amplification efficiency and to relatively quantify mRNAabundance. mRNA abundance was normalized to b-actin levels and expressed aspercentage of the vehicle control (100%) for statistical analysis. Cells receiving onlyDMSO (0.1%) served as a vehicle control. Three independent experiments were per-formed in triplicates.
2.10. In vitro VEGFR2 kinase inhibition assay
The ability of brucine to inhibit VEGFR2 tyrosine kinase activity was assayedusing HTScan� VEGFR2 kinase assay kit (Cell Signaling Technology, Danvers, MA,USA). 4 � reaction cocktail containing VEGFR2 was incubated with brucine (5, 10,20, 40 lM), or DMSO (0.1%) for 5 min at room temperature, and then 2 � ATP/sub-strate peptide cocktail was added to the pre-incubated reaction cocktail/brucine orDMSO (0.1%). After incubation at room temperature for 30 min, the reaction wasstopped and transferred to a 96-well streptavidin-coated plate (PerkinElmer LifeSciences, Boston, MA, USA), and incubated for 1 h at room temperature. Primaryantibody [phosphorylated tyrosine monoclonal antibody [(pTyr-100), 1:1000 inPBS/T with 1% bovine serum albumin (BSA)] was added into per well until the wellswere washed thrice with PBS/T (1 � PBS, 0.05% Tween-20). After incubated at roomtemperature for 1 h, phosphorylation of the substrate was monitored with HRP-la-beled anti-mouse IgG antibody (1:500 in PBS/T with 1% BSA), followed by a chromo-genic reaction. Finally, the VEGFR2 kinase assay was detected at 450 nm withmicroplate reader (Biorad, USA). The reaction processed with only DMSO (0.1%)served as a vehicle control. The results were expressed as percent kinase activityof the vehicle control (100%), and IC50 was defined as the compound concentrationthat resulted in 50% inhibition of enzyme activity. The kinase assay was performedthrice independently.
2.11. Cytokine immunoassays
IL-6, IL-8, TNF-a, IFN-c and VEGF were measured by sandwich ELISA followingthe manufacturer’s instructions (R and D Systems, MN, USA). Briefly, serum starvedHUVECs were treated with brucine (40 lM), before stimulation with VEGF (10 ng/mL), and supernatants were harvested after 24 h for cytokine assays. Absorbancewas determined using a microplate reader (Biorad, USA) at 450 nm.
2.12. Nitric oxide (NO) measurement
The NO levels in the HUVECs were measured with Nitric oxide colorimetric as-say kit (Biovision, USA). Briefly, the HUVECs were cultured in 6-well plates. Afterovernight incubation, the cells were treated with brucine (40 lM) before stimula-tion with VEGF (10 ng/mL). After 24 h, the supernatant was collected, and NO pro-duction was determined following manufacturer’s protocol.
2.13. Western blotting analysis
To determine the effects of brucine on VEGFR2-mediated signaling cascade, HU-VECs were firstly starved in ECGM containing 0.5% FBS for 12 h. After being washedwith fresh medium, cells were treated with DMSO (0.1%) or various concentrationsof brucine (10, 20, 40 lM), for 30 min, followed by the stimulation with 50 ng/mL ofVEGF for 2 min (for VEGFR2 phosphorylation) or 20 min (for AKT/mTOR pathwayphosphorylation and ERK pathway phosphorylation). The whole-cell extracts were
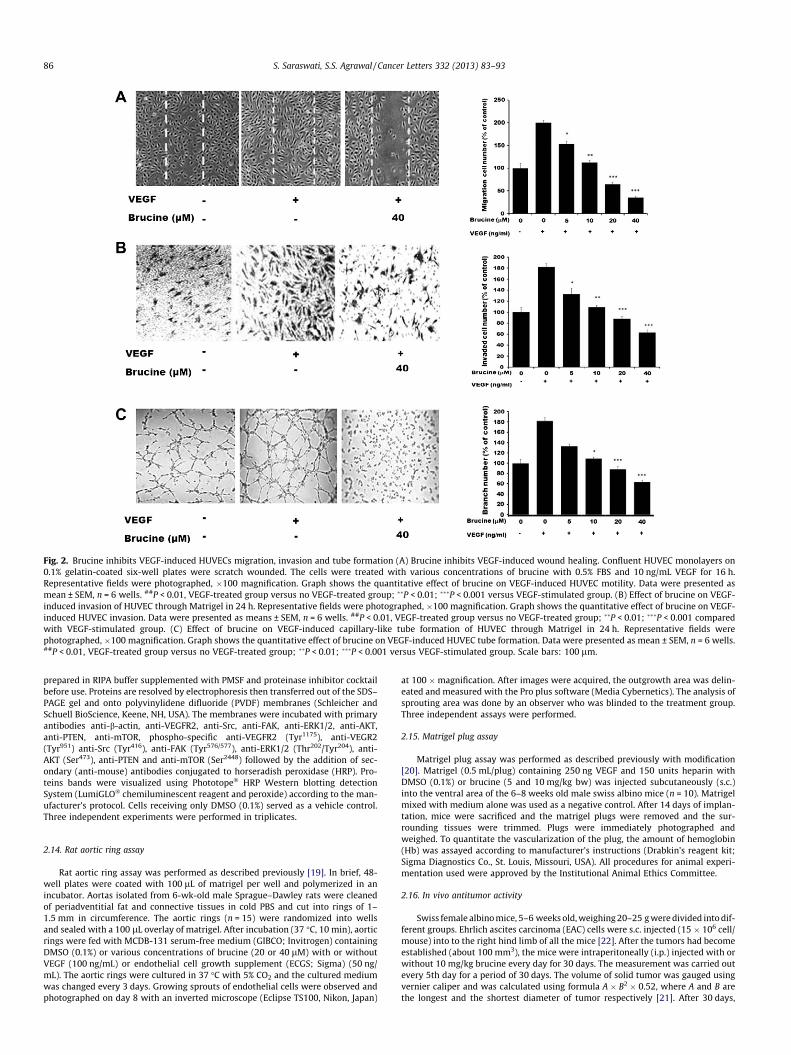
Fig. 2. Brucine inhibits VEGF-induced HUVECs migration, invasion and tube formation (A) Brucine inhibits VEGF-induced wound healing. Confluent HUVEC monolayers on0.1% gelatin-coated six-well plates were scratch wounded. The cells were treated with various concentrations of brucine with 0.5% FBS and 10 ng/mL VEGF for 16 h.Representative fields were photographed, �100 magnification. Graph shows the quantitative effect of brucine on VEGF-induced HUVEC motility. Data were presented asmean ± SEM, n = 6 wells. ##P < 0.01, VEGF-treated group versus no VEGF-treated group; ��P < 0.01; ���P < 0.001 versus VEGF-stimulated group. (B) Effect of brucine on VEGF-induced invasion of HUVEC through Matrigel in 24 h. Representative fields were photographed, �100 magnification. Graph shows the quantitative effect of brucine on VEGF-induced HUVEC invasion. Data were presented as means ± SEM, n = 6 wells. ##P < 0.01, VEGF-treated group versus no VEGF-treated group; ��P < 0.01; ���P < 0.001 comparedwith VEGF-stimulated group. (C) Effect of brucine on VEGF-induced capillary-like tube formation of HUVEC through Matrigel in 24 h. Representative fields werephotographed, �100 magnification. Graph shows the quantitative effect of brucine on VEGF-induced HUVEC tube formation. Data were presented as mean ± SEM, n = 6 wells.##P < 0.01, VEGF-treated group versus no VEGF-treated group; ��P < 0.01; ���P < 0.001 versus VEGF-stimulated group. Scale bars: 100 lm.
86 S. Saraswati, S.S. Agrawal / Cancer Letters 332 (2013) 83–93
prepared in RIPA buffer supplemented with PMSF and proteinase inhibitor cocktailbefore use. Proteins are resolved by electrophoresis then transferred out of the SDS–PAGE gel and onto polyvinylidene difluoride (PVDF) membranes (Schleicher andSchuell BioScience, Keene, NH, USA). The membranes were incubated with primaryantibodies anti-b-actin, anti-VEGFR2, anti-Src, anti-FAK, anti-ERK1/2, anti-AKT,anti-PTEN, anti-mTOR, phospho-specific anti-VEGFR2 (Tyr1175), anti-VEGR2(Tyr951) anti-Src (Tyr416), anti-FAK (Tyr576/577), anti-ERK1/2 (Thr202/Tyr204), anti-AKT (Ser473), anti-PTEN and anti-mTOR (Ser2448) followed by the addition of sec-ondary (anti-mouse) antibodies conjugated to horseradish peroxidase (HRP). Pro-teins bands were visualized using Phototope� HRP Western blotting detectionSystem (LumiGLO� chemiluminescent reagent and peroxide) according to the man-ufacturer’s protocol. Cells receiving only DMSO (0.1%) served as a vehicle control.Three independent experiments were performed in triplicates.
2.14. Rat aortic ring assay
Rat aortic ring assay was performed as described previously [19]. In brief, 48-well plates were coated with 100 lL of matrigel per well and polymerized in anincubator. Aortas isolated from 6-wk-old male Sprague–Dawley rats were cleanedof periadventitial fat and connective tissues in cold PBS and cut into rings of 1–1.5 mm in circumference. The aortic rings (n = 15) were randomized into wellsand sealed with a 100 lL overlay of matrigel. After incubation (37 �C, 10 min), aorticrings were fed with MCDB-131 serum-free medium (GIBCO; Invitrogen) containingDMSO (0.1%) or various concentrations of brucine (20 or 40 lM) with or withoutVEGF (100 ng/mL) or endothelial cell growth supplement (ECGS; Sigma) (50 ng/mL). The aortic rings were cultured in 37 �C with 5% CO2 and the cultured mediumwas changed every 3 days. Growing sprouts of endothelial cells were observed andphotographed on day 8 with an inverted microscope (Eclipse TS100, Nikon, Japan)
at 100 �magnification. After images were acquired, the outgrowth area was delin-eated and measured with the Pro plus software (Media Cybernetics). The analysis ofsprouting area was done by an observer who was blinded to the treatment group.Three independent assays were performed.
2.15. Matrigel plug assay
Matrigel plug assay was performed as described previously with modification[20]. Matrigel (0.5 mL/plug) containing 250 ng VEGF and 150 units heparin withDMSO (0.1%) or brucine (5 and 10 mg/kg bw) was injected subcutaneously (s.c.)into the ventral area of the 6–8 weeks old male swiss albino mice (n = 10). Matrigelmixed with medium alone was used as a negative control. After 14 days of implan-tation, mice were sacrificed and the matrigel plugs were removed and the sur-rounding tissues were trimmed. Plugs were immediately photographed andweighed. To quantitate the vascularization of the plug, the amount of hemoglobin(Hb) was assayed according to manufacturer’s instructions (Drabkin’s reagent kit;Sigma Diagnostics Co., St. Louis, Missouri, USA). All procedures for animal experi-mentation used were approved by the Institutional Animal Ethics Committee.
2.16. In vivo antitumor activity
Swiss female albino mice, 5–6 weeks old, weighing 20–25 g were divided into dif-ferent groups. Ehrlich ascites carcinoma (EAC) cells were s.c. injected (15 � 106 cell/mouse) into to the right hind limb of all the mice [22]. After the tumors had becomeestablished (about 100 mm3), the mice were intraperitoneally (i.p.) injected with orwithout 10 mg/kg brucine every day for 30 days. The measurement was carried outevery 5th day for a period of 30 days. The volume of solid tumor was gauged usingvernier caliper and was calculated using formula A � B2 � 0.52, where A and B arethe longest and the shortest diameter of tumor respectively [21]. After 30 days,
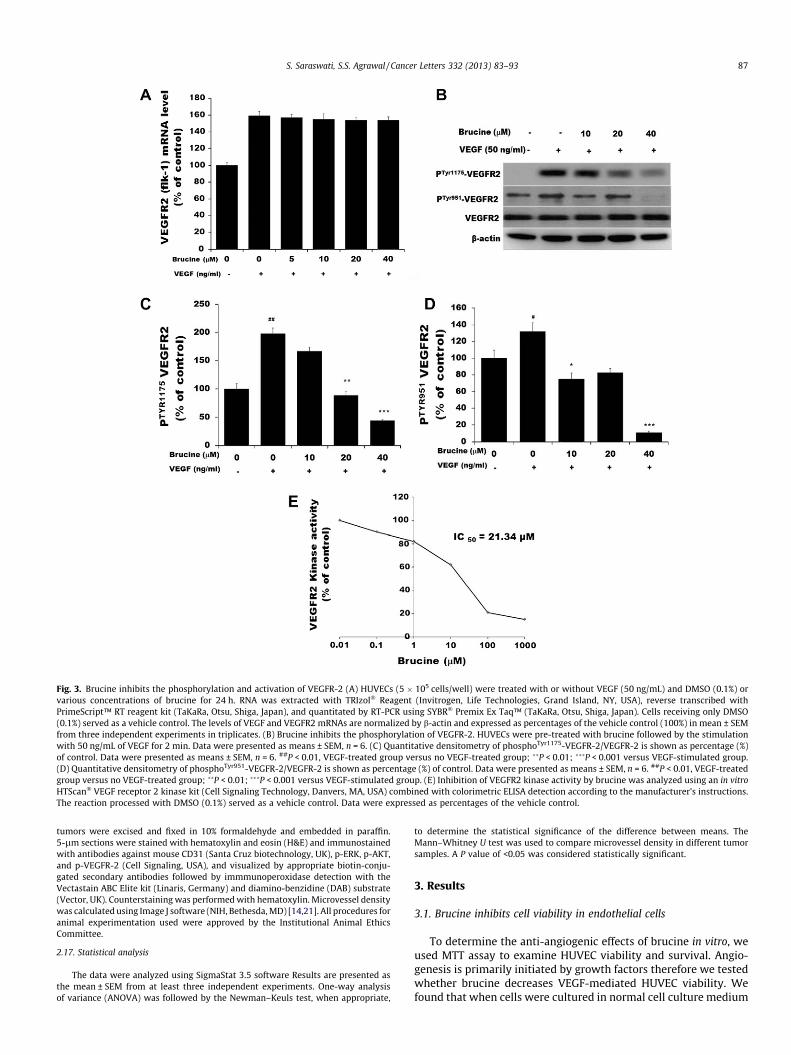
Fig. 3. Brucine inhibits the phosphorylation and activation of VEGFR-2 (A) HUVECs (5 � 105 cells/well) were treated with or without VEGF (50 ng/mL) and DMSO (0.1%) orvarious concentrations of brucine for 24 h. RNA was extracted with TRIzol� Reagent (Invitrogen, Life Technologies, Grand Island, NY, USA), reverse transcribed withPrimeScript™ RT reagent kit (TaKaRa, Otsu, Shiga, Japan), and quantitated by RT-PCR using SYBR� Premix Ex Taq™ (TaKaRa, Otsu, Shiga, Japan). Cells receiving only DMSO(0.1%) served as a vehicle control. The levels of VEGF and VEGFR2 mRNAs are normalized by b-actin and expressed as percentages of the vehicle control (100%) in mean ± SEMfrom three independent experiments in triplicates. (B) Brucine inhibits the phosphorylation of VEGFR-2. HUVECs were pre-treated with brucine followed by the stimulationwith 50 ng/mL of VEGF for 2 min. Data were presented as means ± SEM, n = 6. (C) Quantitative densitometry of phosphoTyr1175-VEGFR-2/VEGFR-2 is shown as percentage (%)of control. Data were presented as means ± SEM, n = 6. ##P < 0.01, VEGF-treated group versus no VEGF-treated group; ��P < 0.01; ���P < 0.001 versus VEGF-stimulated group.(D) Quantitative densitometry of phosphoTyr951-VEGFR-2/VEGFR-2 is shown as percentage (%) of control. Data were presented as means ± SEM, n = 6. ##P < 0.01, VEGF-treatedgroup versus no VEGF-treated group; ��P < 0.01; ���P < 0.001 versus VEGF-stimulated group. (E) Inhibition of VEGFR2 kinase activity by brucine was analyzed using an in vitroHTScan� VEGF receptor 2 kinase kit (Cell Signaling Technology, Danvers, MA, USA) combined with colorimetric ELISA detection according to the manufacturer’s instructions.The reaction processed with DMSO (0.1%) served as a vehicle control. Data were expressed as percentages of the vehicle control.
S. Saraswati, S.S. Agrawal / Cancer Letters 332 (2013) 83–93 87
tumors were excised and fixed in 10% formaldehyde and embedded in paraffin.5-lm sections were stained with hematoxylin and eosin (H&E) and immunostainedwith antibodies against mouse CD31 (Santa Cruz biotechnology, UK), p-ERK, p-AKT,and p-VEGFR-2 (Cell Signaling, USA), and visualized by appropriate biotin-conju-gated secondary antibodies followed by immmunoperoxidase detection with theVectastain ABC Elite kit (Linaris, Germany) and diamino-benzidine (DAB) substrate(Vector, UK). Counterstaining was performed with hematoxylin. Microvessel densitywas calculated using Image J software (NIH, Bethesda, MD) [14,21]. All procedures foranimal experimentation used were approved by the Institutional Animal EthicsCommittee.
2.17. Statistical analysis
The data were analyzed using SigmaStat 3.5 software Results are presented asthe mean ± SEM from at least three independent experiments. One-way analysisof variance (ANOVA) was followed by the Newman–Keuls test, when appropriate,
to determine the statistical significance of the difference between means. TheMann–Whitney U test was used to compare microvessel density in different tumorsamples. A P value of <0.05 was considered statistically significant.
3. Results
3.1. Brucine inhibits cell viability in endothelial cells
To determine the anti-angiogenic effects of brucine in vitro, weused MTT assay to examine HUVEC viability and survival. Angio-genesis is primarily initiated by growth factors therefore we testedwhether brucine decreases VEGF-mediated HUVEC viability. Wefound that when cells were cultured in normal cell culture medium

Fig. 4. Brucine inhibits the activation of VEGFR2-mediated downstream signaling.HUVECs were pre-treated with brucine followed by the stimulation with 50 ng/mLof VEGF for 2 min (for VEGFR2 phosphorylation) or 20 min (for AKT/mTOR pathwayphosphorylation and ERK pathway phosphorylation). Protein was extracted andwestern blot analysis was performed using anti-p-VEGFR-2, VEGFR-2, p-AKT, AKT,p-ERK1/2, ERK1/2, p-mTOR, mTOR, p-FAK, FAK, p-PTEN, PTEN, pSrc, Src. Data werepresented as means ± SEM, n = 6. Three independent experiments were performedin triplicates.
88 S. Saraswati, S.S. Agrawal / Cancer Letters 332 (2013) 83–93
(ECGM supplemented with 20% FBS) in absence of VEGF, brucineinhibits the HUVEC viability in dose dependent manner (Fig. 1B,P < 0.01 and P < 0.001).
Further, it was observed that brucine significantly inhibitsVEGF-mediated HUVEC survival when cells were starved withECGM containing 0.5% FBS for 24 h firstly, followed by the additionof DMSO (0.1%) or various concentrations of brucine (5, 10, 20,40 lM) then stimulated with VEGF (10 ng/mL), and incubated foranother 24 h (Fig. 1C, P < 0.001). As detected by BrdU incorporationassay (Fig. 1D), DNA synthesis of HUVECs is also significantly inhib-ited by brucine in a dose-dependent manner (P < 0.05; P < 0.001).To further examine whether brucine would result in toxic effectsof HUVEC, LDH cytotoxic assay was carried out. As shown inFig. 1E, brucine caused minute toxicity on HUVECs.
3.2. Brucine inhibits VEGF-induced endothelial cell migration andinvasion and capillary structure formation of HUVECs
Cell migration is a key step in angiogenesis [22]; therefore weinvestigated the effects of brucine on the chemotactic motility ofthe endothelial cells by using wound-healing (Fig. 2A) assay. Theresults show that brucine inhibits VEGF-induced HUVECs migra-tion in a concentration-dependent manner with significant inhibi-tion at the dose of 40 lM. We also performed transwell assays todetermine whether brucine affected cell invasion. As shown inFig. 2B, large number of cells migrated to the lower side of mem-brane in the transwell chamber after stimulation with VEGF. How-ever, the number of invasive cells are significantly less in thepresence of brucine (P < 0.001). During the complex process ofangiogenesis, the maturation of migrated endothelial cells into acapillary tube is a critical early step. Therefore, we investigatedwhether it could also inhibit tube formation. When HUVECs wereseeded on the growth factor-reduced matrigel, robust tubular-likestructures were formed in the presence of VEGF. Approximately10 lM brucine inhibits 50% VEGF-induced tube formation of HU-VECs on Matrigel, and 40 lM brucine significantly inhibits VEGF-induced tube formation of HUVECs on Matrigel (Fig. 2C;P < 0.001). These results indicate that brucine may block VEGF-in-duced angiogenesis in vitro by inhibiting the migration, invasionand tubular structure formation of endothelial cells.
3.3. Brucine insignificantly changes VEGF-induced VEGFR2 mRNAsexpression in HUVECs
VEGF is a fundamental mediator of physiological and patholog-ical angiogenesis [23], and acts through two tyrosine kinase recep-tors (VEGFR2 and VEGFR1). VEGFR2 is a major transducer of VEGFsignal in endothelial cells [24] therefore; we examined its effect onVEGF-induced VEGFR2 mRNAs expression in HUVECs. The mRNAswere isolated from different groups and reverse transcribed to asingle-stranded cDNA, and then relative gene expression wasdetermined using quantitative real-time PCR (qRT-PCR). As shownin Fig. 3A, there are not distinct differences in the expression ofVEGFR2 mRNAs between brucine-treated groups and the VEGF-treated control group. The result suggests that the anti-angiogenicactivity of brucine is not owing to down-regulation of VEGF-acti-vated VEGFR2 mRNAs expression in HUVECs.
3.4. Brucine is a potent VEGFR2 kinase inhibitor
Strong evidence shows that blocking the activity of VEGFR2(KDR/flk-1) can limit the ability of most tumors to stimulate theformation of blood vessels [24] and VEGFR inhibitors are a promis-ing class of angiogenesis treatment drugs. Thus, we tested the ef-fect of brucine on VEGF-induced tyrosine phosphorylation ofVEGFR-2, a critical receptor tyrosine kinase (RTK) on endothelial
cell surface. Serum-starved HUVECs were treated with brucine orDMSO (0.1%) for 30 min, followed by stimulation with VEGF for2 min. The phosphorylation state of VEGFR-2 was assessed by wes-tern blot with anti-phospho-VEGFR2 antibody. As shown in Fig. 3B,brucine inhibits VEGF-induced tyrosine phosphorylation of VEG-FR2 in two different phosphorylation sites (Tyr951 and Tyr1175)in a dose-dependent manner. Quantitative densitometry of proteinphosphorylation is shown as percentage (%) of vehicle control(Fig. 3C and D; P < 0.001). The protein levels were normalized tobeta-actin. The total amount of VEGFR2 protein in each sampleof cells remained comparable, suggesting that inhibition of phos-phorylation of VEGFR2 by brucine was not due to reduced VEGFR2expression (Fig. 3B).
To further determine whether the inhibition of tyrosine phos-phorylation of VEGFR2 by brucine is caused by inhibition of the ki-nase activity of VEGFR2, we carried out an ELISA-based in vitrotyrosine kinase assay. Brucine also shows a strong inhibition ofVEGFR2 kinase activity with an IC50 of 21.34 ± 2.14 lM (Fig. 3E;P < 0.001). SU5416, a known inhibitor of VEGFR2, was used as a po-sitive control and showed inhibition of kinase activity with an IC50
of 1.5 lM (data not shown), as described previously [25].
3.5. Brucine inhibits the activation of VEGFR2-mediated signalingpathways
Interaction of VEGFR2 with VEGF led to the activation of variousdownstream signaling molecules responsible for endothelial cellmigration, proliferation and survival [26–28]. To further delineatethe mechanism that underlies the anti-angiogenic effects of bru-cine, we screened some key kinases involved in VEGFR2-mediated

Fig. 5. Brucine down-regulates VEGF-induced (A) IL-6, (B) IL-8, (C) TNF-a, (D) IFN-c and (E) NO. Serum-starved HUVECs were preincubated with DMSO (0.1%) or brucine(40 lM), before stimulation with VEGF (10 ng/mL), and supernatants were harvested after 24 h for cytokine assays. IL-6, IL-8, TNF-a, IFN-c, VEGF were measured by sandwichELISA following the manufacturer’s instructions (R and D systems, USA). Absorbance was determined using a microplate reader (Biorad, USA) at 450 nm. The NO levels in theHUVECs were measured with Nitric oxide colorimetric assay kit (Biovision, USA) following the manufacturer’s instructions. Absorbance was determined using a microplatereader (Biorad, USA) at 540 nm. Cells receiving only DMSO (0.1%) served as a vehicle control. Data are expressed as mean ± SEM from three independent experiments.���P < 0.001 versus VEGF-treated control.
S. Saraswati, S.S. Agrawal / Cancer Letters 332 (2013) 83–93 89
signaling pathway. VEGF induces survival of endothelial cells (ECs)mainly via the activation of AKT [29], whereas activation of ERK1/2MAPKs is thought to be essential for VEGF-induced proliferation[30]. To assess the effect of brucine on these pathways, serum-starved HUVECs were treated with VEGF for 20 min in the presenceor absence of brucine and cell lysates were subjected to immuno-detection using antibodies against either pAKT (Ser473) or pERK1/2.The result shows that pERK1/2 is enhanced by VEGF treatmentwhile the expression level of ERK1/2 remains unchanged. Brucineinhibits the phosphorylation of ERK1/2 at the concentration of40 lM without affecting total ERK1/2 expression level (Fig. 4). A re-cent study suggests that the AKT/mTOR pathways and Hsp90,which are critical for angiogenesis, are phosphorylated or activatedby VEGFR-2 activation in the endothelial cells [31–34]. As shown inFig. 4, expression levels of p-AKT and p-mTOR dramatically in-creases by VEGF treatment, while p-PTEN, a negative regulator ofPI3K/AKT signaling pathway, is not affected by this treatment. Pre-treatment of the cells with brucine significantly inhibits the phos-
phorylation of AKT and mTOR, while the total amount of AKT andmTOR remains unchanged. Moreover, the action of brucine on thephosphorylation of FAK and Src is examined. The result shows thatbrucine inhibits VEGF-induced phosphorylation of FAK at the doseof 20 and 40 lM and Src at the concentration of 40 lM respectively(Fig. 4). Taken together, our result demonstrates that brucine ex-erts its anti-angiogenic effect by selectively targeting certain sig-naling events downstream of VEGFR-2.
3.6. Brucine down-regulates VEGF-induced IL-6, IL-8, TNF-a, IFN-c andNO
In inflammatory conditions VEGFR activation is coupled to cyto-kine release, pro-inflammatory molecules, and leukocyte endothe-lial interactions, which exacerbate the inflammatory response [35].Therefore, we investigated the effect of brucine on endothelial cellcytokine release. As shown in Fig. 5, HUVECs treated for 24 h withVEGF up-regulate the secretion of IL-8 (Fig. 5A), IL-6 (Fig. 5B),

Fig. 6. Brucine inhibits VEGF-induced angiogenesis ex vivo and in vivo. (A) Aortic rings were embedded in Matrigel, treated with various concentrations of brucine in thepresence or absence of VEGF and incubated in supplemented media at 37 �C, 5% CO2, for 8 days. The images shown here are representative of three experiments (�100). Scalebar: 100 lM (B) Bar graphs showing number of microvessel sprouts treated with various concentrations of brucine in the presence or absence of VEGF. Data are expressed asmean ± SEM from three independent experiments. ���P < 0.001 versus VEGF-treated control. (C) Mice were injected with 0.5 mL of Matrigel containing various concentrationof brucine, 250 ng of VEGF, and 150 units of heparin into the ventral area (five mice per group). After 14 d, the skin of mice was pulled back to expose the intact Matrigel plugs.The images shown here are representative of three experiments (�100). (D) Excised matrigel was weighed. Data are expressed as mean ± SEM from three independentexperiments. ���P < 0.001 versus VEGF-treated control. (E) Matrigel was homogenized (Tekmar TR-10, Ohio, USA) in 2.0 mL of Drabkin Reagent (Sigma–Aldrich, USA) andcentrifuged at 12,000 g for 20 min. The supernatants were filtered through a 0.22 lm filter (Millipore). Hemoglobin in the samples was quantified colorimetrically at 540 nmin a spectrophotometer (Shimadzu, Japan). The concentration of hemoglobin was calculated from a known amount of hemoglobin assayed in parallel. The content ofhemoglobin in each implant is expressed as g/dl/per wet tissue. Data are expressed as mean ± SEM from three independent experiments. ���P < 0.001 versus VEGF-treatedcontrol.
90 S. Saraswati, S.S. Agrawal / Cancer Letters 332 (2013) 83–93
TNF-a (Fig. 5C), and IFN-c (Fig. 5D). The increased levels of IL-6, IL-8, TNF-a, and IFN-c observed are found to decrease with brucinetreatment (Fig. 5). Further brucine also significantly inhibits NOlevels (Fig. 5F, P < 0.001).
3.7. Brucine inhibits VEGF-induced angiogenesis ex vivo and in vivo
The effect of brucine on microvessel sprout formation [19] wasdetermined by aortic ring sprout formation assay, a widely usedex vivo anti-angiogenesis assay that mimics several stages of angi-ogenesis, including endothelial cell proliferation, migration andtube formation. Rat aortic rings were embedded in Matrigel andfed with medium containing different concentrations of brucine.The rings were then stimulated with 100 ng/mL VEGF or 50 lg/mL ECGS and sprout formation was examined by microscopy. Asshown in Fig. 6A, VEGF significantly triggers microvessel sprouting,leading to the formation of a complex network of microvesselaround the aortic rings, whereas treatment with brucine at 20and 40 lM antagonizes the sprouting, suggesting that brucine sup-presses VEGF-induced angiogenesis ex vivo (Fig. 6B).
We then evaluated the anti-angiogenic effects of brucine in vivoin mouse matrigel plug model of angiogenesis. Matrigel plug wass.c. implanted with or without VEGF/brucine for 14 days in Swissalbino mice. As shown in Fig. 6C, remarkable microvessel forma-tion is seen in VEGF-supplemented matrigel plug. Brucine stronglysuppresses the VEGF-induced angiogenesis after 14 days of matri-
gel implantation (P < 0.001). We next quantified the level of angio-genesis by determining the weight and hemoglobin content of theplugs. Brucine decreases Hb content by 89% (Fig. 6D; P < 0.001) andplug weight by 48% (Fig. 6E; P < 0.001) as compared with VEGF-treated control. These results strongly suggest the antiangiogeniceffect of brucine in vivo.
3.8. Brucine inhibits tumor growth through inhibiting tumorangiogenesis
Angiogenesis is the key step in tumor growth and metastasis,which provides necessary oxygen and nutrients for the tumor[3]. To investigate the effect of brucine on tumor growth of Ehrlichascites carcinoma tumor model was used with a dose of 10 mg/kgof brucine i.p. daily starting from day 6. As shown in Fig. 7, brucinetreatment significantly inhibits tumor growth and decreases thetumor size. The average tumor volume in the control mice in-creases from 98.63 ± 34.28 mm3 to 1890 ± 289.352 mm3 after30 days, whereas the average tumor volume in the brucine-treatedmice decreases from 96.93 ± 26.70 mm3 to 41.34 ± 31.91 mm3
(Fig. 7A). The body weights of animals corresponded well withthe growth of tumors in respective group of animals (Fig. 7B).Quantitatively weights of tumor lumps treated with brucine arealso found smaller (P < 0.001) as compared to control group(Fig. 7C). The average tumor weight in the control group is10.63 ± 2.6 g; whereas the average tumor weight in the brucine-

Fig. 7. Brucine attenuates tumor growth and VEGFR-2 phosphorylation in EAC tumor model. (A) 15 � 106 EAC cells/mouse were injected s.c. into 5–6 weeks old swiss albinomice. After solid tumors grew to �100 mm3, the mice were i.p. treated with brucine (10 mg/kg bw). Tumor growth was measured with calipers every fifth days using theformula V = A � B2 � 0.52. Data are expressed as mean ± SEM from three independent experiments. P < 0.001 versus control group. (B) Decrease in bodyweight with brucinetreatment at 25 mg/kg bw. Data are expressed as mean ± SEM from three independent experiments. P < 0.001 versus control group. (C) Representative images of solid tumorlump shows brucine treated group is significantly smaller than those in the control group (D) The tumor tissue was removed from mice at 30 days and weighed. (E) Tumor isfixed with 4% paraformaldehyde at 4 �C and embedded in paraffin. Serial sections (5 lm) were processed for IHC (n = 5) with antibodies against p-VEGFR-2, p-ERK, p-AKT andCD31. Counterstaining was performed with hematoxylin. (F) % MVD was determined by selecting the blood vessel (CD31) area per field in selected vascularized areas dividedby the whole area. Data are expressed as mean ± SEM from three independent experiments. ���P < 0.001 versus control group.
S. Saraswati, S.S. Agrawal / Cancer Letters 332 (2013) 83–93 91
treated group was only 2.1 ± 0.98 g (Fig. 7D) indicating that prolif-eration rate of tumor cells in treated mice was greatly inhibited bybrucine.
To further investigate whether brucine inhibits tumor growthby suppressing tumor angiogenesis, immunostaining for CD31was performed (Fig. 7E). Our data shows that the average numberof blood vessels in brucine treated group is 1.5 ± 0.92 blood ves-sels/high power field (HPF) (Fig. 7F) compared with 12.3 ± 3.12blood vessels/HPF in the control group (P < 0.001). Moreover, bru-cine significantly decreases the expression level of phospho-VEG-FR-2, p-ERK and p-AKT, compared to control group (Fig. 7E). Inconclusion, these results suggest that brucine inhibit neovascular-ization and suppress tumor progression through inhibition of theVEGFR-2 signaling pathway.
4. Discussion
In this study, we report the novel biological functions of brucineas an inhibitor of angiogenesis. Brucine inhibits various aspects ofangiogenesis including endothelial cell proliferation, migration andcapillary structure formation in a concentration-dependent man-ner as well as the stimulative effects of endothelial cell death in re-sponse to VEGF in vitro. Brucine significantly inhibits tumorangiogenesis in an Ehrlich ascites carcinoma tumor model, reducesneovascularization of matrigel plug assay in vivo and abrogatesVEGF-induced microvessel sprouting in an ex vivo rat aortic ringassay.
Phosphorylation of VEGFR-2 is critical for VPF/VEGF-mediatedmicrovascular permeability, endothelial cell proliferation, andmigration [36–38]. In the present study, we found that a half-max-
imum inhibitory concentration 21.34 ± 2.14 lM (Fig. 3B) of brucinesignificantly blocks the kinase activity of VEGFR2, via down-regu-lation of VEGF-induced phosphorylation of VEGFR-2 expressionas observed by western blotting in vitro, making brucine a potentVEGFR2 inhibitor.
Phosphorylation of VEGFR2 at Tyr-1175 is required for the acti-vation of AKT and endothelial cell proliferation [39] and its phos-phorylation leads to the activation of downstream signalingevents in endothelial cells [40]. Phosphorylated Tyr1175 of VEGFR2mediates activation of the MAPK/ERK cascade and was shown tocontribute to cell proliferation in endothelial cells. Src family ki-nase is substantially involved in VEGF-induced angiogenesisin vitro and in vivo [41–44]. Other signaling molecules that havebeen involved in VEGF-induced migration through VEGFR2 includeFAK and its substrate paxillin, which are participated in focal adhe-sion during cell migration [45,46]. By interacting between focaladhesion kinase (FAK) and Src, a dual kinase complex FAK-Srcforms, and is activated by multiple integrin-regulated linkages.Activated FAK-Src promotes cell motility, cell cycle progressionand cell survival [43]. AKT/mammalian target of rapamycin(mTOR)/ribosomal protein S6 kinase (p70S6K) signaling has alsobeen identified as a novel, functional mediator in angiogenesis[47,48]. As expected, the level of p-PTEN did not change followingbrucine treatment, indicating that the anti-angiogenic effects ofbrucine are not related to p-PTEN. Taken together, our results indi-cate that brucine modulates VEGF-mediated vascular permeabilityand angiogenesis by inhibiting phosphorylation of FAK, ERK, Src,AKT and mTOR (Fig. 3C) in endothelia cells in vitro.
Tumor-induced angiogenesis is a well-known target in cancercontrol and prevention, therefore, inhibition of proangiogenic sig-nals in cancer cells is an important aspect of this strategy

92 S. Saraswati, S.S. Agrawal / Cancer Letters 332 (2013) 83–93
[49,50]. In this context, brucine strongly inhibits levels of variousproangiogenic factors like VEGF, IL-6, IL-8, TNF-a, and IFN-c in HU-VECs (Fig. 5) in vitro as evident by ELISA. NO can induce VEGFexpression and mediate angiogenic effect of VEGF as it is involvedin EC proliferation, migration, protease release and increased vas-cular permeability, thus important for initiation of angiogenesis.We therefore hypothesized that the reduced levels of VEGF wouldbe associated to low level of NO in vitro.
Furthermore, we evaluated the ex vivo and in vivo antiangio-genic efficacy of brucine using rat aortic ring and matrigel plug as-say respectively. We found that brucine remarkably suppressesVEGF induced neovascularization in both rat aortic assay ex vivoand matrigel plug assay in vivo. Hb content (Fig. 6D) and Plugweight (Fig. 6E) are significantly decreases in brucine treatedgroup. Brucine significantly attenuates tumor growth in mice inoc-ulated with EAC cells (P < 0.001). Brucine effectively suppresses tu-mor volume (Fig. 7A) and tumor weight (Fig. 7D) without anyadverse effects on mice. Previous studies demonstrated that bru-cine downregulates VEGF expression in solid tumor [14]. Theattenuation of tumor growth by brucine was due to the inhibitionof microvessel density and decreased expression of the p-VEGFR-2signaling molecule as identified by immunohistochemistry(Fig. 7E). These results clearly demonstrate that brucine can be uti-lized as anti-cancer drugs through the blocking of VEGF signalingpathways in endothelial cells leading to inhibition of neovesselgrowth.
In summary, we found that brucine inhibits angiogenesis viadownregulating phosphorylation of VEGFR2 tyrosine kinase andsuppressing VEGFR2-mediated signaling pathway which playsmultiple roles in regulating neovascularization. Therefore, our re-sults strongly suggest that brucine may be effective antagonistsof VEGF/VEGFR-2-stimulated angiogenesis and therefore mightbe a potential candidate for developing multifunctional anti-canceragent using its inhibitory activity on several aspects of tumorgrowth and angiogenesis.
References
[1] R. Kerbel, J. Folkman, Clinical translation of angiogenesis inhibitors, Nat. Rev.Cancer 2 (2002) 727–739.
[2] D. Hanahan, R.A. Weinberg, Hallmarks of cancer: the next generation, Cell 144(2011) 646–674.
[3] N. Ferrara, R.S. Kerbel, Angiogenesis as a therapeutic target, Nature 438 (2005)967–974.
[4] N. Ferrara, VEGF and the quest for tumor angiogenesis factors, Nat. Rev. Cancer2 (2002) 795–803.
[5] J. Schlessinger, New roles for Src kinases in control of cell survival andangiogenesis, Cell 100 (2000) 293–296.
[6] J.H. Qi, L. Claesson-Welsh, VEGF-induced activation of phosphoinositide 3-kinase is dependent on focal adhesion kinase, Exp. Cell Res. 263 (2001) 173–182.
[7] C.H. Cho, C.S. Lee, M. Chang, I.H. Jang, S.J. Kim, I. Hwang, et al., Localization ofVEGFR-2 and PLD2 in endothelial caveolae is involved in VEGF-inducedphosphorylation of MEK and ERK, Am. J. Physiol. Heart Circ. Physiol. 286 (2004)H1881–H1888.
[8] B.W. Kim, M. Choi, Y.S. Kim, H. Park, H.R. Lee, C.O. Yun, et al., Vascularendothelial growth factor (VEGF) signaling regulates hippocampal neurons byelevation of intracellular calcium and activation of calcium/calmodulin proteinkinase II and mammalian target of rapamycin, Cell Signal. 20 (2008) 714–725.
[9] J.H. Baek, J.E. Jang, C.M. Kang, H.Y. Chung, N.D. Kim, K.W. Kim, Hypoxia-induced VEGF enhances tumor survivability via suppression of serumdeprivation-induced apoptosis, Oncogene 19 (2000) 4621–4631.
[10] N. Ferrara, Vascular endothelial growth factor as a target for anticancertherapy, Oncologist 9 (2004) 2–10.
[11] J. Holash, S. Davis, N. Papadopoulos, S.D. Croll, L. Ho, M. Russell, et al., VEGF-Trap: a VEGF blocker with potent antitumor effects, Proc. Natl. Acad. Sci. USA99 (2002) 11393–11398.
[12] M.E. Noble, J.A. Endicott, L.N. Johnson, Protein kinase inhibitors: insights intodrug design from structure, Science 303 (2004) 1800–1805.
[13] T. Kamba, D.M. McDonald, Mechanisms of adverse effects of anti-VEGF therapyfor cancer, Br. J. Cancer 96 (2007) 1788–1795.
[14] S.S. Agrawal, S. Saraswati, R. Mathur, M. Pandey, Cytotoxic and antitumoreffects of brucine on Ehrlich ascites tumor and human cancer cell line, Life Sci.89 (2011) 147–158.
[15] B.C. Cai, T.S. Wang, M. Kurokawa, K. Shiraki, M. Hattori, Cytotoxicities ofalkaloids from processed and unprocessed seeds of Strychnos nux-vomica, ActaPharm. Sinic. 19 (1998) 425–428.
[16] P.S. Rao, M. Ramanadham, M.N. Prasad, Anti-proliferative and cytotoxic effectsof Strychnos nux-vomica root extract on human multiple myeloma cell line-RPMI 8226, Food Chem. Toxicol. 47 (2009) 283–288.
[17] S.S. Agrawal, S. Saraswati, R. Mathur, M. Pandey, Brucine, a plant derivedalkaloid inhibits inflammatory angiogenesis in a murine sponge model,Biomed. Prev. Nutr. 1 (2011) 180–185.
[18] J.Y. Tang, S. Li, Z.H. Li, Z.J. Zhang, G. Hu, L.C. Cheang, et al., Calycosin promotesangiogenesis involving estrogen receptor and mitogen-activated proteinkinase (MAPK) signaling pathway in zebrafish and HUVEC, PLoS One 5(2010) e11822.
[19] R.F. Nicosia, A. Ottinetti, Growth of microvessels in serum-free matrix cultureof rat aorta. A quantitative assay of angiogenesis in vitro, Lab. Invest. 63 (1990)115–122.
[20] A. Passaniti, R.M. Taylor, R. Pili, Y. Guo, P.V. Long, J.A. Haney, et al., A simple,quantitative method for assessing angiogenesis and antiangiogenic agentsusing reconstituted basement membrane, heparin, and fibroblast growthfactor, Lab. Invest. 67 (1992) 519–528.
[21] S.S. Agrawal, S. Saraswati, R. Mathur, M. Pandey, Antitumor properties ofBoswellic acid against Ehrlich ascites cells bearing mouse, Food Chem. Toxicol.49 (2011) 1924–1934.
[22] M. Shibuya, Vascular endothelial growth factor (VEGF)-receptor2: itsbiological functions, major signaling pathway, and specific ligand VEGF-E,Endothelium 13 (2006) 63–69.
[23] M. Klagsbrun, P.A. D’Amore, Vascular endothelial growth factor and itsreceptors, Cytokine Growth Factor Rev. 7 (1996) 259–270.
[24] I. Zachary, Signaling mechanisms mediating vascular protective actions ofvascular endothelial growth factor, Am. J. Physiol. Cell Physiol. 280 (2001)C1375–C1386.
[25] P. Carmeliet, L. Moons, A. Luttun, V. Vincenti, V. Compernolle, M. Mol, et al.,Synergism between vascular endothelial growth factor and placental growthfactor contributes to angiogenesis and plasma extravasation in pathologicalconditions, Nat. Med. 7 (2001) 575–583.
[26] N. Ferrara, H.P. Gerber, J. Le Couter, The biology of VEGF and its receptors, Nat.Med. 9 (2003) 669–676.
[27] X. Pang, Z. Yi, J. Zhang, B. Lu, B. Sung, W. Qu, et al., Celastrol suppressesangiogenesis-mediated tumor growth through inhibition of AKT/mammaliantarget of rapamycin pathway, Cancer Res. 70 (2010) 1951–1959.
[28] I. Edirisinghe, S.R. Yang, H. Yao, S. Rajendrasozhan, S. Caito, D. Adenuga, et al.,VEGFR2 inhibition augments cigarette smoke-induced oxidative stress andinflammatory responses leading to endothelial dysfunction, FASEB J. 22 (2008)2297–2310.
[29] A.K. Olsson, VEGF receptor signalling – in control of vascular function, Nat. Rev.Mol. Cell Biol. 7 (2006) 359–371.
[30] H.P. Gerber, A. McMurtrey, J. Kowalski, M. Yan, B.A. Keyt, V. Dixit, et al.,Vascular endothelial growth factor regulates endothelial cell survival throughthe phosphatidylinositol 30-kinase/Akt signal transduction pathway,requirement for Flk-1/KDR activation, J. Biol. Chem. 273 (1998) 30336–30343.
[31] A. Parenti, L. Morbidelli, X.L. Cui, J.G. Douglas, J.D. Hood, H.J. Granger, et al.,Nitric oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal-regulated kinase1/2 activation in postcapillaryendothelium, J. Biol. Chem. 273 (1998) 4220–4226.
[32] H.M. Verheul, B. Salumbides, K. Van Erp, H. Hammers, D.Z. Qian, T. Sanni, et al.,Combination strategy targeting the hypoxia inducible factor-1a withmammalian target of rapamycin and histone deacetylase inhibitors, Clin.Cancer Res. 144 (2008) 3589–3597.
[33] P. García-Maceira, J. Mateo, Silibinin inhibits hypoxia-inducible factor-1alphaand mTOR/p70S6K/4E-BP1 signaling pathway in human cervical and hepatomacancer cells: implications for anticancer therapy, Oncogene 28 (2009) 313–324.
[34] B.G. Wouters, M. Koritzinsky, Hypoxia signaling through mTOR and theunfolded protein response in cancer, Nat. Rev. Cancer 8 (2008) 851–864.
[35] B. Liu, L. Faia, M. Hu, R.B. Nussenblatt, Pro-angiogenic effect of IFNc isdependent on the PI3K/mTOR/translational pathway in human retinalpigmented epithelial cells, Mol. Vis. 16 (2010) 184–193.
[36] J.S. Pober, W.C. Sessa, Evolving functions of endothelial cells in inflammation,Nat. Rev. 7 (2007) 803–815.
[37] H.F. Dvorak, J.A. Nagy, D. Feng, L.F. Brown, A.M. Dvorak, Vascular permeabilityfactor/vascular endothelial growth factor and the significance of microvascularhyperpermeability in angiogenesis, Curr. Top. Microbiol. Immunol. 237 (1999)97–132.
[38] I. Zachary, Signaling transduction mechanisms mediating biological actions ofthe vascular endothelial growth factor family, Cardiovasc. Res. 49 (2001) 568–581.
[39] I. Shiojima, K. Walsh, Role of Akt signaling in vascular homeostasis andangiogenesis, Circ. Res. 90 (2002) 1243–1250.
[40] J. Liu, S. Agarwal, Mechanical signals activate vascular endothelial growthfactor receptor-2 to upregulate endothelial cell proliferation duringinflammation, J. Immunol. 2012 (1855) 1215–1221.
[41] B.P. Eliceiri, X.S. Puente, J.D. Hood, D.G. Stupack, D.D. Schlaepfer, X.Z. Huang,et al., Src-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5in vascular endothelial growth factor signaling, J. Cell Biol. 157 (2002) 149–160.
[42] M.T. Chou, J. Wang, D.J. Fujita, Src kinase becomes preferentially associatedwith the VEGFR, KDR/Flk-1, following VEGF stimulation of vascular endothelialcells, BMC Biochem. 3 (2002) 32.

S. Saraswati, S.S. Agrawal / Cancer Letters 332 (2013) 83–93 93
[43] N. Ali, M. Yoshizumi, Y. Fujita, Y. Izawa, Y. Kanematsu, K. Ishizawa, et al., Anovel Src kinase inhibitor, M475271, inhibits VEGF-induced human umbilicalvein endothelial cell proliferation and migration, J. Pharmacol. Sci. 98 (2005)130–141.
[44] S.K. Mitra, D.D. Schlaepfer, Integrin-regulated FAK-Src signaling in normal andcancer cells, Curr. Opin. Cell Biol. 18 (2006) 516–523.
[45] H.K. Avraham, T.H. Lee, Y. Koh, T.A. Kim, S. Jiang, M. Sussman, et al., Vascularendothelial growth factor regulates focal adhesion assembly in human brainmicrovascular endothelial cells through activation of the focal adhesion kinaseand related adhesion focal tyrosine kinase, J. Biol. Chem. 278 (2003) 36661–36668.
[46] K. Holmqvist, M.J. Cross, C. Rolny, R. Hägerkvist, N. Rahimi, T. Matsumoto,et al., The adaptor protein shb binds to tyrosine 1175 in vascular endothelial
growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellularmigration, J. Biol. Chem. 279 (2004) 22267–22275.
[47] M. Matsuo, S. Yamada, K. Koizumi, H. Sakurai, I. Saiki, Tumor-derived fibroblastgrowth factor-2 exerts lymphangiogenic effects through Akt/mTOR/p70S6kinase pathway in rat lymphatic endothelial cells, Eur. J. Cancer 43(2007) 1748–1754.
[48] W. Li, D. Tan, Z. Zhang, J.J. Liang, R.E. Brown, Activation of Akt-mTOR-p70S6Kpathway in angiogenesis in hepatocellular carcinoma, Oncol. Rep. 20 (2008)713–719.
[49] T.A. Bhat, R.P. Singh, Tumor angiogenesis – a potential target in cancerchemoprevention, Food Chem. Toxicol. 46 (2008) 1334–1345.
[50] Y. Cao, Q. Liu, Therapeutic targets of multiple angiogenic factors for thetreatment of cancer and metastasis, Adv. Cancer Res. 97 (2007) 203–224.





![Supplernentary Notes on the American Species of Strychnos ~Vainfo.cnptia.embrapa.br/digital/bitstream/item/68670/1/IAN-BT12.pdf · Krulcof] and Monachino - Arne1'ican Species ot Struchmos](https://img.pdfslide.us/doc/110x75/60f8d73e3ddace0a747aa712/supplernentary-notes-on-the-american-species-of-strychnos-krulcof-and-monachino.jpg)























